

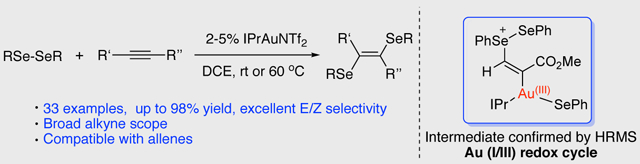
Abstract
Gold-catalyzed alkyne and allene diselenations were developed. Excellent regioselectivity (trans) and good to excellent yields were achieved (up to 98% with 2% catalyst loading) with a wide range of substrates. Mechanistic investigation revealed the formation of a vinyl gold(I) intermediate followed by an intermolecular selenium cation migration, suggesting that a gold (I/III) redox process was successfully implemented under mild conditions.
Keywords: gold redox catalysis, deselenation, mild oxidant, alkyne, allene
Graphical Abstract
The past two decades have witnessed tremendous growth in homogeneous gold catalysis.[1] The unique capability of the gold cation as a carbophilic Lewis acid[2] has rendered it a superior catalyst in nucleophilic addition reactions towards alkenes, allenes, and alkynes.[3] On the other hand, the redox chemistry of gold(I) catalyst is largely neglected, mainly due to the high oxidation potential (AuI/III = 1.4 eV).[4] Thanks to the pioneering work from Zhang, Hashmi, Toste and other groups, this challenge has been successfully overcome by utilizing external strong oxidants such as selecfluor, hypervalent iodine reagents, aryldiazonium salts and even alkynyl bromides.[5] Another significant observation in this regard was reported by Glorius’ group, who first indentified that diazonium salt could be used as a mild oxidant for gold (I) oxidation in the presence of an additional ruthenium-based photoredox catalyst(Scheme 1A).[6] Despite of these progresses, the necessity of using an external oxidant impedes further development of this methodology with only limited reaction scope. More recently, direct oxidative addition at a gold(I) complex emerged.[7] However, the reported examples so far either requires highly-strained starting materials in the case of X-X oxidative addition (X=Si or C) or a delicate catalyst design to activate allyl or aryl halides. Thus, developing a novel and effective approach to accomplish gold redox catalysis is not only important for enhancing the basic understanding of this rapidly-growing field, but also holds promise for generating synthetically valuable building blocks.
Scheme 1.
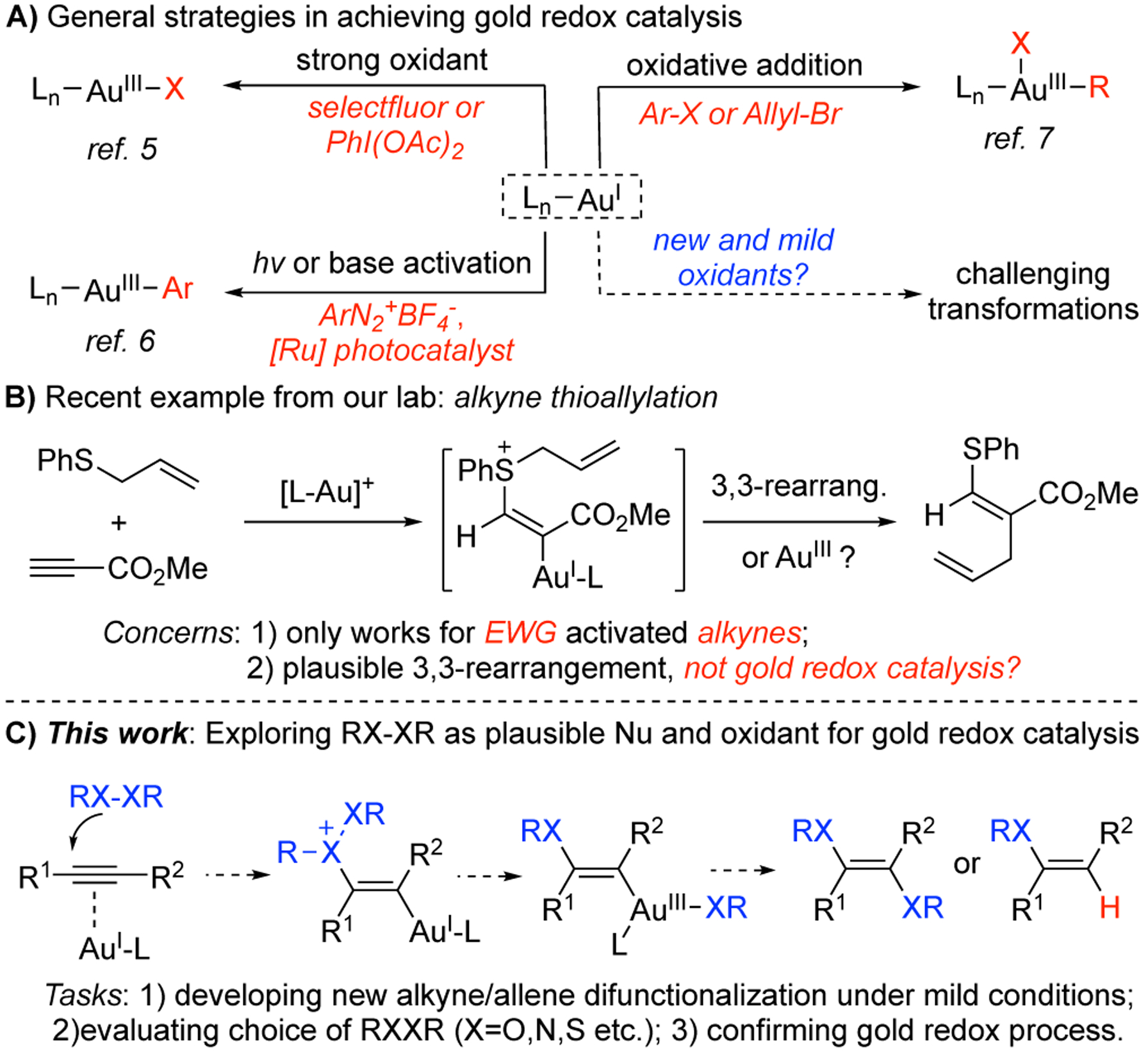
Recent development on gold redox catalysis.
