

Abstract
The ability of skeletal muscle to switch between lipid and glucose oxidation for ATP production during metabolic stress is pivotal for maintaining systemic energy homeostasis, and dysregulation of this metabolic flexibility is a dominant cause of several metabolic disorders. However, the molecular mechanism that governs fuel selection in muscle is not well understood. Here, we report that brain-derived neurotrophic factor (BDNF) is a fasting-induced myokine that controls metabolic reprograming through the AMPK/CREB/PGC-1α pathway in female mice. Female mice with a muscle-specific deficiency in BDNF (MBKO mice) were unable to switch the predominant fuel source from carbohydrates to fatty acids during fasting, which reduced ATP production in muscle. Fasting-induced muscle atrophy was also compromised in female MBKO mice, likely a result of autophagy inhibition. These mutant mice displayed myofiber necrosis, weaker muscle strength, reduced locomotion, and muscle-specific insulin resistance. Together, our results show that muscle-derived BDNF facilitates metabolic adaption during nutrient scarcity in a gender-specific manner and that insufficient BDNF production in skeletal muscle promotes the development of metabolic myopathies and insulin resistance.
INTRODUCTION
As the largest tissue that comprises about 40% of the total body mass, skeletal muscle is a major player in regulating glucose uptake, lipid storage, and energy balance (1). A key metabolic feature of skeletal muscle is its plasticity in adjusting myocellular fuel choice according to different nutritional circumstances, a phenomenon called “metabolic flexibility” (2). To spare the limited amounts of glucose for organs that rely heavily on glucose metabolism for synthesizing ATP during fasting or exercise, skeletal muscle increases fatty acid (FA) uptake and switches from the preferred glycolysis to FA oxidation (FAO) for ATP generation. Loss of this flexibility is tightly associated with metabolic disorders such as insulin resistance and obesity (3). As proposed by Randle et al. (4), the underlying mechanism of this metabolic adaptation is controlled biochemically by the high intracellular concentration of FAO byproducts that allosterically inhibit glycolytic enzymes and thus slow down glycolytic flux. This “glucose-FA cycle” set the foundation for breakthroughs in understanding the adaptation responses; however, regulation of fuel utilization in muscles is now known to be far more complex than originally thought (5). Because skeletal muscle is an endocrine organ that actively produces myokines to regulate systemic energy metabolism in response to various challenges (6), it is reasonable to assume that some myokines may play a role in regulating metabolic plasticity in skeletal muscle.
Brain-derived neurotrophic factor (BDNF) is a member of the neurotrophin family that plays important roles in synaptic plasticity, neuronal survival, development, and differentiation (7). By binding to its cognate receptor TrkB, BDNF triggers TrkB autophosphorylation, which activates several signaling pathways including phosphoinositide 3 kinase (PI3K)/Akt, Ras/extracellular signal-regulated kinase (ERK), and phospholipase Cγ (PLCγ)/cAMP responsive element binding protein (CREB) pathways (8). BDNF is also a metabolic hormone that inhibits food intake (9, 10). Because selective depletion of Bdnf in neurons of ventral medial hypothalamus or dorsomedial hypothalamus is sufficient to induce hyperphagia and increase body weight gain in mice, the hypothalamus is believed to be the major target of BDNF in controlling appetite (11). Therefore, most research on BDNF/TrkB signaling has mainly focused on its role in the central nervous system (CNS) — although Bdnf and Ntrk2 mRNA is readily detected in peripheral tissues (12) and BDNF regulates lipid catabolism in cultured L6 muscle cells (13). Together with the robust increase in Bdnf expression in muscles after exercise (14, 15), these observations prompted us to investigate whether muscle-derived BDNF plays a role in controlling energy metabolism during periods that require metabolic transition. In this report, we revealed that BDNF acts as a myokine and reprograms skeletal muscle to cope with nutrient scarcity.
RESULTS
BDNF is a fasting-induced myokine in female mice
BDNF and its receptor TrkB can be detected in skeletal muscle (12, 16) but their function in this tissue is a long-standing and controversial question in the field. In search of physiological factors that regulate Bdnf expression in skeletal muscle, we found that Bdnf expression was significantly higher in the gastrocnemius muscle of female mice than in male animals (Fig. S1A). Moreover, 24-h fasting elevated Bdnf expression specifically in the gastrocnemius, extensor digitorum longus (EDL), and tibialis anterior (TA) muscles of female mice (Fig. 1A and Fig. S1B). However, we did not detect any changes in Bdnf expression in muscles collected from fasted male mice (Fig. 1A and Fig. S1B). In contrast, fasting upregulated Bdnf expression in the cortex of male and female mice (Fig. 1A), suggesting that Bdnf expression in response to nutrient stress is both sex- and muscle type-specific. Consistent with these findings, increased circulating BDNF level was detected in female mice only after 24 h of fasting (Fig. 1B). To further elucidate the mechanism of fasting-regulated Bdnf expression, we studied the effect of glucose depletion in C2C12 myotubes, a glycolytic muscle cell line that was isolated from a female mouse (17). Both Bdnf expression (Fig. 1C) and secretion (Fig. S1C) were increased when C2C12 myotubes were cultured in glucose-free medium. BDNF secretion from glucose-depleted C2C12 myotubes was further confirmed by immunoblotting (Fig. S1D), which was inhibited when Bdnf expression was depleted (Fig. S1E). We also found that forskolin or the cell-permeable cAMP analogue sp-cAMP stimulated Bdnf expression in C2C12 myotubes (Fig. 1D and Fig. S1F), suggesting that , Bdnf expression was regulated by CREB, the transcription factor that governs hepatic lipid and glucose metabolism during fasting (18, 19). Indeed, C2C12 myotubes overexpressing CREB displayed higher Bdnf expression (Fig. 1E). CREB expression, phosphorylation, and activity were also upregulated in glucose-depleted myotubes and gastrocnemius of fasted mice (Fig. 1, A, F and G). Moreover, CREB tethering to the CRE (5ʹ-TCACGTCA-3ʹ) on the Bdnf promoter was increased in glucose-depleted C2C12 myotubes (Fig. 1H). Our data thus suggest that BDNF is a CREB-regulated myokine in response to fasting.
Fig. 1.
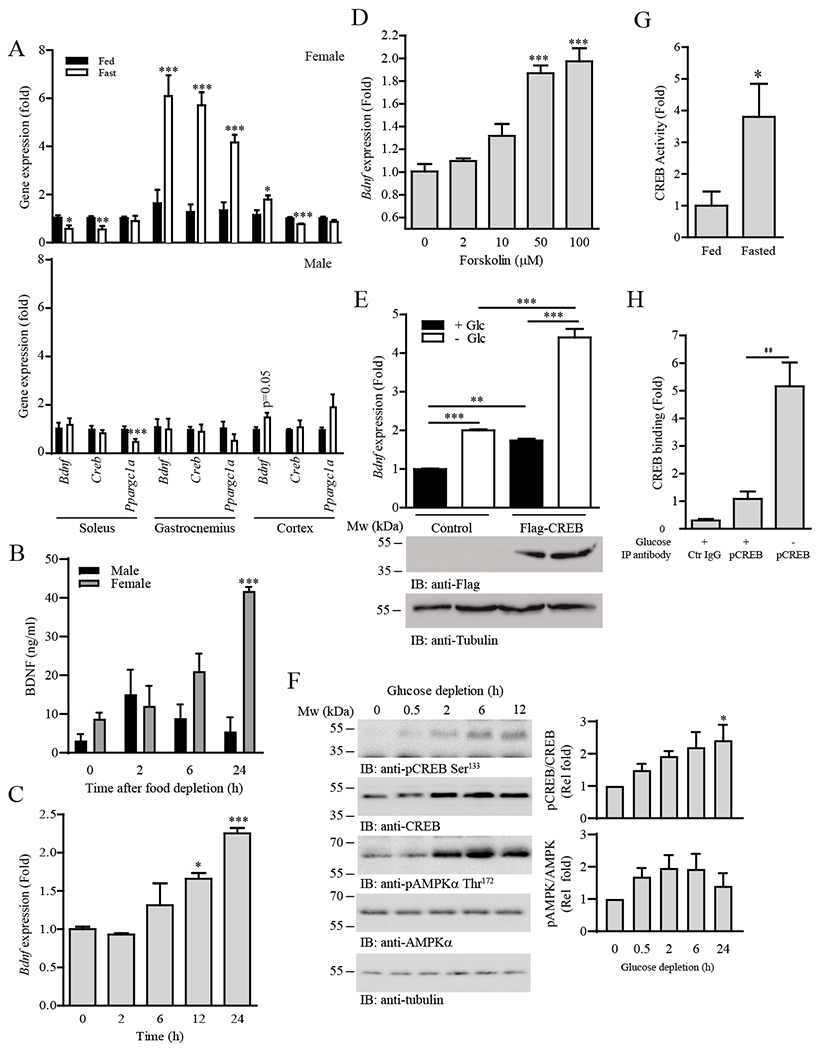
Fasting induces Bdnf expression in glycolytic muscle through CREB signaling.
(A) Real-time PCR of Bdnf, Creb, and Ppargc1a (which encodes PGC-1α) expression in various mouse tissues after fasting (24 h); n = 4–5 mice per group. *P < 0.05, ***P < 0.001 compared to fed; Student’s t-test.
(B) Circulating BDNF concentration in wild-type mice after fasting for various time intervals; n = 5–6 mice per group. ***P < 0.001 compared to 0 h of the same sex; one-way ANOVA.
(C) Bdnf expression in C2C12 myotubes after glucose depletion for various time intervals; n = 4 independent experiments. *P < 0.05, ***P < 0.001 compared to 0 h; one-way ANOVA.
(D) Real-time PCR of Bdnf expression in C2C12 myotubes stimulated with different concentrations of forskolin for 24 h; n = 4 independent experiments. ***P < 0.001 compared to 0 µM; one-way ANOVA.
(E) Real-time PCR (top panel) of Bdnf expression in C2C12 myotubes transfected with empty vector (Control) or FLAG-tagged CREB (Flag-CREB) and cultured in glucose-free medium (24 h). The abundance of FLAG-CREB and tubulin were determined by immunoblotting (middle and bottom panels); n = 3 independent experiments. **P < 0.01, ***P < 0.01; two-way ANOVA.
(F) C2C12 myotubes were cultured in glucose-free medium for the indicated time points and immunoblotted for phosphorylated CREB and AMPK and total CREB, AMPKα, and tubulin. The immunoblots are representative of three independent experiments. Quantification is shown in the right panel. *P < 0.05 compared to 0 h; one-way ANOVA.
(G) Wild-type mice were fasted for 24 h. Gastrocnemius muscles were collected and the DNA binding activity of CREB was examined by ELISA; n = 4 mice per group. *P < 0.05; Student’s t-test.
(H) Genomic DNA and associated proteins were collected from C2C12 myotubes cultured in normal or glucose-free medium for 24 h. ChIP assay was performed using control IgG (Ctr IgG) or anti-CREB, and the associated Bdnf promoter was detected using real-time PCR; n = 4 independent experiments. **P < 0.01; Student’s t-test.
