

Abstract
In this article, the correlation between the copy number of survival motor neuron 2 (SMN2) gene, neuronal apoptosis inhibitory protein (NAIP), and the phenotype of spinal muscular atrophy patients were analyzed.Forty patients with spinal muscular atrophy (SMA) were included in the study at the Department of Medical Genetics of the First People's Hospital and the Department of Neurology of the Second People's Hospital in Yunnan Province from January 2012 to September 2018. Multiplex ligation-dependent probe amplification assay was performed to determine the copy numbers of SMN2 and NAIP genes. Statistical analysis was performed to determine the correlation between copy numbers of the SMN2 and NAIP genes and the clinical phenotypes of SMA.Our results show that among the 40 SMA patients, there were 13 type I cases, 16 type II cases and 11 type III cases. A total of 37 patients possessed a homozygous deletion of SMN1 exons 7 and 8, while the other 3 SMA patients possessed a single copy of SMN1 exon 8. There was no correlation between SMA subtypes and the deletion types of SMN1 exon 7 and 8 (P = .611). The percentage of 2, 3, and 4 copies of SMN2 exon 7 was 25.0%, 62.5%, and 12.5%, respectively. The percentage of 0, 1, and 2 copies of NAIP exon 5 was 10%, 57.5%, and 32.5%, respectively. The distributions of SMN2 and NAIP copy numbers among various SMA types were significantly different (all P < .05). Five combined SMN1-SMN2-NAIP genotypes were detected, of which 0-3-1 genotype had the highest proportion than the others, accounting for 42.5%. The copy number of SMN2 and NAIP gene had synergistic effect on SMA phenotype. The combined SMN1-SMN2-NAIP genotypes with fewer copies were associated with earlier onset age, higher mortality, and smaller average age at death in SMA patients.
Therefore, we conclude that the copy number variance of SMN2 and NAIP is correlated with the SMA phenotype. Analysis of the copy number structure of the SMN1-SMN2-NAIP gene is helpful for SMA typing, disease prognosis prediction, and genetic counseling.
Keywords: clinical phenotype and genotype, multiplex ligation-dependent probe amplification (MLPA), neuronal apoptosis inhibitory protein, spinal muscular atrophy, survival motor neuron gene
1. Introduction
Spinal muscular atrophy (SMA)is an autosomal recessive neuromuscular disease characterized by symmetrical proximal muscle weakness and atrophy due to degeneration of the anterior horn cells of the spinal cord. It affects approximately 1 in 6000 to 10,000 white newborns and is the second most common fatal autosomal recessive disorder after cystic fibrosis.[1] The population carrying rate is about 1/54 in the world,[2,3] and the frequency of SMA carriers in the Chinese population is about 1/42 to 1/62.[4–6]
SMA is classified into types I through IV depending on the age of onset and the clinical course. SMA type I, also known as infantile SMA or Werdnig–Hoffmann disease, is the most common and severe type of SMA. SMA type I manifests as severe muscle weakness and hypotonia with onset in early infancy, and fatal respiratory failure usually before 2 years of age. SMA type II (intermediate form) occurs between 6 and 18 months of age, characterized by an ability to sit but not to walk unaided, and survival beyond 4 years of age. SMA type III (Kugelberg–Welander disease) is typically present with signs of weakness at or after 18 months of age and progresses to a chronic course. SMA type IV is a mild late (adult) onset form that is usually present in the second or third decade of life and has a normal life expectancy similar to SMA type III.[7,8]
SMA is caused by a deletion or mutation in the survival motor neuron 1 (SMN1) gene. SMN1 was discovered in 1995 and is located on chromosome 5q11-q13.1.[9] The SMN gene comprises 9 exons, with a stop codon is present near the end of exon 7. Two almost identical SMN genes are present at 5q13: the telomeric or SMN1 gene and centromeric or SMN2 gene. The coding sequence of SMN2 differs from that of SMN1 by a single nucleotide (840C > T), which does not alter the amino acid but has been shown to be important in splicing.[10]SMN2 copy numbers are a demonstrated phenotypic modifier in the SMA population, in which milder forms of the disease are associated with increased copy numbers of SMN2.[11–14]