Recently, we reported a highly efficient and stereoselective thioallylation of electron-deficient alkynes (Scheme 1B).[8] Mechanistic study suggested that this reaction was initiated by sulfur nucleophilic addition to form a vinyl gold intermediate, which underwent a sequential allyl transfer via Au(I/III) cycle. Notably, we for the first time propose the allyl sulfonium cation functions as a mild internal oxidant to accomplish the Au(I/III) oxidation, which represents a powerful strategy to combine the excellent π-acid reactivity of Au(I) complexes with gold redox catalysis.
Although we have successfully observed Au(III) species via Mass Spectrometry in the thioallylation of alkynes, we could not completely rule out a possible [3,3]-rearrangement pathway[9] which doesn’t involve Au(I) oxidation. Moreover, only electron deficient alkynes (EWG modified alkynes) successfully participated in this transformation, which greatly limited the reaction scope. Other alkynes and allenes, which are typical substrates in gold catalysis, did not give the desired products. Therefore, it is important to conduct a more detailed investigations regarding detailed reaction mechanism and further extend the reaction to a broader scope. Herein, we report our efforts on facilitating RXXR addition toward alkynes and allenes under gold-catalyzed conditions (Scheme 1C). Notably, regioselective gold-catalyzed alkyne and allene diselenations were successfully achieved. The transformation took place under mild conditions with high efficiency, giving desired products in good to excellent yields and excellent regioselectivity. Mechanistic investigations confirmed Au(I/III) redox cycle, which validates the great potential of applying selenium cation as an effective mild oxidant to promote gold redox catalysis in future. What’s more, the diselenation product can be further derived into value-added synthetic intermediates.[10]
As suggested in Scheme 1C, compounds with RXXR moiety could be of interest in performing sequential nucleophilic addition and redox chemistry with a single Au(I) catalyst. Unlike thioallylation, [3,3]-rearrangement path is not available in this alkyne difuntionalization reaction, which provides a simple model for studying gold redox catalytic cycle. Notably, with a relatively weak X-X bond, it has been reported in literature that RXXR type compounds could form RX radical and proceed aforementioned transformation via a radical path.[11] As a result, poor regioselectivity was often observed. The proposed gold catalytic route, if successful, will not only provide an alternative approach for RXXR activation, but also likely accomplish the reaction with much better stereo-outcome. Potential challenges can be 1) reduced [LAu]+ reactivity caused by X-Au coordination and 2) slow C-X reductive elimination (relative to C-C), which could lead to competing protodeauration as the major byproduct. To initiate our investigation, we first attempted using ROOR, RNNR and RSSR as nucleophiles to react with alkynes. The results are summarized in Figure 1.
Figure 1.
