Elena Cortés-Vicente
Elena Cortés-Vicente, MD, PhD
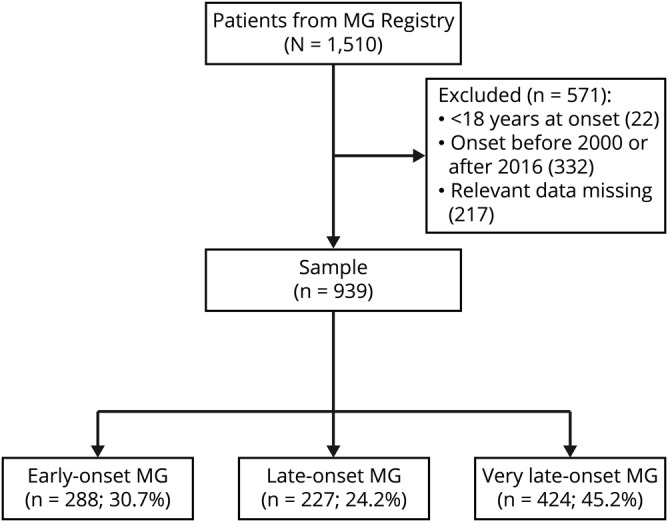
1From the Neuromuscular Diseases Unit, Department of Neurology (E.C.-V., R.Á.-V., R.R.-G., J.D.-M., L.Q., E.G., I.I.), Hospital de la Santa Creu i Sant Pau; Department of Medicine (E.C.-V., R.Á.-V., I.I.), Universitat Autònoma de Barcelona; Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) (E.C.-V., R.Á.-V., S.S., C.C., T.S., R.R.-G., J.D.-M., L.Q., E.G., I.I.) and Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED) (C.P., A.L.d.M., A.L.P.-N.), Instituto de Salud Carlos III, Madrid; Neurology Department (C.P., B.V.), Neuromuscular Disorders Unit, Instituto de Biomedicina de Sevilla, Hospital U. Virgen del Rocío, CSIC, Universidad de Sevilla; Unitat de Neuromuscular (C.C., M.A.A.), Neurology Department, Hospital Universitari de Bellvitge–IDIBELL, l'Hospitalet de Llobregat, Barcelona; Neuromuscular Diseases Unit (A.-G.S., L.G.), Department of Neurology, Institute of Neurosciences, Hospital Universitario Clínico San Carlos, Madrid; Neurology Department (J.P., T.G.-S.), Hospital Clínico, Santiago de Compostela; Neuromuscular Diseases Unit (A.R.-F., A.M.-P.), Department of Neurology, Hospital Germans Trias i Pujol; Department of Medicine (A.R.-F.), Universitat Autònoma de Barcelona, Badalona; Neuromuscular Unit (T.S., A.L.-M.), Neurology Department, Hospital Universitari i Politècnic La Fe; Department of Medicine (T.S.), Universitat de València; Neurology Department (A.L.d.M., A.F.-T.), Donostia University Hospital; Neurosciences Area (A.L.d.M.), Biodonostia Research Institute, University of the Basque Country, San Sebastián; Neurology Department (M.T.G.), Hospital Universitario Reina Sofía, Córdoba; Department of Neurology (I.J.), Complejo Hospitalario de Navarra, Pamplona; Neuromuscular Diseases Unit (G.G.-G.), Department of Neurology, Hospital Universitario Infanta Sofía, Universidad Europea de Madrid, San Sebastián de los Reyes, Madrid; Complejo asistencial hospitalario de Burgos (M.A.M.), Burgos; Hospital Universitario de Gran Canaria Doctor Negrín (M.D.M.), Las Palmas de Gran Canaria; Hospital Central de Asturias (G.M.), Oviedo; and Service of Neurology (A.L.P.-N., Á.C.-A.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, Santander, Spain.
1,
Rodrigo Álvarez-Velasco
Rodrigo Álvarez-Velasco, MD
1From the Neuromuscular Diseases Unit, Department of Neurology (E.C.-V., R.Á.-V., R.R.-G., J.D.-M., L.Q., E.G., I.I.), Hospital de la Santa Creu i Sant Pau; Department of Medicine (E.C.-V., R.Á.-V., I.I.), Universitat Autònoma de Barcelona; Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) (E.C.-V., R.Á.-V., S.S., C.C., T.S., R.R.-G., J.D.-M., L.Q., E.G., I.I.) and Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED) (C.P., A.L.d.M., A.L.P.-N.), Instituto de Salud Carlos III, Madrid; Neurology Department (C.P., B.V.), Neuromuscular Disorders Unit, Instituto de Biomedicina de Sevilla, Hospital U. Virgen del Rocío, CSIC, Universidad de Sevilla; Unitat de Neuromuscular (C.C., M.A.A.), Neurology Department, Hospital Universitari de Bellvitge–IDIBELL, l'Hospitalet de Llobregat, Barcelona; Neuromuscular Diseases Unit (A.-G.S., L.G.), Department of Neurology, Institute of Neurosciences, Hospital Universitario Clínico San Carlos, Madrid; Neurology Department (J.P., T.G.-S.), Hospital Clínico, Santiago de Compostela; Neuromuscular Diseases Unit (A.R.-F., A.M.-P.), Department of Neurology, Hospital Germans Trias i Pujol; Department of Medicine (A.R.-F.), Universitat Autònoma de Barcelona, Badalona; Neuromuscular Unit (T.S., A.L.-M.), Neurology Department, Hospital Universitari i Politècnic La Fe; Department of Medicine (T.S.), Universitat de València; Neurology Department (A.L.d.M., A.F.-T.), Donostia University Hospital; Neurosciences Area (A.L.d.M.), Biodonostia Research Institute, University of the Basque Country, San Sebastián; Neurology Department (M.T.G.), Hospital Universitario Reina Sofía, Córdoba; Department of Neurology (I.J.), Complejo Hospitalario de Navarra, Pamplona; Neuromuscular Diseases Unit (G.G.-G.), Department of Neurology, Hospital Universitario Infanta Sofía, Universidad Europea de Madrid, San Sebastián de los Reyes, Madrid; Complejo asistencial hospitalario de Burgos (M.A.M.), Burgos; Hospital Universitario de Gran Canaria Doctor Negrín (M.D.M.), Las Palmas de Gran Canaria; Hospital Central de Asturias (G.M.), Oviedo; and Service of Neurology (A.L.P.-N., Á.C.-A.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, Santander, Spain.
1,
Sonia Segovia
Sonia Segovia, RN
1From the Neuromuscular Diseases Unit, Department of Neurology (E.C.-V., R.Á.-V., R.R.-G., J.D.-M., L.Q., E.G., I.I.), Hospital de la Santa Creu i Sant Pau; Department of Medicine (E.C.-V., R.Á.-V., I.I.), Universitat Autònoma de Barcelona; Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) (E.C.-V., R.Á.-V., S.S., C.C., T.S., R.R.-G., J.D.-M., L.Q., E.G., I.I.) and Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED) (C.P., A.L.d.M., A.L.P.-N.), Instituto de Salud Carlos III, Madrid; Neurology Department (C.P., B.V.), Neuromuscular Disorders Unit, Instituto de Biomedicina de Sevilla, Hospital U. Virgen del Rocío, CSIC, Universidad de Sevilla; Unitat de Neuromuscular (C.C., M.A.A.), Neurology Department, Hospital Universitari de Bellvitge–IDIBELL, l'Hospitalet de Llobregat, Barcelona; Neuromuscular Diseases Unit (A.-G.S., L.G.), Department of Neurology, Institute of Neurosciences, Hospital Universitario Clínico San Carlos, Madrid; Neurology Department (J.P., T.G.-S.), Hospital Clínico, Santiago de Compostela; Neuromuscular Diseases Unit (A.R.-F., A.M.-P.), Department of Neurology, Hospital Germans Trias i Pujol; Department of Medicine (A.R.-F.), Universitat Autònoma de Barcelona, Badalona; Neuromuscular Unit (T.S., A.L.-M.), Neurology Department, Hospital Universitari i Politècnic La Fe; Department of Medicine (T.S.), Universitat de València; Neurology Department (A.L.d.M., A.F.-T.), Donostia University Hospital; Neurosciences Area (A.L.d.M.), Biodonostia Research Institute, University of the Basque Country, San Sebastián; Neurology Department (M.T.G.), Hospital Universitario Reina Sofía, Córdoba; Department of Neurology (I.J.), Complejo Hospitalario de Navarra, Pamplona; Neuromuscular Diseases Unit (G.G.-G.), Department of Neurology, Hospital Universitario Infanta Sofía, Universidad Europea de Madrid, San Sebastián de los Reyes, Madrid; Complejo asistencial hospitalario de Burgos (M.A.M.), Burgos; Hospital Universitario de Gran Canaria Doctor Negrín (M.D.M.), Las Palmas de Gran Canaria; Hospital Central de Asturias (G.M.), Oviedo; and Service of Neurology (A.L.P.-N., Á.C.-A.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, Santander, Spain.
