

Abstract
With the introduction of combinatory antiretroviral therapy, patients infected with human immunodeficiency virus type 1 (HIV-1) can live much longer than before. However, the identification of HIV-associated neurocognitive disorder (HAND), especially HIV-associated dementia in 15–20% of patients infected with HIV-1, indicates additional complexity. These disorders turn out to be subtype dependent. Recently, many studies are ongoing trying to understand how the virus induces neuronal injury which could lead to neurological dysfunction. Most of these studies are focusing on the HIV-1 release of proteins such as Tat. However, the exact role of these proteins and their involvement in neuronal degeneration remains unidentified; this is especially true since viral proteins from different HIV-1 subtypes differ in their ability to cause neuronal damage. This review describes the role of different HIV-1 subtypes, identifies probable pathways involved in neuronal damage, the contribution of different HIV-1 subtypes to the progression of HAND, and potential treatments for HAND.
Keywords: HIV-1, Subtypes, HIV-associated neurocognitive disorder
Introduction
Patients infected with human immunodeficiency virus type 1 (HIV-1) (35.1 million adults and 1.8 million children [https://www.WHO.int/hiv/data/en/]) including those using the effective but not curative combinatory antiretroviral therapy (cART) suffer from deregulation and impairment of organs such as heart, kidney, and brain1. Studies involving 37,000 HIV-infected patients using cART (from 2003 to 2013) showed that comorbidity increased with age in those patients compared to uninfected patients and having the same age2. AIDS-related deaths have been reduced by 51% since 2004 (the peak of HIV-related deaths) and have further decreased from 1.4 million in 2010 (https://www.hiv.gov/hiv-basics/overview/data-and-trends/global-statistics).
HIV is divided into two types, HIV-1 and HIV-2, among which HIV-1 is the most common. HIV-1 is further separated into three groups: major Group M (major), non-major Group N (non-M, non-O), and Group O (outlier). By far, more than 90% of AIDS cases derive from the infection of H IV-1 Group M (https://www.avert.org/professionals/hiv-science/types-strains#footnote1_gl1pyu4 and References within). Group M is divided into subtypes (clades), among these are the most studied subtypes: A, B, C, and D. Subtype C is the most prevalent subtype in Asia, while subtypes A and D are found mainly in Africa. Subtype B is the predominant type in the developed world, including the United States and European countries. Some of the subtypes are described to be more virulent or more resistant to different drugs3. Epidemiological research found that disease progression is closely related to the different viral subtypes4. Furthermore, the progression and severity of HIV-associated neurocognitive disorder (HAND), which is common in patients even in the cART era, is found to be different in patients infected with different HIV-1 subtypes. This review will discuss the role of these subtypes and their contribution in the progression of HAND.
Impact of HIV-1 subtypes on the progression of neurological disorders
Approximately 30–50% of HIV-infected patients develop some neurological dysfunction, ranging from mild-to-severe symptoms. HAND is now classified into three categories: minor cognitive/motor disorder (impairment in more than one cognitive abilities); mild cognitive/motor complex (CMC) (cognitive abnormality with mild functional disorder), and HIV-associated dementia (HAD). HAD is the most severe HAND identified by severe functional impairment resulting in dramatic reductions in the ability to care for oneself, work efficiency, and quality of life.
HAND symptoms result from neuronal deregulation directly or indirectly brought on by HIV-1 infection in the central nervous systems (CNS). HIV does not infect neurons; however, HIV-1 does infect microglia and brain-resident macrophages. These infected cells produce and release toxic diffusible factors that can lead to neuronal death (Fig. 1). In addition to exerting a toxic effect on neurons through macrophages and microglia, HIV viral proteins are found to cause neuronal dysfunction and neuronal death in recent studies. Tat, one of the six regulatory viral proteins controlling the ability of HIV to infect cells, is detectable in the serum of HAND patients. TAT protein and mRNA levels correlate with the severity of neurological dysfunction5,6. In PSAPP mice, Tat expression in astrocytes induced neurodegeneration, tau phosphorylation, and amyloid deposition in the brain – confirming the neurotoxicity of Tat7. Tat enters neurons by receptor-mediated endocytosis and directly induces apoptosis in both rat and human neurons. This apoptosis is caused by the expression of AMPAR through the release of tumor necrosis factor-α independent of nuclear factor κB activation8. Further studies found that nitric oxide synthase and endolysosomes are also involved in Tat-induced neurotoxicity9,10. Synapse loss brought on by HIV Tat is dependent on calcium influx through N-methyl-D-aspartate receptor (NMDAR) and the activity of mir-128a, which inhibits the expression of the presynaptic protein SNAP2511.
Figure 1.

Summary of HIV-1 proteins’ contribution to the progression of HIV-associated neurocognitive disorder (HAND). The contribution of HIV-1 Tat, Vpr, gp120, and Nef to neuronal apoptosis and HAND.
In addition to Tat, Vpr, a second regulatory viral protein presents in the serum and cerebrospinal fluid (CSF) of HIV patients, causes a depolarization of neurons. Vpr causes an inward current by forming cation-selective ion channels across the cytoplasmic membrane, resulting in hippocampal neuronal death12. The first 40 N-terminal amino acids of Vpr are sufficient to create the ion channel which results in neuron death13. Besides the ability of the N-terminal region to induce neuronal death, the C-terminal fragment (70–96) is also found to cause neuronal apoptosis associated with the activation of caspase-314. Similarly, caspase-8 activation is identified in human neuronal cultures after exposure to Vpr15. In in vivo studies done in neonatal mice, Vpr is injected through the ventricle and induces the loss of neurons and dendritic processes in the cortex, hippocampus, cerebellum, and choroids plexus16. Transgenic mice expressing Vpr in basal ganglia monocytoid cells exhibit behavior deficits. Neuronal injury is also observed in the basal ganglia of these mice, including the activation of caspase-3 as well as loss of synaptophysin, GABAnergic, and cholinergic neurons17.