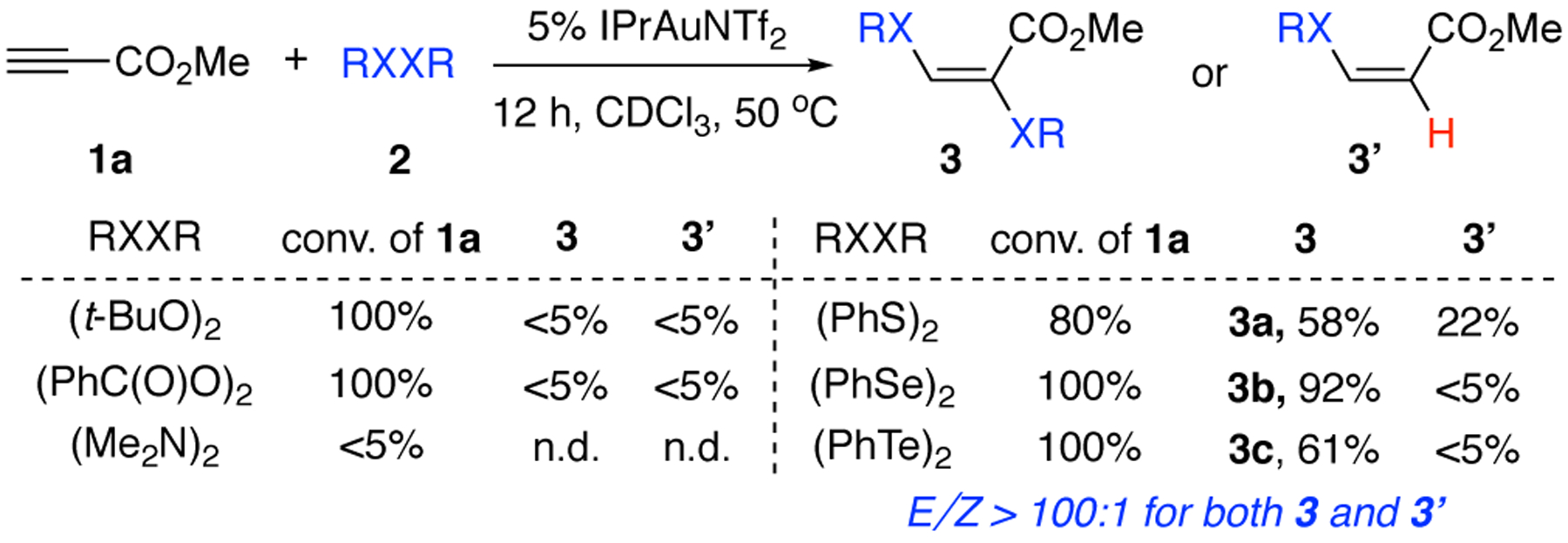
Gold-catalyzed RXXR addition to alkynes
Treating alkyne 1a with peroxide or peracid gave 100% conversion. However, desired product 3 or byproduct 3’ was not obtained based on crude NMR. Reaction with tetramethyl hydrazine gave almost no alkyne conversion, likely due to catalyst poisoning. Interestingly, reaction of disulfide 2a (PhSSPh) gave the desired dithiolation product 3a in 58% yield (80% conversion). As expected, hydrothiolation product 3a’ was observed (22%) as the byproduct. It is important to note that both 3a and 3a’ were observed as single isomer (E/Z > 100:1). The alkene geometry was later confirmed by X-ray crystallography (vide infra). These results are exciting, since they suggested that the proposed combination of gold π-acid activation and redox catalysis is valid.
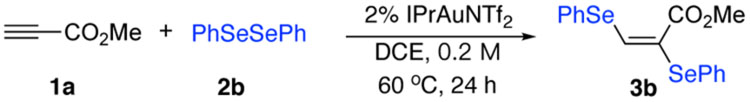
In theory, protodeauration could occur form both gold(I) intermediate (vinyl gold) or gold(III) intermediate.[12] Considering gold(III) reductive elimination is a rather rapid process, we postulate that stronger oxidants may help to avoid protodeauruation by promoting the reaction with faster gold (I) oxidation. Thus, we turned our attention to diselenides based on 1) Se as better nucleophile over S and 2) selenium cation as a stronger oxidant. To our delight, the desired diselenation product 3b was formed in excellent yield (90%). Further catalyst screening revealed that electron-rich gold catalyst IPrAuNTf2 gave the best yield and DCE was identified as the optimal solvent. The catalyst loading can be reduced to 2% without sacrificing the yield or regioselectivity. Other metal catalysts including Pt, Ag, Cu, Fe gave inferior results (see SI for details). Reduced yield was observed when using PhTeTePh (3c), which is likely due to a stronger coordination between Te and gold catalyst, comparing to Se or S, that leads to slower reaction. Comparisons of the optimal conditions with some alternative conditions are summarized in Table 1.
Table 1.
Gold-catalyzed alkyne diselenation: condition screening
 | ||||
|---|---|---|---|---|
| entry | Variation from standard conditions | conv. | 3b (E/Z) | 3b’ |
| 1 | none | 100% | 92% (>100:1) | 5% |
| 2 | 5% Ph3PAuNTf2 | 60% | 46% | - |
| 3 | 5% (PhO)3PAuNTf2 | 60% | 31% | - |
| 4 | 5% XPhosAuNTf2 | 100% | 72% (40:1) | 14% |
| 5 | 2% IPrAuNTf2 (0.5 M) | 100% | 85% (32:1) | 7% |
| 6 | 1% IPrAuNTf2 (0.5 M) | 100% | 85% (20:1) | 8% |
| 7 | Other [Au] catalyst | - | - | - |
| 8 | 10% PtCl2 | 80% | 67% | - |
| 9 | other metal catalysts (Ag, Cu, Fe, etc) | <60% | ||
| 10 | other solvents | <80% | ||
| 11 | 10% HOTf | <5% | nd | - |
| 12 | no catalyst | <5% | nd | - |
Reaction conditions: the catalyst (2%) was added into a DCE solution of alkyne (0.3 mmol) and diselenide (0.2 mmol), and reacted at 60 °C for 24 h. Conversion and yield were determined by 1H NMR spectroscopy with dimethyl sulfone as the internal standard.