1,
Carmen Paradas
Carmen Paradas, MD, PhD
1From the Neuromuscular Diseases Unit, Department of Neurology (E.C.-V., R.Á.-V., R.R.-G., J.D.-M., L.Q., E.G., I.I.), Hospital de la Santa Creu i Sant Pau; Department of Medicine (E.C.-V., R.Á.-V., I.I.), Universitat Autònoma de Barcelona; Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) (E.C.-V., R.Á.-V., S.S., C.C., T.S., R.R.-G., J.D.-M., L.Q., E.G., I.I.) and Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED) (C.P., A.L.d.M., A.L.P.-N.), Instituto de Salud Carlos III, Madrid; Neurology Department (C.P., B.V.), Neuromuscular Disorders Unit, Instituto de Biomedicina de Sevilla, Hospital U. Virgen del Rocío, CSIC, Universidad de Sevilla; Unitat de Neuromuscular (C.C., M.A.A.), Neurology Department, Hospital Universitari de Bellvitge–IDIBELL, l'Hospitalet de Llobregat, Barcelona; Neuromuscular Diseases Unit (A.-G.S., L.G.), Department of Neurology, Institute of Neurosciences, Hospital Universitario Clínico San Carlos, Madrid; Neurology Department (J.P., T.G.-S.), Hospital Clínico, Santiago de Compostela; Neuromuscular Diseases Unit (A.R.-F., A.M.-P.), Department of Neurology, Hospital Germans Trias i Pujol; Department of Medicine (A.R.-F.), Universitat Autònoma de Barcelona, Badalona; Neuromuscular Unit (T.S., A.L.-M.), Neurology Department, Hospital Universitari i Politècnic La Fe; Department of Medicine (T.S.), Universitat de València; Neurology Department (A.L.d.M., A.F.-T.), Donostia University Hospital; Neurosciences Area (A.L.d.M.), Biodonostia Research Institute, University of the Basque Country, San Sebastián; Neurology Department (M.T.G.), Hospital Universitario Reina Sofía, Córdoba; Department of Neurology (I.J.), Complejo Hospitalario de Navarra, Pamplona; Neuromuscular Diseases Unit (G.G.-G.), Department of Neurology, Hospital Universitario Infanta Sofía, Universidad Europea de Madrid, San Sebastián de los Reyes, Madrid; Complejo asistencial hospitalario de Burgos (M.A.M.), Burgos; Hospital Universitario de Gran Canaria Doctor Negrín (M.D.M.), Las Palmas de Gran Canaria; Hospital Central de Asturias (G.M.), Oviedo; and Service of Neurology (A.L.P.-N., Á.C.-A.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, Santander, Spain.
1,
Carlos Casasnovas
Carlos Casasnovas, MD, PhD
1From the Neuromuscular Diseases Unit, Department of Neurology (E.C.-V., R.Á.-V., R.R.-G., J.D.-M., L.Q., E.G., I.I.), Hospital de la Santa Creu i Sant Pau; Department of Medicine (E.C.-V., R.Á.-V., I.I.), Universitat Autònoma de Barcelona; Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) (E.C.-V., R.Á.-V., S.S., C.C., T.S., R.R.-G., J.D.-M., L.Q., E.G., I.I.) and Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED) (C.P., A.L.d.M., A.L.P.-N.), Instituto de Salud Carlos III, Madrid; Neurology Department (C.P., B.V.), Neuromuscular Disorders Unit, Instituto de Biomedicina de Sevilla, Hospital U. Virgen del Rocío, CSIC, Universidad de Sevilla; Unitat de Neuromuscular (C.C., M.A.A.), Neurology Department, Hospital Universitari de Bellvitge–IDIBELL, l'Hospitalet de Llobregat, Barcelona; Neuromuscular Diseases Unit (A.-G.S., L.G.), Department of Neurology, Institute of Neurosciences, Hospital Universitario Clínico San Carlos, Madrid; Neurology Department (J.P., T.G.-S.), Hospital Clínico, Santiago de Compostela; Neuromuscular Diseases Unit (A.R.-F., A.M.-P.), Department of Neurology, Hospital Germans Trias i Pujol; Department of Medicine (A.R.-F.), Universitat Autònoma de Barcelona, Badalona; Neuromuscular Unit (T.S., A.L.-M.), Neurology Department, Hospital Universitari i Politècnic La Fe; Department of Medicine (T.S.), Universitat de València; Neurology Department (A.L.d.M., A.F.-T.), Donostia University Hospital; Neurosciences Area (A.L.d.M.), Biodonostia Research Institute, University of the Basque Country, San Sebastián; Neurology Department (M.T.G.), Hospital Universitario Reina Sofía, Córdoba; Department of Neurology (I.J.), Complejo Hospitalario de Navarra, Pamplona; Neuromuscular Diseases Unit (G.G.-G.), Department of Neurology, Hospital Universitario Infanta Sofía, Universidad Europea de Madrid, San Sebastián de los Reyes, Madrid; Complejo asistencial hospitalario de Burgos (M.A.M.), Burgos; Hospital Universitario de Gran Canaria Doctor Negrín (M.D.M.), Las Palmas de Gran Canaria; Hospital Central de Asturias (G.M.), Oviedo; and Service of Neurology (A.L.P.-N., Á.C.-A.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, Santander, Spain.
1,
Antonio Guerrero-Sola
Antonio Guerrero-Sola, MD
1From the Neuromuscular Diseases Unit, Department of Neurology (E.C.-V., R.Á.-V., R.R.-G., J.D.-M., L.Q., E.G., I.I.), Hospital de la Santa Creu i Sant Pau; Department of Medicine (E.C.-V., R.Á.-V., I.I.), Universitat Autònoma de Barcelona; Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) (E.C.-V., R.Á.-V., S.S., C.C., T.S., R.R.-G., J.D.-M., L.Q., E.G., I.I.) and Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED) (C.P., A.L.d.M., A.L.P.-N.), Instituto de Salud Carlos III, Madrid; Neurology Department (C.P., B.V.), Neuromuscular Disorders Unit, Instituto de Biomedicina de Sevilla, Hospital U. Virgen del Rocío, CSIC, Universidad de Sevilla; Unitat de Neuromuscular (C.C., M.A.A.), Neurology Department, Hospital Universitari de Bellvitge–IDIBELL, l'Hospitalet de Llobregat, Barcelona; Neuromuscular Diseases Unit (A.-G.S., L.G.), Department of Neurology, Institute of Neurosciences, Hospital Universitario Clínico San Carlos, Madrid; Neurology Department (J.P., T.G.-S.), Hospital Clínico, Santiago de Compostela; Neuromuscular Diseases Unit (A.R.-F., A.M.-P.), Department of Neurology, Hospital Germans Trias i Pujol; Department of Medicine (A.R.-F.), Universitat Autònoma de Barcelona, Badalona; Neuromuscular Unit (T.S., A.L.-M.), Neurology Department, Hospital Universitari i Politècnic La Fe; Department of Medicine (T.S.), Universitat de València; Neurology Department (A.L.d.M., A.F.-T.), Donostia University Hospital; Neurosciences Area (A.L.d.M.), Biodonostia Research Institute, University of the Basque Country, San Sebastián; Neurology Department (M.T.G.), Hospital Universitario Reina Sofía, Córdoba; Department of Neurology (I.J.), Complejo Hospitalario de Navarra, Pamplona; Neuromuscular Diseases Unit (G.G.-G.), Department of Neurology, Hospital Universitario Infanta Sofía, Universidad Europea de Madrid, San Sebastián de los Reyes, Madrid; Complejo asistencial hospitalario de Burgos (M.A.M.), Burgos; Hospital Universitario de Gran Canaria Doctor Negrín (M.D.M.), Las Palmas de Gran Canaria; Hospital Central de Asturias (G.M.), Oviedo; and Service of Neurology (A.L.P.-N., Á.C.-A.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, Santander, Spain.
1,
Julio Pardo
Julio Pardo, MD, PhD
1From the Neuromuscular Diseases Unit, Department of Neurology (E.C.-V., R.Á.-V., R.R.-G., J.D.-M., L.Q., E.G., I.I.), Hospital de la Santa Creu i Sant Pau; Department of Medicine (E.C.-V., R.Á.-V., I.I.), Universitat Autònoma de Barcelona; Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) (E.C.-V., R.Á.-V., S.S., C.C., T.S., R.R.-G., J.D.-M., L.Q., E.G., I.I.) and Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED) (C.P., A.L.d.M., A.L.P.-N.), Instituto de Salud Carlos III, Madrid; Neurology Department (C.P., B.V.), Neuromuscular Disorders Unit, Instituto de Biomedicina de Sevilla, Hospital U. Virgen del Rocío, CSIC, Universidad de Sevilla; Unitat de Neuromuscular (C.C., M.A.A.), Neurology Department, Hospital Universitari de Bellvitge–IDIBELL, l'Hospitalet de Llobregat, Barcelona; Neuromuscular Diseases Unit (A.-G.S., L.G.), Department of Neurology, Institute of Neurosciences, Hospital Universitario Clínico San Carlos, Madrid; Neurology Department (J.P., T.G.-S.), Hospital Clínico, Santiago de Compostela; Neuromuscular Diseases Unit (A.R.-F., A.M.-P.), Department of Neurology, Hospital Germans Trias i Pujol; Department of Medicine (A.R.-F.), Universitat Autònoma de Barcelona, Badalona; Neuromuscular Unit (T.S., A.L.-M.), Neurology Department, Hospital Universitari i Politècnic La Fe; Department of Medicine (T.S.), Universitat de València; Neurology Department (A.L.d.M., A.F.-T.), Donostia University Hospital; Neurosciences Area (A.L.d.M.), Biodonostia Research Institute, University of the Basque Country, San Sebastián; Neurology Department (M.T.G.), Hospital Universitario Reina Sofía, Córdoba; Department of Neurology (I.J.), Complejo Hospitalario de Navarra, Pamplona; Neuromuscular Diseases Unit (G.G.-G.), Department of Neurology, Hospital Universitario Infanta Sofía, Universidad Europea de Madrid, San Sebastián de los Reyes, Madrid; Complejo asistencial hospitalario de Burgos (M.A.M.), Burgos; Hospital Universitario de Gran Canaria Doctor Negrín (M.D.M.), Las Palmas de Gran Canaria; Hospital Central de Asturias (G.M.), Oviedo; and Service of Neurology (A.L.P.-N., Á.C.-A.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, Santander, Spain.
