

SUMMARY
Adoptive transfer of genetically-modified immune cells holds great promise for cancer immunotherapy. CRISPR knock-in targeting can improve cell therapies, but more high-throughput methods are needed to test which knock-in gene constructs most potently enhance primary cell functions in vivo. We developed a widely adaptable technology to barcode and track targeted integrations of large non-viral DNA templates, and applied it to perform pooled knock-in screens in primary human T cells. Pooled knock-in of dozens of unique barcoded templates into the TCR-locus revealed gene constructs that enhanced fitness in vitro and in vivo. We further developed Pooled Knock-In Sequencing (PoKI-Seq), combining single-cell transcriptome analysis and pooled knock-in screening to measure cell abundance and cell state ex vivo and in vivo. This platform nominated a novel TGFβR2–41BB chimeric receptor that improved solid tumor clearance. Pooled knock-in screening enables parallelized re-writing of endogenous genetic sequences to accelerate discovery of knock-in programs for cell therapies.
Graphical Abstract
In Brief
Development of a platform to assess the functional effects of pools of knock-in constructs targeting one locus allows discovery of constructs promoting anti-tumor activity in T cells.
INTRODUCTION
Genetically engineered immune cell therapies have proven effective for treatment of some cancer types (Esensten et al., 2017; Fesnak et al., 2016; June and Sadelain, 2018; Sadelain et al., 2017), but they still cannot cure most tumors. Antigen-specific human T cells can be generated ex vivo by non-targeted viral transduction of defined T cell receptors (TCRs), chimeric antigen receptors (CARs), or other synthetic receptors (Esensten et al., 2017). More recent work has revealed potential advantages of targeted integrations of antigen receptor genes into defined genomic loci (Eyquem et al., 2017), which can be achieved with programmable nucleases such as CRISPR-Cas9 along with viral or non-viral knock-in templates (Gwiazda et al., 2016; Eyquem et al., 2017; Roth et al., 2018).
CAR T cells are now approved therapy for some hematological malignancies, but have not proved effective for solid tumors. While engineering the antigen specificity of T cells can target them to tumors, the cells must also maintain their functionality in the face of challenging immunosuppressive tumor microenvironments (Anderson et al., 2017; Binnewies et al., 2018). Suppressive cytokines such as TGFβ can be co-opted by tumors to inhibit T cell function (Dahmani and Delisle, 2018; Thomas and Massagué, 2005). Tregs, myeloid derived suppressor cells (MDSCs), and dendritic and stromal cell populations within the tumor can deprive T cells of necessary co-stimulatory signals or induce inhibitory signals (Chen and Mellman, 2013; Ostrand-Rosenberg et al., 2012). The excessive antigen density and kinetics of antigen exposure in solid tumors can drive exhaustion or dysfunction of T cells (Khan et al., 2019; Wherry, 2011). Next-generation cellular immunotherapies will likely need to be engineered with enhanced function to overcome these challenges presented by solid tumors (Rafiq et al., 2019).
Engineering immune cells that lack inhibitory checkpoint pathways is one strategy that is being widely pursued to enhance cellular therapies. Immune checkpoint inhibitors such as antibodies to PD-1 and CTLA4 have shown dramatic clinical success in re-activating the functionality of endogenous T cells in a subset of patients and tumor types (Weiner et al., 2010). CRISPR-Cas9 genetic ablation of checkpoint targets such as PD-1 in adoptive T cell therapies has also shown increased functionality in certain models and is being persued clinically (Ren et al., 2017; Rupp et al., 2017; Stadtmauer et al., 2020; Su et al., 2016).
CRISPR is not only being used to knockout well-studied checkpoint genes, but CRISPR screens can inform target selection. Genome-scale pooled CRISPR loss-of-function screens now allow forward genetic studies in many mammalian cell types (Wang et al., 2014; Shalem et al., 2014) including primary T cells where they have identified genes that may be targeted in future immunotherapies (Dong et al., 2019; Shifrut et al., 2018; Wei et al., 2019). Coupling pooled CRISPR gene knockouts with single cell RNA sequencing provides a powerful approach to assess the effect of each gene perturbation on cell state (Adamson et al., 2016; Datlinger et al., 2017; Dixit et al., 2016; Jaitin et al., 2016). Loss-of-function selections and transcriptome sequencing can reveal critical gene pathways to target.
While gene ablation can enhance cell therapies, the translational potential for knock-in of large gene cassettes to re-write cell programs is even greater. Next-generation T cell therapies are being engineered with transgenes designed to overcome suppressive signals in the tumor environment (Kloss et al., 2018; Lynn et al., 2019; Yamamoto et al., 2019). Decades of studies of T cell signaling and function, especially in murine models, suggest numerous candidate gene products that may enhance cell therapies. However, to date we have lacked rapid methods to test and directly compare the functions of large DNA knock-in integrations in human primary cells, which would accelerate the development of more effective cell therapies.
Here, we developed a robust new platform to assess the functional effects of pools of knock-in constructs targeting the same locus in parallel and determine which would be most effective at promoting anti-tumor activity. We used nonviral CRISPR targeting to introduce libraries of candidate immunotherapeutic knock-in constructs into a defined genome position in human T cells allowing us to compete the cells against each other in vitro and in vivo to test which constructs enhance T cell function. We designed a 36-member library of barcoded knock-in templates that included published and novel dominant negative receptors, synthetic “switch” receptors with engineered intracellular domains, and heterologous transcription factors, metabolic regulators or receptors. High-throughput pooled screening of targeted cells identified distinct members of the library that promoted T cell fitness under various resting, stimulated and immunosuppressive in vitro conditions. Direct competition among adoptively transferred human T cells targeted with the pool of constructs in immunodeficient mice bearing human melanoma cells revealed a subset of constructs that promoted in vivo accumulation of tumor infiltrating lymphocytes (TILs). Pooled knock-ins combined with single-cell sequencing of template barcodes and transcriptomes independently confirmed the constructs that promoted in vivo tumor accumulation, and also revealed high-dimensional cellular phenotypes induced by each construct ex vivo and in an in vivo tumor microenvironment. A TGFβR2–41BB chimeric receptor enhanced cell fitness in TGFβ-treated cells in vitro and tumor accumulation in vivo and also promoted gene expression of key effector cytokines. The knock-in cassette encoding a defined TCR and TGFβR2–41BB improved clearance of a solid tumor model in vivo, nominating a potential lead construct for further clinical development. Overall, these studies demonstrate the power of pooled knock-in technology to discover and functionally characterize complex synthetic gene constructs that can be written into targeted genome sites to generate more effective cellular therapies.
RESULTS
Pooled non-viral knock-in targeting of barcoded DNA templates
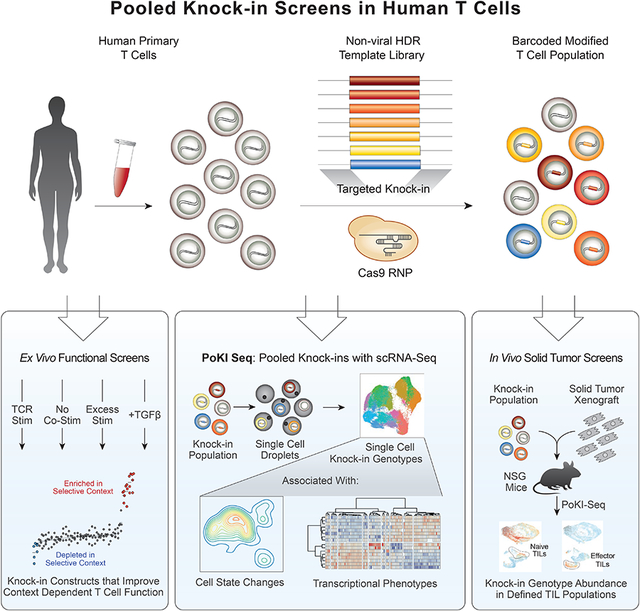
The ability to pool cells with a panel of different large sequence modifications at a specific endogenous genome locus and directly compare the functional consequences would accelerate engineering of cell therapies (Sanjana, 2017). We aimed to develop an adaptable platform to introduce multiple DNA-barcoded templates into a specified genomic target and assess effects on human T cell behavior ex vivo or in vivo with phenotypic screens or single-cell transcriptome analysis (Figure 1A). As a clinically relevant proof-of-concept, we hypothesized that pooled knock-in of an HDR template library, where each member contains a constant defined TCR-α and TCR-β pair (the cancer antigen targeted NY-ESO-1 TCR, clone 1G4, was used throughout the study) along with a unique third gene product, could rapidly identify new polycistronic DNA sequences that modify T cell function in therapeutically-relevant contexts (Figure 1B and Figure S1).
Figure 1. Development of targeted pooled knock-in screening.
(A) Adaptable platform to integrate libraries of multiple DNA-barcoded templates into a specified genomic target and dissect human T cell behavior ex vivo or in vivo with phenotypic screens or single-cell transcriptome analysis.
(B) Strategy for selective amplification and sequencing of successful on-target knock-in DNA construct barcodes. A short region of DNA mismatches is introduced into one homology arm of the HDR templates. The on-target HDR knock-ins that have both primer binding sites will be selectively amplified from genomic DNA (gDNA) or cDNA (Figures S2A).
(C) Percent of amplicon sequencing reads with GFP or RFP barcodes in the indicated sorted populations 7 days after pooled knock-in of a barcoded two-member library with each construct encoding the same TCR specificity (NY-ESO-1 1G4 clone) plus a distinct additional insert (GFP or RFP) and corresponding DNA barcode.
(D) Calculation of knock-in multiplicity in individual cells based on dual GFP / RFP 2-member library knock-in experiments (Figure S2C–E). The predicted percentage (right) of cells with biallelic integrations is estimated to be twice the observed percentage (left) of dual GFP+RFP+ cells as GFP+GFP+ and RFP+RFP+ biallelic cells would be observed in single positive gates.
(E) Quantification of template switching in pooled knock-in experiments. The frequency of template switching at each stage of the protocol (left panel) was estimated by sorting single positive populations of T cells edited with the 2-member barcoded library (NY-ESO-1 TCR+GFP and NY-ESO-1 TCR+RFP) and using the homology arm mismatch priming strategy to selectively amplify the barcodes of on-target integrations. The predicted frequency of template switching in an arbitrarily large N-member library (N>2) can be calculated from the observed template switching for a 2-member library (STAR Methods). The pooled electroporation condition (bold) was used for subsequent pooled knock-in screens.
n=2 (C–E) individual healthy human donors.
Functional assessment of knock-in DNA sequences in pools depends on quantitation of the “on-target” integration of each construct in the cell population before and after selective pressures. We developed a deep sequencing strategy that selectively captures on-target knock-in constructs while avoiding NHEJ-edited or un-edited target genomic sequences, non-integrated HDR templates, or off-target integrations (Figure 1B and Figure S2A). Briefly, the majority of a HDR template’s homology arm sequences are not incorporated with on-target HDR integrations (Paquet et al., 2016) and a short DNA sequence that does not match the target genomic locus can be introduced into one homology arm without a large reduction in knock-in efficiency (Figure 1B and Figure S2B). PCR primers that anneal to the inserted knock-in sequence and to the genomic flanking sequence can then selectively amplify successful on-target HDR events (Figure S2A). Addition of a barcode unique for each insert within degenerate bases of the TCR-α sequence enabled a rapid DNA sequence readout by PCR of the abundance of each individual on-target insert in the pooled population by either of two complementary methods: 1) sequencing directly from genomic DNA (gDNA), or 2) sequencing from reverse transcribed mRNA (cDNA) (Figure 1B).
We first demonstrated that expected DNA barcodes could be selectively sequenced from cells expressing the corresponding knock-in encoded protein products. We performed non-viral genome targeting (Roth et al., 2018, Nguyen and Roth et al., 2019) of a two-member barcoded library encoding GFP and RFP. Sorting cells with successful HDR knock in (based on expression of the NY-ESO-1 TCR) that co-expressed either GFP+ or RFP+ strongly enriched for the appropriate DNA barcode isolated sequenced from either gDNA or cDNA (Figure 1C and Figure S2C–E). Pooled screening approaches have been challenged by template switching that complicate interpretation of DNA barcode readouts (Hanna and Doench, 2018; Hegde et al., 2018; Sack et al., 2016). In addition, multiple barcoded templates introduced in the same cell could complicate interpretation of each one’s effects (Feldman et al., 2018; Roth et al., 2018). We therefore assessed the rates of biallelic integrations and template switching during pooled knock-in experiments using the GFP and RFP duplexed barcoded library. Only ~25% of cells showed evidence of multiple integrations (Figure 1D and Figure S2E). Template switching varied based on the stage of library production when constructs were pooled, and we chose to pool all subsequent knock-in construct libraries at the electroporation stage (Figure 1E). With this pooling strategy only ~10% of cells showed evidence of template switching (e.g. nine in ten knock-in positive cells will have the correct combination of knock-in construct and barcode) (Figure 1E and Figure S2C–J). ~ 90% of barcodes from sorted GFP+ cells were correct (Figure S2J). These results showed that engineered cell phenotypes could be quantitatively tracked in pooled knock-in experiments through DNA barcode sequencing.
Pooled knock-in of a multiplexed library of large DNA constructs
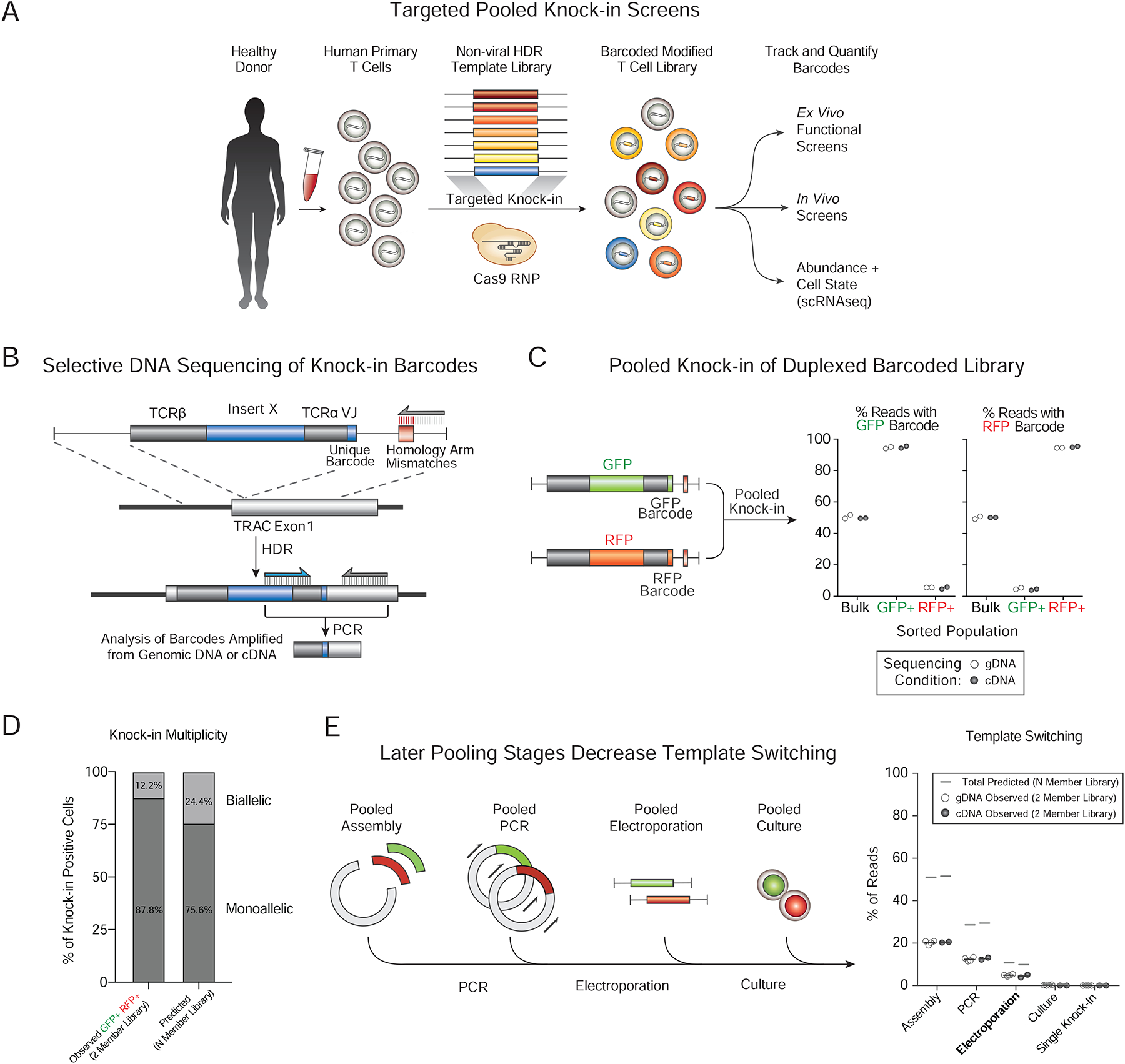
We next examined whether a larger library of 36 pooled templates could also be introduced and tracked by their DNA barcodes. Quantitative barcode sequencing demonstrated that even with the larger library all knock-in constructs were well-represented across multiple pooled knock-in experiments performed in four independent human donors (Figure 2A). The library contained large knock-in constructs ranging from 2 kb to 3 kb and even the largest constructs were well-represented across these biological replicates (Figure 2B). The 36-member library contained the GFP and RFP templates previously tested in the 2-member library, and when gating on knock-in positive cells (by dextramer staining for the introduced NY-ESO-1 TCR), GFP+ and RFP+ cells could be identified (Figure 2C). As expected, the percentage of reads with the GFP or RFP sequencing barcodes closely corresponded to the percentage of GFP or RFP positive cells observed at the protein level across four human donors (Figure 2D).
Figure 2. Pooled knock-in screening of a multiplexed library of large DNA inserts.
(A) Non-viral targeted pooled knock-in of a 36-member construct library into the TRAC locus in primary human T cells and subsequent sequencing of knock-in barcodes 7 days post-electroporation. All construct barcodes in the 36-member library were consistently well-represented with even library distribution (Gini coefficient = 0.048).
(B) A weak negative correlation between knock-in efficiency and insert size was observed (R2 = 0.11). Even the largest library members (~3 kb inserts) were well represented.
(C) Flow cytometry identified all knock-in positive cells that stained for the NY-ESO-1 TCR (introduced to the TRAC locus; off-target integrations should not yield NY-ESO-1 TCR+ cells). The percentage of knock-in positive cells that expressed GFP or RFP protein could be assessed and these cells sorted.
(D) The percentages of knock-in cells that expressed GFP (NY-ESO-1 TCR+GFP+) or RFP (NY-ESO-1 TCR+RFP+) corresponded closely with sequenced frequencies of GFP or RFP template barcodes. ns = not significant (Paired two-way T test).
(E) Validation of homology arm (HA) mismatch priming strategy with a 36-member large knock-in library. Knock-in positive cells were sorted based on NY-ESO-1 TCR expression as well as either GFP+, RFP+ or neither. When sequencing on-target knock-ins using primer matching the genomic sequence (and lacking the mismatches introduced into the homology arms, Figure S2A), the percent of sequenced reads with a GFP or RFP barcode in their respective populations closely matched the predicted percentage after correction for expected template switching and biallelic integrations (Figure S2 and STAR Methods However, as expected, sequencing with a primer binding the template homology arm (containing the mismatch sequences) did not strongly enrich the on-target knock-ins barcodes.