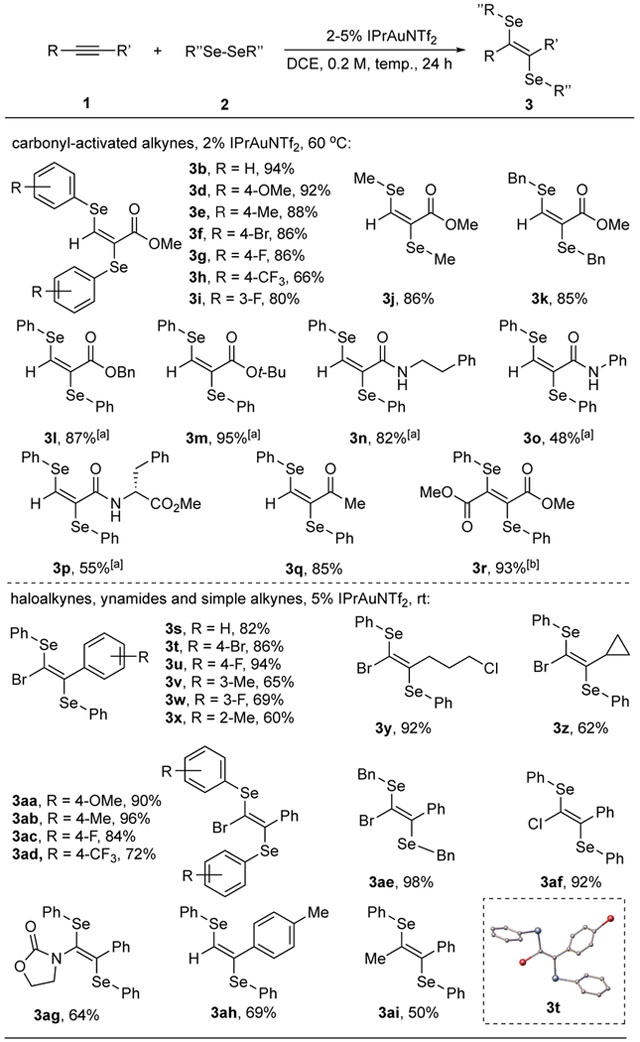
With the optimal conditions in hand, we next explored the reaction scope as shown in Table 2. Most aryl diselenides with either electron-donating or electron-withdrawing substituents proved to be suitable for this transformation, giving good to excellent yields when reacting with methyl propiolate (3a-3g). One exception is para-CF3 substituted aryl deselenides, which only provided 3h with moderate yield due to sluggish nucleophilic addition step. Alkyl diselenides participated in this reaction smoothly as well, realizing 3j and 3k in satisfactory yields. Other carbonyl-activated alkynes, including esters (3l and 3m), ketone (3q) and acetylene diester (3r) all underwent diselenation successfully with excellent yields; amides (3n-3p) on the other hand gave lower yields presumably due to substrate coordination. We next turned our attention to a broader scope of alkynes. First, various bromoalkynes with either aryl or alkyl group connecting to the C-C triple bond reacted with phenyl diselenide and provided desired products in moderate to excellent yields (3s-3z); other diselenides were also compatible with bromoalkynes (3aa-3ae) The fact that no cyclopropyl ring opening product was observed ruled out a radical reaction pathway (3z). Chloroalkyne was a valid substrate for this reaction as expected (3af). We were pleased to find out that electron-rich alkynes such as ynamides underwent diselenation successfully, giving desired products in moderate yields (3aq). Encouraged by this result, we tested both terminal and internal alkynes without electronic biases. We are delighted to find out that desired products 3ah and 3ai were formed in moderate yield under gold-catalyzed conditions, which further showcased the broad scope of this transformation. It is worthwhile to mention that in all cases diselenation products were formed as essentially one alkene isomer, and their E-geometry were ambiguously identified by X-ray crystallography of product 3t.
Table 2.
Substrate scope for diselenation of alkynes
|
Reaction conditions: the catalyst was added into a DCE solution of alkyne (0.3 mmol) and diselenide (0.2 mmol), and reacted at given temperature for 24 h. Isolated yield. [a] 0.2 mmol alkyne and 0.3 mmol diselenide were used for the ease of purification. [b] React at 80 °C.