Both copies of the SMN1 exon 7 are absent in approximately 95% of affected SMA patients. Although SMA patients lack SMN1, they always carry at least 1 copy of SMN2, which is partially functional but unable to compensate for the absence of SMN1. The remaining 5% of affected cases are compound heterozygotes for SMN1 exon 7 deletion and small intragenic mutations.[15] Type IV patients show only 20% to 30% homozygous deletion of SMN1 gene.[16,17]
NAIP, a regulatory gene, shows homology with a baculoviral inhibitor of apoptosis and inhibits apoptosis in mammalian cells.[18] The NAIP gene, spanning 56 kb of genomic sequence with 6.1 kb transcript, consists of 17 exons and is present in multiple and variable copy number on chromosome 5. Deletion of intact NAIP gene is associated with SMA. Homozygous deletion of NAIP exon 5 is more commonly observed in the severe and acute form of SMA than mild or chronic forms. Deletion of NAIP is found in approximately 45% of type I SMA and 18% of type II and III SMA.[19]
The NAIP gene is located in the functional domain near the SMN1 gene and is a potentially better SMA candidate regulatory gene. However, its functional role in the pathogenesis of SMA has not been fully elucidated. Therefore, to elucidate the relationship between SMN2 and NAIP copy number variation and SMA phenotype is important for clinical classification, disease progression, prognosis evaluation, and genetic counseling of SMA patients in this region.
2. Methods
From January 2012 to September 2018, 40 patients with SMA who were admitted to the Department of Medical Genetics of the First Affiliated Hospital of Yunnan Province and the Department of Neurology of the Second People's Hospital of Yunnan Province were selected for clinical classification according to the criteria defined by the 1992 International SMA Association,[20] including 22 males and 18 females. The patient's age ranged from 3 months to 24 years, and the median age was 3 years. The age of onset of patients ranged from 3 months to 6.5 years, with a median age of 3 years. Thirteen patients suffered from type I SMA, 16 patients from type II SMA, and 11 patients from type III SMA. The study was reviewed and approved by the Ethics Review Committee of the First People's Hospital of Yunnan Province (batch number: 2014YXLH055), and all patients and their legal guardians signed the informed consent form for the genetic diagnosis.
The experiments were performed as follows:
-
(1)
Reagents and instruments: The instruments included QIAamp DNA Blood Mini Genomic DNA Extraction Kit (Qiagen, Germany), ABI2720 PCR Amplifier (ABI Applied Biosystems, Foster City, Calif., USA), SALSA MLPA probemix P021-A2 SMA Kit (MRC-Holland, The Netherlands) and ABI- 3130 Genetic Analyzer (ABI), POP4 Gel (ABI). The primers were obtained from Bioengineering (Shanghai, China) Co, Ltd.
-
(2)
Extraction of genomic DNA: 2 mL of venous blood was taken from the subject, and EDTA-K2 was anticoagulated. The genomic DNA of the test subject was extracted with QIAamp DNA Blood Mini reagent, and the DNA concentration was measured by an ultraviolet spectrophotometer. The DNA concentration was then adjusted to 100 ng/μL with sterilized double-distilled water and stored at −20°C.
-
(3)
MLPA technology amplification and detection: SALSA MLPA probemix P021-A2 SMA kit (MRC) was used, strictly in accordance with the SMA-MLPA kit operating instructions. Samples with 2 copies of SMN1, SMN2, and NAIP genes were selected as negative controls, and reference samples were randomly distributed among all samples.
-
(4)
Analysis of MLPA results: CoffalyserNET software (MRC, www.mlpa.com-MLPA) was used to analyze the experimental results. According to the kit instructions, the copy number of exons 7, 8 of SMN1 and SMN2 genes and the copy number of exon 5 of NAIP gene were estimated. The parameters used to determine the copy numbers were listed in Table 1.
-
(5)
Patient survival follow-up: Data collection was performed on the patient's morbidity status and current living status through a combination of telephone, home visit, and in-patient medical case analysis.
-
(6)
Statistical analysis: All data were analyzed using the SPSS software package (version 17, SPSS Inc., Chicago, IL). The measurement data were expressed as mean ± standard deviation, the count data were expressed as the rate, and the comparison was performed by the χ2 test (Fisher exact test). P-value of <.05 was considered statistically significant.
Table 1.
The relationship between the relative ratio and the copy number.

