

Abstract
A fundamental question in advancing Parkinson disease (PD) research is whether it represents one disorder or many. Does each genetic PD inform a common pathobiology or represent a unique entity? Do the similarities between genetic and idiopathic forms of PD outweigh the differences? If aggregates of α-synuclein in Lewy bodies and Lewy neurites are present in most (α-synucleinopathies), are they also etiopathogenically significant in each (α-synuclein pathogenesis)? Does it matter that postmortem studies in PD have demonstrated that mixed protein-aggregate pathology is the rule and pure α-synucleinopathy the exception? Should we continue to pursue convergent biomarkers that are representative of the diverse whole of PD or subtype-specific, divergent biomarkers, present in some but absent in most? Have clinical trials that failed to demonstrate efficacy of putative disease-modifying interventions been true failures (shortcomings of the hypotheses, which should be rejected) or false failures (shortcomings of the trials; hypotheses should be preserved)? Each of these questions reflects a nosologic struggle between the lumper's clinicopathologic model that embraces heterogeneity of one disease and the splitter's focus on a pathobiology-specific set of diseases. Most important, even if PD is not a single disorder, can advances in biomarkers and disease modification be revised to concentrate on pathologic commonalities in large, clinically defined populations? Or should our efforts be reconstructed to focus on smaller subgroups of patients, distinguished by well-defined molecular characteristics, regardless of their phenotypic classification? Will our clinical trial constructs be revised to target larger and earlier, possibly even prodromal, cohorts? Or should our trials efforts be reconstructed to target smaller but molecularly defined presymptomatic or postsymptomatic cohorts? At the Krembil Knowledge Gaps in Parkinson's Disease Symposium, the tentative answers to these questions were discussed, informed by the failures and successes of the fields of breast cancer and cystic fibrosis.
Phenotypic refinements aside, Parkinson disease (PD) remains largely defined as it was clinically >200 years ago and pathologically 100 years ago. Technical advances in the exploration and analysis of pathobiological processes have been used to validate, rather than to question, this clinicopathologic construct. As a result, the whole has become the sum of many and increasing parts. To order the increasing complexity into an etiopathologic whole, bioinformatic tools integrate molecular and genetic discoveries, connecting synaptic dysfunction, failed vesicular trafficking, mitochondrial dysfunction, impaired proteostasis, α-synuclein (α-syn) pathobiology, and neuroinflammation as pieces in a single puzzle.1 In the therapeutic realm, promising molecular interventions have been administered to patients in the absence of relevant measures of target engagement, that is, bioassays of relevant molecular pathways rescued by such interventions. Clinical trials aimed at slowing or halting progressive PD disability are undertaken with the hypothesis that a molecular modification will affect common neurodegeneration pathways in most patients with the clinical diagnosis of PD. Consequently, disease modification has remained elusive during the 4 decades in which we have been trying, with hypotheses and disease constructs largely unaltered.
The fundamental question is whether PD is one disorder with varying clinical manifestations influenced by a number of genetic and environmental factors that need to be revised, thereby allowing current approaches to understanding pathogenesis and advancing disease modification therapies, or reconstructed to frame PD as a syndrome in which pathologic commonalities are subordinate to unique etiologic or pathophysiologic entities, thereby forcing new approaches to advancing our understanding of the disease and developing successful disease modification. Understanding the answer to this question will have far-reaching implications in terms of trial design, disease modeling, biomarker development, therapeutic targeting, and so on. A key question is whether the negative clinical trials of putative disease-modifying interventions have been true failures (failure of the hypothesis; negative outcomes deserve to be trusted) or false failures (failure of the trial methodology; hypotheses deserve another chance).
The Krembil Knowledge Gaps in PD Symposium, held in Toronto from April 24 to 26, 2019, tackled this fundamental conceptual challenge: if PD is not a single disorder, can advances in biomarkers and disease modification be revised to concentrate on commonalities of pathogenic mechanisms in large populations (assuming biologic convergence), or do they need to be reconstructed for application to smaller subgroups of patients, distinguished by well-defined molecular characteristics, with some mechanistic understanding of etiopathogenesis and a relevant bioassay to measure it? This report summarizes the deliberations at that meeting and acknowledges the unresolved tension between the revisionist and the reconstructionist approach to PD and its treatment.
Key knowledge gaps in 2019: Revision or reconstruction?
At the core, the key question is whether PD is one disease with many facets, each contributing to explaining the whole, or many diseases sharing some elements with one another but with distinctive pathogenic elements. As a field, we have lived with this many-diseases/one-disease tension by reasoning that both can be correct (figure 1). The road ahead for biomarker development and clinical trial efforts toward disease modification depends on whether PD is a multinodal disease process connected by α-syn aggregation or a collection of independent pathologic processes that are largely, if not entirely, independent. The former is the more alluring model: molecular genetic forms of parkinsonism are considered pieces of the same puzzle, driving the creation of key research constructs (table 1).
Figure 1. Venn diagrams of the revision vs reconstruction models for PD.
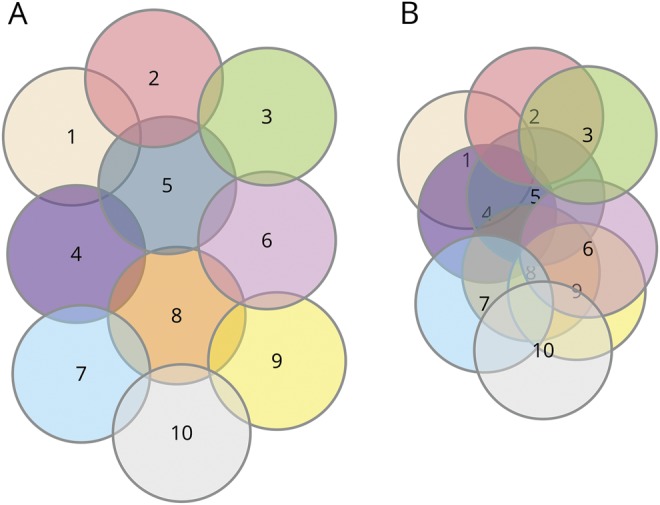
Each numbered circle represents a distinct biological entity or disease. Left, a model warranting reconstruction. While the diseases are related to one another, they are mostly unique; most biological elements are not shared. According to this model, if α-synuclein (α-syn) is common to all or most forms of PD, it cannot be pathogenic to each; genetic Parkinson disease (PD) subtypes inform about those subtypes and not others. Right, a model best served by revision. All diseases have more in common with one another; their uniqueness is mostly theoretical given the large overlap. This model does not discount a potential important role for α-syn in many or all of the diseases; a therapy that might work for a genetic PD subtype may work for others. The model also allows the explanation that PD may involve multiple pathways, and if we can fix one of them, we could delay sporadic PD by years.
Table 1.
Key research constructs: Differences between revision and reconstruction

Knowledge gap 1: Does the heterogeneity of PD represent a disease spectrum with different clinical subtypes and rates of progression or distinct molecular entities, each a separate, definable disease?
The former premise has been a dominant force. Adopting the view that PD is a spectrum supports efforts at increasing or merging ongoing cohorts to maximize scale and deploying improved analytic processes to the accumulating data (revision). However, the data-driven–generated subtypes thus far created are not stable over time,2,3 have not been reproducible,4,5 and do not predict unique patterns of aggregation of α-syn or other pathologies.6,7 Moreover, measurements of putative aggregated protein-based biomarkers have shown substantial signal overlap between patients and controls and between disease phenotypes, and replication of results across cohorts is lacking.8 Combining cohorts anchored on clinical criteria and dependent on confirmation of dopamine deficit is a strategy likely to run into the same validation and replication difficulties because the problem may not be one of statistical power but rather of underlying assumptions. As our biomarker armamentarium broadens, a reversal of the developmental direction from biomarker to phenotype, stratifying on the basis of biological clusters of therapeutic significance rather than on phenotypic classification, will require an overhaul of these efforts (reconstruction).