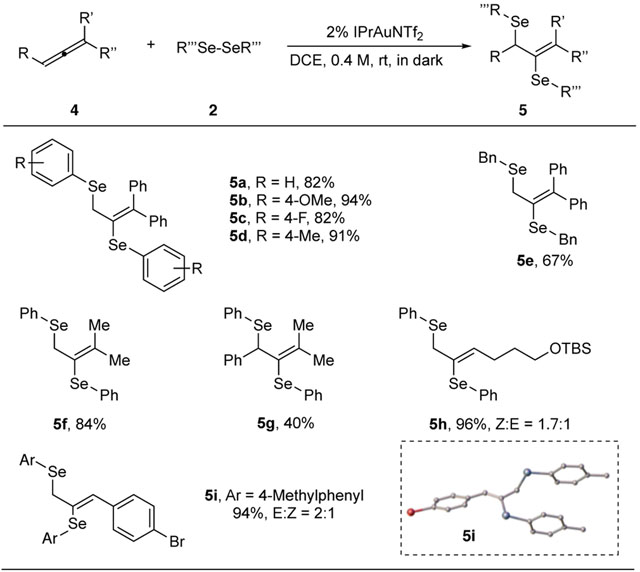
Encouraged by the successful diselenation of alkynes, we wondered if this transformation can be further extended to allenes, which were not suitable substrates in previous thioallylation studies. After detailed reaction optimization, we were pleased to find out that allene diselenation products could also be formed smoothly via this protocol. As shown in Table 3, when treating 1,1-diphenyl allene with different diselenides, desired products were formed in good to excellent yields (5a-5e). 1,1-dimethyl allene could also give the desired products in excellent yield (5f). Trisubstituted allenes underwent diselenation reaction uneventfully, providing desired product 5g in moderate yield. Monosubstituted allenes also provided excellent yields, although only moderate E/Z selectivity were observed (5i-5k). The structure of E/Z isomers were unambigiously assigned by X-ray crystallography (E-5i).
Table 3.
Substrate scope for diselenation of allenes
|
Reaction conditions: the catalyst (2%) was added into a DCE solution of allene (0.6 mmol) and diselenide (0.2 mmol) and reacted at rt for 24 h. Isolated yield.
To better elucidate the mechanism,[13] a series of experiments were carried out. First, cross-over experiment clearly supported an intermolecular selenium transfer mechanism (Figure 2A). In addition, direct oxidative addition of phenyl diselenide with gold(I) catalyst was not observed (see SI). When using more electron-rich styrene, such reaction didn’t occur, which ruled out a Au(I)-assisted selenium cation formation pathway. Based on these results, we propose a mechanism which includes gold-catalyzed nucleophilic addition of diselenides and subsequent selenium transfer via gold redox catalysis, and attempted to trap the key intermediates with Mass spectrometry (Figure 2B). A mass corresponding to vinyl gold intermediate A was clearly detected. In addition, CID experiment of m/z of intermediate A suggested a Au(II)-SePh+ fragment, which most likely came from a Au(III) intermediate B that possesses the same mass as intermediate A. Another Au(III) intermediate C which contains a similar composition as in the thioallylation case was also observed, and CID confirmed the existence of a Au(III) fragment too. The Mass study thus strongly supported that Au(I/III) redox cycle is plausible for this transformation.
Figure 2.
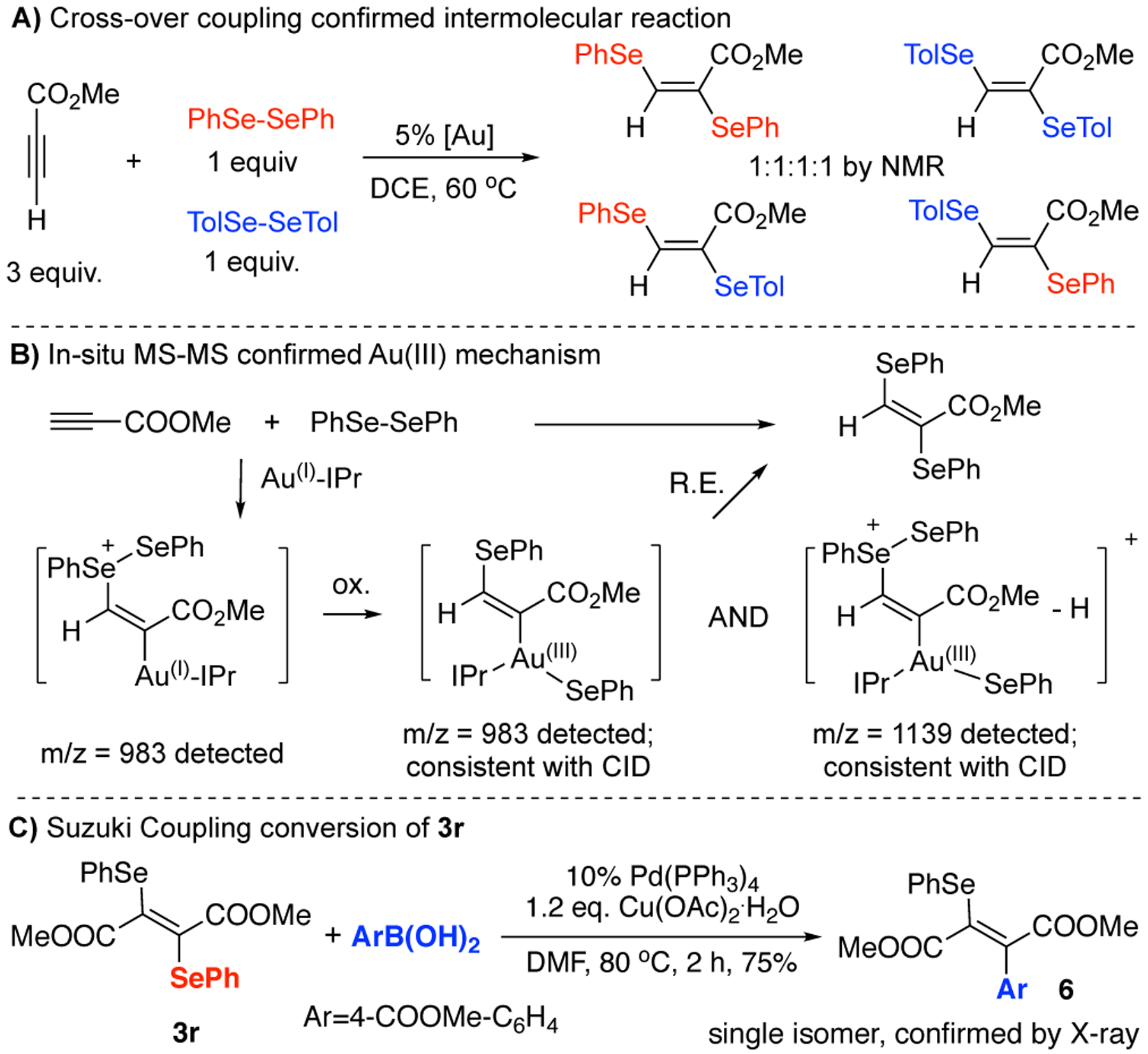
Mechanistic investigations and 3r conversion
While 1,2-diselenide products 3 are interesting building blocks, they can be readily converted into other compounds through simple transformations. As shown in Figure 2C, treating 3r under standard Suzuki coupling conditions gave desired couping product 6 in 75% yield. Interestingly, although excess amount ArB(OH)2 were used, only single isomer with mono-substitution product was obtained with structure confirmed by X-ray crystallography (see SI). This result suggested a Heck-type mechanism might occurred with this substrate instead of direct CSe bond oxidative addition as proposed in literature.[10a] This excellent chemo and stereoselectivity greatly highlighted the potential synthetic utility of these 1,2-diselenide compounds under this new and mild gold redox conditions.
