

Abstract
Insulin resistance is a common feature of many metabolic disorders. The dramatic rise in the incidence of insulin resistance over the past decade has enhanced focus on its developmental origins. Since various developmental insults ranging from maternal disease, stress, over/undernutrition, and exposure to environmental chemicals can all program the development of insulin resistance, common mechanisms may be involved. This review discusses the possibility that increases in maternal androgens associated with these various insults are key mediators in programming insulin resistance. Additionally, the intermediaries through which androgens misprogram tissue insulin sensitivity, such as changes in inflammatory, oxidative, and lipotoxic states, epigenetic, gut microbiome and insulin, as well as data gaps to be filled are also discussed.
Keywords: fetal, steroids, androgen antagonist, insulin sensitizer
1. Introduction
Insulin resistance is a characteristic feature of many metabolic disorders, including the metabolic syndrome, type 2 diabetes mellitus (T2DM), obesity, non-alcoholic fatty liver disease (NAFLD), polycystic ovary syndrome (PCOS), gestational diabetes mellitus, and others. Traditionally, genetics and lifestyle are considered the main contributors to the development of these disorders of insulin resistance (Zheng et al., 2018, Nilsson and Ling, 2017). However, the dramatic rise in incidence of this pathology during the last decades cannot be adequately explained by genetic predisposition and lifestyle changes alone. Accumulating evidence suggests factors acting during early fetal development are additional contributors (Franks et al., 2013, Vaiserman and Lushchak, 2019). As fetal growth and differentiation are largely dependent on the maternal and intrauterine hormonal milieu during early development, hormones and factors that change the hormonal milieu during the fetal ontogeny may have an important role in the development of insulin resistance. Hormonal influences during fetal differentiation are so critical that even mild and transient changes in maternal and intrauterine hormone levels within accepted physiological limits can affect the trajectory of normal fetal growth (Miranda and Sousa, 2018). Of these hormones, steroid hormones play a significant and widespread role in the long term organization of fetal organ systems (Fowden and Forhead, 2004, Seckl and Holmes, 2007).
Steroidal influences on tissue organization stem from the plasticity of the fetus, a feature entrenched in the physiological process of fetal development and differentiation, which is most clearly evident in the process of sexual differentiation. Prior to sexual differentiation primitive gonads and reproductive structures are bipotential, and based on the genetic sex and under the influence of sex hormones, they develop into either male or female gonads and reproductive organs (Jost, 1983, Jost et al., 1973). The presence of the Y chromosome in the male, for example, leads to production of testis-determining factor (TDF/ sex-determining region Y [SRY]), a transcription factor that directs the development of bipotential gonads into testes. The developing testes produce hormones, including Mullerian Inhibiting Substance/Anti-Mullerian Hormone (MIS/AMH) and testosterone. This hormonal milieu induces regression of the Mullerian ducts, which are otherwise destined to form female reproductive structures, and promotes the development of Wolffian ducts into male reproductive structures. The steroid hormone testosterone further promotes maturation of these structures and organization of the reproductive neuroendocrine axis and reproductive behavior. Gonadal sexual differentiation begins early in gestation in humans (Rabinovici and Jaffe, 1990), and during this critical window, inappropriate exposure of the female fetus (XX chromosomal complement) to excess testosterone leads to virilization of reproductive organs and masculinization of reproductive behaviors (Berenbaum, 2002). These observations are embedded in classical physiology and demonstrate the plasticity of the developing fetus and the importance of the maternal and fetal steroid hormone milieu in shaping fetal developmental trajectories (McEwen, 1992).
This plasticity allows the fetus to respond to the prevailing maternal environment, which induces cellular and molecular changes to allow phenotypic adaptations. This process is termed ‘developmental plasticity’, and in theory aids in improving the offspring’s chances for survival in the external environment (Laubach et al., 2018, Lema, 2020). However, the organizational changes that allow short-term survival often prove to be maladaptive in the long term by augmenting risk for non-communicable diseases such as T2DM (Hales and Barker, 1992), metabolic syndrome (Rinaudo and Wang, 2012), and obesity (Breier et al., 2001). This concept has been formalized as the Developmental Origins of Health and Disease (DOHaD) hypothesis and is of relevance to many maternal and environmental insults (Barker, 2004, Barker, 2007, Barouki et al., 2012, Gluckman and Hanson, 2004, Padmanabhan et al., 2010a, Wadhwa et al., 2009). Hormones, particularly steroid hormones, play a critical role in mediating the influence of these internal and external environmental signals and direct either adaptive or maladaptive responses in the developing fetus (Dufty et al., 2002).
Adaptive organizational changes often remain dormant, requiring an activational step to unmask the programmed phenotypic and behavioral changes. An example of such activational processes is evident in reproductive behavior. While the neurocircuitry required for reproductive behaviors is organized during early development, these pathways remain relatively quiescent prior to puberty, requiring subsequent stimulation by sex steroids to induce appropriate adult reproductive behavior (Romeo, 2003). Similarly, the maladaptive organizations induced during early life in metabolic systems may remain dormant until activated by prevailing postnatal environmental cues, with hormones including steroids serving as mediators of this activational process (Puttabyatappa et al., 2016).
The reprogramming associated with fetal exposure to steroids at abnormal concentrations or at inappropriate times are often detrimental, culminating in a diverse array of adult pathologies. Inappropriate exposure to steroids can occur through disease, maternal stress, adverse maternal nutritional states, and environmental stressors, including exposure to chemicals with the potential to disrupt steroid hormone signaling (Padmanabhan et al., 2016). Since adverse conditions and exposures not only impact the maternal hormonal milieu but also the fetal hormonal environment, the placenta serves as a critical intermediary in this developmental process by acting as a conduit between the maternal and fetal compartments while also serving as a steroidogenic organ (Wooding and Burton, 2008, Strauss et al., 1996); thus, the placenta plays a critical role in regulating fetal exposures to maternal and environmental factors. Data from the Helsinki Birth Cohort have shown that abnormal placenta are linked with development of adult onset cardiometabolic diseases in the offspring (Thornburg et al., 2010). Therefore, conditions that compromise placental function either through direct maternal-fetal transfer or through altered placental steroidogenesis (e.g. as disrupted in preeclampsia, gestational diabetes, and endocrine-disrupting chemicals [EDCs]) (Nugent and Bale, 2015) contribute to inappropriate fetal exposure to steroids with potential consequences on fetal development and the long-term health of the offspring.
2. Androgen programming of insulin resistance
Insulin secreted from pancreatic beta-cells maintains postprandial nutrient homeostasis through stimulation of anabolic processes: increasing glucose influx into muscle and adipose tissue, protein and glycogen synthesis in muscle and liver, lipid synthesis and storage in liver and adipose tissue, and through inhibition of fatty acid oxidation, lipolysis, glycogenolysis, and gluconeogenesis (Guo, 2014). A failure to respond to insulin and achieve these physiological effects defines insulin resistance and results in compensatory increases in beta-cell insulin secretion. Lack of responsiveness to insulin occurs at systemic, tissue, and cellular levels as a result of increases in inflammation, oxidative stress, hypoadiponectinemia, and dyslipidemia; all of which negatively affect insulin action and tissue function (Hocking et al., 2013, Burgos-Moron et al., 2019, Iwabu et al., 2019). At the cellular level, reduced insulin response may result from insulin receptor desensitization or reduced expression, suppression of members of the insulin receptor signaling pathway and associated regulatory network, and mitochondrial and endoplasmic reticulum dysfunction (Flamment et al., 2012, Guo, 2014, Gonzalez-Franquesa and Patti, 2017).
Developmental exposure to various insults can lead to programming of insulin resistance as evident from clinical, epidemiological, and experimental studies (Table 1). While steroid hormones can directly influence insulin action as evident from sex-specific differences in glucose homeostasis and adipose distribution (Varlamov et al., 2015, Mauvais-Jarvis, 2015), the evidence that developmental insults can lead to changes in maternal steroids especially androgens (Table 2) and induce insulin resistance (Table 1) point to androgens as potential culprits in the developmental programming of insulin resistance. Examples of conditions that result in excessive maternal androgen exposure include virilizing congenital adrenal hyperplasia (CAH) and the PCOS. In CAH, mutations in the 21-hydroxylase enzyme, which is required for biosynthesis of mineralocorticoids and glucocorticoids, results in androgen excess with consequential fetal exposure (Parsa and New, 2017); importantly, these offspring exhibit postnatal signs of reduced insulin sensitivity (Tamhane et al, 2018). Evidence indicates that during pregnancy women with PCOS have higher androgen levels with associated disruptions in placental steroidogenesis (Sir-Petermann et al., 2002, Maliqueo et al., 2013), which likely combine to result in excess fetal androgen exposure. Daughters of these women show signs of metabolic derangements, including hyperinsulinemia during adolescence (Sir-Petermann et al., 2009). Maternal stress not only increases maternal glucocorticoids levels (Dunkel Schetter, 2011), but it also induces a hyperandrogenic maternal hormonal milieu (Barrett and Swan, 2015). A 30-year follow-up study reported hyperinsulinemia among offspring born to mothers administrated glucocorticoids during pregnancy (Dalziel et al, 2005). Maternal under- and over-nutrition have both been linked with the programming of obesity, T2DM, and insulin resistance (Vaiserman, 2017, Duque-Guimaraes and Ozanne, 2013). In addition to direct effects of the altered nutritional state, these conditions also alter the maternal androgenic milieu (Mossa et al., 2019, Pasquali and Oriolo, 2019). For example, maternal obesity is associated with increased androgen levels (Pasquali and Oriolo, 2019, Arnon et al., 2016, Whyte et al., 2007, Maliqueo et al., 2017), suggesting a potential role for androgens in the programming of offspring phenotypes in such nutritional models.
Table 1:
Developmental insults with consequential insulin resistance in the offspring.
| Maternal Insult | Model | ||||
|---|---|---|---|---|---|
| Humans | Macaques | Ruminants | Rat | Mice | |
| Disease State | Virilizing CAH (Tamhane et al, 2018) PCOS (Sir-Petermann et al., 2009 |
See table 3 for gestational hyperandrogenic experimental models | |||
| Gestational Diabetes (Kubo et al, 2014) | Maternal hyperglycemia (Aerts et al, 1990; Gauguier et al, 1990) | Gestational diabetes in transgenic mouse line TNDM29 (Ma et al, 2004) | |||
| Undernutrition | Dutch Hunger Winter (Painter et al 2005) Chinese Famine (Li et al, 2010) Leningrad Siege study (Stanner et al, 1997) Pune Maternal Nutrition Study (Yajnik et al, 2008) |
Nutrition restricted sheep (Gardner et al, 2005; Ford et al, 2007; Todd et al, 2009; George et al, 2012) | Maternal caloric restriction (Palou et al, 2012); protein restriction (Fernandez-Twinn et al, 2005); micronutrient (Zinc) deficiency (Jou et al, 2010) | Maternal protein restriction (Bringhenti et al, 2011) |
|
| Overnutrition | Obesity during pregnancy (Boney et al, 2005; Zhang et al, 2011) | Overfed baboons, preweaning (Lewis et al, 1986) High fat diet fed rhesus macaques (McCurdy et al, 2009) |
Overfed sheep (Burt et al, 2007; Long et al, 2010) | Maternal high fat diet (Buckley et al, 2005; Cerf et al, 2005; Taylor et al, 2005); Western diet /high fat-high sugar diet (Nivoit et al, 2009); early postnatal overnutrition and catch-up growth (Berends et al, 2013) |
Maternal high fat diet (Murabayashi et al, 2013; Chang et al, 2019) Western diet /high fat high sugar diet (Samuelsson et al, 2008) |
| Stress | Dutch and Chinese Famines and Seize of Leningard (see above); 1998 ice storm (Dancause et al, 2013); Severe hyperemesis gravidarum (Ayyavoo et al, 2013) |
Heat stress (Camacho et al, 2017); Gestational glucocorticoid administration (Gatford et al, 2000; Long et al, 2012; Long et al, 2013) | Gestational glucocorticoid administration (Nyirenda et al, 1998; O’Regan et al, 2004) | ||
| EDC - Bisphenol A (BPA) | NHANES survey (Lang et al, 2008) | Sheep gestational BPA treatment (Veiga-Lopez et al, 2016) | Gestational BPA treatment (Ma et al, 2013; Li et al, 2014) | Gestational BPA treatment (Alonso-Magdalena et al, 2006; Alonso-Magdalena et al, 2010; Angle et al, 2013) | |
| EDC – Phthalates | Korean Ewha Birth and Growth Cohort Study (Han et al, 2019); Young Taiwanese (YOTA) Cohort Study (Chen et al, 2017) NHANES survey (James-Todd et al, 2012) |
Gestational and lactational Di(2-ethylhexyl) phthalate treatment (Lin et al, 2011) | Gestational and lactational Di(2-ethylhexyl) phthalate treatment (Hunt et al, 2017) | ||
PCOS = polycystic ovary syndrome; CAH = congenital adrenal hyperplasia; TNDM29 = Transient neonatal diabetes mellitus locus 29; EDC = endocrine-disrupting chemical; NHANES = National Health and Nutrition Examination Survey
Table 2:
Induction of hyperandrogenemic maternal hormone milieu by developmental insults.
| Maternal Insult | Maternal Androgen Milieu |
|---|---|
Disease State
|
Sir-Petermann et al, 2002 Parsa and New, 2017 Morisset et al, 2013 Acromite et al, 1999 |
| Undernutrition | Mossa et al, 2019 |
| Overnutrition |
Pasquali and Oriolo, 2019 Maliqueo et al, 2017 Arnon et al, 2016 Whyte et al, 2007 |
| Stress | Barrett and Swan, 2015 |
| EDC - Bisphenol A | Rutkowska and Rachon, 2014 Takeuchi et al, 2004 |
| EDC - Phthalates | Sathyanarayana et al, 2017 |
PCOS = polycystic ovary syndrome; CAH = congenital adrenal hyperplasia; EDC = endocrine-disrupting chemical
Environmental endocrine-disrupting chemicals (EDCs), especially those that have the capacity to influence androgen biosynthesis or bioavailability as well as those that modulate androgen receptor signaling are also potential contributors to androgenic programming of metabolic disorders such as insulin resistance (Diamanti-Kandarakis et al., 2009, Gore et al., 2015). Rapid industrialization has led to widespread fetal and offspring exposure to EDCs through maternal-fetal transfer both across the placenta and via lactation, resulting in exposure during critical susceptibility windows (Masuda et al., 1978, Ando et al., 1985, Bowman and Choudhury, 2016). Many naturally-occurring compounds [e.g phytoestrogens, including genistein and coumestrol] as well as anthropogenic chemicals [e.g industrial solvents and lubricants (e.g. polychlorinated biphenyls (PCBs), polybrominated biphenyls, and dioxins), plasticizers (e.g. bisphenol A (BPA)), pesticides (e.g. dichlorodiphenyltrichloroethane (DDT)), fungicides (e.g. vinclozolin), and pharmaceutical agents (e.g. diethylstilbestrol)] (Kabir et al., 2015) are EDCs that can serve as steroid mimics and activate steroid receptor signaling. Meta-analyses conducted by the National Toxicology Program and analyses of the National Health and Nutrition Examination Survey 2003–2004 data have shown strong associations between the incidence of T2DM and persistent organic pollutants (DDT and PCBs) and BPA (Lang et al., 2008, Taylor et al., 2013). Especially concerning is the fact that many of these EDCs are detectable in pregnant women and cord blood (Woodruff et al., 2011, Lee et al., 2017, Goodrich et al., 2019), thus suggesting the potential for these chemicals to disrupt fetal and offspring developmental trajectories. Maternal exposure to BPA and phthalates have been shown to alter maternal androgen milieu (Takeuchi et al., 2004, Sathyanarayana et al., 2017) raising the possibility that the programming effects of these EDCs may be mediated via alterations in the maternal androgenic milieu. Apart from their agonistic/antagonistic actions (De Falco et al., 2015), EDCs can indirectly affect the prevailing steroid milieu by modulating rate-limiting enzymes involved in steroid hormone biosynthesis and/or metabolism (Sanderson, 2006, Tabb and Blumberg, 2006). Evidence that EDCs affect steroid biosynthesis comes from data demonstrating reduced placental aromatase (CYP19) expression in trophoblast cultures treated with BPA (Chu et al., 2018), reduced 3β-hydroxysteroid dehydrogenase and CYP19 in the JEG 3 placental cell line treated with phthalate metabolites (Xu et al., 2016), and increased 7α-hydroxylase/17,20-lyase (CYP17) expression in ovarian cells exposed to BPA (Zhou et al., 2008). In addition, EDC-mediated disruption in hepatic steroid hormone metabolism could influence steroid bioavailability. EDCs have been shown to bind nuclear receptors such as the aryl hydrocarbon receptor (AhR) and pregnane X receptor (PXR) and regulate the expression of hepatic enzymes that metabolize steroid hormones (Sanderson, 2006, Tabb and Blumberg, 2006, Mikamo et al., 2003). For example, AhR binding of PCB or dioxin upregulates expression of CYP1A1, CYP1A2, and CYP1B1 (Kohn et al., 1993, Wakui et al., 2006, Tarnow et al., 2019), while alkylphenols and DDT acting through PXR inhibit hepatic clearance of estradiol in rats (El-Hefnawy et al., 2017). Overall these data indicate the capacity for EDCs to act indirectly as well as directly to influence the maternal and fetal steroidal milieu and contribute to the programming of insulin resistance, a premise requiring further study.
Experimental animal models provide robust direct evidence in support of steroidal programming of insulin resistance (Table 3). For instance, gestational androgen treatment culminates in insulin resistance during adulthood in female offspring of rodents, sheep, and non-human primates (Abbott et al., 2016, Roland et al., 2010, Cardoso and Padmanabhan, 2019). In sheep, days 30–90 of gestation encompass the period of organization of metabolic systems (Padmanabhan and Veiga-Lopez, 2014). The main metabolic defects observed in adult sheep treated prenatally with testosterone from days 30–90 of gestation include peripheral insulin resistance with compensatory hyperinsulinemia and associated dyslipidemia, oxidative stress, and hepatic steatosis (Puttabyatappa and Padmanabhan, 2017, Padmanabhan et al., 2010b, Veiga-Lopez et al., 2013). Similarly, animal models of gestational glucocorticoid administration have documented adverse fetal programming of the hypothalamic-pituitary-adrenal (HPA) axis and long-term effects during adulthood on cardiometabolic function (Moisiadis and Matthews, 2014, Constantinof et al., 2015). Gestational exposure to EDCs in various animal models also provide direct evidence for the role of steroidal disruption in programming insulin resistance. Such evidence is available for a wide range of EDCs, including BPA and phthalate metabolites (Veiga-Lopez et al., 2016, Provvisiero et al., 2016, Strakovsky et al., 2015). The developmental impact of protein restriction (a model of undernutrition) and high fat/western diet feeding (a model of overnutrition) have been studied in both rodents and nonhuman primates (Ong and Ozanne, 2015, Friedman, 2015, Neri and Edlow, 2015, Nicholas et al., 2016). Although all these conditions are associated with offspring insulin resistance with coincident increases in maternal androgens (Table 2), androgen ablation studies are required to ascertain whether androgens are indeed the programmers of insulin resistance in these scenarios.