(F–G) Distribution of library members (barcode frequencies) was consistent throughout T cell expansion over 10 days of post-electroporation ex vivo culture in IL2. IL2RA-encoding construct showed an increased abundance over input. Dotted lines represent maximum and minimum relative abundances of control library members (GFP, RFP, tNGFR). *P < 0.05, ****P < 0.0001 (two-way analysis of variance with Holm–Sidak’s multiple comparisons test).
Unless otherwise indicated, all experiments were analyzed seven days after electroporation of primary T cells from n=4 (A–D, F–G) or n=2 (E) individual healthy human donors.
We next directly validated the homology arm mismatch sequencing strategy (Figure 1B) to selectively amplify on-target barcodes using the larger 36-member pooled knock-in library. For both gDNA and cDNA sequencing conditions, when GFP+ or RFP+ cells were sorted from the cells with successful on-target knock-in (NY-ESO-1 TCR+), the correct barcodes were selectively sequenced when using primers that bound the genomic sequence at the rate predicted when taking multiple integrations and template switching into account (Figure 2E and STAR Methods). However, as expected, primers complementary to the template homology arm mismatches – which should preferentially amplify non-HDR integrated templates – did not show the same enrichment for correct barcodes in the sorted populations (Figure 2E). Finally, we were able to readily track the relative abundances of knock-in construct barcodes over time in an expanding population of T cells cultured in interleukin 2 (IL-2). Barcode abundance was maintained throughout expansion for all constructs, with a notable exception of the knock-in construct encoding IL2RA, the high affinity receptor for the interleukin (IL-2), which enriched over time in culture consistent with its known roles promoting T cell fitness (Figure 2F–G). These results showed large pooled knock-in libraries can be generated and their barcodes can be quantitatively sequenced in a human T cell population.
Pooled knock-in screens identify gene constructs to improve context-dependent fitness of human T cells
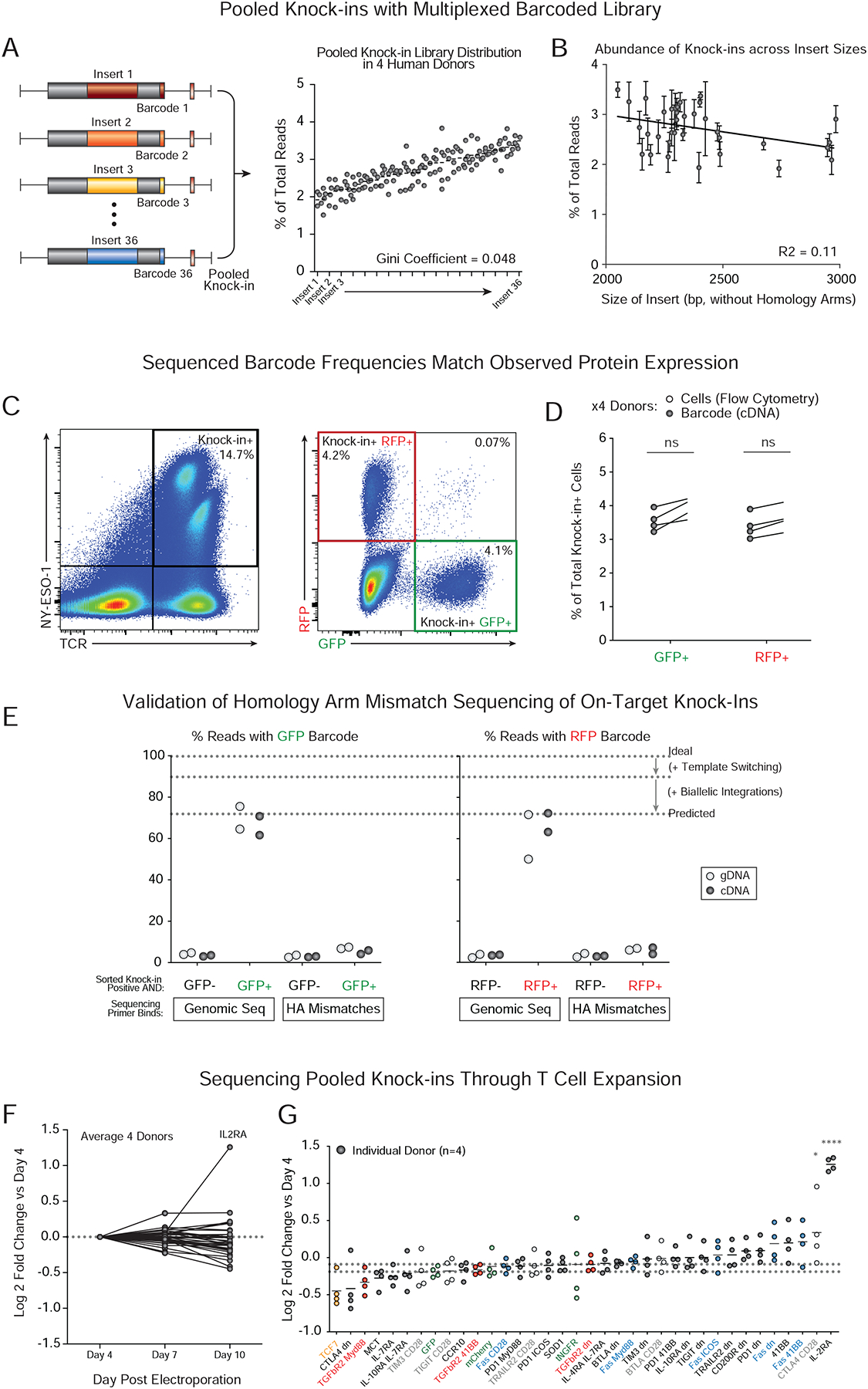
Next, the barcoded knock-in construct library was introduced into a population of cells with various selective pressures applied to identify constructs that could enhance T context-dependent cell fitness. We used this modified T cell library to discover synthetic constructs that can be written into the endogenous TRAC locus in primary human T cells to enhance functions relevant for cancer immunotherapies. The 36-member library of long DNA sequences that we designed included published and novel dominant negative receptors (Chen et al., 2017; Gorelik and Flavell, 2000; Kim et al., 2017; Kloss et al., 2018; Shin et al., 2016), synthetic “switch” receptors with engineered intracellular domains (Arber et al., 2017; Leen et al., 2014; Liu et al., 2016; Oda et al., 2017; Prosser et al., 2012; Shin et al., 2012; Tay et al., 2017; Yamamoto et al., 2019), and heterologous transcription factors, metabolic regulators and receptors (Figure 3A and Figure S3A). We chose to include synthetic knock-in constructs that could alter cellular responses to suppressive or apoptotic signals (e.g. with chimeric synthetic receptors designed with extracellular domains that recognize inhibitory signals linked to intracellular domains from stimulatory receptors), as well as constructs that would overexpress natural gene products associated with cell fitness or activation (such as IL2RA or 41BB). Each member of the library was cloned with self-cleaving peptide sequences between the TCRβ (full-length) and TCRα (V-region only) along with TRAC locus homology arms. Non-viral genome targeting with each member of this HDR template library achieved successful knock-in demonstrated by expression of the newly integrated NY-ESO-1 TCR (Figure S3B–E).
Figure 3. Targeted pooled knock-in screens in primary human T cells to assess context-dependent fitness benefits.
(A) A 36-member pooled knock-in library was designed with dominant negative receptors, synthetic “switch” receptors with engineered intracellular domains, and heterologous transcription factors, metabolic regulators and receptors (details in Figure S3).
(B) Log2 fold change in barcode frequency 5 days after in vitro TCR stimulation relative to the input population. Constructs derived from the apoptotic mediator FAS receptor protein increased in relative frequency across donors (**P < 0.01, ***P < 0.001, ****P < 0.0001, two-way analysis of variance with Holm–Sidak’s multiple comparisons test).
(C) Assessment pooled knock-in constructs in T cells under diverse in vitro selective pressures. Fitness effects are quantified as log2 fold change of barcode frequencies after 5 days of in vitro culture relative to input frequencies. IL2RA heterologous expression preferentially expanded in the absence of stimulation (corresponding to data shown in Figure 2F–G). With excess TCR stimulation, Fas derived receptors (Blue) preferentially expanded, and a polycistron encoding transcription factor TCF7 (Orange) also trended towards increased expansion (Figure S4). Checkpoint receptors engineered with intracellular-CD28 domains (Grey) drove expansion in cells treated with antigen (dextramer) stimulation only (no anti-CD28 co-stimulation). TGFβR2-derived chimeric proteins (Red) drove selective expansion in the presence of exogenous TGFβ. Dots represent mean of experiments from 4 independent healthy donors (see also Figure S4).
(D) Distribution of log2 fold changes over the input for cells stimulated in the presence of exogenous TGFβ. Under these conditions, multiple FAS-derived receptors and TGFβR2-derived receptors increased relative expansion.
**P < 0.01, ****P < 0.0001 (two-way analysis of variance (ANOVA) with Holm–Sidak’s multiple comparisons test).
n=4 (BD) individual healthy human donors
We first tested which DNA sequences inserted into the TRAC locus would enhance T cell expansion upon in vitro stimulation. Following anti-CD3/CD28 stimulation, we observed a striking enrichment of constructs encoding various synthetic versions of the Fas receptor (Figure 3B). Fas is a TNF super family surface receptor that mediates apoptosis through interactions with its ligand, FASL (Strasser et al., 2009). The pooled knock-in screens confirmed the functional effects of a recently described truncated Fas receptor (Yamamoto et al., 2019) included as a positive control, and also identified novel synthetic Fas switch receptors, including one with a 4–1BB intracellular domain, that also promoted T cell fitness in stimulated conditions (Figure 3B). In contrast, a truncated CTLA4 protein decreased T cell fitness in the same conditions across four donors, likely due to competition for CD28 co-stimulatory signaling (Figure 3B). These data established a flexible discovery platform to identify synthetic knock-in programs that promote selectable T cell phenotypes.
Effective adoptive T cell therapies for cancer must function even in immunosuppressive environments. To identify gene modifications that enhance T cell fitness upon various challenges, we repeated the pooled screen in diverse in vitro conditions designed to mimic aspects of tumor micro-environments (Figure 3C and Figure S4A–D). With excessive stimulation, which partially mirrors antigenic abundance in tumor environments, the transcription factor TCF7 (encoding the protein TCF1) slightly increased cell abundance, although this effect did not reach statistical significance (Figure S4B). TCR stimulation without anti-CD28 co-stimulation selected for various chimeric receptors with intracellular CD28 domains such as TIM3-CD28 and CTLA4-CD28 (Figure S4C). Finally, addition of the immunosuppressive cytokine TGFβ1 gave cells expressing engineered TGFβR receptors a selective fitness advantage, including both a previously described truncated TGFβR2 receptor (Gorelik and Flavell, 2000; Kim et al., 2017; Kloss et al., 2018) included as a positive control and a novel chimeric TGFβR2–41BB receptor (Figure 3D and Figure S4D–E). The results of these pooled targeted knock-in screens were reproducible across multiple biological replicates, technical replicates, sorting conditions and pooling conditions (Figure S4F–H). These data from pooled knock-in screens highlight condition-specific functional enhancements conferred by synthetic proteins or forced expression of key regulators.
Pooled knock-in hits individually validated and improved in vitro cancer cell killing
Pooled knock-in screens identified gene constructs that conferred competitive advantages to knock-in cells in the targeted population. We wanted to validate the pooled knock-in screening platform and confirm that the knock-in construct “hits” would similarly improve T cell fitness in individual knock-in experiments (Figure 4A). First, we tested to ensure that the DNA knock-in constructs resulted in expression of the expected protein products. Surface and intracellular staining of cells with eight individual knock-in constructs revealed robust expression for each expected protein, including increased expression of Fas and CD25 (IL2RA) above endogenous expression levels in stimulated T cells (Figure 4B and Figure S5A). We next directly validated for eight individual knock-in constructs the fitness benefits identified in the context dependent in vitro pooled screens (Figure 3C). Indeed, the anti-apoptotic FAS-41BB chimera and anti-suppressive TGFβR2–41BB chimera both promoted context-dependent increased expansion relative to controls (Figure 4C and Figure S5B). Increased cell expansion induced by the TGFβR2–41BB construct correlated with evidence of increased cell proliferation in the presence of TGFβ (measured by CFSE dilution relative to control cells) (Figure 4C and Figure S5B). These findings validated the pooled library-based approach to identify individual knock-in sequences to engineer T cell function.
Figure 4. Functional validation and improved in vitro cancer cell killing with novel gene constructs identified by pooled knock-in screens.
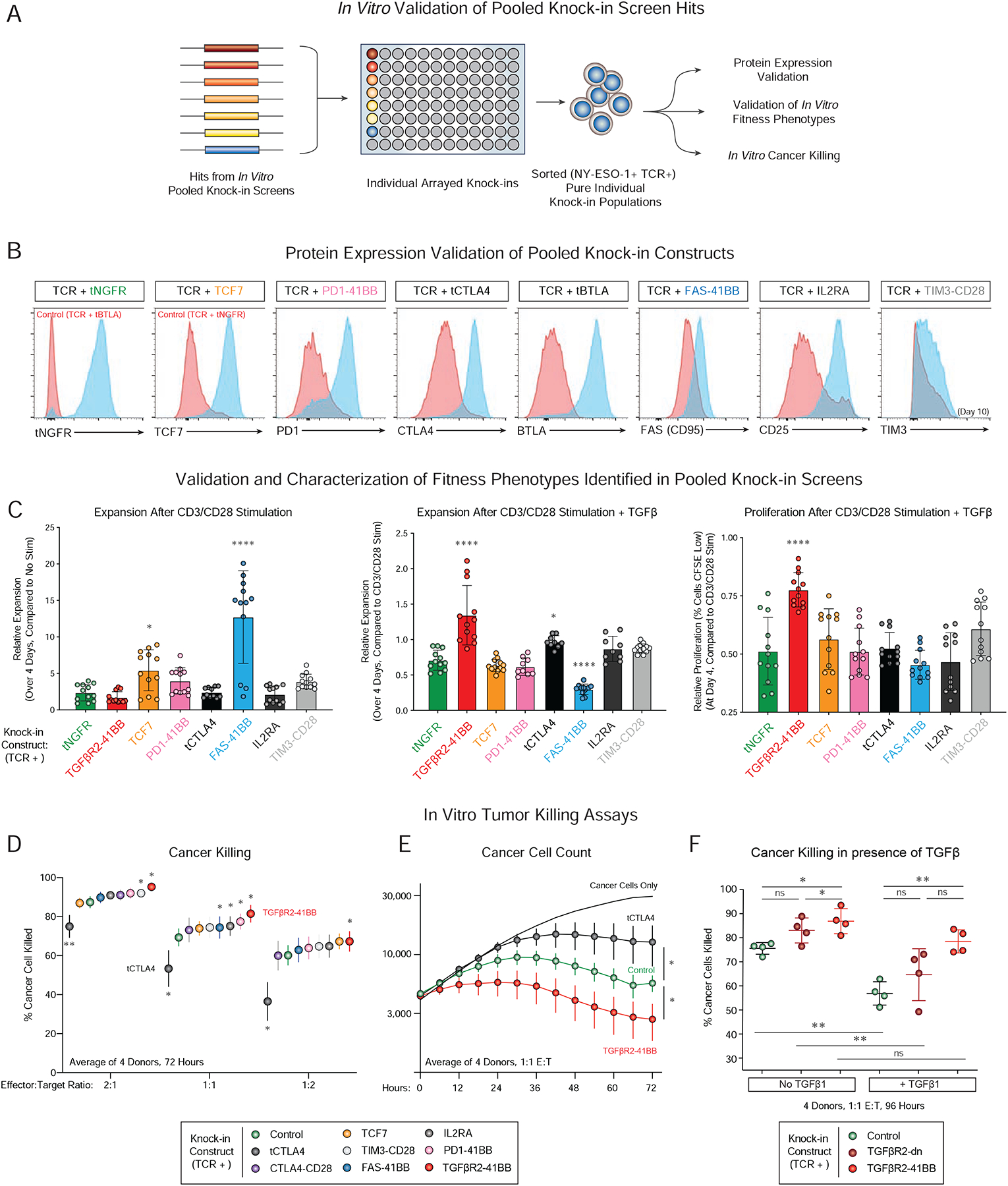
(A) Arrayed knock-in experiments validated the improved context-dependent fitness in pooled knock-in screens for selected library members (FAS-41BB, TGFBR2–41BB, IL2RA, TIM3-CD28, CTLA-CD28) compared to control constructs (tNGFR), neutral constructs that did not cause statistically-significant fitness improvements in the contexts tested (TCF7, PD1–41BB, tBTLA), and a negative hit from the screens (truncated CTLA4; tCTLA4).
(B) Flow cytometry confirmed overexpression of expected protein product encoded in knock-in constructs (Blue) relative to control cells (Red) 7 days post-electroporation of stimulated T cells (TIM3 was assessed on Day 10), gated on knock-in positive cells (NY-ESO-1+, see also Figure S5A).
(C) Relative expansion (compared to input cell number; Left, Middle) and proliferation (CFSE Staining, Right) for eight individual knock-in constructs five days after stimulation (Left). Stimulation + TGFβ (Middle, Right) mirrored the effects seen in pooled screens for the FAS-41BB and TGFβR2–41BB constructs.
(D) In vitro cancer cell killing 72 hours post co-culture of indicated sorted NY-ESO-1+ T knock-in cells with A375 human melanoma target cells. TGFβR2–41BB (Red) significantly improved target cell killing compared to control cells (tNGFR, Green) at multiple effector (T cell) to Target (A375 cancer cell) ratios. In contrast, tCTLA4 (Black), impaired killing (Figure S5C).
(E) Time course data for cancer cell killing data from D, averaged across 4 donors. Y-axis quantifies A375 cancer cell counts per well.
(F) The TGFβR2–41BB knock-in construct enhanced NY-ESO-1+ cancer cell killing in vitro both in the absence and presence of exogenous TGFβ1 compared to knock-in cells with a control tNGFR construct.
Experiments performed in n=4 (B–F) independent human donors. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 (paired two-tailed T test).
See also Figure S5.
We next tested to see if the identified knock-in constructs that promoted context-dependent T cell ex vivo expansion could also enhance in vitro cancer cell killing. Although this was not the phenotype initially tested in the pooled screens, the TGFβR2–41BB increased target in vitro cancer cell killing in T cells from four human blood donors across a range of effector to target cell ratios (Figure 4D–E). In contrast, a truncated CTLA4 construct showed reduced cell killing (Figure 4D–E), consistent with its impaired fitness in the pooled knock-in screens (although diminished proliferation assessed by CFSE was not observed with this individual construct, Figure S5B). Finally, we examined if the TGFβR2–41BB chimeric receptor also showed further context-dependent improvements in in vitro cancer killing. In the presence of exogenous TGFβ, the TGFβR2–41BB construct successfully rescued the impaired cancer cell killing across experiments performed from four healthy human donors (Figure 4F). Although the pooled screens focused on cell-intrinsic effects on T cell fitness, they also successfully identified novel gene constructs that can enhance in vitro anti-cancer cell efficacy.