Gp120, a glycoprotein that forms part of the HIV-1 envelope, interacts with several receptors found in the CNS such as CD4, CCR5, CXCR4, and nAChR18–20. Gp120 kills neurons by stimulating the neurotoxic pathways mediated by nitric oxide, calcium, glutamate, and superoxide anions21,22. Besides these classical neurotoxic pathways, gp120 reduces intracellular furin levels reducing the cleavage of pro-brain-derived neurotrophic factor (BDNF) into mature BDNF. This reduction in cleavage results in the imbalance between antiapoptotic and proapoptotic neurotrophins, contributing to neuronal injury23. Cell death brought by gp120IIIB found in subtype B HIV-1, has a specificity for the CXCR4 receptor, and could also be mediated by the calcium – highly permeable acetylcholine receptor α7-nAChR. The expression of α7-nAChR increases in gp120-treated SH-SY5Y cells and gp120-transgenic mice striatum24. The gp120IIIB-induced neurotoxicity in neurons requires the presence of CXCR4, phosphatase, and tensin homolog on chromosome 10 (PTEN) and p38 MAPK25–28.
Until now, it is a common belief that HIV-1 virus can-not infect neurons but does infect brain macrophages. However, the HIV-Nef gene sequence was detected in hippocampal neurons isolated from postmortem HIV patients29. These findings corroborate with other reports that used in situ PCR and demonstrated the presence of viral proteins in neurons30, suggesting the possibility that HIV-1 can infect neurons. If indeed neurons could be infected by the HIV, there would be additional neuronal damage since Nef has been found to bind directly to calmodulin. Calmodulin is involved in a wide range of cellular calcium-dependent signaling pathways by regulating the activity of many enzymes31. Furthermore, Nef causes an increase in K+ current evoked after membrane depolarization, either by direct binding to the K+ channel or through interacting with receptors regulating the K+ channel32. The possibility that Nef contributes to the neuronal damage reported in HIV-patients is further supported by human neuronal cultures after exposure to Nef that results in nuclear fragmentation and a decrease in cell number33. Furthermore, transgenic mice expressing Nef in astrocytes exhibit impaired spatial and recognition memory, supporting the contribution of Nef to HAND34.
There have been several studies conducted to identify the relationship between HIV-1 subtypes, and the severity of neurocognitive impairments with individuals infected by different subtypes of HIV-1. In one of these studies, researchers found that among those HIV-infected adults in Uganda, HAD is more common in patients with subtype D (which is more CXCR4-tropic) than those infected with subtype A (which is more CCR5-tropic). On the other hand, in HIV-infected children with subtype A, they demonstrated no statistical difference than those infected with subtype D35. Besides the relationship of subtypes A and D with the neurocognitive disorder, subtype C is found to have slower replication kinetics in monocyte-derived macrophages, leading to lower levels of macrophage-mediated neurotoxicity36. The diminished neuroinvasive capacity of subtype C is contributed to the different primary conformation of Tat. Tat from subtype C does not cause neuron injury unless morphine was present and acted through glial cells expressing μ-opioid receptors37. Clinically, patients infected with subtype C show decreased incidence of cognitive deficits38. Severe combined immune deficiency mice injected with macrophages infected by subtype B display more severe cognitive deficits than mice infected with subtype C39. In vitro studies have found that subtype B Tat is more potent than subtype C Tat when inducing neuronal death (Fig. 2)40.