1,
Alba Ramos-Fransi
Alba Ramos-Fransi, MD, PhD
1From the Neuromuscular Diseases Unit, Department of Neurology (E.C.-V., R.Á.-V., R.R.-G., J.D.-M., L.Q., E.G., I.I.), Hospital de la Santa Creu i Sant Pau; Department of Medicine (E.C.-V., R.Á.-V., I.I.), Universitat Autònoma de Barcelona; Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) (E.C.-V., R.Á.-V., S.S., C.C., T.S., R.R.-G., J.D.-M., L.Q., E.G., I.I.) and Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED) (C.P., A.L.d.M., A.L.P.-N.), Instituto de Salud Carlos III, Madrid; Neurology Department (C.P., B.V.), Neuromuscular Disorders Unit, Instituto de Biomedicina de Sevilla, Hospital U. Virgen del Rocío, CSIC, Universidad de Sevilla; Unitat de Neuromuscular (C.C., M.A.A.), Neurology Department, Hospital Universitari de Bellvitge–IDIBELL, l'Hospitalet de Llobregat, Barcelona; Neuromuscular Diseases Unit (A.-G.S., L.G.), Department of Neurology, Institute of Neurosciences, Hospital Universitario Clínico San Carlos, Madrid; Neurology Department (J.P., T.G.-S.), Hospital Clínico, Santiago de Compostela; Neuromuscular Diseases Unit (A.R.-F., A.M.-P.), Department of Neurology, Hospital Germans Trias i Pujol; Department of Medicine (A.R.-F.), Universitat Autònoma de Barcelona, Badalona; Neuromuscular Unit (T.S., A.L.-M.), Neurology Department, Hospital Universitari i Politècnic La Fe; Department of Medicine (T.S.), Universitat de València; Neurology Department (A.L.d.M., A.F.-T.), Donostia University Hospital; Neurosciences Area (A.L.d.M.), Biodonostia Research Institute, University of the Basque Country, San Sebastián; Neurology Department (M.T.G.), Hospital Universitario Reina Sofía, Córdoba; Department of Neurology (I.J.), Complejo Hospitalario de Navarra, Pamplona; Neuromuscular Diseases Unit (G.G.-G.), Department of Neurology, Hospital Universitario Infanta Sofía, Universidad Europea de Madrid, San Sebastián de los Reyes, Madrid; Complejo asistencial hospitalario de Burgos (M.A.M.), Burgos; Hospital Universitario de Gran Canaria Doctor Negrín (M.D.M.), Las Palmas de Gran Canaria; Hospital Central de Asturias (G.M.), Oviedo; and Service of Neurology (A.L.P.-N., Á.C.-A.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, Santander, Spain.
1,
Teresa Sevilla
Teresa Sevilla, MD, PhD
1From the Neuromuscular Diseases Unit, Department of Neurology (E.C.-V., R.Á.-V., R.R.-G., J.D.-M., L.Q., E.G., I.I.), Hospital de la Santa Creu i Sant Pau; Department of Medicine (E.C.-V., R.Á.-V., I.I.), Universitat Autònoma de Barcelona; Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) (E.C.-V., R.Á.-V., S.S., C.C., T.S., R.R.-G., J.D.-M., L.Q., E.G., I.I.) and Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED) (C.P., A.L.d.M., A.L.P.-N.), Instituto de Salud Carlos III, Madrid; Neurology Department (C.P., B.V.), Neuromuscular Disorders Unit, Instituto de Biomedicina de Sevilla, Hospital U. Virgen del Rocío, CSIC, Universidad de Sevilla; Unitat de Neuromuscular (C.C., M.A.A.), Neurology Department, Hospital Universitari de Bellvitge–IDIBELL, l'Hospitalet de Llobregat, Barcelona; Neuromuscular Diseases Unit (A.-G.S., L.G.), Department of Neurology, Institute of Neurosciences, Hospital Universitario Clínico San Carlos, Madrid; Neurology Department (J.P., T.G.-S.), Hospital Clínico, Santiago de Compostela; Neuromuscular Diseases Unit (A.R.-F., A.M.-P.), Department of Neurology, Hospital Germans Trias i Pujol; Department of Medicine (A.R.-F.), Universitat Autònoma de Barcelona, Badalona; Neuromuscular Unit (T.S., A.L.-M.), Neurology Department, Hospital Universitari i Politècnic La Fe; Department of Medicine (T.S.), Universitat de València; Neurology Department (A.L.d.M., A.F.-T.), Donostia University Hospital; Neurosciences Area (A.L.d.M.), Biodonostia Research Institute, University of the Basque Country, San Sebastián; Neurology Department (M.T.G.), Hospital Universitario Reina Sofía, Córdoba; Department of Neurology (I.J.), Complejo Hospitalario de Navarra, Pamplona; Neuromuscular Diseases Unit (G.G.-G.), Department of Neurology, Hospital Universitario Infanta Sofía, Universidad Europea de Madrid, San Sebastián de los Reyes, Madrid; Complejo asistencial hospitalario de Burgos (M.A.M.), Burgos; Hospital Universitario de Gran Canaria Doctor Negrín (M.D.M.), Las Palmas de Gran Canaria; Hospital Central de Asturias (G.M.), Oviedo; and Service of Neurology (A.L.P.-N., Á.C.-A.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, Santander, Spain.
1,
Adolfo López de Munain
Adolfo López de Munain, MD, PhD
1From the Neuromuscular Diseases Unit, Department of Neurology (E.C.-V., R.Á.-V., R.R.-G., J.D.-M., L.Q., E.G., I.I.), Hospital de la Santa Creu i Sant Pau; Department of Medicine (E.C.-V., R.Á.-V., I.I.), Universitat Autònoma de Barcelona; Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) (E.C.-V., R.Á.-V., S.S., C.C., T.S., R.R.-G., J.D.-M., L.Q., E.G., I.I.) and Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED) (C.P., A.L.d.M., A.L.P.-N.), Instituto de Salud Carlos III, Madrid; Neurology Department (C.P., B.V.), Neuromuscular Disorders Unit, Instituto de Biomedicina de Sevilla, Hospital U. Virgen del Rocío, CSIC, Universidad de Sevilla; Unitat de Neuromuscular (C.C., M.A.A.), Neurology Department, Hospital Universitari de Bellvitge–IDIBELL, l'Hospitalet de Llobregat, Barcelona; Neuromuscular Diseases Unit (A.-G.S., L.G.), Department of Neurology, Institute of Neurosciences, Hospital Universitario Clínico San Carlos, Madrid; Neurology Department (J.P., T.G.-S.), Hospital Clínico, Santiago de Compostela; Neuromuscular Diseases Unit (A.R.-F., A.M.-P.), Department of Neurology, Hospital Germans Trias i Pujol; Department of Medicine (A.R.-F.), Universitat Autònoma de Barcelona, Badalona; Neuromuscular Unit (T.S., A.L.-M.), Neurology Department, Hospital Universitari i Politècnic La Fe; Department of Medicine (T.S.), Universitat de València; Neurology Department (A.L.d.M., A.F.-T.), Donostia University Hospital; Neurosciences Area (A.L.d.M.), Biodonostia Research Institute, University of the Basque Country, San Sebastián; Neurology Department (M.T.G.), Hospital Universitario Reina Sofía, Córdoba; Department of Neurology (I.J.), Complejo Hospitalario de Navarra, Pamplona; Neuromuscular Diseases Unit (G.G.-G.), Department of Neurology, Hospital Universitario Infanta Sofía, Universidad Europea de Madrid, San Sebastián de los Reyes, Madrid; Complejo asistencial hospitalario de Burgos (M.A.M.), Burgos; Hospital Universitario de Gran Canaria Doctor Negrín (M.D.M.), Las Palmas de Gran Canaria; Hospital Central de Asturias (G.M.), Oviedo; and Service of Neurology (A.L.P.-N., Á.C.-A.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, Santander, Spain.
1,
Maria Teresa Gómez
Maria Teresa Gómez, MD
1From the Neuromuscular Diseases Unit, Department of Neurology (E.C.-V., R.Á.-V., R.R.-G., J.D.-M., L.Q., E.G., I.I.), Hospital de la Santa Creu i Sant Pau; Department of Medicine (E.C.-V., R.Á.-V., I.I.), Universitat Autònoma de Barcelona; Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) (E.C.-V., R.Á.-V., S.S., C.C., T.S., R.R.-G., J.D.-M., L.Q., E.G., I.I.) and Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED) (C.P., A.L.d.M., A.L.P.-N.), Instituto de Salud Carlos III, Madrid; Neurology Department (C.P., B.V.), Neuromuscular Disorders Unit, Instituto de Biomedicina de Sevilla, Hospital U. Virgen del Rocío, CSIC, Universidad de Sevilla; Unitat de Neuromuscular (C.C., M.A.A.), Neurology Department, Hospital Universitari de Bellvitge–IDIBELL, l'Hospitalet de Llobregat, Barcelona; Neuromuscular Diseases Unit (A.-G.S., L.G.), Department of Neurology, Institute of Neurosciences, Hospital Universitario Clínico San Carlos, Madrid; Neurology Department (J.P., T.G.-S.), Hospital Clínico, Santiago de Compostela; Neuromuscular Diseases Unit (A.R.-F., A.M.-P.), Department of Neurology, Hospital Germans Trias i Pujol; Department of Medicine (A.R.-F.), Universitat Autònoma de Barcelona, Badalona; Neuromuscular Unit (T.S., A.L.-M.), Neurology Department, Hospital Universitari i Politècnic La Fe; Department of Medicine (T.S.), Universitat de València; Neurology Department (A.L.d.M., A.F.-T.), Donostia University Hospital; Neurosciences Area (A.L.d.M.), Biodonostia Research Institute, University of the Basque Country, San Sebastián; Neurology Department (M.T.G.), Hospital Universitario Reina Sofía, Córdoba; Department of Neurology (I.J.), Complejo Hospitalario de Navarra, Pamplona; Neuromuscular Diseases Unit (G.G.-G.), Department of Neurology, Hospital Universitario Infanta Sofía, Universidad Europea de Madrid, San Sebastián de los Reyes, Madrid; Complejo asistencial hospitalario de Burgos (M.A.M.), Burgos; Hospital Universitario de Gran Canaria Doctor Negrín (M.D.M.), Las Palmas de Gran Canaria; Hospital Central de Asturias (G.M.), Oviedo; and Service of Neurology (A.L.P.-N., Á.C.-A.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, Santander, Spain.