Knowledge gap 2: Will past negative clinical trials be replaced by future successes if treatments are delivered to a larger and earlier cohort or to smaller, biologically defined populations, prodromal or manifesting?
The conventional view is that failure of clinical trials to meet efficacy endpoints has 3 primary causes: shortcomings of the clinical trials themselves (e.g., wrong dosage or insufficient blood-brain barrier penetration, inadequate target engagement, insensitive outcome measures, too short a follow-up period), cohorts too advanced for the intervention of interest to be effective (once symptoms appear, it may be too late), and poor translatability from animal models to human trials. Proposals for revision are aimed at studying larger and earlier cohorts and creating better animal models. Proposals for reconstruction are aimed at recruiting more selective cohorts, whether presymptomatic or postsymptomatic, identified by biomarkers reflecting specific disease mechanisms, ensuring that putative therapies are tested only in those most likely to respond.
Lessons from other fields
Other fields of medicine have had their share of failures in biomarker development and clinical trials of disease-modifying interventions. Did they struggle with moving away from a single-disease model? Potentially informative lessons from breast cancer and the monogenic disease cystic fibrosis (CF) were discussed at the Krembil Knowledge Gaps Symposium.
Cystic fibrosis
Despite the discovery of the CF transmembrane conductance regulator (CFTR) gene in 1989, treatments for CF were exclusively symptomatic, targeting the secondary effects of dysfunction in the CFTR protein, involved in the secretion of chloride and bicarbonate and the regulation of water in the lung, sweat glands, pancreas, and gastrointestinal tract. Depending on whether each of the ≈1,900 CFTR mutations impaired protein production, its tridimensional functional shape, ion transport through the channel door, or other aspects of the protein function, CF was split into 6 major classes. The first disease-modifying medication, ivacaftor, was found to depend on the presence of at least 1 G551D CFTR mutation, assayed with epithelial cells in vitro (not animal models), which excluded 95 of every 100 patients with CF. A promising phase 2 trial (n = 39)9 of only these genotyped and assay-positive patients was followed by a successful phase 3 placebo-controlled trial (n = 84 in each of 2 arms).10 The first Food and Drug Administration approval of a disease-modifying intervention for CF occurred 23 years after the discovery of the CF gene. Although it was effective in only 4% to 5% of patients with CF, it represented a proof of concept that genetically subtyped diseases could be modifiable with class-specific therapies. From this small but definite success, the pool of responders has been increased in selected genetic and class subtypes with the use of double- and triple-drug combinations. Critically, the Therapeutic Development Program of the Cystic Fibrosis Foundation was instrumental in derisking pharmaceutical industry high-throughput drug discovery programs and early-phase clinical trials.
Key developmental milestones in CF
The 3 developmental milestones were identification of mutation class-specific response to small molecules, the use of highly predictive cell-based screening and biomarker platforms that avoided animal models of efficacy, and multidrug combination of potentiator and corrector therapies with potential for synergism that was evaluated from the outset. The CF experience provides an example of how even a single-gene disease was successfully tackled once it was divided into identifiable molecular subtypes, mechanistically defined with cell-based biomarkers.
Breast cancer
After a randomized clinical trial in 1981 showed that radical mastectomy yielded no benefits over quadrantectomy (breast-preserving surgery),11,12 breast cancer began to be split into clusters of different pathobiological entities. Individual patient data from randomized trials allowed the recognition that the benefit of tamoxifen was limited only to women with estrogen receptor (ER)–positive tumors, whether premenopausal or postmenopausal.13 Incremental improvements in survival required the development of other drug treatments for ER-positive breast cancers, including aromatase inhibitors, selective ER degraders, CDK4/6 inhibitors, and mammalian target of rapamycin inhibitors.
In parallel, identification of the human epidermal growth factor receptor 2 (HER2) led to successful clinical trials of HER2-targeted drug therapies such as trastuzumab (Herceptin), pertuzumab (Perjeta), and trastuzumab-DM1 (Kadcyla) in the 20% of women with aggressive HER2-positive breast cancers,14 but only after post hoc analyses of initially negative trials revealed the presence of a potential responsive population. Breast cancer is now divided into several pathology-molecular subtypes with distinct response to treatments, relapse patterns, and long-term survival.15
Key developmental milestones in breast cancer
The 3 developmental milestones were recognition of the molecular basis for disease heterogeneity, subtyping based on molecular testing and histopathology, and clinical trials with targeted drug treatments based on molecular subtype. This experience demonstrates that a single negative trial sufficed to reject the hypothesis that breast cancer was a single but heterogeneous disease.
Conceptual roadblocks in the future application of these lessons to PD
Should pathology remain the gold standard for PD diagnosis and biomarker development?
Conceptually, PD is considered an α-synucleinopathy. However, there is no dose-dependent correlation between Lewy pathology and cell loss or clinical features. In fact, studies have found either no16 or negative correlations17 between Braak stage and cell density in the substantia nigra. Furthermore, Lewy pathology is not necessary for nigral degeneration, as demonstrated by the appreciable proportion of leucine-rich repeat kinase 2 LRRK2- and Parkin-related cases with substantia nigra neuron loss and motor deficits consistent with PD but without Lewy bodies.18 Also challenging the primacy of α-syn pathology, PD is usually associated with abnormal accumulation of several different types of misfolded proteins; the exclusive presence of α-syn pathology is the exception, not the rule.19–21 Lastly, some studies suggest that protein aggregates may even be compensatory in a brain under molecular stress and that, with the plausible exception of α-syn–related genetic PD subtypes (SNCA-PD), α-syn aggregation is neither necessary nor sufficient for neurodegeneration or clinical parkinsonism.22
Key roadblocks
Development of biomarkers of diagnostic susceptibility and detection based on α-syn and other aggregated proteins may be insufficient for assessing disease trait or state. Protein aggregates could be related to convergent protective strategies rather than specific pathogenetic mechanisms.23 Definitive evidence of a direct causal relationship between Lewy pathology and neurodegeneration in humans is lacking.
Should pathology be the target of therapeutic development?
If the extent of the contribution of α-syn aggregation to the pathogenesis of most subtypes of PD can be questioned, then targeting it in full length or through specific epitopes of maximum antigenicity prediction24 may be insufficient or irrelevant outside of SNCA-PD. Upregulation of α-syn (and other inflammatory markers such as Toll-like receptor 2, interleukin [IL]-6, and tumor necrosis factor) has been documented in mice or rats in response to infections or inflammation.25,26 Within this framework, α-syn aggregation may behave not unlike an acute-phase reactant, increasing in concentration during an inflammatory state.
Aggregated β-amyloid has been targeted in Alzheimer disease (AD) trials over the past 2 decades. Apart from recent intriguing but unpublished results suggesting efficacy of aducanumab, all 32 phase 2 and 3 antiamyloid clinical trials, including 6 on prodromal cohorts, have been negative despite achieving expected target engagement, that is, demonstrating significant reduction in CSF- or PET-based aggregated β-amyloid. Critically, nearly 40% of the trials (largely, but not exclusively, of ɣ secretase and β-secretase inhibitors) have demonstrated worsening of cognition and, in half of these, acceleration of hippocampal atrophy in the treated group compared to placebo. Fourteen clinical trial participants with pathology-confirmed AD who received β-amyloid42 immunization (AN1792; Elan Pharmaceuticals, Dublin, Ireland) progressed to moderate or severe dementia despite extensive plaque removal in all regions examined at postmortem.27
While β-amyloid aggregation and α-syn aggregation are structurally and functionally different targets, 2 main arguments may predict future failures of targeting α-syn with approaches similar to those used for β-amyloid: both pathologies often co-occur as downstream events in sporadic AD and PD; and proteinopathy burden does not correlate with symptom severity. In sum, these proteins represent pathologic markers but may not themselves be pathogenic (etiologic).
Key roadblocks
α-Syn–based biomarkers of disease could have limited value for the selection of patients for trials of putative disease-modifying interventions. Targeting α-syn aggregation may have beneficial effects in selected subsegments of PD (e.g., α-syn mutations/multiplications) but could be futile or harmful in others.