Figure 2.
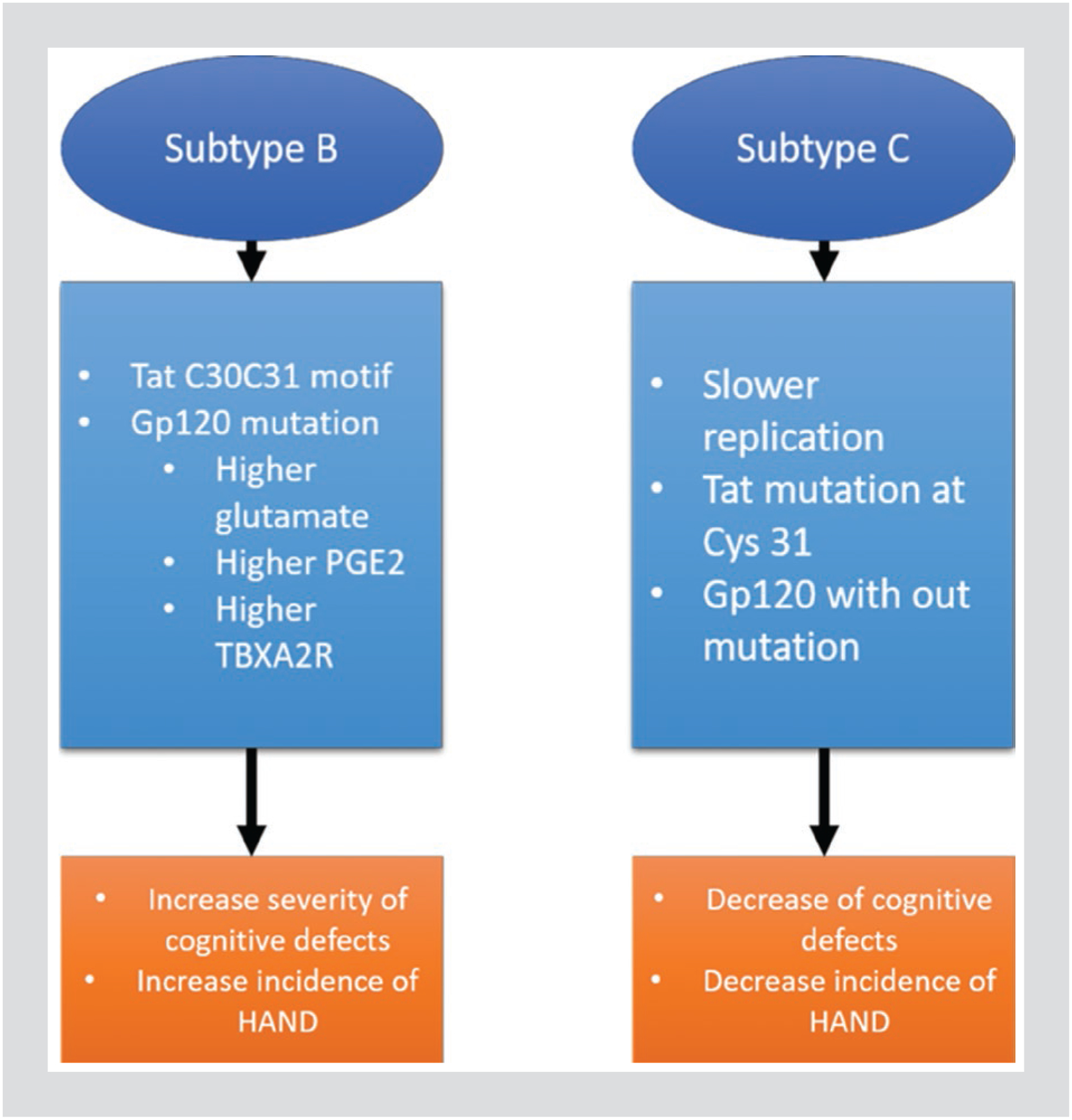
Comparison of subtype B to subtype C. A comparison of HIV-1 subtype B and subtype C elements and their contribution to the severity and incidence of HIV-associated neurocognitive disorder.
Studies in recent years have discovered subtype-specific differences in the neurotoxicity and the induction of Aβ production in neurons by HIV-1 Tat protein from subtypes B and C41. These differences are attributed to the di-cysteine C30–C31 motif found in Tat from subtype B42. The mechanism why subtype C Tat protein is less neurotoxic might be attributed to the natural mutation found at cysteine 31 that is critical in mediating persistent excitation of the NMDAR. This persistent excitation is achieved by disrupting a disulfide bond on the NR1 subunit of the NMDAR that eventually leads to cognitive dysfunctions43.
In addition to the lower toxicity of Tat protein from subtype C, gp120 protein from the same subtype was also found to be less toxic compared to that isolated from subtype B. As indicated in the results from human astrocytes, subtype B gp120 induces higher levels of glutamate, as well as prostaglandin E2 and thromboxane A2 receptor, the neuropathogenic byproducts of cyclooxygenase-2-mediated arachidonic acid metabolism44. Further, the capability of inducing CMC is hypothesized to be determined by the mutations in the gp120 sequences, which means that there exist some variations of gp120 specific for causing CMC45. Speaking of particular mutations in viral proteins responsible for causing cognitive dysfunction, a mutation at amino acid 77 in Vpr distinguishes HIV patients with (77Q) or without (77R) HAND. However, 77Q is dominant in HIV patients with dementia; the Vpr protein with the 77Q mutation is found to induce less neurotoxicity than Vpr with 77R46.
Similar conditions apply to Nef, the three-dimensional structure of Nef in the brain of HAD patients infected with HIV-1 subtype B is found to be more identical to Nef from subtype D and less similar to Nef collected from patients that do not have HAD. In addition, Nef from the brain of HAD patients is structurally different from Nef from the same patient’s peripheral organs indicating the possible existence of specific genetic alterations that change the structure of Nef in the brain, which contributes to cognitive deficit47.
Potential treatments of HAND
At present, there are no specific treatments for HAND. The treatment of HAND usually employs a multidiscipline approach with neurologists, HIV specialists, psychiatrists, and psychologist participating. Early studies identified reversal of brain metabolic abnormalities48 and improvement in motor functions in patients receiving a higher dose of zidovudine (azidothymidine [AZT]), a nucleoside reverse transcriptase inhibitor (NRTI). AZT treatment resulted in lower dementia prevalence49–52, indicating the activity of this antiretroviral drug in preventing HAND. Since AZT generally does not entirely stop HIV replication, only slowing replication down it is usually combined with other antiretroviral drugs. Studies have found that cART was able to reduce the incidence of HAD53 and improve motor speed performance in patients receiving a specific cART combination54. However, some antiretroviral agents such as efavirenz (EFV) and rilpivirine (RPV), which fall in the non-NRTI (NNRTI) class, have CNS side effects that exacerbate neurological dysfunction55,56. Therefore, it is essential for physicians to consider the neurological health of HIV patients when choosing the most appropriate cART drug combination57.