BDNF increases mitochondrial respiration and lipid oxidation in muscle cells
The limited nutrient supply during fasting provokes a compensatory increase in FAO in skeletal muscle to preserve glucose for tissues that rely heavily on glucose metabolism for ATP production (20). Given that Bdnf expression increased during fasting (Fig. 1A), muscle-derived BDNF may serve as a modulator of lipid oxidation in muscles. As expected, BDNF treatment promoted FAO in C2C12 myotubes in a dose-dependent manner (Fig. 2A). This increase in β-oxidation was potentially due to increased mitochondrial content because we detected a higher level of mitochondrial proteins [cytochrome c (Cyto c), succinate dehydrogenase (SDH), and pyruvate dehydrogenase (PDH)] (Fig. 2B) and DNA (mtDNA; Fig. 2C) in BDNF-treated C2C12 myotubes. In agreement, cellular mitochondrial activities including oxygen consumption rate (OCR; Fig. 2D), basal and maximal mitochondrial respiration (Fig. 2, E and F), and ATP production (Fig. 2G) were all elevated in the BDNF-stimulated cells. In contrast, non-mitochondrial respiration and ATP coupling efficiency were not altered after BDNF stimulation (Fig. S2, A and B), further suggesting that increased respiration was caused by an increase in mitochondrial content. Consistent with this notion, BDNF stimulation of C2C12 myotubes increased the activity of AMP-activated protein kinase (AMPK) and the expression of peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α)—key regulators of mitochondrial biogenesis (21) (Fig. 2H). Phosphorylation of acetyl CoA carboxylase (ACC) (Fig. 2H) was increased, further supporting the enhanced FAO in BDNF-stimulated myotubes (Fig. 2A). BDNF also enhanced CREB activity, as evidenced by augmented Ser133 phosphorylation, which correlated with the increased expression of Nur77, a transcription factor that promotes FAO (22) (Fig. 2H). Expression of another key transcriptional regulator that enhances transcription of β-oxidative genes, peroxisome proliferator-activated receptor α (PPARα), was also enhanced in BDNF-treated C2C12 myotubes (Fig. 2H). BDNF did not robustly induce Akt activation in muscle cells (Fig. 2H), in contrast to its effect in neurons (23). These results indicate that compared to neurons, BDNF utilizes distinct signaling cascades in skeletal muscle cells to increase mitochondrial content, promote cellular respiration, and induce FAO.
Fig. 2.
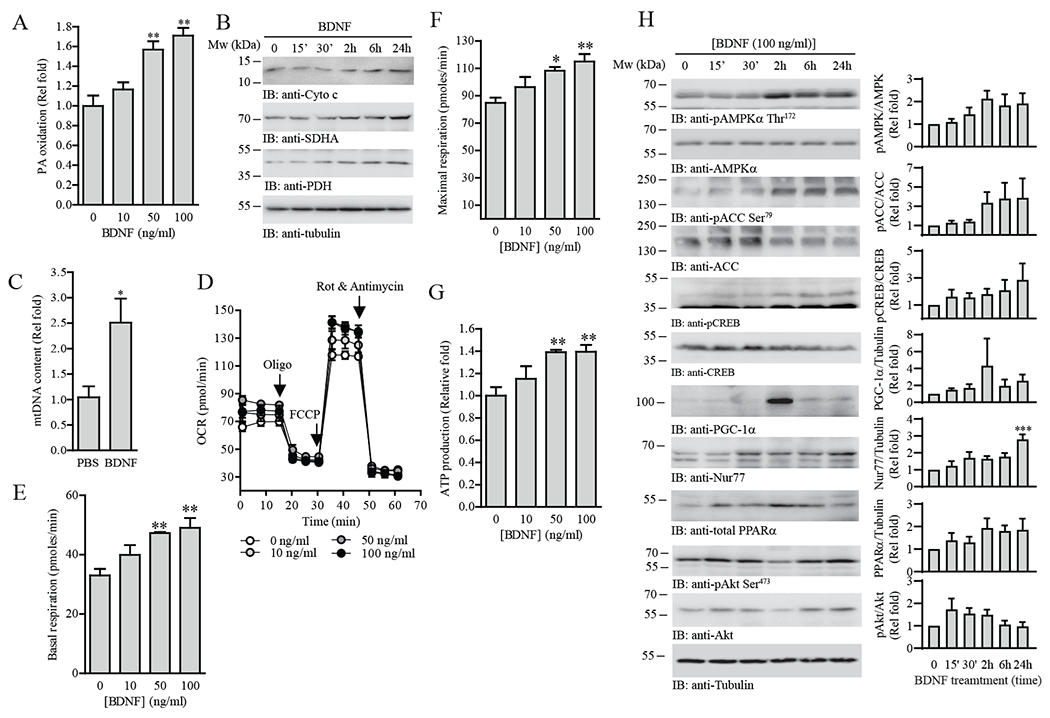
BDNF increases mitochondrial content, cellular respiration, and lipid oxidation in muscle cells.
(A) Palmitic (PA) oxidation in C2C12 myotubes treated with the indicated concentrations of BDNF for 24 h was measured with an extracellular flux analyzer; n = 4 independent experiments. **P < 0.01 compared to 0 ng/mL; one-way ANOVA.
(B) Immunoblotting of the mitochondrial protein content in C2C12 myotubes treated with BDNF (100 ng/mL) for the indicated time points. Immunoblots are representative of two independent experiments.
(C) Real-time PCR of mitochondrial DNA in C2C12 myotubes treated with BDNF (100 ng/mL, 24 h). Results were normalized to genomic DNA content; n = 3 independent experiments. *P < 0.05; Student’s t-test.
(D) OCR traces for BDNF-stimulated (24 h) C2C12 myotubes; n = 4 independent experiments. Oligo, oligomycin. Rot, rotenone. (E) Basal respiration, (F) maximal respiration, and (G) ATP production of C2C12 myotubes treated with different concentrations of BDNF (10–100 ng/mL) for 24 h; n = 4 independent experiments. *P < 0.05, **P < 0.01 compared to 0 ng/mL; one-way ANOVA.
(H) Analysis of various signaling pathways in C2C12 myotubes stimulated by BDNF (100 ng/mL) for the indicated time points. The phosphorylated and total abundance of the indicated proteins was determined by immunoblotting. Immunoblots are representative of three independent experiments. Quantification is shown in the right panel; n = 3 experiments. *P < 0.05 compared to 0 h; one-way ANOVA.
Muscle-specific Bdnf knockout mice display impaired energy metabolism
To confirm the importance of muscle-derived BDNF in energy metabolism, especially during the fed-fasting transition, we generated skeletal muscle-specific Bdnf knockout (MBKO) mice using LoxP-Cre recombination (Fig. S3A). Real-time PCR indicated a significant reduction of Bdnf expression in the skeletal muscles, which was not observed in other tissues in MBKO mice (Fig. S3B). In addition, TrkB phosphorylation in the gastrocnemius muscle, but not in the cortex, was significantly reduced, demonstrating tissue-specific ablation of BDNF/TrkB signaling (Fig. S3C). Although body weight and composition did not differ between male MBKO and control Fl/Fl mice (Fig. S4, A and B), female MBKO mice possessed greater body weight and white adipose tissue (WAT) mass (Fig. 3A–C and Fig. S3D), indicating that muscle-secreted BDNF has a sex-specific effect on body weight regulation. These female mutant mice had normal food intake (Fig. S3E) but decreased systemic metabolism (reduced O2 consumption, CO2 production, and energy expenditure) and physical activity (Fig. 3D–G). Brown adipose tissue (BAT) of female MBKO mice showed greater expression of Dio2 (which encodes iodothyronine deiodinase 2) (Fig. S5A), suggesting a local increase in energy expenditure (24). In contrast, the expression of genes that promote WAT browning – Cidea (which encodes cell death inducing DFFA like effector a), Fabp3 (which encodes fatty acid binding protein 3) and Prdm16 (which encodes PR domain containing 16) – was not altered in MBKO mice (Fig. S5B). During the day time rest period when food intake ceased, the skeletal muscle of Fl/Fl mice showed increased β-oxidation for ATP production (25), which was reflected by a decrease in respiratory exchange ratio (RER; Fig. 3H). However, such a decrease in RER was not detected in female MBKO mice (Fig. 3H), indicating that glycolysis was still the major biochemical reaction for ATP production. In support of this conclusion, the RER of MBKO mice was also higher after food deprivation (Fig. 3I and Fig. S5C). In contrast, male MBKO and Fl/Fl mice displayed comparable locomotion and energy metabolism under both fed and fasted states (Fig. S4C–H). These findings indicate that muscle-derived BDNF is a female-specific myokine that controls metabolic reprograming during fasting. Thus, only female animals were used in subsequent phenotypic and mechanistic analyses.
Fig. 3.
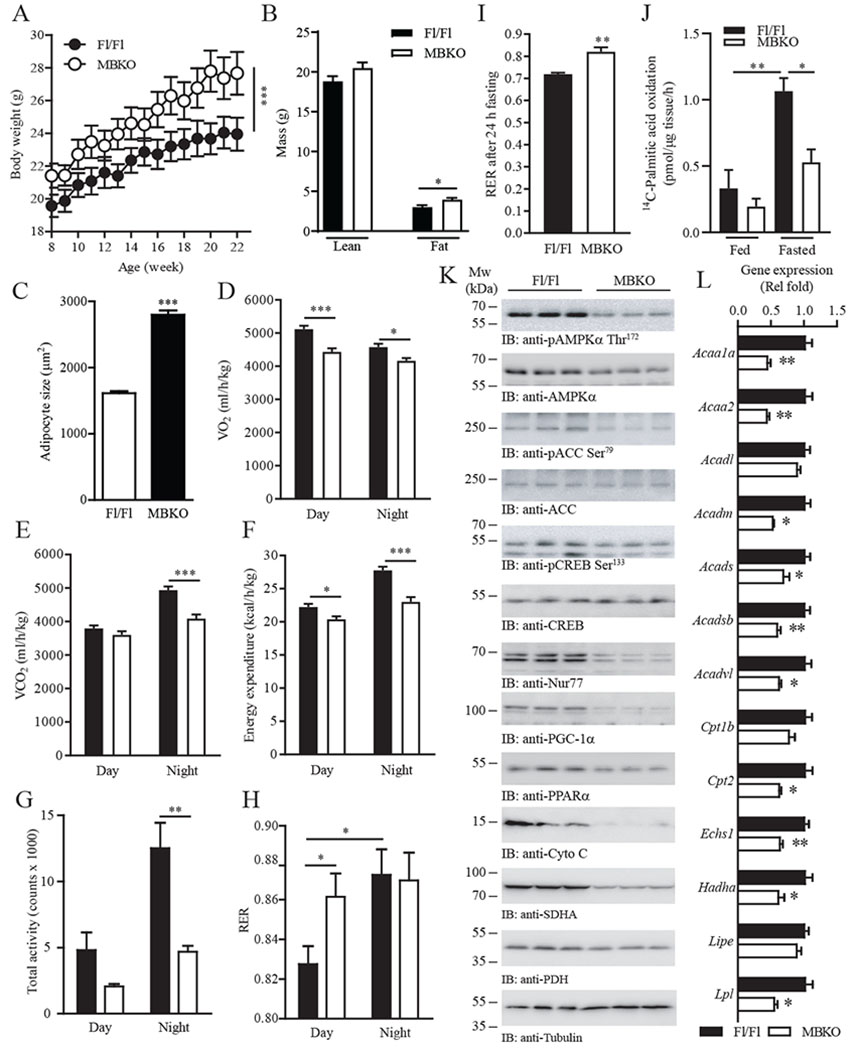
Deficiency in muscle-derived BDNF impairs systemic energy metabolism.
(A) Growth curve of female Fl/Fl and MBKO mice fed a chow diet; n = 10 mice per group. ***P < 0.01; two-way repeated measures ANOVA.
(B) Total lean and fat mass of 28-week-old female Fl/Fl and MBKO mice; n = 6 mice per group. *P < 0.01; Student’s t-test.
(C) Adipocyte size in inguinal WAT collected from 28-week-old female Fl/Fl and MBKO mice; n = 5–6 mice per group. ***P < 0.01; Student’s t-test.
(D to H) Daily oxygen consumption (VO2) (D), daily CO2 production (VCO2) (E), daily energy expenditure (F), daily locomotion (G), and average RER (H) of 28-week-old female MBKO mice; n = 6 mice per group. *P < 0.05, **P < 0.01, ***P < 0.001; Student’s t-test.
(I) RER of 28-week-old female Fl/Fl and MBKO mice after fasting (24 h); n = 5 mice per group. **P < 0.01; Student’s t-test.
(J) FAO in gastrocnemius muscle isolated from 10-month-old female Fl/Fl and MBKO mice that had been fed or fasted for 16 hours; n = 4 mice per group. *P < 0.05, **P < 0.01; two-way ANOVA.