Should distribution of pathology define progression?
If the presence of α-syn aggregates defines PD, its distribution has to define PD progression. The autopsy study quantifying Lewy pathology from individuals with and without PD that led to the Braak hypothesis was anchored on cases with pathology in the dorsal motor nucleus of the vagus nerve and accepted a single Lewy neurite as positive for any region. When considered together, these brains suggested a pattern with a beginning in the peripheral nervous system, specifically the Meissner plexa in the gut and in the olfactory bulb, and an end in the neocortex.28 Although never proved, it has been assumed that there must be a sequential spread over time between these distribution patterns, with support from animal studies showing self-propagation of α-syn from neuron to neuron through permissive templating in a prion-like fashion.29,30 Stage 3 of the 6 hypothesized Braak stages of α-syn progression represents the intersection between the constructs of prodrome and clinically apparent disease, according to current motor-centric diagnostic criteria.
The many exceptions to the hypothesis of stereotypical progression of pathology31 and the fact that symptomatic deficits align with neuronal degeneration rather than with α-syn aggregation32 render problematic the efforts to calculate disease progression clinically (e.g., phenoconversion from prodrome to disease)33 or with longitudinal changes of CSF total α-syn. Similarly, clinical (e.g., rate of change in Movement Disorders Society–sponsored Unified Parkinson's Disease Rating Scale scores) and imaging (e.g., dopamine transporter striatal binding ratio signal loss) markers of progression have not detected meaningful effects on disease progression in trials conducted to date. Moreover, what might define progression on one measure does not on another. Indeed, there is poor concordance of progression between the Unified Parkinson's Disease Rating Scale score and dopamine transporter striatal binding ratios.34
Key roadblock
While convergent dopamine-, clinical-, and α-syn–based biomarkers may have utility in some populations, biomarkers of divergence (present in some, absent in most) will be needed to address the etiologic heterogeneity.
Would testing treatments that failed in early PD be successful in prodromal PD?
Because earlier treatment is better in many medical conditions, the “too late” argument has often been cited as explaining past failures in neurodegenerative diseases. If this is correct, then retesting the interventions of prior trials in prodromal populations may yield the first success. However, if PD subsumes many biological subtypes, what is the relationship of prodromal PD to these different subtypes?
In the absence of molecular/biological identification of prodromal disease, we run the risk of extending the negative trials to prodromal PD, as the AD field has already demonstrated with antiaggregation interventions in prodromal AD (mild cognitive impairment). This may be magnified by the fact that the most common prodromal PD phenotype, REM sleep behavior disorder, can also evolve into dementia with Lewy bodies and multiple system atrophy.35 It is possible that earlier will not be better if we target a large, clinically defined syndrome rather than smaller but more homogeneous biological constructs.
Key roadblocks
Molecular criteria for subtyping are needed for both prodromal PD and manifesting PD.
Tying biomarkers to disease mechanisms
Like the examples of breast cancer and CF, genotype and molecular subtyping will need to replace phenotypic subtyping, which does not predict pathobiology, pathology, or pathophysiology.6,7 Greater granularity in the clinical characterization of patients, based on more sophisticated semiologic, neuroimaging, or wearable technology–based activity sensors, should be of considerable utility in providing quantitative and objective endpoint measures. However, like “putting the cart before the horse,” these approaches are unlikely to succeed alone in guiding the development of biomarkers or selecting patient subtypes for disease-modifying treatments. These ideas lead to 2 formulations:
Revision: fitting data to purpose. Genetic risk profiles have been undertaken with the purpose of enhancing risk prediction for a disease considered polygenic. Under this paradigm, the use of multiple common risk alleles can approach the predictive accuracy of a single gene mutation in a mendelian disease.36 For instance, rare risk variants in the glucocerebrosidase (GBA) and leucine-rich repeat kinase 2 (LRRK2) genes can serve “to distinguish patients with Parkinson's disease from healthy controls.”37 Genetic biomarkers are aimed at enhancing predictive or manifesting (PD/not-PD) diagnostic accuracy in at risk or clinically defined cohorts, respectively, as well as predicting risk of progression to key phenotypic outcomes such as dementia.38 Clinical phenotypes act as the independent variable; genetic and biological measures, the dependent variable.8 Research on extremes of clinical phenotype (e.g., young-onset PD) or genotype (e.g., GBA-PD) inform the larger whole by providing linked pieces to a disease puzzle that can be brought into full view through their study.1
Reconstruction: fitting purpose to data. The exponential increase in significant hits from genome-wide association studies (to date, 90 genetic risk loci for PD) may not reflect contributors to a single phenomenon but expressions of multiple and mostly independent biological mechanisms, which may even be conditional on other unmeasured factors. An examination of these loci as independent variables might reveal a range of phenotypes, potentially including but not restricted to the PD spectrum. Under such a framework, a given GBA mutation explains GBA-PD but may not meaningfully contribute to the whole of (sporadic) PD. According to this approach, genetic/biological measures act as the independent variable; clinical phenotype, the dependent variable. There is not a single disease puzzle but several puzzles, each explaining related but molecularly separate diseases, albeit converging in the selective vulnerability of specific neuronal populations, most notably nigrostriatal neurons.
GBA-PD
Similar to CFTR in CF, different GBA mutations lead to different properties of the encoded glucocerebrosidase (GCase) protein. While some mutations may result in retention of the protein in the endoplasmic reticulum, other mutations can affect the interaction with saposin C (GCase activator) in the lysosome or lead to a complete lack of protein (null mutations).39 Therefore, therapeutic strategies such as chaperones may be beneficial for only specific mutations, while other strategies such as gene therapy may conceivably be beneficial for all mutation carriers. This is further complicated by the fact that different mutation types (e.g., mild p.N370S or severe p.L444P) have differential effects on the progression of the disease.39–41 This differential progression will require stratification by mutation type, even in GBA-specific clinical trials, to avoid a differential rate of progression in 1 arm of the trial due merely to different frequencies of specific mutation types in each group.
Understanding the mechanism for each type of GBA-PD is likely to allow the identification of GBA-specific biomarkers and the development of mechanism-specific drugs. Because a subgroup of patients with sporadic PD have low GCase despite the absence of a GBA mutation,42 recruitment for the ongoing phase II GBA-specific trial (ClinicalTrials.gov identifier: NCT02906020) includes a subset of patients with sporadic (non-GBA) PD with low GCase. If the outcome were to be negative or futile, it would support avoiding further developments in a wider population than biologically appropriate.
Revision
Although overextending the scope of molecular therapies compromises their signal-to-noise ratio, studies encompassing >1 GBA mutation (or sporadic PD with low GCase) may deserve consideration. In reconstruction, subtyping for mutation-specific GBA-PD will reduce the population available for recruitment but correspondingly increase the odds of success (by targeting smaller but better defined subsets).
LRRK2-PD
Certain LRRK2 mutations (e.g., p.G2019S) are associated with increased LRRK2 kinase activity. Preliminary data suggest that LRRK2 kinase activity may also be increased in some PD brains without a LRRK2 mutation.43 Thus, a disease-modifying strategy that aims to inhibit kinase activity is currently being tested. Cancer has seen successes in applying a similar strategy with kinase inhibitors (34 kinase inhibitors were approved by the Food and Drug Administration for cancer by 2017).44 These successes have, in part, resulted from applying the concept of kinase dependency in that (1) kinase inhibitors will be successful only in diseases with upregulated kinase activity and (2) optimum use of kinase inhibitors as therapies can be achieved only if coupled with biomarkers that identify kinase-dependent disease.45 Learning from these successes in cancer, researchers know that the use of LRRK2 kinase inhibitors in PD requires development of diagnostic biomarkers to identify patients with PD with upregulated LRRK2 kinase activity in the CNS (LRRK2 kinase–dependent PD). Therefore, treatment with LRRK2 kinase inhibitors could potentially be extended to other patients with PD with enhanced LRRK2 kinase activity (analogous to patients with PD without GBA mutations but with evidence of reduced GCase activity). Biomarkers to measure LRRK2 kinase inhibition in the CNS by these drugs will also be required. Collectively, these biomarkers will be necessary for clinical trials to test the hypothesis that LRRK2 kinase inhibition slows disease progression in patients with LRRK2 kinase–dependent PD.
