

Abstract
G-quadruplexes (GQs) are highly ordered nucleic acid structures that play fundamental roles in regulating gene expression and maintaining genomic stability. GQs are topologically diverse and enriched in promoter sequences of growth regulatory genes and proto-oncogenes, suggesting they may serve as attractive targets for drug design at the level of transcription rather than inhibiting the activity of the protein products of these genes. The c-kit promoter contains three adjacent GQ-forming sequences that have proposed antagonistic effects on gene expression and thus are promising drug targets for diseases like gastrointestinal stromal tumors, mast cell disease, and leukemia. Since GQ stability is influenced by primary structure, secondary structure, and ion interactions, a greater understanding of GQ structure, dynamics, and ion binding properties is needed to develop novel, GQ-targeting therapeutics. Here, we performed molecular dynamics (MD) simulations to systematically study the c-kit2 and c-kit* GQs, evaluating nonpolarizable and polarizable force fields (FFs) and examining the effects of base substitutions and cation type (K+, Na+, and Li+) on the dynamics of their isolated and linked structures. We found that the Drude polarizable FF outperformed the additive CHARMM36 FF in two-and three-tetrad GQs and solutions of KCl, NaCl, and LiCl. Drude simulations with different cations agreed with the known GQ stabilization preference (K+ > Na+ > Li+) and illustrated that tetrad core-ion coordination differs as a function of cation type. Finally, we showed that differences in primary and secondary structure influence loop sampling, ion binding, and core-ion energetics of GQs.
Keywords: Nucleic acids, Drude oscillator, induced dipoles, nucleic acid-ion interactions
Graphical Abstract
INTRODUCTION
Nucleic acid sequences rich in guanine can fold into G-quadruplexes (GQs), highly ordered DNA or RNA structures that are present in telomeres, gene promoter regions, and 5’- and 3’- untranslated regions of mRNA.1–4 As such, GQs are believed to play functional roles in chromatin structure, chromosomal stability, and regulation of gene expression at transcriptional5 and translational6,7 levels. Interest in GQ structure and stability is growing since studies have demonstrated that stable GQs modulate replication, DNA repair, telomere elongation8 and contribute to neurodegenerative diseases,9 mental retardation,10 premature aging,11 and various types of cancer.12,13 More than 700,000 potential GQ-forming sequences (pGQs) have been identified in the human genome, localized in genomic regions with important cellular functions.14–18 In fact, over half of human genes contain pGQs near promoter regions and their occurrence is even more frequent in regulatory genes and oncogenes.15–18 To date, several topologically diverse promoter GQs have been experimentally identified and studied;19–26 however, much is still unknown regarding their conformational sampling and the interactions governing their stability.
Folded GQs are highly stable and composed of linker regions and a tetrad core, characterized by square planar arrangement of guanines that are stabilized by Hoogsteen hydrogen bonds.27 The carbonyl oxygen (O6) of each guanine faces inward, requiring cation binding to counteract core electronegativity and stabilize the structure.28 Cation binding in the tetrad core is thought to promote GQ folding29 and may contribute more to GQ stability than hydrogen bonding or stacking interactions.30 The dominant cation found in GQ structures is K+, but the preference for K+ stabilization over other cations like Na+ and Li+ is not well understood. Differences in ion interactions, primary structure, base pairing, and base stacking give rise to diverse GQ folded topologies.31,32 This topological diversity, combined with their implications in human disease, make GQs novel drug targets.3,13,33 Towards GQ-specific drug design, more work is needed to characterize the relationship between GQ structure and disease, which has proven to be more complicated than whether or not a GQ structure forms. In some cases, the relationship between GQ structure and function may be linked directly to the their folded topology as mutations and dynamic equilibrium between conformers can modulate gene transcription.34–36 In other cases, adjacent promoter GQs, like those in the c-kit promoter region, may have antagonistic effects on gene expression.37 Therefore, a more complete understanding of GQ structure and dynamics at the atomistic level is needed to specifically target GQs with small molecules. Understanding the role of primary and secondary structure features, as well as direct and auxiliary ion interactions, is critical to this effort.