Table 3:
Developmental Programming of Insulin Resistance by Gestational Androgen
| Animal Model | Insulin Resistance | Remarks | |
|---|---|---|---|
| Species | Treatment | ||
| Rhesus Macaques | Testosterone GD 40–80 (term 160) |
Yes |
Fetal: transient hyperglycemia and hyperinsulinemia (Abbott et al, 2010) Infants: increased basal insulin and beta cell number (Nicol et al, 2014) Adulthood: impaired disposition index, a measure of beta cell function and the ability to dispose glucose load (Eisner et al 2005) |
| Rhesus Macaques | Testosterone GD 100–115 (term 164) |
Yes | Impaired glucose tolerance in adulthood (Eisner et al, 2005) |
| Sheep (Suffolk) |
Testosterone GD 30–90 (term 147) |
Yes |
Juvenile period: decreased insulin sensitivity (Cardoso et al, 2016; Recabarren et al, 2005) Prepubertal period: decreased (Padmanabhan et al, 2010) or normal (Cardoso et al, 2016; Recabarren et al, 2005) insulin sensitivity based on proximity to puberty Postpubertal/ early adulthood: normal (Cardoso et al, 2016; Recabarren et al, 2005) or increased (Veiga-Lopez et al, 2013) insulin sensitivity Adulthood: Reduced insulin sensitivity (Padmanabhan et al, 2010) |
| Sheep (Suffolk) |
DHT GD 30–90 (term 147) |
Yes | Reduced insulin sensitivity at prepubertal age (Padmanabhan et al, 2010) |
| Sheep (Suffolk) |
Testosterone GD 60–90 (term 147) |
Yes | Reduced insulin sensitivity in adulthood (Padmanabhan et al, 2010) |
| Sheep (Scottish Greyface x Texel) |
Testosterone GD 62–102 (term 147) |
Yes | Adult age: normal glucose homeostasis but elevated basal insulin levels (Hogg et al, 2011) |
| Rat (Wister) |
Testosterone GD 20 (term 21) |
Yes | Single injection of testosterone during gestation impaired insulin sensitivity with normal HOMA index and glucose tolerance (Noroozzadeh et al, 2015) |
| Rat (Wistar) |
Testosterone GD 15–19 (term 21) |
Yes | Female offspring were insulin resistant compared male offspring, and this phenotype was prevented by flutamide or tamoxifen co-administration (Hu et al, 2015) |
| Rat (Sprague Dawley) | DHT GD 16–19 (term 21) |
Yes | Insulin resistance observed at pubertal age (Yan et al, 2013) |
| Rat (Wistar) |
Testosterone (5.0mg daily) GD 16–19 (term 21) |
No | Administration of 5 mg testosterone daily did not affect insulin sensitivity, but animals developed lipid disturbances and hepatic steatosis (Sun et al, 2012) |
| Rat (Sprague Dawley) | DHT Day 35–125 postnatal |
Yes | Hyperglycemia and hyperinsulinemia with increased body weight and perirenal fat at the end of the treatment period1 (Yanes et al, 2011) |
| Rat (Wistar) |
DHT Day 21–111 postnatal |
Yes | Decreased insulin sensitivity at the end of 90 day DHT treatment1 (Mannerås et al, 2007) |
| Rat (Wistar) |
Letrazole Day 21–111 postnatal |
No | No effect on insulin sensitivity at the end of 90 day DHT treatment (Mannerås et al, 2007) |
| Rat (Wistar) |
Estradiol Valerate Single injection Day 56 postnatal |
No | No change in insulin sensitivity or androgen levels (Stener-Victorin et al, 2005) |
| Rat (Sprague Dawley) | DHEA Day 21–41 postnatal |
Yes | Higher fasting glucose and insulin1 (Wang et al, 2004) |
| Rat (Sprague Dawley) | Testosterone Day 28–48 postnatal |
Yes | Hyperinsulinemia with reduced rate of glucose uptake1 (Holmäng et al, 1990) |
| Mouse (C57BL/N6) | Letrazole Day 28–88 postnatal |
Yes | Impaired glucose tolerance at unspecified age1 (Kauffman et al, 2015) |
| Mouse (C57Bl/6J) | DHT GD 16–18 (term 20) |
No | No effect on fasting glucose levels or insulin levels at 3 and 16 weeks of age (Caldwell et al, 2014) |
| Mouse (C57Bl/6J) | DHT Day 21–111 postnatal |
No | No effect on fasting glucose levels or insulin levels at 16 weeks of age1 (Caldwell et al, 2014) |
| Mouse (C57Bl/6J) | DHEA Day 21–111 postnatal |
No | No effect on fasting glucose levels or insulin levels at 16 weeks of age1 (Caldwell et al, 2014) |
| Mouse (C57Bl/6J) | Letrozole Day 21–111 postnatal |
No | No effect on fasting glucose levels or insulin levels at 16 weeks of age1 (Caldwell et al, 2014) |
| Mouse (C57Bl/6J) | DHT Day 19–109 postnatal |
Yes | Glucose intolerant at the end of a 90 day DHT treatment1 (van Houten et al, 2012) |
| Mouse (CBB6/F1) | DHT GD 16–18 (term 20) |
Yes | Prepuberty through adulthood: impaired glucose intolerance (Rolland et al, 2010) |
GD = gestational days; DHT = dihydrotestosterone; DHEA = Dehydroepiandrosterone;
Observations made either during treatment or end of treatment period – whether these effects are programmed or due to activational effects is not known.
a. Androgen programming of peripheral insulin resistance
Peripheral insulin resistance is characterized by clinical findings of hyperglycemia, hyperinsulinemia, and impaired glucose tolerance. Evidence supportive of androgen programming of peripheral insulin resistance in the offspring comes from studies in mice, sheep, and rhesus macaques treated with testosterone during gestation (Roland et al., 2010, Cardoso and Padmanabhan, 2019, Abbott et al., 2016). Comparative studies of sheep treated prenatally with testosterone and dihydrotestosterone (DHT, a non-aromatizable and more active metabolite of testosterone) provide direct evidence that insulin resistance is programmed specifically through the androgenic actions of testosterone (Padmanabhan et al., 2010b). The involvement of androgens in the programming of insulin resistance is also supported by evidence of hyperinsulinemia during adolescence in daughters born to women with PCOS (Sir-Petermann et al., 2009) who have higher androgen levels during pregnancy (Sir-Petermann et al., 2002). The insulin resistance evident in offspring from pregnancies of women with CAH (Reisch et al., 2011), maternal disease states such as PCOS and gestational diabetes (Bianco and Josefson, 2019, Sir-Petermann et al., 2009), nutritional deficit/excess (Godfrey et al., 2017, Ong and Guest, 2018), stress (Facchi et al., 2019, Moisiadis and Matthews, 2014), and environmental EDC exposures (Veiga-Lopez et al., 2016, Gore et al., 2015, Sargis and Simmons, 2019) along with the evidence that these conditions induce increased maternal androgen levels (Table 2) (Barrett and Swan, 2015, Takeuchi et al., 2004, Rutkowska and Rachon, 2014, Sathyanarayana et al., 2017, Maliqueo et al., 2017, Arnon et al., 2016, Whyte et al., 2007, Parsa and New, 2017), point to androgens as a likely culprit in the programming of peripheral insulin resistance, a premise that needs to be formally tested using interventional strategies such as androgen antagonism.
b. Androgen programming of tissue-specific insulin resistance
Disruptions in both insulin-responsive tissues (liver, muscle, and adipose tissue) as well as in insulin-producing tissues (pancreas) collaborate in the development of systemic and tissue-level insulin resistance. Tissue effects of insulin involve ligand binding to the insulin receptor (IR) that triggers downstream signaling events, including phosphorylation of protein kinase B (AKT), mitogen-activated protein kinase/extracellular signal–regulated kinase (MAPK/ERK), and the mammalian target of rapamycin (mTOR) (Guo, 2014, Saltiel and Kahn, 2001). Collectively, these signaling events promote glucose uptake as well as lipid and protein anabolic metabolism. Therefore, impairment at any level of the insulin signaling pathway may result in insulin resistance with consequential hyperglycemia (Rask-Madsen and Kahn, 2012). Evidence to date points to the role that developmental steroid exposures, especially androgens, play in programming tissue-specific insulin sensitivity. The following sections address the androgen programming of insulin sensitivity at metabolic targets, namely, liver, muscle, adipose tissue, pancreas, brain, and cardiac tissues.
(i). Liver
Liver is a critical regulator of metabolic physiology, with insulin playing a central role in glucose, protein, and lipid metabolism. In the liver, insulin reduces blood glucose levels by inhibiting gluconeogenesis and glycogenolysis while promoting glycogen synthesis. Gluconeogenesis is inhibited by insulin-stimulated AKT-mediated phosphorylation of the transcription factor forkhead box protein O1 (FOXO1), which downregulates expression of gluconeogenic enzymes, including phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase (G6Pase) (Guo, 2014). Glycogen synthesis is stimulated through AKT-dependent phosphorylation and inhibition of glycogen synthase kinase-3 beta (GSK3B), which promotes the dephosphorylation and activation of glycogen synthase. AKT-dependent inhibition of tuberous sclerosis protein-2 (TSC2) consequently activates mTORC1 and ribosomal protein S6 kinase (S6K), which promote protein synthesis (Inoki et al., 2002) and stimulate transcription of sterol regulatory element binding protein 1 (SREBP1), resulting in increased expression of lipogenic genes (Ruiz et al., 2014).
Studies addressing the programming of offspring hepatic insulin sensitivity by maternal disease state are not available in humans; however, data from animal models of developmental insults show defects at multiple levels of the insulin signaling pathway. For instance, both male and female offspring born to obese sheep manifest decreased protein abundance of IR, phosphorylated AKT, and phosphorylated FOXO1 (Nicholas et al., 2013). Similarly, male offspring of obese mice manifest decreased levels of insulin receptor substrate (IRS) 1, phosphorylated IRS1, and phosphorylated AKT as well as increased levels of GSK3B (Martin-Gronert et al., 2010). Hyperinsulinemia and hyperglycemia observed in male offspring born to pregnant rats treated with dexamethasone, a model of fetal exposure to excess glucocorticoids, is also accompanied by increased expression and activity of the gluconeogenic enzyme PEPCK, suggesting defects in hepatic insulin signaling (Drake et al., 2005). Gestational treatment with BPA in mice also reduced expression of Irs2, Akt2, G6pc, and Srebp1 in the liver of both male and female offspring (Van der Meer, 2015). All of the above discussed maternal insults (obesity, stress, and EDC exposure) provide evidence in support of the hypothesis that gestational insults that program the development of hepatic insulin resistance and systemic hyperinsulinemia are also associated with increases in maternal androgens (Table 2) (Barrett and Swan, 2015, Maliqueo et al., 2017, Arnon et al., 2016, Whyte et al., 2007, Takeuchi et al., 2004, Rutkowska and Rachon, 2014). The commonality of androgen excess in these pathophysiological states suggests that androgens are a likely contributor to the programming of these metabolic defects. Importantly, the exact developmental changes observed after these programming events may vary depending on both the timing of the insult and the life period during which analyses are performed. For example, while gestational testosterone treatment induces higher mTOR protein level and greater phosphorylation of S6K and GSK3B in the livers of female fetal day 90 sheep suggesting an insulin sensitive state (Lu et al., 2016), IR2, IRS2, AKT, and mTOR mRNA are downregulated in adult life (Nada et al., 2010) indicative of an insulin resistant hepatic phenotype. The reduced hepatic insulin sensitivity during adult life is supported by evidence of reduced phosphorylation of AKT under insulin-stimulated conditions in prenatally testosterone-treated female sheep (Lu et al., 2016). Co-treatment with an androgen antagonist prevented the fetal, but not the adult, changes in insulin signaling, indicating that the consequences of androgen programming during adult life may be subject to activational inputs and thus masked or unmasked by the prevailing adult hormone milieu. For example, increases in modulators of insulin sensitivity such as lipid accumulation in the liver leading to hepatic steatosis, inflammation, and oxidative stress during adult life of all animal models discussed above (McCurdy et al., 2009, Ashino et al., 2012, Lynch et al., 2017, Puttabyatappa et al., 2019b, Shimpi et al., 2017, Puttabyatappa et al., 2017) may aid in maintaining the insulin resistant state programmed by gestational insults. Although hepatic lipid accumulation is also observed in offspring of gestationally DHT-treated mice, the mechanisms by which this develops and the disruptions in the insulin signaling cascade remains to be determined (Caldwell et al., 2014).
(ii). Muscle
Muscle is largely responsible for postprandial glucose disposal, with insulin promoting glucose uptake through AKT stimulation of GLUT4 protein translocation to the cell membrane (Dimitriadis et al., 2011). While disease states such as PCOS are associated with aberrant skeletal muscle gene expression and signaling pathways (Nilsson et al., 2018, Hansen et al., 2019, Shen et al., 2019, Stepto et al., 2019), generational studies addressing developmental insults in androgen programming of muscle-specific insulin signaling disruptions is lacking in humans. However, in animal models of maternal obesity, which show increased maternal androgens levels (Maliqueo et al., 2017, Whyte et al., 2007, Pasquali and Oriolo, 2019, Arnon et al., 2016) and offspring insulin resistance, muscle-specific disruptions in insulin signaling and GLUT4 levels are evident. One study of male offspring born to obese pregnant rats found reduced levels of skeletal muscle Glut4 expression (Simar et al., 2012), while another study in rat offspring (of unspecified sex) had reduced expression of Ir and Glut4 in muscle (Bayol et al., 2005). In sheep models of maternal obesity, insulin resistance in the offspring is associated with reduced expression of insulin signaling intermediates when both male and female offspring were assessed together (Yan et al., 2010). Similarly, female mice born to obese dams exhibited reduced IRS1 and PI3K protein levels as well as decreased AKT phosphorylation at serine residue 473 (i.e. site of activation by insulin) in the skeletal muscle (Shelley et al., 2009). The associated increased levels of maternal androgens during pregnancy that occur in both mice and humans with obesity (Maliqueo et al., 2017, Arnon et al., 2016, Whyte et al., 2007, Pasquali and Oriolo, 2019, Arnon et al., 2016) suggest that androgens may be involved in the programming of offspring muscle insulin resistance in the context of maternal obesity. Studies of gestationally testosterone-treated sheep support this premise with the observation of decreased phosphorylated-GSK3B and GLUT4 protein levels in female fetuses at fetal day 90 and impaired insulin stimulation of AKT phosphorylation in adult female offspring (Lu et al., 2016). Although GLUT4 levels have not been examined during adulthood, the reduced AKT phosphorylation is likely to result in reduced GLUT4 membrane translocation in adult sheep as well, likely resulting in increased glucose levels.
(iii). Adipose
As in muscle, adipose tissue glucose uptake is primarily driven by insulin; however, because adipose tissue accounts for only 5–10% of whole body glucose uptake, it has only a minor role in postprandial glucose regulation (Dimitriadis et al., 2011). Despite this fact, adipose tissue influences the insulin sensitivity of other tissues through release of free fatty acids and adipokines (Rosen and Spiegelman, 2006, Tchernof and Despres, 2013); as such, increased adiposity and adipose dysfunction are major risk factors for the development of insulin resistance (Kahn and Flier, 2000). Because of this role of adipose tissue, the majority of studies examining adipose programming by developmental insults such as maternal obesity is focused on the assessment of adipose tissue phenotype.
The hyperandrogenic state of PCOS likely contributes to the insulin resistance in the absence or presence of obesity (Dumesic et al., 2007). Importantly, the daughters born to mothers with PCOS also manifest insulin resistance (Sir-Petermann et al., 2009), suggesting that the maternal hyperandrogenic state of PCOS may contribute to the developmental programming of insulin resistance across generations. At the level of adipose tissue, several studies in women with PCOS, mostly conducted on subcutaneous adipose tissue (SAT), have found adipose tissue to be insulin resistant (Dumesic et al., 2019, Cree-Green et al., 2015, Cree-Green et al., 2017); other studies have found adipose to be more insulin sensitive (Corbould and Dunaif, 2007, Ciaraldi et al., 2009). In studies demonstrating adipose insulin resistance, disruptions are evident at multiple levels of insulin signaling, including: 1) a rightward shift in the insulin dose–response curve for insulin-stimulated glucose uptake in isolated adipocytes (Ciaraldi et al., 1992); 2) reduced phosphorylation of IRS1 and IRS2 (Wang et al., 2005, Qiu et al., 2005); 3) hyperactivation of GSK3B that is resistant to insulin-stimulated down-regulation (Chang et al., 2008); and 4) decreased expression of GLUT4 (Rosenbaum et al., 1993). These findings are supportive of androgens gestationally programming adipose insulin resistance. Considering that the majority of women with PCOS are also obese, the increased adiposity per se may further amplify the programmed insulin sensitivity defects induced by androgen excess. This premise is supported by several observations, including: 1) treatment of female rhesus macaques with testosterone from menarche (a developmental programming window) coupled with a Western diet leads to greater insulin resistance than that observed in either the testosterone-treated or Western diet-fed conditions alone (True et al., 2017); and 2) gestational testosterone treatment coupled with postnatal obesity in female sheep leads to earlier impairments in insulin sensitivity compared to offspring only exposed to testosterone (Padmanabhan et al., 2010c). Interestingly, gestational testosterone treatment promoted preferential accumulation of metabolically deleterious visceral adipose tissue (VAT) in female rhesus macaques (Eisner et al., 2003), indicating that the steroidal programming of metabolism includes disruption of adipocyte insulin signaling and adipose mass and distribution. Male offspring from dams gestationally stressed by diet-induced obesity in mice (Fernandez-Twinn et al., 2014) and caloric restricted rats (Berends et al., 2013) manifest insulin resistance and impaired adipose insulin signaling characterized by reduced levels of Ir2, Irs1, phosphatidylinositol-3-kinases (Pi3k), and Akt. Associated increases in maternal androgen levels in these models (Table 2) (Maliqueo et al., 2017, Mossa et al., 2019) suggest that programming of insulin resistance in adipose tissue may be androgen-dependent. In stark contrast, a lack of change in the phosphorylation states of AKT, mTOR, ERK, GSK3B, and S6K during fetal life as well as increased mRNA expression of IR2, mTOR, AKT, PI3K, and peroxisome proliferator-activated receptor gamma (PPARγ) in the VAT with increased insulin-stimulated phosphorylated-AKT in the VAT and SAT in adult female offspring of gestationally testosterone-treated sheep (Lu et al., 2016, Nada et al., 2010) reflect an insulin sensitive state in adipose tissue. Whether these differences in adipose insulin sensitivity programmed by gestational insults is due to differences in the adipose depot examined, species- or sex-specific differences, developmental time point studied, or differences in adipocyte differentiation [adipocyte hypertrophy in PCOS and offspring of obese mice (Manneras-Holm et al., 2011, Ibanez et al., 2018, Caldwell et al., 2014) vs. adipocyte hypotrophy in gestational testosterone treated sheep (Cardoso et al., 2016)] requires further examination.
(iv). Pancreas
As insulin resistance is often accompanied by compensatory increases in insulin secretion, impairment in pancreatic beta-cells can contribute to compromised insulin sensitivity, potentially through inappropriate insulin secretion (Shanik et al., 2008). In pregnant women hyperandrogenemia during pregnancy (Gozukara et al., 2015) is associated with the development of gestational diabetes leading to the development of insulin resistance in the offspring (Bianco and Josefson, 2019), disruptions in beta-cell function and development of compensatory hyperinsulinemia (Kelstrup et al., 2013, Singh et al., 2006). Similarly, in rat models of maternal high fat feeding, male offspring have disrupted beta-cell gene expression characteristic of impaired function (Agarwal et al., 2019) and impaired insulin signaling (e.g. reduced IRS1 and AKT with increased FOXO1 expression) (Bringhenti et al., 2016). As high fat diet-fed dams exhibit hyperandrogenemia during gestation (Maliqueo et al., 2017, Arnon et al., 2016, Whyte et al., 2007), androgens may program beta-cell disruptions in the offspring. The identification of pancreatic beta-cell defects with associated elevations in basal insulin secretion in female fetuses born to gestationally testosterone-treated sheep support such a notion (Rae et al., 2013). Another study of testosterone administration at gestational days 62 and 82 showed that female fetuses had increased beta-cell numbers at both fetal and adolescent ages with elevated basal insulin secretion (Ramaswamy et al., 2016), suggesting androgen programming of beta-cell dysfunction that leads to hyperinsulinemia. The observation that this phenotype was evident only in animals that received testosterone and not diethylstilbestrol or dexamethasone during gestation imply specific involvement of androgens in the steroidal programming of beta-cell dysfunction (Ramaswamy et al., 2016).