Revision without reconstruction
If LRRK2 kinase activity is a pathogenic driver in most LRRK2-PD and some non–LRRK2-PD, opportunities for therapies may materialize in >1 genotype and perhaps in sporadic kinase-dependent PD subtypes.
Inflammation
Markers of immune activation have been observed in most subtypes of PD. These include peripheral inflammatory cytokines such as IL-6, tumor necrosis factor, IL-1β, IL-2, IL-10, and C-reactive protein.46 A more proinflammatory cytokine profile at diagnosis is associated with faster subsequent motor progression and more cognitive decline.47 However, it is unclear whether these markers represent downstream mediators (overlapping areas in the Venn diagram of figure 1) or upstream etiopathogenic events.
Revision
If causal or contributing, adjunctive anti-inflammatory interventions may lessen disease progression across most disease subtypes. Inflammation or immune activation might be particularly critical at an earlier time than putative interventions can be enacted and influenced by host factors such as the microbiome. Alterations in the composition of gut microbiota have been linked to inflammatory, metabolic, oncologic, and psychiatric disorders.48 The evidence of aggregated α-syn in the intestine early in PD, potentially even in normal individuals, makes the gut-brain connection particularly compelling. In addition, different microbiomes coexist in other body sites such as the nose, mouth, or the skin, and they too can potentially influence the triggering of PD. The microbiome is highly dynamic, influenced by host genome variability, region of residence, age, sex, diet, medications, and PD itself (e.g., secondary to constipation).49 Because it is still unclear how the microbiome might be modulated to affect disease expression, measuring intestinal microbiota in observational and interventional studies is imperative to better define its role across PD subtypes.
Reconstruction
Although epidemiologic evidence suggests that nonsteroidal anti-inflammatory drugs do not influence PD,50 it remains possible that subgroups with different patterns of immune activation could be targeted with selective anti-inflammatory or immunomodulatory strategies.
Challenges to clinical trials of disease-modifying drugs: Matching the what to the who
While many of our explanations for past negative outcomes have emphasized the “how” (inadequate clinical trials) and “when” (patients too advanced), if the “what” (right drug, right target) and “who” (right patients) are perfectly aligned, the larger anticipated effect size may allow imperfections in the how (right clinical trial design and execution) and when (right patient staging).51
Who
Potential therapies known to affect a presumed pathogenic pathway will require a demonstration that the relevant mechanism (biomarker of pathogenesis) is active and pathogenic in the intended recipients. The selection of study participants should be based on mechanism rather than phenotype. As with the experiences in CF and breast cancer, the first successes may materialize in small populations (likely <5% of those with PD). The obvious first population to target will likely be individuals carrying the relatively common risk factor genes, LRRK2 and GBA.
Stratification
Different treatments are best suited to different subgroups. Future studies should ideally bank biosamples (blood and CSF) and cells (e.g., peripheral blood mononuclear cells, induced pluripotent stem cells) to assess for biomarkers of response and failure after study participation. Study consent forms should secure participants’ permission for samples and data to be used in future analyses after their participation in the study is completed. This would admittedly represent a challenge because neuronal cells are more difficult to obtain than tumor samples (although adult somatic cells could be reprogrammed into neurons).
Generalizability
GBA- and LRRK2-targeted therapy trials may not generalize to other forms of PD or, perhaps, to all expressions of GBA and LRRK2 mutations in manifesting individuals, given that different GBA and LRRK2 mutations may result in different pathogenetic mechanisms, as learned from the experience in CF.
What
As noted above, the what should not be considered in absence of the who. Of the recent or ongoing phase II/III trials (inosine, isradipine, deferiprone, and passive α-syn immunization), only one paired the what with the who most likely to benefit (inosine for patients with PD with low serum urate levels in the Study of Urate Elevation in Parkinson's Disease [SURE-PD3]). Regardless of how promising a treatment may have seemed in preclinical studies, it may be possible to translate that promise to a patient population without etiologic enrichment for the targeted pathogenic mechanism and in a manner that demonstrates target engagement.
From single to multiple therapies
Combined or cocktail treatments will potentially be more beneficial by targeting >1 dominant pathogenic mechanism in any given disease subtype.51 However, it is likely that a single intervention will need to be proven effective in a small patient subgroup before the targeted pool can be increased through the addition of a second intervention (as discussed for CF). In addition, a combined approach may be best positioned to address the “triggers, facilitators, and aggravators”52 for each subtype (e.g., LRRK2-PD), according to any changes in the relative dominance or contribution of a pathogenic mechanism over time or if several cellular mechanisms act in parallel rather than in series for that subtype. To add further complexity, defining a therapeutic window for each agent may require considering a linear relationship between duration of treatment and side effects but a potentially nonlinear relationship between duration of treatment and efficacy. Finally, more collaboration to expedite and derisk drug development by industry will also enhance the ability to study combination therapies. Precompetitive collaboration (“co-opetition”) is a business model whereby competitor companies work together in a competitively safe space to solve problems cooperatively to the benefit of all.53
How
The traditional approach to clinical trials tends to be costly and time-consuming, going stepwise from preclinical development to clinical phases 1 through 3. An alternative nimbler and more efficient approach is that of learning and confirming clinical trials.54 The interest in innovative trial designs has given rise to adaptive designs, which allow the review of accumulating information during an ongoing clinical trial to potentially modify trial characteristics. Challenges to the application of these concepts to PD, in contrast to selected other diseases such as breast cancer, include the lower incidence, slow progression, lack of reliable rapid readouts of changes in underlying disease status, and availability of potent symptomatic treatments that mask important changes. Targeting patients whose symptomatic management has been optimized before study entry may mitigate the last issue and address concerns that any disease-modifying effects may in part represent (or be masked by) symptomatic benefits.
Adaptive design
Adaptive design actually means adaptive by design. To properly define the simulations and to preserve the integrity and validity of the trial, properly designed simulations and adaptation rules must be specified in advance (planned adaptation) (figure 2). Adaptive enrichment designs represent a variation that fulfills the desire to target therapies to patients who can benefit the most from treatment. In such designs, a trial would initially consider a broad population (e.g., PD with and without GBA mutations but low GCase). The first study period, a discovery phase, reveals participant groups most likely to benefit from treatment (e.g., only patients with mutation-specific GBA-PD). Participants from this subgroup are then randomized to treatment groups in a validation phase. Hence, the study power is increased despite a reduction in sample size by focusing only on subgroups most likely to show benefit. The adaptive design sample size re-estimation allows adjustment of the sample size based on a review of the interim data. An internal pilot is a sample size re-estimation used to reassess nuisance parameters midstudy. With moderate to large sample sizes, internal pilot designs can be used to make appropriate modifications with minimal inflation of the type I error rate. Thus, there is little reason (statistically) not to apply this design for most clinical trials. However, such trial designs are logistically complicated. Strict attention must be paid to maintaining the blinding and minimizing time gaps during which adaptation decisions are made to ensure the efficiency and integrity of adaptive studies.
Figure 2. Planned adaptation allows learning and confirming in adaptive clinical trials.

Only planned adaptations can be guaranteed to avoid unknown biases resulting from the adaptation (from Kairalla et al.60). GS = Group sequential; SSR = Sample-size re-estimation.
Master protocols
Master protocols are protocols “designed with multiple substudies, which may have different objectives and involve coordinated efforts to evaluate one or more investigational drugs in one or more disease subtypes within the overall trial structure,” according to the Food and Drug Administration Guidance for Industry definition. Master protocols bring efficiency by creating 1 optimized trial infrastructure applying it perpetually to study multiple therapies, using the following: basket trials (test single drug or drug combination in multiple populations or settings), umbrella trials (test multiple investigational drugs in a single population), and platform trials (test multiple therapies in a single disease in a perpetual manner, with therapies allowed to enter or leave the platform according to a decision algorithm) (figure 3).
