

LEDGF/p75 differentially regulates viral transcription during HIV latency and proviral reactivation.
Abstract
Transcriptional status determines the HIV replicative state in infected patients. However, the transcriptional mechanisms for proviral replication control remain unclear. In this study, we show that, apart from its function in HIV integration, LEDGF/p75 differentially regulates HIV transcription in latency and proviral reactivation. During latency, LEDGF/p75 suppresses proviral transcription via promoter-proximal pausing of RNA polymerase II (Pol II) by recruiting PAF1 complex to the provirus. Following latency reversal, MLL1 complex competitively displaces PAF1 from the provirus through casein kinase II (CKII)–dependent association with LEDGF/p75. Depleting or pharmacologically inhibiting CKII prevents PAF1 dissociation and abrogates the recruitment of both MLL1 and Super Elongation Complex (SEC) to the provirus, thereby impairing transcriptional reactivation for latency reversal. These findings, therefore, provide a mechanistic understanding of how LEDGF/p75 coordinates its distinct regulatory functions at different stages of the post-integrated HIV life cycles. Targeting these mechanisms may have a therapeutic potential to eradicate HIV infection.
INTRODUCTION
Development of therapeutics for eradicating latent provirus in infected CD4+ T cells and myeloid cells is the ultimate goal in HIV research. Currently, two paradoxical yet promising therapeutic strategies have been proposed. The “shock and kill” approach aims to purge the latent reservoir by pharmacologically reactivating proviral transcription, leading to clearance of the infected cells by cytolysis and the patient’s immune system. The “block and lock” strategy intends to fully suppress proviral transcription and keep the latent reservoirs permanently in a deep dormant state (1). However, the regulatory mechanisms underlying proviral transcription are poorly understood, which impedes the targeted approaches using either strategy to cure HIV infection.
HIV latent infection features a silent proviral transcription, characterized by RNA polymerase II (Pol II) pausing, whereas productive infection is marked by active transcription in infected cells (2). Clearance of Pol II pausing is indispensable for processive transcriptional elongation of proviral genes during latency reversal (3). However, the molecular mechanisms underlying the establishment and maintenance of Pol II pausing and how it is overcome for productive proviral transcription remain elusive. The viral protein Tat alleviates Pol II pausing at long terminal repeat (LTR) by recruiting super elongation complex (SEC), which catalyzes the serine-2 phosphorylation in the C-terminal domain of the largest subunit of Pol II (4, 5). However, the role of epigenetic status in Tat-dependent SEC recruitment and Pol II pausing release from chromatinized viral promoter during HIV reactivation is still elusive.
Lens epithelium-derived growth factor (LEDGF)/p75 is a chromatin-associated factor that directs the selective integration of HIV virus into transcription units of active genes by binding to the HIV integrase (6). Recent studies have revealed an unexpected role for LEDGF/p75 in HIV quiescence through repressing proviral transcription in latently infected cells (7). However, the molecular mechanisms behind its regulatory function in proviral transcription remain largely unknown. Through its integrase binding domain (IBD), LEDGF/p75 serves different roles depending on the cellular partners it binds (8). For example, LEDGF/p75 contributes to both transcriptional activation and repression of the Hox genes by facilitating differential recruitment of the trithorax and polycomb proteins in leukemic and mouse embryonic fibroblast cells (9–11). The multiple functions of LEDGF/p75 indicated that it may be involved in proviral transcription at different steps of the integrated HIV life cycle as well.
Here, we uncovered the dual functions of LEDGF/p75 in viral gene transcription during HIV latency and proviral reactivation. Mechanistically, LEDGF/p75 associates with and recruits Pol II-associated factor 1 (PAF1) to maintain Pol II pausing on proviral LTR, thereby promoting HIV latency. When HIV provirus is induced for latency reversal, mixed-lineage leukaemia 1 (MLL1) competitively displaces PAF1 from the integrated LTR through casein kinase II (CKII)–dependent phosphorylation of integrase binding motif (IBM) on MLL1. MLL1 replacement of PAF1 at the integrated HIV LTR not only clears the barrier for Pol II elongation but also facilitates SEC recruitment to promote the release of paused Pol II into viral genes, which collectively lead to the productive proviral transcription during latency reversal. Targeting the molecular mechanisms responsible for the regulatory function of LEDGF/p75 in HIV proviral transcription may provide potential therapeutic approaches for execution of either the shock and kill or the block and lock concept to eradicate HIV infection.
RESULTS
LEDGF/p75 has opposing roles in viral gene transcription during HIV latency and proviral reactivation
Recent studies have revealed an unexpected yet important role for LEDGF/p75 in postintegrative silencing of HIV-1 gene expression in latently infected cells (7). To test whether these findings hold true in separate cell models of HIV latency and determine the molecular mechanisms underlying its function in proviral transcription, we depleted LEDGF/p75 expression in the widely used E4 clone of Jurkat T cells that carries a single copy of latent HIV provirus at the CD2AP intron (Fig. 1A and fig. S1A) (12). Consistent with the previous report, both our flow cytometry and quantitative reverse transcription polymerase chain reaction (RT-qPCR) analyses noted a moderate but significant transcriptional reactivation of the latent provirus at days 6 and 9 following LEDGF/p75 depletion (Fig. 1, B and C). The suppressive effect of LEDGF/p75 on proviral transcription is irrelevant to the viral integration site as transcriptional reactivation was also observed in another 2D10 cell model of HIV latency (fig. S1, B to D), in which the second exon of the MSRB1 gene is integrated with the provirus that contains H13L variant of Tat to promote proviral latency by attenuating its affinity for cyclin T1 (fig. S1A) (12, 13). Transcriptional reactivation of the HIV gene upon LEDGF/p75 depletion in distinct cell models of HIV latency indicates that it functions as a maintenance factor for proviral latency.
Fig. 1. LEDGF/p75 is required for proviral transcription during HIV latency and proviral reactivation.
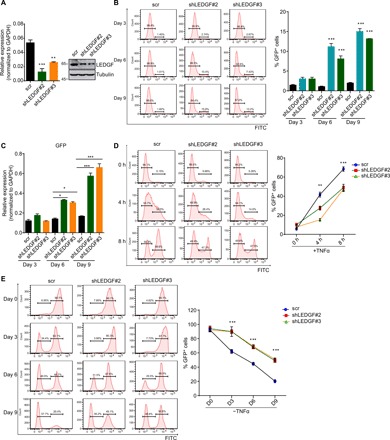
(A) RT-qPCR and Western blot analysis of the knockdown efficiency of LEDGF/p75 short hairpin RNAs (shRNAs) in E4 cells 72 hours after lentiviral transduction. Data are represented as means ± SD (n = 3). (B) Flow cytometry analysis of GFP+ cells at different time points post-LEDGF/p75 depletion in E4 cells. A representative flow histogram and quantification of GFP+ cells (means ± SD; n = 3) are shown. FITC, fluorescein isothiocyanate. (C) RT-qPCR analysis of GFP transcript level in E4 cells depleted for LEDGF/p75 for different days. Transcript was normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and shown as means + SD (n = 3). (D) Effect of LEDGF/p75 depletion on HIV proviral reactivation by flow cytometry analysis. A representative flow histogram of HIV proviral reactivation and their quantification (means ± SD; n = 2) are shown. (E) LEDGF/p75 deficiency restrains HIV reentry into latency. Cells were fully activated with TNFα overnight, and the percentage of GFP+ cells was monitored by flow cytometry at indicated time points after TNFα withdrawal. The percentage of GFP+ cells was quantitated and shown as means + SD (n = 2). *P < 0.05, **P < 0.01, and ***P < 0.001.
LEDGF/p75 is a chromatin-associated protein that primarily targets transcriptionally active regions (14). It acts as a transcriptional coactivator and is implicated in both normal and disease development (11, 15). To explore whether LEDGF/p75 is involved in transcriptional reactivation of the HIV gene during latency reversal, we knocked down LEDGF/p75 expression in E4 and 2D10 cells and activated them to induce proviral reactivation with tumor necrosis factor–α (TNFα). Flow cytometry and RT-qPCR analyses revealed fewer green fluorescent protein–positive (GFP+) cells and lower levels of GFP transcript in LEDGF/p75-depleted cells than in control cells at different time points after induction (Fig. 1D and fig. S1E), demonstrating that robust HIV transcription during proviral reactivation necessitates LEDGF/p75. Latently infected cells require continuous activation signals to sustain viral transcription for active replication, and the replicative provirus will reenter dormancy progressively once the activation signals are withdrawn (12). We found that both the rate and extent that activated provirus reverts to latency are markedly reduced when the stimuli are removed, pointing to a crucial role for LEDGF/p75 in HIV latency establishment as well (Fig. 1E and fig. S1F). Our findings are consistent with a previous study, which reported that LEDGF/p75 renders differential responses of HIV to latency reversing agents by selectively integrating HIV virus (16). Together, these data not only validated the vital role for LEDGF/p75 in the establishment and maintenance of HIV dormancy but also revealed its critical function in proviral transcriptional activation during latency reversal.