3. Results
3.1. Forty cases of SMA patients with SMN1 gene mutation
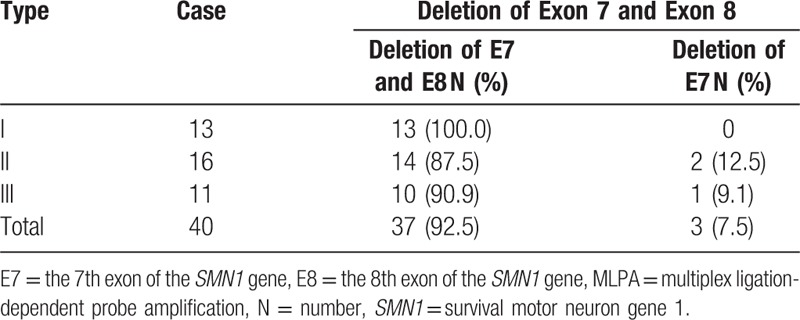
The samples taken from forty patients with SMA were analyzed by MLPA and 37 (92.5%) patients had homozygous deletion of exon 7 and 8 of SMN1. Only 3 patients with exon 7 deletion were present, accounting for 7.5% (3/40) of patients. There was no correlation between different SMA types and the 7th and 8th exon deletion types of SMN1 gene (P = .611). See Table 2.
Table 2.
MLPA technology to detect the deletion of exon 7 and exon 8 in the SMN1 gene.
3.2. The relationship between SMN2 and NAIP copy number and SMA clinical phenotype
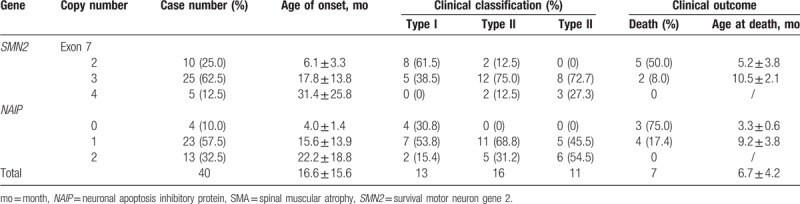
Among the 40 patients with SMA, the copy number 2, 3, and 4 of exon 7 of the SMN2 gene accounted for 25.0% (10/40), 62.5% (25/40), and 12.5% (5/40) of patients, respectively. The copy number 0, 1, and 2 of the 5th exon of the NAIP gene accounted for 10% (4/40), 57.5% (23/40), and 32.5% (13/40) of patients, respectively. The average age of onset for those with 2 copies of SMN2 gene was lower than that of patients with 3 or 4 copies of SMN2. The mortality rate of patients with 2 copies of SMN2 gene was 50% (5/10), which was 6.2 times higher than that of patients with 3 copies of the SMN2 gene, and the average death age of patients with 2 copies of SMN2 gene was less than that with 3 copies of SMN2 gene. There were no deaths in those with 4 copies.
The average age of onset of the NAIP gene in patients with 0 copies was less than that of patients with 1 and 2 copies, and the death toll was 75.0% (3/4), which was 4.3 times higher than that of patients with 1 copy of NAIP gene. The average age of patients with 0 copies was less than that with 1 copy, and there were no deaths in patients with 2 copies of NAIP gene. The results showed that there was a significant difference in the distribution of SMN2 and NAIP gene copy numbers among different types of SMA (P all <.05), and the copy number is related to the age of SMA onset. The smaller the copy number, the earlier the age of onset, the higher the death rate, and the smaller the average age at death. See Table 3.
Table 3.
Relationship between SMN2 and NAIP gene copy number and clinical phenotype of SMA.