Figure 3. Platform trial example.
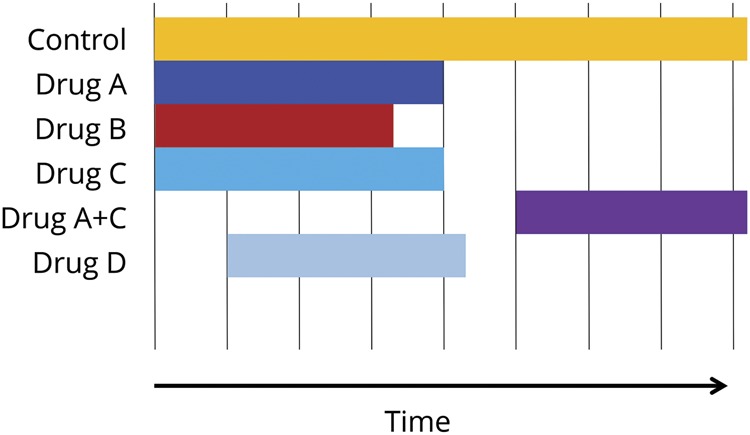
It allows multiple drugs examined from the outset, which can be removed or combined (e.g., A + C). It also permits new drugs (D) to be examined at a later time point in the same trial.
Master protocols use a trial network with shared infrastructure to streamline trial logistics, to improve data quality, and to facilitate data collection and sharing and a common protocol that incorporates innovative statistical approaches to study design and data analysis, enabling a broader set of objectives to be met more efficiently than would be possible in independent trials. The scientific efficiencies of this type of adaptive design include a shared placebo group, sample size savings, the study of multiple drugs and multiple markers, and the flexibility to add/remove agents. Successful examples in which a platform trial has been implemented include the Investigation of Serial Studies to Predict Your Therapeutic Response through Imaging and Molecular Analysis 2 (I-SPY 2) to screen drugs in neoadjuvant breast cancer55 and Dominantly Inherited Alzheimer Network Trials Unit (DIAN-TU) in at-risk or early-onset genetic AD.56 The first Platform Trial Initiative for amyotrophic lateral sclerosis was recently established at the Healey Center for ALS at Massachusetts General, with 5 promising drugs entering evaluation as of September 2019 (massgeneral.org/neurology/als/research/platform-trial). Public-private partnership efforts similar to those of the European Prevention of Alzheimer's Dementia project57 will be required from the PD community to facilitate the needed application of these study designs to PD. Efforts from the Critical Path Institute and the Alzheimer's Association will also be essential in cementing the needed collaboration among industry, academia, and regulatory agencies in developing effective combination therapies for PD, AD, and other neurodegenerative disorders.58 Challenges in the execution of master protocols include ensuring the willingness of all sponsors to share data and matching multiple dosing regimens (frequencies, forms, and routes) within the trial infrastructure.
Special considerations
There is much to be learned from existing cohorts, including deep clinical, genetic, and -omics phenotyping, with emphasis on genetic forms of PD (revision). As argued previously, large population-based aging cohorts, agnostic to phenotype, will have the power to find biological signals of pathogenic mechanisms for which putative therapies already exist and could be repurposed. Proof-of-concept adaptive clinical trials of putative disease-modifying drugs can be embedded within biomarker-development cohorts (reconstruction).
Financial risk/hypothesis certainty
While the mechanism adopted by the CF research community was instrumental in derisking pharmaceutical industry drug discovery and early-phase clinical trials, particularly to manage failures due to safety, drugs can fail on efficacy. Thus, it is imperative to have high confidence in the therapeutic hypothesis being tested in a clinical trial and the likelihood of adequate target engagement. No derisking effort can prevent candidate drugs from failing on efficacy because patients were not selected on the basis of the mechanism of action that the potential drug is purported to have. The complex interplay between the financial risks and the certainty of the therapeutic hypothesis has been elegantly addressed for rare diseases.59 If the fundamentals about drug development for disease modification were to move from a large indication (e.g., sporadic PD), for which a high risk tolerance is mitigated by the potential of future astronomical returns even with very low probability of success (revision), to a small indication (e.g., a molecular PD subtype; reconstruction), the development process would tolerate less risk, and the therapeutic hypothesis would have to be definitive rather than tentative. A rare-disease model would also need to rely on orphan designation for cost and revenue advantages and exploit the potential of drug repurposing.
Animal models in drug development
Animal models cannot be expected to recapitulate the complexity of a human disease that exists in many forms and typically presents late in life. Although some fields (e.g., CF) have abandoned the reliance of therapeutic development on animal models, they could serve to understand specific mechanisms suggested by biospecimens and neuroimaging studies in humans (revision). The need for research applications to demonstrate efficacy first in animal models before prospective trials can proceed in humans will need to be reconsidered (reconstruction).
Conclusions
As recently articulated,51 more critical than the tools of warfare (clinical trial designs, endpoint sensitivity, treatments themselves) may be the reconfiguration of warfare itself. The who/what we are fighting for has not evolved from the days of blunt targeting of early PD, in which sophisticated weapons were deployed on a population without evidence of vulnerability to the effect of those weapons. Some aspects of our approach can benefit from a revision strategy, an immediate evolutionary adaptation, or shortcut. Many areas require reconstruction, which needs not take longer but may be more revolutionary than evolutionary and will certainly be disruptive. The tension between revision and reconstruction may not be solved in the short term (table 2).
Table 2.
Point-counterpoint: Revision vs reconstruction

Revision (shortcut)
Opportunities exist for harnessing existent and accruing genetic cohorts, most meaningfully in GBA-specific clinical trials for GBA-PD subtypes stratified by mutation type and in kinase-dependent PD subtypes selected through biomarkers of upregulated kinase activity for targeting with kinase inhibitors. If LRRK2 kinase activity is a pathogenic driver in non–LRRK2 kinase-dependent PD, treatment success may materialize in >1 kinase-dependent PD genotype/subtype. In addition, ongoing and future clinical trials in genetic and even nongenetic cohorts can gather data to generate markers of response and failure to refine the “who” in subsequent efforts.
Reconstruction (long-cut)
While drug combinations are a future expectation, proving success of individual drugs will require ascertainment of molecular PD subtypes regardless of progress in clinical trial designs. The ongoing phenotype-based biomarker development programs will need to evolve into phenotype-agnostic biology-driven biomarkers, able to distinguish causative mechanisms from nonspecific, late, or common/convergent mechanisms, inappropriate or inadequate as therapeutic targets. The therapeutic paradigm for neuroprotection will have to emulate the lessons from other fields of medicine, tailoring biology-specific/multimechanism strategies to well-defined PD subtypes. Although there will be value in post hoc responder analyses of future large clinical trials, the more biologically enriched the targeted groups for the treatment of interest are, the greater the odds of therapeutic success.
Glossary
- AD
Alzheimer disease
- α-syn
α-synuclein
- CF
cystic fibrosis
- CFTR
CF transmembrane conductance regulator
- DIAN-TU
Dominantly Inherited Alzheimer Network Trials Unit
- ER
estrogen receptor
- GCase
glucocerebrosidase
- HER2
human epidermal growth factor receptor 2
- I-SPY 2
Investigation of Serial Studies to Predict Your Therapeutic Response through Imaging and Molecular Analysis 2
- IL
interleukin
- LRRK2
leucine-rich repeat kinase 2
- PD
Parkinson disease
- SNCA
synuclein
- SURE-PD3
Study of Urate Elevation in Parkinson's Disease
Appendix. Author contributions
Footnotes
Editorial, page 467
Study funding
Major support for the Revision vs Reconstruction meeting leading to this manuscript was provided by The Krembil Foundation, the Ronald Kimel Foundation Trust, the Parkinson's Foundation, the International Parkinson and Movement Disorder Society, and the American Parkinson Disease Association. Additional support was provided by the International Parkinson and Movement Disorders Society, the Parkinson Foundation, and the following pharmaceutical companies: Sunovion, UCB, Valeo Pharma, Acadia PharmaceuXcals Inc, Medtronic of Canada Ltd, ProtoKinetics, Theravance Biopharma US, Inc, and Neurocrine Bioscience. The Revision vs Reconstruction meeting was endorsed by the Michael J. Fox Foundation and the American Parkinson Disease Association. C.H. Williams-Gray holds a Research Councils UK/UK Research Innovation Research Innovation Fellowship awarded by the Medical Research Council (MR/R007446/1); R.A. Barker is supported by the Wellcome Trust/Medical Research Council Stem Cell Institute (203151/Z/16/Z).