The human c-kit gene encodes for a receptor tyrosine kinase, a clinically validated drug target since aberrant c-kit gene expression is associated with diseases like gastrointestinal stromal tumors, mast cell disease, and leukemia.38–42 The c-kit promoter is guanine-rich and contains three GQ-forming sequences, called c-kit1, c-kit2, and c-kit*. These GQs are structurally unique42–45 and have suggested roles in modulating c-kit expression.37–41,45–48 The c-kit GQs are only separated by a few nucleotides (Figure 1A) and may influence each other’s stability and function.37,48–50 The c-kit1 GQ has been shown to decrease expression of c-kit transcripts, whereas the folded c-kit2 and c-kit* GQs increase expression.37,48 While many studies have focused on the c-kit1 and c-kit2 structures,23,41,43,44,51 the recent emergence of the c-kit* structure45 calls for further investigation into its dynamics and the behavior of simultaneously folded c-kit2/c-kit* GQs (Figure 1B), as their interrelation may be critical for biological function.
Figure 1.
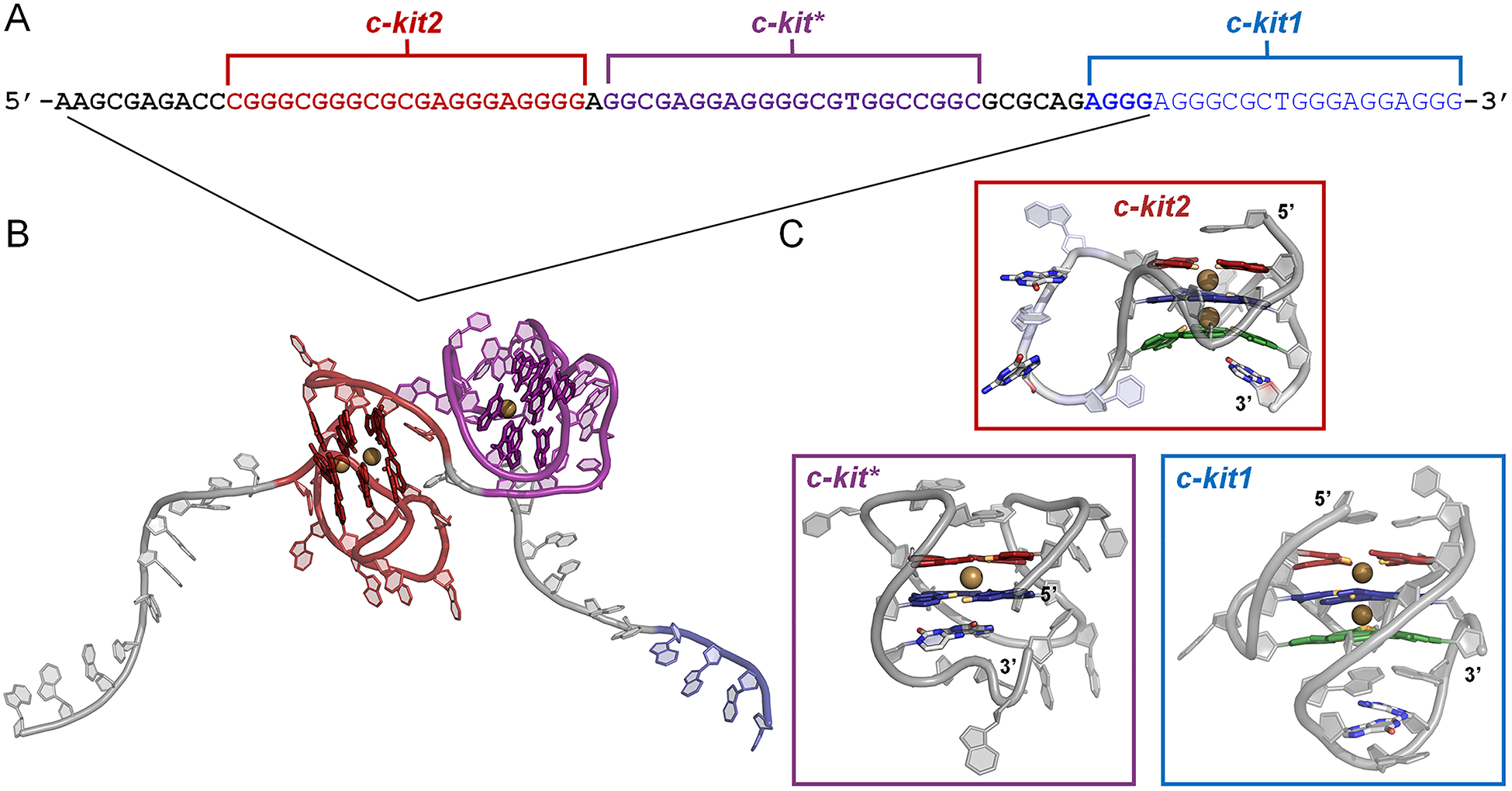
G-rich region of the c-kit promoter region. (A) Primary sequence of the G-rich region, including three GQ-forming sequences (c-kit2: red, c-kit*: purple, and c-kit1: blue). Bolded portions of the primary sequence indicate the sequences used to construct the linked c-kit2/c-kit* complex. (B) Energy-minimized c-kit2/c-kit* structure, rendered in cartoon. Core guanines are rendered as sticks. (C) Cartoon representation of experimental c-kit2, c-kit*, and c-kit1 GQ structures with bound K+ ions (gold). The guanine bases of the GQ core are colored by tetrad (1 – red, 2 – blue, and 3 – green).
