

Abstract
We report here a catalytic method for the modular ring expansion of cyclic aliphatic alcohols. In this work, proton-coupled electron transfer (PCET) activation of an allylic alcohol substrate affords an alkoxy radical intermediate that undergoes subsequent C–C bond cleavage to furnish an enone and a tethered alkyl radical. Recombination of this alkyl radical with the revealed olefin acceptor in turn produces a ring-expanded ketone product. The regioselectivity of this C–C bond-forming event can be reliably controlled via substituents on the olefin substrate, providing a means to convert a simple N-membered ring substrate to either n+1 or n+2 ring adducts in a selective fashion.
Graphical Abstract

Aliphatic rings are key structural elements in functional molecules ranging from pharmaceuticals and natural products to ligands and polymers.1 Accordingly, methods that enable direct interconversion between ring-size isomers are important technologies that can provide useful retrosynthetic disconnections and facilitate the evaluation of structure-function studies (Figure 1a and 1b). While appealing in principle, the development of general catalytic methods for ring-size modulation remains limited in practice, as successful transformations require orchestrating non-degenerate C−C bond-breaking and bond-forming events to occur in a controlled and sequential fashion. Moreover, conversion of readily accessible 5- and 6-membered ring starting materials to either medium- or small-ring compounds is often thermodynamically challenging due to a concomitant increase in ring-strain.2 Significant and enabling advances in this area have been realized by Dong, Zuo, Beckwith, and others,3 which have demonstrated the value of this strategy. However, the discovery and development of complementary methods are necessary to further expand the reach and generality of this approach.
Figure 1.
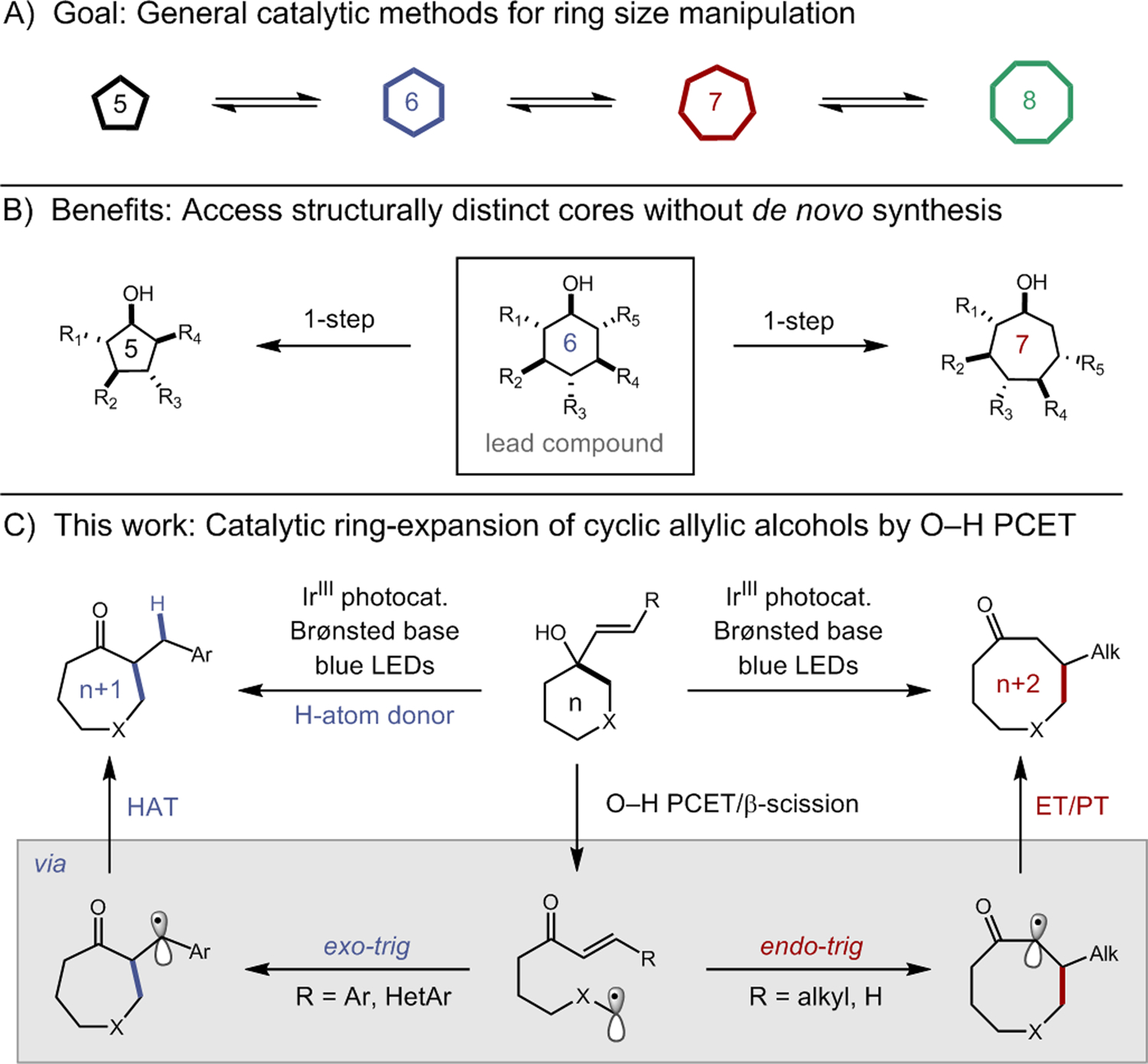
(a) Development of general methods for ring-size modulation; (b) access to structurally distinct cores via ring-size modulation; (c) PCET-enabled ring-expansion methods of cyclic alcohols.