In conclusion, we herein reported a gold-catalyzed diselenation reaction of alkynes and allenes. Excellent yields and stereoselectivity as well as broad reaction scope were achieved for this transformation. Au(I/III) mechanism was proposed based on the cross-over experiment, control experiments and in-situ MS results. Other mild oxidants are currently under investigation in our lab.
Supplementary Material
Acknowledgements
We are grateful to NSF (CHE-1665122 and CHE-1915878) and NIH (1R01GM120240-01) for financial supports.
Footnotes
Publisher's Disclaimer: This manuscript has been accepted after peer review and appears as an Accepted Article online prior to editing, proofing, and formal publication of the final Version of Record (VoR). This work is currently citable by using the Digital Object Identifier (DOI) given below. The VoR will be published online in Early View as soon as possible and may be different to this Accepted Article as a result of editing. Readers should obtain the VoR from the journal website shown below when it is published to ensure accuracy of information. The authors are responsible for the content of this Accepted Article.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- [1].a) Hashmi ASK, Gold Bulletin 2004, 37, 51–65; [Google Scholar]; b) Fürstner A, Davies PW, Angew. Chem. Int. Ed 2007, 46, 3410–3449; [DOI] [PubMed] [Google Scholar]; c) Hashmi ASK, Chem. Rev 2007, 107, 3180–3211; [DOI] [PubMed] [Google Scholar]; d) Jiménez-Núñez E, Echavarren AM, Chem. Rev 2008, 108, 3326–3350; [DOI] [PubMed] [Google Scholar]; e) Gorin DJ, Sherry BD, Toste FD, Chem. Rev 2008, 108, 3351–3378; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Dorel R, Echavarren AM, Chem. Rev 2015, 115, 9028–9072; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Asiri AM, Hashmi ASK, Chem. Soc. Rev 2016, 45, 4471–4503; [DOI] [PubMed] [Google Scholar]; h) Zheng Z, Wang Z, Wang Y, Zhang L, Chem. Soc. Rev 2016, 45, 4448–4458; [DOI] [PubMed] [Google Scholar]; i) Li Y, Li W, Zhang J, Chem. Eur. J 2017, 23, 467–512; [DOI] [PubMed] [Google Scholar]; j) Zhao X, Rudolph M, Hashimi ASK, Chem. Commun, 2019, 55, 12127–12135. [DOI] [PubMed] [Google Scholar]
- [2].a) Praveen C, Coord. Chem. Rev 2019, 392, 1–34; [Google Scholar]; b) Gorin DJ, Toste FD, Nature 2007, 446, 395–403; [DOI] [PubMed] [Google Scholar]; c) Pernpointner M, Hashmi ASK, J. Chem. Theory Comput 2009, 5, 2717–2725. [DOI] [PubMed] [Google Scholar]
- [3].a) Mascareñas JL, Varela I, López F, Acc. Chem. Res 2019, 52, 465–479; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Schmidbaur H, Schier A, Organometallics 2010, 29, 2–23; [Google Scholar]; c) Li Z, Brouwer C, He C, Chem. Rev 2008, 108, 3239–3265; [DOI] [PubMed] [Google Scholar]; d) A. Arcadi, Chem. Rev 2008, 108, 3266–3325; [DOI] [PubMed] [Google Scholar]; e) Hashmi ASK, Hutchings GJ, Angew. Chem. Int. Ed 2006, 45, 7896–7936; [DOI] [PubMed] [Google Scholar]; f) Chiarucci M, Bandini M, Beilstein J. Org. Chem 2013, 9, 2586–2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bratsch SG, J. Phys. Chem. Ref. Data 1989, 18, 1–21. [Google Scholar]