3.3. Relationship between SMN1-SMN2-NAIP genotype and clinical phenotype of SMA
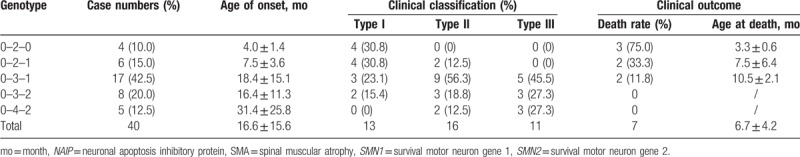
Five SMN1-SMN2-NAIP genotypes were detected in 40 patients with SMA, of which 0-3-1 was the highest, accounting for 42.5% (17/40) of patients (Table 4). Type 0-2-0 and 0-2-1 were the most common type I patients, accounting for 30.8% (4/13) of patients. Types 0-3-1 were the most common in type II and type III patients, accounting for 56.3% (9/16) and 45.5% (5/11), respectively. However, the proportion of 0-3-2 and 0-4-2 in type III patients was higher than that in type I and type II patients, accounting for 27.3% (3/13) of patients. There was a significant difference in the distribution of SMN1-SMN2-NAIP genotypes between different types of SMA (P = .026). SMN2 and NAIP genes had synergistic modification effects on SMA phenotype. The smaller the copy number, the earlier the patient's onset age, the higher the death rate, and the smaller the average age at death.
Table 4.
Relationship between SMN1-SMN2-NAIP genotype and clinical phenotype of SMA.
4. Discussion
SMA is a more common autosomal recessive disorder in childhood. The SMN1 gene located on chromosome 5q11.2-q13.2 is mainly associated with development of SMA, and about 80% to 95% of children with SMA have homozygous SMN1 gene. Deletion of a small number (∼5%) of complex heterozygous mutations may ultimately lead to a decrease in the expression of SMN protein.[7] The modified SMN2 gene of SMA is highly homologous to the SMN1 gene, with 5 base differences, 1 in intron 6, 1 in exon 7, 2 in intron 7, and 1 in exon 8.[4] The most pivotal gene modification is located at the 6th base in the 7th exon of SMN1 gene is C, while that of SMN2 gene is T. Because the large variations in the region of splicing enhancer in the SMN2 gene exon, about 90% of the exon 7 in SMN2 pre-mRNA is incorrectly spliced out, and finally encoded an unstable, easily degraded truncated protein (SMNΔ7).[21,22] However, the SMN2 gene can still express 10% to 20% of full-length functional SMN proteins. The more gene copy number, the greater the production of full-length transcripts, which can partially compensate for the deficiency of SMN protein and reduce the severity of SMA phenotype. The SMN2 gene copy number is inversely related to the severity of SMA.[7]
In addition to the SMN gene, the NAIP gene is also located in the functional domain near chromosome 5q13.2 and the distribution of the NAIP domain in the central nervous system is closely related to the degradation of SMA neural selection. The NAIP gene is a closely-associated SMA gene, and its copy number variation is related to the degree of SMA disease. When the copy number of NAIP gene is smaller, and the patient's condition is more serious.[23,24] The molecular genetic basis of SMA pathogenesis is potentially due to the deletion of exon 5. Therefore, clarifying the copy number and gene sequence structure of SMN1, SMN2, and NAIP genes is of great significance for analyzing the molecular mechanism and clinical diagnosis of SMA pathogenesis.[14]
MLPA technology is a relatively quantitative analysis technique. The concentration of DNA template used does not need to be accurately quantified. There are multiple internal reference probes for different chromosomes in the system, which constitute an internal control system of the detection system, effectively avoiding false positive and false negative results. In recent years, MLPA technology has been widely used in the diagnosis of SMA, especially its 1-time detection, which can simultaneously analyze the copy number variation of multiple genes, providing important support for SMA typing, disease prognosis, prenatal diagnosis, and genetic counseling.[4,5,25,26] In this study, we used MLPA technique to analyze SMN1 gene deletion in 40 patients with SMA and detected 37 cases of simultaneous deletion of exon 7 and exon 8 of SMN1, accounting for 92.5% (37/40) of patients, and 3 cases of a single copy of SMN1 exon 8, accounting for 7.5% (3/40) of patients. There was no correlation between SMA subtypes and the deletion types of SMN1 exon 7 and 8 (P = .611), indicating that the SMN1 gene is the molecular genetic basis of pathogenesis, with no correlation of SMA phenotype.[13]