LEDGF/p75-associated PAF1 complex establishes and maintains HIV proviral latency
LEDGF/p75 is implicated in multiple cellular and biological processes, including splicing, MLL-rearranged leukemogenesis, and stress response through physical association with various proteins (8, 11, 17). To elucidate the underlying molecular mechanisms with which LEDGF/p75 suppresses HIV transcription for latent infection, cellular factors were affinity purified in human embryonic kidney (HEK) 293FRT cells to identify proteins specifically interacting with LEDGF/p75 and distinct fragments of its associated MLL2 protein (Fig. 2A). In addition to the previously reported imitation-switch 1 (ISW 1) (7), multidimensional protein identification technology (MudPIT) analysis also revealed a specific interaction of LEDGF/p75 with most components of PAF1 complex, which is composed of PAF1, cell division cycle 73 (CDC73), Cln3-requiring 9 (Ctr9), LEO1, and restores TBP function 1 (RTF1) subunits (Fig. 2B). This interaction appears direct and bona fide as it occurs for both the exogenously expressed LEDGF/p75 and PAF1 in bacteria and HEK293T cells, as well as for the endogenous LEDGF/p75 and PAF1 complex in the HIV latently infected E4 cells (Fig. 2, C and D, and fig. S2A). Further truncation analysis showed that the IBD domain of LEDGF/p75 and the C-terminal region of PAF1 are required for mediating their association (fig. S2, B and C).
Fig. 2. PAF1 complex associates with LEDGF/p75 and is required for both the establishment and maintenance of HIV latency.
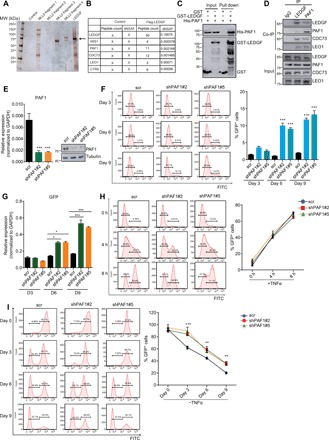
(A) Visualization of factors associating with LEDGF/p75 in silver stain gel. Arrow denotes the Flag-tagged LEDGF/p75 in the silver stain gel. MW, molecular weight. (B) Identification of factors associating with LEDGF/p75 by MudPIT analysis. Normalized spectral abundance factors (NSAF) are shown for the candidate factors. (C) Direct interaction between PAF1 and LEDGF/p75 demonstrated by glutathione S-transferase (GST) pull-down assay. Both input and pull-down samples were subjected to immunoblotting with indicated antibodies. (D) Association between the endogenous LEDGF/p75 and PAF1 complex in E4 cells revealed by reciprocal coimmunoprecipitation (Co-IP) assay. IgG, immunoglobulin G. (E) PAF1 transcript and protein level in E4 cells were assessed by RT-qPCR and Western blot 72 hours after PAF1 depletion with two independent shRNAs. PAF1 mRNA level was normalized to GAPDH and represented as means + SD (n = 3). (F) Effect of PAF1 knockdown on HIV latency by flow cytometric analysis. A representative flow histogram and quantification of GFP+ cells (means + SD; n = 3) were shown. (G) GFP transcript level in E4 cells depleted for PAF1 for different number of days was determined by RT-qPCR and shown as means + SD (n = 3). (H) Representative flow histograms and quantification of GFP+ cells after distinct hours of TNFα stimulation in E4 cells depleted for PAF1 are shown as means + SD (n = 3). (I) Control and PAF1 knockdown 2D10 cells were fully activated by overnight stimulation with TNFα, and percentage of GFP+ cells was monitored by flow cytometry at indicated time points after TNFα removal. Quantitation is shown as means + SD (n = 3). *P < 0.05, **P < 0.01, and ***P < 0.001
PAF1, which was initially identified as an elongation factor to promote Pol II transcription in yeast, was subsequently found to participate in the regulation of all stages of Pol II transcriptional cycle in various organisms (18, 19). The physical interaction between LEDGF/p75 and PAF1 prompted us to test a potential regulatory role for PAF1 in control of HIV transcription during proviral latency and reactivation. Both our flow cytometry and RT-qPCR analyses showed more GFP+ cells and higher levels of GFP transcript in PAF1-depleted cells than in control E4 cells (Fig. 2, E to G). This phenomenon was also noted in 2D10 cells (fig. S2, D to F), which together indicate that PAF1 functions like its associated LEDGF/p75 to suppress proviral transcription to sustain HIV latency. We next examined proviral transcription in latently infected cells stimulated to reverse proviral latency and found that the percentage of GFP+ cells and the GFP transcript level were not affected by PAF1 depletion (Fig. 2H and fig. S2G), demonstrating that PAF1 is dispensable for proviral transcription during HIV latency reversal. Intriguingly, we found that the rate and extent for reversion of the activated provirus to latency were markedly decreased in the absence of PAF1 (Fig. 2I and fig. S2H), which is reminiscent of the critical role for LEDGF/p75 in latency reentry (Fig. 1E and fig. S1F). Together, these results demonstrate that the LEDGF/p75-associated PAF1 complex also participates in the establishment and maintenance of HIV latency through suppressing proviral transcription.
Codependence between LEDGF/p75 and PAF1 complex to silence proviral transcription for the establishment and maintenance of HIV latency
The increased population of GFP+ cells and elevated level of GFP transcript in the latently infected cells lacking either LEDGF/p75 or PAF1 imply their direct roles in restricting the transcriptional activity of HIV LTR. Our luciferase reporter assay revealed that forced expression of either LEDGF/p75 or PAF1 attenuates both basal and Tat-induced activity of HIV LTR in a dosage-dependent manner (Fig. 3A and fig. S3A). In addition, we observed a synergistic inhibitory effect on HIV LTR activity when LEDGF/p75 and PAF1 are simultaneously expressed (Fig. 3A and fig. S3A). Conversely, depletion of either LEDGF/p75 or the respective subunit of PAF1 complex like CDC73 and LEO1 augments both the basal and Tat-induced activity of HIV LTR (Fig. 3B and fig. S3, B to G). Nevertheless, LTR activity is not further markedly elevated in the absence of both LEDGF/p75 and PAF1 complex as compared to their individual depletion, suggesting that they act in the same pathway to repress LTR activity (Fig. 3B and fig. S3B). To further confirm the direct and important roles of LEDGF/p75 and PAF1 in suppression of LTR activity during HIV latency, we used the Gal4-tethering system to target them either individually or simultaneously to a luciferase reporter that is driven by Gal4 binding site containing HIV LTR and found that the basal and Tat-induced HIV LTR activity are reduced, in a dose-dependent manner, by expression of Gal4-fused LEDGF/p75 or PAF1 (Fig. 3C and fig. S3, H and I). Moreover, a combined suppressive effect on the basal and Tat-induced HIV LTR activity was also observed in this system when Gal4-fused LEDGF/p75 and PAF1 were simultaneously expressed at a moderate level (Fig. 3C and fig. S3J).
Fig. 3. LEDGF/p75 suppresses proviral transcription during HIV latency by recruiting PAF1 to maintain RNA Pol II pausing at the provirus.
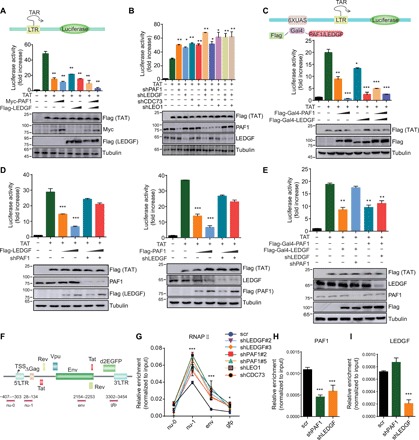
(A) LEDGF/p75 and PAF1 repress Tat-dependent activation of HIV transcription. 293 T cells were transfected with the indicated constructs and the luciferase activity was shown as relative to that in cells only transfected with reporter plasmid (means + SD; n = 3; arbitrarily set to 1). (B) Knockdown of LEDGF/p75 or PAF1 complex elevates the Tat-induced transcriptional activity of the HIV LTR. Luciferase activity in each cellular extract was measured and shown similarly as in (A). (C) Targeting LEDGF/p75 and PAF1 by Gal4-UAS (upstream activation sequence) system suppresses the Tat-induced transcriptional activity of the HIV LTR. (D) Interdependence between LEDGF/p75 and PAF1 on suppression of the Tat-induced transcriptional activity of the HIV LTR. (E) Luciferase reporter assay to examine the activity of Gal4-fused PAF1 or LEDGF/p75 on HIV LTR upon depletion of endogenous LEDGF/p75 or PAF1. (F) Schematic illustration for the location of primers used for the ChIP-qPCR analyses. TSS, transcription start site. (G) ChIP-qPCR analysis of Pol II distribution in E4 cells depleted for the indicated protein for 9 days. Pol II enrichment was normalized to input and shown as means ± SD (n = 3). RNAP, RNA polymerase. (H) PAF1 enrichment at the nu-1 region was determined by ChIP-qPCR, normalized to input, and shown as means + SD (n = 3). (I) LEDGF/p75 enrichment at the nu-1 region was normalized to input and shown as means + SD (n = 3). *P < 0.05, **P < 0.01, and ***P < 0.001.