- [5].a) Hofer M, de Haro T, Gómez-Bengoa E, Genoux A, Nevado C, Chem. Sci 2019, 10, 8411–8420; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yang Y, Antoni P, Zimmer M, Sekine K, Mulks FF, Hu L, Zhang L, Rudolph M, Rominger F, Hashmi ASK, Angew. Chem. Int. Ed 2019, 131, 5129–5133; [DOI] [PubMed] [Google Scholar]; c) Jimoh AA, Hosseyni S, Ye X, Wojtas L, Hu Y, Shi X, Chem. Commun 2019, 55, 8150–8153; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Xie J, Sekine K, Witzel S, Krämer P, Rudolph M, Rominger F, Hashmi ASK, Angew. Chem. Int. Ed 2018, 57, 16648–16653; [DOI] [PubMed] [Google Scholar]; e) Shaikh AC, Ranade DS, Rajamohanan PR, Kulkarni PP, Patil NT, Angew. Chem. Int. Ed 2017, 56, 757–761; [DOI] [PubMed] [Google Scholar]; f) Peng H, Cai R, Xu C, Chen H, Shi X, Chem. Sci 2016, 7, 6190–6196; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Kim S, Rojas-Martin J, Toste FD, Chem. Sci 2016, 7, 85–88; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) He Y, Wu H, Toste FD, Chem. Sci 2015, 6, 1194–1198; [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Peng H, Xi Y, Ronaghi N, Dong B, Akhmedov NG, Shi X, J. Am. Chem. Soc 2014, 136, 13174–13177; [DOI] [PubMed] [Google Scholar]; j) Ball LT, Lloyd-Jones GC, Russell CA, J. Am. Chem. Soc 2014, 136, 254–264; [DOI] [PubMed] [Google Scholar]; k) Yu Y, Yang W, Pflästerer D, Hashmi ASK, Angew. Chem. Int. Ed 2014, 53, 1144–1147; [DOI] [PubMed] [Google Scholar]; l) Pažický M, Loos A, Ferreira MJ, Serra D, Vinokurov N, Rominger F, Jäkel C, Hashmi ASK, Limbach M, Organometallics 2010, 29, 4448–4458; [Google Scholar]; m) Hashmi ASK, Ramamurthi TD, Rominger F, J. Organomet. Chem 2009, 694, 592–597; [Google Scholar]; n) Zhang G, Peng Y, Cui L, Zhang L, Angew. Chem. Int. Ed 2009, 48, 3112–3115; [DOI] [PubMed] [Google Scholar]; o) Taschinski S, Döpp R, Ackermann M, Rominger F, de Vries F, Menger MFSJ, Rudolph M, Hashmi ASK, Klein JEMN, Angew. Chem. Int. Ed 2019, 58, 16988–16993; [DOI] [PMC free article] [PubMed] [Google Scholar]; p) Yang Y, Eberle L, Mulks FF, Wunsch JF, Zimmer M, Rominger F, Rudolph M, Hashimi ASK, J. Am. Chem. Soc 2019, 141, 17414–17420; [DOI] [PubMed] [Google Scholar]; q) Yang Y, Schießl J, Zallouz S, Göker V, Gross J, Rudolph M, Rominger F, Hashmi ASK, Chem. Eur. J 2019, 25, 9624–9628; [DOI] [PubMed] [Google Scholar]; r) Huang L, Rominger F, Rudolph M, Hashimi ASK, Chem. Commun 2016, 52, 6435–6438; [DOI] [PubMed] [Google Scholar]; s) Huang L, Rudolph M, Rominger F, Hashmi ASK, Angew. Chem. Int. Ed 2016, 55, 4808–4813.d [DOI] [PubMed] [Google Scholar]
- [6].a) Sahoo B, Hopkinson MN, Glorius F, J. Am. Chem. Soc 2013, 135, 5505–5508; [DOI] [PubMed] [Google Scholar]; b) Tlahuext-Aca A, Hopkinson MN, Garza-Sanchez RA, Glorius F, Chem. Eur. J 2016, 22, 5909–5913; [DOI] [PubMed] [Google Scholar]; c) Cai R, Lu M, Aguilera EY, Xi Y, Akhmedov NG, Petersen JL, Chen H, Shi X, Angew. Chem. Int. Ed 2015, 54, 8772–8776. [DOI] [PubMed] [Google Scholar]