In this context, we became interested in developing new ring-expansion methods based on our recent work in the ring-opening isomerization of cyclic alcohols.4a,4b In these reactions, proton-coupled electron transfer (PCET) activation of the strong O–H bond in an aliphatic alcohol substrate is jointly catalyzed by an excited-state oxidant and weak Brønsted base to form a key alkoxy radical intermediate. This O-centered radical can then mediate the β-scission of an adjacent C−C bond to furnish a new carbonyl group and a tethered alkyl radical, which can either be reduced by a thiol H-atom donor catalyst or trapped by a variety of other stoichiometric reagents. We questioned whether a similar series of elementary steps might be adapted for use in a ring-expansion protocol utilizing readily accessible cyclic allylic alcohol-based substrates derived from the addition of vinyl nucleophiles to simple ketones (Figure 1c). In this scheme, the alkoxy radical formed via excited-state PCET would undergo ring-opening β-scission to reveal a new α,β-unsaturated ketone and a tethered alkyl radical. The alkyl radical could then recombine with the revealed enone to form a new C−C bond within the framework of a ring-expanded carbocycle. By varying the regioselectivity of this olefin addition step, such a strategy could provide access to homologated products incorporating either both olefinic carbons (n+2 expansion) or only a single carbon (n+1 expansion) into the new ring structure. Such methods would provide a useful complement to classical radical ring expansion reactions developed by Beckwith, Dowd and Saegusa.3m–3q Here we report the successful development of both variants, providing a modular means to take simple and readily accessible cyclic alcohols directly to medium-ring ketones through a common PCET-based mechanism. The design, optimization, and scope of these processes are presented below.
As depicted in Figure 1c, we envisioned a prospective mechanism for the n+2 ring expansion wherein excited-state O–H PCET and subsequent ring-opening β-scission would result in the formation of a nascent enone acceptor and tethered alkyl radical. Radical 1,4-conjugate addition between these partners would lead to the formation of a new ring structure that incorporates both carbons of the exocyclic olefin. Following C−C bond formation, the resulting α-acyl radical can be reduced by the Ir(II) state of the photocatalyst to furnish an enolate intermediate and then protonated by the conjugate acid of the Brønsted base catalyst to close the cycle.