Several studies have shown that the copy number of SMN2 and NAIP is related to the disease type. Watihayati et al[27] found that 25% of type I patients carried 1 copy of SMN2, while the remaining 71% carried 2 copies. Among the type II and type III SMA patients, 29% of cases carried 2 copies of the gene, while 71% carried 3 or 4 copies of SMN2. Deletion analysis of NAIP showed that 50% of type I SMA patients had a homozygous deletion of exon 5 of this gene and that only 10% of type II SMA cases carried a homozygous deletion, while all type III patients carried intact copies of the NAIP gene. Qu et al[13] found that in 232 Chinese patients with SMA, the copy number of exon 7 of SMN2 gene was 2, 3, and 4, accounting for 28.5% (66/232), 65.9% (153/232), and 5.6% (13/232) of patients, respectively, but 0% in those with 0 or 1 copy. The copy number of exon 5 of NAIP gene was 0, 1, and 2 copies, accounted for 15.1% (35/232), 62.5% (145/232), and 22.4% (52/232), respectively. Fang et al[14] found that in 42 Chinese patients with SMA, the copy number 1, 2, 3, and 4 of exon 7 of SMN2 gene accounted for 4.8% (2/42), 33.3% (14/42), 57.1% (24/42), and 4.8% (2/42) of patients, respectively, with none in those with 0 copies. The copy number 0, 1, and 2 of exon 5 of NAIP gene accounted for 9.5% (4/42), 61.9% (26/42), and 28.6% (12/42) of patients respectively. Ahn et al[24] found that 6 of 33 SMA patients had NAIP deletions and showed a more-severe phenotype of SMA. The proportion of patients who had died or received ventilator support was also higher in those with an NAIP deletion. Similar to the above research results, in 40 patients with SMA in this study, the copy number of exon 7 of SMN2 gene was 2, 3, and 4 copies, accounting for 25.0% (10/40), 62.5% (25/40), and 12.5% (5/40) of patients, respectively. The copy number of exon 5 of NAIP gene was 0, 1, and 2 copies, accounting for 10.0% (4/40), 57.5% (23/40), and 32.5% (13/40) of patients, respectively. The above results indicate that the SMN2 gene of Chinese SMA patients is mostly 2 or 3 copies, and the NAIP gene is mostly 1 copy. The data of this group indicated that the distribution of SMN2 and NAIP gene copy number was significantly different among different types of SMA (P < .05), and the copy number was correlated with the age of SMA. The smaller the copy number, the earlier of onset, the higher the proportion of death. It can be seen from Table 3 that the proportion of 3 copies of SMN2 gene in patients with type I SMA is 38.5%, while the proportion of 0 copies of NAIP gene is 30.8%, lower than that reported by others.[27,28] The higher proportions of 3 copies of SMN2 gene and lower proportions of 0 copies of NAIP gene indicate that the survival status of Chinese patients with type I SMA is better than that of Caucasians. This finding is similar to that reported by Qu et al.[13]
In this study, 5 SMN1-SMN2-NAIP genotypes were detected in 40 patients with SMA, of which 0-3-1 was the highest, accounting for 42.5% (17/40) of patients. Type 0-2-0 and 0-2-1 patients were the most common type I patients, accounting for 30.8% (4/13). There was a significant difference in the distribution of SMN1-SMN2-NAIP genotypes among different types of SMA (P < .05). SMN2 and NAIP genes had synergistic modification effects on SMA phenotype. The smaller the copy number was, the earlier the age of onset. In addition, the higher the death rate was, the smaller the average age at death. A recent report suggests that the dose effect of SMN2 is much higher than that of NAIP in terms of the age, mortality and survival rate of SMA patients.