(K) Metabolic signaling in female Fl/Fl and MBKO (28-week-old) gastrocnemius muscle after fasting (24 h) as determined by immunoblotting. Each lane represents a different mouse.
(L) Real-time PCR of FAO genes in the gastrocnemius muscle of fasted (24 h) female MBKO (28-week-old) mice; n = 4 mice per group. *P < 0.05, **P < 0.01; Student’s t-test. AcAA1a, encodes acetyl-CoA acyltransferase 1a; AcAA2, encodes acetyl-CoA acyltransferase 2; Acadl, encodes acyl-CoA dehydrogenase, long chain; Acadm, encodes acyl-CoA dehydrogenase, medium chain; Acads, encodes acyl-CoA dehydrogenase, short chain; Acadsb, encodes acyl-CoA dehydrogenase, short/branched chain; Acadvl, encodes acyl-CoA dehydrogenase, very long chain; Cpt1b, encodes carnitine palmitoyltransferase 1b; Cpt1b, encodes carnitine palmitoyltransferase 2; Echs1, encodes enoyl Co-A hydratase short chain 1; Hadha, encodes enoyl-CoA hydratase; Lipe, encodes hormone-sensitive lipase; Lpl, encodes lipoprotein lipase.
Consistent with the indirect calorimetry study, muscle from fasted MBKO mice displayed lower FAO (Fig. 3J). Immunoblotting analysis revealed impaired AMPK/ACC signaling (Fig. 3K and Fig. S5D), suggesting compromised FAO. The phosphorylation or abundance of the transcription regulators CREB, Nur77, PPARα, and PGC-1α was decreased (Fig. 3K and Fig. S5D), which correlated with the reduced expression of their target genes in FA catabolism [AcAA (which encodes acetyl CoA acyltransferases), Acad (which encodes acyl CoA dehydrogenases), Echs (which encodes enoyl CoA hydratase), Hadha (which encodes hydroxyacyl-CoA dehydrogenase), Cpt2 (which encodes carnitine palmitoyltransferase 2), and Lpl (which encodes lipoprotein lipase)] (Fig. 3L). FGF21 expression in the gastrocnemius was higher in MBKO mice than in Fl/Fl control mice (Fig. S6A). Downregulation of AMPK and PGC-1α also lowered mitochondrial content in MBKO muscle as evidenced by the decreased abundance of Cyto c, SDH, and PDH (Fig. 3K and Fig. S5D). On the other hand, the abundance of the transcription factors involved in β-oxidation was not altered in the muscle of fed female MBKO mice (Fig. S5E), indicating that BDNF is involved only in fasting-induced metabolic reprogramming.
The skeletal muscle of MBKO mice has lower energy content but higher lipid metabolite accumulation
If β-oxidation is dysregulated in MBKO mice under nutrient stress, lipid metabolism in MBKO muscle should be impeded, leading to lipid accumulation. Surprisingly, triglyceride (TG) content in the gastrocnemius was lower in fasted female MBKO mice than in Fl/Fl mice (Fig. 4A) but the expression of key genes involved in FA synthesis [Fasn (which encodes fatty acid synthase)], FA uptake (Cd36), and lipolysis [Atgl (which encodes adipose triglyceride lipase, the rate limiting enzyme of lipolysis)] was increased (Fig. S6B), suggesting that the reduced intramyocellular TG content was a result of enhanced lipolysis but not impaired FA uptake or synthesis. Consistent with this notion, FA content was elevated in the gastrocnemius of fasted MBKO mice (Fig. 4A). Levels of acylcarnitines and ceramides were also higher in the MBKO gastrocnemius (Fig. 4A). These results indicate that TG and FA catabolism in MBKO muscles were uncoupled and that increased lipolysis depleted TG content but that reduction in mitochondrial number (Fig. 3K) and β-oxidation (Fig. 3H) led to the accumulation of FA, acylcarnitines, and ceramides. Because soleus muscles do not switch from oxidative metabolism during fasting (20), it was consistent that Bdnf expression (Fig. 1A) and lipid metabolite content were not altered in the soleus muscle of fasted MBKO mice (Fig. 4A).
Fig. 4.
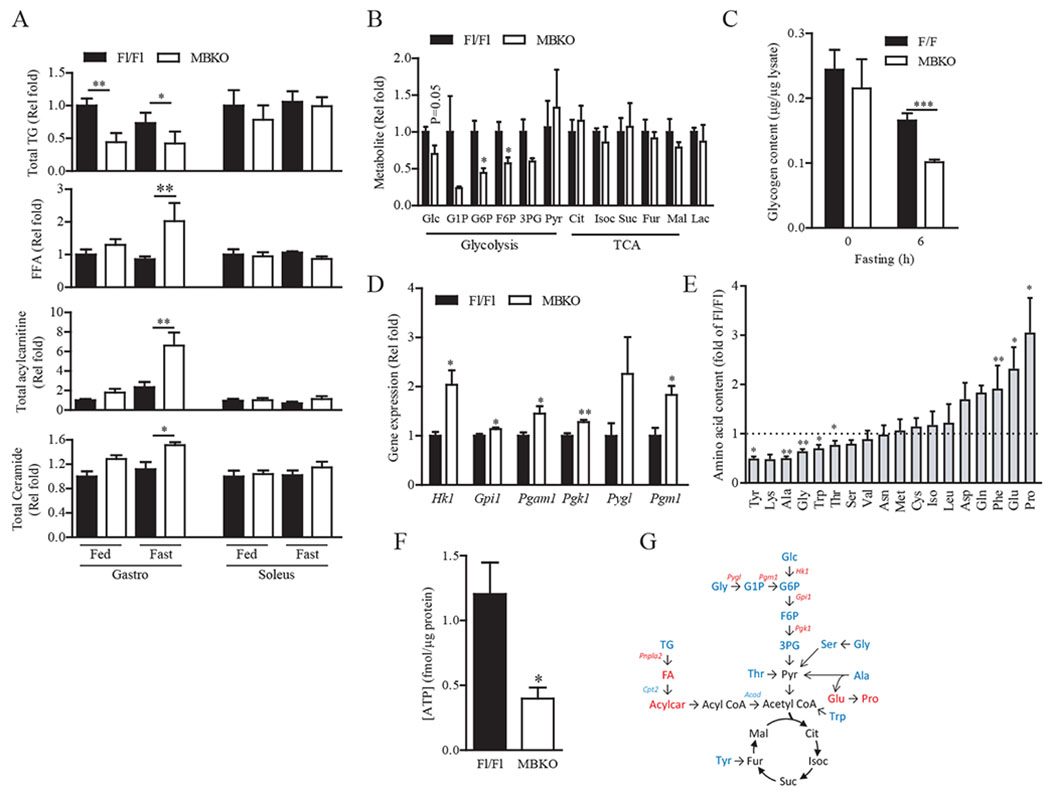
The skeletal muscle of MBKO mice has reduced energy content but higher lipid metabolite accumulation.
(A) Total triglyceride (TG), free fatty acid (FFA), and acylcarnitine content in the gastrocnemius and soleus muscles of female MBKO mice as determined by LC/MS; n = 4 mice per group.
(B) Metabolites of glycogenolysis, glycolysis, and tricarboxylic acid (TCA) cycle in the gastrocnemius muscle of female Fl/Fl and MBKO mice after 24-h fasting; n = 4–5 mice per group. Glc, glucose; G1P, glucose-1-phosphate; G6P, glucose-6-phosphate; F6P, fructose-6-phosphate; 3PG, 3-phosphoglycerate; Pyr, pyruvate; Cit, citrate; Isoc, isocitrate; Suc, succinate; Fur, fumarate; Mal, malate; Lac, lactate.
(C) Glycogen content in the gastrocnemius muscle of female Fl/Fl and MBKO mice after fasting; n = 6–7 mice per group.
(D) Expression of glycogenolytic and glycolytic genes in the gastrocnemius muscle of female Fl/Fl and MBKO mice after 24-h fasting as determined by RNA sequencing; n = 4 mice per group. Hk1, encodes hexokinase 1; Gpi1, encodes glucose phosphate isomerase 1; Pgam1, encodes phosphoglycerate mutase 1; Pgk1, encodes phosphoglycerate kinase 1; Pyg1, encodes glycogen phosphrylase 1; Pgm1, encodes phosphoglucomutase 1.
(E) AA content in the gastrocnemius muscle from female Fl/Fl and MBKO mice fasted for 24 h; n = 5 mice per group.
(F) ATP content in the gastrocnemius muscle of female MBKO mice after 24-h fasting; n = 5 mice per group.
(G) Summary of the biochemical pathways of glucose and lipid metabolism affected by Bdnf ablation in the gastrocnemius during fasting. Upregulated metabolites or genes are indicated in red and the downregulated metabolites or genes are indicated in blue. *P < 0.05, **P < 0.01; Student’s t-test (A, B, D–F), two-way ANOVA (C).
In addition to incomplete β-oxidation, glycogen and glycolysis metabolites were depleted in the gastrocnemius of fasted MBKO mice (Fig. 4, B and C). However, expression of glycoenolytic and glycolytic genes was increased (Fig. 4D), suggesting that the reduced glycolytic intermediate content was due to increased glycogenolysis and glycolysis, which was possibly to compensate for the insufficient ATP generation resulting from defects in β-oxidation. The concentrations of pyruvate and citric acid cycle intermediates were not changed in MBKO muscle, which was possibly caused by enhanced amino acid (AA)-pyruvate conversion because the concentrations of AAs that can be converted to pyruvate (alanine, glycine, and threonine) were significantly reduced in the MBKO gastrocnemius (Fig. 4E). Moreover, the concentrations of alanine catabolism byproducts, glutamate and proline, were significantly higher in the MBKO gastrocnemius (Fig. 4E), further supporting the conclusion of accelerated alanine breakdown. Total ATP content in the gastrocnemius was also lower in fasted MBKO mice than in Fl/Fl mice (Fig. 4F), although the concentration of pyruvate was comparable between the two genotypes, supporting the notion that MBKO muscles have insufficient mitochondrial number (Fig. 3K) to catabolize pyruvate. Consequently, alanine export from the muscle to the liver for gluconeogenesis (26) was reduced, resulting in a lower circulating level of alanine in fasted MBKO mice (Table 1). Together, our results indicate that a deficiency of muscle-derived BDNF impairs FAO during fasting, leading to the accumulation of lipid metabolites and inadequate ATP production, which elevates glycolysis and enhances AA catabolism as compensatory responses (Fig. 4G).
Table 1.
Blood metabolite analysis of MBKO mice
| Fed | Fasted | |||
|---|---|---|---|---|
| Fl/Fl | MBKO | Fl/Fl | MBKO | |
| Glucose (mg/dL) | 128.4 ± 8.2 | 140.7 ± 6.8 | 92.1 ± 3.8b | 119.9 ± 10.2* |
| Insulin (ng/ml) | 0.63 ± 0.08 | 0.88 ± 0.08 | 0.35 ± 0.06a | 0.74 ± 0.09* |
| TG (mM) | 1.03 ± 0.08 | 1.51 ± 0.20* | 3.46 ± 0.54c | 5.90 ± 0.71*, c |
| FFA (mM) | 0.70 ± 0.08 | 1.04 ± 0.16 | 1.09 ± 0.25 | 1.63 ± 0.14a |
| BDNF (ng/ml) | 5.49 ± 2.83 | 3.61 ± 0.93 | 42.46 ± 5.42c | 14.69 ± 5.64* |
| Alanine (nM) | 0.33 ± 0.02 | 0.30 ± 0.02 | 0.67 ± 0.07b | 0.39 ± 0.01**, b |
| CK activity (nmol/min/ml) | 39.36 ± 20.27 | 53.19 ± 9.74 | 37.10 ± 18.32 | 98.47 ± 1.05**, b |
Sera were collected from mice (6-month-old; n = 4–8 mice per group) fed ad libitum or fasted for 24 h. Results are expressed as the mean ± SEM
P < 0.05
P < 0.01
P < 0.001 compared to Fl/Fl mice of the same treatment group.