PoKI-Seq: Pooled knock-ins with single-cell transcriptome analysis to assess abundance and cell state
Pooled screening approaches reveal modifications that affect cellular abundance in a population. However, cell abundance measures only one aspect of cellular function, and an ideal screening methodology would allow systematic assessment of modified cell states as well. Recently, novel barcoding strategies have overcome this and allowed pooled populations of CRISPR knock-out cells to be assessed by scRNA-seq (Adamson et al., 2016; Datlinger et al., 2017; Dixit et al., 2016; Jaitin et al., 2016), including primary human T cells (Shifrut et al. 2018). We next tested whether pooled knock-in screening could similarly be coupled with scRNA-seq to generate high-dimensional phenotypic information on modified cell states while also recording each cell’s specific knock-in construct barcodes, a method we term PoKI-Seq (Pooled Knock-In Sequencing) (Figure 5A). Briefly, the mRNA library converted to cDNA following single cell droplet isolation (10X) was split, with part used for scRNA-seq and the other for targeted amplicon sequencing of the knock-in barcode (Figure S6A).
Figure 5. PoKI-Seq combines pooled knock-in screening with single-cell RNA sequencing.
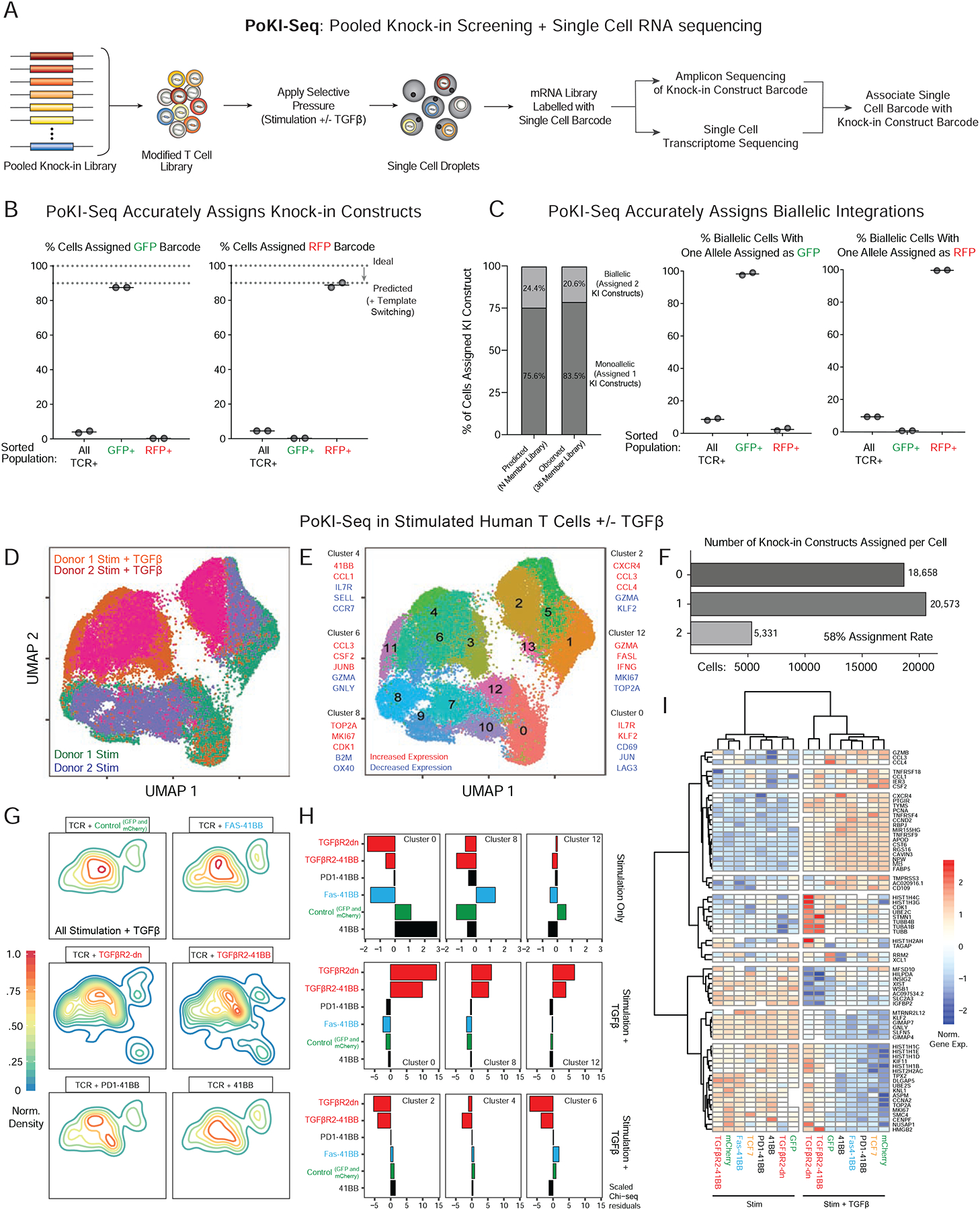
(A) Design of pooled knock-in experiments paired with single cell RNA-sequencing, termed PoKI-seq. This platform provides high-dimensional assessment of cell phenotypes caused by each knock-in construct’s genotype (Figure S6A).
(B) To validate the molecular assignment of knock-in construct barcodes to each individual cell, bulk knock-in positive cells expressing the integrated NY-ESO-1 TCR were sorted, as were NY-ESO-1+ cells that also expressed either GFP+ or RFP+. In the sorted GFP+ and RFP+ populations, the vast majority of template barcodes corresponded to the expression of the expected protein product (Figure 1E).
(C) PoKI-seq identified cells with multiple integrations. The frequency of observed cells with multiple knock-in barcodes likely to due to biallelic integrations closely matched those predicted based on 2-member GFP/RFP library knock-in experiments (Figure 1D). As expected, in sorted GFP+ or RFP+ cells with biallelic integrations, one of the barcodes corresponded to GFP or RFP respectively.
(D) UMAP representation of all single cell states identified in vitro with pooled knock-in T cell populations. 7 days following pooled knock-in of a 35-member library (all constructs in Figure S3A except tNGFR), sorted knock-in positive T cells (NY-ESO-1 TCR+) were stimulated at a 1:1 ratio with CD3/CD28 beads in the presence or absence of exogenous TGFβ and PoKI-seq was performed 4 days later.
(E) Nearest neighbor clustering (Louvain) overlaid on the UMAP representation revealed single cell populations corresponding to distinct cell states and associated hallmark genes.
(F) Assignment of knock-in constructs for each single cell in D. Over 58% of cells were assigned a knock-in construct. Approximately 3.4% of cells were assigned 3 or greater knock-in barcodes, potentially due to sequencing cell doublets, rare imperfect integration of multiple templates or template switching.
(G) Density plots (in the UMAP representation of single cell states) for cells with indicated knock-in constructs in TGFβ-treated conditions.
(H) Over-representation analysis for cells with select knock-in constructs in defined single cell clusters as measured by observed vs. expected frequencies (Chi-square residuals). In the context of stimulation only (top row), the FAS-41BB construct enriched in the proliferative cluster 8. With the addition of exogenous suppressive cytokine TGFβ, cells with TGFβR2-derived knock-in constructs showed strong enrichment in clusters corresponding to proliferative (cluster 8) and effector states (cluster 12), and depletion from the clusters associated with response to TGFβ (clusters 2, 4, 6).
(I) Gene expression heatmap for select knock-in constructs in PoKI-seq experiment. Gene list was generated from genes in the clusters examined in H with absolute log fold change of >0.8 compared to all other clusters. Transcriptional effects of TGFβR2-derived constructs strongly correlated with each other in the presence of exogenous TGFβ but not in the stimulation-only condition. TGFβR2-derived constructs altered the transcriptional response to TGFβ, maintaining expression of genes otherwise associated with the stimulation-only condition, such as proliferative markers MKI67 and TOP2A.
Experiments performed in n=2 (B–I) independent human donors.
See also Figure S6.
First, to validate that the fidelity of PoKI-Seq template barcodes is maintained throughout the experimental pipeline, we repeated GFP and RFP sorting experiments with the single cell platform. Sorting GFP+ or RFP+ cells from the pooled knock-in positive population (NY-ESO-1 TCR+) strongly enriched for the expected template barcodes, confirming the ability of PoKI-Seq to accurately assign specific knock-in constructs to cells (Figure 5B). One advantage of PoKI-Seq over bulk amplicon sequencing of pooled knock-ins is the ability to discriminate cells with a single knock-in construct from those that have received more than one (most likely from biallelic integrations). The overall proportion of cells assigned two knock-in constructs (Figure 5C) closely corresponded with predicted frequencies based on GFP/RFP 2-member library experiments (Figure 1D). As expected, when sorted GFP or RFP positive cells were assigned two knock-in constructs, at least one of the knock-in constructs assigned was almost always either GFP or RFP respectively (Figure 5C). Droplet doublets could also appear as multiple barcode calls, but the predicted doublet rate of ~2% (Zheng et al., 2017) would account for only a fraction of the cells with more than one barcode. Overall, these experiments confirmed the molecular biology of PoKI-seq and also demonstrated its ability to phenotype individual cells with either single or multiple knock-in constructs.
We next used PoKI-Seq to assign template barcodes to single cell transcriptomes in a large population of cells with the full pooled knock-in library. We performed PoKI-Seq on cells from two human blood donors following ex vivo stimulation in the presence or absence of exogenous TGFβ. Distinct clusters of cell states emerged, especially with addition of exogenous TGFβ (Figure 5D–E). Over 40,000 individual cells were sequenced, of which ~58% were successfully assigned one (monoallelic) or two (biallelic) barcodes (Figure 5F). Quality control metrics following PoKI-Seq confirmed robust rates of gene and UMI identification per cell and an average >130X coverage of cells assigned to each knock-in construct (per blood donor and per ex vivo condition tested) (Figure S6B–C). Single cell template barcode assignments were confirmed further as transcripts encoded by corresponding knock-in constructs tended to be expressed at increased levels, similar to what had been observed at the protein level with delivery of individual knock-in constructs (Figure 4B and Figure S6D). These metrics together established the fidelity of barcode assignment to single cell transcriptomes for the PoKI-Seq platform.
Next, we examined whether PoKI-Seq could measure cell state changes in ex vivo human T cells caused by specific knock-in constructs. Each knock-in construct caused distinct enrichment patterns in individual cell clusters in both TCR stimulation and stimulation plus TGFβ conditions that broadly corresponded to results from the in vitro pooled knock-in screens (Figure S6E). Density plots for individual knock-in constructs revealed significant cell state differences in T cells modified with TGFβR2-derived constructs compared to control and non-TGFBR2-derived constructs in the stimulation plus TGFβ culture condition (Figure 5G). Specifically, cells possessing TGFβR2-derived constructs were more likely to be found in clusters otherwise associated with the stimulation-only condition rather than the stimulation + TGFβ condition, including cluster 8, characterized by genes associated with cell proliferation, and cluster 12, characterized by genes associated with cell killing (Figures 5E, H and S6E–F). Similarly, the TGFβR2-derived knock-in constructs were relatively depleted from cells in the clusters otherwise promoted by TGFβ treatment (Clusters 2, 4 and 6) (Figure 5H). Clustering of knock-in constructs across all genes differentially expressed in the identified single cell clusters showed strong similarity between the TGFβR2-derived constructs in the presence of TGFβ and revealed downstream target genes that are modulated by the receptors. Hierarchical clustering revealed that exposure to TGFβ drove the greatest transcriptional changes (Figure 5I and Figure S6G). For example, in the presence of exogenous TGFβ, TGFβR2-derived chimeric receptors promoted continued expression of various hallmark proliferative genes such as MKI67 and TOP2A, while these genes were inhibited by TGFβ in cells expressing other KI constructs. PoKI-Seq confirmed the shared biological effects of multiple TGFβR2-derived receptors, which convert a suppressive signal into a signal that promotes cell proliferation and an effector cell state. More generally PoKI-Seq was able to reveal target gene pathways and context-dependent changes in cell state modulated by individual knock-in constructs.
In vivo PoKI-seq of engineered human T cells in a solid tumor microenvironment
Despite the flexibility of in vitro assays, certain aspects of cancer immunobiology can only be assessed in vivo. We therefore tested if PoKI-Seq could also be used to assess in vivo fitness of engineered human T cells in a solid tumor model (Figure 6A). We performed in vivo pooled knock-in screen using an antigen specific human melanoma xenograft model (Roth et al., 2018), followed by either bulk amplicon sequencing (Figure 1B) or PoKI-Seq with combined single cell amplicon and RNA-sequencing (Figure 4A). A pooled modified T cell population was transferred into immunodeficient NSG mice bearing human melanoma cells expressing the NY-ESO-1 antigen, and T cells were extracted from the tumor five days later (Figure S7A). Due to the lower number of human T cells isolatable from individual solid tumor xenografts (generally 10,000–20,000 cells per mouse), greater variance between replicates and donors was observed in the in vivo screens than in the in vitro screens (Figure S7B–C). Nonetheless, across four individual human donors, several knock-in constructs promoted statistically significant in vivo accumulation of engineered T cells in the tumor environment (Figure 6B), including a construct not identified in the in vitro screens. The TCF7 knock-in construct that only showed mild effects in vitro (not reaching statistical significance) promoted accumulation of T cells in the tumor microenviroment across four donors (Figure 6B). These results underscore the power of complementary in vitro and in vivo assays to assess synthetic gene functions.
Figure 6. In vivo PoKI-seq in primary human T cells captures cell abundance and cell state in solid tumor microenvironment.
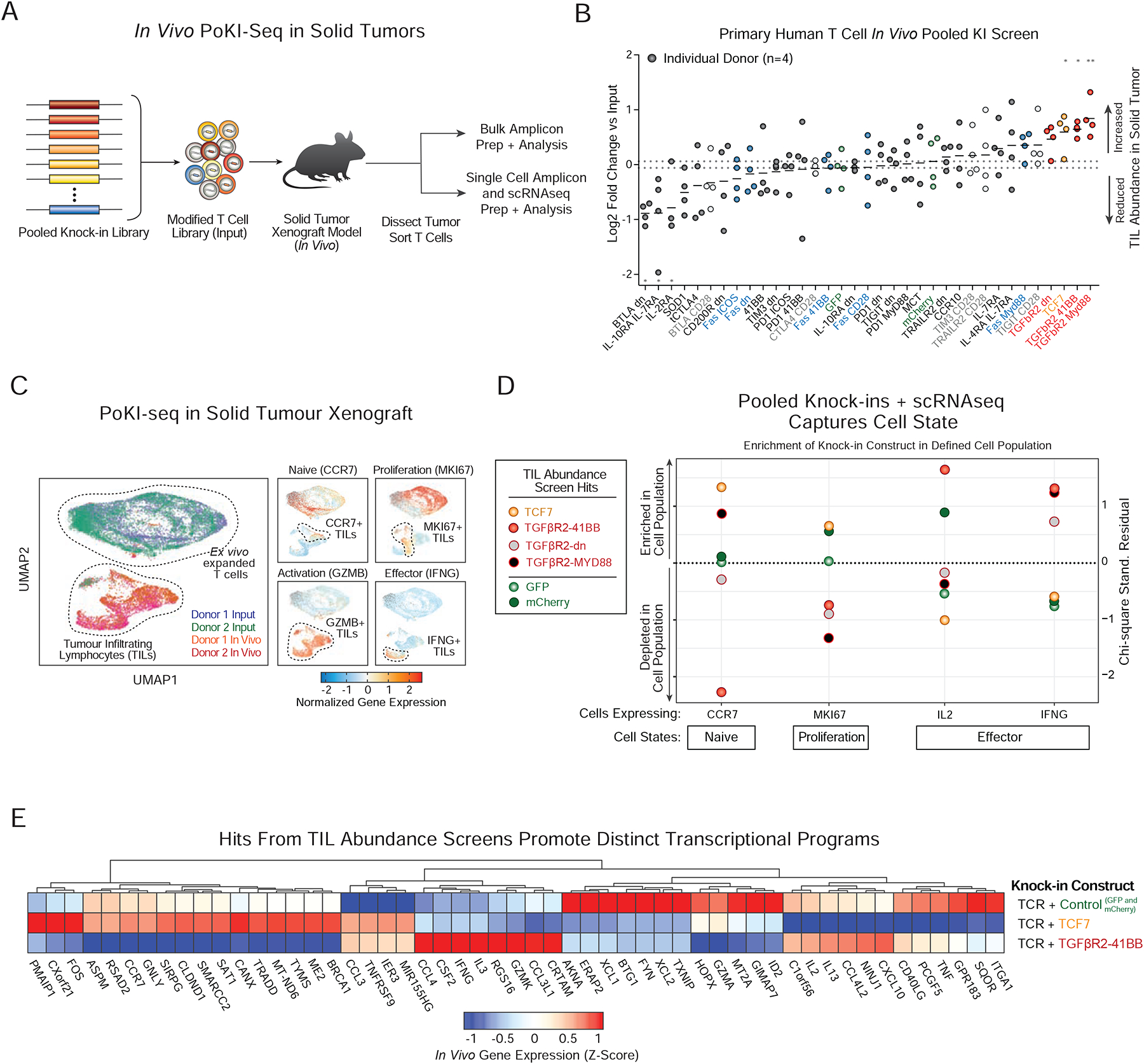
(A) Layout of in vivo pooled knock-in screens in a solid tumor xenograft model (A375 cells) of human melanoma. After either ex vivo expansion only (Input), or 5 days after adoptive transfer of 10 million T cells (~1 million knock-in positive cells) into tumor bearing NSG mice, live TCR+ T cells were sorted for analysis. Either bulk amplicon sequencing of knock-in barcodes was performed to capture cell abundance, or single cell droplets were generated and PoKI-Seq was performed to capture both cell abundance and cell state.
(B) Average log2 fold change in barcode abundances within the TILs compared to input cells is shown for each donor (horizontal lines represent max and min values for control constructs). The transcription factor TCF7 and synthetic TGFβR2-derived receptors enriched in abundance within tumor infiltrating T cells. *P < 0.05, **P<0.001 (two-way analysis of variance with Fischer’s LSD test).
n=4 donors (2 bulk amplicon sequencing, 2 single cell PoKI-seq) (n=2 donors).
(C) UMAP representation of all single cell states identified ex vivo and in vivo with pooled knock-in cell populations from two human blood donors. Normalized gene expression (Z-Score) on the UMAP representation revealed differences in expression within the in vivo population in markers of differentiation/proliferation status (CCR7 and MK167) and effector function (GZMB and IFNG).
(D) Subpopulations in vivo could be defined by scRNAseq using hallmark genes for cell states such as naïve-like T cells (CCR7), proliferation (MK167) and effectors (IL2 and IFNG). Subpopulations were defined as the top 10% of cells expressing each gene. Chi-squared residuals are displayed (n=2 independent donors).
(E) In vivo transcriptional signatures observed in tumor-infiltrating T cells with polycistronic constructs encoding NY-ESO-1 TCR plus control, TCF7, or TGFβR2–41BB. All cells assigned a specific knock-in construct were pseudo-bulked and differentially expressed genes with absolute value of log2 fold-change of >0.8 are displayed.
See also Figure S7.