Protease inhibitors (PIs) are a class of antiretroviral drugs that inhibit the activity of a protease that cleaves nascent proteins for assembly of new virions. Among the series of PIs developed for HIV patients, some were tested for the effect on β-amyloid deposition in vitro and in vivo, a hallmark of dementia. Atazanavir, ritonavir, and saquinavir (SQV) were found to modestly inhibit Aβ degradation while lopinavir, nelfinavir (NFV), and ritonavir enhanced secretion of undigested Aβ after phagocytosis. Lopinavir, NFV, ritonavir, and SQV were found to inhibit Aβ40 production from primary human cortical neurons58. Indinavir, another HIV PI, alleviated memory deficits induced by celecoxib or streptozotocin by inhibiting brain AChE activity, as well as thiobarbituric acid reactive species levels and restoring reduced glutathione levels59. Ritonavir, in addition to slightly inhibiting Aβ degradation, induces the expression of P-glycoprotein, a blood–brain barrier (BBB) drug transporter, resulting in restricted entry of HIV PIs into the brain, leading to neurological dysfunction60. On the contrary, inhibition of P-glycoprotein at the BBB is found to increase the brain distribution of an HIV PI, NFV, supporting P-glycoprotein as a potential target for HAND61.
Several factors limit the distribution of the drugs to the brain. First, cART can bound the plasma proteins and therefore be less available for the CNS62. Second, the BBB has a complex structure which is a major obstacle for an effective drug delivery and creates a poor pharmacokinetic profile63. Different approaches are developed to deliver cART through the barrier, including biotechnologies, prodrugs based, nanogels, liposomes, or chemical modification for CNS delivery64.
NRTI has a low molecular weight and low protein binding and therefore reaches a good CSF concentration. Raltegravir (RGV) exhibits a good passage into CSF. Nevirapine (NVP) had the highest CSF/plasma penetration compared to other drugs. In the opposite, the penetrability of EFV in CSF is limited. Moreover, the PIs have, in general, a limited penetrability in the CSF due to the presence of efflux mechanisms and high plasma protein binding. However, their penetrability can be boosted by low-dose ritonavir. Some PIs like SQV, NFV, tipranavir, and atazanavir do not even reach therapeutical concentrations in CSF and can be were below the detection limit65.
The dolutegravir resistance is more common in subtype B versus non-B clades (18.7% vs. 22%), due to a G140S substitution. In all the non-B, two nucleotides are required, raising the genetic barrier to the emergence of G140S mutations66. In the RGV population, HIV-1 subtype B develops the mutations N155H, Q148H/R/K, and Y143R/C/H. The mutation Q148H/R/K can be found with 13% of prevalence in all B subtypes versus 0% in non-B. However, some studies showed the same susceptibility among the integrase inhibitors, RGV, elvitegravir, and MK-2408 with HIV-1 subtype B and C, resulting in similar outcomes67.
Despite having a low genetic barrier, EFV and NVP have been widely used. A single mutation in the binding pocket of the NNRTIs is sufficient to initiate a clinical failure68. The NNRTI resistance is caused by two mechanisms: the mutations K103N and E138K affecting the entry of the NNRTI from the binding pocket69,70 or is altering the pocket geometry through the mutations Y181C and Y188L71,72. In tissue culture, under EFV pressure but not NVP or delavirdine, the HIV-1 clade C develops the V160M mutation, conferring high-level resistance to all NNRTIs73. There is no significant difference of V106M prevalence in patients receiving NVP between subtypes B and C74. V106M and Y181L are the major NNRTI drug-resistant mutations75. The prevalence of V106M is 14% in HIV-infected patients carrying subtype C, whereas a small percentage of subtype B-infected patients carries this mutation76.
The prevalence of the other major mutation Y181C is 21% for the subtype B and only 12% for the subtype C, both B and C are likely to develop the Y181C mutation77. Some subtype C-infected patients carry mutation such as E138K/Q/R, M230I/L, and Y188L is resistant to etravirine or RPV, respectively, than those infected with subtype B. Moreover, RPV is associated with the E138A substitution occurring more frequently in subtypes C (5.9–7.5%) than B (0–2.3%)78.
Besides antiretroviral drugs, other therapies are exhibiting promising effects toward reducing HAND. Memantine and ifenprodil, two GluN2B-preferring NMDAR antagonists, promoted the recovery from Tat-induced synaptic loss and cell survival through inhibiting Tat-induced activation of cell death pathways79. Furthermore, estrogen was found to delay Tat-induced neuronal apoptosis by inhibiting a mitochondrial apoptotic signaling pathway in an endoplasmic reticulum-sensitive manner80 and decreases the neurotoxicity of factors released by Gp120-treated microglia81–83. Meth-ylphenidate (Ritalin), the psychostimulant employed to alleviate the neuropsychiatric symptoms, was found to improve cognitive performance in patients diagnosed with HAND84. A suggested treatment for HAND is to promote the synaptic concentration of dopamine through inhibiting the neurotransmitter transporters involved in the uptake of dopamine in HIV patients with dopaminergic dysfunction84–86.
Another obstacle in reducing HAND is the CNS hosting latent viruses as a reservoir in children and adults, contributing to the virus persistence in the brain. The CNS reservoir might reactivate after cART cessation and spread to other compartments. Some latency-reversing agents are available but present neurotoxic effects and BBB penetrability issues such as cART87.
Conclusion
With the introduction of highly active antiretroviral therapy, the life span of HIV patients has been dramatically prolonged. However, the prevalence of HAND has also increased, especially the mild-to-moderate neurological dysfunction form. HAND is possibly caused by HIV-1 viral proteins resulting in CNS toxicity. The onset and progression of the neurological disorder vary with the age of patients and the different subtype of HIV-1 virus. Recent discoveries have demonstrated how different subtypes result in different through the viral proteins specific structures and sequences and results in different incidence and severity of HAND. More studies are required to understand better the mechanisms of how viral proteins cause HAND, as well as to explore the similarities and differences between different subtypes and degree of HAND. In addition, more research should be done to better identify the differences between HAND and neurodegenerative and neuropsychiatric diseases not caused by HIV infections. Equal importance should be given to the development of appropriate treatments for HAND, either with improved regimens or neuroprotective and psychostimulant compounds or novel specific therapies for HAND.