P < 0.05
P < 0.01
P < 0.001 compared to the same genotype; Student’s t-test. CK, creatine kinase; FFA, free fatty acid; TG, triglyceride.
Muscle-specific deficiency of BDNF abolishes fasting-induced autophagy and promotes muscle weakness
Negative energy balance increases whole-body proteolysis and intracellular AA content (27). The reduced total AA concentration in the gastrocnemius of fasted MBKO mice (Fig. S7A) suggested that fasting-provoked protein catalysis was suppressed. Indeed, lean mass reduction was decreased to a lesser extent in female MBKO mice after food deprivation (Fig. 5A), which was not observed in male MBKO mice (Fig. S4I). In contrast, fasting-induced adipose tissue atrophy was exaggerated in MBKO mice (Fig. 5A), which was associated with higher circulating free FA levels (Table 1). Total protein ubiquitination and the abundance of ubiquitin ligases (MuRF1 and atrogin 1) were not changed in the gastrocnemius of fasted MBKO mice (Fig. S7B), suggesting that the ubiquitin/proteasome system was not involved in the abnormal muscle physiology of MBKO mice. Because the activity of AMPK, the master regulator of nutrient stress-induced autophagy (28), was reduced in the gastrocnemius of fasted MBKO mice (Fig. 3K), we suspected that fasting-induced autophagy was blunted. Indeed, AMPK-mediated Ser555 phosphorylation of Unc-51 like kinase-1 (ULK1), a marker of autophagy initiation (29), was significantly reduced in the fasted MBKO gastrocnemius (Fig. 5B), which was associated with decreased LC3 lipidation and p62 degradation (Fig. 5B). In addition, immunofluorescence staining showed fewer LC3 punta in the gastrocnemius muscle of fasted MBKO (Fig. 5C). Total expression and activity of the autophagy suppressor mTOR was elevated in MBKO muscle (Fig. 5B), which was associated with phosphorylation of Ser757 in ULK1, an inhibitory phosphorylation event (30, 31) (Fig. 5B). The EDL and TA of MBKO mice also displayed altered autophagy signaling (Fig. S7C). Histological analysis of gastrocnemius from fasted MBKO mice revealed several characteristics of muscle degeneration including central nuclei, phagocytosis, and fiber necrosis, which persisted after refeeding (Fig. 5D). No obvious structural defects were detected in male MBKO muscles (Fig. S4, J and K). Consistent with the tissue damage detected by histological analysis, serum creatine kinase (CK) levels were significantly higher in female MBKO mice (Table 1), which was further elevated during fasting, suggesting that nutrient depletion exaggerated myocellular damage. Female MBKO mice also exhibited weaker muscle strength even when fed (Fig. 5E), which was probably a result of defective metabolism and tissue structure. Gastrocnemius muscle from female mice also displayed lower tetanic force and higher stiffness during ex vivo assessment, although specific twitch force and fatigue rate of gastrocnemius muscle were comparable between the two genotypes (Fig. 5F). Exercise endurance is another metabolic challenge that requires the selection of the appropriate carbon substrate (32), and this parameter was also compromised in fed MBKO mice (Fig. 5G), further demonstrating that MBKO mice were metabolically inflexible.
Fig. 5.
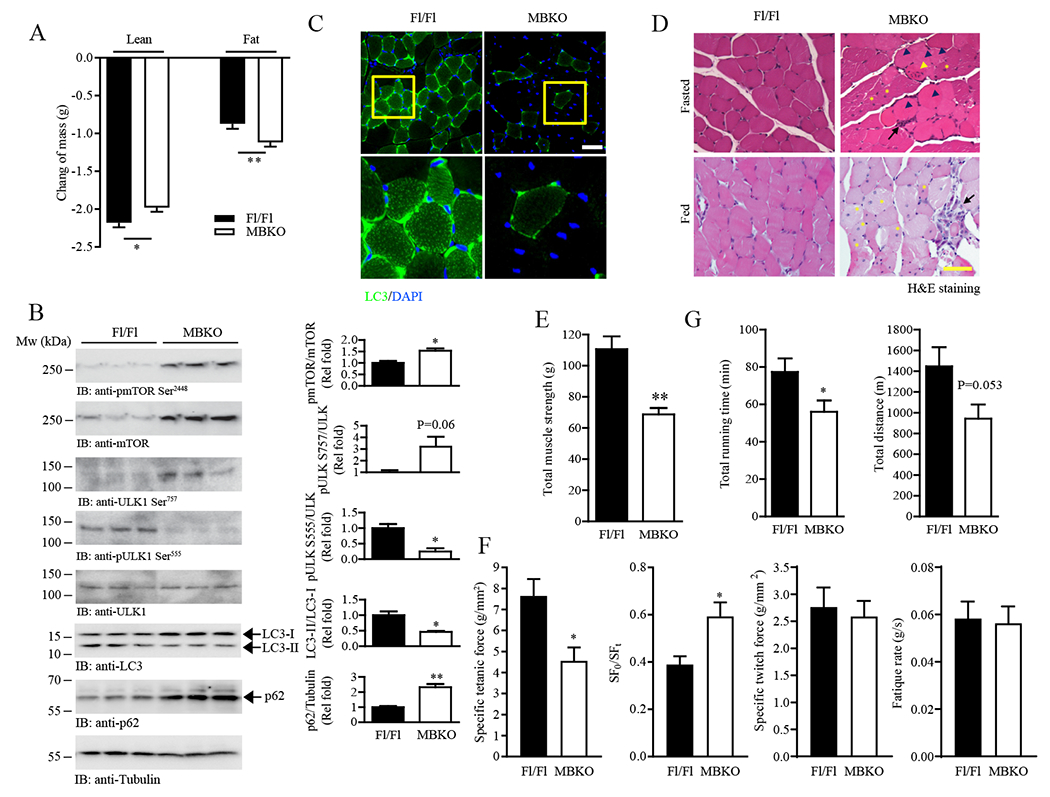
Diminished fasting-induced autophagy in MBKO muscle.
(A) Changes in total lean and fat mass of female Fl/Fl and MBKO mice after 24-h fasting; n = 10–12 mice per group.
(B) Autophagy signaling in the gastrocnemius muscle of female Fl/Fl and MBKO mice after 24 hours of fasting as determined by immunoblotting. Each lane represents a different mouse. Quantification is also shown; n = 3 mice per group.
(C) Representative LC3 immunofluorescent images of gastrocnemius muscle sections from fasted (24 h) female Fl/Fl and MBKO mice (upper panel). Scale bar, 50 µm. Magnified images of the selected regions (yellow boxes) are shown in the lower panel. n = 3 mice per group.
(D) Representative H&E staining of gastrocnemius muscle sections from fed or fasted female Fl/Fl and MBKO mice. Centronuclear myopathy (yellow asterisks), necrosis (black arrow heads), myositis (black arrows) and focal invasion of non-necrotic muscle fibers by inflammatory cells (yellow arrowhead) are indicated. Scale bar, 50 µm. n = 3 mice per group.
(E) Total muscle strength of fed female Fl/Fl and MBKO mice (6-month-old) as determined by the grip-strength test; n = 6 mice per group.
(F) The specific tetanic force, stiffness (SF0/SFt), specific twitch fore, and fatigue rate of gastrocnemius muscles isolated from fed female Fl/Fl and MBKO mice (6-month-old) as determined by ex vivo functional assessment; n = 6 mice per group.
(G) Total time and distance run by fed female Fl/Fl and MBKO mice (6-month-old) on a treadmill; n = 6 mice per group. *P < 0.05, **P < 0.01; Students t-test.
Muscle-specific deficiency in BDNF leads to the development of insulin resistance
Accumulation of intramuscular acylcarnitines or inefficient mitochondrial activity in skeletal muscle results in systemic insulin resistance (33, 34). These two changes were detected in the gastrocnemius of MBKO mice (Fig. 3K and 4A) and accordingly, MBKO mice displayed fasting hyperglycemia and hyperinsulinemia (Table 1). Insulin resistance in MBKO mice was further confirmed by poor glucose tolerance (Fig. 6, A and B). Glucose infusion (Fig. 6C) and glucose turnover (Fig. 6D) rates were also significantly lower in MBKO mice than in Fl/Fl animals as evidenced by hyperinsulinemic-euglycemic clamp assays. Moreover, insulin-induced glucose uptake was reduced in gastrocnemius but not in WAT in MBKO mice (Fig. 6, E and F). Insulin injection (35) induced the phosphorylation of Akt and AS160 in gastrocnemius muscle to a lesser extent in MBKO mice (Fig. 6G), an effect that was consistent with the reduction in glucose uptake (Fig. 6E). In contrast to the permanent depletion of Bdnf expression that disrupted lipid metabolism in MBKO muscle, a transient knockdown of BDNF levels in C2C12 myotubes did not impair insulin signaling (Fig. 6H), indicating that long-term Bdnf depletion is required to establish metabolic dysfunction and insulin resistance in skeletal muscle. Insulin-suppressed hepatic glucose production was not significantly altered in MBKO mice (Fig. 6I) but hepatic glycogen content in MBKO mice was lower than in Fl/Fl control mice (Fig. 6J). Together, our results suggest that depletion of muscle-derived BDNF specifically reduces the insulin sensitivity of gastrocnemius muscle.
Fig. 6.
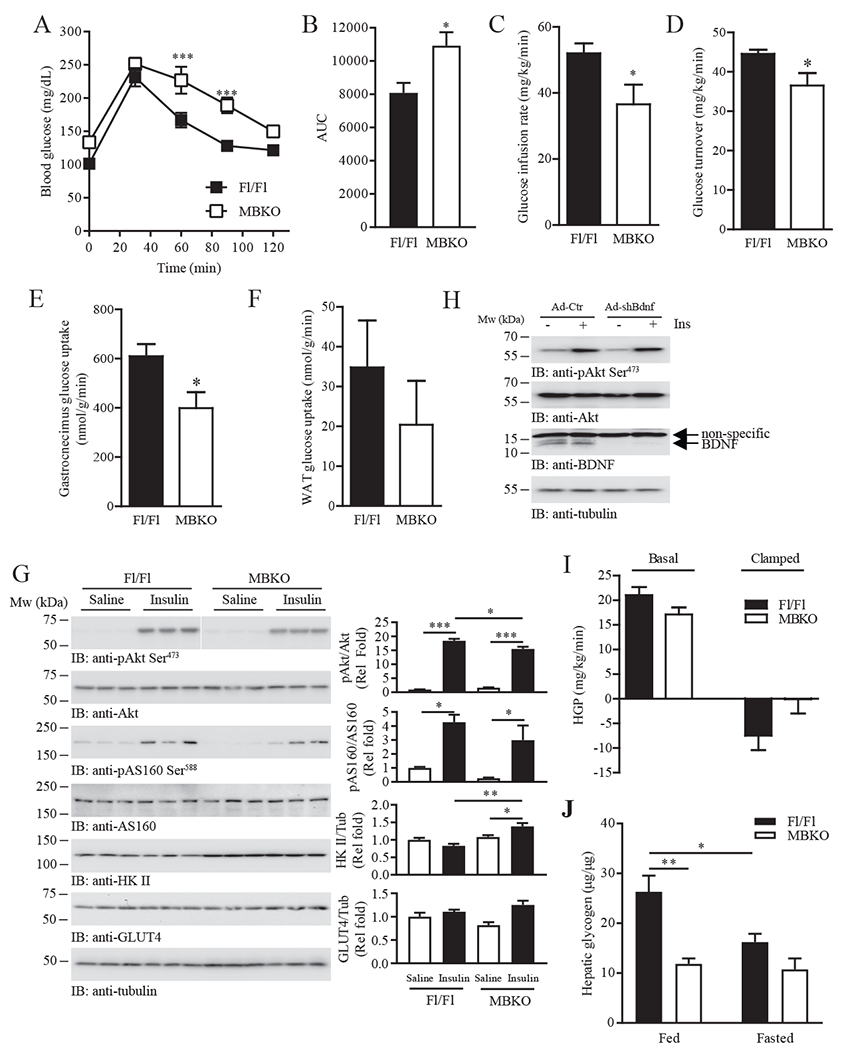
MBKO mice display muscle-specific insulin resistance.