Disclosure
A.J. Espay has received grant support from the NIH and the Michael J. Fox Foundation; personal compensation as a consultant/scientific advisory board member for Abbvie, Adamas, Acadia, Acorda, Neuroderm, Impax, Sunovion, Lundbeck, Osmotica Pharmaceutical, and USWorldMeds; publishing royalties from Lippincott Williams & Wilkins, Cambridge University Press, and Springer; and honoraria from USWorldMeds, Lundbeck, Acadia, Sunovion, the American Academy of Neurology, and the Movement Disorders Society. L.V. Kalia holds a Canadian Institutes of Health Research (CIHR) Clinician-Scientist Award; receives research support from CIHR, Michael J. Fox Foundation for Parkinson's Research, Natural Sciences and Engineering Research Council of Canada, Ontario Brain Institute, Parkinson Canada, and Toronto General & Western Hospital Foundation; holds contracts with ApoPharma; and received educational grants from Allergan and honoraria from Pfizer, Shire, and the NIH. Z. Gan-Or has served as an advisor for Lysosomal Therapeutics, Inc, Idorsia, Prevail Therapeutics, Inception Sciences (now Ventus), and Denali; received grants from the Michael J. Fox Foundation, the Canadian Consortium on Neurodegeneration in Aging, the Canadian Glycomics Network, the Canada First Research Excellence Fund, awarded to McGill University for the Healthy Brains for Healthy Lives program, and the Fonds d'accélération des collaborations en santé, granted by MESI/CQDM; and is supported by Fonds de recherche du Québec-Santé Chercheurs-boursiers award with Parkinson's Quebec and by the Young Investigator Award by Parkinson's Canada. C.H. Williams-Gray has received grants from the Michael J. Fox Foundation, the Rosetrees Trust, the Evelyn Trust, Addenbrooke's Charitable Trust, Parkinson's UK, the Academy of Medical Sciences UK, the Stevenage Biosciences Catalyst, and the Patrick Berhoud Trust; and is supported by the National Institute for Health Research (NIHR) Cambridge Biomedical Research Centre grant 146281 (the views expressed are those of the author[s] and not necessarily those of the NIHR or the Department of Health and Social Care); she has received honoraria from Lundbeck and consultancy fees from Modus Outcomes. P.L. Bedard has received grant support (to his institution) from Bristol-Myers Squibb, Sanofi, AstraZeneca, Genentech/Roche, Servier, GlaxoSmithKline, Novartis, SignalChem, PTC Therapeutics, Nektar, Merck, Seattle Genetics, Mersana, Immunomedics, and Lilly. S.M. Rowe has received grant support from the NIH, the Cystic Fibrosis Foundation, Vertex Pharmaceuticals Inc, Bayer, Forest Research Institute, AstraZeneca, N30/Nivalis, Novartis, Galapagos/AbbVie, Proteostasis, Eloxx, Celtaxsys, PTC Therapeutics, and Vertex Pharmaceuticals Inc; and personal honoraria from Bayer, Novartis, Renovion, and Vertex Pharmaceuticals Inc. F. Morgante received honoraria from UCB Pharma, BIAL, Chiesi, Medtronic, Zambon, Chiesi, Abbvie, and Merz; she receives royalties from Springer for the book Disorders of Movement. A. Fasano received honoraria for consultancies from Abbvie, Abbott, BrainLab, Boston Scientific, Chiesi farmaceutici, Ipsen, Medtronic, Sunovion, and UCB; honoraria for participation in advisory boards from Abbvie, Boston Scientific, and Ipsen; and research grants from Abbvie, Boston Scientific, Cummings Foundation, Dystonia Medical Research Foundation Canada, Michael J. Fox Foundation, Medtronic, University of Toronto, and Weston Foundation. B. Stecher is a patient and Parkinson advocate. He writes on tmrwedition.com/. He has received honoraria from Zambon, Biogen, Abbvie, Roche, the Buck Institute, McGill, and the Tanenbaum Open Science Institute. M.A. Kauffman is an employee of the CONICET. He has received grant support from Ministry of Science and Technology of Argentina and Ministry of Health of Buenos Aires. He has received honoraria payments for educational activities from Janssen Pharmaceuticals and Bago Pharmaceuticals. M.J. Farrer has received research grant support from the Canadian Federal Government, British Columbia (Leadership Fund-Life Labs, Genome BC, & the Province), and the Weston Foundation. He has intellectual property rights, including US patent WO 2005/004794 A2 & JP 2011-30292 International Publication No. WO 2006/045392 A2, Method of Treating Neurodegenerative Diseases, International Publication No. WO 2006/068492 A1, US Publication No. US-2008-0009454-A1, and Norwegian patent 323175 “Identification of LRRK2 in parkinsonism, including mutation c.6055G>A (p.G2019S) and mouse models.” He has received royalties from Mayo Foundation, Athena Diagnostics & H. Lundbeck A/S. C.S. Coffey has received grant funding from the Michael J. Fox Foundation, National Heart, Lung, and Blood Institute, and National Institute of Neurological Disorders and Stroke (NINDS) and consulting funds from Michael J. Fox Foundation. M.A. Schwarzschild is an employee of Massachusetts General Hospital and a member of the faculty of Harvard Medical School and is supported by research funding (from the NIH, Michael J. Fox Foundation, the Parkinson Foundation, Target ALS, The Maximilian E. & Marion O. Hoffman Foundation Biotie/Accorda, and the US Department of Defense), clinical practice compensation, an endowment, and university funds. He serves on the scientific advisory boards of the Michael J. Fox Foundation, CBD Solutions, and Cure Parkinson's Trust. He has served as a consultant for New ß Innovation. He has received royalties from Massachusetts General Hospital for licensing of adenosine A2A receptor knockout mice. T. Sherer is the chief executive officer of the Michael J. Fox Foundation for Parkinson's Research. R.B. Postuma reports grants and personal fees from Fonds de la Recherche en Sante, the CIHR, Parkinson Canada, the Weston‐Garfield Foundation, the Michael J. Fox Foundation, and the Webster Foundation, as well as personal fees from Takeda, Roche/Prothena, Teva Neurosciences, Novartis Canada, Biogen, Boehringer Ingelheim, Theranexus, GE HealthCare, Jazz Pharmaceuticals, AbbVie, Janssen, and Otsuko. A.P. Strafella has received funding from Canada Research Chair Program, CIHR, National Parkinson Foundation, Parkinson Disease Foundation, Parkinson Canada, Ontario Gambling Association, Tourette Syndrome Association, Brain Canada, and Weston Brain Institute, as well as honoraria from GE Healthcare Canada LTD. A.B. Singleton has nothing to disclose. R.A. Barker has served as an advisor to UCB, Cellino, Sana Biotherapeutics, BlueRock Therapeutics, Oxford Biomedica, LCT, Novo Nordisk, and Fujifilm Cellular Dynamics International. He has received grant support from the Medical Research Council, Wellcome Trust; Parkinson's UK; Cure Parkinson's Trust; Rosetrees Trust; EU; Evelyn Trust; John Black Charitable Trust; and CHDI and is supported by the NIHR Cambridge Biomedical Research Centre (the views expressed are those of the author[s] and not necessarily those of the NIHR or the Department of Health and Social Care). He has received royalties from Wiley and Springer-Nature. K. Kieburtz has received support as consultant from Clintrex LLC, Roche/Genentech, Novartis, and Blackfynn; grant support from the NIH (NINDS, National Center for Advancing Translational Sciences), and Michael J. Fox Foundation; and support from ownership of Clintrex LLC, Hoover Brown LLC, and Safe Therapeutics LLC. C.W. Olanow has served as a consultant to Fuji, Lundbeck, Newron, Teva, UCB, and Zambon. He owns stock in Clintrex, which provides consulting services for