(v). Heart
Cardiac tissue uses free fatty acids as its primary source of energy; however, it also utilizes glucose during fetal life and periods of stress (Abel, 2004). As in skeletal muscle, insulin influences glucose uptake and storage in the myocardium. Maternal weight gain and obesity are associated with the development of cardiovascular defects (Gaillard, 2015). In sheep models of maternal obesity, offspring manifest hyperinsulinemia with increased activation of AKT, ERK, and mTOR in cardiac tissues with cardiac hypertrophy (Fernandez-Twinn et al., 2012). As gestational obesity is associated with increases in maternal androgens (Table 2) (Maliqueo et al., 2017, Arnon et al., 2016, Whyte et al., 2007) these cardiac changes may be programmed by androgens. In support of this, gestationally testosterone-treated sheep also exhibit elevated expression of mTORC1 and increased phosphorylation of PI3K, AKT, and mTOR with associated cardiac hypertrophy (Vyas et al., 2016).
(vi). Brain
The function of insulin in the brain is not confined to regulation of glucose availability from the periphery. Insulin influences neuronal function, and disruptions in brain insulin action is thought to be a basis for neurodegenerative and mood disorders as well as the metabolic syndrome (Kleinridders et al., 2014). In addition, there is crosstalk between insulin and appetite regulators, including orexigens such as neuropeptide Y (NPY) and anorexigens such as proopiomelanocortin (POMC) (Vogt and Bruning, 2013, Breton, 2013), which influence systemic tissue-level insulin sensitivity. Gestational insults that program peripheral insulin resistance (e.g. diet-induced maternal obesity) also induce insulin resistance in the male rat hippocampus with reduced levels of Ir2, Pi3k, and Akt levels (Gomes et al., 2018). Similar changes have also been observed in female offspring of gestationally testosterone-treated sheep with reduced IR2 levels in the hypothalamic arcuate nucleus, an effect that was reversed with gestational androgen antagonist treatment (Cernea et al., 2016). This data coupled with the fact that maternal androgens are elevated in animal models of diet-induced obesity (Whyte et al., 2007, Arnon et al., 2016, Maliqueo et al., 2017) indicate that insulin resistance in these neurons may be programmed by androgens. Surprisingly, male offspring of gestationally estradiol-treated mice had decreased hypothalamic IR mRNA and protein expression associated with the development of insulin resistance (Wang et al., 2018), indicating the potential for estrogenic mediation of insulin resistance as well. However, the finding that androgen receptor antagonists reversed the decrease in IR expression in the hypothalamic arcuate nucleus in prenatal testosterone-treated sheep effectively rules out estrogen as the mediator in this model and point to androgens as the culprits.
3. Mediators of androgen programming of insulin resistance
Steroid hormones elicit their actions through direct actions and via intermediaries to reorganize pathways involved in insulin production and function. Direct actions may involve signaling through their nuclear or membrane receptors with consequential alterations in gene transcription and signal transduction that regulate metabolic functions (Faulds et al., 2012). Indirect actions may be facilitated via alterations in the inflammatory, oxidative, or lipotoxic state as well as through the involvement of epigenetic modifications. More recently, the role of the gut microbiome in the developmental programming and pathogenesis of insulin resistance has also been suggested.
a. Inflammation
Inflammation is a complex process that involves the production of cytokines and other inflammatory mediators as well as the activation and invasion of immune cells; these effects are associated with the metabolic dysfunction associated with obesity and T2DM (Esser et al., 2014). While physiological inflammatory modulation of processes regulated by adrenocortical steroids are involved in the processes of lung maturation and parturition (Keelan, 2018) as well as in the sex steroid programming of behavioral sexual dimorphism (McCarthy et al., 2017), maternal disease states (e.g. gestational diabetes (Pantham et al., 2015), preeclampsia (Gomez-Lopez et al., 2019, Tenorio et al., 2019), maternal stress, obesity, and exposure to EDCs) that lead to the development of offspring insulin resistance (Godfrey et al., 2017, Facchi et al., 2019, Sargis and Simmons, 2019) are also associated with pathophysiological maternal inflammation (Ingvorsen et al., 2015, Schmatz et al., 2010, Bansal et al., 2018, Dewi et al., 2017, Ozias et al., 2015, Alfaradhi et al., 2016, Dudele et al., 2017, Zota et al., 2018, Kelley et al., 2019a, Gu et al., 2018, Song et al., 2019, Desplats et al., 2019) and an altered androgen milieu (Table 2) (Barrett and Swan, 2015, Maliqueo et al., 2017, Arnon et al., 2016, Whyte et al., 2007, Takeuchi et al., 2004, Rutkowska and Rachon, 2014, Vejrazkova et al., 2014, Acromite et al., 1999, Morisset et al., 2013, Sathyanarayana et al., 2017) in both humans and animals. These findings implicate androgens and inflammatory processes in the programming of insulin resistance. Strong associations exist between inflammatory biomarkers and cortisol in mothers with major depressive disorder (Osborne et al., 2018) as well as placental inflammation following maternal exposure to BPA in the sheep (Song et al., 2019). As these states are associated with an increased androgenic maternal milieu (Barrett and Swan, 2015, Takeuchi et al., 2004, Rutkowska and Rachon, 2014) it is possible that inflammatory state in these situations also involve androgens. The finding that gestational treatment with testosterone leads to placental inflammation in sheep (Kelley et al., 2019b), and the female offspring of these animals develop insulin resistance (Padmanabhan et al., 2010b) lend support to the possibility that gestational androgen-mediated programming of offspring insulin resistance may involve inflammatory pathways. Further studies are needed to establish the absolute contribution of inflammatory processes to programmed metabolic dysfunction and to determine if inflammation is the key mediator of the androgenic programming of insulin resistance.
b. Oxidative stress
Physiological levels of reactive oxygen species (ROS) aid in placentation and fetal development by virtue of their role in cell signaling and gene transcription. However, breakdown of antioxidant defense mechanisms and accumulation of ROS lead to oxidative stress (Lushchak, 2014), with consequential damage to cellular components, including proteins, lipids, DNA, and cellular organelles (Valko et al., 2007). Developmental stress arising from maternal disease states (Tenorio et al., 2019, Zhu et al., 2015), maternal obesity (Valko et al., 2007), and stress (Lorigooini et al., 2019, Venkatesh et al., 2019), as well as from environmental chemical exposures (Veiga-Lopez et al., 2015, Watkins et al., 2015, Ferguson et al., 2014, Puttabyatappa et al., 2019a, Neier et al., 2015) are associated with maternal oxidative stress (Valko et al., 2007, Luo et al., 2006), a known contributor to the developmental origins of insulin resistance (Rodriguez-Rodriguez et al., 2018, Thompson and Al-Hasan, 2012). Because these conditions are also associated with changes in the maternal androgen milieu (Table 2) (Sir-Petermann et al., 2002, Morisset et al., 2013, Acromite et al., 1999, Barrett and Swan, 2015, Maliqueo et al., 2017, Arnon et al., 2016, Whyte et al., 2007, Takeuchi et al., 2004, Rutkowska and Rachon, 2014, Sathyanarayana et al., 2017), the fetal programming of insulin resistance likely involve crosstalk between androgen signaling and oxidative stress pathways. Evidence for involvement of oxidative stress in the androgen programming of insulin resistance also comes from strong associations between steroid hormone-disrupting EDCs (e.g. phenols, parabens, and phthalate metabolites) and maternal and fetal oxidative stress in epidemiologic studies (Veiga-Lopez et al., 2015, Watkins et al., 2015, Ferguson et al., 2014, Puttabyatappa et al., 2019a) along with the induction of offspring insulin resistance after gestational exposure to these same EDCs in animal models (Veiga-Lopez et al., 2016, Rajesh and Balasubramanian, 2014, Angle et al., 2013). Apart from their steroid potential, EDCs such as BPA and phthalates are also associated with increases in maternal androgen levels (Takeuchi et al., 2004, Rutkowska and Rachon, 2014, Sathyanarayana et al., 2017) implicating androgens as potential mediators in inducing a pro-oxidant state. Other studies have shown that high fat diet induces obesity (Hariri and Thibault, 2010), placental oxidative stress (Lin et al., 2011), and insulin resistance in offspring (Matsuzawa-Nagata et al., 2008) and antioxidant supplementation during pregnancy ameliorates hyperinsulinemia in offspring (Sen and Simmons, 2010). These findings suggest a direct involvement of maternal oxidative stress in the programming of offspring insulin resistance. Since maternal obesity is associated with increases in maternal androgen levels (Maliqueo et al., 2017, Arnon et al., 2016, Whyte et al., 2007), androgens may be involved in increasing the oxidative stress. This premise is supported by the observation that gestational exposure to testosterone in sheep leads to increases in placental oxidative stress (Kelley et al., 2019b) and induction of insulin resistance in the female offspring (Padmanabhan et al., 2010b). However, key questions remain regarding the relative contribution of androgen excess and oxidative stress and their interactions in the programming of offspring insulin resistance.
c. Lipotoxicity
Excess accumulation of triglycerides in non-adipose tissues results in both acute and chronic cellular dysfunction, a process termed lipotoxicity (Weinberg, 2006). These effects may be due either to direct lipotoxic effects or indirect induction of inflammation and oxidative stress as a result of lipid peroxide production. Observations in women with gestational diabetes and obesity as well as in animal models mimicking these conditions show increased maternal, placental, and fetal lipid accumulation (Diamant et al., 1982, Dong et al., 2013, Jeve et al., 2015, Li et al., 2011, McCurdy et al., 2009, Pruis et al., 2014, Saben et al., 2014, Visiedo et al., 2013). Since these conditions also result in offspring insulin resistance (Bianco and Josefson, 2019, Matsuzawa-Nagata et al., 2008), lipotoxicity may be a contributing factor in the developmental programming of insulin action. Since these maternal conditions are also associated with increased maternal androgens (Table 2) (Maliqueo et al., 2017, Arnon et al., 2016, Whyte et al., 2007, Morisset et al., 2013), androgens may also play a role in the development of lipotoxicity. The observation in sheep models that gestational testosterone treatment increases placental lipid accumulation (Kelley et al., 2019b) supports such a possibility. Similar increases in placental lipotoxicity are also observed in sheep models of gestational BPA treatment (Song et al., 2019). As BPA exposure during pregnancy can increase maternal androgens (Takeuchi et al., 2004, Rutkowska and Rachon, 2014) and are known to induce insulin resistance (Veiga-Lopez et al., 2016), maternal androgens might contribute to development of later-life insulin resistance through lipotoxicity.
d. Epigenetics
Another mechanism by which steroids program metabolic pathways in offspring involves epigenetic alterations. Epigenetic changes include changes in DNA methylation, histone modifications, and expression of noncoding RNAs, all of which can lead to alterations in gene expression. Considering steroids are known to modulate developmentally-programmed epigenetic changes (Forger, 2018), androgen-induced epigenetic modifications of genes involved in insulin action in insulin target tissues or insulin production from pancreatic beta-cells are likely contributors to the developmental origins of insulin resistance (Kerr et al., 2019, Sharma et al., 2017, Ebrahimi et al., 2019).
Epigenetic modulation via DNA methylation involves addition of a methyl group to cytosines by DNA methyltransferases (DNMTs), resulting in changes in gene transcription (Ciccarone et al., 2018). DNA methylation is involved in the developmental programming of insulin resistance (Davegardh et al., 2018, Zhou et al., 2018), including in conditions associated with maternal disease states (Kamrani et al., 2019, Elliott et al., 2019), obesity (Fernandez-Twinn et al., 2019), stress (Cao-Lei et al., 2016), and following exposure to EDCs (Rahmani et al., 2018, Stel and Legler, 2015). Additional support comes from observations of 1) DNA methylation of hepatic genes involved in insulin and glucocorticoid receptor signaling in mice with fetal exposure to glucocorticoids under conditions of maternal food restriction (Ogawa et al., 2014); 2) the strong association that exists between gestational BPA levels and genome-wide DNA methylation and transcriptome changes in human fetal liver (Faulk et al., 2015); and 3) hypermethylation of DNA in the pancreas and liver of offspring from gestationally BPA-treated mice and rats, respectively (Bansal et al., 2017, Rajesh and Balasubramanian, 2014). Because all of these conditions are also associated with increases in maternal androgen levels (Table 2) (Morisset et al., 2013, Sir-Petermann et al., 2002, Barrett and Swan, 2015, Maliqueo et al., 2017, Arnon et al., 2016, Whyte et al., 2007, Takeuchi et al., 2004, Rutkowska and Rachon, 2014), the possibility exists that DNA methylation changes underlying the programming of insulin resistance in these scenarios may be a consequence of increased maternal androgens. Confirmation of the involvement of androgen-mediated DNA methylation in the programming of insulin resistance requires ablation studies with androgen antagonists and prevention of DNA methylation changes via maternal folic acid supplementation (Ly et al., 2016).
Epigenetic regulation through histone modifications includes acetylation, methylation, ubiquitination, phosphorylation, and sumoylation, all of which affect transcriptional activity through modification of the structure and conformation of chromatin and/or by the differential recruitment of transcriptional co-repressors or co-activators (Bannister and Kouzarides, 2011). Histone alterations are brought about by enzymes that include “writers” such as histone acetyltransferases (HATs) and histone methyltransferases (HMTs) that modify histones as well as “erasers” such as histone deacetylases (HDACs) and lysine demethylases (KDMs) that remove these modifications (Hyun et al., 2017). The impact of developmental insults on the programming of insulin resistance through histone modifications is understudied (Vaiserman and Lushchak, 2019). Evidence of a possible role of steroid-mediated histone modifications in fetal programming comes from the reprogramming of stress responses by fetal glucocorticoid exposures that induce transcriptional changes of genes involved in hypothalamic-pituitary-adrenal axis function through histone modifications (Dirven et al., 2017). Altered expression of the pancreatic beta-cell differentiation gene PDX1 in gestationally protein-restricted and BPA-treated rats (Chang et al., 2016, Park et al., 2008) as well as altered hepatic gene expression in gestationally high fat diet-fed macaques and BPA-treated mice (Aagaard-Tillery et al., 2008, Strakovsky et al., 2015) are both associated with histone modifications. Of interest, these gestational manipulations lead to increases in maternal androgen levels (Barrett and Swan, 2015, Maliqueo et al., 2017, Arnon et al., 2016, Whyte et al., 2007, Takeuchi et al., 2004, Rutkowska and Rachon, 2014) and offspring insulin resistance (Rinaudo and Wang, 2012). Gestational testosterone treatment in sheep also increased expression of PDX1 in female fetuses (Rae et al., 2013), lending support to the possibility that androgen programming of insulin homeostasis may occur via histone modifications, a premise that needs formal testing.
The epigenetic regulation of insulin resistance may also be mediated via changes in noncoding RNAs (De Rosa et al., 2018, Desai et al., 2015), which include microRNAs (miRNAs), short (<18–25 nucleotide), long (exceeding 200 nucleotides), and circular RNAs, of which miRNAs are the best studied. The evidence that steroidal programming involves miRNAs comes from sexual differentiation of neonatal mice brain (Morgan and Bale, 2017). The role of miRNAs in androgen programming of insulin resistance is supported by observations that miRNA expression is increased after maternal insults that promote an androgenic maternal hormone milieu (Table 2) and program insulin resistance, such as maternal disease states (Hemmatzadeh et al., 2019, Poirier et al., 2017), diet-induced obesity (Benatti et al., 2014, Puppala et al., 2018, Mendez-Mancilla et al., 2018), growth restriction (Zheng et al., 2017, Hromadnikova et al., 2019), stress (Cao-Lei et al., 2016, Wu et al., 2016), and BPA exposure (Lin et al., 2017, Martinez-Ibarra et al., 2019) in both humans and animal models. The observation that gestational testosterone treatment increases expression of miRNAs that target insulin signaling, albeit in fetal ovaries (Luense et al., 2011), along with findings that their offspring develop insulin resistance during adulthood (Padmanabhan et al., 2010b), raise the possibility that reprogramming of insulin resistance by androgens may also involve changes in expression of miRNA associated with insulin signaling genes in metabolic tissues.
e. Microbiome
The identification of bacteria and/or their DNA in amniotic fluid, fetal membranes, umbilical cord blood, placenta, and meconium indicate that gut colonization begins during in utero life (Li et al., 2014). Gut microbiota are required for host metabolism and fetal development, while reduced microbial diversity and dysbiosis is associated with gastrointestinal and systemic disorders, including obesity and insulin resistance (Wang et al., 2016). This is confirmed by studies in germ-free animals that consume more calories, become overweight, and exhibit insulin resistance and dyslipidemia (Laugerette et al., 2011). This association of dysbiosis with the development of insulin resistance (Wang et al., 2016, Codagnone et al., 2019) coupled with the observation that infants born to mothers with obesity or stress have altered gut microbiota (Collado et al., 2010, Zijlmans et al., 2015, Codagnone et al., 2019) and increased maternal androgens (Table 2) with consequential programing of offspring insulin resistance collectively suggest the potential for steroidal programming of insulin resistance through alterations in the microbiome. Although the influence of steroids on the gut microbiota have been explored (Huang et al., 2015, Tetel et al., 2018), the role of the microbiome in androgen programming is not established. There is evidence that gestational exposure to stress in mice (Gur et al., 2017), treatment of pregnant rats with the native steroid testosterone (Gur et al., 2017), or exposure of pregnant mice to EDCs such as BPA or synthetic estrogens (Javurek et al., 2016) all lead to altered gut microbiota. Since these gestational insults are associated with increased maternal androgens (Table 2) and can program insulin resistance, it is possible that androgen-induced gut microbial changes may be involved in androgen reprogramming of insulin responsiveness and development of insulin resistance. The finding that transplantation of gut microbiota from woman with PCOS into mice resulted in insulin resistance (Qi et al., 2019) indicates that further studies using fecal microbiota transplantation can be used to assess the relative contribution of gut microbiota in androgen programming of insulin resistance.
f. Insulin
An intriguing hypothesis set forth by published data and evident in clinical practice is the capacity of insulin to drive the development of insulin resistance (Erion and Corkey, 2017, Gavin et al., 1974, Rizza et al., 1985, Shanik et al., 2008). In the context of developmental programming, it is interesting to note that gestational hyperinsulinemia observed in gestational diabetes mellitus and obesity are associated with the development of insulin resistance-associated conditions in the offspring later in life (Ruchat et al., 2013, Bellamy et al., 2009, Nicholas et al., 2016). Importantly, a major hormonal change associated with increased maternal androgens is hyperinsulinemia as androgen-mediated increases in insulin levels during pregnancy is evident in hyperandrogenic pregnant women with PCOS (Radon et al., 1999, Sir-Petermann et al., 2007) and pregnant sheep treated with testosterone (Abi Salloum et al., 2015). As such, programming of offspring insulin resistance by gestational testosterone excess may be mediated via increased levels of insulin itself. Indeed, since multiple conditions are also associated with increase in maternal androgens (Table 2) (Maliqueo et al., 2017, Arnon et al., 2016, Whyte et al., 2007, Morisset et al., 2013), the possibility androgen-mediated increases in maternal insulin levels contribute to programming of insulin resistance cannot be excluded. Moreover, it is interesting to speculate how such a scenario could establish a potential positive feedback loop in which persistent or even transient androgen excess increases insulin levels that then increase insulin resistance prompting further insulin release and potentiation of the effect. Intriguingly, insulin’s potential role as a mediator of androgen-induced insulin resistance is supported by evidence that the metabolic disruptions observed in prenatally testosterone-treated sheep can be ameliorated by co-treatment with an insulin sensitizer (Puttabyatappa et al., 2017). Importantly, while suggesting a potential route by which androgens induce insulin resistance, such a mechanism would also provide support for potential interventions, including insulin sensitizers as well as exercise.