- [7].a) Akram MO, Das A, Chakrabarty I, Patil NT, Org. Lett 2019, 21, 8101–8105; [DOI] [PubMed] [Google Scholar]; b) Harper MJ, Arthur CJ, Crosby J, Emmett EJ, Falconer RL, Fensham-Smith AJ, Gates PJ, Leman T, McGrady JE, Bower JF, Russell CA, J. Am. Chem. Soc 2018, 140, 4440–4445; [DOI] [PubMed] [Google Scholar]; c) Akram MO, Mali PS, Patil NT, Org. Lett 2017, 19, 3075–3078; [DOI] [PubMed] [Google Scholar]; d) Chahdoura F, Lassauque N, Bourissou D, Amgoune A, Org. Chem. Front 2016, 3, 856–860; [Google Scholar]; e) Guenther J, Mallet-Ladeira S, Estevez L, Miqueu K, Amgoune A, Bourissou D, J. Am. Chem. Soc 2014, 136, 1778–1781; [DOI] [PubMed] [Google Scholar]; f) Serra J, Whiteoak CJ, Acuña-Parés F, Font M, Luis JM, Lloret-Fillol J, Ribas X, J. Am. Chem. Soc 2015, 137, 13389–13397; [DOI] [PubMed] [Google Scholar]; g) Serra J, Parella T, Ribas X, Chem. Sci 2017, 8, 946–952; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Joost M, Zeineddine A, Estévez L, Mallet-Ladeira S, Miqueu K, Amgoune A, Bourissou D, J. Am. Chem. Soc 2014, 136, 14654–14657; [DOI] [PubMed] [Google Scholar]; i) Zeineddine A, Estévez L, Mallet-Ladeira S, Miqueu K, Amgoune A, Bourissou D, Nat. Commun 2017, 8, 565; [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Levin MD, Toste FD, Angew. Chem. Int. Ed 2014, 53, 6211–6215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Wang J, Zhang S, Xu C, Wojtas L, Akhmedov NG, Chen H, Shi X, Angew. Chem. Int. Ed 2018, 57, 6915–6920; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ye X, Zhao P, Zhang S, Zhang Y, Wang Q, Shan C, Wojtas L, Guo H, Chen H, Shi X, Angew. Chem. Int. Ed, 2019, 58, 17226–17230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Ackermann M, Bucher J, Rappold M, Graf K, Romingere F, Hashimi ASK, Chem. Asian J 2013, 32, 1786–1794; [DOI] [PubMed] [Google Scholar]; b) Hashimi ASK, Graf K, Ackermann M, Rominger F, ChemCatChem 2013, 5, 1200–1204. [Google Scholar]
- [10].a) Stein AL, Bilheri FN, Zeni G, Chem. Commun 2015, 51, 15522–15525; [DOI] [PubMed] [Google Scholar]; b) DOnofrio F, Parlanti L, Piancatelli G, Synlett, 1996, 1, 63–64. [Google Scholar]
- [11].a) Ogawa A, Doi M, Ogawa I, Hirao T, Angew. Chem. Int. Ed 1999, 38, 2027–2029; [DOI] [PubMed] [Google Scholar]; b) Ogawa A, Ogawa I, Sonoda N, J. Org. Chem 2000, 65, 7682–7685; [DOI] [PubMed] [Google Scholar]; c) Ogawa A, Obayashi R, Doi M, Sonoda N, Hirao T, J. Org. Chem 1998, 63, 4277–4281; [DOI] [PubMed] [Google Scholar]; d) Ogawa A, Obayashi R, Ine H, Tsuboi Y, Sonoda N, Hirao T, J. Org. Chem 1998, 63, 881–884; [DOI] [PubMed] [Google Scholar]; e) Ogawa A, Ogawa I, Obayashi R, Umezu K, Doi M, Hirao T, J. Org. Chem 1999, 64, 86–92; [DOI] [PubMed] [Google Scholar]; f) Mugesh G, du Mont W-W, Sies H, Chem. Rev 2001, 101, 2125–2180; [DOI] [PubMed] [Google Scholar]; g) Nogueira CW, Zeni G, Rocha JBT, Chem. Rev 2004, 104, 6255–6286. [DOI] [PubMed] [Google Scholar]
- [12].dos Santos Comprido L. Nunes, Klein JEMN, Knizia G, Kästner J, Hashmi ASK, Chem. Eur. J 2017, 23, 10901–10905. [DOI] [PubMed] [Google Scholar]
- [13].Hashmi ASK, Angew. Chem. Int. Ed 2010, 49, 5232–5241. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.