1,
Ivonne Jericó
Ivonne Jericó, MD, PhD
1From the Neuromuscular Diseases Unit, Department of Neurology (E.C.-V., R.Á.-V., R.R.-G., J.D.-M., L.Q., E.G., I.I.), Hospital de la Santa Creu i Sant Pau; Department of Medicine (E.C.-V., R.Á.-V., I.I.), Universitat Autònoma de Barcelona; Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) (E.C.-V., R.Á.-V., S.S., C.C., T.S., R.R.-G., J.D.-M., L.Q., E.G., I.I.) and Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED) (C.P., A.L.d.M., A.L.P.-N.), Instituto de Salud Carlos III, Madrid; Neurology Department (C.P., B.V.), Neuromuscular Disorders Unit, Instituto de Biomedicina de Sevilla, Hospital U. Virgen del Rocío, CSIC, Universidad de Sevilla; Unitat de Neuromuscular (C.C., M.A.A.), Neurology Department, Hospital Universitari de Bellvitge–IDIBELL, l'Hospitalet de Llobregat, Barcelona; Neuromuscular Diseases Unit (A.-G.S., L.G.), Department of Neurology, Institute of Neurosciences, Hospital Universitario Clínico San Carlos, Madrid; Neurology Department (J.P., T.G.-S.), Hospital Clínico, Santiago de Compostela; Neuromuscular Diseases Unit (A.R.-F., A.M.-P.), Department of Neurology, Hospital Germans Trias i Pujol; Department of Medicine (A.R.-F.), Universitat Autònoma de Barcelona, Badalona; Neuromuscular Unit (T.S., A.L.-M.), Neurology Department, Hospital Universitari i Politècnic La Fe; Department of Medicine (T.S.), Universitat de València; Neurology Department (A.L.d.M., A.F.-T.), Donostia University Hospital; Neurosciences Area (A.L.d.M.), Biodonostia Research Institute, University of the Basque Country, San Sebastián; Neurology Department (M.T.G.), Hospital Universitario Reina Sofía, Córdoba; Department of Neurology (I.J.), Complejo Hospitalario de Navarra, Pamplona; Neuromuscular Diseases Unit (G.G.-G.), Department of Neurology, Hospital Universitario Infanta Sofía, Universidad Europea de Madrid, San Sebastián de los Reyes, Madrid; Complejo asistencial hospitalario de Burgos (M.A.M.), Burgos; Hospital Universitario de Gran Canaria Doctor Negrín (M.D.M.), Las Palmas de Gran Canaria; Hospital Central de Asturias (G.M.), Oviedo; and Service of Neurology (A.L.P.-N., Á.C.-A.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, Santander, Spain.
1,
Gerardo Gutiérrez-Gutiérrez
Gerardo Gutiérrez-Gutiérrez, MD
1From the Neuromuscular Diseases Unit, Department of Neurology (E.C.-V., R.Á.-V., R.R.-G., J.D.-M., L.Q., E.G., I.I.), Hospital de la Santa Creu i Sant Pau; Department of Medicine (E.C.-V., R.Á.-V., I.I.), Universitat Autònoma de Barcelona; Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) (E.C.-V., R.Á.-V., S.S., C.C., T.S., R.R.-G., J.D.-M., L.Q., E.G., I.I.) and Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED) (C.P., A.L.d.M., A.L.P.-N.), Instituto de Salud Carlos III, Madrid; Neurology Department (C.P., B.V.), Neuromuscular Disorders Unit, Instituto de Biomedicina de Sevilla, Hospital U. Virgen del Rocío, CSIC, Universidad de Sevilla; Unitat de Neuromuscular (C.C., M.A.A.), Neurology Department, Hospital Universitari de Bellvitge–IDIBELL, l'Hospitalet de Llobregat, Barcelona; Neuromuscular Diseases Unit (A.-G.S., L.G.), Department of Neurology, Institute of Neurosciences, Hospital Universitario Clínico San Carlos, Madrid; Neurology Department (J.P., T.G.-S.), Hospital Clínico, Santiago de Compostela; Neuromuscular Diseases Unit (A.R.-F., A.M.-P.), Department of Neurology, Hospital Germans Trias i Pujol; Department of Medicine (A.R.-F.), Universitat Autònoma de Barcelona, Badalona; Neuromuscular Unit (T.S., A.L.-M.), Neurology Department, Hospital Universitari i Politècnic La Fe; Department of Medicine (T.S.), Universitat de València; Neurology Department (A.L.d.M., A.F.-T.), Donostia University Hospital; Neurosciences Area (A.L.d.M.), Biodonostia Research Institute, University of the Basque Country, San Sebastián; Neurology Department (M.T.G.), Hospital Universitario Reina Sofía, Córdoba; Department of Neurology (I.J.), Complejo Hospitalario de Navarra, Pamplona; Neuromuscular Diseases Unit (G.G.-G.), Department of Neurology, Hospital Universitario Infanta Sofía, Universidad Europea de Madrid, San Sebastián de los Reyes, Madrid; Complejo asistencial hospitalario de Burgos (M.A.M.), Burgos; Hospital Universitario de Gran Canaria Doctor Negrín (M.D.M.), Las Palmas de Gran Canaria; Hospital Central de Asturias (G.M.), Oviedo; and Service of Neurology (A.L.P.-N., Á.C.-A.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, Santander, Spain.
1,
Ana Lara Pelayo-Negro
Ana Lara Pelayo-Negro, MD
1From the Neuromuscular Diseases Unit, Department of Neurology (E.C.-V., R.Á.-V., R.R.-G., J.D.-M., L.Q., E.G., I.I.), Hospital de la Santa Creu i Sant Pau; Department of Medicine (E.C.-V., R.Á.-V., I.I.), Universitat Autònoma de Barcelona; Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) (E.C.-V., R.Á.-V., S.S., C.C., T.S., R.R.-G., J.D.-M., L.Q., E.G., I.I.) and Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED) (C.P., A.L.d.M., A.L.P.-N.), Instituto de Salud Carlos III, Madrid; Neurology Department (C.P., B.V.), Neuromuscular Disorders Unit, Instituto de Biomedicina de Sevilla, Hospital U. Virgen del Rocío, CSIC, Universidad de Sevilla; Unitat de Neuromuscular (C.C., M.A.A.), Neurology Department, Hospital Universitari de Bellvitge–IDIBELL, l'Hospitalet de Llobregat, Barcelona; Neuromuscular Diseases Unit (A.-G.S., L.G.), Department of Neurology, Institute of Neurosciences, Hospital Universitario Clínico San Carlos, Madrid; Neurology Department (J.P., T.G.-S.), Hospital Clínico, Santiago de Compostela; Neuromuscular Diseases Unit (A.R.-F., A.M.-P.), Department of Neurology, Hospital Germans Trias i Pujol; Department of Medicine (A.R.-F.), Universitat Autònoma de Barcelona, Badalona; Neuromuscular Unit (T.S., A.L.-M.), Neurology Department, Hospital Universitari i Politècnic La Fe; Department of Medicine (T.S.), Universitat de València; Neurology Department (A.L.d.M., A.F.-T.), Donostia University Hospital; Neurosciences Area (A.L.d.M.), Biodonostia Research Institute, University of the Basque Country, San Sebastián; Neurology Department (M.T.G.), Hospital Universitario Reina Sofía, Córdoba; Department of Neurology (I.J.), Complejo Hospitalario de Navarra, Pamplona; Neuromuscular Diseases Unit (G.G.-G.), Department of Neurology, Hospital Universitario Infanta Sofía, Universidad Europea de Madrid, San Sebastián de los Reyes, Madrid; Complejo asistencial hospitalario de Burgos (M.A.M.), Burgos; Hospital Universitario de Gran Canaria Doctor Negrín (M.D.M.), Las Palmas de Gran Canaria; Hospital Central de Asturias (G.M.), Oviedo; and Service of Neurology (A.L.P.-N., Á.C.-A.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, Santander, Spain.
1,
María Asunción Martín
María Asunción Martín, MD
1From the Neuromuscular Diseases Unit, Department of Neurology (E.C.-V., R.Á.-V., R.R.-G., J.D.-M., L.Q., E.G., I.I.), Hospital de la Santa Creu i Sant Pau; Department of Medicine (E.C.-V., R.Á.-V., I.I.), Universitat Autònoma de Barcelona; Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) (E.C.-V., R.Á.-V., S.S., C.C., T.S., R.R.-G., J.D.-M., L.Q., E.G., I.I.) and Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED) (C.P., A.L.d.M., A.L.P.-N.), Instituto de Salud Carlos III, Madrid; Neurology Department (C.P., B.V.), Neuromuscular Disorders Unit, Instituto de Biomedicina de Sevilla, Hospital U. Virgen del Rocío, CSIC, Universidad de Sevilla; Unitat de Neuromuscular (C.C., M.A.A.), Neurology Department, Hospital Universitari de Bellvitge–IDIBELL, l'Hospitalet de Llobregat, Barcelona; Neuromuscular Diseases Unit (A.-G.S., L.G.), Department of Neurology, Institute of Neurosciences, Hospital Universitario Clínico San Carlos, Madrid; Neurology Department (J.P., T.G.-S.), Hospital Clínico, Santiago de Compostela; Neuromuscular Diseases Unit (A.R.-F., A.M.-P.), Department of Neurology, Hospital Germans Trias i Pujol; Department of Medicine (A.R.-F.), Universitat Autònoma de Barcelona, Badalona; Neuromuscular Unit (T.S., A.L.-M.), Neurology Department, Hospital Universitari i Politècnic La Fe; Department of Medicine (T.S.), Universitat de València; Neurology Department (A.L.d.M., A.F.-T.), Donostia University Hospital; Neurosciences Area (A.L.d.M.), Biodonostia Research Institute, University of the Basque Country, San Sebastián; Neurology Department (M.T.G.), Hospital Universitario Reina Sofía, Córdoba; Department of Neurology (I.J.), Complejo Hospitalario de Navarra, Pamplona; Neuromuscular Diseases Unit (G.G.-G.), Department of Neurology, Hospital Universitario Infanta Sofía, Universidad Europea de Madrid, San Sebastián de los Reyes, Madrid; Complejo asistencial hospitalario de Burgos (M.A.M.), Burgos; Hospital Universitario de Gran Canaria Doctor Negrín (M.D.M.), Las Palmas de Gran Canaria; Hospital Central de Asturias (G.M.), Oviedo; and Service of Neurology (A.L.P.-N., Á.C.-A.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, Santander, Spain.