(A) Glucose tolerance test (GTT) of 5 month old female Fl/Fl and MBKO mice fasted for 16 hours; n = 6 mice per group.
(B) Area under curve (AUC) for the GTT shown in Fig. 6A; n = 6 mice per group.
(C) Glucose infusion rate for 16 h-fasted female Fl/Fl and MBKO (28-week-old) mice during hyperinsulinemic-euglycemic clamping; n = 6–8 mice per group.
(D) Whole body glucose turnover in female Fl/Fl and MBKO (28-week-old) mice during hyperinsulinemic-euglycemic clamping; n = 6–8 mice per group.
(E) Insulin-stimulated glucose uptake by the gastrocnemius muscle of female Fl/Fl and MBKO (28-week-old) mice during hyperinsulinemic-euglycemic clamping; n = 6 mice per group.
(F) Insulin-stimulated glucose uptake by WAT of female Fl/Fl and MBKO (28-week-old) mice during hyperinsulinemic-euglycemic clamping; n = 6 mice per group.
(G) Insulin-stimulated signaling in gastrocnemius muscle isolated from 16 h-fasted female Fl/Fl and MBKO mice (6-month-old). Each lane represents a different mouse. Quantification is shown in the right panels; n = 3 mice per group.
(H) Differentiated C2C12 myotubes infected for 48 h with control adenovirus (Ad-Ctr) or adenovirus expressing shBdnf (ad-shBdnf) were stimulated with human insulin (100 nM, 30 mins) and immunoblotted as indicated. Each lane represents a different set of cells.
(I) Hepatic glucose production (HGP) of Fl/Fl and MBKO (28-week-old) mice during hyperinsulinemic-euglycemic clamping; n = 6 mice per group.
(J) Hepatic glycogen content in fed or 24 h-fasted female Fl/Fl and MBKO mice (6-month-old); n = 4 mice per group. *P < 0.05, **P < 0.01, ***P < 0.001; two-way repeated measures ANOVA (A), Student’s t-test (B–E), two-way ANOVA (G, J).
DISCUSSION
Skeletal muscle is a structurally and metabolically plastic tissue that maintains systemic energy homeostasis in response to various metabolic stresses. It is an important site for glucose disposal during the postprandial period and performs metabolic changes to utilize FA for glucose sparing when nutrient supply is limited. Skeletal muscle is also the largest protein reservoir to provide AA for compensatory energy production during catabolic periods. Thus, it is not surprising to find that metabolic inflexibility in muscles is a dominant cause of various metabolic disorders (36). In this study, we found that skeletal muscle-generated BDNF coordinated metabolic reprograming in muscle during fasting in females. Our data suggest that when extracellular glucose dropped, BDNF production in glycolytic muscle fibers was increased, which activated CREB. By promoting the activity of signaling pathways involving AMPK, PPARα, PGC-1α, and Nur77, muscle-generated BDNF acted as an autocrine factor to facilitate mitochondrial transportation of FA and expression of genes involved in FAO. Because these signaling cascades are also important to mitochondrial biogenesis and fasting-induced autophagy, their disruption in MBKO muscles led to lower ATP production from FAO and reduced AA release from autophagy during fasting, which resulted in myofiber necrosis. Consequently, the damaged myofibers were associated with weaker muscle strength, reduced physical locomotion, low overall metabolic rate, and enhanced adiposity.
Increased intramyocellular BDNF content and circulating BDNF levels after exercise has been reported in humans (13). However, kinetics of the exercise-induced elevation in serum BDNF failed to correlate with BDNF content in muscle, suggesting that BDNF in muscles may act only as an autocrine or paracrine factor (13). Indeed, it has been suggested that neuronal tissues and blood platelets are the major source of circulating BDNF (37, 38). In contrast to the human studies, our findings in MBKO mice demonstrated that BDNF production by muscle is important for increasing the surge in BDNF levels during fasting. Several mechanisms may explain this discrepancy. First, direct BDNF export from muscles into circulation may be a rodent-specific response. Second, the final destination for BDNF exported from muscles in response to fasting and exercise may be different. Third, muscle-produced BDNF may act on the neurons that link muscle and brain to induce BDNF production from the CNS. Thus, further studies using transgenic animals expressing tagged BDNF in skeletal muscle are warranted to delineate the specific role of tissue-produced BDNF in the overall hormone content in circulation. Although BDNF has not always been successfully detected in mouse serum (39), BDNF serum levels are decreased in streptozotocin-induced diabetic mice and increased in a mouse model of acute asthma (40, 41), suggesting that BDNF is detectable in mouse circulation. Our current studies also support the notion that BDNF is present in mouse blood. Presumably, the variation in detection methods accounts for the discrepancy in results.
Because physical inactivity has been suggested as a primary cause of metabolic inflexibility (42), it is reasonable to suspect that the defective phenotypes observed in MBKO mice may result from their low physical activity (Fig. 3G). However, some metabolic characteristics that occur after bed rest or restricted activity were not phenocopied in MBKO mice. In particular, restricted activity results in diminished expression of CD36 and increased TG accumulation in skeletal muscle (43, 44), but MBKO muscle exhibited enhanced Cd36 expression (Fig. S6B) and reduced TG levels (Fig. 4A), indicating that physical inactivity and BDNF deficiency in skeletal muscle results in differential metabolic outcomes. Our experiments with cultured myotubes also showed that BDNF has a prominent role in metabolic regulation (Fig. 2A). Furthermore, Bdnf expression in skeletal muscle of wild-type mice increased significantly after fasting (Fig. 1A), suggesting that BDNF production may help muscles cope with nutrient stress.
Gender has a profound impact on metabolism. Compared with men, women rely more on lipids during fasting and exercise (45). We found that fasting significantly increased Bdnf expression in female mouse muscles only (Fig. 1A). Consequently, metabolic dysfunction in fasted MBKO mice was sexually dimorphic, which suggests that elevated Bdnf expression in fasted muscle is a response unique to female mice for regulating lipid metabolism. BDNF functions in the nervous system are also sex-dependent; for instance, BDNF alleviates acetic acid-induced pain only in male rats (46), whereas BDNF is critical in anxiety- and depression-associated behaviors in female rats (47). We also found that the BDNF mimic 7,8-dihydroxyflavone counters obesity only in female mice (48), which further supports the notion that the sexual-dimorphism of BDNF can be applied to systemic energy metabolism. The molecular mechanism underlying the sex-specific response to BDNF remains unclear but sex hormones, in particular estrogen, have been suggested to be important because they regulate Bdnf expression and crosstalk with the TrkB cascade in the nervous system (49). Estrogen production is also induced by fasting in female mice (50) and depletion of the estrogen receptor α (ERα) in skeletal muscle dysregulates glucose and lipid metabolism, leading to the development of obesity (51). Furthermore, estrogen activates CREB activity and expression (52). Thus, it is tempting to hypothesize that estrogen, BDNF, and CREB may form a network that regulates skeletal muscle metabolism in female mice. However, the functional interaction between these factors has not yet been examined in skeletal muscle, warranting further investigation.
During β-oxidation, FA is shuttled into mitochondria by the carnitine palmitoyltransferase (CPT) system, in which acyl CoA is catalyzed to acylcarnitine in the outer mitochondrial membrane by CPT1, which is then converted back into acyl CoA by CPT2 in the inner mitochondrial membrane. The production of acyl CoA from acylcarnitine was impaired in MBKO muscle, leading to the accumulation of acylcarnitines (Fig. 4A), which is consistent with the reduction in Cpt2 expression and the levels of its upstream regulators PPARα and PGC-1α (Fig. 3K). This phenotype resembles the pathological features of metabolic myopathies induced by CPT2 deficiency in human patients (53), which includes elevated lipid storage, central nuclei in myocytes, myocyte necrosis, muscle weakness, and enhanced glycolysis during metabolic stress (54, 55). The clinical symptoms of metabolic myopathies are usually subtle under normal circumstances but become obvious when patients are subjected to energy stresses like prolonged exercise or fasting (56), which are also consistent with our findings that the aberrant phenotypes of MBKO mice were detected only during nutrient depletion. Because the accumulation of FA or its metabolites such as acylcarnitine is a major cause of insulin resistance in skeletal muscle, the high intracellular concentration of these molecules in MBKO muscle thus interferes with insulin-induced glucose uptake, leading to hyperglycemia. In agreement with this finding, insulin resistance and fasting hyperinsulinemia have been reported in a patient with a CPT2 mutation (55). Because few studies have been performed to examine the effect of lipid accumulation in CPT2-deficient muscle, it remains unknown if insulin resistance is a general characteristic of the disease. Nevertheless, our data indicate that BDNF signaling is crucial for balancing lipid and glucose metabolism in skeletal muscle and that impairment of this pathway leads to insulin resistance.
Balanced autophagy flux in skeletal muscle is critical for the overall health of an organism. Although maintaining basal autophagy flux is essential to clear damaged organelles or recycle macromolecules in muscles during metabolic stress (57), excess autophagy promotes extensive muscle wasting that affects tissue mass, muscle strength, myofiber regeneration, and favorable metabolism (58). We found that BDNF production by muscle was essential for autophagy-induced muscular protein degradation during fasting. BDNF signaling in cultured hippocampal neurons has been suggested to activate mTOR to support neuronal survival primarily through inhibiting autophagy flux (59). However, BDNF has also been reported to suppress the activity of the Akt/mTOR/p70S6K axis to enhance neuronal autophagy (60). In line with this finding, BDNF upregulates autophagic activity in streptozotocin-induced diabetic mice to prevent Purkinje cell loss (61). Due to the limited studies on the role and effect of BDNF on autophagy, it remains unknown whether these discrepant findings were caused by differences in cell model used, type of stimulation, or experimental procedures. Our results showed that BDNF promoted autophagy progression in skeletal muscle, consistent with impaired AMPK signaling. Indeed, several key phenotypes including decreased circulating alanine levels, LC3 lipidation, and phosphorylation of Ser555 in ULK1, combined with increased mTOR-dependent phosphorylation of Ser757 in ULK1, and a lack of changes in atrogin 1 and MuRF1 abundance are common to both MBKO and muscle-specific AMPK-knockout (AMPK-MKO) mice (28) during fasting, suggesting that AMPK may be involved in defective autophagy in MBKO muscle. On the other hand, substantial decreases in FAO and muscle glycogen content were detected in MBKO mice but not in AMPK-MKO mice. Moreover, AMPK-MKO mice display hypoglycemia, whereas MBKO mice exhibited hyperglycemia during fasting. Presumably, AMPK is an important but not the sole effector of BDNF in regulating muscle metabolism in response to fasting. Ablating Atg7 in muscles protects against obesity development by inducing FGF21 (62). Similar to this report, the impaired autophagy in MBKO muscle was associated with increased Fgf21 expression (Fig. S6A); however, the body weight of MBKO mice was higher than that of Fl/Fl control mice. Further investigation is needed to fully explore the complex physiological outcomes in muscles without Bdnf expression.