Given the physical interaction between LEDGF/p75 and PAF1 complex and their similar role in the regulation of HIV LTR activity, we tested whether they are codependent to achieve their inhibitory function in HIV transcription. Although ectopic expression of LEDGF/p75 or PAF1 suppresses HIV LTR activity in a dose-dependent manner, their inhibitory effect was abolished when endogenous PAF1 or LEDGF/p75 was depleted (Fig. 3D), suggesting that an interdependence exists for them to acquire their ability to suppress the LTR activity. To dissect the molecular details underlying their codependence, we measured the impact of endogenous PAF1 and LEDGF/p75 on the suppressive function of either Gal4-fused LEDGF/p75 or PAF1 in HIV LTR activity and found that endogenous PAF1 is indispensable for the repression of LTR activity by Gal4-fused LEDGF/p75, while the suppressive function of Gal4-fused PAF1 is not affected by endogenous LEDGF/p75 depletion (Fig. 3E). The combined data suggest that LEDGF/p75 acts upstream of PAF1 and may help recruit PAF1 to suppress LTR activity during HIV latency.
Transcriptional pausing by Pol II is a major determinant for silencing proviral transcription during HIV latency (20, 21). Recent studies revealed an unexpected role for PAF1 in transcriptional repression of many cellular genes in higher eukaryotes by maintaining Pol II pausing at their promoter-proximal region (22, 23). These findings imply that defects in Pol II pausing may underlie the transcriptional reactivation of the HIV provirus upon depletion of LEDGF/p75 or PAF1 in the latently infected cells. As hypothesized, we observed a remarkable change in the Pol II distribution profile across the HIV genome, with a significant increase of its occupancy at regions encoding env and GFP following depletion of LEDGF/p75 or subunits of PAF1 complex as compared to that in control cells (Fig. 3, F and G). LEDGF/p75 depletion also results in a marked reduction of PAF1 association with LTR, while LEDGF/p75 binding is barely affected by PAF1 depletion in the latently infected cells (Fig. 3, H and I), which is in line with their functional interdependence observed in our luciferase reporter assays. These data, therefore, support a model in which LEDGF/p75 interacts and recruits PAF1 to establish and maintain HIV dormancy by control of Pol II transcriptional pausing in latently infected cells.
LEDGF/p75 recruits MLL1 complex to promote transcriptional reactivation of HIV provirus during latency reversal
We next sought to identify the molecular mechanisms for which LEDGF/p75 activates proviral transcription during HIV latency reversal. LEDGF/p75 is a cofactor of the MLL1 and MLL2 branches of COMPASS (complex proteins associated with Set1)–like complexes and plays a vital role in the activation of transcriptional programs through its recruitment of either complex and the subsequent trimethylation of H3K4 (H3K4me3) (9, 24). In line with the close relationship of H3K4me3 with transcriptional potency, we observed a marked increase of H3K4me3 level at the proviral LTR when latently infected cells were induced to reverse HIV latency (Fig. 4A). To identify the molecular machinery for H3K4me3 implementation at the HIV LTR during latency reversal, we first compared the expression status of the six H3K4 methyltransferases reported in mammalian cells and found that SET1A, SET1B, MLL1, and MLL4 are robustly expressed, while MLL2 and MLL3 are only marginally detectable in the latently HIV-infected cells (Fig. 4B). Intriguingly, only MLL1 depletion, but not ablation of SET1A, SET1B, or MLL4, decreased proviral reactivation (Fig. 4, C and D, and fig. S4, A to D), indicating that MLL1 acts as the predominant H3K4me3 methyltransferase at the proviral LTR and may contribute to its transcriptional activity during latency reversal. Loss of MLL1 expression largely abrogated the increase of H3K4me3 at proviral LTR during latency reversal (Fig. 4E). Unlike LEDGF/p75 and PAF1, MLL1 is dispensable for the maintenance of proviral latency (fig. S4E). However, MLL1 depletion accelerates reentry of the activated provirus into a latent state, further supporting its role in activating proviral transcription during latency reversal (Fig. 4F and fig. S4F).
Fig. 4. LEDGF/p75 recruits MLL1 complex to boost HIV transcription during proviral reactivation.
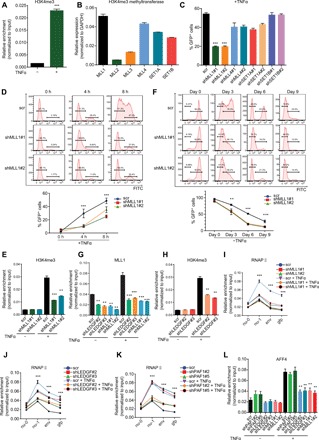
(A) H3K4me3 level at the LTR was normalized to input and shown as means + SD (n = 3). (B) Expression of H3K4 methyltransferases in E4 cells was determined by RT-qPCR and shown as means + SD (n = 3). (C) Flow cytometry to examine viral transcription in E4 cells depleted for indicated H3K4 methyltransferases and quantitation is presented as means + SD (n = 2). (D) Representative flow histograms of proviral reactivation in control- and MLL1- depleted E4 cells stimulated with TFNα for indicated times. Quantitations are shown as means ± SD (n = 2). (E) H3K4me3 level at the LTR in control and MLL1-depleted E4 cells stimulated with TNFα for 4 hours is shown as means + SD (n = 3). (F) Representative flow histograms for the kinetics of latency reversion and their quantitation (means ± SD; n = 3) in control- and MLL1-depleted E4 cells at different time points after TNFα removal are shown. (G) MLL1 occupancy at the LTR in E4 cells depleted for LEDGF/p75 or MLL1 is shown as means + SD (n = 3). (H) H3K4me3 occupancy at the LTR in control- and LEDGF/p75-depleted E4 cells was shown as means + SD (n = 3). (I and J) E4 cells were transduced with shRNAs against MLL1 (I) or LEDGF/p75 (J) and treated with or without TNFα for 4 hours. Pol II distribution over the HIV genome was measured by ChIP-qPCR with the indicated primers and shown as means ± SD (n = 3). (K) Pol II distribution over the proviral genome in control- and PAF1-depelted E4 cells with or without 4 hours of TNFα treatment was shown as means ± SD (n = 3). (L) AFF4 (AF4/FMR2 family member 4) binding at the LTR in E4 cells depleted for the indicated protein and treated with or without TNFα for 4 hours was shown as means ± SD (n = 3). **P < 0.01 and ***P < 0.001.
Considering the physical association of LEDGF/p75 with MLL1 complex, their similar roles in proviral reactivation and a critical role for LEDGF/p75 in targeting MLL1 complex to leukemic genes (10, 11), we then assessed a potential role for LEDGF/p75 in MLL1-dependent proviral transcription during latency reversal. Our chromatin immunoprecipitation–qPCR (ChiP-qPCR) analysis revealed that MLL1 is markedly recruited to the integrated LTR during latency reversal, and loss of LEDGF/p75 largely abolished its recruitment and the subsequent H3K4me3 implementation (Fig. 4, G and H). In line with previous reports (20), we observed Pol II pausing at LTR as a characteristic feature of the integrated HIV provirus in the latently infected cells, and TNFα stimulation induces a marked increase in the amount of Pol II at the promoter-proximal site and downstream coding regions (Fig. 4I), indicating that both transcriptional initiation and elongation are elevated to allow for processive proviral transcription during latency reversal. However, TNFα-induced increase of Pol II initiation and elongation on the HIV provirus was reduced when the expression of MLL1 or LEDGF/p75 was depleted (Fig. 4, I and J). Loss of PAF1 expression barely affects the increased initiation and elongation of Pol II at the provirus during latency reversal, which is consistent with its dispensable role during this process (Fig. 4K and Fig. 2H). Different from the depletion of LEDGF/p75 or PAF1 in unstimulated cells, Pol II pausing at proviral LTR remains unchanged in the absence of MLL1, further supporting it being unnecessary for the maintenance of HIV latency (fig. S4E).