Increased abundance may not always correspond to functional efficacy of TILs. scRNA-seq also revealed each knock-in construct’s effect on the transcriptome in both the input and tumor-infiltrating T cells. We next used PoKI-Seq to compare the transcriptomes of ex vivo expanded T cells with pooled knock-in templates and sorted pooled knock-in T cells from the in vivo tumor microenvironment (Figure 6C and Figure S7C–F). A UMAP representation of the single cell transcriptomes showed a high degree of overlap between cells from two human donors and marked differences between the ex vivo (input) cell population and tumor infiltrating (in vivo) T cells including differential expression of genes associated with proliferation or effector function (Figure 6C). We next examined whether the knock-in programs that increased TIL abundance enriched or depleted in subsets of the TIL population expressing hallmark genes for naïve, effector, and other cell populations (Figure 6D). While both the transcription factor TCF7 and TGFβR2-derived synthetic receptors increased T cell abundance in solid tumors, they induced different cell states. Strikingly, the TCF7 construct was relatively enriched in CCR7+ TILs, while TGFβR2–41BB was relatively enriched in both IL-2+ and IFNG+ TIL populations (Figure 6D). These data suggest that TCF7 promotes a more naïve or quiescent state, whereas the TGFβR2–41BB construct promotes accumulation of cells expressing key effector cytokine genes. Consistent with this model, in mouse chronic viral infection and syngeneic tumor models, TCF7+ cells have increased proliferative potential and less differentiated phenotypes during immunotherapies (Kurtulus et al., 2019; Siddiqui et al., 2019; Wu et al., 2016), but TCF7 expression was lost upon differentiation into effector cells (Siddiqui et al., 2019). To further investigate the differences in cell state between these two cell abundance hits, we compared the in vivo transcriptional programs induced by TCF7, the switch receptor TGFβR2–41BB, and controls (GFP and mCherry) using pseudo-bulked transcriptional data from all cells with each knock-in construct barcode (Figure 6E). While both TCF7 and TGFβR2–41BB promoted expression of genes such as TNFSFR9 (4–1BB) relative to control cells, the TGFβR2–41BB knock-in induced a distinct transcriptional program compared to TCF7 knock-in cells, with higher expression of effector cytokines such as IFN-ɣ that could enhance tumor clearance.
A novel TGFβR2–41BB chimeric receptor nominated by in vitro and in vivo pooled knock-in screens improves solid tumor clearance by primary human T cells
Finally, we examined in vivo the functional capacity in a solid tumor xenograft model (Figure 7A) of two hits from TIL abundance screens which promoted distinct cell states, TCF7, which enriched in CCR7+ TIL sub-populations, and TGFβR2–41BB, which enriched in IL-2+ and IFNG+ TIL sub-populations (Figure 6D). T cells engineered with a polycistronic knock-in construct expressing a NY-ESO-1 specific TCR (1G4 clone) along with either a control construct (tNGFR), the transcription factor TCF7, or the chimeric TGFβR2–41BB receptor all showed statistically significant reductions in tumor size relative to vehicle only injections of media (Figure 7B). Consistent with TCF7 promoting TIL abundance, but not promoting mature effector cytokine expressing cells, the TCF7 cell population did not enhance tumor control relative to tNGFR controls (Figure 7B). In contrast, the TGFβR2–41BB receptor construct that promoted abundance and effector cytokine gene expression in TILs markedly reduced tumor sizes (P < 0.0001 at day 33 vs TCR+tNGFR). Cells with the TGFβR2–41BB construct were the only ones that successfully cleared the solid tumor model at the doses tested here, an effect that was reproducible in many of the mice tested with modified T cells from 4 human blood donors (Figure 7B–C). Taken together, pooled knock-in screening combined with scRNA-seq can rapidly reveal new DNA sequences with potential to enhance therapeutic T cell functions.
Figure 7. A novel TGFβR2–41BB chimeric receptor identified by pooled knock-in screening improved clearance of an in vivo solid tumor model.
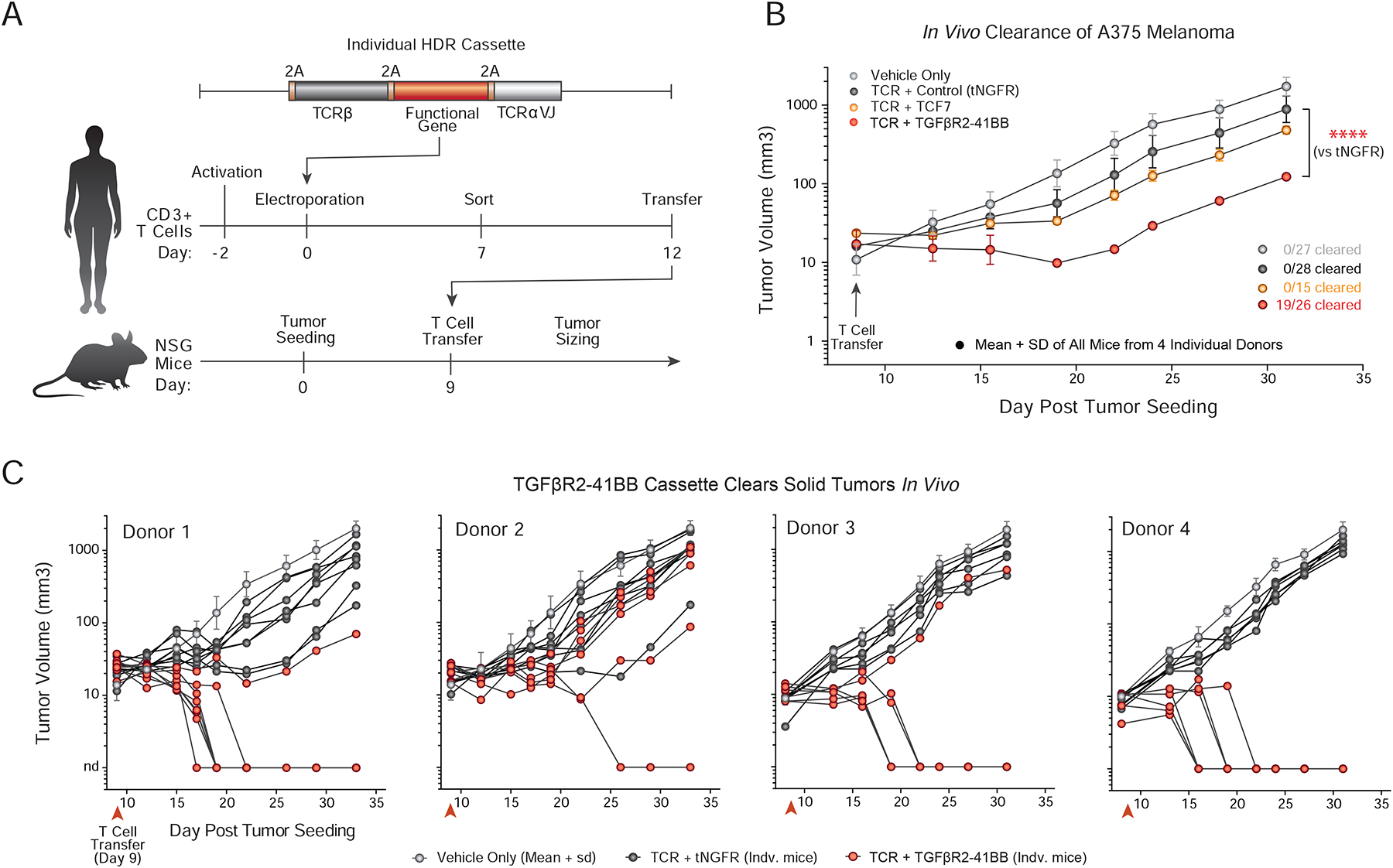
(A) Individual constructs nominated by pooled knock-in screens and scRNAseq analyses were adoptively transferred into NSG mice implanted with A375 melanoma tumors.
(B) Tumor sizing after adoptive transfer of vehicle alone (saline, Grey) or T cells targeted with a various polycistrons encoding NY-ESO-1 antigen specificity and potential therapeutic constructs. Engineered cells with any NY-ESO-1 TCR constructs showed statistically significant reductions in tumor size compared to vehicle (media only) alone. TGFβR2–41BB construct performed significantly better than the tNGFR control, and was the only construct that resulted in complete tumor clearance (19 out of 26 tumors). Mean + SD from n=4 (tNGFR, TGFβR2–41BB) or n=2 (TCF7) unique healthy human donors. ****P < 0.0001 (two-way analysis of variance with Holm–Sidak’s multiple comparisons test).
(C) Individual in vivo tumor growth curves of A375 melanoma xenograft mode are shown for mice treated with T cells with TGFβR2–41BB and NY-ESO-1 TCR knock-in and controls, as in (B). While variability was observed across the four donors tested, TGFβR2–41BB cells showed statistically significant reductions in tumor burden. In 19 out of the 26 mice tested with T cells from four different human blood donors, the TGFβR2–41BB knock-in cells cleared the tumor, which was not observed at these doses in any of the control mice.
DISCUSSION
Genetically engineered T cell therapies – including CAR T cells and TCR-modified T cells – are approved treatments for some hematological cancers and under active clinical development for a wide range of diseases. Mounting evidence suggests that for many clinical applications engineering antigen specificity will not be sufficient to cure disease. For example, T cell function is often suppressed by tumors and further engineering of antigen-specific T cells will be required to promote more effective cell states. Loss-of-function studies in T cells have revealed genes that can be ablated to enhance T cell responses (Dong et al., 2019; Shifrut et al., 2018; Wei et al., 2019). Many groups now are testing candidate transcription factors (Lynn et al., 2019) or synthetic cell receptors (Kloss et al., 2018; Yamamoto et al., 2019) that can be overexpressed to reprogram CAR T cells, prevent exhaustion, or convert suppressive extracellular signals to activating signals. The field needs a systematic approach to compare complex transgene constructs head-to-head and determine which are most effective for specific disease indications. The pooled knock-in platform presented here offers a new opportunity to screen systematically for large gene constructs that can be added to a T cell to modulate critical functions for effective cell therapies.
Targeted non-viral pooled knock-in screens have notable advantages over other systems that potentially could be used to discover effects of gene gain-of-function. Most over-expression studies have been performed with lentiviral or retroviral vectors. Although these viral vectors could be pooled, several important technical factors would limit their applications. These vectors have variable integration copy numbers and integrate at semi-random genome sites, which can cause variability in transgene expression and altered cell phenotypes due to insertional mutagenesis (Cavazzana-Calvo et al., 2010; Romero et al., 2013). These sources of variability could complicate interpretation of pooled screens, introducing both false positives and false negatives. In addition, lentiviral and retroviral library preparation is laborious and can lead to significant barcode swapping due to viral recombination (Sack et al., 2016). In contrast to lentiviral and retroviral transgenesis, pooled non-viral HDR templates allow for targeted integration at a defined site with relatively consistent transgene expression levels and low rates of barcode mis-assignment without requiring viral packaging steps.
Pooled, non-viral knock-in screens add to a growing constellation of CRISPR technologies to enable scalable gain-of-function screening, especially CRISPR-activation (CRISPRa) approaches to increase expression of endogenous genes (Chavez et al., 2015; Gilbert et al., 2013, 2014). Technical challenges have limited CRISPRa applications in therapeutically relevant primary human cell types. Furthermore, CRISPRa can only induce natural gene products encoded in the genome and does so with variable efficiency. Gain-of-function screens can be accomplished in cell lines at targeted genomic sites with CRISPR-mediated saturating mutagenesis (Hess et al., 2016; Ma et al., 2016). CRISPR-based pooled knock-in of short oligos and the functional assessment of single nucleotide variants is possible at an endogenous locus in a cell line (Findlay et al., 2014; Findlay et al., 2018). In contrast to these technologies that have shown promise for screening small mutations in immortalized cells lines, we now present a new CRISPR platform to test large knock-in candidate therapeutic constructs in primary human cells, which should accelerate the development of improved cellular therapies.
Our integrated pooled knock-in platform nominated a lead hit (TGFβR2–41BB) that increased T cell fitness in vivo and promoted key effector cytokines including IFNɣ. TGFβ is a key suppressive cytokine normally produced by immune cells but which can be co-opted by tumors to suppress effective immune responses (Batlle and Massagué, 2019; Gigante et al., 2012). Knock-in of large genetic constructs enables the engineering of synthetic receptors to re-wire the signal T cells receive when exposed to TGFβ in the tumor microenvironment, including switching the suppressive TGFβ signal to a stimulatory 41BB signal. This lead construct indeed showed markedly enhanced clearance of an in vivo solid tumor model. The preclinical success of a novel construct nominated by pooled knock-in screens highlights the power of the platform. The pooled knock-in platform will also enable follow-up screens to optimize the lead construct’s efficacy and safety.
Barcoded knock-in libraries have even broader potential to study functional effects of sequence variation at any selected site in the genome. A major challenge to quantitative tracking of targeted genomic integrations of pooled knock-in constructs has been the abundance of non-integrated DNA templates (viral or non-viral) in targeted cells and the risk of template switching during molecular steps of pooled library production, integration, and sequencing. Here we overcame these technical limitations with a widely adaptable technology to barcode and track on-target integrations of HDR templates in a pooled library. The HDR knock-in modifications all occur at the same genome locus and are tested in cells by head-to-head competition against each other. Each knock-in construct can be tracked in the targeted cell population under diverse in vitro and in vivo conditions to reveal the most effective modifications. Effective barcoding of targeted knock-in sequences also allowed single-cell transcriptome readouts in a pool of modified cells to assess the effects of each knock-in construct on cell state, a method we term Pooled Knock-In Sequencing (PoKI-seq). This allowed us not only to screen for constructs that increased cell abundance with different in vitro and in vivo selective pressures, but also to perform high dimensional analysis of the cellular phenotypes resulting from each large knock-in DNA cassette. Perturb-seq technologies have been transformative for systematic assessment of gene ablations. PoKI-seq offers a complementary single-cell approach to study large knock-in perturbations in pooled experiments. Barcodes could be sequenced from either cDNA or genomic DNA, which suggests future pooled phenotypic screens will be possible to assess function of coding and non-coding regions of the genome. Furthermore, multiplexed knock-in targeting is achievable (Roth et al., 2018), so future studies could examine the combinatorial effects of DNA inserts at multiple loci in the same cell. We will be able to scale knock-in construct size and the number of members in each library as we continue to improve knock-in efficiency and reduce costs of DNA synthesis.
Overall, the ability to rapidly integrate diverse libraries of new genetic sequences at defined sites in the genome will open many new avenues for understanding the functional rules of the human genome code and for reprogramming enhanced cell therapies. Barcoded pools of non-viral HDR templates can now be used to explore the in vitro and in vivo functions of diverse sequence variants at targeted coding and non-coding elements in the genomes of primary human cells. Future studies will be able to extend similar approaches to additional genomic loci, additional panels of candidate knock-in constructs, additional selectable in vitro and in vivo phenotypes and additional cell types. Engineered cell therapies are an emerging “drug” class, alongside small molecules and biologics (Fischbach et al., 2013). Non-viral pooled knock-in screening will accelerate the discovery and development of synthetic DNA sequences to reprogram the specificity and function of adoptive cellular therapies.
STAR Methods
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed and will be fulfilled by the lead contact, Alexander Marson (alexander.marson@ucsf.edu). Plasmids generated in this study have been deposited to Addgene (Table S1) or are available upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Isolation and culture of human primary T cells
Primary human T cells were isolated from either fresh whole blood or residuals from leukoreduction chambers after Trima Apheresis (Blood Centers of the Pacific, San Francisco, CA) from healthy donors. Peripheral blood mononuclear cells (PBMCs) were isolated from whole blood samples by Lymphoprep centrifugation (STEMCELL) using SepMate tubes (STEMCELL). T cells were isolated from PBMCs from all cell sources by magnetic negative selection using an EasySep Human T Cell Isolation Kit (STEMCELL). Isolated T cells were either used immediately following isolation for electroporation experiments or frozen down in Bambanker freezing medium (Bulldog Bio) per manufacturer’s instructions for later use. Freshly isolated T cells were stimulated as described below. Previously frozen T cells were thawed, cultured in media without stimulation for 1 day, and then stimulated and handled as described for freshly isolated samples. Fresh blood was taken from healthy human donors under a protocol approved by the UCSF Committee on Human Research (CHR #13–11950).
XVivo15 medium (Lonza) supplemented with 5% fetal bovine serum, 50 μM 2-mercaptoethanol, and 10 mM N-acetyl L-cystine was used to culture primary human T cells. In preparation for electroporation, T cells were stimulated for 2 days at a starting density of approximately 1 million cells per mL of media with anti-human CD3/CD28 Dynabeads (ThermoFisher), at a bead to cell ratio of 1:1, and cultured in XVivo15 media containing IL-2 (500 U ml−1; UCSF Pharmacy), IL-7 (5 ng ml−1; ThermoFisher), and IL-15 (5 ng ml−1; Life Tech). Following electroporation, T cells were cultured in XVivo15 media containing IL-2 (500 U ml−1) and maintained at approximately 1 million cells per mL of media. Every 2–3 days, electroporated T cells were topped up, with or without splitting, with additional media along with additional fresh IL-2 (final concentration of 500 U ml−1). When necessary, T cells were transferred to larger culture vessels.
In Vivo Mouse Solid Tumor Xenograft
An NY-ESO-1 melanoma tumor xenograft model was used as previously described (Roth et al., 2018). All mouse experiments were completed under a UCSF Institutional Animal Care and Use Committee protocol. We used 8 to 12 week old NOD/SCID/IL-2Rɣ-null (NSG) male mice (Jackson Laboratory) for all experiments. For pooled knock-in experiments (Figure 4), mice were seeded with tumors by subcutaneous injection into a shaved right flank of 1×106 A375 human melanoma cells (ATCC CRL-1619). The length and width of the tumor was measured using electronic calipers and volume was calculated as v = 1/6 * π * length * width * (length + width) / 2. Indicated numbers of T cells with the pooled knock-in library were resuspended in 100 μl of serum-free RPMI and injected retro-orbitally. A bulk edited T cell population (10×106) containing at least 10% NY-ESO-1 knock-in positive cells was transferred. Five days after T cell transfer, single-cell suspensions from tumors and spleens were produced by mechanical dissociation of the tissue through a 70 μm filter, and T cells (CD45+ TCR+) were sorted from bulk tumorcytes by FACS. For individual in vivo validation experiments (Figure 6B–C), mice were seeded with 0.5×106 A375 cells. At nine days post tumor seeding, 1.5×106 sorted NY-ESO-1+ T cells from the indicated edited populations were adoptively transferred. Investigators were blinded to experimental treatment group during sizing measurements. All animal experiments were performed in compliance with relevant ethical regulations per an approved IACUC protocol (UCSF), including a tumor size limit of 2.0 cm in any dimension.
METHOD DETAILS
RNP production
RNPs were produced by complexing a two-component gRNA to Cas9. The two-component gRNA consisted of a crRNA and a tracrRNA, both chemically synthesized (Dharmacon or IDT) and lyophilized. Upon arrival, lyophilized RNA was resuspended in 10 mM Tris-HCL (7.4 pH) with 150 mM KCl at a concentration of 160 μM and stored in aliquots at −80 °C. Poly(L-glutamic acid) (PGA) MW 15–50 kDa (Sigma) was resuspended to 100mg/mL in water, sterile filtered, and stored in aliquots at −80C. Cas9-NLS (QB3 Macrolab) was recombinantly produced, purified, and stored at 40 μM in 20 mM HEPES-KOH, pH 7.5, 150 mM KCl, 10% glycerol, 1 mM DTT.
To produce RNPs, the crRNA and tracrRNA aliquots were thawed, mixed 1:1 by volume, and annealed by incubation at 37 °C for 30 min to form an 80 μM gRNA solution. Next, PGA mixed with freshly-prepared gRNA at 0.8:1 volume ratio prior to complexing with Cas9 protein for final volume ratio gRNA:PGA:Cas9 of 1:0.8:1 (2:1 gRNA to Cas9 molar ratio) and incubated at 37 °C for 15 min to form a 14.3 μM RNP solution (Nguyen et al., 2019; Roth et al., 2018). RNPs were electroporated immediately after complexing.