4. Conclusions and Future Directions
The ‘thrifty phenotype hypothesis’ proposed by Hales and Barker first explained how malnutrition during pregnancy leads to poor fetal growth and the programming of maladaptive metabolic responses in the developing fetus (Barker, 2005). One of the key metabolic reprogramming events is the establishment of insulin responsiveness, which leads to the development of insulin resistance. Since then this hypothesis has evolved into the DOHaD hypothesis, which captures the impact of wide ranging insults occurring during critical in utero and postnatal developmental windows on the reprogramming of metabolic functions. Critically, the mechanisms by which these developmental insults program later-life insulin resistance are still unresolved. As discussed above, a commonality among the various insults promoting insulin resistance (e.g. maternal disease states, stress, over/undernutrition, lifestyle, and exposure to EDCs) is alterations in the maternal steroid hormone milieu with specific increases in androgen levels (Figure 1) (Table 2) (Barrett and Swan, 2015, Maliqueo et al., 2017, Arnon et al., 2016, Whyte et al., 2007, Takeuchi et al., 2004, Rutkowska and Rachon, 2014). Since steroids, and especially androgens, play a major role in establishing the program of sexual differentiation at both the gonadal and brain levels, exposure to excess androgens during critical periods of organization of metabolic functions may also influence the programming of metabolic tissues, potentially resulting in maladaptive changes that manifest as adult onset metabolic dysfunction, including insulin resistance. Human observations of insulin resistance among offspring born to mothers with high testosterone levels during pregnancy (e.g. women with PCOS) (Sir-Petermann et al., 2009) as well as in animal models of gestational treatment with androgens (Table 3) (Abbott et al., 2016, Roland et al., 2010, Cardoso and Padmanabhan, 2019) support this conjecture. Despite this evidence, however, further studies are needed to confirm the relative contribution of androgens in programming insulin resistance due to other maternal insults through the use of specific androgen receptor antagonists or other molecular approaches.
Figure 1:
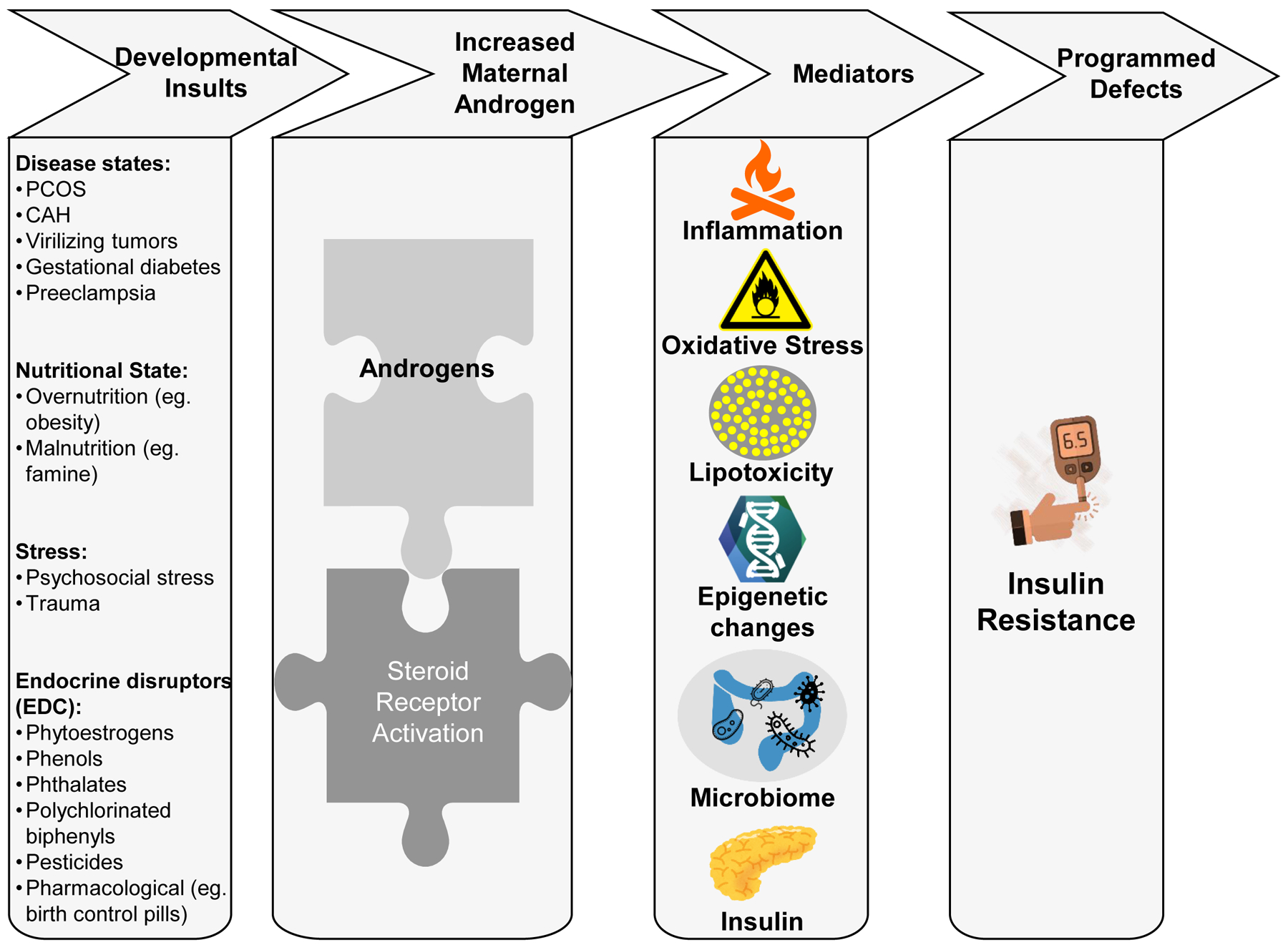
Schematic showing the potential link between developmental insults and increase in maternal androgen levels that program the onset of insulin resistance in the offspring. The increase in maternal androgens probably utilizes various mediators including changes in inflammatory state, oxidative stress, lipotoxicity, epigenetic, gut microbiome and insulin to bring about metabolic reprogramming leading to development of insulin resistance. Images used were sourced from open-source resources: www.pixabay.com, www.openclipart.org, and www.cleanpng.com.
Another key observation arising from this discussion is that many of the studies conducted thus far have been confined to one sex of the offspring. Studies using rodent models have also grouped both sexes together to interpret findings. As it is now established that developmental insults have sex-specific effects (Dearden et al., 2018), future studies in both humans and animal models are required to be performed in both sexes. As sexual dimorphism is also apparent in placental function (Kalisch-Smith et al., 2017), studying the sex-based differences in this key mediator of maternal-fetal transport are also required. Another challenge to consider is the sensitivity of specific developmental periods to respective in utero or postnatal insults since the same exposure at different times during prenatal life can lead to a wide spectrum of effects (Selevan et al., 2000). Additionally, as studies in animal models allow longitudinal and direct assessment of relevant target tissues to empower investigation of potential inter- and trans-generational transfer of traits, the comparability of the most sensitive window(s) for exposures in animal models are essential to provide comparison data across species, which in turn will identify those critical windows of vulnerability to be targeted for policy development and risk management. Considering pregnancies associated with hyperandrogenic state such as PCOS, preeclampsia, obesity, and stress are common in clinical practice, the establishment of androgens as mediators driving adverse metabolic programming in offspring will aid in the development of targeted clinical interventions to overcome the long-term effects of developmental insults that drive insulin resistance by restoring and supporting a normal maternal steroid milieu. Developing such interventions may hold immense potential for addressing the early-life origins of metabolic dysfunction that are epidemic across the globe.
Funding support:
This work was supported by NIH: P01 HD44232 and P30 ES017885 (VP) and P30 ES027792 (RMS). MP’s effort was supported by Ruth L. Kirschstein Institutional Training Grant T32 ES007062.
Footnotes
Disclosure statement: Authors have nothing to disclose
References
- AAGAARD-TILLERY KM, GROVE K, BISHOP J, KE X, FU Q, MCKNIGHT R & LANE RH 2008. Developmental origins of disease and determinants of chromatin structure: maternal diet modifies the primate fetal epigenome. J Mol Endocrinol, 41, 91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ABBOTT DH, BRUNS CR, BARNETT DK, DUNAIF A, GOODFRIEND TL, DUMESIC DA & TARANTAL AF 2010. Experimentally induced gestational androgen excess disrupts glucoregulation in rhesus monkey dams and their female offspring. Am J Physiol Endocrinol Metab, 299, E741–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ABBOTT DH, LEVINE JE & DUMESIC DA 2016. Translational Insight Into Polycystic Ovary Syndrome (PCOS) From Female Monkeys with PCOS-like Traits. Curr Pharm Des, 22, 5625–5633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ABEL ED 2004. Glucose transport in the heart. Front Biosci, 9, 201–15. [DOI] [PubMed] [Google Scholar]
- ABI SALLOUM B, VEIGA-LOPEZ A, ABBOTT DH, BURANT CF & PADMANABHAN V 2015. Developmental programming: exposure to testosterone excess disrupts steroidal and metabolic environment in pregnant sheep. Endocrinology, 156, 2323–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ACROMITE MT, MANTZOROS CS, LEACH RE, HURWITZ J & DOREY LG 1999. Androgens in preeclampsia. Am J Obstet Gynecol, 180, 60–3. [DOI] [PubMed] [Google Scholar]
- AERTS L, HOLEMANS K & VAN ASSCHE FA 1990. Maternal diabetes during pregnancy: consequences for the offspring. Diabetes Metab Rev, 6, 147–67. [DOI] [PubMed] [Google Scholar]
- AGARWAL P, BRAR N, MORRISEAU TS, KERELIUK SM, FONSECA MA, COLE LK, JHA A, XIANG B, HUNT KL, SESHADRI N, et al. 2019. Gestational Diabetes Adversely Affects Pancreatic Islet Architecture and Function in the Male Rat Offspring. Endocrinology, 160, 1907–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ALFARADHI MZ, KUSINSKI LC, FERNANDEZ-TWINN DS, PANTALEAO LC, CARR SK, FERLAND-MCCOLLOUGH D, YEO GS, BUSHELL M & OZANNE SE 2016. Maternal Obesity in Pregnancy Developmentally Programs Adipose Tissue Inflammation in Young, Lean Male Mice Offspring. Endocrinology, 157, 4246–4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ALONSO-MAGDALENA P, MORIMOTO S, RIPOLL C, FUENTES E & NADAL A 2006. The estrogenic effect of bisphenol A disrupts pancreatic beta-cell function in vivo and induces insulin resistance. Environ Health Perspect, 114, 106–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ALONSO-MAGDALENA P, VIEIRA E, SORIANO S, MENES L, BURKS D, QUESADA I & NADAL A 2010. Bisphenol A exposure during pregnancy disrupts glucose homeostasis in mothers and adult male offspring. Environ Health Perspect, 118, 1243–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ANDO M, SAITO H & WAKISAKA I 1985. Transfer of polychlorinated biphenyls (PCBs) to newborn infants through the placenta and mothers’ milk. Arch Environ Contam Toxicol, 14, 51–7. [DOI] [PubMed] [Google Scholar]
- ANGLE BM, DO RP, PONZI D, STAHLHUT RW, DRURY BE, NAGEL SC, WELSHONS WV, BESCH-WILLIFORD CL, PALANZA P, PARMIGIANI S, et al. 2013. Metabolic disruption in male mice due to fetal exposure to low but not high doses of bisphenol A (BPA): evidence for effects on body weight, food intake, adipocytes, leptin, adiponectin, insulin and glucose regulation. Reprod Toxicol, 42, 256–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ARNON L, HAZUT N, TABACHNIK T, WELLER A & KOREN L 2016. Maternal testosterone and reproductive outcome in a rat model of obesity. Theriogenology, 86, 1042–1047. [DOI] [PubMed] [Google Scholar]
- ASHINO NG, SAITO KN, SOUZA FD, NAKUTZ FS, ROMAN EA, VELLOSO LA, TORSONI AS & TORSONI MA 2012. Maternal high-fat feeding through pregnancy and lactation predisposes mouse offspring to molecular insulin resistance and fatty liver. J Nutr Biochem, 23, 341–8. [DOI] [PubMed] [Google Scholar]
- AYYAVOO A, DERRAIK JG, HOFMAN PL, BIGGS J, BLOOMFIELD FH, CORMACK BE, STONE P & CUTFIELD WS 2013. Severe hyperemesis gravidarum is associated with reduced insulin sensitivity in the offspring in childhood. J Clin Endocrinol Metab, 98, 3263–8. [DOI] [PubMed] [Google Scholar]
- BANNISTER AJ & KOUZARIDES T 2011. Regulation of chromatin by histone modifications. Cell Res, 21, 381–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BANSAL A, HENAO-MEJIA J & SIMMONS RA 2018. Immune System: An Emerging Player in Mediating Effects of Endocrine Disruptors on Metabolic Health. Endocrinology, 159, 32–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BANSAL A, RASHID C, XIN F, LI C, POLYAK E, DUEMLER A, VAN DER MEER T, STEFANIAK M, WAJID S, DOLIBA N, et al. 2017. Sex- and Dose-Specific Effects of Maternal Bisphenol A Exposure on Pancreatic Islets of First- and Second-Generation Adult Mice Offspring. Environ Health Perspect, 125, 097022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BARKER DJ 2004. The developmental origins of adult disease. J Am Coll Nutr, 23, 588S–595S. [DOI] [PubMed] [Google Scholar]
- BARKER DJ 2005. The developmental origins of insulin resistance. Horm Res, 64 Suppl 3, 2–7. [DOI] [PubMed] [Google Scholar]
- BARKER DJ 2007. The origins of the developmental origins theory. J Intern Med, 261, 412–7. [DOI] [PubMed] [Google Scholar]
- BAROUKI R, GLUCKMAN PD, GRANDJEAN P, HANSON M & HEINDEL JJ 2012. Developmental origins of non-communicable disease: implications for research and public health. Environ Health, 11, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BARRETT ES & SWAN SH 2015. Stress and Androgen Activity During Fetal Development. Endocrinology, 156, 3435–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAYOL SA, SIMBI BH & STICKLAND NC 2005. A maternal cafeteria diet during gestation and lactation promotes adiposity and impairs skeletal muscle development and metabolism in rat offspring at weaning. J Physiol, 567, 951–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BELLAMY L, CASAS JP, HINGORANI AD & WILLIAMS D 2009. Type 2 diabetes mellitus after gestational diabetes: a systematic review and meta-analysis. Lancet, 373, 1773–9. [DOI] [PubMed] [Google Scholar]
- BENATTI RO, MELO AM, BORGES FO, IGNACIO-SOUZA LM, SIMINO LAP, MILANSKI M, VELLOSO LA, TORSONI MA & TORSONI AS 2014. Maternal high-fat diet consumption modulates hepatic lipid metabolism and microRNA-122 (miR-122) and microRNA-370 (miR-370) expression in offspring. British Journal of Nutrition, 111, 2112–2122. [DOI] [PubMed] [Google Scholar]
- BERENBAUM SA 2002. Prenatal Androgens and Sexual Differentiation of Behavior In: EUGSTER EA & PESCOVITZ OH (eds.) Developmental Endocrinology: From Research to Clinical Practice. Totowa, NJ: Humana Press. [Google Scholar]
- BERENDS LM, FERNANDEZ-TWINN DS, MARTIN-GRONERT MS, CRIPPS RL & OZANNE SE 2013. Catch-up growth following intra-uterine growth-restriction programmes an insulin-resistant phenotype in adipose tissue. Int J Obes (Lond), 37, 1051–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BIANCO ME & JOSEFSON JL 2019. Hyperglycemia During Pregnancy and Long-Term Offspring Outcomes. Curr Diab Rep, 19, 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BONEY CM, VERMA A, TUCKER R & VOHR BR 2005. Metabolic syndrome in childhood: association with birth weight, maternal obesity, and gestational diabetes mellitus. Pediatrics, 115, e290–6. [DOI] [PubMed] [Google Scholar]
- BOWMAN JD & CHOUDHURY M 2016. Phthalates in neonatal health: friend or foe? J Dev Orig Health Dis, 7, 652–664. [DOI] [PubMed] [Google Scholar]
- BREIER BH, VICKERS MH, IKENASIO BA, CHAN KY & WONG WP 2001. Fetal programming of appetite and obesity. Mol Cell Endocrinol, 185, 73–9. [DOI] [PubMed] [Google Scholar]
- BRETON C 2013. The hypothalamus-adipose axis is a key target of developmental programming by maternal nutritional manipulation. J Endocrinol, 216, R19–31. [DOI] [PubMed] [Google Scholar]
- BRINGHENTI I, ORNELLAS F, MANDARIM-DE-LACERDA CA & AGUILA MB 2016. The insulin-signaling pathway of the pancreatic islet is impaired in adult mice offspring of mothers fed a high-fat diet. Nutrition, 32, 1138–43. [DOI] [PubMed] [Google Scholar]
- BUCKLEY AJ, KESERU B, BRIODY J, THOMPSON M, OZANNE SE & THOMPSON CH 2005. Altered body composition and metabolism in the male offspring of high fat-fed rats. Metabolism, 54, 500–7. [DOI] [PubMed] [Google Scholar]
- BURGOS-MORON E, ABAD-JIMENEZ Z, MARANON AM, IANNANTUONI F, ESCRIBANO-LOPEZ I, LOPEZ-DOMENECH S, SALOM C, JOVER A, MORA V, ROLDAN I, et al. 2019. Relationship Between Oxidative Stress, ER Stress, and Inflammation in Type 2 Diabetes: The Battle Continues. J Clin Med, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BURT BE, HESS BW, NATHANIELSZ PW & FORD SP 2007. Flock differences in the impact of maternal dietary restriction on offspring growth and glucose tolerance in female offspring. Soc Reprod Fertil Suppl, 64, 411–24. [DOI] [PubMed] [Google Scholar]
- CAMACHO LE, CHEN X, HAY WW JR. & LIMESAND SW 2017. Enhanced insulin secretion and insulin sensitivity in young lambs with placental insufficiency-induced intrauterine growth restriction. Am J Physiol Regul Integr Comp Physiol, 313, R101–R109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CALDWELL AS, MIDDLETON LJ, JIMENEZ M, DESAI R, MCMAHON AC, ALLAN CM, HANDELSMAN DJ & WALTERS KA 2014. Characterization of reproductive, metabolic, and endocrine features of polycystic ovary syndrome in female hyperandrogenic mouse models. Endocrinology, 155, 3146–59. [DOI] [PubMed] [Google Scholar]
- CAO-LEI L, LAPLANTE DP & KING S 2016. Prenatal Maternal Stress and Epigenetics: Review of the Human Research. Current Molecular Biology Reports, 2, 16–25. [Google Scholar]
- CARDOSO RC & PADMANABHAN V 2019. Prenatal Steroids and Metabolic Dysfunction: Lessons from Sheep. Annu Rev Anim Biosci, 7, 337–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CARDOSO RC, VEIGA-LOPEZ A, MOELLER J, BECKETT E, PEASE A, KELLER E, MADRIGAL V, CHAZENBALK G, DUMESIC D & PADMANABHAN V 2016. Developmental Programming: Impact of Gestational Steroid and Metabolic Milieus on Adiposity and Insulin Sensitivity in Prenatal Testosterone-Treated Female Sheep. Endocrinology, 157, 522–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CERF ME, WILLIAMS K, NKOMO XI, MULLER CJ, DU TOIT DF, LOUW J & WOLFE-COOTE SA 2005. Islet cell response in the neonatal rat after exposure to a high-fat diet during pregnancy. Am J Physiol Regul Integr Comp Physiol, 288, R1122–8. [DOI] [PubMed] [Google Scholar]
- CERNEA M, PHILLIPS R, PADMANABHAN V, COOLEN LM & LEHMAN MN 2016. Prenatal testosterone exposure decreases colocalization of insulin receptors in kisspeptin/neurokinin B/dynorphin and agouti-related peptide neurons of the adult ewe. Eur J Neurosci, 44, 2557–2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHANG E, HAFNER H, VARGHESE M, GRIFFIN C, CLEMENTE J, ISLAM M, CARLSON Z, ZHU A, HAK L, ABRISHAMI S, et al. 2019. Programming effects of maternal and gestational obesity on offspring metabolism and metabolic inflammation. Sci Rep, 9, 16027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHANG H, WANG D, XIA W, PAN X, HUO W, XU S & LI Y 2016. Epigenetic disruption and glucose homeostasis changes following low-dose maternal bisphenol A exposure. Toxicol Res (Camb), 5, 1400–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHANG W, GOODARZI MO, WILLIAMS H, MAGOFFIN DA, PALL M & AZZIZ R 2008. Adipocytes from women with polycystic ovary syndrome demonstrate altered phosphorylation and activity of glycogen synthase kinase 3. Fertil Steril, 90, 2291–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN SY, HWANG JS, SUNG FC, LIN CY, HSIEH CJ, CHEN PC & SU TC 2017. Mono-2-ethylhexyl phthalate associated with insulin resistance and lower testosterone levels in a young population. Environ Pollut, 225, 112–117. [DOI] [PubMed] [Google Scholar]
- CHU PW, YANG ZJ, HUANG HH, CHANG AA, CHENG YC, WU GJ & LAN HC 2018. Low-dose bisphenol A activates the ERK signaling pathway and attenuates steroidogenic gene expression in human placental cells. Biol Reprod, 98, 250–258. [DOI] [PubMed] [Google Scholar]