Productive transcription of HIV provirus during latency reversal requires the disruption of Pol II pausing at LTR and its rapid entry into the downstream transcribing region, which is partially accomplished through SEC-mediated serine 2 phosphorylation of the C-terminal domain of the largest subunit of Pol II (4, 5, 25). As expected, we noticed a remarkable increase of SEC recruitment to the proviral LTR after TNFα stimulation of the latently infected cells. However, the recruitment of SEC was largely compromised when either LEDGF/p75 or MLL1 was depleted (Fig. 4L and fig. S4G), suggesting that defects in transcriptional elongation by Pol II and the inefficient proviral transcription of the provirus are at least, in part, resulting from the compromised SEC recruitment. Together, these data suggest a cascade that underlies proviral transcriptional activation during HIV latency reversal: LEDGF/p75 associates and recruits MLL1 complex to implement the H3K4me3 epigenetic mark and therefore promotes SEC binding at the proviral LTR, which, in turn, stimulates the initiation and elongation of Pol II and proviral transcription.
MLL1 phosphorylation by CKII empowers its competition with PAF1 for binding to LEDGF/p75 and proviral LTR
Our protein truncation analysis revealed that PAF1 associates with the IBD of LEDGF/p75, which overlaps with the binding site of MLL1 reported previously (fig. S2B) (26, 27). These findings suggest that MLL1 may compete with PAF1 for binding to LEDGF/p75 during HIV latency reversal. To test this assumption, we first compared their interaction with LEDGF/p75 in HIV latency and proviral reactivation and found that these interactions are dynamic, with a marked reduction or increase of interaction between LEDGF/p75 and PAF1 or MLL1 during the transition from HIV latency to proviral reactivation (Fig. 5A and fig. S5A). Forced expression of MLL1 dissociates PAF1 binding from LEDGF/p75 and vice versa (Fig. 5B and fig. S5B). Moreover, deletion of the IBD motif in LEDGF/p75 disrupted its association with both MLL1 and PAF1 to the basal level and abolished its increased binding to MLL1 during HIV latency reversal (Fig. 5C and fig. S5C). Since Pol II pausing at proviral LTR needs to be resolved to allow for processive transcriptional elongation, we examined PAF1 binding at proviral LTR and found that its level is significantly decreased when latently infected cells were activated to induce latency reversal (Fig. 5D). However, PAF1 dissociation from the proviral LTR was abolished when MLL1 was depleted (Fig. 5D). The level of LEDGF/p75 on the proviral LTR remains constant, and PAF1 loss does not affect MLL1 recruitment to the LTR during proviral reactivation (Fig. 5E and fig. S5D). Collectively, these results indicate that competitive binding for LEDGF/p75 between MLL1 and PAF1 occurs on the proviral LTR, and MLL1 binding to LEDGF/p75 is indispensible for PAF1 to fall off the HIV provirus to enable transcriptional elongation during latency reversal.
Fig. 5. MLL1 phosphorylation by CSNK2-A2 is required for its competitive association with LEDGF/p75 and recruitment to the proviral LTR.
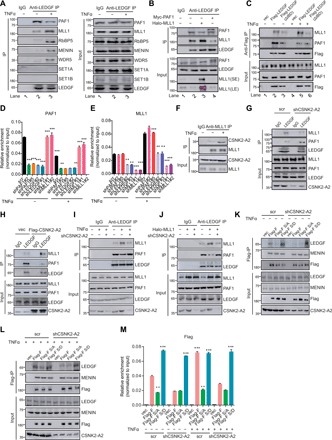
(A) TNFα stimulation decreases PAF1 but enhances MLL’s association with LEDGF/p75 in E4 cells, assessed by immunopreciptation and immunoblotting with the indicated antibodies. (B) PAF1 and MLL1 compete for LEDGF/p75 binding in 293 T cells. (C) The IBD of LEDGF/p75 mediates the increased association of LEDGF/p75 with MLL1 in E4 cells stimulated with TNFα for 4 hours. (D) PAF1 occupancy at the LTR in E4 cells depleted for indicated protein and treated with or without TNFα for 4 hours was normalized to input and shown as means + SD (n = 3). (E) MLL1 occupancy in E4 cells depleted for indicated protein and treated with or without TNFα for 4 hours was determined and shown as in (D). (F) Association of CSNK2-A2 with MLL1 in E4 cells stimulated with or without TNFα for 4 hours. (G) Association of LEDGF/p75 with MLL1 and PAF1 in E4 cells depleted for CSNK2-A2. (H) Association of LEDGF/p75 with MLL1 and PAF1 in E4 cells overexpressing exogenous CSNK2-A2. (I) Association of LEDGF/p75 with MLL1 and PAF1 in control- and CSNK2-A2–depleted E4 cells treated with or without TNFα for 4 hours. (J) Association of LEDGF/p75 with MLL1 and PAF1 in control- and CSNK2-A2– depleted 293 T cells overexpressing Halo-MLL1. (K and L) Lentiviral constructs encoding wild-type, nonphosphomimetic (S136, S142, and S153A), or phosphomimetic (S136, S142, and S153D) deletion mutant (1 to 1406) of MLL1 were transduced into E4 cells that had been depleted for CSNK2-A2 for 3 days. The transduced cells were either untreated (K) or stimulated with TNFα (L) for 4 hours and then used for immunoprecipitation and immunobloting with the indicated antibodies. (M) E4 cells were treated as in (K) and (L), and the level of N-terminal wild-type or mutant MLL1 fragment on HIV LTR was determined by Flag ChIP and shown as means + SD (n = 3). **P < 0.01 and ***P < 0.001. SE, short exposure; LE, long exposure.
As the association of LEDGF/p75 with PAF1 and MLL1 is mutually exclusive, we next sought to elucidate the underlying mechanisms that determine the switch for association of LEDGF/p75 with PAF1 and MLL1 during HIV latency reversal. A recent elegant study revealed, from structural perspective, that the LEDGF/p75 interactome is regulated by phosphorylation of the structurally conserved IBM on its binding partners, including MLL1 (28). The MLL1 IBM is located from amino acids 125 to 155 and contains three serines (serine-136, serine-142, and serine-153) that all can be phosphorylated in vivo based on mass spectrometry datasets integrated in the PhophoSitePlus. Substituting two of the three serine residues with alanine largely abolished the interaction between LEDGF/p75 and MLL in in vitro biochemical studies and alleviated the leukemogenic capacity of MLL1 fusion proteins in mouse hematopoietic stem cells (28). In light of these findings, we reasoned that kinase-dependent phosphorylation of IBM on MLL increases its binding affinity for LEDGF/p75 and competitively displaces PAF1 from the proviral LTR during latency reversal.
To identify kinases for MLL1 IBM phosphorylation, we analyzed the IBM sequence with an online bioinformatic tool, NetPhos 3.1 server and also looked into the MLL1 interactome in a published mass spectrometry dataset (28). CKII stood out as a strong candidate kinase for MLL1 IBM phosphorylation in these two sets of analysis (data not shown). CKII is a heterotetrameric complex composed of two β regulatory subunits and homo- or heterodimers of catalytic subunit α or α′, encoded, respectively, by the CSNK2A1 or CSNK2A2 gene in human cells (29). To confirm and determine which catalytic subunit of CKII is involved in the MLL1 IBM phosphorylation, we first examined their association with MLL1 and observed a specific binding of CSNK2-A2, but not CSNK2-A1, to MLL1 (fig. S5, E and F). The association of MLL1 with CSNK2-A2 is remarkably increased upon TNFα stimulation (Fig. 5F and fig. S5F), suggesting that CSNK2-A2 may be implicated in MLL1-dependent dissociation of PAF1 from LEDGF/p75 and the proviral LTR. The interaction between MLL1 and LEDGF/p75 is markedly decreased, while the association of PAF1 with LEDGF/p75 is elevated in CSNK2-A2–depleted cells as compared to the control (Fig. 5G and fig. S5G). This interaction change was reversed when cells were forced to express exogenous CSNK2-A2 (Fig. 5H). Moreover, the increased or decreased interaction of LEDGF/p75 with MLL1 or PAF1 in cases of HIV latency reversal and ectopic MLL1 expression were largely abrogated when CSNK2-A2 is depleted or inhibited (Fig. 5, I and J, and fig. S5, H and I), indicating CSNK2-A2, together with its kinase activity, are important for the competitive binding of MLL1 to LEDGF/p75.