AstraZeneca, Acorda/Civitas/Biotie, Blackthorne, Britannia, Cynapsus, Dart, Denali, EMD Serono, Inhibikase, Intec, Ipsen, Jazz, Kyowa‐Kirin, Lundbeck, Michael J. Fox Foundation, Monosol, Neuraly, Neurocrine, Neuroderm, Otsuka/INC, Pfizer, Pharma Two B, PhotoPharmics, Sanofi/Genzyme, Sarepta, Serina, Sunovion, SynAgile, Takeda, Teva, Ultragenyx, US WorldMeds, Vaccinex, Voyager, vTv, and Weston Foundation. He has served on the boards of the Michael J. Fox Foundation, the National Space Biomedical Research Institute, UCB, and Zambon. A. Lozano is a consultant for Medtronic, Boston Scientific, and Abbott. J.H. Kordower is a paid consultant to Clintrex Inc, NsGeneinc, Axovant Inc, Fuji-CDI Inc, Inhibikase Inc, Abbvie, and the Michael J. Fox Foundation. J.M. Cedarbaum is an independent consultant. He is former employee of and shareholder in Biogen. P. Brundin has received commercial support as a consultant from Axial Biotherapeutics, CuraSen, Fujifilm-Cellular Dynamics International, IOS Press Partners, LifeSci Capital LLC, Lundbeck A/S, and Living Cell Technologies LTD. He has received commercial support for grants/research from Lundbeck A/S and Roche. He has ownership interests in Acousort AB and Axial Biotherapeutics and is on the steering committee of the Nilotinib in Parkinson's Disease trial. D.G. Standaert is a member of the faculty of the University of Alabama at Birmingham and is supported by endowment and university funds. Dr. Standaert is an investigator in studies funded by Abbvie, Inc, Avid Radiopharmaceuticals, the American Parkinson Disease Association, the Michael J. Fox Foundation for Parkinson Research, Alabama Department of Commerce, the Department of Defense, and NIH grants P01NS087997, P50NS108675, R25NS079188, P2CHD086851, P30NS047466, and T32NS095775. He has a clinical practice and is compensated for these activities through the University of Alabama Health Services Foundation. In addition, since January 1, 2019, he has served as a consultant for or received honoraria from Axovant Sciences, Inc, Censa Pharmaceuticals, Abbvie Inc, Grey Matter Technologies, Theravance, Inc, the Kennedy Krieger Institute, McGraw Hill Publishers, Sanofi-Aventis, RTI Consultants, Cerevance, Inc, Yale University, and Michigan State University. A.E. Lang has served as an advisor for Abbvie, Allon Therapeutics, Avanir Pharmaceuticals, Biogen Idec, Boerhinger-Ingelheim, Ceregene, Lilly, Medtronic, Merck, Novartis, NeuroPhage Pharmaceuticals, Teva, and UCB; received honoraria from Medtronic, Teva, UCB, AbbVie; received grants from Brain Canada, CIHR, Edmond J. Safra Philanthropic Foundation, Michael J. Fox Foundation, the Ontario Brain Institute, National Parkinson Foundation, Parkinson Society Canada, Tourette Syndrome Association, and W. Garfield Weston Foundation; received publishing royalties from Saunders, Wiley-Blackwell, Johns Hopkins Press, and Cambridge University Press; and has served as an expert witness in cases related to the welding industry. Go to Neurology.org/N for full disclosures.
References
- 1.Fujita KA, Ostaszewski M, Matsuoka Y, et al. Integrating pathways of Parkinson's disease in a molecular interaction map. Mol Neurobiol 2014;49:88–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Simuni T, Caspell-Garcia C, Coffey C, Lasch S, Tanner C, Marek K. How stable are Parkinson's disease subtypes in de novo patients: analysis of the PPMI cohort? Parkinsonism Relat Disord 2016;28:62–67. [DOI] [PubMed] [Google Scholar]
- 3.Greenland JC, Williams-Gray CH, Barker RA. The clinical heterogeneity of Parkinson's disease and its therapeutic implications. Eur J Neurosci 2019;49:328–338. [DOI] [PubMed] [Google Scholar]
- 4.Marras C, Lang A. Parkinson's disease subtypes: lost in translation? J Neurol Neurosurg Psychiatry 2013;84:409–415. [DOI] [PubMed] [Google Scholar]
- 5.Mestre TA, Eberly S, Tanner C, et al. Reproducibility of data-driven Parkinson's disease subtypes for clinical research. Parkinsonism Relat Disord 2018;56:102–106. [DOI] [PubMed] [Google Scholar]
- 6.De Pablo-Fernandez E, Lees AJ, Holton JL, Warner TT. Prognosis and neuropathologic correlation of clinical subtypes of Parkinson disease. JAMA Neurol 2019;76:470–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Espay AJ, Marras C. Clinical Parkinson disease subtyping does not predict pathology. Nat Rev Neurol 2019;15:189–190. [DOI] [PubMed] [Google Scholar]
- 8.Espay AJ, Schwarzschild MA, Tanner CM, et al. Biomarker-driven phenotyping in Parkinson's disease: a translational missing link in disease-modifying clinical trials. Mov Disord 2017;32:319–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Accurso FJ, Rowe SM, Clancy JP, et al. Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N Engl J Med 2010;363:1991–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ramsey BW, Davies J, McElvaney NG, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med 2011;365:1663–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Veronesi U, Saccozzi R, Del Vecchio M, et al. Comparing radical mastectomy with quadrantectomy, axillary dissection, and radiotherapy in patients with small cancers of the breast. N Engl J Med 1981;305:6–11. [DOI] [PubMed] [Google Scholar]
- 12.Veronesi U, Cascinelli N, Mariani L, et al. Twenty-year follow-up of a randomized study comparing breast-conserving surgery with radical mastectomy for early breast cancer. N Engl J Med 2002;347:1227–1232. [DOI] [PubMed] [Google Scholar]
- 13.Early Breast Cancer Trialists' Collaborative Group. Tamoxifen for early breast cancer: an overview of the randomised trials. Lancet 1998;351:1451–1467. [PubMed] [Google Scholar]
- 14.Pegram MD, Pauletti G, Slamon DJ. HER-2/neu as a predictive marker of response to breast cancer therapy. Breast Cancer Res Treat 1998;52:65–77. [DOI] [PubMed] [Google Scholar]
- 15.Malhotra GK, Zhao X, Band H, Band V. Histological, molecular and functional subtypes of breast cancers. Cancer Biol Ther 2010;10:955–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parkkinen L, O'Sullivan SS, Collins C, et al. Disentangling the relationship between Lewy bodies and nigral neuronal loss in Parkinson's disease. J Parkinsons Dis 2011;1:277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Patterson L, Rushton SP, Attems J, Thomas AJ, Morris CM. Degeneration of dopaminergic circuitry influences depressive symptoms in Lewy body disorders. Brain Pathol 2019;29:544–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kalia LV, Lang AE, Hazrati LN, et al. Clinical correlations with Lewy body pathology in LRRK2-related Parkinson disease. JAMA Neurol 2015;72:100–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Irwin DJ, Grossman M, Weintraub D, et al. Neuropathological and genetic correlates of survival and dementia onset in synucleinopathies: a retrospective analysis. Lancet Neurol 2017;16:55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buchman AS, Yu L, Wilson RS, et al. Progressive parkinsonism in older adults is related to the burden of mixed brain pathologies. Neurology 2019;92:e1821–e1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Visanji NP, Lang AE, Kovacs GG. Beyond the synucleinopathies: alpha synuclein as a driving force in neurodegenerative comorbidities. Translational neurodegeneration 2019;8:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Espay AJ, Vizcarra JA, Marsili L, et al. Revisiting protein aggregation as pathogenic in sporadic Parkinson and Alzheimer diseases. Neurology 2019;92:329–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De