Acknowledgments
Supported by NIH grants (R01-NS059327; R01-NS076401, R01-MH093331, and R01-AG054411) awarded to BES.
Footnotes
Conflicts of interest
None.
References
- 1.Serrano-Villar S, Sainz T, Lee SA, Hunt PW, Sinclair E, Shacklett BL, et al. HIV-infected individuals with low CD4/CD8 ratio despite effective antiretroviral therapy exhibit altered T cell subsets, heightened CD8+ T cell activation, and increased risk of non-AIDS morbidity and mortality. PLoS Pathog. 2014;10:e1004078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hsue P, Shreay S, Song X, Meyer N. A Longitudinal Analysis of Comorbidities among Human Immunodeficiency Virus (HIV) Patients and Matched Non-HIV Controls in the USA. ID Week, New Orleans, Abstract 950; 2016. [Google Scholar]
- 3.Spira S, Wainberg MA, Loemba H, Turner D, Brenner BG. Impact of clade diversity on HIV-1 virulence, antiretroviral drug sensitivity and drug resistance. J Antimicrob Chemother. 2003;51:229–40. [DOI] [PubMed] [Google Scholar]
- 4.Kuritzkes DR. HIV-1 subtype as a determinant of disease progression. J Infect Dis. 2008;197:638–9. [DOI] [PubMed] [Google Scholar]
- 5.McRae M. HIV and viral protein effects on the blood brain barrier. Tissue Barriers. 2016;4:e1143543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bachani M, Sacktor N, McArthur JC, Nath A, Rumbaugh J. Detection of anti-tat antibodies in CSF of individuals with HIV-associated neurocognitive disorders. J Neurovirol. 2013;19:82–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Giunta B, Hou H, Zhu Y, Rrapo E, Tian J, Takashi M, et al. HIV-1 tat contributes to Alzheimer’s disease-like pathology in PSAPP mice. Int J Clin Exp Pathol. 2009;2:433–43. [PMC free article] [PubMed] [Google Scholar]
- 8.Nookala AR, Shah A, Noel RJ, Kumar A. HIV-1 tat-mediated induction of CCL5 in astrocytes involves NF-κB, AP-1, C/EBPα and C/EBPγ transcription factors and JAK, PI3K/Akt and p38 MAPK signaling pathways. PLoS One. 2013;8:e78855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ru W, Tang SJ. HIV-associated synaptic degeneration. Mol Brain. 2017;10:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hui L, Chen X, Haughey NJ, Geiger JD. Role of endolysosomes in HIV-1 tat-induced neurotoxicity. ASN Neuro. 2012;4:243–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eletto D, Russo G, Passiatore G, Del Valle L, Giordano A, Khalili K, et al. Inhibition of SNAP25 expression by HIV-1 tat involves the activity of mir-128a. J Cell Physiol. 2008;216:764–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.González ME. The HIV-1 vpr protein: a multifaceted target for therapeutic intervention. Int J Mol Sci. 2017;18:E126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Piller SC, Ewart GD, Jans DA, Gage PW, Cox GB. The amino-terminal region of vpr from human immunodeficiency virus Type 1 forms ion channels and kills neurons. J Virol. 1999;73:4230–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Godet AN, Guergnon J, Croset A, Cayla X, Falanga PB, Colle JH, et al. PP2A1 binding, cell transducing and apoptotic properties of vpr(77–92): a new functional domain of HIV-1 vpr proteins. PLoS One. 2010;5:e13760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leymarie O, Lepont L, Berlioz-Torrent C. Canonical and non-canonical autophagy in HIV-1 replication cycle. Viruses. 2017;9:E270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheng X, Mukhtar M, Acheampong EA, Srinivasan A, Rafi M, Pomerantz RJ, et al. HIV-1 vpr potently induces programmed cell death in the CNS in vivo. DNA Cell Biol. 2007;26:116–31. [DOI] [PubMed] [Google Scholar]
- 17.Thaney VE, Sanchez AB, Fields JA, Minassian A, Young JW, Maung R, et al. Transgenic mice expressing HIV-1 envelope protein gp120 in the brain as an animal model in neuro AIDS research. J Neurovirol. 2018;24:156–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Capó-Vélez CM, Delgado-Vélez M, Báez-Pagán CA, Lasalde-Dominicci JA. Nicotinic acetylcholine receptors in HIV: possible roles during HAND and inflammation. Cell Mol Neurobiol. 2018;38:1335–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hayasaka H, Kobayashi D, Yoshimura H, Nakayama EE, Shioda T, Miyasaka M, et al. The HIV-1 gp120/CXCR4 axis promotes CCR7 ligand-dependent CD4 T cell migration: CCR7 homo-and CCR7/CXCR4 hetero-oligomer formation as a possible mechanism for up-regulation of functional CCR7. PLoS One. 2015;10:e0117454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berger EA, Murphy PM, Farber JM. Chemokine receptors as HIV-1 coreceptors: roles in viral entry, tropism, and disease. Annu Rev Immunol. 1999;17:657–700. [DOI] [PubMed] [Google Scholar]