In summary, we demonstrated that BDNF linked insulin responsiveness, autophagy, and metabolic flexibility in skeletal muscle during fasting, and that deficiency in BDNF production led to the development of metabolic myopathy and insulin resistance in female mice. These findings underscore that BDNF is not only a critical neurotrophic factor in the CNS, but also an essential metabolic regulator in peripheral tissues
MATERIALS AND METHODS
Chemicals and reagents
C2C12 cells were purchased from the American Type Culture Collection (ATCC; Manassas, VA) and maintained as instructed by ATCC. Transfection of C2C12 cells was performed using DharmaFECT1 (Thermo Fisher Scientific, Waltham. MA) according to manufacturer’s instructions. Recombinant human BDNF and anti-BDNF antibody were purchased from Abcam (Cambridge, UK). Human insulin was obtained from Elli Lily and Company (Indianapolis, IN). All other chemicals were purchased from Sigma-Aldrich (St. Louis MO). Antibodies against pAkt S473 (#9271), pAMPKα T172 (#2535), AMPKα (#2603), pACC S79 (#3661), ACC (#3676), pCREB S133 (#9198), CREB (#4820), and TrkB (#4603) were purchased from Cell Signaling Technology (Danvers, MA). All autophagy and mTOR signaling pathway antibodies were also obtained from Cell Signaling Technology. Anti-Akt1 (sc5298), anti-IRβ (sc53742), and anti-pTrkB Y706 (sc135645) antibodies were obtained from Santa Cruz Biotechnology (Dallas, TX). Anti-PPARα (#8934) and PGC-1α (#54481) were obtained from Abcam while anti-tubulin antibody (T6074) was obtained from Sigma-Aldrich. [1-14C]-Palmitic acid was purchased from PerkinElmer (Waltham, MA). Adenoviruses that express shBdnf were synthesized by Cyagen (Santa Clara, CA) based on a previously reported sequence (63).
Animal experiments
MBKO mice were generated by crossing BDNF Flox/Flox mice (cat #004339; Jackson Laboratory, Bar Harbor, ME) with transgenic mice carrying the human α-skeletal actin-promoter-driven Cre (HSA-Cre; cat # 006149; Jackson Laboratory, USA). Because of the sex-specific response in obesity development, all in vivo experiments were performed using female mice. Genotyping was performed via PCR using genomic DNA extracted from the tail and using primers suggested by the Jackson Laboratory. Mice were housed in environmentally controlled conditions under a 12-h light/dark cycle with ad libitum access to standard rodent pellet food and water. All in vivo assays were performed using 8-week-old female mice due to the sex-dimorphic effect and were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Oklahoma Health Sciences Center and Committee on the Use of Live Animals in Teaching and Research (CULATR) of the University of Hong Kong.
Blood glucose levels were measured using the ACCU-CHEK Advantage Blood Glucose Meter (Roche, Basel, Switzerland). Serum insulin, FGF21, and BDNF were measured by ELISA (Crystal Chem, Elk Grove Village, IL; Abcam; and Abnova, Taipei, Taiwan, respectively). Serum triglyceride and free FA levels were measured using the Triglyceride Quantification Colorimetric Kit and the Free Fatty Acid Quantification Colorimetric Kit, respectively (BioVision, Milpitas, CA). Alanine and β-hydroxybutyrate serum concentrations and creatine kinase activities were detected using commercially available kits (Cayman Chemical, Ann Arbor, MI). Hepatic glycogen content was determined by the Glycogen Assay Kit (Cayman Chemical). The glucose tolerance test (GTT) was performed after intraperitoneally injecting D-glucose (2 g/kg of body weight) into female mice that had fasted overnight fasted (16 h).
Tissue CREB activities were determined by CREB TFact DNA-binding ELISA (Assay Biotechnology Company Inc., Fremont, CA). Muscle glycogen and ATP levels were measured enzymatically using commercially available kits (Cayman Chemical). AMP concentration determination and profiling of various lipid and glucose metabolites were performed at the West Coast Metabolomics Center at the University of California Davis Genome Center (USA) using liquid chromatography-mass spectrometry (LC/MS). LC/quadrupole/time-of-flight (QTOF)MS analyses were performed using an Agilent 1290 Infinity LC system (Santa Clara, CA) coupled to either an Agilent 6530 (positive ion mode) or an Agilent 6550 mass spectrometer equipped with an ion funnel (iFunnel; negative ion mode). Lipids were separated on an Acquity UPLC CSH C18 column (Waters, Milford, MA) maintained at 65 °C at a flow rate of 0.6 mL/min. The mobile phases consisted of 60:40 acetonitrile:water with 10 mM ammonium formate and 0.1% formic acid (A) and 90:10 propan-2-ol:acetonitrile with 10 mM ammonium formate and 0.1% formic acid. The gradient was as follows: 0 min 85% (A); 0–2 min 70% (A); 2–2.5 min 52% (A); 2.5–11 min 18% (A); 11–11.5 min 1% (A); 11.5–12 min 1% (A); 12–12.1 min 85% (A); and 12.1–15 min 85% (A). A sample volume of 3 µL was used for the injection and sample temperature was maintained at 4 °C in the autosampler. The QTOF mass spectrometers were operated with electrospray ionization (ESI) performing full scans in the mass range m/z 65–1700 in positive (Agilent 6530, equipped with a JetStreamSource) and negative (Agilent 6550, equipped with a dual JetStream Source) modes producing both unique and complementary spectra. Instrument parameters were as follows (positive mode): gas temp 325 °C, gas flow 8 l/min, nebulizer 35 psig, sheath gas 350 °C, sheath gas flow 11, capillary voltage 3500 V, nozzle voltage 1000 V, fragmentor 120 V, and skimmer 65 V. Data (both profile and centroid) were collected at a rate of 2 scans per second. In negative ion mode, the parameters were identical to positive ion mode except for the following: gas temp 200 °C, gas flow 14 l/min, and fragmentor 175 V. For 6530 QTOF, a reference solution generating ions of 121.050 and 922.007 m/z in positive mode and 119.036 and 966.0007 m/z in negative mode was used for continuous mass correction. For 6550 QTOF, the reference solution was introduced by a dual spray ESI, with the same ions and continuous mass correction.
Cell culture
C2C12 myoblasts were cultured in DMEM supplemented with 5% FBS, 15% calf serum, 100 IU/mL penicillin and 100 μg/mL streptomycin (Invitrogen, Carlsbad, CA). Differentiation of myoblasts into myotubes was performed by incubating 100% confluent myoblast with differentiating medium (2% horse serum, 100 IU/mL penicillin, and 100 μg/mL streptomycin) for 4 days. C2C12 differentiation was confirmed by morphological changes as previously described (64).
Immunoblotting
Tissue or cell extracts were prepared by homogenizing tissues in lysis buffer (50 mM Tris pH 7.4, 40 mM NaCl, 1 mM EDTA, 0.5% Triton X-100, 1.5 mM Na3VO4, 50 mM NaF, 10 mM Na4P2O7, 10 mM sodium β-glycerol phosphate, and protease inhibitor cocktail). Cell debris was removed by centrifugation and the supernatants were collected for further analysis. Protein concentration of the lysates was determined by the Bio-Rad Protein Assay (Bio-Rad Laboratories, Hercules, CA), after which an equal amount of protein lysate from each sample was loaded for SDS-PAGE. SDS-PAGE and blotting were performed using the mini-PROTEAN Tetra (Bio-Rad Laboratories) and protein transfer was performed using the Trans-Blot Turbo Transfer System (Bio-Rad Laboratories). Immunoblotting signals within the linear detection range were detected with a G:Box Chemi XRQ imager (Syngene, Cambridge, UK) and analyzed using ImageJ (NIH, Bethesda, MD). Representative results from two or more independent experiments are shown.
Histology and immunofluorescent staining
Immunofluorescent staining was performed using paraffin-embedded sections and hematoxylin and eosin (H&E) staining was performed as previously described (65). Representative results from at least three animals are shown.
Determination of mitochondrial content, mitochondrial respiration, and lipid oxidation
Total DNA was isolated using the DNAeasy Kit (Qiagen, Hilden, Germany). The amount of mitochondrial DNA was determined by real-time PCR using the primers 5ʹ-CCCAGCTACTACCATCATTCAAGT-3ʹ (forward) and 5ʹ-GATGGGTTTGGGAGATTGGTTGATGT-3ʹ (reverse). β-actin levels were used for normalization.
Mitochondrial respiration of C2C12 myotubes was determined by the XFe 96 Extracellular Flux Analyzer using the XF Mito Stress Test Kit (Seahorse Bioscience, Billerica, MA). The concentrations of oligomycin, carbonyl cyanide-p-trifluoromethoxyphenyl-hydrazon (FCCP), antimycin A, and rotenone used were 100 μm, 100 μm, 100 μm, and 50 μm respectively. The OCR and extracellular acidification rate were recorded and non-mitochondrial respiration was determined as the lowest OCR detected after antimycin A and rotenone injection. Basal respiration was determined as the difference between the OCR before compounds were injected and the non-mitochondrial respiration rate. Maximal respiration was determined as the difference between the highest OCR recorded after FCCP injection and the non-mitochondrial respiration rate. OCR from exogenous lipid oxidation of C2C12 myotubes was determined by Extracellular Flux Analyzer using the XF Palmitate-BSA FAO Substrate (Seahorse Bioscience). Additionally, 1 mM BSA-coupled palmitic acid and 40 µM Etomoxir were used in the analysis.
FAO in muscle lysates was determined as previously described (66). Briefly, gastrocnemius muscle collected from fasted (16 h) mice were homogenized and incubated with a reaction mixture containing 14C-palmitatic acid (0.4 µCi per reaction). After percholic acid precipitation, the 14C-incorporated CO2 was captured by a strong base (1M NaOH) in the Whatman paper disc. The radioactivity was then measured by scintillation counting.
Gene expression analysis and RNA sequencing
Total RNA was extracted by Trizol Isolation Reagent (Invitrogen). First-strand cDNA from total RNA was synthesized using Superscript III reverse transcriptase (Invitrogen) and Oligo-dT17 as primer. Lipid metabolism gene expression was determined using the Mouse Fatty Acid Metabolism RT2 Profiler PCR Array (Qiagen). Primers used for real-time PCR were as follows: BDNF, 5ʹ-ATGTCTATGAGGGTTCGGCG-3ʹ (forward) and 5ʹ-GCGAGTTCCAGTGCCTTTG-3 (reverse); actin, 5ʹ-CTGTCGAGTCGCGTCCA-3ʹ (forward) and 5ʹ-ACCCATTCCCACCATCACAC-3ʹ (reverse); β-actin, AACCGTGAAAAGATGACCCAGAT (forward) and CACAGCCTGGATGGCTACGT (reverse). Real-time PCR was performed using 5 PRIME™ RealMasterMix SYBR ROX (Thermo Fisher Scientific) on an ABI7500 Real-time PCR System (Applied Biosystems, Foster City, CA).
mRNA sequencing was performed using the Illumina NextSeq v2 High SR75 (Illumina, San Diego, CA). Adapters were trimmed and low-quality bases were removed from raw reads using Trimmomatic. Processed reads were then mapped to the mouse reference genome (NCBI build 37.2) using TopHat and Cufflinks. Differential gene expression was evaluated using Cuffdiff.
Chromatin immunoprecipitation (ChIP) analysis
Binding of CREB to the Bdnf promoter was analyzed using a ChIP kit from Merck Millipore (Burlington, MA) using a CREB antibody (Cell Signaling Technology). Real-time PCR was performed using the following primers: 5ʹ-TTCGAGGCAGAGGAGGTATC-3ʹ (forward) and 5ʹ-GGCTGGGAGATTTCATGCTA-3ʹ (reverse).
Hyperinsulinemic-euglycemic clamp, metabolic cages, and body composition analysis
Insulin sensitivity and glucose metabolism, including glucose infusion rate, whole body glucose turnover, tissue-specific glucose uptake, and hepatic glucose production, were measured in partially restrained, awake mice using a 2-h hyperinsulinemic-euglycemic clamp (67). Briefly, human insulin was infused into overnight fasted mice (16 h) at a rate of 15 pmol/kg/min to raise plasma insulin within the physiological range. Blood samples were collected at 10–20 min intervals for the immediate measurement of plasma glucose, and 20% glucose was infused at variable rates to maintain basal glucose levels. Insulin-stimulated whole-body glucose metabolism was estimated with a continuous infusion of 3H-glucose throughout the insulin clamp (0.1 µCi/min). To estimate insulin-stimulated glucose uptake in individual organs, 2-[1-14C]deoxy-D-glucose (2-[14C]DG) was administered as a bolus (10 µCi) at 75 min after the start of the insulin clamp. Blood samples were drawn at 80, 85, 90, 100, 110, and 120 min for measuring plasma 3H-glucose, 3H2O, and 2-[14C]DG concentrations. At the end of the insulin clamp, mice were anesthetized and tissue samples were taken for biochemical and molecular analyses.