Franceschi G, Fecchio C, Sharon R, et al. Alpha-synuclein structural features inhibit harmful polyunsaturated fatty acid oxidation, suggesting roles in neuroprotection. J Biol Chem 2017;292:6927–6937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shen N, Song G, Yang H, et al. Identifying the pathological domain of alpha- synuclein as a therapeutic for Parkinson's disease. Int J Mol Sci 2019;20:E2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kishimoto Y, Zhu W, Hosoda W, Sen JM, Mattson MP. Chronic mild gut inflammation accelerates brain neuropathology and motor dysfunction in alpha-synuclein mutant mice. Neuromolecular Med 2019;21:239–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen SG, Stribinskis V, Rane MJ, et al. Exposure to the functional bacterial amyloid protein curli enhances alpha-synuclein aggregation in aged Fischer 344 rats and Caenorhabditis elegans. Sci Rep 2016;6:34477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nicoll JAR, Buckland GR, Harrison CH, et al. Persistent neuropathological effects 14 years following amyloid-beta immunization in Alzheimer's disease. Brain 2019;142:2113–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 2003;24:197–211. [DOI] [PubMed] [Google Scholar]
- 29.Visanji NP, Brooks PL, Hazrati LN, Lang AE. The prion hypothesis in Parkinson's disease: Braak to the future. Acta Neuropathol Commun 2013;1:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luk KC, Lee VM. Modeling Lewy pathology propagation in Parkinson's disease. Parkinsonism Relat Disord 2014;20(suppl 1):S85–S87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burke RE, Dauer WT, Vonsattel JP. A critical evaluation of the Braak staging scheme for Parkinson's disease. Ann Neurol 2008;64:485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nuber S, Petrasch-Parwez E, Winner B, et al. Neurodegeneration and motor dysfunction in a conditional model of Parkinson's disease. J Neurosci 2008;28:2471–2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berg D, Postuma RB, Adler CH, et al. MDS research criteria for prodromal Parkinson's disease. Mov Disord 2015;30:1600–1611. [DOI] [PubMed] [Google Scholar]
- 34.Simuni T, Siderowf A, Lasch S, et al. Longitudinal change of clinical and biological measures in early Parkinson's disease: Parkinson's progression markers initiative cohort. Mov Disord 2018;33:771–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Postuma RB, Iranzo A, Hu M, et al. Risk and predictors of dementia and parkinsonism in idiopathic REM sleep behaviour disorder: a multicentre study. Brain 2019;142:744–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Khera AV, Chaffin M, Aragam KG, et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet 2018;50:1219–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nalls MA, McLean CY, Rick J, et al. Diagnosis of Parkinson's disease on the basis of clinical and genetic classification: a population-based modelling study. Lancet Neurol 2015;14:1002–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Collins LM, Williams-Gray CH. The genetic basis of cognitive impairment and dementia in Parkinson's disease. Front Psychiatry 2016;7:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gan-Or Z, Liong C, Alcalay RN. GBA-associated Parkinson's disease and other synucleinopathies. Curr Neurol Neurosci Rep 2018;18:44. [DOI] [PubMed] [Google Scholar]
- 40.Gan-Or Z, Amshalom I, Kilarski LL, et al. Differential effects of severe vs mild GBA mutations on Parkinson disease. Neurology 2015;84:880–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cilia R, Tunesi S, Marotta G, et al. Survival and dementia in GBA-associated Parkinson's disease: the mutation matters. Ann Neurol 2016;80:662–673. [DOI] [PubMed] [Google Scholar]
- 42.Alcalay RN, Levy OA, Waters CC, et al. Glucocerebrosidase activity in Parkinson's disease with and without GBA mutations. Brain 2015;138:2648–2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Di Maio R, Hoffman EK, Rocha EM, et al. LRRK2 activation in idiopathic Parkinson's disease. Sci Transl Med 2018;10:eaar5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ferguson FM, Gray NS. Kinase inhibitors: the road ahead. Nat Rev Drug Discov 2018;17:353–377. [DOI] [PubMed] [Google Scholar]
- 45.Sawyers CL. Opportunities and challenges in the development of kinase inhibitor therapy for cancer. Genes Dev 2003;17:2998–3010. [DOI] [PubMed] [Google Scholar]
- 46.Qin XY, Zhang SP, Cao C, Loh YP, Cheng Y. Aberrations in peripheral inflammatory cytokine levels in Parkinson disease: a systematic review and meta-analysis. JAMA Neurol 2016;73:1316–1324. [DOI] [PubMed] [Google Scholar]
- 47.Williams-Gray CH, Wijeyekoon R, Yarnall AJ, et al. Serum immune markers and disease progression in an incident Parkinson's disease cohort (ICICLE-PD). Mov Disord 2016;31:995–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Falony G, Joossens M, Vieira-Silva S, et al. Population-level analysis of gut microbiome variation. Science 2016;352:560–564. [DOI] [PubMed] [Google Scholar]
- 49.Hill-Burns EM, Debelius JW, Morton JT, et al. Parkinson's disease and Parkinson's disease medications have distinct signatures of the gut microbiome. Mov Disord 2017;32:739–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ren L, Yi J, Yang J, Li P, Cheng X, Mao P. Nonsteroidal anti-inflammatory drugs use and risk of Parkinson disease: a dose-response meta-analysis. Medicine 2018;97:e12172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lang AE, Espay AJ. Disease modification in Parkinson's disease: current approaches, challenges, and future considerations. Mov Disord 2018;33:660–677. [DOI] [PubMed] [Google Scholar]
- 52.Johnson ME, Stecher B, Labrie V, Brundin L, Brundin P. Triggers, facilitators, and aggravators: redefining Parkinson's disease pathogenesis. Trends Neurosci 2019;42:4–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stephenson D, Perry D, Bens C, et al. Charting a path toward combination therapy for Alzheimer's disease. Expert Rev Neurother 2015;15:107–113. [DOI] [PubMed] [Google Scholar]
- 54.Cedarbaum JM. Elephants, Parkinson's disease, and proof-of-concept clinical trials. Mov Disord 2018;33:697–700. [DOI] [PubMed] [Google Scholar]
- 55.Messmer MF, Wilhelm EE, Shoulson I. I-SPY 2 breast cancer trial as a model for innovation in Alzheimer disease therapies. JAMA Neurol 2017;74:1027–1028. [DOI] [PubMed] [Google Scholar]
- 56.Bateman RJ, Benzinger TL, Berry S, et al. The DIAN-TU Next Generation Alzheimer's Prevention Trial: adaptive design and disease progression model. Alzheimers Demen 2017;13:8–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ritchie CW, Molinuevo JL, Truyen L, Satlin A, Van der Geyten S, Lovestone S. Development of interventions for the secondary prevention of Alzheimer's dementia: the European Prevention of Alzheimer's Dementia (EPAD) project. Lancet Psychiatry 2016;3:179–186. [DOI] [PubMed] [Google Scholar]
- 58.Perry D, Sperling R, Katz R, et al. Building a roadmap for developing combination therapies for Alzheimer's disease. Expert Rev Neurother 2015;15:327–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Minikel EV. How pharmaceutical industry financial modelers think about your rare disease [online]. Available at: cureffi.org/2019/04/29/financial-modeling-in-rare-disease/. Accessed June 22, 2019. [Google Scholar]
- 60.Kairalla JA, Coffey CS, Thomann MA, Muller KE. Adaptive trial designs: a review of barriers and opportunities. Trials 2012;13:145. [DOI] [PMC free article] [PubMed] [Google Scholar]