[13] As can be seen from Table 3 and Table 4, the patient mortality rate was 17.4% (4/23) when the NAIP gene was 1 copy, and the patient mortality rate increased to 33.3% (2/6) when the 0-2-1 genotype was used. The 0-3-1 genotype was 11.8% (2/17), which further supports the above conclusions. Our combined SMN1-SMN2-NAIP genotype analyses implied that the phenotype-modifier effect was more significant when the copy number increased for both SMN2 and NAIP genes than when there was an increase in either individual gene. Thus, identification of only SMN2 copies would be incomplete for genetic counseling and prognosis assessment for a SMA family, as NAIP copy number also has a certain modifier effect on the survival status of the SMA patients.[13] Furthermore, our data revealed that only confirming the gene copy numbers might not be sufficient to the clinical diagnosis, prenatal diagnosis and phenotype evaluation of SMA. The abnormal gene structures should be considered in the clinical diagnosis of SMA. Therefore, the analyses of SMA-related gene structures were also important to the molecular diagnostics of SMA patients.[14]
Due to the small sample size, the conclusions of this study have certain limitations. For example, the limited number of cases sometimes may affect the conclusion for the disease outcome of children with SMA due to larger standard deviation of the statistical results. In addition, the interactions of SMA with the regulatory genes may influence the phenotype of SMA. There are also several modified genes such as human 4F5 gene (H4F5), general transcription factor IIH polypeptide 2 gene (GTF2H2) and plastin 3 (PLS3) in the vicinity of the SMN1 gene in the 5q13 region.[29–32] Because of their proximity to the SMN1 gene, H4F5, and GTF2H2 are thought to be disease-modifying factors. In previous studies, the absence of H4F5 or GTF2H2 was highly associated with the severe form of SMA type I, although no direct association between these gene deficits and SMA phenotypes has been observed.[33] Beyond the region 5q13, the effect of PLS3, coronin-like actin-binding protein 1C (CORO1C) and neurocalcin delta (NCALD) on SMA have been confirmed in vivo and in vitro recently. Through upregulation of PLS3 and CORO1C genes and downregulation of NCALD gene, endocytosis of cells damaged by SMN gene defect can be repaired, synaptic function can be restored, which affects SMA prognosis.[29,31–34] As a result, the SMN1 gene conversion, SMN1 subtle mutations, SMN2 copy number, the extent of deletion in the 5q13 region, and the other non-5q13 molecular modifiers should all be considered in the genotype-phenotype analysis of SMA.
Author contributions
Conceptualization: Yinhong Zhang, Jing He, Yunqian Zhang, Li Li, Xinhua Tang, Lei Wang, Chanchan Jin, Sean Tighe, Yingting Zhu, Baosheng Zhu.
Data curation: Yinhong Zhang, Jing He, Yunqian Zhang, Li Li, Xinhua Tang, Lei Wang, Jingjing Guo, Chanchan Jin, Sean Tighe, Yingting Zhu, Baosheng Zhu.
Formal analysis: Yinhong Zhang, Jing He, Yunqian Zhang, Li Li, Xinhua Tang, Lei Wang, Jingjing Guo, Chanchan Jin, Sean Tighe, Yuan Zhang, Yingting Zhu, Baosheng Zhu.
Funding acquisition: Yinhong Zhang, Jing He.
Investigation: Yinhong Zhang, Jing He, Yunqian Zhang, Li Li, Xinhua Tang, Lei Wang, Jingjing Guo, Chanchan Jin, Sean Tighe, Yingting Zhu, Baosheng Zhu.
Methodology: Yinhong Zhang, Jing He, Li Li, Xinhua Tang, Chanchan Jin, Yuan Zhang, Yingting Zhu, Baosheng Zhu.
Project administration: Yinhong Zhang, Chanchan Jin, Sean Tighe, Yingting Zhu, Baosheng Zhu.
Resources: Yinhong Zhang, Jing He, Yunqian Zhang, Li Li, Xinhua Tang, Lei Wang, Jingjing Guo, Chanchan Jin, Sean Tighe, Yingting Zhu, Baosheng Zhu.
Software: Yinhong Zhang, Jing He, Yunqian Zhang, Li Li, Xinhua Tang, Lei Wang, Jingjing Guo, Chanchan Jin, Sean Tighe, Yuan Zhang, Yingting Zhu, Baosheng Zhu.
Supervision: Yingting Zhu, Baosheng Zhu.
Validation: Yinhong Zhang, Jing He, Yunqian Zhang, Li Li, Xinhua Tang, Lei Wang, Jingjing Guo, Chanchan Jin, Sean Tighe, Yingting Zhu, Baosheng Zhu.
Visualization: Yinhong Zhang, Jing He, Yunqian Zhang, Li Li, Xinhua Tang, Lei Wang, Jingjing Guo, Chanchan Jin, Sean Tighe, Yingting Zhu, Baosheng Zhu.
Writing – original draft: Yinhong Zhang, Jing He, Yunqian Zhang, Li Li, Xinhua Tang, Lei Wang, Sean Tighe.
Writing – review and editing: Yinhong Zhang, Sean Tighe, Yuan Zhang, Yingting Zhu, Baosheng Zhu.