Whole body fat and lean mass of the animals were determined by 1H-MRS (Echo Medical Systems, Oceanside, New York). Computerized metabolic cages (TSE Systems, Bad Homburg, Germany) were used to simultaneously measure VO2 consumption, VCO2 production, energy expenditure rates, respiratory quotient, physical activity, and food/water intake in awake mice over 3 days. Labmaster cages that were the most similar to facility home cages were used, thereby allowing the use of bedding in the cage and minimizing any animal anxiety during the experimental period. Energy expenditure rates were normalized to the lean mass of the animals. Energy metabolism of mice used in the food depletion experiment was measured by the Comprehensive Lab Animal Monitoring System (CLAMS; Columbus Instruments, Columbus, OH).
Insulin injection into mice
Experiments were performed with female mice (6-month-old) that were fasted for 16 h. After anesthetization by intraperitoneal administration of sodium pentobarbital (50 mg/kg of body weight), saline or 5 U human insulin (Elli Lily and Company) was injected into the mouse through the inferior vena cava. After 5 min, the gastrocnemius muscles were removed and frozen in liquid nitrogen.
Muscle strength and exercise endurance
Exercise phenotyping of the animals was performed using a 6-lane treadmill with a variable speed motor. Three and two days before the experiment, the 6-month-old female mice were placed on the non-operating treadmill to acclimate them to the sight and smell of the apparatus and the exercise training room. One day before the experiment, the mice were allowed to walk or run slowly (~5 m/min) for 15 min. On the day of the test, the mice were placed on the unmoving treadmill for 10 min, after which the treadmill was turned on at the lowest speed for 10 min. After this warmup, the treadmill was turned up to a speed of 7 m/min. The mice were exercised to fatigue by increasing the speed by 1 m/min every 3 min until the animal refused to continue moving on the treadmill belt for more than 10 seconds.
The muscle strength of the animals (6-month-old females) was tested using the grip strength meter (Bioseb, Pinellas Park, FL) according to the manufacturer’s instructions. Briefly, mice were held by the tail, allowed to grasp the metal mesh of the meter with their four limbs, and then were pulled backward in the horizontal plane. The force applied to the mesh at the moment the grasp was released was recorded as the peak tension. The test was repeated five consecutive times within the same session, and the mean of all trials was presented as the grip strength of the animal.
Under general anesthesia, the gastrocnemius from the left hind limb was isolated from 6-month-old female mice, then mounted on a holder vertically to the dual-mode muscle lever arm system (Aurora Scientific Inc., Ontario, Canada). Muscle functional tests were performed according to our previously established protocol (68). The whole muscle was incubated in the organ bath of the ex vivo muscle functional test system (800A; Aurora Scientific Inc.) containing mammalian Ringer solution (121 mmol/L NaCl, 5.4 mmol/L KCl, 1.2 mmol/L MgSO4·7H2O, 25 mmol/L NaHCO3, 5 mmol/L HEPES, 11.5 mmol/L glucose, and 2.5 mmol/L CaCl2) maintained at room temperature and continuously pumped with a gaseous mixture containing 95% O2 and 5% CO2. A 15-min stabilization period was needed after mounting. The optimal length (Lo) of the muscle was measured after two tetanic contractions (1A, 300 ms duration, 150 Hz stimulation frequency) with 5 min intervals. Under the Lo, the muscle was electronically stimulated two more times by a single stimulus with a 1 min interval to evaluate the twitch characteristic (twitch force, F0). A continuous stimulus was given three times for 300 ms at 80 Hz with 5 min rest to evaluate the tetanic contraction ability (tetanic force, Ft). The contraction strength, contraction time, and half-relaxation time were acquired directly. After the functional test, muscle mass was determined. The muscle cross-sectional area (MCSA) was calculated by dividing the muscle mass by the muscle optimal length (Lo) and the density of mammalian skeletal muscle (1.06 mg/mm3). Normalized to MCSA, the specific twitch force (SF0) and specific tetanic force (SFt) were obtained according to our previously established protocol (68).
Statistical analysis
Results are expressed as the mean ± SEM and were considered significant when P < 0.05. Statistical tests performed were Student’s t-test, z-test, one-way ANOVA, or two-way ANOVA followed by Tukey’s multiple comparison test or Bonferroni post-tests. Analyses were performed using GraphPad Prism (GraphPad Software, La Jolla, CA).
Supplementary Material
Fig S1 Regulation of Bdnf expression and secretion.
Fig S2 Effect of BDNF on cellular metabolism.
Fig S3 Generation of MBKO mice.
Fig S4 Metabolic characteristics of male MBKO mice.
Fig S5 Analysis of β-oxidation in gastrocnemius muscle from female MBKO mice.
Fig S6 Gene expression analysis in gastrocnemius muscle from female MBKO mice.
Fig S7 The ubiquitin-proteasome system is normal in the gastrocnemius muscle of female MBKO mice.
ACKNOWLEDGMENTS
We would like to thank the Oklahoma Medical Research Foundation (OMRF) for performing RNA sequencing analysis; the National Mouse Metabolic Phenotyping Center (MMPC) at University of Massachusetts Medical School for their assistance in performing metabolic studies including hyperinsulinemic-euglycemic clamp, metabolic cages, 1H-MRS body composition analysis, and treadmill exercise phenotyping; the West Coast Metabolomics Center at UC Davis for tissue metabolites analysis; and Dr. Helen Zhi (Biostatistics and Clinical Research Unit) at the University of Hong Kong for statistical analysis consultation.
FUNDING:
This work is supported by grants from the Presbyterian Health Foundation (PHF) Seed Grant Program, HKU Research Committee Seed Funding Programme for Basic Research (201606159006), and the Hong Kong Government Research Grant Council (GRF17113817) to CBC; the National Natural Science Foundation of China (NSFC 81470159), CAMS Initiative for Innovative Medicine (CAMS-I2M 2016-I2M-3-007), and the Drug Innovation Major Project (2018ZX09711001-003-005) to XY; and the National Institute of Health (5U2C-DK093000) to JK.
Footnotes
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
DATA and MATERIALS AVAILABILITY
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplemental Materials.
REFERENCES AND NOTES
- 1.Zurlo F, Larson K, Bogardus C, Ravussin E, Skeletal muscle metabolism is a major determinant of resting energy expenditure. J Clin Invest 86, 1423–1427 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goodpaster BH, Sparks LM, Metabolic Flexibility in Health and Disease. Cell Metab 25, 1027–1036 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gao AW, Canto C, Houtkooper RH, Mitochondrial response to nutrient availability and its role in metabolic disease. EMBO Mol Med 6, 580–589 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Randle PJ, Garland PB, Hales CN, Newsholme EA, The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1, 785–789 (1963). [DOI] [PubMed] [Google Scholar]
- 5.Hue L, Taegtmeyer H, The Randle cycle revisited: a new head for an old hat. Am J Physiol Endocrinol Metab 297, E578–591 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rai M, Demontis F, Systemic Nutrient and Stress Signaling via Myokines and Myometabolites. Annu Rev Physiol 78, 85–107 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Huang EJ, Reichardt LF, Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci 24, 677–736 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Numakawa T, Suzuki S, Kumamaru E, Adachi N, Richards M, Kunugi H, BDNF function and intracellular signaling in neurons. Histol Histopathol 25, 237–258 (2010). [DOI] [PubMed] [Google Scholar]
- 9.Pelleymounter MA, Cullen MJ, Wellman CL, Characteristics of BDNF-induced weight loss. Exp Neurol 131, 229–238 (1995). [DOI] [PubMed] [Google Scholar]
- 10.Kernie SG, Liebl DJ, Parada LF, BDNF regulates eating behavior and locomotor activity in mice. EMBO J 19, 1290–1300 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Unger TJ, Calderon GA, Bradley LC, Sena-Esteves M, Rios M, Selective deletion of Bdnf in the ventromedial and dorsomedial hypothalamus of adult mice results in hyperphagic behavior and obesity. J Neurosci 27, 14265–14274 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shelton DL, Sutherland J, Gripp J, Camerato T, Armanini MP, Phillips HS, Carroll K, Spencer SD, Levinson AD, Human trks: molecular cloning, tissue distribution, and expression of extracellular domain immunoadhesins. J Neurosci 15, 477–491 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matthews VB, Astrom MB, Chan MH, Bruce CR, Krabbe KS, Prelovsek O, Akerstrom T, Yfanti C, Broholm C, Mortensen OH, Penkowa M, Hojman P, Zankari A, Watt MJ, Bruunsgaard H, Pedersen BK, Febbraio MA, Brain-derived neurotrophic factor is produced by skeletal muscle cells in response to contraction and enhances fat oxidation via activation of AMP-activated protein kinase. Diabetologia 52, 1409–1418 (2009). [DOI] [PubMed] [Google Scholar]
- 14.Cuppini R, Sartini S, Agostini D, Guescini M, Ambrogini P, Betti M, Bertini L, Vallasciani M, Stocchi V, Bdnf expression in rat skeletal muscle after acute or repeated exercise. Arch Ital Biol 145, 99–110 (2007). [PubMed] [Google Scholar]
- 15.Ogborn DI, Gardiner PF, Effects of exercise and muscle type on BDNF, NT-4/5, and TrKB expression in skeletal muscle. Muscle Nerve 41, 385–391 (2010). [DOI] [PubMed] [Google Scholar]
- 16.Saka Y, Yoshimura O, Tahara H, Takeda Y, Moriyama H, Maejima H, Tobimatsu Y, The mRNA expression of neurotrophins in different skeletal muscles of young rats. Hiroshima J Med Sci 56, 23–28 (2007). [PubMed] [Google Scholar]
- 17.Slack JP, Grupp IL, Ferguson DG, Rosenthal N, Kranias EG, Ectopic expression of phospholamban in fast-twitch skeletal muscle alters sarcoplasmic reticulum Ca2+ transport and muscle relaxation. J Biol Chem 272, 18862–18868 (1997). [DOI] [PubMed] [Google Scholar]
- 18.Herzig S, Hedrick S, Morantte I, Koo SH, Galimi F, Montminy M, CREB controls hepatic lipid metabolism through nuclear hormone receptor PPAR-gamma. Nature 426, 190–193 (2003). [DOI] [PubMed] [Google Scholar]
- 19.Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ, Greenberg ME, Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron 20, 709–726 (1998). [DOI] [PubMed] [Google Scholar]
- 20.Samec S, Seydoux J, Russell AP, Montani JP, Dulloo AG, Skeletal muscle heterogeneity in fasting-induced upregulation of genes encoding UCP2, UCP3, PPARgamma and key enzymes of lipid oxidation. Pflugers Arch 445, 80–86 (2002). [DOI] [PubMed] [Google Scholar]
- 21.Jornayvaz FR, Shulman GI, Regulation of mitochondrial biogenesis. Essays Biochem 47, 69–84 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chao LC, Wroblewski K, Ilkayeva OR, Stevens RD, Bain J, Meyer GA, Schenk S, Martinez L, Vergnes L, Narkar VA, Drew BG, Hong C, Boyadjian R, Hevener AL, Evans RM, Reue K, Spencer MJ, Newgard CB, Tontonoz P, Skeletal muscle Nur77 expression enhances oxidative metabolism and substrate utilization. J Lipid Res 53, 2610–2619 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chan CB, Liu X, Pradoldej S, Hao C, An J, Yepes M, Luo HR, Ye K, Phosphoinositide 3-kinase enhancer regulates neuronal dendritogenesis and survival in neocortex. J Neurosci 31, 8083–8092 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Christoffolete MA, Linardi CC, de Jesus L, Ebina KN, Carvalho SD, Ribeiro MO, Rabelo R, Curcio C, Martins L, Kimura ET, Bianco AC, Mice with targeted disruption of the Dio2 gene have cold-induced overexpression of the uncoupling protein 1 gene but fail to increase brown adipose tissue lipogenesis and adaptive thermogenesis. Diabetes 53, 577–584 (2004). [DOI] [PubMed] [Google Scholar]