- 21.Royal W 3rd, Zhang L, Guo M, Jones O, Davis H, Bryant JL, et al. Immune activation, viral gene product expression and neurotoxicity in the HIV-1 transgenic rat. J Neuroimmunol. 2012;247:16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pandhare J, Dash S, Jones B, Villalta F, Dash C. A novel role of proline oxidase in HIV-1 envelope glycoprotein-induced neuronal autophagy. J Biol Chem. 2015;290:25439–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bachis A, Avdoshina V, Zecca L, Parsadanian M, Mocchetti I. Human immunodeficiency virus Type 1 alters brain-derived neurotrophic factor processing in neurons. J Neurosci. 2012;32:9477–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Capó-Vélez CM, Morales-Vargas B, García-González A, Grajales-Reyes JG, Delgado-Vélez M, Madera B, et al. The alpha7-nicotinic receptor contributes to gp120-induced neurotoxicity: implications in HIV-associated neurocognitive disorders. Sci Rep. 2018;8:1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thaney VE, Kaul M. Type I interferons in neuroHIV. Viral Immunol. 2019;32:7–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu J, Xu C, Chen L, Xu P, Xiong H. Involvement of kv1.3 and p38 MAPK signaling in HIV-1 glycoprotein 120-induced microglia neurotoxicity. Cell Death Dis. 2012;3:e254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zou S, El-Hage N, Podhaizer EM, Knapp PE, Hauser KF. PTEN gene silencing prevents HIV-1 gp120(IIIB)-induced degeneration of striatal neurons. J Neurovirol. 2011;17:41–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Singh IN, El-Hage N, Campbell ME, Lutz SE, Knapp PE, Nath A, et al. Differential involvement of p38 and JNK MAP kinases in HIV-1 tat and gp120-induced apoptosis and neurite degeneration in striatal neurons. Neuroscience. 2005;135:781–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Torres-Muñoz J, Stockton P, Tacoronte N, Roberts B, Maronpot RR, Petito CK, et al. Detection of HIV-1 gene sequences in hippocampal neurons isolated from postmortem AIDS brains by laser capture micro-dissection. J Neuropathol Exp Neurol. 2001;60:885–92. [DOI] [PubMed] [Google Scholar]
- 30.Bagasra O, Lavi E, Bobroski L, Khalili K, Pestaner JP, Tawadros R, et al. Cellular reservoirs of HIV-1 in the central nervous system of infected individuals: identification by the combination of in situ polymerase chain reaction and immunohistochemistry. AIDS. 1996;10:573–85. [DOI] [PubMed] [Google Scholar]
- 31.Hayashi N, Matsubara M, Jinbo Y, Titani K, Izumi Y, Matsushima N, et al. Nef of HIV-1 interacts directly with calcium-bound calmodulin. Protein Sci. 2002;11:529–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mangino G, Famiglietti M, Capone C, Veroni C, Percario ZA, Leone S, et al. HIV-1 myristoylated nef treatment of murine microglial cells activates inducible nitric oxide synthase, NO2 production and neurotoxic activity. PLoS One. 2015;10:e0130189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saribas AS, Cicalese S, Ahooyi TM, Khalili K, Amini S, Sariyer IK, et al. HIV-1 nef is released in extracellular vesicles derived from astrocytes: evidence for nef-mediated neurotoxicity. Cell Death Dis. 2017;8:e2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chompre G, Cruz E, Maldonado L, Rivera-Amill V, Porter JT, Noel RJ Jr., et al. Astrocytic expression of HIV-1 nef impairs spatial and recognition memory. Neurobiol Dis. 2013;49:128–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ruiseñor-Escudero H, Sikorskii A, Familiar-Lopez I, Persaud D, Ziemniak C, Nakasujja N, et al. Neruodevelopmental outcomes in pre-school children living with HIV-1 subtypes A and D in uganda. Pediatr Infect Dis J. 2018;37:e298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Santos da Silva E, Mulinge M, Lemaire M, Masquelier C, Beraud C, Rybicki A, et al. The envelope cytoplasmic tail of HIV-1 subtype C contributes to poor replication capacity through low viral infectivity and cell-to-cell transmission. PLoS One. 2016;11:e0161596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim S, Hahn YK, Podhaizer EM, McLane VD, Zou S, Hauser KF, et al. A central role for glial CCR5 in directing the neuropathological interactions of HIV-1 tat and opiates. J Neuroinflammation. 2018;15:285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ranga U, Shankarappa R, Siddappa NB, Ramakrishna L, Nagendran R, Mahalingam M, et al. Tat protein of human immunodeficiency virus Type 1 subtype C strains is a defective chemokine. J Virol. 2004;78:2586–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rao VR, Sas AR, Eugenin EA, Siddappa NB, Bimonte-Nelson H, Berman JW, et al. HIV-1 clade-specific differences in the induction of neuropathogenesis. J Neurosci. 2008;28:10010–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Campbell GR, Watkins JD, Loret EP, Spector SA. Differential induction of rat neuronal excitotoxic cell death by human immunodeficiency virus Type 1 clade B and C tat proteins. AIDS Res Hum Retroviruses. 2011;27:647–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aralaguppe SP, Sharma S, Menon M, Prasad VR, Saravanan S, Murugavel KG, et al. The evolving profile of the signature amino acid residues in HIV-1 subtype C tat. AIDS Res Hum Retroviruses. 2016;32:503–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aksenov MY, Aksenova MV, Mactutus CF, Booze RM. HIV-1 protein-mediated amyloidogenesis in rat hippocampal cell cultures. Neurosci Lett. 2010;475:174–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li W, Huang Y, Reid R, Steiner J, Malpica-Llanos T, Darden TA, et al. NMDA receptor activation by HIV-tat protein is clade dependent. J Neurosci. 