1,
María Dolores Mendoza
María Dolores Mendoza, MD
1From the Neuromuscular Diseases Unit, Department of Neurology (E.C.-V., R.Á.-V., R.R.-G., J.D.-M., L.Q., E.G., I.I.), Hospital de la Santa Creu i Sant Pau; Department of Medicine (E.C.-V., R.Á.-V., I.I.), Universitat Autònoma de Barcelona; Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) (E.C.-V., R.Á.-V., S.S., C.C., T.S., R.R.-G., J.D.-M., L.Q., E.G., I.I.) and Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED) (C.P., A.L.d.M., A.L.P.-N.), Instituto de Salud Carlos III, Madrid; Neurology Department (C.P., B.V.), Neuromuscular Disorders Unit, Instituto de Biomedicina de Sevilla, Hospital U. Virgen del Rocío, CSIC, Universidad de Sevilla; Unitat de Neuromuscular (C.C., M.A.A.), Neurology Department, Hospital Universitari de Bellvitge–IDIBELL, l'Hospitalet de Llobregat, Barcelona; Neuromuscular Diseases Unit (A.-G.S., L.G.), Department of Neurology, Institute of Neurosciences, Hospital Universitario Clínico San Carlos, Madrid; Neurology Department (J.P., T.G.-S.), Hospital Clínico, Santiago de Compostela; Neuromuscular Diseases Unit (A.R.-F., A.M.-P.), Department of Neurology, Hospital Germans Trias i Pujol; Department of Medicine (A.R.-F.), Universitat Autònoma de Barcelona, Badalona; Neuromuscular Unit (T.S., A.L.-M.), Neurology Department, Hospital Universitari i Politècnic La Fe; Department of Medicine (T.S.), Universitat de València; Neurology Department (A.L.d.M., A.F.-T.), Donostia University Hospital; Neurosciences Area (A.L.d.M.), Biodonostia Research Institute, University of the Basque Country, San Sebastián; Neurology Department (M.T.G.), Hospital Universitario Reina Sofía, Córdoba; Department of Neurology (I.J.), Complejo Hospitalario de Navarra, Pamplona; Neuromuscular Diseases Unit (G.G.-G.), Department of Neurology, Hospital Universitario Infanta Sofía, Universidad Europea de Madrid, San Sebastián de los Reyes, Madrid; Complejo asistencial hospitalario de Burgos (M.A.M.), Burgos; Hospital Universitario de Gran Canaria Doctor Negrín (M.D.M.), Las Palmas de Gran Canaria; Hospital Central de Asturias (G.M.), Oviedo; and Service of Neurology (A.L.P.-N., Á.C.-A.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, Santander, Spain.
1,
Germán Morís
Germán Morís, MD
1From the Neuromuscular Diseases Unit, Department of Neurology (E.C.-V., R.Á.-V., R.R.-G., J.D.-M., L.Q., E.G., I.I.), Hospital de la Santa Creu i Sant Pau; Department of Medicine (E.C.-V., R.Á.-V., I.I.), Universitat Autònoma de Barcelona; Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) (E.C.-V., R.Á.-V., S.S., C.C., T.S., R.R.-G., J.D.-M., L.Q., E.G., I.I.) and Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED) (C.P., A.L.d.M., A.L.P.-N.), Instituto de Salud Carlos III, Madrid; Neurology Department (C.P., B.V.), Neuromuscular Disorders Unit, Instituto de Biomedicina de Sevilla, Hospital U. Virgen del Rocío, CSIC, Universidad de Sevilla; Unitat de Neuromuscular (C.C., M.A.A.), Neurology Department, Hospital Universitari de Bellvitge–IDIBELL, l'Hospitalet de Llobregat, Barcelona; Neuromuscular Diseases Unit (A.-G.S., L.G.), Department of Neurology, Institute of Neurosciences, Hospital Universitario Clínico San Carlos, Madrid; Neurology Department (J.P., T.G.-S.), Hospital Clínico, Santiago de Compostela; Neuromuscular Diseases Unit (A.R.-F., A.M.-P.), Department of Neurology, Hospital Germans Trias i Pujol; Department of Medicine (A.R.-F.), Universitat Autònoma de Barcelona, Badalona; Neuromuscular Unit (T.S., A.L.-M.), Neurology Department, Hospital Universitari i Politècnic La Fe; Department of Medicine (T.S.), Universitat de València; Neurology Department (A.L.d.M., A.F.-T.), Donostia University Hospital; Neurosciences Area (A.L.d.M.), Biodonostia Research Institute, University of the Basque Country, San Sebastián; Neurology Department (M.T.G.), Hospital Universitario Reina Sofía, Córdoba; Department of Neurology (I.J.), Complejo Hospitalario de Navarra, Pamplona; Neuromuscular Diseases Unit (G.G.-G.), Department of Neurology, Hospital Universitario Infanta Sofía, Universidad Europea de Madrid, San Sebastián de los Reyes, Madrid; Complejo asistencial hospitalario de Burgos (M.A.M.), Burgos; Hospital Universitario de Gran Canaria Doctor Negrín (M.D.M.), Las Palmas de Gran Canaria; Hospital Central de Asturias (G.M.), Oviedo; and Service of Neurology (A.L.P.-N., Á.C.-A.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, Santander, Spain.
1,
Ricard Rojas-Garcia
Ricard Rojas-Garcia, MD, PhD
1From the Neuromuscular Diseases Unit, Department of Neurology (E.C.-V., R.Á.-V., R.R.-G., J.D.-M., L.Q., E.G., I.I.), Hospital de la Santa Creu i Sant Pau; Department of Medicine (E.C.-V., R.Á.-V., I.I.), Universitat Autònoma de Barcelona; Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) (E.C.-V., R.Á.-V., S.S., C.C., T.S., R.R.-G., J.D.-M., L.Q., E.G., I.I.) and Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED) (C.P., A.L.d.M., A.L.P.-N.), Instituto de Salud Carlos III, Madrid; Neurology Department (C.P., B.V.), Neuromuscular Disorders Unit, Instituto de Biomedicina de Sevilla, Hospital U. Virgen del Rocío, CSIC, Universidad de Sevilla; Unitat de Neuromuscular (C.C., M.A.A.), Neurology Department, Hospital Universitari de Bellvitge–IDIBELL, l'Hospitalet de Llobregat, Barcelona; Neuromuscular Diseases Unit (A.-G.S., L.G.), Department of Neurology, Institute of Neurosciences, Hospital Universitario Clínico San Carlos, Madrid; Neurology Department (J.P., T.G.-S.), Hospital Clínico, Santiago de Compostela; Neuromuscular Diseases Unit (A.R.-F., A.M.-P.), Department of Neurology, Hospital Germans Trias i Pujol; Department of Medicine (A.R.-F.), Universitat Autònoma de Barcelona, Badalona; Neuromuscular Unit (T.S., A.L.-M.), Neurology Department, Hospital Universitari i Politècnic La Fe; Department of Medicine (T.S.), Universitat de València; Neurology Department (A.L.d.M., A.F.-T.), Donostia University Hospital; Neurosciences Area (A.L.d.M.), Biodonostia Research Institute, University of the Basque Country, San Sebastián; Neurology Department (M.T.G.), Hospital Universitario Reina Sofía, Córdoba; Department of Neurology (I.J.), Complejo Hospitalario de Navarra, Pamplona; Neuromuscular Diseases Unit (G.G.-G.), Department of Neurology, Hospital Universitario Infanta Sofía, Universidad Europea de Madrid, San Sebastián de los Reyes, Madrid; Complejo asistencial hospitalario de Burgos (M.A.M.), Burgos; Hospital Universitario de Gran Canaria Doctor Negrín (M.D.M.), Las Palmas de Gran Canaria; Hospital Central de Asturias (G.M.), Oviedo; and Service of Neurology (A.L.P.-N., Á.C.-A.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, Santander, Spain.
1,
Jordi Díaz-Manera
Jordi Díaz-Manera, MD, PhD
1From the Neuromuscular Diseases Unit, Department of Neurology (E.C.-V., R.Á.-V., R.R.-G., J.D.-M., L.Q., E.G., I.I.), Hospital de la Santa Creu i Sant Pau; Department of Medicine (E.C.-V., R.Á.-V., I.I.), Universitat Autònoma de Barcelona; Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) (E.C.-V., R.Á.-V., S.S., C.C., T.S., R.R.-G., J.D.-M., L.Q., E.G., I.I.) and Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED) (C.P., A.L.d.M., A.L.P.-N.), Instituto de Salud Carlos III, Madrid; Neurology Department (C.P., B.V.), Neuromuscular Disorders Unit, Instituto de Biomedicina de Sevilla, Hospital U. Virgen del Rocío, CSIC, Universidad de Sevilla; Unitat de Neuromuscular (C.C., M.A.A.), Neurology Department, Hospital Universitari de Bellvitge–IDIBELL, l'Hospitalet de Llobregat, Barcelona; Neuromuscular Diseases Unit (A.-G.S., L.G.), Department of Neurology, Institute of Neurosciences, Hospital Universitario Clínico San Carlos, Madrid; Neurology Department (J.P., T.G.-S.), Hospital Clínico, Santiago de Compostela; Neuromuscular Diseases Unit (A.R.-F., A.M.-P.), Department of Neurology, Hospital Germans Trias i Pujol; Department of Medicine (A.R.-F.), Universitat Autònoma de Barcelona, Badalona; Neuromuscular Unit (T.S., A.L.-M.), Neurology Department, Hospital Universitari i Politècnic La Fe; Department of Medicine (T.S.), Universitat de València; Neurology Department (A.L.d.M., A.F.-T.), Donostia University Hospital; Neurosciences Area (A.L.d.M.), Biodonostia Research Institute, University of the Basque Country, San Sebastián; Neurology Department (M.T.G.), Hospital Universitario Reina Sofía, Córdoba; Department of Neurology (I.J.), Complejo Hospitalario de Navarra, Pamplona; Neuromuscular Diseases Unit (G.G.-G.), Department of Neurology, Hospital Universitario Infanta Sofía, Universidad Europea de Madrid, San Sebastián de los Reyes, Madrid; Complejo asistencial hospitalario de Burgos (M.A.M.), Burgos; Hospital Universitario de Gran Canaria Doctor Negrín (M.D.M.), Las Palmas de Gran Canaria; Hospital Central de Asturias (G.M.), Oviedo; and Service of Neurology (A.L.P.-N., Á.C.-A.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, Santander, Spain.