Double-stranded HDR DNA Template Production
Each double-stranded homology directed repair DNA template (HDRT) contained a novel/synthetic DNA insert flanked by homology arms. We used Gibson Assemblies to construct plasmids containing the HDRT sequences and then used these plasmids as templates for high-output PCR amplification (Kapa Hot Start polymerase). The resulting PCR amplicons/HDRTs were SPRI purified (1.0×) and eluted into H2O. The concentrations of eluted HDRTs were determined, using a 1:20 dilution, by NanoDrop and then normalized to 1 μg/μL. The size of the amplified HDRT was confirmed by gel electrophoresis in a 1.0% agarose gel. All HDR DNA template sequences used in the study are listed in Table S1.
Primary T cell electroporation
For all electroporation experiments, primary T cells were prepared and cultured as described above. After stimulation for 48–56 hours, T cells were collected from their culture vessels and the anti-human CD3/anti-CD28 Dynabeads were magnetically separated from the T cells. Immediately before electroporation, de-beaded cells were centrifuged for 10 min at 90g, aspirated, and resuspended in the Lonza electroporation buffer P3. Each experimental condition received a range of 750,000 – 1 million activated T cells resuspended in 20 uL of P3 buffer, and all electroporation experiments were carried out in 96 well format.
All electroporations were done using a Lonza 4D 96-well electroporation system with pulse code EH115. Unless otherwise indicated, 3.5 μl RNPs (comprising 50 pmol of total RNP) were electroporated, along with 1–3 μl HDR Template at 1 μg μl−1 (1–3 μg HDR template total). Immediately after all electroporations, 80 μl of pre-warmed media (without cytokines) was added to each well, and cells were allowed to rest for 15 min at 37 °C in a cell culture incubator while remaining in the electroporation cuvettes. After 15 min, cells were moved to final culture vessels and media supplemented with 500 U ml−1 IL-2 was added.
Flow cytometry and cell sorting
All flow cytometric analyses were performed on an Attune NxT Acoustic Focusing Cytometer (ThermoFisher). FACS was performed on the FACSAria platform (BD). Cell surface staining for flow cytometry and cell sorting was performed by pelleting and resuspending in 25 uL of FACS buffer (2% FBS in PBS) with antibodies diluted accordingly (STAR Methods) for 20 min at RT in the dark. Cells were washed once in FACS buffer before resuspension and analysis.
In Vitro Proliferation Assay
Non-virally edited T-cells were expanded in independent cultures prior to the assay. The unsorted, edited populations were pooled after approximately two weeks of expansion (with 500 U/mL of IL-2 supplemented every 2–3 days) for a competitive mixed proliferation assay.
For the NY-ESO-1 competitive mixed proliferation assay, we pooled unsorted samples with either 1G4+dnTGFβR2+ or 1G4+tNGFR+ T cells. To determine the input number of each population, we took into account the number of viable 1G4+TCR+ in either population (knock-in% * total viable cell count), as determined by flow cytometry analysis. The pooled sample was then distributed into round bottom 96 well plates at a total starting cell count of 50,000. The distributed samples were then cultured without stimulation or with Immunocult (CD3/CD28/CD2). All samples were cultured in XVivo15 media supplemented with IL-2 (500 U/mL) with or without the addition of TGFβ1. After 5 days in culture, samples were analyzed on flow cytometry and the relative outgrowth of 1G4+dnTGFβR2+ and 1G4+tNGFR+ subpopulations was quantified.
In Vitro Cytokine Release Assay
T cells were stimulated with CD3/CD28 Dynabeads (1:1 ratio) or Dynabeads plus 25 ng/ml TGFβ (Miltenyi Biotec, Bergisch Gladbach, Germany). 24 hours later eBioscience Brefeldin A Solution (1000X) (ThermoFisher Scientific) was added and incubated for 4 hours. Cells were stained with extracellular antibodies 7-AAD, CD8-APC/Cy7, CD4-FITC (BioLegend) and intracellular antibodies IFN-γ-PE, IL-2-APC (BD Biosciences) and TNF-α-PacificBlue (BioLegend) using FIX & PERM Cell Fixation & Cell Permeabilization Kit (ThermoFisher Scientific) according to the manufacturer’s information.
In Vitro Antigen Specific Killing Assay
A375-nRFP (NY-ESO-1+ HLA-A*0201+) melanoma cell lines stably transduced to express nuclear RFP (Zaretsky 2016 NEJM) were seeded approximately 24 h before starting the co-culture (~1,500 cells seeded per well). Modified T cells were added at the indicated E:T ratios. The killing assay was performed in cRPMI with IL-2 (50 U/mL) and glucose, and for indicated assays TGFβ1 (25 ng/mL). RFP time lapse imaging using an IncuCyte ZOOM (Essen) was performed for 4–5 days. Cancer cell clearance (“A375 Clearance”) was defined as: (# RFP+ cells in A375 only wells - # RFP+ cells in indicated co-culture well) / (# RFP+ cells in A375 only wells) relative to a control T cell population (e.g. TCR + tNGFR). The control T cell population clearance value was divided by the experimental T cell population clearance value such that an A375 clearance value greater than 1 indicates more cancer cell killing relative to control T cells, while a value less than 1 indicates decreased cancer cell killing relative to control T cells.
Pooled Knock-in Screening
Targeted pooled knock-in screening was performed using the non-viral genome targeting method as described, except with ~10bps of DNA mismatches introduced into the 3’ homology arm of the TRAC exon 1 targeting HDR template used to replace the endogenous TCR (Roth et al., 2018). A barcode unique for each member of the knock-in library was also introduced into ~6 degenerate bases at the 3’ end of the TCRαVJ region of the HDR template (Figure 1B). The 36 constructs included in the pooled knock-in library (Table S1) were designed using the Benchling DNA sequence editor, commercially synthesized as a dsDNA geneblock (IDT), and individually cloned using Gibson Assemblies into a pUC19 plasmid containing the NY-ESO-1 TCR replacement HDR sequence (except for pooled assembly conditions, whereas all geneblocks in the library were pooled prior to assembly). The library was pooled at various stages as described in figure legends (Figure S3), but unless otherwise noted HDR templates were pooled prior to electroporation.
The modified T cell libraries generated by pooled knock-ins were electroporated, cultured, and expanded as described, before being subjected to a variety of in vitro assays beginning at day 7 post electroporation and ending at day 12 post electroporation. In stimulation assays, the modified T cell library was stimulated with CD3/CD28 dynabeads at a 1:1 bead to cell ratio, and at a 5:1 bead to cell ratio for the excessive stimulation condition. In the TGFβ assay, 25 ng/mL of human TGFβ was added to the culture media. For the CD3 stimulation only condition, a 1G4 TCR (NY-ESO-1 specific) binding dextramer (Immudex) was bound to cells at 1:50 dilution in 50 uL (500,000 cells total) for 12 minutes at room temperature, prior to return to culture media. All in vitro assays began with 500,000 sorted NY-ESO-1+ T cells unless otherwise described.
At the conclusion of the in vitro or in vivo assays, T cells were pelleted and either genomic DNA was extracted (QuickExtract) or mRNA was stabilized in Trizol. mRNA was purified using a Zymo Direct-zol spin column according to manufacturer’s instructions, and converted to cDNA using a Maxima H RT First Strand cDNA Synthesis (Thermo) according to manufacturer’s instructions. Unless otherwise stated, libraries were made from isolated mRNA/cDNA. A two-step PCR was performed on the isolated genomic DNA or cDNA. The first PCR (PCR1) included a forward primer binding in the TCRαVJ region of the insert and a reverse primer binding in the genomic region overlapping the site of the mismatches in the 3’ homology arm (Figure S2 and Table S1), and used Kapa Hifi Hotstart polymerase for 12 cycles, followed by a 1.0X SPRI purification. The second PCR used NEB Next Ultra II Q5 polymerase for 10 cycles to append P5 and P7 Illumina sequencing adaptors and sample-specific barcodes, followed again by 1.0X SPRI purification. Normalized libraries were pooled across samples and sequenced on an Illumina Mini-Seq with a 2X150 bp reads run mode.
PoKI-Seq: Pooled Knock-in Screening plus Single-Cell RNA Sequencing
Single-cell RNA sequencing was performed on 8 separate samples (2 donors, 2 recipients per donor, matched pre- and post-implantation cells) with the Chromium Single Cell 3′ Reagent Kit, v3 chemistry (10x Genomics, PN-1000092) following the manufacturer’s protocol. Briefly, TCR-positive cells were sorted by FACS (BD FACS Aria) and resuspended at 1000 cells/ul in PBS + 1% FBS for a targeted recovery of 6000 cells per condition. We performed 11 cycles of PCR for cDNA amplification after GEM recovery, and 25% of each cDNA sample was carried into transcriptome library preparation. We performed 13 cycles of PCR to introduce Chromium i7 multiplex indices (10× Genomics, PN-120262). cDNA was diluted 1:5 in Buffer EB and quantified by Bioanalyzer DNA High Sensitivity (Agilent, 5067–4626) and/or Qubit dsDNA High Sensitivity (Thermo Fisher, Q32854) reagents. Samples were pooled equally and sequenced on a NovaSeq S4 flow cell (Illumina) using read parameters 28×8×91. Raw fastq files were mapped to the human transcriptome (GRCh38) using Cell Ranger (10× Genomics, version 3.0.2) and further analyzed using Seurat (version 3.0.1).
After the initial 11 cycle cDNA amplification step described above, 50% (20ul) of each cDNA sample was loaded into a KAPA HIFI 2× PCR reaction using 1uM p5 forward primer (AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTC) and 1uM of a custom TCRa-read2 reverse primer (GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTGAGGAACCAGCCTTATTGTTCA TCCGTA) and run with the following parameters: 98C for 45”, [98C for 20”, 67C for 30”, 72C for 30”] × 10, 72C for 60”, hold at 4C. The PCR products were purified with 0.8× AMPure XP (Beckman Coulter, A63882) and eluted in 45ul Buffer EB (Qiagen, 1014608). We performed 9 cycles of PCR to introduce Chromium i7 multiplex indices (10× Genomics, PN-120262). cDNA was diluted 1:5 in Buffer EB and quantified by Tapestation DNA High Sensitivity (Agilent, 5067–5593) and/or Qubit dsDNA High Sensitivity (Thermo Fisher, Q32854) reagents. Samples were pooled equally and sequenced on a NovaSeq SP flow cell (Illumina) with 25% PhiX using read parameters 28×8×98.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Analysis
Statistical details for all experiments, including value and definition of n, error bars, and significance thresholds can be found in the Figure Legends.
Predicted Amount of Template Switching
An estimation of the total amount of template switching (barcode switching) during a large pooled experiment where there is some distance between a DNA barcode and the functional DNA sequence with which the barcode is associated can be inferred using the observed amount of template switching from a small library (Figure 1E). A simple model requires two assumptions.
Assumption 1: Template switching is random, and any barcode that is swapped is equally likely to switch to any other barcode.
Assumption 2: A template that has switched once is equally likely to switch again.
Using a two-member library where DNA barcodes are associated with observable functional DNA sequences (e.g. encoding a detectable surface marker or fluorescent protein), cells that are confirmed to express the functional DNA sequence can be isolated from all other cells that express the other (e.g. sorting for GFP+ cells) and their DNA barcodes sequenced. The percentage of the sequenced DNA barcodes that does not match the barcode associated with the functional DNA sequence is thus the observed amount of template switching, represented as a ratio.
X = Observed rate of template switching
Y = Actual rate of template switching
In a two-member library half of the actual template switching will occur with another template that contains the same barcode, and is thus not detected. Therefore, the observed amount of switching must be multiplied by 2.
| 1) |
However a template that has switched once is equally likely to switch again, and therefore additional rounds of switching after the first can occur.
| 2) |
Simplifying the equation yields:
| 3) |
| 4) |
| 5) |
| 6) |
| 7) |
Thus, a general equation for total template switching (Y) can be derived from the observed amount of template switching (X) in a two-member library. For example, if the observed template switching rate was 0.2 (20% of total reads contained an incorrect barcode), then the actual predicted rate of template switching would be 0.5 (50% of the reads are predicted to have switched, but because many switched to a template containing the same barcode, they were not observed). In a library of arbitrarily large size, there will be no significant switching with templates containing the same barcode, and thus the observed and actual amounts of template switching will converge.
With the parameters of the pooled knock-in library design (~400 bps between unique library insert and its corresponding barcode, separated by the new knocked in TCR-α specificity) described here (Figure 1B), the amount of predicted template switching with pooled assembly reactions was ~50%, and with a pooled electroporation condition was only ~10%.
Predicted Correct Knock-in Barcode Frequency
Experiments using a two member GFP and RFP pooled knock-in library enabled direct measurements of the rate of bi-allelic integrations and template switching. Biallelic knock-in rate was estimated to be ~25% of total knock-in positive cells (Figure 1D and Figure S2E), while the rate of template switching in the pooled electroporation condition used throughout the study was determined to be ~10% (Figure 1E). Using these values, the predicted frequency of sequenced barcodes containing the correct knock-in barcode for an individual construct that has been sorted from an arbitrarily large knock-in population (e.g. sorted GFP+ cells from the 36-member knock-in library) can be calculated.
Given 1000 sorted knock-in positive cells, 75% will possess a monoallelic knock-in (750 cells, 750 knock-in alleles), while 25% will possess biallelic knock-ins (250 cells, 500 knock-in alleles).
Half of the alleles in biallelic knock-in cells will have a different knock-in construct/barcode than the sorted (GFP) knock-in construct/barcode, assuming an arbitrarily large knock-in library.
Therefore, out of 1250 total alleles, 1000 would be predicted to contain the correct knock-in construct/barcode before accounting for template switching.
Assuming a 10% rate of template switching, 100 out of 1000 alleles with the correct knock-in construct (GFP) will contain a barcode for a different construct.
In total then, 900 out of 1250 alleles, or 72% of alleles, will contain the correct knock-in barcode.
Analysis of Pooled Knock-in Screens
To identify negative and positive hits in our screens, abundance of guides was first determined in R using PDict on the raw sequenced fastq files using exact matching for knock-in construct specific barcodes (Table S1). Log2 fold change (LFC) was calculated for each knock-in construct, defined throughout as the log2 fold change over indicated input population. All donor replicates in each screen were grouped for statistical analysis to account for biological noise.
Analysis of PoKI-Seq Screens
Pre-processing of the Illumina sequencing results from the 10X Genomics V2 libraries was performed with CellRanger software, version 2.0.0. This pipeline produces sparse numerical matrices for each sample, with gene-level counts of unique molecular identifiers (UMI) identified for all single cells passing default quality control metrics. These gene expression matrices were processed with Seurat R package (Butler et al., 2018), as described elsewhere (https://satijalab.org/seurat/pbmc3k_tutorial.html). Only cells with more than 500 genes identified were used for downstream analyses. Using Seurat, counts were log normalized, regressing out total UMI counts per cell and percent of mitochondrial genes detected per cell, and scaled to obtain gene level z-scores. Of note, all samples were processed simultaneously in the same experiment and thus sample origin was not regressed out (Butler et al., 2018). We then applied principal component analysis (PCA), using the 1,000 most variable genes across cells. The first 30 PCA components were used to construct a uniform manifold approximation and projection (UMAP) to visualize single cells in a two dimensional plot, as in Figure 5D. Clustering in Figure 5E, was performed by the Louvain algorithm on the shared nearest neighbor graph, as implemented by the FindClusters command from the Seurat R package. For synthetic bulk differential gene expression in Figure 5I and Figure 6E, UMI counts per gene were summed for all cells with control knock-ins in each sample, and the DESeq2 R package was used to determine differentially expressed genes. For gene list enrichment analysis in cell clusters, the REACTOME database (Fabregat et al., 2018) was used as reference to generate Figure S6E.
To associate guides with identified cell barcodes, we processed both fastq files from the 10X libraries and from the separately amplified cDNA PCR. The read2 files were matched to the knock-in construct library using matchPattern as implemented in the ShortRead R package (Morgan et al., 2009). The pattern used was the 3’ end of the sequence of the inserted TCR alpha gene containing the specific knock-in barcode within degenerate bases, allowing for 1 mismatch total. The mate Read1 pairs for reads with matched knock-in construct barcodes were used to determine the cell barcode and UMI assignment. We filtered out reads appearing less than twice and cells with more than two assigned knock-in barcodes. The Chi-square test was used to determine over-representation of cells with guides for the same gene target across cell-state driven clusters. Standardized residuals from the chi-square test were scaled and used to generate Figure 5H. Additional specific details for the generation of individual plots and analysis are contained in the associated figure legends in Figure 5 and Figure S6.
DATA AND CODE AVAILABILITY
PoKI-Seq and scRNAseq data are available on GEO (GSE143417). Amplicon sequencing data from pooled knock-in screens, annotated DNA sequences for all constructs, and all custom scripts used for amplicon sequencing data processing, single cell RNA sequencing data filtering, and for generation of the presented analysis have not been deposited in a public repository but are available from the corresponding author on request.
Supplementary Material
Figure S1. Knock-in of polycistronic gene cassettes encoding TCR specificity and a new functional product at the human TRAC locus, Related to Figure 1. (A) Schematic of a polycistronic therapeutic knock-in cassette that enables simultaneous reprogramming of TCR specificity and integration of a co-regulated protein of interest at the endogenous TCR-α locus. The cassette includes a 2A sequence, a new full-length TCR-β chain, a second 2A sequence, the sequence for a protein of interest, a third 2A sequence, and the sequence of the new variable region of the TCR-α chain. Successful knock-in yields a polycistronic mRNA encoding new TCR-β and TCR-α chains as well as a third protein (“Insert X”).
(B) Flow cytometry shows successful knock-in of a NY-ESO-1 TCR plus tNGFR three gene polycistronic knock-in cassette. Cells that remained TCR positive but not NY-ESO-1 dextramer positive likely represent unedited cells expressing their endogenous TCR (Roth et al., 2018).
(C) TCR+Insert X knock-in strategy at the endogenous TRAC locus leads to coordinated gene expression of a novel TCR-β chain and TCR-α chain, and a protein of interest. TCR stimulation causes TCR internalization, and after 24 hours of stimulation, we observed decreased NY-ESO-1 TCR expression by dextramer staining. In contrast, tNGFR protein expression remained high after 24 hours of stimulation as expected.
(D) Schematic representation of a four gene polycistronic knock-in cassette containing a new replacement TCR specificity and two additional co-regulated proteins of interest at the endogenous TCR-α locus.
(E) Flow cytometry shows successful knock-in of a TCR+tNGFR+GFP four gene polycistronic knock-in cassette. NY-ESO-1+ cells almost all express GFP and tNGFR concordantly (top right flow plot) whereas NY-ESO-1− TCR- cells largely do not (bottom left flow plot). A relatively small percentage of TCR+ NY-ESO-1− cells express both GFP and tNGFR, and even fewer expressed either alone (bottom right flow plot).
(F) Direct comparison between two different knock-in gene constructs can be performed by separate individual knock-ins and pooling of the resulting cells, as long as the knock-in constructs can be distinguished in some way, such as at the protein level by flow cytometric staining for reporter gene expression (e.g. tNGFR).
(G) Gating strategy to determine relative expansion of NY-ESO-1 TCR+ dnTGFβR2+ T-cells over NY-ESO-1 TCR+ tNGFR+ after four days in an in vitro competitive growth assay.
(H) A previously described dominant negative TGFβ receptor 2 (dnTGFβR2) (Kloss et al., 2018) at the “Insert X” site provided resistance to the suppressive effects of exogenous TGFβ. NY-ESO-1 TCR+ dnTGFβR2+ T cells expanded preferentially relative to NY-ESO-1+ tNGFR+ T cells when stimulated in the presence of TGFβ1. ***P<0.001 (two-way analysis of variance (ANOVA) with Holm–Sidak’s multiple comparisons test).