2008;28:12190–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Samikkannu T, Agudelo M, Gandhi N, Reddy PV, Saiyed ZM, Nwankwo D, et al. Human immunodeficiency virus Type 1 clade B and C gp120 differentially induce neurotoxin arachidonic acid in human astrocytes: implications for neuroAIDS. J Neurovirol. 2011;17:230–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Donnelly MR, Ciborowski P. Proteomics, biomarkers, and HIV-1: a current perspective. Proteomics Clin Appl. 2016;10:110–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Na H, Acharjee S, Jones G, Vivithanaporn P, Noorbakhsh F, McFarlane N, et al. Interactions between human immunodeficiency virus (HIV)-1 vpr expression and innate immunity influence neurovirulence. Retrovirology. 2011;8:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lamers SL, Fogel GB, Liu ES, Barbier AE, Rodriguez CW, Singer EJ, et al. Brain-specific HIV nef identified in multiple patients with neurological disease. J Neurovirol. 2018;24:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brunetti A, Berg G, Di Chiro G, Cohen RM, Yarchoan R, Pizzo PA, et al. Reversal of brain metabolic abnormalities following treatment of AIDS dementia complex with 3’-azido-2’,3’-dideoxythymidine (AZT, zidovudine): a PET-FDG study. J Nucl Med. 1989;30:581–90. [PubMed] [Google Scholar]
- 49.Baldeweg T, Catalan J, Gazzard BG. Risk of HIV dementia and opportunistic brain disease in AIDS and zidovudine therapy. J Neurol Neurosurg Psychiatry. 1998;65:34–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chiesi A, Agresti MG, Dally LG, Zaccarelli M, Tomino C, Floridia M, et al. Decrease in notifications of AIDS dementia complex in 1989–1990 in Italy: possible role of the early treatment with zidovudine. Medicina (Firenze). 1990;10:415–6. [PubMed] [Google Scholar]
- 51.Eggers C, Arendt G, Hahn K, Husstedt IW, Maschke M, Neuen-Jacob E, et al. HIV-1-associated neurocognitive disorder: epidemiology, pathogenesis, diagnosis, and treatment. J Neurol. 2017;264:1715–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sánchez-Portocarrero J, Jiménez-Escrig A, Pérez-Cecilia E, Ayuso-Mateos JL, Roca V, Yague MR, et al. AIDS dementia complex: incidence, clinical profile and impact of zidovudine treatment. Eur J Neurol. 1996;3:191–7. [DOI] [PubMed] [Google Scholar]
- 53.Del Palacio M, Alvarez S, Muñoz-Fernández MÁ. HIV-1 infection and neurocognitive impairment in the current era. Rev Med Virol. 2012; 22:33–45. [DOI] [PubMed] [Google Scholar]
- 54.Sacktor NC, Skolasky RL, Lyles RH, Esposito D, Selnes OA, McArthur JC, et al. Improvement in HIV-associated motor slowing after antiretroviral therapy including protease inhibitors. J Neurovirol. 2000;6:84–8. [DOI] [PubMed] [Google Scholar]
- 55.De Almeida TB, de Azevedo MC, Pinto JF, Ferry FR, da Silva GA, de Castro IJ, et al. Drug metabolism and transport gene polymorphisms and efavirenz adverse effects in Brazilian HIV-positive individuals. J Antimicrob Chemother. 2018;73:2460–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ciccarelli N, Fabbiani M, Di Giambenedetto S, Fanti I, Baldonero E, Bracciale L, et al. Efavirenz associated with cognitive disorders in otherwise asymptomatic HIV-infected patients. Neurology. 2011; 76:1403–9. [DOI] [PubMed] [Google Scholar]
- 57.Raines C, Radcliffe O, Treisman GJ. Neurologic and psychiatric complications of antiretroviral agents. J Assoc Nurses AIDS Care. 2005; 16:35–48. [DOI] [PubMed] [Google Scholar]
- 58.Lan X, Kiyota T, Hanamsagar R, Huang Y, Andrews S, Peng H, et al. The effect of HIV protease inhibitors on amyloid-β peptide degradation and synthesis in human cells and Alzheimer’s disease animal model. J Neuroimmune Pharmacol. 2012;7:412–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sharma B, Singh N, Singh M, Jaggi AS. Exploitation of HIV protease inhibitor indinavir as a memory restorative agent in experimental dementia. Pharmacol Biochem Behav. 2008;89:535–45. [DOI] [PubMed] [Google Scholar]
- 60.Perloff MD, von Moltke LL, Fahey JM, Greenblatt DJ. Induction of P-glycoprotein expression and activity by ritonavir in bovine brain microvessel endothelial cells. J Pharm Pharmacol. 2007;59:947–53. [DOI] [PubMed] [Google Scholar]
- 61.Kaddoumi A, Choi SU, Kinman L, Whittington D, Tsai CC, Ho RJ, et al. Inhibition of P-glycoprotein activity at the primate blood-brain barrier increases the distribution of nelfinavir into the brain but not into the cerebrospinal fluid. Drug Metab Dispos. 2007;35:1459–62. [DOI] [PubMed] [Google Scholar]
- 62.Yilmaz A, Price RW, Gisslén M. Antiretroviral drug treatment of CNS HIV-1 infection. J Antimicrob Chemother. 2012;67:299–311. [DOI] [PubMed] [Google Scholar]
- 63.Das MK, Sarma A, Chakraborty T. Nano-ART and neuroAIDS. Drug Deliv Transl Res. 2016;6:452–72. [DOI] [PubMed] [Google Scholar]
- 64.Nair M, Jayant RD, Kaushik A, Sagar V. Getting into the brain: potential of nanotechnology in the management of neuroAIDS. Adv Drug Deliv Rev. 2016;103:202–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ene L, Duiculescu D, Ruta SM. How much do antiretroviral drugs penetrate into the central nervous system? J Med Life. 2011;4:432–9. [PMC free article] [PubMed] [Google Scholar]