Our initial efforts in n+2 ring expansion focused on piperidine-based alcohol substrate 1. Adapting previously established O−H PCET conditions, we were pleased to find that treatment of 1 with 3 mol% of [Ir(dF(CF3)ppy)2(5,5′-d(CF3)bpy)]PF6 (A) and 40 mol% of a diphenyl phosphate base under blue light irradiation in CH2Cl2 directly afforded the 8-membered ring ketone 1a in 27% yield (Table 1, entry 1).4b Further optimization revealed that aromatic solvents (entries 4–7) were generally more effective than non-aromatic solvents (entries 1–3), with α,α,α-trifluorotoluene and toluene proving optimal (entries 6, 7). In addition, we found that the use of trifluoroacetate as the Brønsted base for the PCET event (entry 8) was more effective than the use of diaryl or dialkyl phosphates despite their comparable basicities (entries 7, 9, 10).5 Interestingly, the counter-cation of the anionic base was also found to influence reaction performance, with PBu4+(PhO)2P(O)O– (entry 7) and PBu4+CF3CO2– (entry 8) outperforming NBu4+(PhO)2P(O)O– (entry 11) and NBu4+CF3CO2– (entry 12), respectively – an outcome that may be due to the improved solubility of the phosphonium salt in toluene. Finally, control experiments indicated that there was no conversion in the absence of photocatalyst or visible light irradiation (entries 13, 14) while low conversion (15%) was observed in the absence of the Brønsted base co-catalyst (entry 15). Notably, 1a is the sole product isomer detected by 1H-NMR analysis of the crude reaction mixture, indicating that both β-scission of the alkoxy radical and the subsequent 1,4-conjugate addition step are highly regioselective.
Table 1.
Optimization studiesa

|
Optimization reactions were performed on 0.05 mmol scale.
Internal temperature of the reaction mixture under LED irradiation (see SI for details).
Yields were determined by 1H NMR analysis of crude reaction mixtures relative to an internal standard.
With these optimized conditions in hand, we next set out to investigate the generality of the n+2 ring expansion (Table 2). On preparative scale, a variety of alkene partners, including terminal olefins (1), 1,2- disubstituted olefins (2, 3, and 4), trisubstituted olefins (5, 6), and allenes (9) were all well tolerated. Similarly, various ring substitution patterns were also accommodated, including 4-methylated piperidine (7, 8), hexahydropyrimidine (10), N-Cbz piperidine (11), 2-azabicyclo[2.2.1]heptane (12), azepane (13), 1,4-diazepane (14), and 2-aminocyclohexanol (15, 16). Notably, all the products presented in Table 2 are formed as single regioisomers, consistent with a selective β-scission event to form more stabilized α-amino radical intermediates. In reactions forming diastereomeric products, (5, 6, and 7) high stereoselectivities were observed in enolate protonation (d.r. >20:1) – a likely consequence of the strong conformational preferences of 8-membered ring ketone enolates as originally suggested by Still.6 In the case of allene derivative 9, non-conjugated olefin 9a was the only product detected, suggesting a highly α-regioselective protonation of the intermediate 1,3-dienolate, in accord with literature precedent.7 While this reaction functions well with aza-heterocyclic substrates, n+2 ring expansions of analogous oxygen heterocycles and simple carbocyclic alcohols proved less efficient. As prior work indicates that the nitrogen substituent is not required for efficient β-scission,4a this observation may suggest that these aza-cyclic substrates are privileged as a result of the enhanced nucleophilicity of the α-amino radical in the conjugate addition step. In terms of applications, the aminoketone heterocycles produced in this reaction have been used previously as starting points for the synthesis of pyrrolizidine and indolizidine structures via transannular reductive amination.8 As such, we are optimistic that these methods will provide a simple and modular approach to the syntheses of this prominent family of alkaloid targets.9
Table 2.
Scope of n+2 ring expansiona

|
Reactions were run on 0.5 mmol scale. Reported yields are for isolated and purified material and represent the average of two experiments. Diastereomeric ratios were determined by 1H NMR of the crude reaction mixtures.
PhMe was used as the reaction solvent.
PhCF3 was used as the reaction solvent.
Relative configuration was determined by X-ray crystallographic analysis (see SI for details).
5 mol% photocatalyst and 0.1 M concentration were used.
PBu4+ (PhO)2P(O)O– (25 mol%) was used as the base.
We next questioned whether we could adapt this method to achieve a complementary n+1 type ring expansion. Based on recent work from Sparling and Lovett, we anticipated that the regioselectivity of the C–C bond forming step might be reversed upon incorporation of an aryl group at the terminal position of the substrate olefin (Figure 2a).10 These authors demonstrated that intermolecular addition of α-amino radicals to cinnamates occurs regioselectively at the olefinic carbon proximal to the carbonyl to form stabilized benzylic radical intermediates. Accordingly, we reasoned that reactions employing cinnamyl analogs of 1 might alter the preferred cyclization mode from 8-endo to 7-exo, delivering a 7-membered cyclic ketone product through a formal n+1 ring expansion.
Figure 2.

(a) Unconventional regioselectivity in radical additions to cinnamates. (b) Results of competition experiments.