We next sought to determine whether phosphorylation of the three serine residues in IBM is involved in the CSNK2-A2–dependent competitive association of MLL1 with LEDGF/p75. To this end, we substituted the three residues with alanine (A) or aspartic acid (D) in the N-terminal fragment of MLL1, which was truncated at the chromosomal break point of MLL-AF9 (ALL1 fused gene from chromosome 9) in leukemic cells, to prevent or mimic phosphorylation and expressed these mutants in parallel with the wild-type control into latently HIV-infected cells (fig. S5J). Our coimmunoprecipitation assays revealed that S/A mutations abolished, while S/D substitutions markedly increased the interaction of MLL1 with LEDGF/p75 (Fig. 5K). In a stark contrast to the wild-type MLL1 fragment, activation of latently infected cells failed to induce the enhanced interaction of LEDGF/p75 with the S/A mutant (Fig. 5L). Loss of CSNK2-A2 or pharmacological inhibition of its kinase activity lowered the association of LEDGF/p75 with the wild-type MLL1 fragment to the level comparable to that found in the S/A mutant, while strong association with the S/D mutant remained unaltered in both HIV latency and proviral reactivation (Fig. 5, K and L, and fig. S5, K and L). Furthermore, S/A mutations decreased the basal binding of ectopic MLL1 fragment to the proviral LTR during HIV latency and abolished its further recruitment during proviral reactivation (Fig. 5M). Nevertheless, an exogenous MLL1 fragment with S/D mutations becomes constitutively bound to the proviral LTR at a level much higher than its wild-type counterpart and is not affected by CSNK2-A2 depletion both in HIV latency and proviral reactivation (Fig. 5M). Collectively, these data indicated that phosphorylation of the serine residues in IBM is required for competitive binding of MLL1 to LEDGF/p75 and the proviral LTR during HIV latency reversal.
CKII is important for proviral transcription during HIV latency reversal
The observation that activation of the latently HIV-infected cells promotes the interaction between MLL1 and CSNK2-A2 intrigued us to elucidate the underlying molecular basis (Fig. 5F). We occasionally noted an elevated protein level of CSNK2-A2 during TNFα-induced proviral reactivation (Fig. 5, F and I). This raised the possibility that CSNK2-A2 may serve as one of the direct transcriptional targets of the TNFα signaling pathway. We observed an elevated transcript level of both catalytic subunits of CKII kinase, with a higher induction of CSNK2-A2 than CSNK2-A1, in the latently infected cells stimulated with TNFα (Fig. 6A). The transcription factor nuclear factor κB (NF-κB) is one of the major downstream mediators of the TNFα signaling, and its prototype is heterodimerically composed of RelA/p65 and NF-κB1/p50 subunits, which recognize and bind to the consensus κB sequence (GGGRNNYYCC; R is purine, Y is pyrimidine, and N is any base) present in the promoter or enhancer of their target genes (30). Bioinformatic analysis of the 2000–base pair (bp) DNA sequence upstream of the transcription start site of CSNK2-A1 and CSNK2-A2 by JASPAR, an online tool for predicting the binding site of transcription factors, revealed multiple κB sequence or its variants within these regions (data not shown). As predicted, a basal binding of p50 and p65 was noticed on the promoters of CSNK2-A1 and CSNK2-A2, and their level were further elevated when HIV latently infected cells underwent TNFα stimulation (Fig. 6B and fig. S6A).
Fig. 6. CSNK2-A2 is crucial for productive proviral transcription during latency reversal.
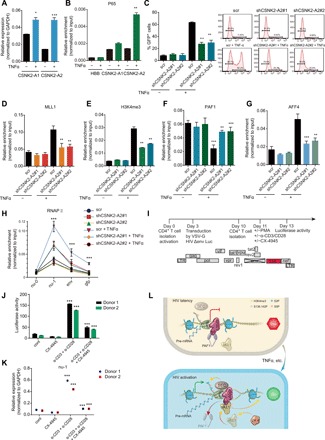
(A) CSNK2-A2 expression (means + SD; n = 3) in E4 cells stimulated with or without TNFα for 4 hours. (B) p65 occupancy at promoters of the CSNK2-A1, CSNK2-A1, and the negative control HBB gene in E4 cells treated with or without TNFα for 8 hours was normalized to input and presented as means + SD (n = 3). (C) The percentage of GFP+ cells in E4 cells depleted for CSNK2-A2 and treated with TNFα for 8 hours was determined by flow cytometry and shown as means + SD (n = 2; left). Representative flow histograms are shown on the right. (D and E) MLL1 (D) and H3K4me3 (E) level at LTR in E4 cells depleted for CSNK2-A2 and treated with TNFα for 8 hours was normalized to input and shown as means + SD (n = 3). (F and G) PAF1 (F) and AFF4 (G) level at LTR in E4 cells depleted for CSNK2-A2 and treated with TNFα for 8 hours was normalized to input and shown as means + SD (n = 3). (H) Pol II distribution across the HIV genome in E4 cells depleted for CSNK2-A2 and treated with TNFα for 8 hours was normalized to input and shown as means + SD (n = 3). (I) Schematic diagram of the NL4.3-deltaEnv-Luc-Nef construct and the experimental outline to analyze HIV gene expression and reactivation in primary CD4+ T cells upon CSNK2-A2 inhibition. (J and K) Primary CD4+ T cells from two donors were latently infected with VSV-G pseudotyped HIV virus and stimulated with or without α-CD3/CD28 antibodies in the presence or absence CX-4945. Luciferase activities (J) and HIV transcript level (K) (means + SD; n = 3) were shown in the indicated treatment conditions. (L) Schematic diagram of the molecular mechanisms underlying the opposing function of LEDGF/p75 in viral transcription during latency and proviral reactivation after HIV integration. *P < 0.05, **P < 0.01, and ***P < 0.001.
As the competitive binding of MLL1 to LEDGF/p75 was CSNK2-A2 dependent, we found that the induction of proviral transcription was impaired by depletion or pharmacological inhibition of CSNK2-A2 (Fig. 6C and fig. S6B). In addition, the recruitment of MLL1, the H3K4me3 deposition, and the PAF1 dissociation from the proviral LTR were all markedly reduced upon loss of CSNK2-A2 or its kinase activity as compared to the controls (Fig. 6, D to F, and fig. S6, C to E). Moreover, SEC recruitment to the proviral LTR was also impaired during latency reversal in cells lacking either expression or kinase activity of CSNK2-A2 (Fig. 6G and fig. S6F). However, the binding of LEDGF/p75 to the proviral LTR was constant and not dependent on CSNK2-A2 or its kinase activity (fig. S6, G and H). A decreased initiation and elongation of Pol II on the HIV provirus were noted during latency reversal when CSNK2-A2 was depleted or pharmacologically inhibited (Fig. 6H and fig. S6I). Hence, these results collectively indicate that CSNK2-A2 and its kinase activity are required for competitive displacement of PAF1 by MLL1 from proviral LTR and revealed its critical role in regulation of transcriptional initiation and elongation of the HIV provirus during latency reversal.
To further assess the roles of CSNK2-A2 during HIV latency reversal, we chose a primary cell model of HIV latency in which CD4+ T cells were infected with the vesicular stomatitis virus glycoprotein (VSVG) pseudotyped NL4.3-deltaEnv-Luc-Nef HIV virus. Because of the existence of Nef and Vpu in the virus, CD4 expression is suppressed in the actively infected cells, whereas the uninfected or latently infected cells continue to express CD4, which can be isolated by positive selection with anti-CD4 conjugated beads (Fig. 6I) (31). We found that inhibition of CSNK2-A2 by CX-4945 markedly reduced the luciferase activities and comprised the proviral transcription during latency reversal of the pseudotyped virus in cells stimulated with either phorbol 12-myristate 13-acetate (PMA) or the combination of anti-CD3 and anti-CD28 monoclonal antibodies (Fig. 6, J and K, and fig. S6, J and K). Thus, CSNK2-A2 and its kinase activity are required for the full reactivation of the latent HIV provirus in primary CD4+ T cells.
DISCUSSION
The present study reveals a previously unidentified paradox role for LEDGF/p75 in transcriptional regulation of the postintegrative HIV latency and proviral reactivation. In case of proviral latency, LEDGF/p75 physically interacts with PAF1 and tethers it to the HIV LTR to establish and maintain Pol II pausing at the viral promoter-proximal region, leading to proviral transcriptional silencing for latent infection. However, when induced to reverse HIV latency, LEDGF/p75 switches its transcriptionally repressive function to act as a transcriptional activator for proviral transcription. Mechanistically, induction of proviral reactivation transcriptionally increases the expression of the CSNK2-A2 kinase, which phosphorylates the IBM on MLL1. Phosphorylated MLL1, competitively displaces PAF1 from the proviral LTR through LEDGF/p75 binding, deposits H3K4me3, and facilitates SEC recruitment to the proviral LTR, which consequently increases the initiation and elongation of Pol II for processive transcription of the integrated HIV provirus during latency reversal (Fig. 6L).
In this study, we observed two contrasting roles for LEDGF/p75 in transcriptional regulation of the postintegrative HIV life cycle: proviral latency and proviral reactivation. HIV proviral latency is concomitant with the transcriptional repression of the viral genome, and multiple cellular factors have been identified to set up and maintain the proviral transcriptional silence state. Although originally identified as a chromatin-associated transcriptional coactivator (32), later functional characterization of LEDGF/p75 revealed its participation in both transcriptional activation and repression of cellular target genes (9, 11). Once integrated into host cells, the HIV viral genes become an integral part of host genome and replicates through cell division. Thus, it is conceivable that HIV genes can behave like cellular genes and hijack the pleiotropic function of LEDGF/p75 to transcriptionally regulate the proviral fate after integration. Our study provides direct evidence that the integrated HIV exploits LEDGF/p75 to establish and maintain proviral latency, and to reverse it, through its transcriptional repressor or activator function, respectively.