Footnotes
Abbreviations: CORO1C = coronin-like actin-binding protein 1C, GTF2H2 = general transcription factor IIH polypeptide 2 gene, H4F5 = human 4F5 gene, MLPA = multiplex ligation-dependent probe amplification, NAIP = neuronal apoptosis inhibitory protein, NCALD = neurocalcin delta, PLS3 = plastin 3, SMA = spinal muscular atrophy, SMN = survival motor neuron gene.
How to cite this article: Zhang Y, He J, Zhang Y, Li L, Tang X, Wang L, Guo J, Jin C, Tighe S, Zhang Y, Zhu Y, Zhu B. The analysis of the association between the copy numbers of survival motor neuron gene 2 and neuronal apoptosis inhibitory protein genes and the clinical phenotypes in 40 patients with spinal muscular atrophy: Observational study. Medicine. 2020;99:3(e18809).
This work is supported by the Cooperation Fund of Kunming Medical University and Science and Technology, Department of Yunnan Province (Grant # 2015FB096), by the Medical Academic Leader of Yunnan Province (Grant # D-201643), and by the Medical Reserve Talents of Yunnan Province (Grant # H-201617).
The authors have no conflicts of interest to disclose.
References
- [1].Pearn J. Incidence, prevalence, and gene frequency studies of chronic childhood spinal muscular atrophy. J Med Genet 1978;15:409–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kolb SJ, Kissel JT. Spinal muscular atrophy. Neurol Clin 2015;33:831–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Sugarman EA, Nagan N, Zhu H, et al. Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis of >72,400 specimens. Eur J Hum Genet 2012;20:27–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Sheng-Yuan Z, Xiong F, Chen YJ, et al. Molecular characterization of SMN copy number derived from carrier screening and from core families with SMA in a Chinese population. Eur J Hum Genet 2010;18:978–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Su YN, Hung CC, Lin SY, et al. Carrier screening for spinal muscular atrophy (SMA) in 107,611 pregnant women during the period 2005-2009: a prospective population-based cohort study. PloS One 2011;6:e17067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chan V, Yip B, Yam I, et al. Carrier incidence for spinal muscular atrophy in southern Chinese. J Neurol 2004;251:1089–93. [DOI] [PubMed] [Google Scholar]
- [7].Verhaart IEC, Robertson A, Wilson IJ, et al. Prevalence, incidence and carrier frequency of 5q-linked spinal muscular atrophy - a literature review. Orphanet J Rare Dis 2017;12:124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Butchbach ME. Copy number variations in the survival motor neuron genes: implications for spinal muscular atrophy and other neurodegenerative diseases. Front Mol Biosci 2016;3:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lefebvre S, Burglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995;80:155–65. [DOI] [PubMed] [Google Scholar]
- [10].Burglen L, Lefebvre S, Clermont O, et al. Structure and organization of the human survival motor neurone (SMN) gene. Genomics 1996;32:479–82. [DOI] [PubMed] [Google Scholar]
- [11].Wirth B, Brichta L, Schrank B, et al. Mildly affected patients with spinal muscular atrophy are partially protected by an increased SMN2 copy number. Hum Genet 2006;119:422–8. [DOI] [PubMed] [Google Scholar]
- [12].Prior TW, Swoboda KJ, Scott HD, et al. Homozygous SMN1 deletions in unaffected family members and modification of the phenotype by SMN2. Am J Med Genet A 2004;130A:307–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Qu YJ, Ge XS, Bai JL, et al. Association of copy numbers of survival motor neuron gene 2 and neuronal apoptosis inhibitory protein gene with the natural history in a Chinese spinal muscular atrophy cohort. J Child Neurol 2015;30:429–36. [DOI] [PubMed] [Google Scholar]
- [14].Fang P, Li L, Zeng J, et al. Molecular characterization and copy number of SMN1, SMN2 and NAIP in Chinese patients with spinal muscular atrophy and unrelated healthy controls. BMC Musculoskelet Disord 2015;16:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Prior TW, Snyder PJ, Rink BD, et al. Newborn and carrier screening for spinal muscular atrophy. Am J Med Genet A 2010;152A:1608–16. [DOI] [PubMed] [Google Scholar]