- CIARALDI TP, ARODA V, MUDALIAR S, CHANG RJ & HENRY RR 2009. Polycystic ovary syndrome is associated with tissue-specific differences in insulin resistance. J Clin Endocrinol Metab, 94, 157–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CIARALDI TP, EL-ROEIY A, MADAR Z, REICHART D, OLEFSKY JM & YEN SS 1992. Cellular mechanisms of insulin resistance in polycystic ovarian syndrome. J Clin Endocrinol Metab, 75, 577–83. [DOI] [PubMed] [Google Scholar]
- CICCARONE F, TAGLIATESTA S, CAIAFA P & ZAMPIERI M 2018. DNA methylation dynamics in aging: how far are we from understanding the mechanisms? Mech Ageing Dev, 174, 3–17. [DOI] [PubMed] [Google Scholar]
- CODAGNONE MG, SPICHAK S, O’MAHONY SM, O’LEARY OF, CLARKE G, STANTON C, DINAN TG & CRYAN JF 2019. Programming Bugs: Microbiota and the Developmental Origins of Brain Health and Disease. Biol Psychiatry, 85, 150–163. [DOI] [PubMed] [Google Scholar]
- COLLADO MC, ISOLAURI E, LAITINEN K & SALMINEN S 2010. Effect of mother’s weight on infant’s microbiota acquisition, composition, and activity during early infancy: a prospective follow-up study initiated in early pregnancy. Am J Clin Nutr, 92, 1023–30. [DOI] [PubMed] [Google Scholar]
- CONSTANTINOF A, MOISIADIS VG & MATTHEWS SG 2015. Programming of stress pathways: A transgenerational perspective. J Steroid Biochem Mol Biol. [DOI] [PubMed] [Google Scholar]
- CORBOULD A & DUNAIF A 2007. The adipose cell lineage is not intrinsically insulin resistant in polycystic ovary syndrome. Metabolism, 56, 716–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CREE-GREEN M, NEWCOMER BR, COE G, NEWNES L, BAUMGARTNER A, BROWN MS, PYLE L, REUSCH JE & NADEAU KJ 2015. Peripheral insulin resistance in obese girls with hyperandrogenism is related to oxidative phosphorylation and elevated serum free fatty acids. Am J Physiol Endocrinol Metab, 308, E726–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CREE-GREEN M, RAHAT H, NEWCOMER BR, BERGMAN BC, BROWN MS, COE GV, NEWNES L, GARCIA-REYES Y, BACON S, THURSTON JE, et al. 2017. Insulin Resistance, Hyperinsulinemia, and Mitochondria Dysfunction in Nonobese Girls With Polycystic Ovarian Syndrome. J Endocr Soc, 1, 931–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DALZIEL SR, WALKER NK, PARAG V, MANTELL C, REA HH, RODGERS A & HARDING JE 2005. Cardiovascular risk factors after antenatal exposure to betamethasone: 30-year follow-up of a randomised controlled trial. Lancet, 365, 1856–62. [DOI] [PubMed] [Google Scholar]
- DANCAUSE KN, VERU F, ANDERSEN RE, LAPLANTE DP & KING S 2013. Prenatal stress due to a natural disaster predicts insulin secretion in adolescence. Early Hum Dev, 89, 773–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAVEGARDH C, GARCIA-CALZON S, BACOS K & LING C 2018. DNA methylation in the pathogenesis of type 2 diabetes in humans. Mol Metab, 14, 12–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE FALCO M, FORTE M & LAFORGIA V 2015. Estrogenic and anti-androgenic endocrine disrupting chemicals and their impact on the male reproductive system. Frontiers in Environmental Science, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE ROSA S, ARCIDIACONO B, CHIEFARI E, BRUNETTI A, INDOLFI C & FOTI DP 2018. Type 2 Diabetes Mellitus and Cardiovascular Disease: Genetic and Epigenetic Links. Front Endocrinol (Lausanne), 9, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEARDEN L, BOURET SG & OZANNE SE 2018. Sex and gender differences in developmental programming of metabolism. Mol Metab, 15, 8–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DESAI M, JELLYMAN JK & ROSS MG 2015. Epigenomics, gestational programming and risk of metabolic syndrome. Int J Obes (Lond), 39, 633–41. [DOI] [PubMed] [Google Scholar]
- DESPLATS P, GUTIERREZ AM, ANTONELLI MC & FRASCH MG 2019. Microglial memory of early life stress and inflammation: Susceptibility to neurodegeneration in adulthood. Neurosci Biobehav Rev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEWI M, CARLSON SE, GUSTAFSON KM, SULLIVAN DK, WICK JA & HULL HR 2017. Programming of infant neurodevelopment by maternal obesity: potential role of maternal inflammation and insulin resistance. Asia Pac J Clin Nutr, 26, S36–S39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DIAMANT YZ, METZGER BE, FREINKEL N & SHAFRIR E 1982. Placental lipid and glycogen content in human and experimental diabetes mellitus. Am J Obstet Gynecol, 144, 5–11. [DOI] [PubMed] [Google Scholar]
- DIAMANTI-KANDARAKIS E, BOURGUIGNON JP, GIUDICE LC, HAUSER R, PRINS GS, SOTO AM, ZOELLER RT & GORE AC 2009. Endocrine-disrupting chemicals: an Endocrine Society scientific statement. Endocr Rev, 30, 293–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DIMITRIADIS G, MITROU P, LAMBADIARI V, MARATOU E & RAPTIS SA 2011. Insulin effects in muscle and adipose tissue. Diabetes Res Clin Pract, 93 Suppl 1, S52–9. [DOI] [PubMed] [Google Scholar]
- DIRVEN BCJ, HOMBERG JR, KOZICZ T & HENCKENS M 2017. Epigenetic programming of the neuroendocrine stress response by adult life stress. J Mol Endocrinol, 59, R11–R31. [DOI] [PubMed] [Google Scholar]
- DONG M, ZHENG Q, FORD SP, NATHANIELSZ PW & REN J 2013. Maternal obesity, lipotoxicity and cardiovascular diseases in offspring. J Mol Cell Cardiol, 55, 111–6. [DOI] [PubMed] [Google Scholar]
- DRAKE AJ, WALKER BR & SECKL JR 2005. Intergenerational consequences of fetal programming by in utero exposure to glucocorticoids in rats. Am J Physiol Regul Integr Comp Physiol, 288, R34–8. [DOI] [PubMed] [Google Scholar]
- DUDELE A, HOUGAARD KS, KJOLBY M, HOKLAND M, WINTHER G, ELFVING B, WEGENER G, NIELSEN AL, LARSEN A, NOHR MK, et al. 2017. Chronic maternal inflammation or high-fat-feeding programs offspring obesity in a sex-dependent manner. Int J Obes (Lond), 41, 1420–1426. [DOI] [PubMed] [Google Scholar]
- DUFTY AM, CLOBERT J & MØLLER AP 2002. Hormones, developmental plasticity and adaptation. Trends in Ecology & Evolution, 17, 190–196. [Google Scholar]
- DUMESIC DA, ABBOTT DH & PADMANABHAN V 2007. Polycystic ovary syndrome and its developmental origins. Rev Endocr Metab Disord, 8, 127–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUMESIC DA, PHAN JD, LEUNG KL, GROGAN TR, DING X, LI X, HOYOS LR, ABBOTT DH & CHAZENBALK GD 2019. Adipose Insulin Resistance in Normal-Weight Women With Polycystic Ovary Syndrome. J Clin Endocrinol Metab, 104, 2171–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUNKEL SCHETTER C 2011. Psychological science on pregnancy: stress processes, biopsychosocial models, and emerging research issues. Annu Rev Psychol, 62, 531–58. [DOI] [PubMed] [Google Scholar]
- DUQUE-GUIMARAES DE & OZANNE SE 2013. Nutritional programming of insulin resistance: causes and consequences. Trends Endocrinol Metab, 24, 525–35. [DOI] [PubMed] [Google Scholar]
- EBRAHIMI R, BAHIRAEE A, NIAZPOUR F, EMAMGHOLIPOUR S & MESHKANI R 2019. The role of microRNAs in the regulation of insulin signaling pathway with respect to metabolic and mitogenic cascades: A review. J Cell Biochem, 120, 19290–19309. [DOI] [PubMed] [Google Scholar]
- EISNER JR, DUMESIC DA, KEMNITZ JW & ABBOTT DH 2000. Timing of prenatal androgen excess determines differential impairment in insulin secretion and action in adult female rhesus monkeys. J Clin Endocrinol Metab, 85, 1206–10. [DOI] [PubMed] [Google Scholar]
- EISNER JR, DUMESIC DA, KEMNITZ JW, COLMAN RJ & ABBOTT DH 2003. Increased adiposity in female rhesus monkeys exposed to androgen excess during early gestation. Obes Res, 11, 279–86. [DOI] [PubMed] [Google Scholar]
- EL-HEFNAWY T, HERNANDEZ C & STABILE LP 2017. The endocrine disrupting alkylphenols and 4,4’-DDT interfere with estrogen conversion and clearance by mouse liver cytosol. Reprod Biol, 17, 185–192. [DOI] [PubMed] [Google Scholar]
- ELLIOTT HR, SHARP GC, RELTON CL & LAWLOR DA 2019. Epigenetics and gestational diabetes: a review of epigenetic epidemiology studies and their use to explore epigenetic mediation and improve prediction. Diabetologia, 62, 2171–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ERION KA & CORKEY BE 2017. Hyperinsulinemia: a Cause of Obesity? Curr Obes Rep, 6, 178–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FACCHI JC, DE LIMA TAL, DE OLIVEIRA LR, DE OLIVEIRA COSTERMANI H, DE SOUZA MIRANDA GD & DE OLIVEIRA JC 2019. Perinatal programming of metabolic diseases: the role of glucocorticoids. Metabolism, 154047. [DOI] [PubMed] [Google Scholar]
- FAULDS MH, ZHAO C, DAHLMAN-WRIGHT K & GUSTAFSSON JA 2012. The diversity of sex steroid action: regulation of metabolism by estrogen signaling. J Endocrinol, 212, 3–12. [DOI] [PubMed] [Google Scholar]
- FAULK C, KIM JH, JONES TR, MCEACHIN RC, NAHAR MS, DOLINOY DC & SARTOR MA 2015. Bisphenol A-associated alterations in genome-wide DNA methylation and gene expression patterns reveal sequence-dependent and non-monotonic effects in human fetal liver. Environ Epigenet, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FERGUSON KK, CANTONWINE DE, RIVERA-GONZALEZ LO, LOCH-CARUSO R, MUKHERJEE B, ANZALOTA DEL TORO LV, JIMENEZ-VELEZ B, CALAFAT AM, YE X, ALSHAWABKEH AN, et al. 2014. Urinary phthalate metabolite associations with biomarkers of inflammation and oxidative stress across pregnancy in Puerto Rico. Environ Sci Technol, 48, 7018–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FERNANDEZ-TWINN DS, ALFARADHI MZ, MARTIN-GRONERT MS, DUQUE-GUIMARAES DE, PIEKARZ A, FERLAND-MCCOLLOUGH D, BUSHELL M & OZANNE SE 2014. Downregulation of IRS-1 in adipose tissue of offspring of obese mice is programmed cell-autonomously through post-transcriptional mechanisms. Mol Metab, 3, 325–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FERNANDEZ-TWINN DS, BLACKMORE HL, SIGGENS L, GIUSSANI DA, CROSS CM, FOO R & OZANNE SE 2012. The programming of cardiac hypertrophy in the offspring by maternal obesity is associated with hyperinsulinemia, AKT, ERK, and mTOR activation. Endocrinology, 153, 5961–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FERNANDEZ-TWINN DS, HJORT L, NOVAKOVIC B, OZANNE SE & SAFFERY R 2019. Intrauterine programming of obesity and type 2 diabetes. Diabetologia, 62, 1789–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FERNANDEZ-TWINN DS, WAYMAN A, EKIZOGLOU S, MARTIN MS, HALES CN & OZANNE SE 2005. Maternal protein restriction leads to hyperinsulinemia and reduced insulin-signaling protein expression in 21-mo-old female rat offspring. Am J Physiol Regul Integr Comp Physiol, 288, R368–73. [DOI] [PubMed] [Google Scholar]
- FLAMMENT M, HAJDUCH E, FERRE P & FOUFELLE F 2012. New insights into ER stress-induced insulin resistance. Trends Endocrinol Metab, 23, 381–90. [DOI] [PubMed] [Google Scholar]
- FORD SP, HESS BW, SCHWOPE MM, NIJLAND MJ, GILBERT JS, VONNAHME KA, MEANS WJ, HAN H & NATHANIELSZ PW 2007. Maternal undernutrition during early to mid-gestation in the ewe results in altered growth, adiposity, and glucose tolerance in male offspring. J Anim Sci, 85, 1285–94. [DOI] [PubMed] [Google Scholar]
- FORGER NG 2018. Past, present and future of epigenetics in brain sexual differentiation. J Neuroendocrinol, 30. [DOI] [PubMed] [Google Scholar]
- FOWDEN AL & FORHEAD AJ 2004. Endocrine mechanisms of intrauterine programming. Reproduction, 127, 515–26. [DOI] [PubMed] [Google Scholar]
- FRANKS PW, PEARSON E & FLOREZ JC 2013. Gene-environment and gene-treatment interactions in type 2 diabetes: progress, pitfalls, and prospects. Diabetes Care, 36, 1413–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FRIEDMAN JE 2015. Obesity and Gestational Diabetes Mellitus Pathways for Programming in Mouse, Monkey, and Man-Where Do We Go Next? The 2014 Norbert Freinkel Award Lecture. Diabetes Care, 38, 1402–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GAILLARD R 2015. Maternal obesity during pregnancy and cardiovascular development and disease in the offspring. Eur J Epidemiol, 30, 1141–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GARDNER DS, TINGEY K, VAN BON BW, OZANNE SE, WILSON V, DANDREA J, KEISLER DH, STEPHENSON T & SYMONDS ME 2005. Programming of glucose-insulin metabolism in adult sheep after maternal undernutrition. Am J Physiol Regul Integr Comp Physiol, 289, R947–54. [DOI] [PubMed] [Google Scholar]
- GATFORD KL, WINTOUR EM, DE BLASIO MJ, OWENS JA & DODIC M 2000. Differential timing for programming of glucose homoeostasis, sensitivity to insulin and blood pressure by in utero exposure to dexamethasone in sheep. Clin Sci (Lond), 98, 553–60. [DOI] [PubMed] [Google Scholar]
- GAUGUIER D, BIHOREAU MT, KTORZA A, BERTHAULT MF & PICON L 1990. Inheritance of diabetes mellitus as consequence of gestational hyperglycemia in rats. Diabetes, 39, 734–9. [DOI] [PubMed] [Google Scholar]
- GAVIN JR 3RD, ROTH J, NEVILLE DM JR., DE MEYTS P & BUELL DN 1974. Insulin-dependent regulation of insulin receptor concentrations: a direct demonstration in cell culture. Proc Natl Acad Sci U S A, 71, 84–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GEORGE LA, ZHANG L, TUERSUNJIANG N, MA Y, LONG NM, UTHLAUT AB, SMITH DT, NATHANIELSZ PW & FORD SP 2012. Early maternal undernutrition programs increased feed intake, altered glucose metabolism and insulin secretion, and liver function in aged female offspring. Am J Physiol Regul Integr Comp Physiol, 302, R795–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GLUCKMAN PD & HANSON MA 2004. The developmental origins of the metabolic syndrome. Trends Endocrinol Metab, 15, 183–7. [DOI] [PubMed] [Google Scholar]
- GODFREY KM, REYNOLDS RM, PRESCOTT SL, NYIRENDA M, JADDOE VW, ERIKSSON JG & BROEKMAN BF 2017. Influence of maternal obesity on the long-term health of offspring. Lancet Diabetes Endocrinol, 5, 53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOMES RM, BUENO FG, SCHAMBER CR, DE MELLO JCP, DE OLIVEIRA JC, FRANCISCO FA, MOREIRA VM, JUNIOR MDF, PEDRINO GR, DE FREITAS MATHIAS PC, et al. 2018. Maternal diet-induced obesity during suckling period programs offspring obese phenotype and hypothalamic leptin/insulin resistance. J Nutr Biochem, 61, 24–32. [DOI] [PubMed] [Google Scholar]
- GOMEZ-LOPEZ N, MOTOMURA K, MILLER D, GARCIA-FLORES V, GALAZ J & ROMERO R 2019. Inflammasomes: Their Role in Normal and Complicated Pregnancies. J Immunol, 203, 2757–2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GONZALEZ-FRANQUESA A & PATTI ME 2017. Insulin Resistance and Mitochondrial Dysfunction. Adv Exp Med Biol, 982, 465–520. [DOI] [PubMed] [Google Scholar]
- GOODRICH JM, INGLE ME, DOMINO SE, TREADWELL MC, DOLINOY DC, BURANT C, MEEKER JD & PADMANABHAN V 2019. First trimester maternal exposures to endocrine disrupting chemicals and metals and fetal size in the Michigan Mother-Infant Pairs study. J Dev Orig Health Dis, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GORE AC, CHAPPELL VA, FENTON SE, FLAWS JA, NADAL A, PRINS GS, TOPPARI J & ZOELLER RT 2015. EDC-2: The Endocrine Society’s Second Scientific Statement on Endocrine-Disrupting Chemicals. Endocr Rev, er20151010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOZUKARA YM, AYTAN H, ERTUNC D, TOK EC, DEMIRTURK F, SAHIN S & AYTAN P 2015. Role of first trimester total testosterone in prediction of subsequent gestational diabetes mellitus. J Obstet Gynaecol Res, 41, 193–8. [DOI] [PubMed] [Google Scholar]
- GU W, WANG Y, QIU Z, DONG J, WANG Y & CHEN J 2018. Maternal exposure to nonylphenol during pregnancy and lactation induces microglial cell activation and pro-inflammatory cytokine production in offspring hippocampus. Sci Total Environ, 634, 525–533. [DOI] [PubMed] [Google Scholar]
- GUO S 2014. Insulin signaling, resistance, and the metabolic syndrome: insights from mouse models into disease mechanisms. J Endocrinol, 220, T1–T23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GUR TL, SHAY L, PALKAR AV, FISHER S, VARALJAY VA, DOWD S & BAILEY MT 2017. Prenatal stress affects placental cytokines and neurotrophins, commensal microbes, and anxiety-like behavior in adult female offspring. Brain Behav Immun, 64, 50–58. [DOI] [PubMed] [Google Scholar]
- HALES CN & BARKER DJ 1992. Type 2 (non-insulin-dependent) diabetes mellitus: the thrifty phenotype hypothesis. Diabetologia, 35, 595–601. [DOI] [PubMed] [Google Scholar]
- HAN H, LEE HA, PARK B, PARK B, HONG YS, HA EH & PARK H 2019. Associations of phthalate exposure with lipid levels and insulin sensitivity index in children: A prospective cohort study. Sci Total Environ, 662, 714–721. [DOI] [PubMed] [Google Scholar]
- HANSEN SL, SVENDSEN PF, JEPPESEN JF, HOEG LD, ANDERSEN NR, KRISTENSEN JM, NILAS L, LUNDSGAARD AM, WOJTASZEWSKI JFP, MADSBAD S et al. 2019. Molecular Mechanisms in Skeletal Muscle Underlying Insulin Resistance in Women Who Are Lean With Polycystic Ovary Syndrome. J Clin Endocrinol Metab, 104, 1841–1854. [DOI] [PubMed] [Google Scholar]
- HARIRI N & THIBAULT L 2010. High-fat diet-induced obesity in animal models. Nutr Res Rev, 23, 270–99. [DOI] [PubMed] [Google Scholar]
- HEMMATZADEH M, SHOMALI N, YOUSEFZADEH Y, MOHAMMADI H, GHASEMZADEH A & YOUSEFI M 2019. MicroRNAs: Small molecules with a large impact on pre-eclampsia. J Cell Physiol. [DOI] [PubMed] [Google Scholar]
- HOCKING S, SAMOCHA-BONET D, MILNER KL, GREENFIELD JR & CHISHOLM DJ 2013. Adiposity and insulin resistance in humans: the role of the different tissue and cellular lipid depots. Endocr Rev, 34, 463–500. [DOI] [PubMed] [Google Scholar]