The c-kit GQs are proposed to be in dynamic equilibrium between three states, (1) folded c-kit1 and unfolded c-kit2 and c-kit* GQs, (2) folded c-kit2 and unfolded c-kit1 and c-kit* GQs, and (3) folded, linked c-kit2/c-kit* and unfolded c-kit1 GQs.37 All three GQs are not believed to fold simultaneously.37 For this study, we focused on the dynamics of the folded c-kit2 and c-kit* structures. The parallel c-kit2 GQ has three associated solution NMR structures (Figure 2A). These solved structures have one or two point mutations in which loop guanines are substituted with adenine or thymine. However, there is no solved structure for the wild-type (WT) sequence. PDB entry 2KQH44 has a G21T substitution and will be referred to here as T21, PDB entry 2KQG44 has G10A and G21T substitutions and will be referred to as A10/T21, and PDB entry 2KYP43 has G12T and G21T substitutions and will be referred to as T12/T21. Differences in these solved structures provide opportunity to examine the effect of primary structure and various base-base interactions on the overall conformational ensemble of the c-kit2 GQ.
Figure 2.
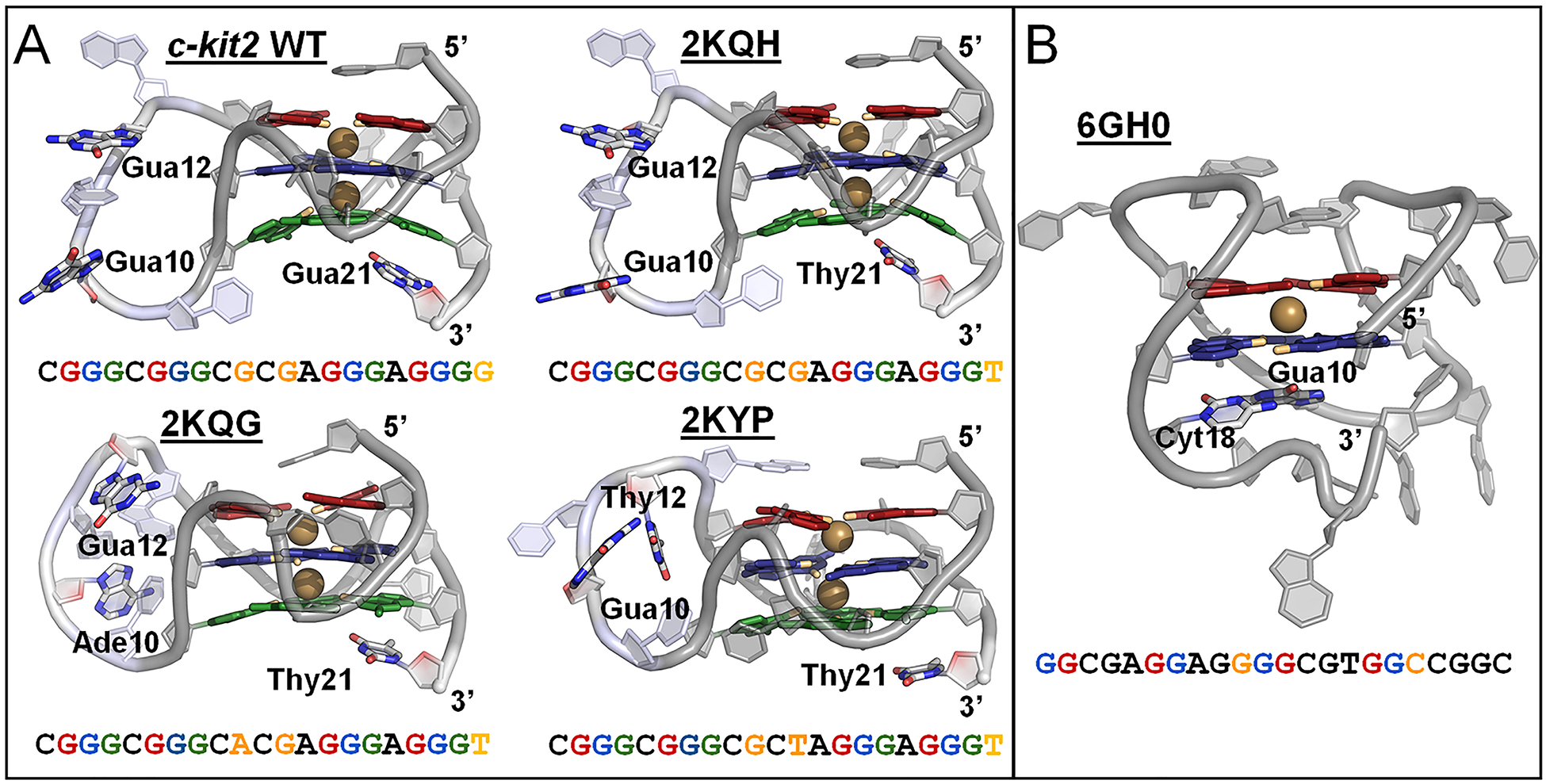
Structure and sequence of the c-kit2 and c-kit* GQs. (A) Cartoon representation of starting c-kit2 structures, highlighting bound K+ ions (colored gold) and nucleotides of the long propeller loop (residues 9–13; colored light blue). Residues 10, 12, and 21 vary in the c-kit2 