However, the viability of this approach was complicated by the fact that the Ir(II) state of photocatalyst A is insufficiently reducing to transfer an electron to the benzylic radical (E1/2 [IrIII/IrII] = −1.07 V vs Fc+/Fc in MeCN; E1/2 [PhCH2•/PhCH2–] = −1.92 V vs Fc+/Fc in MeCN) and close the catalytic cycle.4a,11 To overcome this issue, we proposed to introduce an aryl thiol co-catalyst to the reaction, which could reduce the benzylic radical intermediate via H-atom transfer to deliver the desired n+1 product. The resulting aryl thiyl radical could then be reduced to its corresponding thiolate by the Ir(II) state of A (E1/2[ArS•/ArS–] = −0.22 V vs Fc+/Fc in MeCN; E1/2[IrIII/IrII] = −1.07 V vs Fc+/Fc in MeCN).12 This aryl thiolate could then be protonated by the conjugate acid of the Brønsted base to return the active forms of all three catalysts.4c
The success of this proposal hinged on the ability of the proposed exo-trig cyclization to kinetically outcompete bimolecular HAT reduction of the alkyl radical initially formed in the β-scission event by the thiophenol co-catalyst – a typically rapid elementary step (k ~ 108 M–1s–1) (Figure 2b).13 To probe this question directly, we conducted a competition experiment using our n+2 model substrate 1 in the presence of 25 mol% 2,4,6-triisopropyl thiophenol (TRIP-SH) under otherwise standard conditions, wherein the ratio of cyclized to uncyclized products (1a and 1b, Figure 2b) would report on the kinetic partitioning of the key alkyl radical intermediate. Gratifyingly, under standard conditions cyclized product 1a was obtained in 60% yield, while the product resulting from HAT reduction of the alkyl radical intermediate (1b) was formed in 23% yield. Formation of the acyclic product could be further suppressed by lowering the loading of TRIP-SH to 5 mol% to provide 1a in 88% yield along with 11% of 1b.
Adapting these findings to the proposed n+1 ring expansion, we were pleased to find that treatment of styrene derivative 17 under standard n+2 conditions (Table 1, entry 8) with the addition of 5 mol% of TRIP-SH provided the desired n+1 product 17a in 80% yield and with only trace amounts of the acyclic side product resulting from premature HAT (Table 3, entry 18). This encouraging result led us to examine the scope of n+1 ring expansion in greater detail. On preparative scale, we found that 5- to 7-membered N-heterocycles underwent efficient ring expansion (17–22), and the reaction tolerates aryl substituents with varying electronic character (17–19). Spirocyclic ketone 21a could be accessed from 2-indenyl derivative 21 in good yield. Meanwhile, olefin substrates bearing β-electron withdrawing groups, including ketones (23) and electron deficient N-heterocycles, such as 2-pyridyl (24) and 2-pyrimidinyl (25), were also found to be efficient for n+1 ring expansion. In these cases, the intrinsic selectivity of ring closure favors 7-exo over 8-endo, and the HAT reagent is not required as direct reduction of the radical intermediate by the Ir(II) state of photocatalyst A is viable. This n+1 protocol also tolerates both 5- and 6-membered cyclic ether substrates (26, 27) as well as a variety of carbocyclic substrates (28–31). Of particular note, we discovered that simple derivatives of (+)-camphor (30) and trans-androsterone (31) underwent highly regioselective and diastereoselective n+1 expansion, suggesting that this method may prove useful in the preparation of non-natural terpenoid derivatives that might prove more difficult to access by other means.14 However, unsubstituted 1-alkenylcycloalkanols are not efficient substrates in this chemistry, presumably as β-scission is not kinetically competitive with charge recombination between the alkoxy radical and the reduced Ir(II) state of the photocatalyst. Lastly, during the development of this n+1 process, we observed that reaction efficiency was sensitive to the identity of the solvent, the reaction concentration, the structure and loading of the Brønsted base and HAT co-catalysts, and reaction temperature. As such, any variation from the standard protocol are listed in the footnotes to Table 3.
Table 3.
Scope of n+1 Ring Expansiona

|
Reactions run on 0.5 mmol scale. Reported yields are for isolated and purified material and are the average of two experiments. Diastereomeric ratios were determined by 1H NMR of the crude reaction mixtures.
PhCF3 was used as the reaction solvent.