- HOGG K, WOOD C, MCNEILLY AS & DUNCAN WC 2011. The in utero programming effect of increased maternal androgens and a direct fetal intervention on liver and metabolic function in adult sheep. PLoS One, 6, e24877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOLMANG A, SVEDBERG J, JENNISCHE E & BJORNTORP P 1990. Effects of testosterone on muscle insulin sensitivity and morphology in female rats. Am J Physiol, 259, E555–60. [DOI] [PubMed] [Google Scholar]
- HROMADNIKOVA I, DVORAKOVA L, KOTLABOVA K & KROFTA L 2019. The Prediction of Gestational Hypertension, Preeclampsia and Fetal Growth Restriction via the First Trimester Screening of Plasma Exosomal C19MC microRNAs. Int J Mol Sci, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HU M, RICHARD JE, MALIQUEO M, KOKOSAR M, FORNES R, BENRICK A, JANSSON T, OHLSSON C, WU X, SKIBICKA KP et al. 2015. Maternal testosterone exposure increases anxiety-like behavior and impacts the limbic system in the offspring. Proc Natl Acad Sci U S A, 112, 14348–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUNT BG, WANG YL, CHEN MS, WANG SC & WALTZ SE 2017. Maternal diethylhexyl phthalate exposure affects adiposity and insulin tolerance in offspring in a PCNA-dependent manner. Environ Res, 159, 588–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUANG EY, INOUE T, LEONE VA, DALAL S, TOUW K, WANG Y, MUSCH MW, THERIAULT B, HIGUCHI K, DONOVAN S, et al. 2015. Using corticosteroids to reshape the gut microbiome: implications for inflammatory bowel diseases. Inflamm Bowel Dis, 21, 963–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HYUN K, JEON J, PARK K & KIM J 2017. Writing, erasing and reading histone lysine methylations. Exp Mol Med, 49, e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IBANEZ CA, VAZQUEZ-MARTINEZ M, LEON-CONTRERAS JC, REYES-CASTRO LA, RODRIGUEZ-GONZALEZ GL, BAUTISTA CJ, NATHANIELSZ PW & ZAMBRANO E 2018. Different Statistical Approaches to Characterization of Adipocyte Size in Offspring of Obese Rats: Effects of Maternal or Offspring Exercise Intervention. Front Physiol, 9, 1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- INGVORSEN C, BRIX S, OZANNE SE & HELLGREN LI 2015. The effect of maternal Inflammation on foetal programming of metabolic disease. Acta Physiol (Oxf), 214, 440–9. [DOI] [PubMed] [Google Scholar]
- INOKI K, LI Y, ZHU T, WU J & GUAN KL 2002. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol, 4, 648–57. [DOI] [PubMed] [Google Scholar]
- IWABU M, OKADA-IWABU M, YAMAUCHI T & KADOWAKI T 2019. Adiponectin/AdipoR Research and Its Implications for Lifestyle-Related Diseases. Front Cardiovasc Med, 6, 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JAMES-TODD T, STAHLHUT R, MEEKER JD, POWELL SG, HAUSER R, HUANG T & RICH-EDWARDS J 2012. Urinary phthalate metabolite concentrations and diabetes among women in the National Health and Nutrition Examination Survey (NHANES) 2001–2008. Environ Health Perspect, 120, 1307–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JAVUREK AB, SPOLLEN WG, JOHNSON SA, BIVENS NJ, BROMERT KH, GIVAN SA & ROSENFELD CS 2016. Effects of exposure to bisphenol A and ethinyl estradiol on the gut microbiota of parents and their offspring in a rodent model. Gut Microbes, 7, 471–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JEVE YB, KONJE JC & DOSHANI A 2015. Placental dysfunction in obese women and antenatal surveillance strategies. Best Pract Res Clin Obstet Gynaecol, 29, 350–64. [DOI] [PubMed] [Google Scholar]
- JOST A 1983. Genetic and hormonal factors in sex differentiation of the brain. Psychoneuroendocrinology, 8, 183–93. [DOI] [PubMed] [Google Scholar]
- JOST A, VIGIER B, PREPIN J & PERCHELLET JP 1973. Studies on sex differentiation in mammals. Recent Prog Horm Res, 29, 1–41. [DOI] [PubMed] [Google Scholar]
- JOU MY, PHILIPPS AF & LONNERDAL B 2010. Maternal zinc deficiency in rats affects growth and glucose metabolism in the offspring by inducing insulin resistance postnatally. J Nutr, 140, 1621–7. [DOI] [PubMed] [Google Scholar]
- KABIR ER, RAHMAN MS & RAHMAN I 2015. A review on endocrine disruptors and their possible impacts on human health. Environ Toxicol Pharmacol, 40, 241–58. [DOI] [PubMed] [Google Scholar]
- KAHN BB & FLIER JS 2000. Obesity and insulin resistance. J Clin Invest, 106, 473–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KALISCH-SMITH JI, SIMMONS DG, DICKINSON H & MORITZ KM 2017. Review: Sexual dimorphism in the formation, function and adaptation of the placenta. Placenta, 54, 10–16. [DOI] [PubMed] [Google Scholar]
- KAMRANI A, ALIPOURFARD I, AHMADI-KHIAVI H, YOUSEFI M, ROSTAMZADEH D, IZADI M & AHMADI M 2019. The role of epigenetic changes in preeclampsia. Biofactors, 45, 712–724. [DOI] [PubMed] [Google Scholar]
- KAUFFMAN AS, THACKRAY VG, RYAN GE, TOLSON KP, GLIDEWELL-KENNEY CA, SEMAAN SJ, POLING MC, IWATA N, BREEN KM, DULEBA AJ, et al. 2015. A Novel Letrozole Model Recapitulates Both the Reproductive and Metabolic Phenotypes of Polycystic Ovary Syndrome in Female Mice. Biol Reprod, 93, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KEELAN JA 2018. Intrauterine inflammatory activation, functional progesterone withdrawal, and the timing of term and preterm birth. J Reprod Immunol, 125, 89–99. [DOI] [PubMed] [Google Scholar]
- KELLEY AS, BANKER M, GOODRICH JM, DOLINOY DC, BURANT C, DOMINO SE, SMITH YR, SONG PXK & PADMANABHAN V 2019a. Early pregnancy exposure to endocrine disrupting chemical mixtures are associated with inflammatory changes in maternal and neonatal circulation. Sci Rep, 9, 5422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KELLEY AS, PUTTABYATAPPA M, CIARELLI JN, ZENG L, SMITH YR, LIEBERMAN R, PENNATHUR S & PADMANABHAN V 2019b. Prenatal Testosterone Excess Disrupts Placental Function in a Sheep Model of Polycystic Ovary Syndrome. Endocrinology, 160, 2663–2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KELSTRUP L, DAMM P, MATHIESEN ER, HANSEN T, VAAG AA, PEDERSEN O & CLAUSEN TD 2013. Insulin resistance and impaired pancreatic beta-cell function in adult offspring of women with diabetes in pregnancy. J Clin Endocrinol Metab, 98, 3793–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KERR AG, SINHA I, DADVAR S, ARNER P & DAHLMAN I 2019. Epigenetic regulation of diabetogenic adipose morphology. Mol Metab, 25, 159–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KLEINRIDDERS A, FERRIS HA, CAI W & KAHN CR 2014. Insulin action in brain regulates systemic metabolism and brain function. Diabetes, 63, 2232–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOHN MC, LUCIER GW, CLARK GC, SEWALL C, TRITSCHER AM & PORTIER CJ 1993. A mechanistic model of effects of dioxin on gene expression in the rat liver. Toxicol Appl Pharmacol, 120, 138–54. [DOI] [PubMed] [Google Scholar]
- KUBO A, FERRARA A, WINDHAM GC, GREENSPAN LC, DEARDORFF J, HIATT RA, QUESENBERRY CP JR., LAURENT C, MIRABEDI AS & KUSHI LH 2014. Maternal hyperglycemia during pregnancy predicts adiposity of the offspring. Diabetes Care, 37, 2996–3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LANG IA, GALLOWAY TS, SCARLETT A, HENLEY WE, DEPLEDGE M, WALLACE RB & MELZER D 2008. Association of urinary bisphenol A concentration with medical disorders and laboratory abnormalities in adults. JAMA, 300, 1303–10. [DOI] [PubMed] [Google Scholar]
- LAUBACH ZM, PERNG W, DOLINOY DC, FAULK CD, HOLEKAMP KE & GETTY T 2018. Epigenetics and the maintenance of developmental plasticity: extending the signalling theory framework. Biol Rev Camb Philos Soc, 93, 1323–1338. [DOI] [PubMed] [Google Scholar]
- LAUGERETTE F, VORS C, PERETTI N & MICHALSKI MC 2011. Complex links between dietary lipids, endogenous endotoxins and metabolic inflammation. Biochimie, 93, 39–45. [DOI] [PubMed] [Google Scholar]
- LEE WC, FISHER M, DAVIS K, ARBUCKLE TE & SINHA SK 2017. Identification of chemical mixtures to which Canadian pregnant women are exposed: The MIREC Study. Environ Int, 99, 321–330. [DOI] [PubMed] [Google Scholar]
- LEMA SC 2020. Hormones, developmental plasticity, and adaptive evolution: Endocrine flexibility as a catalyst for ‘plasticity-first’ phenotypic divergence. Mol Cell Endocrinol, 502, 110678. [DOI] [PubMed] [Google Scholar]
- LEWIS DS, BERTRAND HA, MCMAHAN CA, MCGILL HC JR., CAREY KD & MASORO EJ 1986. Preweaning food intake influences the adiposity of young adult baboons. J Clin Invest, 78, 899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI M, SLOBODA DM & VICKERS MH 2011. Maternal obesity and developmental programming of metabolic disorders in offspring: evidence from animal models. Exp Diabetes Res, 2011, 592408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI M, WANG M & DONOVAN SM 2014. Early development of the gut microbiome and immune-mediated childhood disorders. Semin Reprod Med, 32, 74–86. [DOI] [PubMed] [Google Scholar]
- LI G, CHANG H, XIA W, MAO Z, LI Y & XU S 2014. F0 maternal BPA exposure induced glucose intolerance of F2 generation through DNA methylation change in Gck. Toxicol Lett, 228, 192–9. [DOI] [PubMed] [Google Scholar]
- LI Y, HE Y, QI L, JADDOE VW, FESKENS EJ, YANG X, MA G & HU FB 2010. Exposure to the Chinese famine in early life and the risk of hyperglycemia and type 2 diabetes in adulthood. Diabetes, 59, 2400–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIN Y, WEI J, LI Y, CHEN J, ZHOU Z, SONG L, WEI Z, LV Z, CHEN X, XIA W et al. 2011. Developmental exposure to di(2-ethylhexyl) phthalate impairs endocrine pancreas and leads to long-term adverse effects on glucose homeostasis in the rat. Am J Physiol Endocrinol Metab, 301, E527–38. [DOI] [PubMed] [Google Scholar]
- LIN Y, DING D, HUANG Q, LIU Q, LU H, LU Y, CHI Y, SUN X, YE G, ZHU H, et al. 2017. Downregulation of miR-192 causes hepatic steatosis and lipid accumulation by inducing SREBF1: Novel mechanism for bisphenol A-triggered non-alcoholic fatty liver disease. Biochim Biophys Acta Mol Cell Biol Lipids, 1862, 869–882. [DOI] [PubMed] [Google Scholar]
- LIN Y, HAN XF, FANG ZF, CHE LQ, NELSON J, YAN TH & WU D 2011. Beneficial effects of dietary fibre supplementation of a high-fat diet on fetal development in rats. Br J Nutr, 106, 510–8. [DOI] [PubMed] [Google Scholar]
- LONG NM, GEORGE LA, UTHLAUT AB, SMITH DT, NIJLAND MJ, NATHANIELSZ PW & FORD SP 2010. Maternal obesity and increased nutrient intake before and during gestation in the ewe results in altered growth, adiposity, and glucose tolerance in adult offspring. J Anim Sci, 88, 3546–53. [DOI] [PubMed] [Google Scholar]
- LONG NM, SHASA DR, FORD SP & NATHANIELSZ PW 2012. Growth and insulin dynamics in two generations of female offspring of mothers receiving a single course of synthetic glucocorticoids. Am J Obstet Gynecol, 207, 203 e1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LONG NM, SMITH DT, FORD SP & NATHANIELSZ PW 2013. Elevated glucocorticoids during ovine pregnancy increase appetite and produce glucose dysregulation and adiposity in their granddaughters in response to ad libitum feeding at 1 year of age. Am J Obstet Gynecol, 209, 353 e1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LORIGOOINI Z, SADEGHI DEHSAHRAEI K, BIJAD E, HABIBIAN DEHKORDI S & AMINI-KHOEI H 2019. Trigonelline through the Attenuation of Oxidative Stress Exerts Antidepressant- and Anxiolytic-Like Effects in a Mouse Model of Maternal Separation Stress. Pharmacology, 1–11. [DOI] [PubMed] [Google Scholar]
- LU C, CARDOSO RC, PUTTABYATAPPA M & PADMANABHAN V 2016. Developmental Programming: Prenatal Testosterone Excess and Insulin Signaling Disruptions in Female Sheep. Biol Reprod, 94, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LUENSE LJ, VEIGA-LOPEZ A, PADMANABHAN V & CHRISTENSON LK 2011. Developmental programming: gestational testosterone treatment alters fetal ovarian gene expression. Endocrinology, 152, 4974–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LUO Z, FRASER W, JULIEN P, DEAL C, AUDIBERT F, SMITH G, XIONG X & WALKER M 2006. Tracing the origins of “fetal origins” of adult diseases: programming by oxidative stress? Medical hypotheses, 66, 38–44. [DOI] [PubMed] [Google Scholar]
- LUSHCHAK VI 2014. Free radicals, reactive oxygen species, oxidative stress and its classification. Chem Biol Interact, 224, 164–75. [DOI] [PubMed] [Google Scholar]
- LY A, ISHIGURO L, KIM D, IM D, KIM SE, SOHN KJ, CROXFORD R & KIM YI 2016. Maternal folic acid supplementation modulates DNA methylation and gene expression in the rat offspring in a gestation period-dependent and organ-specific manner. J Nutr Biochem, 33, 103–10. [DOI] [PubMed] [Google Scholar]
- LYNCH C, CHAN CS & DRAKE AJ 2017. Early life programming and the risk of non-alcoholic fatty liver disease. J Dev Orig Health Dis, 8, 263–272. [DOI] [PubMed] [Google Scholar]
- MA D, SHIELD JP, DEAN W, LECLERC I, KNAUF C, BURCELIN RR, RUTTER GA & KELSEY G 2004. Impaired glucose homeostasis in transgenic mice expressing the human transient neonatal diabetes mellitus locus, TNDM. J Clin Invest, 114, 339–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MA Y, XIA W, WANG DQ, WAN YJ, XU B, CHEN X, LI YY & XU SQ 2013. Hepatic DNA methylation modifications in early development of rats resulting from perinatal BPA exposure contribute to insulin resistance in adulthood. Diabetologia, 56, 2059–67. [DOI] [PubMed] [Google Scholar]
- MALIQUEO M, CRUZ G, ESPINA C, CONTRERAS I, GARCIA M, ECHIBURU B & CRISOSTO N 2017. Obesity during pregnancy affects sex steroid concentrations depending on fetal gender. Int J Obes (Lond), 41, 1636–1645. [DOI] [PubMed] [Google Scholar]
- MALIQUEO M, LARA HE, SANCHEZ F, ECHIBURU B, CRISOSTO N & SIR-PETERMANN T 2013. Placental steroidogenesis in pregnant women with polycystic ovary syndrome. Eur J Obstet Gynecol Reprod Biol, 166, 151–5. [DOI] [PubMed] [Google Scholar]
- MANNERAS L, CAJANDER S, HOLMANG A, SELESKOVIC Z, LYSTIG T, LONN M & STENER-VICTORIN E 2007. A new rat model exhibiting both ovarian and metabolic characteristics of polycystic ovary syndrome. Endocrinology, 148, 3781–91. [DOI] [PubMed] [Google Scholar]
- MANNERAS-HOLM L, LEONHARDT H, KULLBERG J, JENNISCHE E, ODEN A, HOLM G, HELLSTROM M, LONN L, OLIVECRONA G, STENER-VICTORIN E et al. 2011. Adipose tissue has aberrant morphology and function in PCOS: enlarged adipocytes and low serum adiponectin, but not circulating sex steroids, are strongly associated with insulin resistance. J Clin Endocrinol Metab, 96, E304–11. [DOI] [PubMed] [Google Scholar]
- MARTIN-GRONERT MS, FERNANDEZ-TWINN DS, POSTON L & OZANNE SE 2010. Altered hepatic insulin signalling in male offspring of obese mice. J Dev Orig Health Dis, 1, 184–91. [DOI] [PubMed] [Google Scholar]
- MARTINEZ-IBARRA A, MARTINEZ-RAZO LD, VAZQUEZ-MARTINEZ ER, MARTINEZ-CRUZ N, FLORES-RAMIREZ R, GARCIA-GOMEZ E, LOPEZ-LOPEZ M, ORTEGA-GONZALEZ C, CAMACHO-ARROYO I & CERBON M 2019. Unhealthy Levels of Phthalates and Bisphenol A in Mexican Pregnant Women with Gestational Diabetes and Its Association to Altered Expression of miRNAs Involved with Metabolic Disease. Int J Mol Sci, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MASUDA Y, KAGAWA R, KUROKI H, KURATSUNE M, YOSHIMURA T, TAKI I, KUSUDA M, YAMASHITA F & HAYASHI M 1978. Transfer of polychlorinated biphenyls from mothers to foetuses and infants. Food Cosmet Toxicol, 16, 543–6. [DOI] [PubMed] [Google Scholar]
- MATSUZAWA-NAGATA N, TAKAMURA T, ANDO H, NAKAMURA S, KURITA S, MISU H, OTA T, YOKOYAMA M, HONDA M, MIYAMOTO K et al. 2008. Increased oxidative stress precedes the onset of high-fat diet-induced insulin resistance and obesity. Metabolism, 57, 1071–7. [DOI] [PubMed] [Google Scholar]
- MAUVAIS-JARVIS F 2015. Sex differences in metabolic homeostasis, diabetes, and obesity. Biology of sex differences, 6, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCCARTHY MM, NUGENT BM & LENZ KM 2017. Neuroimmunology and neuroepigenetics in the establishment of sex differences in the brain. Nat Rev Neurosci, 18, 471–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCCURDY CE, BISHOP JM, WILLIAMS SM, GRAYSON BE, SMITH MS, FRIEDMAN JE & GROVE KL 2009. Maternal high-fat diet triggers lipotoxicity in the fetal livers of nonhuman primates. J Clin Invest, 119, 323–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCEWEN BS 1992. Steroid hormones: effect on brain development and function. Horm Res, 37 Suppl 3, 1–10. [DOI] [PubMed] [Google Scholar]
- MENDEZ-MANCILLA A, LIMA-ROGEL V, TORO-ORTIZ JC, ESCALANTE-PADRON F, MONSIVAIS-URENDA AE, NOYOLA DE & SALGADO-BUSTAMANTE M 2018. Differential expression profiles of circulating microRNAs in newborns associated to maternal pregestational overweight and obesity. Pediatr Obes, 13, 168–174. [DOI] [PubMed] [Google Scholar]
- MIKAMO E, HARADA S, NISHIKAWA J & NISHIHARA T 2003. Endocrine disruptors induce cytochrome P450 by affecting transcriptional regulation via pregnane X receptor. Toxicol Appl Pharmacol, 193, 66–72. [DOI] [PubMed] [Google Scholar]
- MIRANDA A & SOUSA N 2018. Maternal hormonal milieu influence on fetal brain development. Brain Behav, 8, e00920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOISIADIS VG & MATTHEWS SG 2014. Glucocorticoids and fetal programming part 1: Outcomes. Nat Rev Endocrinol, 10, 391–402. [DOI] [PubMed] [Google Scholar]
- MORGAN CP & BALE TL 2017. Sex differences in microRNA-mRNA networks: examination of novel epigenetic programming mechanisms in the sexually dimorphic neonatal hypothalamus. Biol Sex Differ, 8, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MORISSET AS, DUBE MC, DROLET R, PELLETIER M, LABRIE F, LUU-THE V, TREMBLAY Y, ROBITAILLE J, JOHN WEISNAGEL S & TCHERNOF A 2013. Androgens in the maternal and fetal circulation: association with insulin resistance. J Matern Fetal Neonatal Med, 26, 513–9. [DOI] [PubMed] [Google Scholar]