- [16].Brahe C, Servidei S, Zappata S, et al. Genetic homogeneity between childhood-onset and adult-onset autosomal recessive spinal muscular atrophy. Lancet (London, England) 1995;346:741–2. [DOI] [PubMed] [Google Scholar]
- [17].Chang JG, Jong YJ, Lin SP, et al. Molecular analysis of survival motor neuron (SMN) and neuronal apoptosis inhibitory protein (NAIP) genes of spinal muscular atrophy patients and their parents. Hum Genet 1997;100:577–81. [DOI] [PubMed] [Google Scholar]
- [18].Kesari A, Misra UK, Kalita J, et al. Study of survival of motor neuron (SMN) and neuronal apoptosis inhibitory protein (NAIP) gene deletions in SMA patients. J Neurol 2005;252:667–71. [DOI] [PubMed] [Google Scholar]
- [19].Shin S, Park SS, Hwang YS, et al. Deletion of SMN and NAIP genes in Korean patients with spinal muscular atrophy. J Korean Med Sci 2000;15:93–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Munsat TL, Davies KE. International SMA consortium meeting. (26-28 June 1992, Bonn, Germany). Neuromuscul Disord 1992;2:423–8. [DOI] [PubMed] [Google Scholar]
- [21].Chaytow H, Huang YT, Gillingwater TH, et al. The role of survival motor neuron protein (SMN) in protein homeostasis. Cell Mol Life Sci 2018;75:3877–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Gray KM, Kaifer KA, Baillat D, et al. Self-oligomerization regulates stability of survival motor neuron protein isoforms by sequestering an SCF (Slmb) degron. Mol Biol Cell 2018;29:96–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Omrani O, Bonyadi M, Barzgar M. Molecular analysis of the SMN and NAIP genes in Iranian spinal muscular atrophy patients. Pediatr Int 2009;51:193–6. [DOI] [PubMed] [Google Scholar]
- [24].Ahn EJ, Yum MS, Kim EH, et al. Genotype-phenotype correlation of SMN1 and NAIP deletions in Korean patients with spinal muscular atrophy. J Clin Neurol 2017;13:27–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Zeng J, Lin Y, Yan A, et al. Establishment of a molecular diagnostic system for spinal muscular atrophy experience from a clinical laboratory in china. J Mol Diagn 2011;13:41–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Parks M, Court S, Bowns B, et al. Non-invasive prenatal diagnosis of spinal muscular atrophy by relative haplotype dosage. Eur J Hum Genet 2017;25:416–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Watihayati MS, Fatemeh H, Marini M, et al. Combination of SMN2 copy number and NAIP deletion predicts disease severity in spinal muscular atrophy. Brain Dev 2009;31:42–5. [DOI] [PubMed] [Google Scholar]
- [28].Wozniacka A, Wieczorkowska M, Gebicki J, et al. Topical application of 1-methylnicotinamide in the treatment of rosacea: a pilot study. Clin Exp Dermatol 2005;30:632–5. [DOI] [PubMed] [Google Scholar]
- [29].Oprea GE, Krober S, McWhorter ML, et al. Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science 2008;320:524–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].He J, Zhang QJ, Lin QF, et al. Molecular analysis of SMN1, SMN2, NAIP, GTF2H2, and H4F5 genes in 157 Chinese patients with spinal muscular atrophy. Gene 2013;518:325–9. [DOI] [PubMed] [Google Scholar]
- [31].Nishio H. PLS3 expression and SMA phenotype: a commentary on correlation of PLS3 expression with disease severity in children with spinal muscular atrophy. J Hum Genet 2014;59:64–5. [DOI] [PubMed] [Google Scholar]
- [32].Strathmann EA, Peters M, Hosseinibarkooie S, et al. Evaluation of potential effects of Plastin 3 overexpression and low-dose SMN-antisense oligonucleotides on putative biomarkers in spinal muscular atrophy mice. PloS One 2018;13:e0203398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hosseinibarkooie S, Peters M, Torres-Benito L, et al. The power of human protective modifiers: PLS3 and CORO1C unravel impaired endocytosis in spinal muscular atrophy and rescue SMA phenotype. Am J Hum Genet 2016;99:647–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Riessland M, Kaczmarek A, Schneider S, et al. Neurocalcin delta suppression protects against spinal muscular atrophy in humans and across species by restoring impaired endocytosis. Am J Hum Genet 2017;100:297–315. [DOI] [PMC free article] [PubMed] [Google Scholar]