- 66.Doyle T, Dunn DT, Ceccherini-Silberstein F, De Mendoza C, Garcia F, Smit E, et al. Integrase inhibitor (INI) genotypic resistance in treatment-naive and raltegravir-experienced patients infected with diverse HIV-1 clades. J Antimicrob Chemother. 2015;70:3080–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bar-Magen T, Sloan RD, Faltenbacher VH, Donahue DA, Kuhl BD, Oliveira M, et al. Comparative biochemical analysis of HIV-1 subtype B and C integrase enzymes. Retrovirology. 2009;6:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Antinori A, Zaccarelli M, Cingolani A, Forbici F, Rizzo MG, Trotta MP, et al. Cross-resistance among nonnucleoside reverse transcriptase inhibitors limits recycling efavirenz after nevirapine failure. AIDS Res Hum Retroviruses. 2002;18:835–8. [DOI] [PubMed] [Google Scholar]
- 69.Kohlstaedt LA, Wang J, Friedman JM, Rice PA, Steitz TA. Crystal structure at 3.5 A resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science. 1992;256:1783–90. [DOI] [PubMed] [Google Scholar]
- 70.Spence RA, Kati WM, Anderson KS, Johnson KA. Mechanism of inhibition of HIV-1 reverse transcriptase by nonnucleoside inhibitors. Science. 1995;267:988–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ren J, Esnouf R, Garman E, Somers D, Ross C, Kirby I, et al. High resolution structures of HIV-1 RT from four RT-inhibitor complexes. Nat Struct Biol. 1995;2:293–302. [DOI] [PubMed] [Google Scholar]
- 72.Das K, Arnold E. HIV-1 reverse transcriptase and antiviral drug resistance. Part 1. Curr Opin Virol. 2013;3:111–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Brenner B, Turner D, Oliveira M, Moisi D, Detorio M, Carobene M, et al. A V106M mutation in HIV-1 clade C viruses exposed to efavirenz confers cross-resistance to non-nucleoside reverse transcriptase inhibitors. AIDS. 2003;17:F1–5. [DOI] [PubMed] [Google Scholar]
- 74.Grossman Z, Istomin V, Averbuch D, Lorber M, Risenberg K, Levi I, et al. Genetic variation at NNRTI resistance-associated positions in patients infected with HIV-1 subtype C. AIDS. 2004;18:909–15. [DOI] [PubMed] [Google Scholar]
- 75.Reuman EC, Rhee SY, Holmes SP, Shafer RW. Constrained patterns of covariation and clustering of HIV-1 non-nucleoside reverse transcriptase inhibitor resistance mutations. J Antimicrob Chemother. 2010;65:1477–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rhee SY, Kantor R, Katzenstein DA, Camacho R, Morris L, Sirivichayakul S, et al. HIV-1 pol mutation frequency by subtype and treatment experience: extension of the HIVseq program to seven non-B subtypes. AIDS. 2006;20:643–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Basson AE, Rhee SY, Parry CM, El-Khatib Z, Charalambous S, De Oliveira T, et al. Impact of drug resistance-associated amino acid changes in HIV-1 subtype C on susceptibility to newer nonnucleoside reverse transcriptase inhibitors. Antimicrob Agents Chemother. 2015;59:960–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sluis-Cremer N, Jordan MR, Huber K, Wallis CL, Bertagnolio S, Mellors JW, et al. E138A in HIV-1 reverse transcriptase is more common in subtype C than B: implications for rilpivirine use in resource-limited settings. Antiviral Res. 2014;107:31–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shin AH, Kim HJ, Thayer SA. Subtype selective NMDA receptor antagonists induce recovery of synapses lost following exposure to HIV-1 tat. Br J Pharmacol. 2012;166:1002–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Adams SM, Aksenova MV, Aksenov MY, Mactutus CF, Booze RM. ER-β mediates 17β-estradiol attenuation of HIV-1 tat-induced apoptotic signaling. Synapse. 2010;64:829–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Paris JJ, Zou S, Hahn YK, Knapp PE, Hauser KF 5α-reduced progestogens ameliorate mood-related behavioral pathology, neurotoxicity, and microgliosis associated with exposure to HIV-1 tat. Brain Behav Immun. 2016;55:202–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Corasaniti MT, Amantea D, Russo R, Piccirilli S, Leta A, Corazzari M, et al. 17beta-estradiol reduces neuronal apoptosis induced by HIV-1 gp120 in the neocortex of rat. Neurotoxicology. 2005;26:893–903. [DOI] [PubMed] [Google Scholar]
- 83.Russo R, Navarra M, Maiuolo J, Rotiroti D, Bagetta G, Corasaniti MT. 17β-estradiol protects SH-SY5Y cells against HIV-1 gp120-induced cell death: evidence for a role of estrogen receptors. NeuroToxicology. 2005;26:905–13. [DOI] [PubMed] [Google Scholar]
- 84.McLaurin KA, Li H, Booze RM, Fairchild AJ, Mactutus CF. Unraveling individual differences in the HIV-1 transgenic rat: therapeutic efficacy of methylphenidate. Sci Rep. 2018;8:136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Frain JA, Chen L. Examining the effectiveness of a cognitive intervention to improve cognitive function in a population of older adults living with HIV: a pilot study. Ther Adv Infect Dis. 2018;5:19–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Prommer E Methylphenidate: established and expanding roles in symptom management. Am J Hosp Palliat Care. 2012;29:483–90. [DOI] [PubMed] [Google Scholar]
- 87.Veenhuis RT, Clements JE, Gama L. HIV eradication strategies: implications for the central nervous system. Curr HIV/AIDS Rep. 2019;16:96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]