All experiments performed in n=2 independent healthy donors.
Figure S2. Selective sequencing of on-target knock-ins and template switching analysis in pooled knock-in screens. Related to Figure 1. (A) DNA sequencing of HDR outcomes is complicated by the large amount of HDRT introduced into cells that remains non-integrated. Introduction of mismatches into the homology enables a one-step PCR to amplify the on-target locus using one primer complementary to the inserted sequence (and thus unable to prime off a genomic locus without knock-in), and a second primer complementary to the genomic sequence at the site where the HDRT homology arm has a mismatch. Only the on-target genomic knock-in sites should possess both primer binding sites and these will be selectively amplified.
(B) Knock-in of ~2.5kb NY-ESO-1 TCR+tNGFR gene construct was slightly less efficient with the homology arm mismatches compared to unaltered homology arms. Mean of three technical replicates in n=2 independent donors. *P < 0.05 (paired two-tailed T test).
(C) Pooling of samples can occur at each distinct step of a non-viral genome targeting protocol: dsDNA fragments containing the unique members of a pooled knock-in library can be pooled prior to assembly into DNA plasmids already containing constant elements such as homology arms (“Pooled Assembly”); DNA plasmids containing the entire HDRT sequence for each unique library member can be pooled prior to a PCR reaction to generate large amounts of dsDNA HDR template (“Pooled PCR”); dsDNA HDR templates for each unique library member can be pooled prior to electroporation into the final cells (“Pooled Electroporation”); or, cells separately electroporated with each unique library member can be pooled following electroporation but before a final readout (“Pooled Culture”).
(D) Successful knock-in of a two member GFP and RFP duplexed barcoded library across pooling conditions. Knock-in positive cells were identified based on expression of the new TCR specificity (assessed by NY-ESO-1 TCR specific dextramer staining) introduced along with the GFP and RFP.
(E) Knock-in positive cells were analyzed for GFP and RFP expression by flow cytometry. Cells with either GFP or RFP templates alone only showed expression of each respective fluorescent protein, while the Pooled Culture condition showed equal populations of GFP and RFP positive cells exclusively, without GFP+RFP+ cells. Pooling conditions prior to the electroporation step (Pooled Assembly, Pooled PCR, or Pooled Electroporation) all showed both single GFP+ or RFP+ cells, as well as dual GFP+RFP+ cells likely due to bi-allelic knock-in at the TRAC locus (Roth et al., 2018). Multiple populations were sorted for barcode sequencing, including knock-in negative cells (NY-ESO-1−), bulk knock-in positive cells (NY-ESO-1 TCR+), and individual populations of RFP+GFP− or RFP−GFP+ cells, followed by either gDNA or cDNA isolation and barcode sequencing.
(F) Knock-in positive cells (NY-ESO-1 TCR+) showed preferential amplification of the knocked-in sequence with the barcode relative to the bulk edited population, while knock-in negative cells showed almost no successful amplification, consistent with expected selectivity for amplifying and sequencing on-target knock-ins relative to non-integrated HDRT sequence or off-target integrations.
(G) Knock-in positive and negative cells were sorted and barcode sequencing performed from either isolated genomic DNA or cDNA. As expected the percentage of sequenced reads containing a valid knock-in was over 90% barcode in sorted knock-in positive cells in both conditions. Sorted knock-in negative cells showed almost no sequencing reads with a knock-in barcode in the cDNA condition, but did show some false positive reads in the genomic DNA condition, potentially due to higher rates of incorrect barcode calling due to greater opportunities for template switching when non-integrated HDR templates are not degraded (in contrast to the DNA digestion step present during mRNA isolation). Sequencing from gDNA would have advantages as template tracking would not be confounded by alterations in transcript levels and eventually pooled knock-in applications could be extended to non-transcribed genome regions, but sequencing from cDNA has advantages as more barcode copies are present per cell, potentially enabling experiments with lower cell numbers.
(H) In knock-in positive cells, the degree to which alleles without a knock-in were amplified during the barcode sequencing PCR was analyzed across pooling stages and comparing isolated cDNA vs genomic DNA (gDNA).
(I) The percentage of sequenced reads (from either genomic DNA or cDNA amplification) that contained the GFP HDR template’s barcode corresponded with the observed percentage of cells expressing GFP protein by flow cytometry across pooling conditions.
(J) The percentage of sequenced barcodes in sorted GFP+ or RFP+ cells that contained the correct barcode is displayed across pooling conditions when sequencing from amplified genomic DNA. Knock-in of GFP or RFP templates individually yielded 100% of reads containing the correct barcode, and pooled culture of cells after electroporation yielded >99% correct barcodes. However, pooling at earlier experimental stages produced a consistent increase in the amount of template switching across donors in either GFP+ (left) or RFP+ (right) sorted cells. The consistent and reproducible levels of template switching observed indicated that this template switching could occur during the molecular assembly stage, during PCR production of the HDR templates, and during actual homology directed repair events within the cell, potentially due to crossing over events between multiple HDR templates.
All experiments performed in n=2 unique healthy donors.
Figure S3. Design of a 36-member pooled knock-in library to alter T cell function, Related to Figure 3. (A) A pooled knock-in library of 36 potentially therapeutic genes was constructed that could be integrated along with a new TCR specificity (NY-ESO-1) using a single HDR template (Table S1). The library was designed to contain both previously published and novel members that potentially modified T cell function in a variety of broad classes: immune checkpoints with their intracellular domains either truncated (“tPD1” or “tCTLA4”) or replaced with an “activating” domain (chimeric switch receptors, “CTLA4-CD28”); apoptotic mediators similarly truncated or with intracellular domains switched; genes involved in cell proliferation; chemokine receptors; transcription factors; genes involved in metabolic pathways associated with survival in tumor environments; and suppressive cytokine receptors either as truncated/dominant negative receptors (“dnTGFβR2”) or with intracellular domains switched. Previously described synthetic genes shown to improve T cell function in various contexts are bolded and references are included in Table S1.
(B) All 36 constructs were synthesized and placed into a TCR insertion cassette that would replace the endogenous T cell receptor with a new specificity (NY-ESO-1 TCR) as well as drive expression of the new gene that potentially modifies T cell function with endogenous TCR-α gene regulation. Each library member was individually tested in an arrayed knock-in screen and assayed for percent knock-in as well as MFI of the surface expressed TCR to assay any potential effects of the individual inserts on TCR expression in cells from two human blood donors.
(C) All 36 constructs successfully showed functional TCR expression as analyzed by surface dextramer staining for the new NY-ESO-1 TCR.
(D) The total insert sizes ranged from ~2,000–3,000 bps (not including the homology arm sequences), and little correlation was observed between template size and knock-in efficiency.
(E) Consistent TCR protein expression levels (assessed as MFI of NY-ESO-1 TCR) were observed following arrayed knock-in of each library member. We did note that the TCR MFI was lower with the truncated (tCTLA4) construct. If this construct produces less transcript it could potentially affect barcode abundance readouts from cDNA (although barcodes are compared to input cell populations in pooled screens to minimize these potential effects).
Complete design strategy, amino acid sequences, and DNA sequences for all 36 library members are contained in Table S1.
Figure S4. Pooled knock-in screening identifies context dependent hits across in vitro selective pressures mimicking aspects of tumor environments, Related to Figure 3. (A) Pooled electroporation of a 36-member library of DNA sequences encoding potential function modifying proteins along with a defined TCR specificity (NY-ESO-1 antigen) generated a pooled library of modified primary human T cells. Various in vitro selective pressures mimicking aspects tumor environments could then be applied and the distribution of unique barcodes in the population of modified T cells assayed relative to input control populations.
(B) Distribution of library construct abundances after in vitro culture for 5 days, represented as a ranked list of log2 fold changes. In the context of excessive amounts of TCR stimulation (anti-CD3/CD28 5:1 bead:cell ratio instead of a standard 1:1 ratio), Fas receptor-derived constructs showed increased relative abundance when compared to the input population. When comparing the excessive stimulation population to standard stimulation, the Fas constructs however showed greater relative inhibition with excessive stimulation. In contrast, a construct expressing the transcription factor TCF7 trended towards a greater relative advantage in cells with excessive anti-CD3/CD28 stimulation.
(C) Stimulation of the modified T cell population through the TCR only (with an NY-ESO-1 specific dextramer) without the presence of a CD28-engaging co-stimulatory signal showed selective increased abundance of some, but not all, CD28 chimeric switch receptors. Constructs encoding CTLA4-CD28, TIM3-CD28, and BTLA-CD28 conferred a selective advantage with TCR stimulation alone, relative to culture with anti-CD3/CD28 stimulation.
(D) When 25 ng/mL of exogenous TGFβ was added to the culture media, Fas derived receptors showed a relative decrease in abundance (but still an absolute increase, Figure 3D) demonstrating potentially enhanced susceptibility to TGFβ-mediated suppression. TGFβR2 derived receptors in contrast showed by far the greatest relative expansion in the presence of TGFβ. A previously published dominant negative TGFβR2 receptor (dnTGFβR2) (Kloss et al., 2018) and a novel chimeric TGFβR2 (41BB intracellular domain) switch receptor both drove strong T cell expansion in TGFβ.
(E) Control comparisons of in vitro screen hits FAS-41BB and TGFβR2–41BB with a third 41BB chimeric receptor (PD1–41BB) as well as heterologous overexpression of 41BB itself confirmed the functional specificity and context dependent increases in cellular fitness for these two constructs. All four constructs possessed a 41BB intracellular domain, but improved context-dependent fitness was determined by the knock-in construct’s extracellular domain.
(F) Pooled knock-in screening could be reproducibly performed on both sorted knock-in positive cells (here sorted on NY-ESO-1 dextramer stained cells) as well as an unsorted bulk population of edited cells (R2 = 0.87).
(G) Large numbers of modified T cells could be generated after pooled knock-in of a 36-member library. Given 1 million primary human T cells at isolation, an average of ~0.5 million knock-in positive cells were recovered by four days post-electroporation (average overall knock-in efficiencies were ~10% – 20%), and these cells continued to expand robustly over additional days in culture without re-stimulation across four healthy human donors, providing enough cells for large in vivo experiments.
(H) The reproducibility of pooled knock-in screen results across technical replicates and pooling stages. Technical replicates of the TCR stimulation screen in the same biologic donor showed strong correlation (R2 = 0.99). The correlation between Pooled Assembly and Pooled Electroporation conditions was also strong, albeit less so (R2 = 0.88).The correlation between technical replicates of Pooled Assembly conditions was similarly slightly weaker (R2 = 0.89). This could be due to the more frequent template switching observed when library pooling occurs at earlier stages (Figure 1E).
N=2 (F–H) or mean of n=4 unique healthy donors (B–D) is displayed and was used to rank the constructs. Dotted lines represent maximum and minimum abundance of control library members. Unless otherwise noted, all graphs display log 2-fold change compared to modified T cell library input, or relative log 2-fold change compared to CD3/CD28 stimulation. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 (two-way analysis of variance (ANOVA) with Holm–Sidak’s multiple comparisons test, B–E).
Figure S5. Arrayed in vitro validation of pooled knock-in screen hits, Related to Figure 4. (A) Time course of protein expression for each indicated knock-in construct at 5, 7 and 10 days post-electroporation compared to control knock-in (NY-ESO-1 TCR + tNGFR for all constructs except tNGFR itself, where a TCR + tBTLA construct was used as a control) in gated NY-ESO-1TCR+ cells. Expression of some endogenous gene products (Fas, IL2RA, Tim-3) was detected, but increased expression was observed with the addition of the knock-in constructs at all time points except day 5 and 7 for TIM3-CD28. At day 10, TIM3-CD28 construct expression was observed above endogenous levels, likely due to consistent high expression off of the TCR promoter relative to activation dependent expression of endogenous Tim-3.
(B) Additional viability (% live cell staining in total lymphocyte population), proliferation (% CFSE Low), and expansion (total cell number compared to input) assays with individual knock-in constructs in sorted cells as in Figure 4C. The Fas-41BB chimeric receptor showed the highest viability following stimulation, as well as the greatest amount of proliferation as measured by CFSE dilution staining 4 days after stimulation. When only CD3 stimulation was provided, a TIM3-CD28 chimeric receptor showed the greatest amount of proliferation, similar to the pooled knock-in screen (Figure S4C). In the context of excessive stimulation (5:1 CD3/CD28 bead to cell ratio), the Fas-41BB chimeric receptor again showed the greatest relative expansion (Figure S4B). Three technical replicates in n=4 independent healthy donors shown.
(C) Sorted NY-ESO-1 TCR+ T cells were co-cultured with target RFP+ A375 melanoma at the indicated effector to target cell ratios beginning 9 days after electroporation and imaged for 72 hours by Incucyte timelapse microscopy. The percentage of target cell killing for each of eight individual knock-in constructs tested is shown against control (average of TCR+tNGFR and TCR+GFP constructs similarly tested). The average + SEM for three technical replicates in each of n=4 independent healthy donors is shown.
Figure S6. PoKI-Seq molecular pipeline, quality control metrics, and single cell phenotypes of pooled knock-in constructs, Related to Figure 5. (A) Diagram of molecular sequencing pipeline to associate a cell’s transcriptome with its knock-in construct using PoKI-Seq. The barcode for the specific knock-in construct (“Knock-in Barcode”) in a cell is encoded in degenerate bases of the coding region of the integrated TCRαVJ region. After transcription and single cell isolation in droplets, the TCR + Gene X mRNA transcripts from the individual cell are bound to a bead containing poly(dT) primers along with a unique cell barcode. Following reverse transcription, a primer binding immediately upstream of the knock-in barcode creates an amplicon containing both the knock-in barcode as well as the cell-barcode. Next-generation sequencing from both ends of this amplicon yields a matched pair of knock-in barcode and cell-barcode, along with a universal molecular identifier (UMI). Only a portion of cDNA isolated during the droplet-based polyA pulldown is required to generate single-cell transcriptomes (25%) and the remainder of the cDNA (75%) can be used for sequencing of the knock-in barcodes.
(B) Quality control metrics from PoKI-Seq in ex vivo primary human T cells. A large number of unique genes and unique UMIs were called per cell. Notably single cells with transcriptomes assigned through Cell Ranger (10X) for which a knock-in construct was not assigned (“0”) showed markedly poorer QC metrics. Cells that did not have a knock-in barcode assigned had fewer total unique genes, fewer total UMIs, and a greater variance in percentage of mitochondrial reads, all which may indicate that these may not have been true live cells. Dying cells, lysed cells, or other non-desired droplet contents may contribute both to poor QC metrics as well as lack of an assigned knock-in construct barcode. Within each of the two donors and two conditions tested (Stim +/− TGFβ), the average coverage (number of individual cells with a monoallelic integration of each knock-in construct) was ~136X. At least 3 UMIs all containing the same knock-in barcode were used to assign a cell to a specific knock-in construct, with the majority of cells possessing many more than 3.
(C) Variance in the percentage of corresponding assignment between single cells with a knock-in construct barcode and single cells with a transcriptome passing filter (Figure S6B). A unique UMI threshold of 3 or more was chosen to assign a knock-in construct to an individual cell.
(D) Heatmap of normalized gene expression values of transcripts containing the knocked-in sequence for selected knock-in constructs. The knock-in constructs are driven by the endogenous TCR promoter, generating a higher expression level than the endogenous genes containing portions of the knock-in construct’s sequence (e.g., Fas-41BB driven off the TCR promoter is expressed at higher levels than endogenous Fas, see Figure 4B and Figure S5A). Transcripts are fragmented during 10X library preparation making it impossible to discriminate transcripts from endogenous genes from those produced from the knock-in constructs. Increased abundance of the expected mRNA associated was observed for many of the knock-in constructs, similar to was seen for expected protein products in Figure 4B and Figure S5A,
(E) Enrichment (Chi-square residual) of each knock-in construct examined using PoKI-Seq within the indicated single cell clusters. TGFβR2 switch receptors or dominant negative receptor showed strong enrichment in specific clusters in the presence of exogenous TGFβ, consistent with their context-dependent specific biological effects on cell states. Color indicates the chi-square residual value and size indicates the chisquare residual’s magnitude.
(F) GO term enrichment analysis within each defined single cell cluster. GO terms further supported the functional interpretation of individual cell-state clusters. The color is the average log fold change for the gene set associated with the indicated GO term within the specified cluster compared to all other clusters. Size is the p-value of the hypergeometric enrichment test.
(G) Pairwise Pearson correlation of the average expression for all differentially expressed genes identified in any of the single cell clusters, calculated for the indicated knock-in constructs and control in both stimulation only and stimulation + TGFβ in vitro conditions. The dominant transcriptional differences were driven by exposure to TGFβ, but within the stimulation condition the knock-in constructs that promoted the greatest proliferative advantages (Fas switch receptors and IL2RA, but notably not the Fas-CD28 construct, see Figure 3A) showed the most similar transcriptional profiles. In contrast, in the presence of TGFβ, all three TGFβR2-derived receptors showed more correlated transcriptional changes with each other than with the other knock-in constructs.
Figure S7. PoKI-Seq within solid tumors in vivo, Related to Figure 6. (A) After pooled knock-in and expansion for 10 days in vitro, a modified population of human T cells electroporated with the 36-member TRAC polycistronic knock-in library was adoptively transferred into immunodeficient NSG mice bearing a human melanoma tumor xenograft (A375 melanoma cells expressing the target peptide/MHC for the NY-ESO-1 TCR) injected subcutaneously 7 days before transfer. A bulk population of 10 million T cells (~1 million knock-in positive NY-ESO-1 TCR+ cells) was transferred. After 5 days of in vivo selective pressure in the solid tumor environment the tumors were dissected, T cells were sorted, and the abundance of barcodes relative to input population was analyzed by DNA sequencing.
(B) Biologic replicates of the in vivo solid tumor pooled knock-in screen with cells from different human blood donors showed greater variance across the library than in vitro pooled knock-in screens (Figure 3B), but consistently showed the same top library hits. Technical replicates of the in vivo pooled knock-in screen with cells from the same blood donor similarly showed greater variance than in vitro pooled knock-in screens (Figure S4H).
(C) The enriched abundance of knock-in constructs from the library observed in vivo in TILs (relative to input population) computed based on single-cell knock-in barcodes correlated (R2 = 0.39) with the enriched abundance observed in independent knock-in screens conducted based on bulk barcode abundance (Figure 4B) (n=2 individual donors for bulk condition and n=2 separate individual donors for single cell condition).
(D) Histogram of the number of knock-in barcodes associated with each sequenced cell barcode. As expected, the vast majority of cell barcodes had only a single knock-in barcode associated with them. Cell barcodes that had two associated knock-in barcodes could represent real biallelic knock-ins or results from template switching during library preparation. Cell with greater than two associated knock-in barcodes were rare, and likely represent template switching events. The minority of cell barcodes with two or more associated knock-ins were filtered from further analysis.
(E) Histogram of the number of unique molecular identifiers (UMIs) associated with each sequenced combination of knock-in barcode and cell barcode. UMIs are added during the reverse transcription step and each represents a unique mRNA transcript. Knock-in barcode/cell barcode combinations with only a single UMI were filtered from further analysis.