- MOSSA F, LATHAM KE, IRELAND JJ & VEIGA-LOPEZ A 2019. Undernutrition and hyperandrogenism during pregnancy: Role in programming of cardiovascular disease and infertility. Mol Reprod Dev, 86, 1255–1264. [DOI] [PubMed] [Google Scholar]
- MURABAYASHI N, SUGIYAMA T, ZHANG L, KAMIMOTO Y, UMEKAWA T, MA N & SAGAWA N 2013. Maternal high-fat diets cause insulin resistance through inflammatory changes in fetal adipose tissue. Eur J Obstet Gynecol Reprod Biol, 169, 39–44. [DOI] [PubMed] [Google Scholar]
- NADA SE, THOMPSON RC & PADMANABHAN V 2010. Developmental programming: differential effects of prenatal testosterone excess on insulin target tissues. Endocrinology, 151, 5165–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NEIER K, MARCHLEWICZ EH, DOLINOY DC & PADMANABHAN V 2015. Assessing human health risk to endocrine disrupting chemicals: a focus on prenatal exposures and oxidative stress. Endocrine Disruptors, 3, e1069916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NERI C & EDLOW AG 2015. Effects of Maternal Obesity on Fetal Programming: Molecular Approaches. Cold Spring Harb Perspect Med, 6, a026591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NICOL LE, O’BRIEN TD, DUMESIC DA, GROGAN T, TARANTAL AF & ABBOTT DH 2014. Abnormal infant islet morphology precedes insulin resistance in PCOS-like monkeys. PLoS One, 9, e106527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NICHOLAS LM, MORRISON JL, RATTANATRAY L, ZHANG S, OZANNE SE & MCMILLEN IC 2016. The early origins of obesity and insulin resistance: timing, programming and mechanisms. Int J Obes (Lond), 40, 229–38. [DOI] [PubMed] [Google Scholar]
- NICHOLAS LM, RATTANATRAY L, MACLAUGHLIN SM, OZANNE SE, KLEEMANN DO, WALKER SK, MORRISON JL, ZHANG S, MUHLHAUSLER BS, MARTIN-GRONERT MS et al. 2013. Differential effects of maternal obesity and weight loss in the periconceptional period on the epigenetic regulation of hepatic insulin-signaling pathways in the offspring. FASEB J, 27, 3786–96. [DOI] [PubMed] [Google Scholar]
- NILSSON E & LING C 2017. DNA methylation links genetics, fetal environment, and an unhealthy lifestyle to the development of type 2 diabetes. Clin Epigenetics, 9, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NILSSON E, BENRICK A, KOKOSAR M, KROOK A, LINDGREN E, KALLMAN T, MARTIS MM, HOJLUND K, LING C & STENER-VICTORIN E 2018. Transcriptional and Epigenetic Changes Influencing Skeletal Muscle Metabolism in Women With Polycystic Ovary Syndrome. J Clin Endocrinol Metab, 103, 4465–4477. [DOI] [PubMed] [Google Scholar]
- NIVOIT P, MORENS C, VAN ASSCHE FA, JANSEN E, POSTON L, REMACLE C & REUSENS B 2009. Established diet-induced obesity in female rats leads to offspring hyperphagia, adiposity and insulin resistance. Diabetologia, 52, 1133–42. [DOI] [PubMed] [Google Scholar]
- NOROOZZADEH M, RAMEZANI TEHRANI F, SEDAGHAT K, GODINI A & AZIZI F 2015. The impact of prenatal exposure to a single dose of testosterone on insulin resistance, glucose tolerance and lipid profile of female rat’s offspring in adulthood. J Endocrinol Invest, 38, 489–95. [DOI] [PubMed] [Google Scholar]
- NUGENT BM & BALE TL 2015. The omniscient placenta: Metabolic and epigenetic regulation of fetal programming. Front Neuroendocrinol, 39, 28–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NYIRENDA MJ, LINDSAY RS, KENYON CJ, BURCHELL A & SECKL JR 1998. Glucocorticoid exposure in late gestation permanently programs rat hepatic phosphoenolpyruvate carboxykinase and glucocorticoid receptor expression and causes glucose intolerance in adult offspring. J Clin Invest, 101, 2174–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OGAWA T, SHIBATO J, RAKWAL R, SAITO T, TAMURA G, KUWAGATA M & SHIODA S 2014. Seeking genes responsible for developmental origins of health and disease from the fetal mouse liver following maternal food restriction. Congenit Anom (Kyoto), 54, 195–219. [DOI] [PubMed] [Google Scholar]
- ONG TP & GUEST PC 2018. Nutritional Programming Effects on Development of Metabolic Disorders in Later Life. Methods Mol Biol, 1735, 3–17. [DOI] [PubMed] [Google Scholar]
- ONG TP & OZANNE SE 2015. Developmental programming of type 2 diabetes: early nutrition and epigenetic mechanisms. Curr Opin Clin Nutr Metab Care, 18, 354–60. [DOI] [PubMed] [Google Scholar]
- OSBORNE S, BIAGGI A, CHUA TE, DU PREEZ A, HAZELGROVE K, NIKKHESLAT N, PREVITI G, ZUNSZAIN PA, CONROY S & PARIANTE CM 2018. Antenatal depression programs cortisol stress reactivity in offspring through increased maternal inflammation and cortisol in pregnancy: The Psychiatry Research and Motherhood - Depression (PRAM-D) Study. Psychoneuroendocrinology, 98, 211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’REGAN D, KENYON CJ, SECKL JR & HOLMES MC 2004. Glucocorticoid exposure in late gestation in the rat permanently programs gender-specific differences in adult cardiovascular and metabolic physiology. Am J Physiol Endocrinol Metab, 287, E863–70. [DOI] [PubMed] [Google Scholar]
- OZIAS MK, LI S, HULL HR, BROOKS WM & CARLSON SE 2015. Relationship of circulating adipokines to body composition in pregnant women. Adipocyte, 4, 44–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PADMANABHAN V, CARDOSO RC & PUTTABYATAPPA M 2016. Developmental Programming, a Pathway to Disease. Endocrinology, 157, 1328–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PADMANABHAN V, SARMA HN, SAVABIEASFAHANI M, STECKLER TL & VEIGA-LOPEZ A 2010a. Developmental reprogramming of reproductive and metabolic dysfunction in sheep: native steroids vs. environmental steroid receptor modulators. Int J Androl, 33, 394–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PADMANABHAN V & VEIGA-LOPEZ A 2014. Reproduction Symposium: developmental programming of reproductive and metabolic health. J Anim Sci, 92, 3199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PADMANABHAN V, VEIGA-LOPEZ A, ABBOTT D, RECABARREN S & HERKIMER C 2010b. Developmental programming: impact of prenatal testosterone excess and postnatal weight gain on insulin sensitivity index and transfer of traits to offspring of overweight females. Endocrinology, 151, 595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PADMANABHAN V, VEIGA-LOPEZ A, ABBOTT DH, RECABARREN SE & HERKIMER C 2010c. Developmental programming: impact of prenatal testosterone excess and postnatal weight gain on insulin sensitivity index and transfer of traits to offspring of overweight females. Endocrinology, 151, 595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PAINTER RC, ROSEBOOM TJ & BLEKER OP 2005. Prenatal exposure to the Dutch famine and disease in later life: an overview. Reprod Toxicol, 20, 345–52. [DOI] [PubMed] [Google Scholar]
- PALOU M, KONIECZNA J, TORRENS JM, SANCHEZ J, PRIEGO T, FERNANDES ML, PALOU A & PICO C 2012. Impaired insulin and leptin sensitivity in the offspring of moderate caloric-restricted dams during gestation is early programmed. J Nutr Biochem, 23, 1627–39. [DOI] [PubMed] [Google Scholar]
- PANTHAM P, AYE IL & POWELL TL 2015. Inflammation in maternal obesity and gestational diabetes mellitus. Placenta, 36, 709–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PARK JH, STOFFERS DA, NICHOLLS RD & SIMMONS RA 2008. Development of type 2 diabetes following intrauterine growth retardation in rats is associated with progressive epigenetic silencing of Pdx1. J Clin Invest, 118, 2316–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PARSA AA & NEW MI 2017. Steroid 21-hydroxylase deficiency in congenital adrenal hyperplasia. J Steroid Biochem Mol Biol, 165, 2–11. [DOI] [PubMed] [Google Scholar]
- PASQUALI R & ORIOLO C 2019. Obesity and Androgens in Women. Front Horm Res, 53, 120–134. [DOI] [PubMed] [Google Scholar]
- POIRIER C, DESGAGNE V, GUERIN R & BOUCHARD L 2017. MicroRNAs in Pregnancy and Gestational Diabetes Mellitus: Emerging Role in Maternal Metabolic Regulation. Curr Diab Rep, 17, 35. [DOI] [PubMed] [Google Scholar]
- PROVVISIERO DP, PIVONELLO C, MUSCOGIURI G, NEGRI M, DE ANGELIS C, SIMEOLI C, PIVONELLO R & COLAO A 2016. Influence of Bisphenol A on Type 2 Diabetes Mellitus. Int J Environ Res Public Health, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PRUIS MG, VAN EWIJK PA, SCHRAUWEN-HINDERLING VB & PLOSCH T 2014. Lipotoxicity and the role of maternal nutrition. Acta Physiol (Oxf), 210, 296–306. [DOI] [PubMed] [Google Scholar]
- PUPPALA S, LI C, GLENN JP, SAXENA R, GAWRIEH S, QUINN A, PALARCZYK J, DICK EJ JR., NATHANIELSZ PW & COX LA 2018. Primate fetal hepatic responses to maternal obesity: epigenetic signalling pathways and lipid accumulation. J Physiol, 596, 5823–5837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PUTTABYATAPPA M, ANDRIESSEN V, MESQUITTA M, ZENG L, PENNATHUR S & PADMANABHAN V 2017. Developmental Programming: Impact of Gestational Steroid and Metabolic Milieus on Mediators of Insulin Sensitivity in Prenatal Testosterone-Treated Female Sheep. Endocrinology, 158, 2783–2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PUTTABYATAPPA M, BANKER M, ZENG L, GOODRICH JM, DOMINO SE, DOLINOY DC, MEEKER JD, PENNATHUR S, SONG PXK & PADMANABHAN V 2019a. Maternal Exposure to Environmental Disruptors and Sexually-Dimorphic Changes in Maternal and Neonatal Oxidative Stress. J Clin Endocrinol Metab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PUTTABYATAPPA M, CARDOSO RC & PADMANABHAN V 2016. Effect of maternal PCOS and PCOS-like phenotype on the offspring’s health. Mol Cell Endocrinol, 435, 29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PUTTABYATAPPA M, MARTIN JD, ANDRIESSEN V, STEVENSON M, ZENG L, PENNATHUR S & PADMANABHAN V 2019b. Developmental programming: Changes in mediators of insulin sensitivity in prenatal bisphenol A-treated female sheep. Reprod Toxicol, 85, 110–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PUTTABYATAPPA M & PADMANABHAN V 2017. Prenatal Testosterone Programming of Insulin Resistance in the Female Sheep. Adv Exp Med Biol, 1043, 575–596. [DOI] [PubMed] [Google Scholar]
- QI X, YUN C, SUN L, XIA J, WU Q, WANG Y, WANG L, ZHANG Y, LIANG X, WANG L, et al. 2019. Gut microbiota-bile acid-interleukin-22 axis orchestrates polycystic ovary syndrome. Nat Med, 25, 1225–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QIU HY, CHU YL, LI M, SUN YY & LI HF 2005. [Tyrosine phosphorylation and protein expression of insulin receptor substrate-2 in the adipose tissue from patients with polycystic ovary syndrome]. Zhonghua Fu Chan Ke Za Zhi, 40, 116–9. [PubMed] [Google Scholar]
- RABINOVICI J & JAFFE RB 1990. Development and regulation of growth and differentiated function in human and subhuman primate fetal gonads. Endocr Rev, 11, 532–57. [DOI] [PubMed] [Google Scholar]
- RADON PA, MCMAHON MJ & MEYER WR 1999. Impaired glucose tolerance in pregnant women with polycystic ovary syndrome. Obstet Gynecol, 94, 194–7. [DOI] [PubMed] [Google Scholar]
- RAE M, GRACE C, HOGG K, WILSON LM, MCHAFFIE SL, RAMASWAMY S, MACCALLUM J, CONNOLLY F, MCNEILLY AS & DUNCAN C 2013. The pancreas is altered by in utero androgen exposure: implications for clinical conditions such as polycystic ovary syndrome (PCOS). PLoS One, 8, e56263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAHMANI S, POUR KHALILI N, KHAN F, HASSANI S, GHAFOUR-BOROUJERDI E & ABDOLLAHI M 2018. Bisphenol A: What lies beneath its induced diabetes and the epigenetic modulation? Life Sci, 214, 136–144. [DOI] [PubMed] [Google Scholar]
- RAJESH P & BALASUBRAMANIAN K 2014. Phthalate exposure in utero causes epigenetic changes and impairs insulin signalling. J Endocrinol, 223, 47–66. [DOI] [PubMed] [Google Scholar]
- RAMASWAMY S, GRACE C, MATTEI AA, SIEMIENOWICZ K, BROWNLEE W, MACCALLUM J, MCNEILLY AS, DUNCAN WC & RAE MT 2016. Developmental programming of polycystic ovary syndrome (PCOS): prenatal androgens establish pancreatic islet alpha/beta cell ratio and subsequent insulin secretion. Sci Rep, 6, 27408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RASK-MADSEN C & KAHN CR 2012. Tissue-specific insulin signaling, metabolic syndrome, and cardiovascular disease. Arterioscler Thromb Vasc Biol, 32, 2052–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RECABARREN SE, PADMANABHAN V, CODNER E, LOBOS A, DURAN C, VIDAL M, FOSTER DL & SIR-PETERMANN T 2005. Postnatal developmental consequences of altered insulin sensitivity in female sheep treated prenatally with testosterone. Am J Physiol Endocrinol Metab, 289, E801–6. [DOI] [PubMed] [Google Scholar]
- REISCH N, ARLT W & KRONE N 2011. Health problems in congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Horm Res Paediatr, 76, 73–85. [DOI] [PubMed] [Google Scholar]
- RINAUDO P & WANG E 2012. Fetal programming and metabolic syndrome. Annu Rev Physiol, 74, 107–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RIZZA RA, MANDARINO LJ, GENEST J, BAKER BA & GERICH JE 1985. Production of insulin resistance by hyperinsulinaemia in man. Diabetologia, 28, 70–5. [DOI] [PubMed] [Google Scholar]
- RODRIGUEZ-RODRIGUEZ P, RAMIRO-CORTIJO D, REYES-HERNANDEZ CG, LOPEZ DE PABLO AL, GONZALEZ MC & ARRIBAS SM 2018. Implication of Oxidative Stress in Fetal Programming of Cardiovascular Disease. Front Physiol, 9, 602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROLAND AV, NUNEMAKER CS, KELLER SR & MOENTER SM 2010. Prenatal androgen exposure programs metabolic dysfunction in female mice. J Endocrinol, 207, 213–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROMEO RD 2003. Puberty: a period of both organizational and activational effects of steroid hormones on neurobehavioural development. J Neuroendocrinol, 15, 1185–92. [DOI] [PubMed] [Google Scholar]
- ROSEN ED & SPIEGELMAN BM 2006. Adipocytes as regulators of energy balance and glucose homeostasis. Nature, 444, 847–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROSENBAUM D, HABER RS & DUNAIF A 1993. Insulin resistance in polycystic ovary syndrome: decreased expression of GLUT-4 glucose transporters in adipocytes. Am J Physiol, 264, E197–202. [DOI] [PubMed] [Google Scholar]
- RUCHAT SM, HIVERT MF & BOUCHARD L 2013. Epigenetic programming of obesity and diabetes by in utero exposure to gestational diabetes mellitus. Nutr Rev, 71 Suppl 1, S88–94. [DOI] [PubMed] [Google Scholar]
- RUIZ R, JIDEONWO V, AHN M, SURENDRAN S, TAGLIABRACCI VS, HOU Y, GAMBLE A, KERNER J, IRIMIA-DOMINGUEZ JM, PUCHOWICZ MA, et al. 2014. Sterol regulatory element-binding protein-1 (SREBP-1) is required to regulate glycogen synthesis and gluconeogenic gene expression in mouse liver. J Biol Chem, 289, 5510–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RUTKOWSKA A & RACHON D 2014. Bisphenol A (BPA) and its potential role in the pathogenesis of the polycystic ovary syndrome (PCOS). Gynecol Endocrinol, 30, 260–5. [DOI] [PubMed] [Google Scholar]
- SABEN J, LINDSEY F, ZHONG Y, THAKALI K, BADGER TM, ANDRES A, GOMEZ-ACEVEDO H & SHANKAR K 2014. Maternal obesity is associated with a lipotoxic placental environment. Placenta, 35, 171–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SALTIEL AR & KAHN CR 2001. Insulin signalling and the regulation of glucose and lipid metabolism. Nature, 414, 799–806. [DOI] [PubMed] [Google Scholar]
- SAMUELSSON AM, MATTHEWS PA, ARGENTON M, CHRISTIE MR, MCCONNELL JM, JANSEN EH, PIERSMA AH, OZANNE SE, TWINN DF, REMACLE C, et al. 2008. Diet-induced obesity in female mice leads to offspring hyperphagia, adiposity, hypertension, and insulin resistance: a novel murine model of developmental programming. Hypertension, 51, 383–92. [DOI] [PubMed] [Google Scholar]
- SANDERSON JT 2006. The steroid hormone biosynthesis pathway as a target for endocrine-disrupting chemicals. Toxicol Sci, 94, 3–21. [DOI] [PubMed] [Google Scholar]
- SARGIS RM & SIMMONS RA 2019. Environmental neglect: endocrine disruptors as underappreciated but potentially modifiable diabetes risk factors. Diabetologia, 62, 1811–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SATHYANARAYANA S, BUTTS S, WANG C, BARRETT E, NGUYEN R, SCHWARTZ SM, HAALAND W, SWAN SH & TEAM T 2017. Early Prenatal Phthalate Exposure, Sex Steroid Hormones, and Birth Outcomes. J Clin Endocrinol Metab, 102, 1870–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHMATZ M, MADAN J, MARINO T & DAVIS J 2010. Maternal obesity: the interplay between inflammation, mother and fetus. J Perinatol, 30, 441–6. [DOI] [PubMed] [Google Scholar]
- SECKL JR & HOLMES MC 2007. Mechanisms of disease: glucocorticoids, their placental metabolism and fetal ‘programming’ of adult pathophysiology. Nat Clin Pract Endocrinol Metab, 3, 479–88. [DOI] [PubMed] [Google Scholar]
- SELEVAN SG, KIMMEL CA & MENDOLA P 2000. Identifying critical windows of exposure for children’s health. Environ Health Perspect, 108 Suppl 3, 451–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SEN S & SIMMONS RA 2010. Maternal Antioxidant Supplementation Prevents Adiposity in the Offspring of Western Diet–Fed Rats. Diabetes, 59, 3058–3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHANIK MH, XU Y, SKRHA J, DANKNER R, ZICK Y & ROTH J 2008. Insulin resistance and hyperinsulinemia: is hyperinsulinemia the cart or the horse? Diabetes Care, 31 Suppl 2, S262–8. [DOI] [PubMed] [Google Scholar]