Our affinity purification and mass spectrometry study revealed a specific physical interaction between LEDGF/p75 and PAF1 complex. PAF1 was first reported to be important for Tat transactivation through affinity purification and mass spectrometry analysis of the Tat-associated factors in mammalian cells (25). Subsequently, PAF1 complex was shown as a restriction factor for HIV reverse transcription and viral integration (33). Increased expression of PAF1 renders cells refractory to infection by the incoming HIV virus and is strongly associated with the reduced levels of proviral transcripts during antiretroviral therapy (33, 34). In line with PAF1 being a restriction factor, we observed that PAF1 depletion phenocopies the transcriptional reactivation of the HIV provirus in latently infected cells lacking LEDGF/p75 expression, suggesting that it acts as a transcriptional repressor and that a functional codependence may exist between PAF1 and LEDGF/p75 in limiting proviral transcription. Our results clearly demonstrate that PAF1 has inhibitory activity for integrated HIV transcription and its binding to viral LTR relies on LEDGF/p75, while the transcriptionally repressive function of LEDGF/p75 requires its recruitment of PAF1 to the proviral LTR. Although originally isolated as a Pol II elongation factor, recent studies identified that PAF1 can also promote promoter proximal pausing of Pol II and suppress transcription of certain cellular genes in various organisms and cell types (22, 23, 35). In line with these findings, we found that depletion of either LEDGF/p75 or PAF1 led to Pol II escape from promoter proximal regions on the proviral LTR, which may explain the transcriptional reactivation of the HIV gene upon their loss in the latently infected cells. However, it remains to be determined how HIV provirus hijacks the specific transcriptional inhibitory function of PAF1 for HIV latency and how PAF1 prevents release of paused Pol II into the proviral coding region in the latently infected cells.
LEDGF/p75 interacts with the N terminus of wild-type MLL1 or leukemic MLL1 fusion protein both indirectly through association with the Menin subunit of COMPASS complex and directly via its IBD that overlaps with the sites bound by other partners, including PAF1 (11, 26, 27). We found a direct competition between MLL1 and PAF1 for LEDGF/p75 binding, and this competition is crucial for MLL1 recruitment and the subsequent implementation of its enzymatic product H3K4me3 on the proviral LTR and the PAF1 dissociation from the HIV provirus during latency reversal. Our results are in agreement with the previous biochemical and structural findings showing that the cellular and retroviral partners of LEDGF/p75 are in direct competition with each other for binding to its IBD (36, 37).
A recent elegant structural study reported that the LEDGF/p75 protein interaction network is regulated by phosphorylation of the structurally conserved IBMs on LEDGF/p75 binding partners, which controls the interaction affinity between LEDGF/p75 and its binding partners. This elaborate regulatory mechanism allows for switch of various partners bound to LEDGF/p75 and accounts for the pleiotropic function of LEDGF/p75 in distinct cellular processes (28). In support of this notion, we found CSNK2-A2–mediated phosphorylation of IBM on MLL1 is indispensable for its competitive binding to LEDGF/p75, PAF1 dissociation from the proviral LTR, and the efficient transcription of the HIV provirus during latency reversal. However, it is currently unknown why CSNK2-A2, but not its paralog CSNK2-A1, was chosen for MLL1 IBM phosphorylation during HIV latency reversal.
CKII has long been known for its role in transcriptional regulation of the HIV provirus (38), but the mechanisms remain unexplored. We found that CSNK2-A2 regulates the transcriptional initiation and elongation of Pol II on the HIV provirus through facilitating MLL1 recruitment and PAF1 dissociation from the proviral LTR. Therefore, our studies not only prove the regulatory functions of CKII in HIV proviral transcription but also provide a mechanistic understanding of how CKII is coupled to transcriptional regulation of the HIV provirus. CKII has also been previously shown to regulate the transcriptional elongation of cellular genes in yeast and mammals through phosphorylation of Tyr57 on the globular domain of H2A (39). Further studies are required to determine whether Tyr57 phosphorylation of H2A underlies its role in proviral transcription during HIV latency reversal as well.
MATERIALS AND METHODS
Cell culture and reactivation
HEK293T cells were obtained from the American Type Culture Collection and maintained in Dulbecco’s modified Eagle’s medium (DMEM; Biological Industries, 06-1055-57-1ACS) and 10% fetal bovine serum (FBS; Biological Industries, 04-001-1ACS). E4 and 2D10 clones of Jurkat T cells were cultivated in RPMI 1640 (Biological Industries, 01-100-1ACS) supplemented with 10% FBS (Biological Industries, 04-001-1ACS), 2.0 mM l-glutamine, and 1% penicillin/streptomycin.
E4 and 2D10 cells depleted for indicated protein for 3 days were stimulated with TNFα (10 ng/ml) for indicated times. Activation of proviral transcription was examined by flow cytometry or RT-qPCR analyses of d2EGFP expression. For shutdown of active proviral transcription, 2D10 cells were treated with 10 ng/ml overnight to achieve full activation of proviral transcription. Cells were then washed twice with phosphate-buffered saline (PBS) and resuspended in RPMI 1640 supplemented with 10% FBS medium. d2EGFP expression was analyzed at the indicated time points following TNFα withdrawal.
Lentivirus packaging and infection
For preparation of lentivirus, pLKO-based RNA interference constructs or pSin-based expression vectors were cotransfected into HEK293T cells with the psPAX2 and pMD2.G plasmids at a ratio of 2:2:1 by Lipofectamine 2000 (Thermo Fisher Scientific, 11668027) following the manufacturer’s instructions. The virus particles were harvested at 48 and 96 hours after transfection, filtered by 0.45-μM filter unit (Millipore), and then stored at −80°C in aliquots.
Cells were infected with lentivirus in the presence of polybrene (4 μg/ml; Sigma-Aldrich) for 24 hours and selected with puromycin (1 μg/ml) for 48 hours to eliminate nontransduced cells before collection for analysis at indicated time points.
Expression constructs and site-directed mutagenesis
Complementary DNAs (cDNAs) encoding the indicated protein or their truncated variants were cloned into the corresponding eukaryotic or bacterial expression vectors following the standard molecular cloning technique. For site-directed substitution of the three serine residues MLL1 IBM, the N-terminal of MLL1 (amino acids 1 to 1406) harboring desirable mutations were generated through assembly of the two overlapping fragments that were mutated during PCR amplification using Gibson Assembly approach (New England Biolabs, E2611S). Detailed information on vector backbone, PCR primers, and restriction enzymes can be found in the table S1.
Flag purification and MudPIT analyses
LEDGF/p75 was doxycycline inducibly expressed in HEK293FRT cells following the manufacturer’s instructions (Thermo Fisher Scientific, R75007), and the nuclear extract was prepared according to the Dignam method. LEDGF/p75 bound proteins were purified on anti-Flag (M2) agarose beads in the presence of Benzonase (Sigma, E1014), separated on SDS–polyacrylamide gel electrophoresis gel, and visualized by silver staining. Trichloroacetic acid–precipitated protein mixtures from the Flag purifications were digested with endoproteinase Lys-C and trypsin (Roche, 11058533103) and analyzed by MudPIT as previously described (40).
HIV reactivation from latently infected primary CD4+ T cells
Pseudotyped HIV viruses were generated by cotransfection of HEK293T cells with pNL4.3-deltaENV-Luc-2ANef and pCMV-VSV-G, and the supernatant was collected, filtered through a 0.22-μM filter, aliquoted, and stored at −80°C until infection of CD4+ T cells.
Whole blood of anonymous healthy donors was purchased from Tianjin Blood Center, and its use in basic medical research has been approved by the Research Committee at Tianjin Medical University. Peripheral blood mononuclear cells (PBMCs) in whole blood were isolated by a density gradient centrifugation over the Ficoll medium (TBDScience, LTS1077). CD4+ T cells were isolated from PBMCs using negative selection by the EasySep Kit (STEMCELL Technologies, 17952) following the manufacturer’s instructions. Isolated CD4+ T cells were seeded at a density of 1 × 106 per well in 150 μl of RPMI 1640 medium and 10% FBS in a 96-well flat-bottom plate and activated with ImmunoCult Human CD3/CD28 T Cell Activator (STEMCELL Technologies, 10791) and 200 IU of interleukin-2 (Sino Biological, GMP-11848-HNAE) for 72 hours followed by transduction with pseudotyped HIV viruses. At day 10, latently infected and uninfected CD4+ cells were isolated using the EasySep Human CD4 Positive Selection Kit II (STEMCELL Technologies, 17852) and activated with PMA (10 ng/ml) or ImmunoCult Human CD3/CD28 T Cell Activator (STEMCELL, 10970) in the presence or absence of 1 μM CX-4945 for 72 hours. Reactivation of HIV provirus was determined by examining the luciferase activity and the HIV transcript level.