PBu4+ CF3CO2– (25 mol%) as the base.
PhMe was used as the reaction solvent.
20 mol% of TRIP-SH was used.
Collidine (3 equiv.) as the base.
Without TRIP-SH.
Relative or absolute configuration was determined by NOESY analysis (see SI for details).
Relative configuration was determined by X-ray crystallographic analysis (see SI for details).
See SI for detailed reaction conditions.
In conclusion, we have developed a new method for the construction of valuable medium-sized ring ketones from readily accessible cyclic allylic alcohols through catalytic ring expansion. The key C–C bond-breaking β-scission event is enabled by excited-state PCET activation of a strong alcohol O−H bond, while the subsequent ring closure can selectively deliver either n+1 or n+2 ring-expanded products by varying the substitution pattern of the olefin acceptor. We note that the outcomes presented here stand in contrast to several previous studies on alkoxy radicals derived from 1-vinylcycloalkanols, where spiroepoxide products are formed predominantly.15 Future efforts will seek to understand the factors leading to this unexpected selectivity for β-scission. These reactions further highlight the ability of excited-state redox events to drive challenging isomerization chemistry under mild catalytic conditions and provide further support for the synthetic utility of PCET-based C–C bond functionalization.
Supplementary Material
ACKNOWLEDGMENT
Funding for the work was provided by the NIH (NIGMS R01 113105) and Bristol-Myers Squibb. K.Y. thanks the Uehara Memorial Foundation for a postdoctoral fellowship.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Experimental details, characterization data, and spectra (PDF). Crystallographic data for 5a (CIF).
Crystallographic data for 6a (CIF).
Crystallographic data for 7a (CIF).
Crystallographic data for 31a (CIF).
The authors declare no competing financial interests. Crystallographic data are deposited with the Cambridge Crystallographic Data Centre (CCDC) under the following accession numbers: 5a (1900667), 6a (1900665), 7a (1900666), and 31a (1900668).
REFERENCES
- (1).(a) Stephen JC; Cutler HG; Biologically Active Natural Products: Pharmaceuticals; CRC Press: Boca Raton, FL, 2000. [Google Scholar]; (b) Mander LN Comprehensive Natural Products II: Chemistry and Biology; Elsevier: Amsterdam, 2010. [Google Scholar]; (c) Allen G; Bevington JC; Booth C; Price C Comprehensive Polymer Science: The Synthesis, Characterization, Reactions & Applications of Polymers; Pergamon: Oxford, 1989. [Google Scholar]
- (2).Galli C; Mandolini L The Role of Ring Strain on the Ease of Ring Closure of Bifunctional Chain Molecules. Eur. J. Org. Chem 2000, 3117–3125. [Google Scholar]
- (3).(a) Sun T; Zhang Y; Qiu B; Wang Y; Qin Y; Dong G; Xu T Rhodium(I)-Catalyzed Carboacylation/Aromatization Cascade Initiated by Regioselective C−C Activation of Benzocyclobutenones. Angew. Chem. Int. Ed 2018, 57, 2859–2863. [DOI] [PubMed] [Google Scholar]; (b) Deng L; Jin L; Dong G Fused-Ring Formation via an Intramolecular “Cut-and-Sew” Reaction between Cyclobutanones and Alkynes. Angew. Chem. Int. Ed 2018, 57, 2702–2706. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhou X; Dong G Nickel-Catalyzed Chemo- and Enantioselective Coupling between Cyclobutanones and Allenes: Rapid Synthesis of [3.2.2] Bicycles. Angew. Chem. Int. Ed 2016, 55, 15091–15095. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Xia Y; Lu G; Liu P; Dong G Catalytic Activation of Carbon–carbon Bonds in Cyclopentanones. Nature, 2016, 539, 546–550. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Deng L; Xu T; Li H; Dong G Enantioselective Rh-catalyzed Carboacylation of C=N Bonds via C−C Activation of Benzocyclobutenones. J. Am. Chem. Soc 2016, 138, 369–374. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Xu T; Dong G Rh-Catalyzed Regioselective Carboacylation of Olefins: A C−C Activation Approach to Access Fused-Ring Systems.” Angew. Chem., Int. Ed 2012, 51, 7567–7571. [DOI] [PubMed] [Google Scholar]; (g) Hu A; Chen Y; Guo J; Yu N; An Q, Zuo Z Cerium-Catalyzed Formal Cycloaddition of Cycloalkanols with Alkenes through Dual Photoexcitation. J. Am. Chem. Soc 2018, 140, 13580–13585. [DOI] [PubMed] [Google Scholar]; (h) Jin S; Nguyen VT; Dang HT; Nguyen DP; Arman HD; Larionov OV Photoinduced Carboborative Ring Contraction Enables Regio- and Stereoselective Synthesis of Multiply Substituted Five-Membered Carbocycles and Heterocycles. J. Am. Chem. Soc 2017, 139, 11365–11368. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Wang N; Gu Q; Li Z; Guo Y; Kiu X Angew. Chem., Int. Ed 2018, 57, 14225–14229. [DOI] [PubMed] [Google Scholar]; (j) Chen P; Billett BA; Tsukamoto T; Dong G “Cut and Sew” Transformations via Transition-Metal-Catalyzed Carbon–Carbon Bond Activation. ACS Catal. 2017, 7, 1340–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Fumagalli G, Stanton S, Bower JF, Recent Methodologies That Exploit C−C Single-Bond Cleavage of Strained Ring Systems by Transition Metal Complexes. Chem. Rev 2017, 117, 9404–9432. [DOI] [PubMed] [Google Scholar]; (l) Donald JR, Unsworth WP, Ring-Expansion Reactions in the Synthesis of Macrocycles and Medium-Sized Rings. Chem. Eur. J 2017, 23, 8780–8799. [DOI] [PubMed] [Google Scholar]; (m) Dowd P; Choi S-C,. A New Tri-n-butyltin Hydride Based Rearrangement of Bromomethyl β-Keto Esters. A Synthetically Useful Ring Expansion to γ-Keto Esters. J. Am. Chem. Soc, 1987, 109, 3493–3494. [Google Scholar]; (n) Dowd P; Choi S-C Free Radical Ring Expansion by Three and Four Carbons. J. Am. Chem. Soc 1987, 109, 6548–6549. [Google Scholar]; (o) Beckwith ALJ; O’Shea DM; Wetswood SW Rearrangement of Suitably Constituted Aryl, Alkyl, or Vinyl Radicals by Acyl or Cyano Group Migration. J. Am. Chem. Soc 1988, 110, 2565–2572. [Google Scholar]; (p) Ito Y; Fujii S; Saegusa T Reaction of 1-Silyloxybicyclo[n.1.0] alkanes with Iron(III) Chlorides. A Facile Synthesis of 2-Cycloalkenones via Ring Enlargement of Cyclic Ketones. J. Org. Chem 1976, 41, 2073–2074. [Google Scholar]; (q) Dowd P; Zhang W Free Radical-Mediated Ring Expansion and Related Annulations. Chem. Rev 1993, 93, 2091–2115. [Google Scholar]; (r) Song Z; Fan C; Tu Y Semipinacol Rearrangement in Natural Product Synthesis. Chem. Rev 2011, 111, 7523–7556. [DOI] [PubMed] [Google Scholar]
- (4).(a) Ota E; Wang H; Frye NL; Knowles RR A Redox Strategy for Light-Driven, Out-of-Equilibrium Isomerizations and Application to Catalytic C–C Bond Cleavage Reactions. J. Am. Chem. Soc 2019, 141, 1457–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yayla HG; Wang H; Tarantino KT; Orbe HS; Knowles RR Catalytic Ring-Opening of Cyclic Alcohols Enabled by PCET Activation of Strong O–H Bonds. J. Am. Chem. Soc 2016, 138, 10794–10797. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhu Q; Graff DE; Knowles RR Intermolecular Anti-Markovnikov Hydroamination of Unactivated Alkenes with Sulfonamides Enabled by Proton-Coupled Electron Transfer. J. Am. Chem. Soc, 2018, 140, 741–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).pKa (trifluoroacetic acid) = 12.65 in MeCN; pKa (BINOL/diphenyl phosphoric acid) = 12–13 in MeCN, see:; (a) Muckerman JT; Skone JH; Ning M;. Wasada-Tsutsui, Y. Toward the accurate calculation of pKa values in water and acetonitrile. Biochimica et Biophysica Acta. 2013, 1827, 882–891. [DOI] [PubMed] [Google Scholar]; (b) Rueping M; Nachtsheim BJ; Ieawsuwan W; Atodiresei I Modulating the Acidity: Highly Acidic Brønsted Acids in Asymmetric Catalysis. Angew. Chem., Int. Ed, 2011, 50, 6706–6720. [DOI] [PubMed] [Google Scholar]