- SHARMA S, KRIEBEL J & GRALLERT H 2017. Epigenetic regulation of glucose metabolism. Curr Opin Clin Nutr Metab Care, 20, 266–271. [DOI] [PubMed] [Google Scholar]
- SHELLEY P, MARTIN-GRONERT MS, ROWLERSON A, POSTON L, HEALES SJ, HARGREAVES IP, MCCONNELL JM, OZANNE SE & FERNANDEZ-TWINN DS 2009. Altered skeletal muscle insulin signaling and mitochondrial complex II-III linked activity in adult offspring of obese mice. Am J Physiol Regul Integr Comp Physiol, 297, R675–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHEN Q, BI H, YU F, FAN L, ZHU M, JIA X & KANG J 2019. Nontargeted metabolomic analysis of skeletal muscle in a dehydroepiandrosterone-induced mouse model of polycystic ovary syndrome. Mol Reprod Dev, 86, 370–378. [DOI] [PubMed] [Google Scholar]
- SHIMPI PC, MORE VR, PARANJPE M, DONEPUDI AC, GOODRICH JM, DOLINOY DC, RUBIN B & SLITT AL 2017. Hepatic Lipid Accumulation and Nrf2 Expression following Perinatal and Peripubertal Exposure to Bisphenol A in a Mouse Model of Nonalcoholic Liver Disease. Environ Health Perspect, 125, 087005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SIMAR D, CHEN H, LAMBERT K, MERCIER J & MORRIS MJ 2012. Interaction between maternal obesity and post-natal over-nutrition on skeletal muscle metabolism. Nutr Metab Cardiovasc Dis, 22, 269–76. [DOI] [PubMed] [Google Scholar]
- SINGH R, PEARSON E, AVERY PJ, MCCARTHY MI, LEVY JC, HITMAN GA, SAMPSON M, WALKER M & HATTERSLEY AT 2006. Reduced beta cell function in offspring of mothers with young-onset type 2 diabetes. Diabetologia, 49, 1876–80. [DOI] [PubMed] [Google Scholar]
- SIR-PETERMANN T, CODNER E, PEREZ V, ECHIBURU B, MALIQUEO M, LADRON DE GUEVARA A, PREISLER J, CRISOSTO N, SANCHEZ F, CASSORLA F et al. 2009. Metabolic and reproductive features before and during puberty in daughters of women with polycystic ovary syndrome. J Clin Endocrinol Metab, 94, 1923–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SIR-PETERMANN T, MALIQUEO M, ANGEL B, LARA HE, PEREZ-BRAVO F & RECABARREN SE 2002. Maternal serum androgens in pregnant women with polycystic ovarian syndrome: possible implications in prenatal androgenization. Hum Reprod, 17, 2573–9. [DOI] [PubMed] [Google Scholar]
- SIR-PETERMANN T, ECHIBURU B, MALIQUEO MM, CRISOSTO N, SANCHEZ F, HITSCHFELD C, CARCAMO M, AMIGO P & PEREZ-BRAVO F 2007. Serum adiponectin and lipid concentrations in pregnant women with polycystic ovary syndrome. Hum Reprod, 22, 1830–6. [DOI] [PubMed] [Google Scholar]
- SONG W, PUTTABYATAPPA M, ZENG L, VAZQUEZ D, PENNATHUR S & PADMANABHAN V 2019. Developmental programming: Prenatal bisphenol A treatment disrupts mediators of placental function in sheep. Chemosphere, 243, 125301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STEL J & LEGLER J 2015. The Role of Epigenetics in the Latent Effects of Early Life Exposure to Obesogenic Endocrine Disrupting Chemicals. Endocrinology, 156, 3466–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STEPTO NK, MORENO-ASSO A, MCILVENNA LC, WALTERS KA & RODGERS RJ 2019. Molecular Mechanisms of Insulin Resistance in Polycystic Ovary Syndrome: Unraveling the Conundrum in Skeletal Muscle? J Clin Endocrinol Metab, 104, 5372–5381. [DOI] [PubMed] [Google Scholar]
- STANNER SA, BULMER K, ANDRES C, LANTSEVA OE, BORODINA V, POTEEN VV & YUDKIN JS 1997. Does malnutrition in utero determine diabetes and coronary heart disease in adulthood? Results from the Leningrad siege study, a cross sectional study. BMJ, 315, 1342–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STENER-VICTORIN E, PLOJ K, LARSSON BM & HOLMANG A 2005. Rats with steroid-induced polycystic ovaries develop hypertension and increased sympathetic nervous system activity. Reprod Biol Endocrinol, 3, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STRAKOVSKY RS, WANG H, ENGESETH NJ, FLAWS JA, HELFERICH WG, PAN YX & LEZMI S 2015. Developmental bisphenol A (BPA) exposure leads to sex-specific modification of hepatic gene expression and epigenome at birth that may exacerbate high-fat diet-induced hepatic steatosis. Toxicol Appl Pharmacol, 284, 101–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STRAUSS JF 3RD, MARTINEZ F & KIRIAKIDOU M 1996. Placental steroid hormone synthesis: unique features and unanswered questions. Biol Reprod, 54, 303–11. [DOI] [PubMed] [Google Scholar]
- SUN M, MALIQUEO M, BENRICK A, JOHANSSON J, SHAO R, HOU L, JANSSON T, WU X & STENER-VICTORIN E 2012. Maternal androgen excess reduces placental and fetal weights, increases placental steroidogenesis, and leads to long-term health effects in their female offspring. Am J Physiol Endocrinol Metab, 303, E1373–85. [DOI] [PubMed] [Google Scholar]
- SYMONDS ME, SEBERT SP, HYATT MA & BUDGE H 2009. Nutritional programming of the metabolic syndrome. Nat Rev Endocrinol, 5, 604–10. [DOI] [PubMed] [Google Scholar]
- TABB MM & BLUMBERG B 2006. New modes of action for endocrine-disrupting chemicals. Mol Endocrinol, 20, 475–82. [DOI] [PubMed] [Google Scholar]
- TAKEUCHI T, TSUTSUMI O, IKEZUKI Y, TAKAI Y & TAKETANI Y 2004. Positive relationship between androgen and the endocrine disruptor, bisphenol A, in normal women and women with ovarian dysfunction. Endocr J, 51, 165–9. [DOI] [PubMed] [Google Scholar]
- TAMHANE S, RODRIGUEZ-GUTIERREZ R, IQBAL AM, PROKOP LJ, BANCOS I, SPEISER PW & MURAD MH 2018. Cardiovascular and Metabolic Outcomes in Congenital Adrenal Hyperplasia: A Systematic Review and Meta-Analysis. J Clin Endocrinol Metab, 103, 4097–4103. [DOI] [PubMed] [Google Scholar]
- TARNOW P, TRALAU T & LUCH A 2019. Chemical activation of estrogen and aryl hydrocarbon receptor signaling pathways and their interaction in toxicology and metabolism. Expert Opin Drug Metab Toxicol, 15, 219–229. [DOI] [PubMed] [Google Scholar]
- TAYLOR PD, MCCONNELL J, KHAN IY, HOLEMANS K, LAWRENCE KM, ASARE-ANANE H, PERSAUD SJ, JONES PM, PETRIE L, HANSON MA et al. 2005. Impaired glucose homeostasis and mitochondrial abnormalities in offspring of rats fed a fat-rich diet in pregnancy. Am J Physiol Regul Integr Comp Physiol, 288, R134–9. [DOI] [PubMed] [Google Scholar]
- TAYLOR KW, NOVAK RF, ANDERSON HA, BIRNBAUM LS, BLYSTONE C, DEVITO M, JACOBS D, KOHRLE J, LEE DH, RYLANDER L, et al. 2013. Evaluation of the association between persistent organic pollutants (POPs) and diabetes in epidemiological studies: a national toxicology program workshop review. Environ Health Perspect, 121, 774–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TCHERNOF A & DESPRES JP 2013. Pathophysiology of human visceral obesity: an update. Physiol Rev, 93, 359–404. [DOI] [PubMed] [Google Scholar]
- TENORIO MB, FERREIRA RC, MOURA FA, BUENO NB, DE OLIVEIRA ACM & GOULART MOF 2019. Cross-Talk between Oxidative Stress and Inflammation in Preeclampsia. Oxid Med Cell Longev, 2019, 8238727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TETEL MJ, DE VRIES GJ, MELCANGI RC, PANZICA G & O’MAHONY SM 2018. Steroids, stress and the gut microbiome-brain axis. J Neuroendocrinol, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- THOMPSON LP & AL-HASAN Y 2012. Impact of oxidative stress in fetal programming. J Pregnancy, 2012, 582748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- THORNBURG KL, O’TIERNEY PF & LOUEY S 2010. Review: The placenta is a programming agent for cardiovascular disease. Placenta, 31 Suppl, S54–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TODD SE, OLIVER MH, JAQUIERY AL, BLOOMFIELD FH & HARDING JE 2009. Periconceptional undernutrition of ewes impairs glucose tolerance in their adult offspring. Pediatr Res, 65, 409–13. [DOI] [PubMed] [Google Scholar]
- TRUE CA, TAKAHASHI DL, BURNS SE, MISHLER EC, BOND KR, WILCOX MC, CALHOUN AR, BADER LA, DEAN TA, RYAN ND, et al. 2017. Chronic combined hyperandrogenemia and western-style diet in young female rhesus macaques causes greater metabolic impairments compared to either treatment alone. Hum Reprod, 32, 1880–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VAISERMAN A & LUSHCHAK O 2019. Developmental origins of type 2 diabetes: Focus on epigenetics. Ageing Res Rev, 55, 100957. [DOI] [PubMed] [Google Scholar]
- VAISERMAN AM 2017. Early-Life Nutritional Programming of Type 2 Diabetes: Experimental and Quasi-Experimental Evidence. Nutrients, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VALKO M, LEIBFRITZ D, MONCOL J, CRONIN MT, MAZUR M & TELSER J 2007. Free radicals and antioxidants in normal physiological functions and human disease. The international journal of biochemistry & cell biology, 39, 44–84. [DOI] [PubMed] [Google Scholar]
- VAN DER MEER T 2015. Multigenerational effects of maternal Bisphenol A exposure on hepatic gene expression in mice., University of Groningen. [Google Scholar]
- VAN HOUTEN EL, KRAMER P, MCLUSKEY A, KARELS B, THEMMEN AP & VISSER JA 2012. Reproductive and metabolic phenotype of a mouse model of PCOS. Endocrinology, 153, 2861–9. [DOI] [PubMed] [Google Scholar]
- VARLAMOV O, BETHEA CL & ROBERTS CT JR 2015. Sex-specific differences in lipid and glucose metabolism. Frontiers in endocrinology, 5, 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VEIGA-LOPEZ A, MOELLER J, PATEL D, YE W, PEASE A, KINNS J & PADMANABHAN V 2013. Developmental programming: impact of prenatal testosterone excess on insulin sensitivity, adiposity, and free fatty acid profile in postpubertal female sheep. Endocrinology, 154, 1731–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VEIGA-LOPEZ A, MOELLER J, SREEDHARAN R, SINGER K, LUMENG C, YE W, PEASE A & PADMANABHAN V 2016. Developmental programming: interaction between prenatal BPA exposure and postnatal adiposity on metabolic variables in female sheep. Am J Physiol Endocrinol Metab, 310, E238–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VEIGA-LOPEZ A, PENNATHUR S, KANNAN K, PATISAUL HB, DOLINOY DC, ZENG L & PADMANABHAN V 2015. Impact of gestational bisphenol A on oxidative stress and free fatty acids: Human association and interspecies animal testing studies. Endocrinology, 156, 911–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VEJRAZKOVA D, VCELAK J, VANKOVA M, LUKASOVA P, BRADNOVA O, HALKOVA T, KANCHEVA R & BENDLOVA B 2014. Steroids and insulin resistance in pregnancy. J Steroid Biochem Mol Biol, 139, 122–9. [DOI] [PubMed] [Google Scholar]
- VENKATESH KK, MEEKER JD, CANTONWINE DE, MCELRATH TF & FERGUSON KK 2019. Association of antenatal depression with oxidative stress and impact on spontaneous preterm birth. J Perinatol, 39, 554–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VISIEDO F, BUGATTO F, SANCHEZ V, COZAR-CASTELLANO I, BARTHA JL & PERDOMO G 2013. High glucose levels reduce fatty acid oxidation and increase triglyceride accumulation in human placenta. Am J Physiol Endocrinol Metab, 305, E205–12. [DOI] [PubMed] [Google Scholar]
- VOGT MC & BRUNING JC 2013. CNS insulin signaling in the control of energy homeostasis and glucose metabolism - from embryo to old age. Trends Endocrinol Metab, 24, 76–84. [DOI] [PubMed] [Google Scholar]
- VYAS AK, HOANG V, PADMANABHAN V, GILBREATH E & MIETELKA KA 2016. Prenatal programming: adverse cardiac programming by gestational testosterone excess. Sci Rep, 6, 28335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WADHWA PD, BUSS C, ENTRINGER S & SWANSON JM 2009. Developmental origins of health and disease: brief history of the approach and current focus on epigenetic mechanisms. Semin Reprod Med, 27, 358–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WAKUI S, YOKOO K, TAKAHASHI H, MUTO T, SUZUKI Y, KANAI Y, HANO H, FURUSATO M & ENDOU H 2006. Prenatal 3,3’,4,4’,5-pentachlorobiphenyl exposure modulates induction of rat hepatic CYP 1A1, 1B1, and AhR by 7,12-dimethylbenz[a]anthracene. Toxicol Appl Pharmacol, 210, 200–11. [DOI] [PubMed] [Google Scholar]
- WANG Y, SUN Y & QIU H 2004. Expression of resistin mRNA in adipose tissue of rat model with polycystic ovarian syndrome and its implication. J Huazhong Univ Sci Technolog Med Sci, 24, 621–4. [DOI] [PubMed] [Google Scholar]
- WANG HH, ZHOU CL, LV M, YANG Q, LI JX, HOU M, LIN J, LIU XM, WU YT, SHENG JZ et al. 2018. Prenatal High Estradiol Exposure Induces Sex-Specific and Dietarily Reversible Insulin Resistance Through Decreased Hypothalamic INSR. Endocrinology, 159, 465–476. [DOI] [PubMed] [Google Scholar]
- WANG M, MONACO MH & DONOVAN SM 2016. Impact of early gut microbiota on immune and metabolic development and function. Semin Fetal Neonatal Med, 21, 380–387. [DOI] [PubMed] [Google Scholar]
- WANG ZC, GU XF, YANG DZ & KUANG JQ 2005. [Expression of insulin receptor substrate 1 and phosphorylation of tyrosine in adipose tissue of polycystic ovary syndrome]. Zhonghua Fu Chan Ke Za Zhi, 40, 120–3. [PubMed] [Google Scholar]
- WATKINS DJ, FERGUSON KK, ANZALOTA DEL TORO LV, ALSHAWABKEH AN, CORDERO JF & MEEKER JD 2015. Associations between urinary phenol and paraben concentrations and markers of oxidative stress and inflammation among pregnant women in Puerto Rico. Int J Hyg Environ Health, 218, 212–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WEINBERG JM 2006. Lipotoxicity. Kidney Int, 70, 1560–6. [DOI] [PubMed] [Google Scholar]
- WHYTE JJ, ALEXENKO AP, DAVIS AM, ELLERSIECK MR, FOUNTAIN ED & ROSENFELD CS 2007. Maternal diet composition alters serum steroid and free fatty acid concentrations and vaginal pH in mice. J Endocrinol, 192, 75–81. [DOI] [PubMed] [Google Scholar]
- WOODING P & BURTON G 2008. Comparative placentation: structures, functions and evolution, Springer Science & Business Media. [Google Scholar]
- WOODRUFF TJ, ZOTA AR & SCHWARTZ JM 2011. Environmental chemicals in pregnant women in the United States: NHANES 2003–2004. Environ Health Perspect, 119, 878–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WU L, LU Y, JIAO Y, LIU B, LI S, LI Y, XING F, CHEN D, LIU X, ZHAO J, et al. 2016. Paternal Psychological Stress Reprograms Hepatic Gluconeogenesis in Offspring. Cell Metab, 23, 735–43. [DOI] [PubMed] [Google Scholar]
- XU RA, MAO B, LI S, LIU J, LI X, LI H, SU Y, HU G, LIAN QQ & GE RS 2016. Structure-activity relationships of phthalates in inhibition of human placental 3beta-hydroxysteroid dehydrogenase 1 and aromatase. Reprod Toxicol, 61, 151–61. [DOI] [PubMed] [Google Scholar]
- YAJNIK CS, DESHPANDE SS, JACKSON AA, REFSUM H, RAO S, FISHER DJ, BHAT DS, NAIK SS, COYAJI KJ, JOGLEKAR CV, et al. 2008. Vitamin B12 and folate concentrations during pregnancy and insulin resistance in the offspring: the Pune Maternal Nutrition Study. Diabetologia, 51, 29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAN X, DAI X, WANG J, ZHAO N, CUI Y & LIU J 2013. Prenatal androgen excess programs metabolic derangements in pubertal female rats. J Endocrinol, 217, 119–29. [DOI] [PubMed] [Google Scholar]
- YAN X, ZHU MJ, XU W, TONG JF, FORD SP, NATHANIELSZ PW & DU M 2010. Up-regulation of Toll-like receptor 4/nuclear factor-kappaB signaling is associated with enhanced adipogenesis and insulin resistance in fetal skeletal muscle of obese sheep at late gestation. Endocrinology, 151, 380–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANES LL, ROMERO DG, MOULANA M, LIMA R, DAVIS DD, ZHANG H, LOCKHART R, RACUSEN LC & RECKELHOFF JF 2011. Cardiovascular-renal and metabolic characterization of a rat model of polycystic ovary syndrome. Gend Med, 8, 103–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG S, RATTANATRAY L, MORRISON JL, NICHOLAS LM, LIE S & MCMILLEN IC 2011. Maternal obesity and the early origins of childhood obesity: weighing up the benefits and costs of maternal weight loss in the periconceptional period for the offspring. Exp Diabetes Res, 2011, 585749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHENG J, XIAO X, ZHANG Q, WANG T, YU M & XU J 2017. Maternal Low-Protein Diet Modulates Glucose Metabolism and Hepatic MicroRNAs Expression in the Early Life of Offspring dagger. Nutrients, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHENG Y, LEY SH & HU FB 2018. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat Rev Endocrinol, 14, 88–98. [DOI] [PubMed] [Google Scholar]
- ZHOU Z, SUN B, LI X & ZHU C 2018. DNA methylation landscapes in the pathogenesis of type 2 diabetes mellitus. Nutr Metab (Lond), 15, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHOU W, LIU J, LIAO L, HAN S & LIU J 2008. Effect of bisphenol A on steroid hormone production in rat ovarian theca-interstitial and granulosa cells. Mol Cell Endocrinol, 283, 12–8. [DOI] [PubMed] [Google Scholar]
- ZHU C, YANG H, GENG Q, MA Q, LONG Y, ZHOU C & CHEN M 2015. Association of oxidative stress biomarkers with gestational diabetes mellitus in pregnant women: a case-control study. PLoS One, 10, e0126490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZIJLMANS MA, KORPELA K, RIKSEN-WALRAVEN JM, DE VOS WM & DE WEERTH C 2015. Maternal prenatal stress is associated with the infant intestinal microbiota. Psychoneuroendocrinology, 53, 233–45. [DOI] [PubMed] [Google Scholar]
- ZOTA AR, GELLER RJ, ROMANO LE, COLEMAN-PHOX K, ADLER NE, PARRY E, WANG M, PARK JS, ELMI AF, LARAIA BA et al. 2018. Association between persistent endocrine-disrupting chemicals (PBDEs, OH-PBDEs, PCBs, and PFASs) and biomarkers of inflammation and cellular aging during pregnancy and postpartum. Environ Int, 115, 9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]