Immunoprecipitation
Cells were washed with ice-cold PBS twice, resuspended in Dignam hypotonic buffer [10 mM Hepes (pH 7.9), 1.5 mM MgCl2, and 10 mM KCl], incubated on ice for 10 min, followed by brief centrifugation for nuclei precipitation. The nuclei were resuspended in radio immunoprecipitation assay (RIPA) buffer [20 mM tris-HCl (pH 7.4), 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS, and 1 mM dithiothreitol], supplemented with 1 mM MgCl2 and proteinase inhibitors (Sigma-Aldrich), and lysed with gentle agitation for 30 min at 4°C. After centrifugation at 13,000 rpm for 30 min, the supernatant was incubated with the Flag agarose (Sigma, A2220) or indicated antibodies and Protein A/G PLUS agarose (Santa Cruz Biotechnology, sc-2003) in the presence of Benzonase (1 U/ml; Novagen, 70664) at 4°C overnight with gentle rotation. The beads were spun down and washed three times with a wash buffer [10 mM Hepes (pH 7.4), 1 mM MgCl2, 300 mM NaCl, 10 mM KCl, and 0.2% Triton X-100) before boiling in SDS loading buffer.
Glutathione S-transferase pull down
Glutathione S-transferase (GST) or GST-LEDGF/p75 proteins were expressed in Rosetta bacteria and immobilized on glutathione-sepharose 4B beads (Sigma, G0924), washed three times with PBS ,and then resuspended in RIPA buffer supplemented with 1 mM MgCl2 and proteinase inhibitors (Sigma-Aldrich). GST or GST-LEDGF/p75 bound beads (1 ml) were incubated for 3 hours at 4°C with His-tagged PAF1 protein, which was expressed in Rosetta and purified with Ni-NTA resins (QIAGEN, 30310) according to the manufacturer’s instructions. Beads were washed with RIPA buffer three times, and the bound proteins were eluted in SDS loading buffer, followed by Western blotting with indicated antibodies.
Flow cytometry
Cells were washed twice with ice-cold PBS, pelleted by centrifugation, resuspended, and then incubated on ice for 10 min in precooled fluorescence-activated cell sorting (FACS) buffer (PBS and 3% FBS) supplemented with of 4′,6-diamidino-2-phenylindole (0.1 μg/ml). d2EGFP fluorescent signal in E4 or 2D10 cells with or without treatment were measured on FACSCalibur flow cytometer (BD Biosciences) and analyzed by FlowJo X software.
Luciferase reporter assay
For luciferase reporter assays, 293 T cells were cotransfected with reporter vector expressing firefly luciferase under the control of HIV LTR or 6XUAS-LTR, which contains six repeats of the Gal4 upstream activation sequence (UAS) inserted upstream of the Sp1-binding site in HIV LTR, LacZ internal control plasmid, and constructs encoding the indicated protein by Lipofectamine 2000. The total amount of transfected plasmids per well of transfection was adjusted to be equal. Thirty-six hours after transfection, cells were harvested and lysed for quantitation of firefly luciferase and LacZ activities on Synergy HT platform (BioTek).
Chromatin immunoprecipitation
ChIP assays were performed as previously described. Briefly, 1 × 107 to 3 × 107 treated or untreated cells were cross-linked with 1% paraformaldehyde at room temperature for 10 min and quenched by glycine. Cells were sonicated or generate chromatin fragments of 200 to 600 bp with a Sonics Vibra-Cell sonicator (Sonics & Materials) followed by immunoprecipitation with the indicated antibodies. The resulting DNA was analyzed using SYBR Green Mix on the CFX connect Real-Time PCR detection System (Bio-Rad) and normalized to input.
Quantitative reverse transcription polymerase chain reaction
Total RNA was isolated using TRIzol, treated with deoxyribonuclease I (New England Biolabs, M0303S), and reverse transcribed using SuperScript III and random primers (Invitrogen). Resulting cDNA was analyzed by quantitative PCR using Green SuperMix for iQ (VVR, 01414-144) on a 7500 Fast Real-Time PCR system (Thermo Fisher Scientific) or on a CFX ConnectTM Real-Time PCR Detection System (Bio-Rad).
Quantitation and statistical analysis
Quantitative data were processed using GraphPad Prism software and represented as means ± SD, with Student’s t test used to determine the statistical significance for indicated comparison.
Supplementary Material
Acknowledgments
We are grateful to J. Karn at Case Western Reserve University for providing E4 and 2D10 cell lines and Z. Qiang at University of California, Berkeley for sharing 6XUAS-LTR luciferase reporter construct. We thank the core facility of Tianjin Medical University for the assistance with flow cytometry and F. Tao at Children’s Research Institute for proofreading and language editing. Funding: This study was primarily supported by starting funds of Tianjin Medical University, the National Natural Science Foundation of China (31872825), Tianjin Natural Science Foundation (18JCYBJC42400), funding from the State Key Laboratory of Experimental Hematology (157-Zk19-03 to D.H.), and the CAMS Initiative for Innovative Medicine (2018-I2M-AI-010 to X.G.). Author contributions: D.H. and X.G. conceptualized and supervised the project, analyzed the data, and wrote the manuscript. R.G. performed most of the experiment, analyzed the data, and prepared the figures. J.B. conducted all nonphosphomimetic and phosphomimetic MLL1 mutant-related experiment and GST pull-down assay. Z.X.L. conducted coimmunoprecipitation assay. L.X., S.H., and J.C. expressed and purified TNFα. H.Y. reviewed all original data and drew the working model. B.T. edited the language of the manuscript. W.Q., H.N., and R.Z. provided assistance in molecular cloning and tissue culture. Y.C., K.Z., X.W., and Z.L. provided constructive suggestions for the project. All authors commented on figures and read and edited the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/20/eaaz8411/DC1
REFERENCES AND NOTES
- 1.Abner E., Jordan A., HIV “shock and kill” therapy: In need of revision. Antiviral. Res. 166, 19–34 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Feinberg M. B., Baltimore D., Frankel A. D., The role of Tat in the human immunodeficiency virus life cycle indicates a primary effect on transcriptional elongation. Proc. Natl. Acad. Sci. U.S.A. 88, 4045–4049 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang Z., Klatt A., Gilmour D. S., Henderson A. J., Negative elongation factor NELF represses human immunodeficiency virus transcription by pausing the RNA polymerase II complex. J. Biol. Chem. 282, 16981–16988 (2007). [DOI] [PubMed] [Google Scholar]
- 4.He N., Liu M., Hsu J., Xue Y., Chou S., Burlingame A., Krogan N. J., Alber T., Zhou Q., HIV-1 Tat and host AFF4 recruit two transcription elongation factors into a bifunctional complex for coordinated activation of HIV-1 transcription. Mol. Cell 38, 428–438 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lin C., Smith E. R., Takahashi H., Lai K. C., Martin-Brown S., Florens L., Washburn M. P., Conaway J. W., Conaway R. C., Shilatifard A., AFF4, a component of the ELL/P-TEFb elongation complex and a shared subunit of MLL chimeras, can link transcription elongation to leukemia. Mol. Cell 37, 429–437 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ciuffi A., Llano M., Poeschla E., Hoffmann C., Leipzig J., Shinn P., Ecker J. R., Bushman F., A role for LEDGF/p75 in targeting HIV DNA integration. Nat. Med. 11, 1287–1289 (2005). [DOI] [PubMed] [Google Scholar]
- 7.Gerard A., Ségéral E., Naughtin M., Abdouni A., Charmeteau B., Cheynier R., Rain J. C., Emiliani S., The integrase cofactor LEDGF/p75 associates with Iws1 and Spt6 for postintegration silencing of HIV-1 gene expression in latently infected cells. Cell Host Microbe 17, 107–117 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Singh P. K., Plumb M. R., Ferris A. L., Iben J. R., Wu X., Fadel H. J., Luke B. T., Esnault C., Poeschla E. M., Hughes S. H., Kvaratskhelia M., Levin H. L., LEDGF/p75 interacts with mRNA splicing factors and targets HIV-1 integration to highly spliced genes. Genes Dev. 29, 2287–2297 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pradeepa M. M., Grimes G. R., Taylor G. C., Sutherland H. G., Bickmore W. A., Psip1/Ledgf p75 restrains Hox gene expression by recruiting both trithorax and polycomb group proteins. Nucleic Acids Res. 42, 9021–9032 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.El Ashkar S., Schwaller J., Pieters T., Goossens S., Demeulemeester J., Christ F., Van Belle S., Juge S., Boeckx N., Engelman A., Van Vlierberghe P., Debyser Z., De Rijck J., LEDGF/p75 is dispensable for hematopoiesis but essential for MLL-rearranged leukemogenesis. Blood 131, 95–107 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yokoyama A., Cleary M. L., Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer Cell 14, 36–46 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pearson R., Kim Y. K., Hokello J., Lassen K., Friedman J., Tyagi M., Karn J., Epigenetic silencing of human immunodeficiency virus (HIV) transcription by formation of restrictive chromatin structures at the viral long terminal repeat drives the progressive entry of HIV into latency. J. Virol. 