- (6).Still WC; Galynker I Chemical consequences of conformation in macrocyclic compounds: An effective approach to remote asymmetric induction. Tetrahedron 1981, 37, 3981–3996. [Google Scholar]
- (7).For α-regioselective dienolate electrophilic reactions, see:; (a) Cargill RL; Bushey DF; Good JJ Alkylation of 1-cyanocyclohexene. J. Org. Chem, 1979, 44, 300–301. [Google Scholar]; (b) Gesson J; Jacuesy J; Mondon M Regioselective dienolate formation and alkylation of alkyl 2-methyl cyclohexene-1 carboxylate. Tetrahedron Lett. 1980, 21, 2509–2512. [Google Scholar]; (c) Davies SG; Easton RJC; Sutton KH; Walker JC; Jones RH Chiral Dienolates: Formation and Stereoselective α-Alkylation of the Lithium Dienolate Derived from (Z)-[(η5-C5H5)Fe(CO)(PPh3)COCH=CHMe]. X-Ray Crystal Structure of (RS)-(Z)-[(η5-C5H5)Fe(CO)(PPh3)COCH=CH Me]. J. Chem. Soc. Perkin Trans 1 1987, 489–493. [Google Scholar]
- (8).(a) Garst ME; Bonfiglio JN; Marks J Hydroboration-carbon monoxide insertion of bis-olefinic amine derivatives. Synthesis of .delta.-coniceine, pyrrolizidine, (±)-heliotridane, and (±)-pseudoheliotridane. J. Org. Chem 1982, 47, 1494–1500. [Google Scholar]; (b) Garst ME; Bonfiglio JN Hydroboration-carbonylation of bisolefinic amines: a facile synthesis of δ-coniceine. Tetrahedron Lett., 1981, 22, 2075–2076. [Google Scholar]
- (9).(a) Roberston J; Stevens K Pyrrolizidine alkaloids. Nat. Prod. Rep, 2014, 31, 1721–1788. [DOI] [PubMed] [Google Scholar]; (b) Michael JP Indolizidine and Quinolizidine Alkaloids. Nat. Prod. Rep, 2008, 25, 139–165. [DOI] [PubMed] [Google Scholar]
- (10).Lovett GH; Sparling BA Decarboxylative Anti-Michael Addition to Olefins Mediated by Photoredox Catalysis. Org. Lett, 2016, 18, 3494–3497. [DOI] [PubMed] [Google Scholar]
- (11).Sim BA; Griller D; Wayner DDM Reduction potentials for substituted benzyl radicals: pKa values for the corresponding toluenes. J. Am. Chem. Soc, 1989, 111, 754–755. [Google Scholar]
- (12).Romero N; Nicewicz DA Mechanistic Insight into the Photoredox Catalysis of Anti-Markovnikov Alkene Hydrofunctionalization Reactions. J. Am. Chem. Soc, 2014, 136, 17024–17035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Dénès F; Pichowicz M; Povie G; Renaud P Thiyl Radicals in Organic Synthesis. Chem. Rev 2014, 114, 2587–2693. [DOI] [PubMed] [Google Scholar]
- (14).Urabe D; Asaba T; Inoue M Convergent Strategies in Total Syntheses of Complex Terpenoids. Chem. Rev 2015, 115, 9207–9231. [DOI] [PubMed] [Google Scholar]
- (15).(a) Galatsis P; Millan SD; Faber T 1-Alkenylcycloalkoxy Radical Chemistry. A Two-Carbon Ring Expansion Methodology. J. Org. Chem 1993, 58, 1215–1220. [Google Scholar]; (b) Galatsis P; Millan SD Use of Iodobenzene Diacetate for the Synthesis of α-Iodoepoxides. Tetrehedron. 1991, 51, 7493–7496. [Google Scholar]; (c) Aureliano Antunes CS; Bietti M; Lanzalunga O; Salamone M Photolysis of 1-Alkylcycloalkanols in the Presence of (Diacetoxyiodo)benzene and I2. Intramolecular Selectivity in the β-Scission Reactions of the Intermediate 1-Alkylcycloalkoxyl Radicals. J. Org. Chem 2004, 69, 5281–5289. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.