(F) Over 75% of cell barcodes that were assigned a knock-in barcode also had single cell transcriptomes that passed quality filters (STAR Methods). A larger number of cell barcodes that had sequenced transcriptomes but did not have a knock-in barcode assigned could be due to inefficiencies in the library preparation process, cells with biallelic knock-ins being filtered out, or cells without an on-target knock-in being present in the sorted and sequenced samples.
Table S1. Related to Figure 3 and S4 Sequence and design information from 36-member library of polycistronic constructs used in pooled knock-in screens (Figure 3), including gRNA sequences and DNA HDRT sequences.
HIGHLIGHTS.
Method for pooled knock-in screens of large DNA sequences at targeted genomic loci
Rapid discovery of novel synthetic constructs to enhance primary human T cell fitness
PoKI-Seq combines pooled knock-ins with single-cell RNA sequencing in vitro and in vivo
Novel chimeric TGFβR2–41BB receptor hit promotes solid tumor clearance
ACKNOWLEDGEMENTS
We thank members of the Marson Lab, Chris Jeans (QB3 MacroLab), Eric Chow (UCSF Center for Advanced Technology), Ryan Wagner and Reuben Hogan (Parnassus CAT), Vinh Nguyen (UCSF Flow Cytometry Core, P30 DK063720 and NIHS10 1S10OD021822-01), Kevin Lou, Julia Carnevale, Sarah Pyle, Antoni Ribas, Fyodor Urinov and E. John Wherry for their helpful suggestions and generous assistance. T.L.R. was supported by the UCSF Medical Scientist Training Program (T32GM007618), the UCSF Endocrinology Training Grant (T32 DK007418), and the NIDDK (F30DK120213). FB was supported by the Care-for-Rare Foundation and the German Research Foundation (DFG). A.M. holds a Career Award for Medical Scientists from the Burroughs Wellcome Fund, is an investigator at the Chan Zuckerberg Biohub and has received funding from the Innovative Genomics Institute (IGI), the American Endowment Foundation, the Cancer Research Institute (CRI), the Lloyd J. Old STAR award, a gift from the Jordan Family, a gift from Barbara Bakar, and is a member of the Parker Institute for Cancer Immunotherapy (PICI). This research was supported by NIH/NIGMS funding for the HIV Accessory & Regulatory Complexes (HARC) Center (P50AI150476; AM)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare competing financial interests: T.L.R., K.R., and A.M. are cofounders of Arsenal Biosciences. T.L.R. served as the CSO of Arsenal Biosciences from March through December 2019. E.S. and D.G. served as advisors for Arsenal Biosciences. A.M. is a co-founder of Spotlight Therapeutics. K.R. was a founder of Cell Design Labs. J.A.B. is a founder of Sonoma Biotherapeutics. A.M. serves as on the scientific advisory board of PACT Pharma, is an advisor to Trizell, and was a former advisor to Juno Therapeutics. A.M. owns equity in Arsenal Biosciences, Spotlight Therapeutics and PACT Pharma. J.A.B. is a consultant for Juno, a Celgene company; a stock holder and member of the Board of Directors of Provention and Rheos Medicines; a member of the Scientific Advisory Board of Pfizer Center for Therapeutic Innovation; a member of the Scientific Advisory Board and stock holder for Vir Therapeutics, Arcus Biotherapeutics, Quentis Therapeutics, Solid Biosciences, NeoCept (Founder) and Celsius Therapeutics (Founder). JAB owns stock in MacroGenics Inc., Provention, Viacyte Inc., and Kadmon Holdings. The Marson Laboratory has received sponsored research support from Juno Therapeutics, Epinomics, Sanofi and a gift from Gilead. Patents have been filed based on the findings described here.
REFERENCES
- Adamson B, Norman TM, Jost M, Cho MY, Nuñez JK, Chen Y, Villalta JE, Gilbert LA, Horlbeck MA, Hein MY, et al. (2016). A Multiplexed Single-Cell CRISPR Screening Platform Enables Systematic Dissection of the Unfolded Protein Response. Cell [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson KG, Stromnes IM, and Greenberg PD (2017). Obstacles Posed by the Tumor Microenvironment to T cell Activity: A Case for Synergistic Therapies. Cancer Cell [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arber C, Young M, and Barth P (2017). Reprogramming cellular functions with engineered membrane proteins. Curr. Opin. Biotechnol [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batlle E, and Massagué J (2019). Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, Coussens LM, Gabrilovich DI, Ostrand-Rosenberg S, Hedrick CC, et al. (2018). Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavazzana-Calvo M, Payen E, Negre O, Wang G, Hehir K, Fusil F, Down J, Denaro M, Brady T, Westerman K, et al. (2010). Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez A, Scheiman J, Vora S, Pruitt BW, Tuttle M, P R Iyer E, Lin S, Kiani S, Guzman CD, Wiegand DJ, et al. (2015). Highly efficient Cas9-mediated transcriptional programming. Nat. Methods [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen DS, and Mellman I (2013). Oncology meets immunology: The cancer-immunity cycle. Immunity [DOI] [PubMed] [Google Scholar]
- Chen N, Morello A, Tano Z, and Adusumilli PS (2017). CAR T-cell intrinsic PD-1 checkpoint blockade: A two-in-one approach for solid tumor immunotherapy. Oncoimmunology [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahmani A, and Delisle JS (2018). TGF-β in T cell biology: Implications for cancer immunotherapy. Cancers (Basel) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datlinger P, Rendeiro AF, Schmidl C, Krausgruber T, Traxler P, Klughammer J, Schuster LC, Kuchler A, Alpar D, and Bock C (2017). Pooled CRISPR screening with single-cell transcriptome readout. Nat. Methods [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit A, Parnas O, Li B, Chen J, Fulco CP, Jerby-Arnon L, Marjanovic ND, Dionne D, Burks T, Raychowdhury R, et al. (2016). Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens. Cell [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong M, Wang G, Chow R, Ye L, Zhu L, Dai X, Park J, Kim H, Errami Y, Guzman C, et al. (2019). Systematic Immunotherapy Target Discovery Using Genome-Scale In Vivo CRISPR Screens in CD8 T Cells. Cell [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esensten JH, Bluestone JA, and Lim WA (2017). Engineering Therapeutic T Cells: From Synthetic Biology to Clinical Trials. Annu. Rev. Pathol. Mech. Dis 12, 305–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabregat A, Jupe S, Matthews L, Sidiropoulos K, Gillespie M, Garapati P, Haw R, Jassal B, Korninger F, May B, et al. (2018). The Reactome Pathway Knowledgebase. Nucleic Acids Res [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman D, Singh A, Garrity AJ, and Blainey PC (2018). Lentiviral co-packaging mitigates the effects of intermolecular recombination and multiple integrations in pooled genetic screens. BioRxiv [Google Scholar]
- Fesnak AD, June CH, and Levine BL (2016). Engineered T cells: the promise and challenges of cancer immunotherapy. Nat. Rev. Cancer 16, 566–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischbach MA, Bluestone JA, and Lim WA (2013). Cell-based therapeutics: The next pillar of medicine. Sci. Transl. Med [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gigante M, Gesualdo L, and Ranieri E (2012). TGF-Beta: a Master Switch in Tumor Immunity. Curr. Pharm. Des [DOI] [PubMed] [Google Scholar]
- Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, Stern-Ginossar N, Brandman O, Whitehead EH, Doudna JA, et al. (2013). CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154, 442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, et al. (2014). Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorelik L, and Flavell RA (2000). Abrogation of TGFβ signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity [DOI] [PubMed] [Google Scholar]
- Hanna RE, and Doench JG (2018). A case of mistaken identity. Nat. Biotechnol [DOI] [PubMed] [Google Scholar]
- Hegde M, Strand C, Hanna RE, and Doench JG (2018). Uncoupling of sgRNAs from their associated barcodes during PCR amplification of combinatorial CRISPR screens. PLoS One [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess GT, Frésard L, Han K, Lee CH, Li A, Cimprich KA, Montgomery SB, and Bassik MC (2016). Directed evolution using dCas9-targeted somatic hypermutation in mammalian cells. Nat. Methods [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaitin DA, Weiner A, Yofe I, Lara-Astiaso D, Keren-Shaul H, David E, Salame TM, Tanay A, van Oudenaarden A, and Amit I (2016). Dissecting Immune Circuits by Linking CRISPR-Pooled Screens with Single-Cell RNA-Seq. Cell [DOI] [PubMed] [Google Scholar]
- June CH, and Sadelain M (2018). Chimeric Antigen Receptor Therapy. N. Engl. J. Med [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan O, Giles JR, McDonald S, Manne S, Ngiow SF, Patel KP, Werner MT, Huang AC, Alexander KA, Wu JE, et al. (2019). TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SK, Barron L, Hinck CS, Petrunak EM, Cano KE, Thangirala A, Iskra B, Brothers M, Vonberg M, Leal B, et al. (2017). An engineered transforming growth factor β (TGF-β) monomer that functions as a dominant negative to block TGF-β signaling. J. Biol. Chem [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloss CC, Lee J, Zhang A, Chen F, Melenhorst JJ, Lacey SF, Maus MV, Fraietta JA, Zhao Y, and June CH (2018). Dominant-Negative TGF-β Receptor Enhances PSMA-Targeted Human CAR T Cell Proliferation And Augments Prostate Cancer Eradication. Mol. Ther [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtulus S, Madi A, Escobar G, Klapholz M, Nyman J, Christian E, Pawlak M, Dionne D, Xia J, Rozenblatt-Rosen O, et al. (2019). Checkpoint Blockade Immunotherapy Induces Dynamic Changes in PD-1 − CD8 + Tumor-Infiltrating T Cells. Immunity [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leen AM, Sukumaran S, Watanabe N, Mohammed S, Keirnan J, Yanagisawa R, Anurathapan U, Rendon D, Heslop HE, Rooney CM, et al. (2014). Reversal of Tumor Immune Inhibition Using a Chimeric Cytokine Receptor. Mol. Ther [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Ranganathan R, Jiang S, Fang C, Sun J, Kim S, Newick K, Lo A, June CH, Zhao Y, et al. (2016). A chimeric switch-receptor targeting PD1 augments the efficacy of second-generation CAR T cells in advanced solid tumors. Cancer Res [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynn RC, Weber EW, Sotillo E, Gennert D, Xu P, Good Z, Anbunathan H, Lattin J, Jones R, Tieu V, et al. (2019). c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature 576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Zhang J, Yin W, Zhang Z, Song Y, and Chang X (2016). Targeted AID-mediated mutagenesis (TAM) enables efficient genomic diversification in mammalian cells. Nat. Methods [DOI] [PubMed] [Google Scholar]
- Morgan M, Anders S, Lawrence M, Aboyoun P, Pagès H, and Gentleman R (2009). ShortRead: A bioconductor package for input, quality assessment and exploration of high-throughput sequence data. Bioinformatics [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen DN, Roth TL, Li J, Chen PA, Mamedov MR, Vo LT, Tobin V, Apathy RA, Goodman D, Shifrut E, et al. (2019). A Cas9 nanoparticle system with truncated Cas9 target sequences on DNA repair templates enhances genome targeting in diverse human immune cell types. BioRxiv [Google Scholar]
- Oda SK, Daman AW, Garcia NM, Wagener F, Schmitt TM, Tan X, Chapuis AG, and Greenberg PD (2017). A CD200R-CD28 fusion protein appropriates an inhibitory signal to enhance T-cell function and therapy of murine leukemia. Blood [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrand-Rosenberg S, Sinha P, Beury DW, and Clements VK (2012). Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin. Cancer Biol [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquet D, Kwart D, Chen A, Sproul A, Jacob S, Teo S, Olsen KM, Gregg A, Noggle S, and Tessier-Lavigne M (2016). Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature [DOI] [PubMed] [Google Scholar]
- Prosser ME, Brown CE, Shami AF, Forman SJ, and Jensen MC (2012). Tumor PD-L1 co-stimulates primary human CD8+ cytotoxic T cells modified to express a PD1: CD28 chimeric receptor. Mol. Immunol [DOI] [PubMed] [Google Scholar]
- Rafiq S, Hackett CS, and Brentjens RJ (2019). Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J, Liu X, Fang C, Jiang S, June CH, and Zhao Y (2017). Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin. Cancer Res [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero Z, Urbinati F, Geiger S, Cooper AR, Wherley J, Kaufman ML, Hollis RP, De Assin RR, Senadheera S, Sahagian A, et al. (2013). β-globin gene transfer to human bone marrow for sickle cell disease. J. Clin. Invest [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth TL, Puig-Saus C, Yu R, Shifrut E, Carnevale J, Li PJ, Hiatt J, Saco J, Krystofinski P, Li H, et al. (2018). Reprogramming human T cell function and specificity with non-viral genome targeting. Nature [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupp LJ, Schumann K, Roybal KT, Gate RE, Ye CJ, Lim WA, and Marson A (2017). CRISPR/Cas9-mediated PD-1 disruption enhances anti-Tumor efficacy of human chimeric antigen receptor T cells. Sci. Rep 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sack LM, Davoli T, Xu Q, Li MZ, and Elledge SJ (2016). Sources of error in mammalian genetic screens. G3 Genes, Genomes, Genet [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadelain M, Rivière I, and Riddell S (2017). Therapeutic T cell engineering. Nature 545, 423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE (2017). Genome-scale CRISPR pooled screens. Anal. Biochem [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shifrut E, Carnevale J, Tobin V, Roth TL, Woo JM, Bui CT, Li PJ, Diolaiti ME, Ashworth A, and Marson A (2018). Genome-wide CRISPR Screens in Primary Human T Cells Reveal Key Regulators of Immune Function. Cell [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JH, Park HB, Oh YM, Lim DP, Lee JE, Seo HH, Lee SJ, Eom HS, Kim IH, Lee SH, et al. (2012). Positive conversion of negative signaling of CTLA4 potentiates antitumor efficacy of adoptive T-cell therapy in murine tumor models. Blood [DOI] [PubMed] [Google Scholar]
- Shin JH, Park HB, and Choi K (2016). Enhanced anti-tumor reactivity of cytotoxic T lymphocytes expressing PD-1 decoy. Immune Netw [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco SA, Calderon-Copete S, Pais Ferreira D, Carmona SJ, Scarpellino L, Gfeller D, Pradervand S, et al. (2019). Intratumoral Tcf1 + PD-1 + CD8 + T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity [DOI] [PubMed] [Google Scholar]
- Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, Mangan PA, Kulikovskaya I, Gupta M, Chen F, et al. (2020). CRISPR-engineered T cells in patients with refractory cancer. Science (80-. ) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasser A, Jost PJ, and Nagata S (2009). The Many Roles of FAS Receptor Signaling in the Immune System. Immunity [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su S, Hu B, Shao J, Shen B, Du J, Du Y, Zhou J, Yu L, Zhang L, Chen F, et al. (2016). CRISPR-Cas9 mediated efficient PD-1 disruption on human primary T cells from cancer patients. Sci. Rep 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay JC, Zha S, and Wang S (2017). Chimeric switch receptor: Switching for improved adoptive T-cell therapy against cancers. Immunotherapy [DOI] [PubMed] [Google Scholar]
- Thomas DA, and Massagué J (2005). TGF-β directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell [DOI] [PubMed] [Google Scholar]
- Wei J, Long L, Zheng W, Dhungana Y, Lim SA, Guy C, Wang Y, Wang Y-D, Qian C, Xu B, et al. (2019). Targeting REGNASE-1 programs long-lived effector T cells for cancer therapy. Nature 576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner LM, Surana R, and Wang S (2010). Monoclonal antibodies: Versatile platforms for cancer immunotherapy. Nat. Rev. Immunol [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ (2011). T cell exhaustion. Nat. Immunol [DOI] [PubMed] [Google Scholar]
- Wu T, Ji Y, Ashley Moseman E, Xu HC, Manglani M, Kirby M, Anderson SM, Handon R, Kenyon E, Elkahloun A, et al. (2016). The TCF1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci. Immunol [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto TN, Lee P-H, Vodnala SK, Gurusamy D, Kishton RJ, Yu Z, Eidizadeh A, Eil R, Fioravanti J, Gattinoni L, et al. (2019). T cells genetically engineered to overcome death signaling enhance adoptive cancer immunotherapy. J. Clin. Invest [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng GXY, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, et al. (2017). Massively parallel digital transcriptional profiling of single cells. Nat. Commun [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Knock-in of polycistronic gene cassettes encoding TCR specificity and a new functional product at the human TRAC locus, Related to Figure 1. (A) Schematic of a polycistronic therapeutic knock-in cassette that enables simultaneous reprogramming of TCR specificity and integration of a co-regulated protein of interest at the endogenous TCR-α locus. The cassette includes a 2A sequence, a new full-length TCR-β chain, a second 2A sequence, the sequence for a protein of interest, a third 2A sequence, and the sequence of the new variable region of the TCR-α chain. Successful knock-in yields a polycistronic mRNA encoding new TCR-β and TCR-α chains as well as a third protein (“Insert X”).
(B) Flow cytometry shows successful knock-in of a NY-ESO-1 TCR plus tNGFR three gene polycistronic knock-in cassette. Cells that remained TCR positive but not NY-ESO-1 dextramer positive likely represent unedited cells expressing their endogenous TCR (Roth et al., 2018).
(C) TCR+Insert X knock-in strategy at the endogenous TRAC locus leads to coordinated gene expression of a novel TCR-β chain and TCR-α chain, and a protein of interest. TCR stimulation causes TCR internalization, and after 24 hours of stimulation, we observed decreased NY-ESO-1 TCR expression by dextramer staining. In contrast, tNGFR protein expression remained high after 24 hours of stimulation as expected.
(D) Schematic representation of a four gene polycistronic knock-in cassette containing a new replacement TCR specificity and two additional co-regulated proteins of interest at the endogenous TCR-α locus.
(E) Flow cytometry shows successful knock-in of a TCR+tNGFR+GFP four gene polycistronic knock-in cassette. NY-ESO-1+ cells almost all express GFP and tNGFR concordantly (top right flow plot) whereas NY-ESO-1− TCR- cells largely do not (bottom left flow plot). A relatively small percentage of TCR+ NY-ESO-1− cells express both GFP and tNGFR, and even fewer expressed either alone (bottom right flow plot).
(F) Direct comparison between two different knock-in gene constructs can be performed by separate individual knock-ins and pooling of the resulting cells, as long as the knock-in constructs can be distinguished in some way, such as at the protein level by flow cytometric staining for reporter gene expression (e.g. tNGFR).
(G) Gating strategy to determine relative expansion of NY-ESO-1 TCR+ dnTGFβR2+ T-cells over NY-ESO-1 TCR+ tNGFR+ after four days in an in vitro competitive growth assay.
(H) A previously described dominant negative TGFβ receptor 2 (dnTGFβR2) (Kloss et al., 2018) at the “Insert X” site provided resistance to the suppressive effects of exogenous TGFβ. NY-ESO-1 TCR+ dnTGFβR2+ T cells expanded preferentially relative to NY-ESO-1+ tNGFR+ T cells when stimulated in the presence of TGFβ1. ***P<0.001 (two-way analysis of variance (ANOVA) with Holm–Sidak’s multiple comparisons test).
All experiments performed in n=2 independent healthy donors.
Figure S2. Selective sequencing of on-target knock-ins and template switching analysis in pooled knock-in screens. Related to Figure 1. (A) DNA sequencing of HDR outcomes is complicated by the large amount of HDRT introduced into cells that remains non-integrated. Introduction of mismatches into the homology enables a one-step PCR to amplify the on-target locus using one primer complementary to the inserted sequence (and thus unable to prime off a genomic locus without knock-in), and a second primer complementary to the genomic sequence at the site where the HDRT homology arm has a mismatch. Only the on-target genomic knock-in sites should possess both primer binding sites and these will be selectively amplified.