82, 12291–12303 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yukl S., Pillai S., Li P., Chang K., Pasutti W., Ahlgren C., Havlir D., Strain M., Gunthard H., Richman D., Rice A. P., Daar E., Little S., Wong J. K., Latently-infected CD4+ T cells are enriched for HIV-1 Tat variants with impaired transactivation activity. Virology 387, 98–108 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pradeepa M. M., Sutherland H. G., Ule J., Grimes G. R., Bickmore W. A., Psip1/Ledgf p52 binds methylated histone H3K36 and splicing factors and contributes to the regulation of alternative splicing. PLOS Genet. 8, e1002717 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sutherland H. G., Newton K., Brownstein D. G., Holmes M. C., Kress C., Semple C. A., Bickmore W. A., Disruption of Ledgf/Psip1 results in perinatal mortality and homeotic skeletal transformations. Mol. Cell. Biol. 26, 7201–7210 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vranckx L. S., Demeulemeester J., Saleh S., Boll A., Vansant G., Schrijvers R., Weydert C., Battivelli E., Verdin E., Cereseto A., Christ F., Gijsbers R., Debyser Z., LEDGIN-mediated inhibition of integrase-LEDGF/p75 interaction reduces reactivation of residual latent HIV. EBioMedicine 8, 248–264 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sharma P., Fatma N., Kubo E., Shinohara T., Chylack L. T. Jr., Singh D. P., Lens epithelium-derived growth factor relieves transforming growth factor-β1-induced transcription repression of heat shock proteins in human lens epithelial cells. J. Biol. Chem. 278, 20037–20046 (2003). [DOI] [PubMed] [Google Scholar]
- 18.Squazzo S. L., Costa P. J., Lindstrom D. L., Kumer K. E., Simic R., Jennings J. L., Link A. J., Arndt K. M., Hartzog G. A., The Paf1 complex physically and functionally associates with transcription elongation factors in vivo. EMBO J. 21, 1764–1774 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van Oss S. B., Cucinotta C. E., Arndt K. M., Emerging insights into the roles of the Paf1 complex in gene regulation. Trends Biochem. Sci. 42, 788–798 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jadlowsky J. K., Wong J. Y., Graham A. C., Dobrowolski C., Devor R. L., Adams M. D., Fujinaga K., Karn J., Negative elongation factor is required for the maintenance of proviral latency but does not induce promoter-proximal pausing of RNA polymerase II on the HIV long terminal repeat. Mol. Cell. Biol. 34, 1911–1928 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Natarajan M., Schiralli Lester G. M., Lee C., Missra A., Wasserman G. A., Steffen M., Gilmour D. S., Henderson A. J., Negative elongation factor (NELF) coordinates RNA polymerase II pausing, premature termination, and chromatin remodeling to regulate HIV transcription. J. Biol. Chem. 288, 25995–26003 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen F. X., Woodfin A. R., Gardini A., Rickels R. A., Marshall S. A., Smith E. R., Shiekhattar R., Shilatifard A., PAF1, a molecular regulator of promoter-proximal pausing by RNA Polymerase II. Cell 162, 1003–1015 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen F. X., Xie P., Collings C. K., Cao K., Aoi Y., Marshall S. A., Rendleman E. J., Ugarenko M., Ozark P. A., Zhang A., Shiekhattar R., Smith E. R., Zhang M. Q., Shilatifard A., PAF1 regulation of promoter-proximal pause release via enhancer activation. Science 357, 1294–1298 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu D., Shilatifard A., Epigenetics of hematopoiesis and hematological malignancies. Genes Dev. 30, 2021–2041 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sobhian B., Laguette N., Yatim A., Nakamura M., Levy Y., Kiernan R., Benkirane M., HIV-1 Tat assembles a multifunctional transcription elongation complex and stably associates with the 7SK snRNP. Mol. Cell 38, 439–451 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang J., Gurung B., Wan B., Matkar S., Veniaminova N. A., Wan K., Merchant J. L., Hua X., Lei M., The same pocket in menin binds both MLL and JUND but has opposite effects on transcription. Nature 482, 542–546 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Murai M. J., Pollock J., He S., Miao H., Purohit T., Yokom A., Hess J. L., Muntean A. G., Grembecka J., Cierpicki T., The same site on the integrase-binding domain of lens epithelium-derived growth factor is a therapeutic target for MLL leukemia and HIV. Blood 124, 3730–3737 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sharma S., Čermáková K., De Rijck J., Demeulemeester J., Fábry M., El Ashkar S., Van Belle S., Lepšik M., Tesina P., Duchoslav V., Novak P., Hubalek M., Srb P., Christ F., Rezacova P., Hodges H. C., Debyser Z., Veverka V., Affinity switching of the LEDGF/p75 IBD interactome is governed by kinase-dependent phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 115, E7053–E7062 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ahmed K., Gerber D. A., Cochet C., Joining the cell survival squad: an emerging role for protein kinase CK2. Trends Cell Biol. 12, 226–230 (2002). [DOI] [PubMed] [Google Scholar]
- 30.Hayden M. S., Ghosh S., Signaling to NF-κB. Genes Dev. 18, 2195–2224 (2004). [DOI] [PubMed] [Google Scholar]
- 31.Martins L. J., Bonczkowski P., Spivak A. M., De Spiegelaere W., Novis C. L., DePaula-Silva A. B., Malatinkova E., Trypsteen W., Bosque A., Vanderkerckhove L., Planelles V., Modeling HIV-1 latency in primary T cells using a replication-competent virus. AIDS Res. Hum. Retroviruses 32, 187–193 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ge H., Si Y., Roeder R. G., Isolation of cDNAs encoding novel transcription coactivators p52 and p75 reveals an alternate regulatory mechanism of transcriptional activation. EMBO J. 17, 6723–6729 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu L., Oliveira N. M., Cheney K. M., Pade C., Dreja H., Bergin A. M., Borgdorff V., Beach D. H., Bishop C. L., Dittmar M. T., McKnight A., A whole genome screen for HIV restriction factors. Retrovirology 8, 94 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abdel-Mohsen M., Wang C., Strain M. C., Lada S. M., Deng X., Cockerham L. R., Pilcher C. D., Hecht F. M., Liegler T., Richman D. D., Deeks S. G., Pillai S. K., Select host restriction factors are associated with HIV persistence during antiretroviral therapy. AIDS 29, 411–420 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jaenicke L. A., von Eyss B., Carstensen A., Wolf E., Xu W., Greifenberg A. K., Geyer M., Eilers M., Popov N., Ubiquitin-dependent turnover of MYC antagonizes MYC/PAF1C complex accumulation to drive transcriptional elongation. Mol. Cell 61, 54–67 (2016). [DOI] [PubMed] [Google Scholar]
- 36.Tesina P., Čermáková K., Hořejší M., Procházková K., Fábry M., Sharma S., Christ F., Demeulemeester J., Debyser Z., Rijck J., Veverka V., Řezáčová P., Multiple cellular proteins interact with LEDGF/p75 through a conserved unstructured consensus motif. Nat. Commun. 6, 7968 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Bartholomeeusen K., Christ F., Hendrix J., Rain J. C., Emiliani S., Benarous R., Debyser Z., Gijsbers R., De Rijck J., Lens epithelium-derived growth factor/p75 interacts with the transposase-derived DDE domain of PogZ. J. Biol. Chem. 284, 11467–11477 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Critchfield J. W., Coligan J. E., Folks T. M., Butera S. T., Casein kinase II is a selective target of HIV-1 transcriptional inhibitors. Proc. Natl. Acad. Sci. U.S.A. 94, 6110–6115 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Basnet H., Su X. B., Tan Y., Meisenhelder J., Merkurjev D., Ohgi K. A., Hunter T., Pillus L., Rosenfeld M. G., Tyrosine phosphorylation of histone H2A by CK2 regulates transcriptional elongation. Nature 516, 267–271 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hu D., Smith E. R., Garruss A. S., Mohaghegh N., Varberg J. M., Lin C., Jackson J., Gao X., Saraf A., Florens L., Washburn M. P., Eissenberg J. C., Shilatifard A., The little elongation complex functions at initiation and elongation phases of snRNA gene transcription. Mol. Cell 51, 493–505 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/20/eaaz8411/DC1