- 25.McCarthy JJ, Andrews JL, McDearmon EL, Campbell KS, Barber BK, Miller BH, Walker JR, Hogenesch JB, Takahashi JS, Esser KA, Identification of the circadian transcriptome in adult mouse skeletal muscle. Physiol Genomics 31, 86–95 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Felig P, Pozefsky T, Marliss E, Cahill GF Jr., Alanine: key role in gluconeogenesis. Science 167, 1003–1004 (1970). [DOI] [PubMed] [Google Scholar]
- 27.Carbone JW, McClung JP, Pasiakos SM, Skeletal muscle responses to negative energy balance: effects of dietary protein. Adv Nutr 3, 119–126 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bujak AL, Crane JD, Lally JS, Ford RJ, Kang SJ, Rebalka IA, Green AE, Kemp BE, Hawke TJ, Schertzer JD, Steinberg GR, AMPK activation of muscle autophagy prevents fasting-induced hypoglycemia and myopathy during aging. Cell Metab 21, 883–890 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, Asara JM, Fitzpatrick J, Dillin A, Viollet B, Kundu M, Hansen M, Shaw RJ, Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331, 456–461 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim J, Kundu M, Viollet B, Guan KL, AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13, 132–141 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nave BT, Ouwens M, Withers DJ, Alessi DR, Shepherd PR, Mammalian target of rapamycin is a direct target for protein kinase B: identification of a convergence point for opposing effects of insulin and amino-acid deficiency on protein translation. Biochem J 344 Pt 2, 427–431 (1999). [PMC free article] [PubMed] [Google Scholar]
- 32.Egan B, Zierath JR, Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab 17, 162–184 (2013). [DOI] [PubMed] [Google Scholar]
- 33.Aguer C, McCoin CS, Knotts TA, Thrush AB, Ono-Moore K, McPherson R, Dent R, Hwang DH, Adams SH, Harper ME, Acylcarnitines: potential implications for skeletal muscle insulin resistance. FASEB J 29, 336–345 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kelley DE, He J, Menshikova EV, Ritov VB, Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 51, 2944–2950 (2002). [DOI] [PubMed] [Google Scholar]
- 35.Chan CB, Liu X, Jung DY, Jun JY, Luo HR, Kim JK, Ye K, Deficiency of phosphoinositide 3-kinase enhancer protects mice from diet-induced obesity and insulin resistance. Diabetes 59, 883–893 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Galgani JE, Moro C, Ravussin E, Metabolic flexibility and insulin resistance. Am J Physiol Endocrinol Metab 295, E1009–1017 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rasmussen P, Brassard P, Adser H, Pedersen MV, Leick L, Hart E, Secher NH, Pedersen BK, Pilegaard H, Evidence for a release of brain-derived neurotrophic factor from the brain during exercise. Exp Physiol 94, 1062–1069 (2009). [DOI] [PubMed] [Google Scholar]
- 38.Seifert T, Brassard P, Wissenberg M, Rasmussen P, Nordby P, Stallknecht B, Adser H, Jakobsen AH, Pilegaard H, Nielsen HB, Secher NH, Endurance training enhances BDNF release from the human brain. Am J Physiol Regul Integr Comp Physiol 298, R372–377 (2010). [DOI] [PubMed] [Google Scholar]
- 39.Radka SF, Holst PA, Fritsche M, Altar CA, Presence of brain-derived neurotrophic factor in brain and human and rat but not mouse serum detected by a sensitive and specific immunoassay. Brain Res 709, 122–301 (1996). [DOI] [PubMed] [Google Scholar]
- 40.Chen C, Wang Y, Zhang J, Ma L, Gu J, Ho G, Contribution of neural cell death to depressive phenotypes of streptozotocin-induced diabetic mice. Dis Model Mech 7, 723–730 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chang RS, Wang SD, Wang YC, Lin LJ, Kao ST, Wang JY, Xiao-Qing-Long-Tang shows preventive effect of asthma in an allergic asthma mouse model through neurotrophin regulation. BMC Complement Altern Med 13, 220 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bergouignan A, Rudwill F, Simon C, Blanc S, Physical inactivity as the culprit of metabolic inflexibility: evidence from bed-rest studies. J Appl Physiol (1985) 111, 1201–1210 (2011). [DOI] [PubMed] [Google Scholar]
- 43.Bergouignan A, Trudel G, Simon C, Chopard A, Schoeller DA, Momken I, Votruba SB, Desage M, Burdge GC, Gauquelin-Koch G, Normand S, Blanc S, Physical inactivity differentially alters dietary oleate and palmitate trafficking. Diabetes 58, 367–376 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bey L, Hamilton MT, Suppression of skeletal muscle lipoprotein lipase activity during physical inactivity: a molecular reason to maintain daily low-intensity activity. J Physiol 551, 673–682 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hedrington MS, Davis SN, Sexual Dimorphism in Glucose and Lipid Metabolism during Fasting, Hypoglycemia, and Exercise. Front Endocrinol (Lausanne) 6, 61 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li F, Zhang JW, Wei R, Luo XG, Zhang JY, Zhou XF, Li CQ, Dai RP, Sex-differential modulation of visceral pain by brain derived neurotrophic factor (BDNF) in rats. Neurosci Lett 478, 184–187 (2010). [DOI] [PubMed] [Google Scholar]
- 47.Monteggia LM, Luikart B, Barrot M, Theobold D, Malkovska I, Nef S, Parada LF, Nestler EJ, Brain-derived neurotrophic factor conditional knockouts show gender differences in depression-related behaviors. Biol Psychiatry 61, 187–197 (2007). [DOI] [PubMed] [Google Scholar]
- 48.Chan CB, Tse MC, Liu X, Zhang S, Schmidt R, Otten R, Liu L, Ye K, Activation of muscular TrkB by its small molecular agonist 7,8-dihydroxyflavone sex-dependently regulates energy metabolism in diet-induced obese mice. Chem Biol 22, 355–368 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carbone DL, Handa RJ, Sex and stress hormone influences on the expression and activity of brain-derived neurotrophic factor. Neuroscience 239, 295–303 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kumar S, Kaur G, Intermittent fasting dietary restriction regimen negatively influences reproduction in young rats: a study of hypothalamo-hypophysial-gonadal axis. PLoS One 8, e52416 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ribas V, Drew BG, Zhou Z, Phun J, Kalajian NY, Soleymani T, Daraei P, Widjaja K, Wanagat J, de Aguiar Vallim TQ, Fluitt AH, Bensinger S, Le T, Radu C, Whitelegge JP, Beaven SW, Tontonoz P, Lusis AJ, Parks BW, Vergnes L, Reue K, Singh H, Bopassa JC, Toro L, Stefani E, Watt MJ, Schenk S, Akerstrom T, Kelly M, Pedersen BK, Hewitt SC, Korach KS, Hevener AL, Skeletal muscle action of estrogen receptor alpha is critical for the maintenance of mitochondrial function and metabolic homeostasis in females. Sci Transl Med 8, 334ra354 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhou J, Zhang H, Cohen RS, Pandey SC, Effects of estrogen treatment on expression of brain-derived neurotrophic factor and cAMP response element-binding protein expression and phosphorylation in rat amygdaloid and hippocampal structures. Neuroendocrinology 81, 294–310 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bonnefont JP, Demaugre F, Prip-Buus C, Saudubray JM, Brivet M, Abadi N, Thuillier L, Carnitine palmitoyltransferase deficiencies. Mol Genet Metab 68, 424–440 (1999). [DOI] [PubMed] [Google Scholar]
- 54.Anichini A, Fanin M, Vianey-Saban C, Cassandrini D, Fiorillo C, Bruno C, Angelini C, Genotype-phenotype correlations in a large series of patients with muscle type CPT II deficiency. Neurol Res 33, 24–32 (2011). [DOI] [PubMed] [Google Scholar]
- 55.Haap M, Thamer C, Machann J, Tschritter O, Loblein K, Kellerer M, Schick F, Jacob S, Haring HU, Stumvoll M, Metabolic characterization of a woman homozygous for the Ser113Leu missense mutation in carnitine palmitoyl transferase II. J Clin Endocrinol Metab 87, 2139–2143 (2002). [DOI] [PubMed] [Google Scholar]
- 56.Berardo A, DiMauro S, Hirano M, A diagnostic algorithm for metabolic myopathies. Curr Neurol Neurosci Rep 10, 118–126 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Joseph AM, Adhihetty PJ, Leeuwenburgh C, Beneficial effects of exercise on age-related mitochondrial dysfunction and oxidative stress in skeletal muscle. J Physiol 594, 5105–5123 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jokl EJ, Blanco G, Disrupted autophagy undermines skeletal muscle adaptation and integrity. Mamm Genome 27, 525–537 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Smith ED, Prieto GA, Tong L, Sears-Kraxberger I, Rice JD, Steward O, Cotman CW, Rapamycin and interleukin-1beta impair brain-derived neurotrophic factor-dependent neuron survival by modulating autophagy. J Biol Chem 289, 20615–20629 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen A, Xiong LJ, Tong Y, Mao M, Neuroprotective effect of brain-derived neurotrophic factor mediated by autophagy through the PI3K/Akt/mTOR pathway. Mol Med Rep 8, 1011–1016 (2013). [DOI] [PubMed] [Google Scholar]
- 61.Bak DH, Zhang E, Yi MH, Kim DK, Lim K, Kim JJ, Kim DW, High omega3-polyunsaturated fatty acids in fat-1 mice prevent streptozotocin-induced Purkinje cell degeneration through BDNF-mediated autophagy. Sci Rep 5, 15465 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim KH, Jeong YT, Oh H, Kim SH, Cho JM, Kim YN, Kim SS, Kim DH, Hur KY, Kim HK, Ko T, Han J, Kim HL, Kim J, Back SH, Komatsu M, Chen H, Chan DC, Konishi M, Itoh N, Choi CS, Lee MS, Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat Med 19, 83–92 (2013). [DOI] [PubMed] [Google Scholar]
- 63.Jeanblanc J, Logrip ML, Janak PH, Ron D, BDNF-mediated regulation of ethanol consumption requires the activation of the MAP kinase pathway and protein synthesis. Eur J Neurosci 37, 607–612 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Burattini S, Ferri P, Battistelli M, Curci R, Luchetti F, Falcieri E, C2C12 murine myoblasts as a model of skeletal muscle development: morpho-functional characterization. Eur J Histochem 48, 223–233 (2004). [PubMed] [Google Scholar]
- 65.Chan CB, Liu X, Ensslin MA, Dillehay DL, Ormandy CJ, Sohn P, Serra R, Ye K, PIKE-A is required for prolactin-mediated STAT5a activation in mammary gland development. EMBO J 29, 956–968 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huynh FK, Green MF, Koves TR, Hirschey MD, Measurement of fatty acid oxidation rates in animal tissues and cell lines. Methods Enzymol 542, 391–405 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kim JK, Hyperinsulinemic-euglycemic clamp to assess insulin sensitivity in vivo. Methods Mol Biol 560, 221–238 (2009). [DOI] [PubMed] [Google Scholar]
- 68.Zhang N, Chow SKH, Leung KS, Lee HH, Cheung WH, An animal model of co-existing sarcopenia and osteoporotic fracture in senescence accelerated mouse prone 8 (SAMP8). Exp Gerontol 97, 1–8 (2017). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1 Regulation of Bdnf expression and secretion.
Fig S2 Effect of BDNF on cellular metabolism.
Fig S3 Generation of MBKO mice.
Fig S4 Metabolic characteristics of male MBKO mice.
Fig S5 Analysis of β-oxidation in gastrocnemius muscle from female MBKO mice.
Fig S6 Gene expression analysis in gastrocnemius muscle from female MBKO mice.
Fig S7 The ubiquitin-proteasome system is normal in the gastrocnemius muscle of female MBKO mice.