1,
Luis Querol
Luis Querol, MD, PhD
1From the Neuromuscular Diseases Unit, Department of Neurology (E.C.-V., R.Á.-V., R.R.-G., J.D.-M., L.Q., E.G., I.I.), Hospital de la Santa Creu i Sant Pau; Department of Medicine (E.C.-V., R.Á.-V., I.I.), Universitat Autònoma de Barcelona; Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) (E.C.-V., R.Á.-V., S.S., C.C., T.S., R.R.-G., J.D.-M., L.Q., E.G., I.I.) and Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED) (C.P., A.L.d.M., A.L.P.-N.), Instituto de Salud Carlos III, Madrid; Neurology Department (C.P., B.V.), Neuromuscular Disorders Unit, Instituto de Biomedicina de Sevilla, Hospital U. Virgen del Rocío, CSIC, Universidad de Sevilla; Unitat de Neuromuscular (C.C., M.A.A.), Neurology Department, Hospital Universitari de Bellvitge–IDIBELL, l'Hospitalet de Llobregat, Barcelona; Neuromuscular Diseases Unit (A.-G.S., L.G.), Department of Neurology, Institute of Neurosciences, Hospital Universitario Clínico San Carlos, Madrid; Neurology Department (J.P., T.G.-S.), Hospital Clínico, Santiago de Compostela; Neuromuscular Diseases Unit (A.R.-F., A.M.-P.), Department of Neurology, Hospital Germans Trias i Pujol; Department of Medicine (A.R.-F.), Universitat Autònoma de Barcelona, Badalona; Neuromuscular Unit (T.S., A.L.-M.), Neurology Department, Hospital Universitari i Politècnic La Fe; Department of Medicine (T.S.), Universitat de València; Neurology Department (A.L.d.M., A.F.-T.), Donostia University Hospital; Neurosciences Area (A.L.d.M.), Biodonostia Research Institute, University of the Basque Country, San Sebastián; Neurology Department (M.T.G.), Hospital Universitario Reina Sofía, Córdoba; Department of Neurology (I.J.), Complejo Hospitalario de Navarra, Pamplona; Neuromuscular Diseases Unit (G.G.-G.), Department of Neurology, Hospital Universitario Infanta Sofía, Universidad Europea de Madrid, San Sebastián de los Reyes, Madrid; Complejo asistencial hospitalario de Burgos (M.A.M.), Burgos; Hospital Universitario de Gran Canaria Doctor Negrín (M.D.M.), Las Palmas de Gran Canaria; Hospital Central de Asturias (G.M.), Oviedo; and Service of Neurology (A.L.P.-N., Á.C.-A.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, Santander, Spain.
1,
Eduard Gallardo
Eduard Gallardo, PhD
1From the Neuromuscular Diseases Unit, Department of Neurology (E.C.-V., R.Á.-V., R.R.-G., J.D.-M., L.Q., E.G., I.I.), Hospital de la Santa Creu i Sant Pau; Department of Medicine (E.C.-V., R.Á.-V., I.I.), Universitat Autònoma de Barcelona; Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) (E.C.-V., R.Á.-V., S.S., C.C., T.S., R.R.-G., J.D.-M., L.Q., E.G., I.I.) and Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED) (C.P., A.L.d.M., A.L.P.-N.), Instituto de Salud Carlos III, Madrid; Neurology Department (C.P., B.V.), Neuromuscular Disorders Unit, Instituto de Biomedicina de Sevilla, Hospital U. Virgen del Rocío, CSIC, Universidad de Sevilla; Unitat de Neuromuscular (C.C., M.A.A.), Neurology Department, Hospital Universitari de Bellvitge–IDIBELL, l'Hospitalet de Llobregat, Barcelona; Neuromuscular Diseases Unit (A.-G.S., L.G.), Department of Neurology, Institute of Neurosciences, Hospital Universitario Clínico San Carlos, Madrid; Neurology Department (J.P., T.G.-S.), Hospital Clínico, Santiago de Compostela; Neuromuscular Diseases Unit (A.R.-F., A.M.-P.), Department of Neurology, Hospital Germans Trias i Pujol; Department of Medicine (A.R.-F.), Universitat Autònoma de Barcelona, Badalona; Neuromuscular Unit (T.S., A.L.-M.), Neurology Department, Hospital Universitari i Politècnic La Fe; Department of Medicine (T.S.), Universitat de València; Neurology Department (A.L.d.M., A.F.-T.), Donostia University Hospital; Neurosciences Area (A.L.d.M.), Biodonostia Research Institute, University of the Basque Country, San Sebastián; Neurology Department (M.T.G.), Hospital Universitario Reina Sofía, Córdoba; Department of Neurology (I.J.), Complejo Hospitalario de Navarra, Pamplona; Neuromuscular Diseases Unit (G.G.-G.), Department of Neurology, Hospital Universitario Infanta Sofía, Universidad Europea de Madrid, San Sebastián de los Reyes, Madrid; Complejo asistencial hospitalario de Burgos (M.A.M.), Burgos; Hospital Universitario de Gran Canaria Doctor Negrín (M.D.M.), Las Palmas de Gran Canaria; Hospital Central de Asturias (G.M.), Oviedo; and Service of Neurology (A.L.P.-N., Á.C.-A.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, Santander, Spain.
1,
Beatriz Vélez
Beatriz Vélez, MD
1From the Neuromuscular Diseases Unit, Department of Neurology (E.C.-V., R.Á.-V., R.R.-G., J.D.-M., L.Q., E.G., I.I.), Hospital de la Santa Creu i Sant Pau; Department of Medicine (E.C.-V., R.Á.-V., I.I.), Universitat Autònoma de Barcelona; Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) (E.C.-V., R.Á.-V., S.S., C.C., T.S., R.R.-G., J.D.-M., L.Q., E.G., I.I.) and Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED) (C.P., A.L.d.M., A.L.P.-N.), Instituto de Salud Carlos III, Madrid; Neurology Department (C.P., B.V.), Neuromuscular Disorders Unit, Instituto de Biomedicina de Sevilla, Hospital U. Virgen del Rocío, CSIC, Universidad de Sevilla; Unitat de Neuromuscular (C.C., M.A.A.), Neurology Department, Hospital Universitari de Bellvitge–IDIBELL, l'Hospitalet de Llobregat, Barcelona; Neuromuscular Diseases Unit (A.-G.S., L.G.), Department of Neurology, Institute of Neurosciences, Hospital Universitario Clínico San Carlos, Madrid; Neurology Department (J.P., T.G.-S.), Hospital Clínico, Santiago de Compostela; Neuromuscular Diseases Unit (A.R.-F., A.M.-P.), Department of Neurology, Hospital Germans Trias i Pujol; Department of Medicine (A.R.-F.), Universitat Autònoma de Barcelona, Badalona; Neuromuscular Unit (T.S., A.L.-M.), Neurology Department, Hospital Universitari i Politècnic La Fe; Department of Medicine (T.S.), Universitat de València; Neurology Department (A.L.d.M., A.F.-T.), Donostia University Hospital; Neurosciences Area (A.L.d.M.), Biodonostia Research Institute, University of the Basque Country, San Sebastián; Neurology Department (M.T.G.), Hospital Universitario Reina Sofía, Córdoba; Department of Neurology (I.J.), Complejo Hospitalario de Navarra, Pamplona; Neuromuscular Diseases Unit (G.G.-G.), Department of Neurology, Hospital Universitario Infanta Sofía, Universidad Europea de Madrid, San Sebastián de los Reyes, Madrid; Complejo asistencial hospitalario de Burgos (M.A.M.), Burgos; Hospital Universitario de Gran Canaria Doctor Negrín (M.D.M.), Las Palmas de Gran Canaria; Hospital Central de Asturias (G.M.), Oviedo; and Service of Neurology (A.L.P.-N., Á.C.-A.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, Santander, Spain.