(B) Knock-in of ~2.5kb NY-ESO-1 TCR+tNGFR gene construct was slightly less efficient with the homology arm mismatches compared to unaltered homology arms. Mean of three technical replicates in n=2 independent donors. *P < 0.05 (paired two-tailed T test).
(C) Pooling of samples can occur at each distinct step of a non-viral genome targeting protocol: dsDNA fragments containing the unique members of a pooled knock-in library can be pooled prior to assembly into DNA plasmids already containing constant elements such as homology arms (“Pooled Assembly”); DNA plasmids containing the entire HDRT sequence for each unique library member can be pooled prior to a PCR reaction to generate large amounts of dsDNA HDR template (“Pooled PCR”); dsDNA HDR templates for each unique library member can be pooled prior to electroporation into the final cells (“Pooled Electroporation”); or, cells separately electroporated with each unique library member can be pooled following electroporation but before a final readout (“Pooled Culture”).
(D) Successful knock-in of a two member GFP and RFP duplexed barcoded library across pooling conditions. Knock-in positive cells were identified based on expression of the new TCR specificity (assessed by NY-ESO-1 TCR specific dextramer staining) introduced along with the GFP and RFP.
(E) Knock-in positive cells were analyzed for GFP and RFP expression by flow cytometry. Cells with either GFP or RFP templates alone only showed expression of each respective fluorescent protein, while the Pooled Culture condition showed equal populations of GFP and RFP positive cells exclusively, without GFP+RFP+ cells. Pooling conditions prior to the electroporation step (Pooled Assembly, Pooled PCR, or Pooled Electroporation) all showed both single GFP+ or RFP+ cells, as well as dual GFP+RFP+ cells likely due to bi-allelic knock-in at the TRAC locus (Roth et al., 2018). Multiple populations were sorted for barcode sequencing, including knock-in negative cells (NY-ESO-1−), bulk knock-in positive cells (NY-ESO-1 TCR+), and individual populations of RFP+GFP− or RFP−GFP+ cells, followed by either gDNA or cDNA isolation and barcode sequencing.
(F) Knock-in positive cells (NY-ESO-1 TCR+) showed preferential amplification of the knocked-in sequence with the barcode relative to the bulk edited population, while knock-in negative cells showed almost no successful amplification, consistent with expected selectivity for amplifying and sequencing on-target knock-ins relative to non-integrated HDRT sequence or off-target integrations.
(G) Knock-in positive and negative cells were sorted and barcode sequencing performed from either isolated genomic DNA or cDNA. As expected the percentage of sequenced reads containing a valid knock-in was over 90% barcode in sorted knock-in positive cells in both conditions. Sorted knock-in negative cells showed almost no sequencing reads with a knock-in barcode in the cDNA condition, but did show some false positive reads in the genomic DNA condition, potentially due to higher rates of incorrect barcode calling due to greater opportunities for template switching when non-integrated HDR templates are not degraded (in contrast to the DNA digestion step present during mRNA isolation). Sequencing from gDNA would have advantages as template tracking would not be confounded by alterations in transcript levels and eventually pooled knock-in applications could be extended to non-transcribed genome regions, but sequencing from cDNA has advantages as more barcode copies are present per cell, potentially enabling experiments with lower cell numbers.
(H) In knock-in positive cells, the degree to which alleles without a knock-in were amplified during the barcode sequencing PCR was analyzed across pooling stages and comparing isolated cDNA vs genomic DNA (gDNA).
(I) The percentage of sequenced reads (from either genomic DNA or cDNA amplification) that contained the GFP HDR template’s barcode corresponded with the observed percentage of cells expressing GFP protein by flow cytometry across pooling conditions.
(J) The percentage of sequenced barcodes in sorted GFP+ or RFP+ cells that contained the correct barcode is displayed across pooling conditions when sequencing from amplified genomic DNA. Knock-in of GFP or RFP templates individually yielded 100% of reads containing the correct barcode, and pooled culture of cells after electroporation yielded >99% correct barcodes. However, pooling at earlier experimental stages produced a consistent increase in the amount of template switching across donors in either GFP+ (left) or RFP+ (right) sorted cells. The consistent and reproducible levels of template switching observed indicated that this template switching could occur during the molecular assembly stage, during PCR production of the HDR templates, and during actual homology directed repair events within the cell, potentially due to crossing over events between multiple HDR templates.
All experiments performed in n=2 unique healthy donors.
Figure S3. Design of a 36-member pooled knock-in library to alter T cell function, Related to Figure 3. (A) A pooled knock-in library of 36 potentially therapeutic genes was constructed that could be integrated along with a new TCR specificity (NY-ESO-1) using a single HDR template (Table S1). The library was designed to contain both previously published and novel members that potentially modified T cell function in a variety of broad classes: immune checkpoints with their intracellular domains either truncated (“tPD1” or “tCTLA4”) or replaced with an “activating” domain (chimeric switch receptors, “CTLA4-CD28”); apoptotic mediators similarly truncated or with intracellular domains switched; genes involved in cell proliferation; chemokine receptors; transcription factors; genes involved in metabolic pathways associated with survival in tumor environments; and suppressive cytokine receptors either as truncated/dominant negative receptors (“dnTGFβR2”) or with intracellular domains switched. Previously described synthetic genes shown to improve T cell function in various contexts are bolded and references are included in Table S1.
(B) All 36 constructs were synthesized and placed into a TCR insertion cassette that would replace the endogenous T cell receptor with a new specificity (NY-ESO-1 TCR) as well as drive expression of the new gene that potentially modifies T cell function with endogenous TCR-α gene regulation. Each library member was individually tested in an arrayed knock-in screen and assayed for percent knock-in as well as MFI of the surface expressed TCR to assay any potential effects of the individual inserts on TCR expression in cells from two human blood donors.
(C) All 36 constructs successfully showed functional TCR expression as analyzed by surface dextramer staining for the new NY-ESO-1 TCR.
(D) The total insert sizes ranged from ~2,000–3,000 bps (not including the homology arm sequences), and little correlation was observed between template size and knock-in efficiency.
(E) Consistent TCR protein expression levels (assessed as MFI of NY-ESO-1 TCR) were observed following arrayed knock-in of each library member. We did note that the TCR MFI was lower with the truncated (tCTLA4) construct. If this construct produces less transcript it could potentially affect barcode abundance readouts from cDNA (although barcodes are compared to input cell populations in pooled screens to minimize these potential effects).
Complete design strategy, amino acid sequences, and DNA sequences for all 36 library members are contained in Table S1.
Figure S4. Pooled knock-in screening identifies context dependent hits across in vitro selective pressures mimicking aspects of tumor environments, Related to Figure 3. (A) Pooled electroporation of a 36-member library of DNA sequences encoding potential function modifying proteins along with a defined TCR specificity (NY-ESO-1 antigen) generated a pooled library of modified primary human T cells. Various in vitro selective pressures mimicking aspects tumor environments could then be applied and the distribution of unique barcodes in the population of modified T cells assayed relative to input control populations.
(B) Distribution of library construct abundances after in vitro culture for 5 days, represented as a ranked list of log2 fold changes. In the context of excessive amounts of TCR stimulation (anti-CD3/CD28 5:1 bead:cell ratio instead of a standard 1:1 ratio), Fas receptor-derived constructs showed increased relative abundance when compared to the input population. When comparing the excessive stimulation population to standard stimulation, the Fas constructs however showed greater relative inhibition with excessive stimulation. In contrast, a construct expressing the transcription factor TCF7 trended towards a greater relative advantage in cells with excessive anti-CD3/CD28 stimulation.
(C) Stimulation of the modified T cell population through the TCR only (with an NY-ESO-1 specific dextramer) without the presence of a CD28-engaging co-stimulatory signal showed selective increased abundance of some, but not all, CD28 chimeric switch receptors. Constructs encoding CTLA4-CD28, TIM3-CD28, and BTLA-CD28 conferred a selective advantage with TCR stimulation alone, relative to culture with anti-CD3/CD28 stimulation.
(D) When 25 ng/mL of exogenous TGFβ was added to the culture media, Fas derived receptors showed a relative decrease in abundance (but still an absolute increase, Figure 3D) demonstrating potentially enhanced susceptibility to TGFβ-mediated suppression. TGFβR2 derived receptors in contrast showed by far the greatest relative expansion in the presence of TGFβ. A previously published dominant negative TGFβR2 receptor (dnTGFβR2) (Kloss et al., 2018) and a novel chimeric TGFβR2 (41BB intracellular domain) switch receptor both drove strong T cell expansion in TGFβ.
(E) Control comparisons of in vitro screen hits FAS-41BB and TGFβR2–41BB with a third 41BB chimeric receptor (PD1–41BB) as well as heterologous overexpression of 41BB itself confirmed the functional specificity and context dependent increases in cellular fitness for these two constructs. All four constructs possessed a 41BB intracellular domain, but improved context-dependent fitness was determined by the knock-in construct’s extracellular domain.
(F) Pooled knock-in screening could be reproducibly performed on both sorted knock-in positive cells (here sorted on NY-ESO-1 dextramer stained cells) as well as an unsorted bulk population of edited cells (R2 = 0.87).
(G) Large numbers of modified T cells could be generated after pooled knock-in of a 36-member library. Given 1 million primary human T cells at isolation, an average of ~0.5 million knock-in positive cells were recovered by four days post-electroporation (average overall knock-in efficiencies were ~10% – 20%), and these cells continued to expand robustly over additional days in culture without re-stimulation across four healthy human donors, providing enough cells for large in vivo experiments.
(H) The reproducibility of pooled knock-in screen results across technical replicates and pooling stages. Technical replicates of the TCR stimulation screen in the same biologic donor showed strong correlation (R2 = 0.99). The correlation between Pooled Assembly and Pooled Electroporation conditions was also strong, albeit less so (R2 = 0.88).The correlation between technical replicates of Pooled Assembly conditions was similarly slightly weaker (R2 = 0.89). This could be due to the more frequent template switching observed when library pooling occurs at earlier stages (Figure 1E).
N=2 (F–H) or mean of n=4 unique healthy donors (B–D) is displayed and was used to rank the constructs. Dotted lines represent maximum and minimum abundance of control library members. Unless otherwise noted, all graphs display log 2-fold change compared to modified T cell library input, or relative log 2-fold change compared to CD3/CD28 stimulation. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 (two-way analysis of variance (ANOVA) with Holm–Sidak’s multiple comparisons test, B–E).
Figure S5. Arrayed in vitro validation of pooled knock-in screen hits, Related to Figure 4. (A) Time course of protein expression for each indicated knock-in construct at 5, 7 and 10 days post-electroporation compared to control knock-in (NY-ESO-1 TCR + tNGFR for all constructs except tNGFR itself, where a TCR + tBTLA construct was used as a control) in gated NY-ESO-1TCR+ cells. Expression of some endogenous gene products (Fas, IL2RA, Tim-3) was detected, but increased expression was observed with the addition of the knock-in constructs at all time points except day 5 and 7 for TIM3-CD28. At day 10, TIM3-CD28 construct expression was observed above endogenous levels, likely due to consistent high expression off of the TCR promoter relative to activation dependent expression of endogenous Tim-3.
(B) Additional viability (% live cell staining in total lymphocyte population), proliferation (% CFSE Low), and expansion (total cell number compared to input) assays with individual knock-in constructs in sorted cells as in Figure 4C. The Fas-41BB chimeric receptor showed the highest viability following stimulation, as well as the greatest amount of proliferation as measured by CFSE dilution staining 4 days after stimulation. When only CD3 stimulation was provided, a TIM3-CD28 chimeric receptor showed the greatest amount of proliferation, similar to the pooled knock-in screen (Figure S4C). In the context of excessive stimulation (5:1 CD3/CD28 bead to cell ratio), the Fas-41BB chimeric receptor again showed the greatest relative expansion (Figure S4B). Three technical replicates in n=4 independent healthy donors shown.
(C) Sorted NY-ESO-1 TCR+ T cells were co-cultured with target RFP+ A375 melanoma at the indicated effector to target cell ratios beginning 9 days after electroporation and imaged for 72 hours by Incucyte timelapse microscopy. The percentage of target cell killing for each of eight individual knock-in constructs tested is shown against control (average of TCR+tNGFR and TCR+GFP constructs similarly tested). The average + SEM for three technical replicates in each of n=4 independent healthy donors is shown.
Figure S6. PoKI-Seq molecular pipeline, quality control metrics, and single cell phenotypes of pooled knock-in constructs, Related to Figure 5. (A) Diagram of molecular sequencing pipeline to associate a cell’s transcriptome with its knock-in construct using PoKI-Seq. The barcode for the specific knock-in construct (“Knock-in Barcode”) in a cell is encoded in degenerate bases of the coding region of the integrated TCRαVJ region. After transcription and single cell isolation in droplets, the TCR + Gene X mRNA transcripts from the individual cell are bound to a bead containing poly(dT) primers along with a unique cell barcode. Following reverse transcription, a primer binding immediately upstream of the knock-in barcode creates an amplicon containing both the knock-in barcode as well as the cell-barcode. Next-generation sequencing from both ends of this amplicon yields a matched pair of knock-in barcode and cell-barcode, along with a universal molecular identifier (UMI). Only a portion of cDNA isolated during the droplet-based polyA pulldown is required to generate single-cell transcriptomes (25%) and the remainder of the cDNA (75%) can be used for sequencing of the knock-in barcodes.
(B) Quality control metrics from PoKI-Seq in ex vivo primary human T cells. A large number of unique genes and unique UMIs were called per cell. Notably single cells with transcriptomes assigned through Cell Ranger (10X) for which a knock-in construct was not assigned (“0”) showed markedly poorer QC metrics. Cells that did not have a knock-in barcode assigned had fewer total unique genes, fewer total UMIs, and a greater variance in percentage of mitochondrial reads, all which may indicate that these may not have been true live cells. Dying cells, lysed cells, or other non-desired droplet contents may contribute both to poor QC metrics as well as lack of an assigned knock-in construct barcode. Within each of the two donors and two conditions tested (Stim +/− TGFβ), the average coverage (number of individual cells with a monoallelic integration of each knock-in construct) was ~136X. At least 3 UMIs all containing the same knock-in barcode were used to assign a cell to a specific knock-in construct, with the majority of cells possessing many more than 3.
(C) Variance in the percentage of corresponding assignment between single cells with a knock-in construct barcode and single cells with a transcriptome passing filter (Figure S6B). A unique UMI threshold of 3 or more was chosen to assign a knock-in construct to an individual cell.
(D) Heatmap of normalized gene expression values of transcripts containing the knocked-in sequence for selected knock-in constructs. The knock-in constructs are driven by the endogenous TCR promoter, generating a higher expression level than the endogenous genes containing portions of the knock-in construct’s sequence (e.g., Fas-41BB driven off the TCR promoter is expressed at higher levels than endogenous Fas, see Figure 4B and Figure S5A). Transcripts are fragmented during 10X library preparation making it impossible to discriminate transcripts from endogenous genes from those produced from the knock-in constructs. Increased abundance of the expected mRNA associated was observed for many of the knock-in constructs, similar to was seen for expected protein products in Figure 4B and Figure S5A,
(E) Enrichment (Chi-square residual) of each knock-in construct examined using PoKI-Seq within the indicated single cell clusters. TGFβR2 switch receptors or dominant negative receptor showed strong enrichment in specific clusters in the presence of exogenous TGFβ, consistent with their context-dependent specific biological effects on cell states. Color indicates the chi-square residual value and size indicates the chisquare residual’s magnitude.
(F) GO term enrichment analysis within each defined single cell cluster. GO terms further supported the functional interpretation of individual cell-state clusters. The color is the average log fold change for the gene set associated with the indicated GO term within the specified cluster compared to all other clusters. Size is the p-value of the hypergeometric enrichment test.
(G) Pairwise Pearson correlation of the average expression for all differentially expressed genes identified in any of the single cell clusters, calculated for the indicated knock-in constructs and control in both stimulation only and stimulation + TGFβ in vitro conditions. The dominant transcriptional differences were driven by exposure to TGFβ, but within the stimulation condition the knock-in constructs that promoted the greatest proliferative advantages (Fas switch receptors and IL2RA, but notably not the Fas-CD28 construct, see Figure 3A) showed the most similar transcriptional profiles. In contrast, in the presence of TGFβ, all three TGFβR2-derived receptors showed more correlated transcriptional changes with each other than with the other knock-in constructs.
Figure S7. PoKI-Seq within solid tumors in vivo, Related to Figure 6. (A) After pooled knock-in and expansion for 10 days in vitro, a modified population of human T cells electroporated with the 36-member TRAC polycistronic knock-in library was adoptively transferred into immunodeficient NSG mice bearing a human melanoma tumor xenograft (A375 melanoma cells expressing the target peptide/MHC for the NY-ESO-1 TCR) injected subcutaneously 7 days before transfer. A bulk population of 10 million T cells (~1 million knock-in positive NY-ESO-1 TCR+ cells) was transferred. After 5 days of in vivo selective pressure in the solid tumor environment the tumors were dissected, T cells were sorted, and the abundance of barcodes relative to input population was analyzed by DNA sequencing.
(B) Biologic replicates of the in vivo solid tumor pooled knock-in screen with cells from different human blood donors showed greater variance across the library than in vitro pooled knock-in screens (Figure 3B), but consistently showed the same top library hits. Technical replicates of the in vivo pooled knock-in screen with cells from the same blood donor similarly showed greater variance than in vitro pooled knock-in screens (Figure S4H).
(C) The enriched abundance of knock-in constructs from the library observed in vivo in TILs (relative to input population) computed based on single-cell knock-in barcodes correlated (R2 = 0.39) with the enriched abundance observed in independent knock-in screens conducted based on bulk barcode abundance (Figure 4B) (n=2 individual donors for bulk condition and n=2 separate individual donors for single cell condition).
(D) Histogram of the number of knock-in barcodes associated with each sequenced cell barcode. As expected, the vast majority of cell barcodes had only a single knock-in barcode associated with them. Cell barcodes that had two associated knock-in barcodes could represent real biallelic knock-ins or results from template switching during library preparation. Cell with greater than two associated knock-in barcodes were rare, and likely represent template switching events. The minority of cell barcodes with two or more associated knock-ins were filtered from further analysis.
(E) Histogram of the number of unique molecular identifiers (UMIs) associated with each sequenced combination of knock-in barcode and cell barcode. UMIs are added during the reverse transcription step and each represents a unique mRNA transcript. Knock-in barcode/cell barcode combinations with only a single UMI were filtered from further analysis.
(F) Over 75% of cell barcodes that were assigned a knock-in barcode also had single cell transcriptomes that passed quality filters (STAR Methods). A larger number of cell barcodes that had sequenced transcriptomes but did not have a knock-in barcode assigned could be due to inefficiencies in the library preparation process, cells with biallelic knock-ins being filtered out, or cells without an on-target knock-in being present in the sorted and sequenced samples.
Table S1. Related to Figure 3 and S4 Sequence and design information from 36-member library of polycistronic constructs used in pooled knock-in screens (Figure 3), including gRNA sequences and DNA HDRT sequences.
Data Availability Statement
PoKI-Seq and scRNAseq data are available on GEO (GSE143417). Amplicon sequencing data from pooled knock-in screens, annotated DNA sequences for all constructs, and all custom scripts used for amplicon sequencing data processing, single cell RNA sequencing data filtering, and for generation of the presented analysis have not been deposited in a public repository but are available from the corresponding author on request.