1,
María Antonia Albertí
María Antonia Albertí, MD
1From the Neuromuscular Diseases Unit, Department of Neurology (E.C.-V., R.Á.-V., R.R.-G., J.D.-M., L.Q., E.G., I.I.), Hospital de la Santa Creu i Sant Pau; Department of Medicine (E.C.-V., R.Á.-V., I.I.), Universitat Autònoma de Barcelona; Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) (E.C.-V., R.Á.-V., S.S., C.C., T.S., R.R.-G., J.D.-M., L.Q., E.G., I.I.) and Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED) (C.P., A.L.d.M., A.L.P.-N.), Instituto de Salud Carlos III, Madrid; Neurology Department (C.P., B.V.), Neuromuscular Disorders Unit, Instituto de Biomedicina de Sevilla, Hospital U. Virgen del Rocío, CSIC, Universidad de Sevilla; Unitat de Neuromuscular (C.C., M.A.A.), Neurology Department, Hospital Universitari de Bellvitge–IDIBELL, l'Hospitalet de Llobregat, Barcelona; Neuromuscular Diseases Unit (A.-G.S., L.G.), Department of Neurology, Institute of Neurosciences, Hospital Universitario Clínico San Carlos, Madrid; Neurology Department (J.P., T.G.-S.), Hospital Clínico, Santiago de Compostela; Neuromuscular Diseases Unit (A.R.-F., A.M.-P.), Department of Neurology, Hospital Germans Trias i Pujol; Department of Medicine (A.R.-F.), Universitat Autònoma de Barcelona, Badalona; Neuromuscular Unit (T.S., A.L.-M.), Neurology Department, Hospital Universitari i Politècnic La Fe; Department of Medicine (T.S.), Universitat de València; Neurology Department (A.L.d.M., A.F.-T.), Donostia University Hospital; Neurosciences Area (A.L.d.M.), Biodonostia Research Institute, University of the Basque Country, San Sebastián; Neurology Department (M.T.G.), Hospital Universitario Reina Sofía, Córdoba; Department of Neurology (I.J.), Complejo Hospitalario de Navarra, Pamplona; Neuromuscular Diseases Unit (G.G.-G.), Department of Neurology, Hospital Universitario Infanta Sofía, Universidad Europea de Madrid, San Sebastián de los Reyes, Madrid; Complejo asistencial hospitalario de Burgos (M.A.M.), Burgos; Hospital Universitario de Gran Canaria Doctor Negrín (M.D.M.), Las Palmas de Gran Canaria; Hospital Central de Asturias (G.M.), Oviedo; and Service of Neurology (A.L.P.-N., Á.C.-A.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, Santander, Spain.
1,
Lucía Galán
Lucía Galán, MD, PhD
1From the Neuromuscular Diseases Unit, Department of Neurology (E.C.-V., R.Á.-V., R.R.-G., J.D.-M., L.Q., E.G., I.I.), Hospital de la Santa Creu i Sant Pau; Department of Medicine (E.C.-V., R.Á.-V., I.I.), Universitat Autònoma de Barcelona; Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) (E.C.-V., R.Á.-V., S.S., C.C., T.S., R.R.-G., J.D.-M., L.Q., E.G., I.I.) and Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED) (C.P., A.L.d.M., A.L.P.-N.), Instituto de Salud Carlos III, Madrid; Neurology Department (C.P., B.V.), Neuromuscular Disorders Unit, Instituto de Biomedicina de Sevilla, Hospital U. Virgen del Rocío, CSIC, Universidad de Sevilla; Unitat de Neuromuscular (C.C., M.A.A.), Neurology Department, Hospital Universitari de Bellvitge–IDIBELL, l'Hospitalet de Llobregat, Barcelona; Neuromuscular Diseases Unit (A.-G.S., L.G.), Department of Neurology, Institute of Neurosciences, Hospital Universitario Clínico San Carlos, Madrid; Neurology Department (J.P., T.G.-S.), Hospital Clínico, Santiago de Compostela; Neuromuscular Diseases Unit (A.R.-F., A.M.-P.), Department of Neurology, Hospital Germans Trias i Pujol; Department of Medicine (A.R.-F.), Universitat Autònoma de Barcelona, Badalona; Neuromuscular Unit (T.S., A.L.-M.), Neurology Department, Hospital Universitari i Politècnic La Fe; Department of Medicine (T.S.), Universitat de València; Neurology Department (A.L.d.M., A.F.-T.), Donostia University Hospital; Neurosciences Area (A.L.d.M.), Biodonostia Research Institute, University of the Basque Country, San Sebastián; Neurology Department (M.T.G.), Hospital Universitario Reina Sofía, Córdoba; Department of Neurology (I.J.), Complejo Hospitalario de Navarra, Pamplona; Neuromuscular Diseases Unit (G.G.-G.), Department of Neurology, Hospital Universitario Infanta Sofía, Universidad Europea de Madrid, San Sebastián de los Reyes, Madrid; Complejo asistencial hospitalario de Burgos (M.A.M.), Burgos; Hospital Universitario de Gran Canaria Doctor Negrín (M.D.M.), Las Palmas de Gran Canaria; Hospital Central de Asturias (G.M.), Oviedo; and Service of Neurology (A.L.P.-N., Á.C.-A.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, Santander, Spain.
1,
Tania García-Sobrino
Tania García-Sobrino, MD
1From the Neuromuscular Diseases Unit, Department of Neurology (E.C.-V., R.Á.-V., R.R.-G., J.D.-M., L.Q., E.G., I.I.), Hospital de la Santa Creu i Sant Pau; Department of Medicine (E.C.-V., R.Á.-V., I.I.), Universitat Autònoma de Barcelona; Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) (E.C.-V., R.Á.-V., S.S., C.C., T.S., R.R.-G., J.D.-M., L.Q., E.G., I.I.) and Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED) (C.P., A.L.d.M., A.L.P.-N.), Instituto de Salud Carlos III, Madrid; Neurology Department (C.P., B.V.), Neuromuscular Disorders Unit, Instituto de Biomedicina de Sevilla, Hospital U. Virgen del Rocío, CSIC, Universidad de Sevilla; Unitat de Neuromuscular (C.C., M.A.A.), Neurology Department, Hospital Universitari de Bellvitge–IDIBELL, l'Hospitalet de Llobregat, Barcelona; Neuromuscular Diseases Unit (A.-G.S., L.G.), Department of Neurology, Institute of Neurosciences, Hospital Universitario Clínico San Carlos, Madrid; Neurology Department (J.P., T.G.-S.), Hospital Clínico, Santiago de Compostela; Neuromuscular Diseases Unit (A.R.-F., A.M.-P.), Department of Neurology, Hospital Germans Trias i Pujol; Department of Medicine (A.R.-F.), Universitat Autònoma de Barcelona, Badalona; Neuromuscular Unit (T.S., A.L.-M.), Neurology Department, Hospital Universitari i Politècnic La Fe; Department of Medicine (T.S.), Universitat de València; Neurology Department (A.L.d.M., A.F.-T.), Donostia University Hospital; Neurosciences Area (A.L.d.M.), Biodonostia Research Institute, University of the Basque Country, San Sebastián; Neurology Department (M.T.G.), Hospital Universitario Reina Sofía, Córdoba; Department of Neurology (I.J.), Complejo Hospitalario de Navarra, Pamplona; Neuromuscular Diseases Unit (G.G.-G.), Department of Neurology, Hospital Universitario Infanta Sofía, Universidad Europea de Madrid, San Sebastián de los Reyes, Madrid; Complejo asistencial hospitalario de Burgos (M.A.M.), Burgos; Hospital Universitario de Gran Canaria Doctor Negrín (M.D.M.), Las Palmas de Gran Canaria; Hospital Central de Asturias (G.M.), Oviedo; and Service of Neurology (A.L.P.-N., Á.C.-A.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, Santander, Spain.
1,
Alicia Martínez-Piñeiro
Alicia Martínez-Piñeiro, MD
1From the Neuromuscular Diseases Unit, Department of Neurology (E.C.-V., R.Á.-V., R.R.-G., J.D.-M., L.Q., E.G., I.I.), Hospital de la Santa Creu i Sant Pau; Department of Medicine (E.C.-V., R.Á.-V., I.I.), Universitat Autònoma de Barcelona; Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) (E.C.-V., R.Á.-V., S.S., C.C., T.S., R.R.-G., J.D.-M., L.Q., E.G., I.I.) and Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED) (C.P., A.L.d.M., A.L.P.-N.), Instituto de Salud Carlos III, Madrid; Neurology Department (C.P., B.V.), Neuromuscular Disorders Unit, Instituto de Biomedicina de Sevilla, Hospital U. Virgen del Rocío, CSIC, Universidad de Sevilla; Unitat de Neuromuscular (C.C., M.A.A.), Neurology Department, Hospital Universitari de Bellvitge–IDIBELL, l'Hospitalet de Llobregat, Barcelona; Neuromuscular Diseases Unit (A.-G.S., L.G.), Department of Neurology, Institute of Neurosciences, Hospital Universitario Clínico San Carlos, Madrid; Neurology Department (J.P., T.G.-S.), Hospital Clínico, Santiago de Compostela; Neuromuscular Diseases Unit (A.R.-F., A.M.-P.), Department of Neurology, Hospital Germans Trias i Pujol; Department of Medicine (A.R.-F.), Universitat Autònoma de Barcelona, Badalona; Neuromuscular Unit (T.S., A.L.-M.), Neurology Department, Hospital Universitari i Politècnic La Fe; Department of Medicine (T.S.), Universitat de València; Neurology Department (A.L.d.M., A.F.-T.), Donostia University Hospital; Neurosciences Area (A.L.d.M.), Biodonostia Research Institute, University of the Basque Country, San Sebastián; Neurology Department (M.T.G.), Hospital Universitario Reina Sofía, Córdoba; Department of Neurology (I.J.), Complejo Hospitalario de Navarra, Pamplona; Neuromuscular Diseases Unit (G.G.-G.), Department of Neurology, Hospital Universitario Infanta Sofía, Universidad Europea de Madrid, San Sebastián de los Reyes, Madrid; Complejo asistencial hospitalario de Burgos (M.A.M.), Burgos; Hospital Universitario de Gran Canaria Doctor Negrín (M.D.M.), Las Palmas de Gran Canaria; Hospital Central de Asturias (G.M.), Oviedo; and Service of Neurology (A.L.P.-N., Á.C.-A.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, Santander, Spain.
1,
Ana Lozano-Veintimilla
Ana Lozano-Veintimilla, MD
1From the Neuromuscular Diseases Unit, Department of Neurology (E.C.-V., R.Á.-V., R.R.-G., J.D.-M., L.Q., E.G., I.I.), Hospital de la Santa Creu i Sant Pau; Department of Medicine (E.C.-V., R.Á.-V., I.I.), Universitat Autònoma de Barcelona; Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) (E.C.-V., R.Á.-V., S.S., C.C., T.S., R.R.-G., J.D.-M., L.Q., E.G., I.I.) and Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED) (C.P., A.L.d.M., A.L.P.-N.), Instituto de Salud Carlos III, Madrid; Neurology Department (C.P., B.V.), Neuromuscular Disorders Unit, Instituto de Biomedicina de Sevilla, Hospital U. Virgen del Rocío, CSIC, Universidad de Sevilla; Unitat de Neuromuscular (C.C., M.A.A.), Neurology Department, Hospital Universitari de Bellvitge–IDIBELL, l'Hospitalet de Llobregat, Barcelona; Neuromuscular Diseases Unit (A.-G.S., L.G.), Department of Neurology, Institute of Neurosciences, Hospital Universitario Clínico San Carlos, Madrid; Neurology Department (J.P., T.G.-S.), Hospital Clínico, Santiago de Compostela; Neuromuscular Diseases Unit (A.R.-F., A.M.-P.), Department of Neurology, Hospital Germans Trias i Pujol; Department of Medicine (A.R.-F.), Universitat Autònoma de Barcelona, Badalona; Neuromuscular Unit (T.S., A.L.-M.), Neurology Department, Hospital Universitari i Politècnic La Fe; Department of Medicine (T.S.), Universitat de València; Neurology Department (A.L.d.M., A.F.-T.), Donostia University Hospital; Neurosciences Area (A.L.d.M.), Biodonostia Research Institute, University of the Basque Country, San Sebastián; Neurology Department (M.T.G.), Hospital Universitario Reina Sofía, Córdoba; Department of Neurology (I.J.), Complejo Hospitalario de Navarra, Pamplona; Neuromuscular Diseases Unit (G.G.-G.), Department of Neurology, Hospital Universitario Infanta Sofía, Universidad Europea de Madrid, San Sebastián de los Reyes, Madrid; Complejo asistencial hospitalario de Burgos (M.A.M.), Burgos; Hospital Universitario de Gran Canaria Doctor Negrín (M.D.M.), Las Palmas de Gran Canaria; Hospital Central de Asturias (G.M.), Oviedo; and Service of Neurology (A.L.P.-N., Á.C.-A.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, Santander, Spain.
1,
Roberto Fernández-Torrón
Roberto Fernández-Torrón, MD
1From the Neuromuscular Diseases Unit, Department of Neurology (E.C.-V., R.Á.-V., R.R.-G., J.D.-M., L.Q., E.G., I.I.), Hospital de la Santa Creu i Sant Pau; Department of Medicine (E.C.-V., R.Á.-V., I.I.), Universitat Autònoma de Barcelona; Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) (E.C.-V., R.Á.-V., S.S., C.C., T.S., R.R.-G., J.D.-M., L.Q., E.G., I.I.) and Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED) (C.P., A.L.d.M., A.L.P.-N.), Instituto de Salud Carlos III, Madrid; Neurology Department (C.P., B.V.), Neuromuscular Disorders Unit, Instituto de Biomedicina de Sevilla, Hospital U. Virgen del Rocío, CSIC, Universidad de Sevilla; Unitat de Neuromuscular (C.C., M.A.A.), Neurology Department, Hospital Universitari de Bellvitge–IDIBELL, l'Hospitalet de Llobregat, Barcelona; Neuromuscular Diseases Unit (A.-G.S., L.G.), Department of Neurology, Institute of Neurosciences, Hospital Universitario Clínico San Carlos, Madrid; Neurology Department (J.P., T.G.-S.), Hospital Clínico, Santiago de Compostela; Neuromuscular Diseases Unit (A.R.-F., A.M.-P.), Department of Neurology, Hospital Germans Trias i Pujol; Department of Medicine (A.R.-F.), Universitat Autònoma de Barcelona, Badalona; Neuromuscular Unit (T.S., A.L.-M.), Neurology Department, Hospital Universitari i Politècnic La Fe; Department of Medicine (T.S.), Universitat de València; Neurology Department (A.L.d.M., A.F.-T.), Donostia University Hospital; Neurosciences Area (A.L.d.M.), Biodonostia Research Institute, University of the Basque Country, San Sebastián; Neurology Department (M.T.G.), Hospital Universitario Reina Sofía, Córdoba; Department of Neurology (I.J.), Complejo Hospitalario de Navarra, Pamplona; Neuromuscular Diseases Unit (G.G.-G.), Department of Neurology, Hospital Universitario Infanta Sofía, Universidad Europea de Madrid, San Sebastián de los Reyes, Madrid; Complejo asistencial hospitalario de Burgos (M.A.M.), Burgos; Hospital Universitario de Gran Canaria Doctor Negrín (M.D.M.), Las Palmas de Gran Canaria; Hospital Central de Asturias (G.M.), Oviedo; and Service of Neurology (A.L.P.-N., Á.C.-A.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, Santander, Spain.
1,
Ángel Cano-Abascal
Ángel Cano-Abascal, MD
1From the Neuromuscular Diseases Unit, Department of Neurology (E.C.-V., R.Á.-V., R.R.-G., J.D.-M., L.Q., E.G., I.I.), Hospital de la Santa Creu i Sant Pau; Department of Medicine (E.C.-V., R.Á.-V., I.I.), Universitat Autònoma de Barcelona; Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) (E.C.-V., R.Á.-V., S.S., C.C., T.S., R.R.-G., J.D.-M., L.Q., E.G., I.I.) and Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED) (C.P., A.L.d.M., A.L.P.-N.), Instituto de Salud Carlos III, Madrid; Neurology Department (C.P., B.V.), Neuromuscular Disorders Unit, Instituto de Biomedicina de Sevilla, Hospital U. Virgen del Rocío, CSIC, Universidad de Sevilla; Unitat de Neuromuscular (C.C., M.A.A.), Neurology Department, Hospital Universitari de Bellvitge–IDIBELL, l'Hospitalet de Llobregat, Barcelona; Neuromuscular Diseases Unit (A.-G.S., L.G.), Department of Neurology, Institute of Neurosciences, Hospital Universitario Clínico San Carlos, Madrid; Neurology Department (J.P., T.G.-S.), Hospital Clínico, Santiago de Compostela; Neuromuscular Diseases Unit (A.R.-F., A.M.-P.), Department of Neurology, Hospital Germans Trias i Pujol; Department of Medicine (A.R.-F.), Universitat Autònoma de Barcelona, Badalona; Neuromuscular Unit (T.S., A.L.-M.), Neurology Department, Hospital Universitari i Politècnic La Fe; Department of Medicine (T.S.), Universitat de València; Neurology Department (A.L.d.M., A.F.-T.), Donostia University Hospital; Neurosciences Area (A.L.d.M.), Biodonostia Research Institute, University of the Basque Country, San Sebastián; Neurology Department (M.T.G.), Hospital Universitario Reina Sofía, Córdoba; Department of Neurology (I.J.), Complejo Hospitalario de Navarra, Pamplona; Neuromuscular Diseases Unit (G.G.-G.), Department of Neurology, Hospital Universitario Infanta Sofía, Universidad Europea de Madrid, San Sebastián de los Reyes, Madrid; Complejo asistencial hospitalario de Burgos (M.A.M.), Burgos; Hospital Universitario de Gran Canaria Doctor Negrín (M.D.M.), Las Palmas de Gran Canaria; Hospital Central de Asturias (G.M.), Oviedo; and Service of Neurology (A.L.P.-N., Á.C.-A.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, Santander, Spain.
1,
Isabel Illa
Isabel Illa, MD, PhD
1From the Neuromuscular Diseases Unit, Department of Neurology (E.C.-V., R.Á.-V., R.R.-G., J.D.-M., L.Q., E.G., I.I.), Hospital de la Santa Creu i Sant Pau; Department of Medicine (E.C.-V., R.Á.-V., I.I.), Universitat Autònoma de Barcelona; Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) (E.C.-V., R.Á.-V., S.S., C.C., T.S., R.R.-G., J.D.-M., L.Q., E.G., I.I.) and Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED) (C.P., A.L.d.M., A.L.P.-N.), Instituto de Salud Carlos III, Madrid; Neurology Department (C.P., B.V.), Neuromuscular Disorders Unit, Instituto de Biomedicina de Sevilla, Hospital U. Virgen del Rocío, CSIC, Universidad de Sevilla; Unitat de Neuromuscular (C.C., M.A.A.), Neurology Department, Hospital Universitari de Bellvitge–IDIBELL, l'Hospitalet de Llobregat, Barcelona; Neuromuscular Diseases Unit (A.-G.S., L.G.), Department of Neurology, Institute of Neurosciences, Hospital Universitario Clínico San Carlos, Madrid; Neurology Department (J.P., T.G.-S.), Hospital Clínico, Santiago de Compostela; Neuromuscular Diseases Unit (A.R.-F., A.M.-P.), Department of Neurology, Hospital Germans Trias i Pujol; Department of Medicine (A.R.-F.), Universitat Autònoma de Barcelona, Badalona; Neuromuscular Unit (T.S., A.L.-M.), Neurology Department, Hospital Universitari i Politècnic La Fe; Department of Medicine (T.S.), Universitat de València; Neurology Department (A.L.d.M., A.F.-T.), Donostia University Hospital; Neurosciences Area (A.L.d.M.), Biodonostia Research Institute, University of the Basque Country, San Sebastián; Neurology Department (M.T.G.), Hospital Universitario Reina Sofía, Córdoba; Department of Neurology (I.J.), Complejo Hospitalario de Navarra, Pamplona; Neuromuscular Diseases Unit (G.G.-G.), Department of Neurology, Hospital Universitario Infanta Sofía, Universidad Europea de Madrid, San Sebastián de los Reyes, Madrid; Complejo asistencial hospitalario de Burgos (M.A.M.), Burgos; Hospital Universitario de Gran Canaria Doctor Negrín (M.D.M.), Las Palmas de Gran Canaria; Hospital Central de Asturias (G.M.), Oviedo; and Service of Neurology (A.L.P.-N., Á.C.-A.), University Hospital “Marqués de Valdecilla (IDIVAL),” University of Cantabria, Santander, Spain.
1