structures and are represented as colored sticks and orange text. (B) Cartoon representation of starting c-kit* structure, highlighting a bound K+ ion (colored gold) and structurally important linker nucleotides (Gua10 and Cyt18) in colored stick representations and orange text. The guanine bases of the GQ cores are colored by tetrad: 1 – red, 2 – blue, and 3 – green (in c-kit2 GQs). O6 atoms pointing inward to coordinate K+ are colored orange.
At physiologically relevant temperature and salt concentration, the c-kit* sequence folds into a single, antiparallel GQ with two guanine tetrads (Figure 2B).45 This GQ is sometimes referred to as the SP GQ in literature because it is a validated binding site for SP1 and AP2 transcription factors.21,23,52 Interactions between folded c-kit2 and c-kit* are thought to stabilize their GQ forms, impairing reversion to duplex form, and function antagonistically to the c-kit1 GQ.37,53 Interplay between the c-kit GQs necessitates targeted drug design that produces therapeutic molecules able to distinguish between specific GQs, which calls for more precise knowledge of their dynamics.37
Molecular dynamics (MD) simulations are well suited to provide atomistic detail on important interactions within GQs. To date, GQs have been the subject of numerous theoretical investigations with MD simulations,54–67 which have made valuable contributions to our understanding of their dynamic behavior. However, most of these simulations relied on fixed-charge, pairwise-additive force fields (FFs) that approximate polarization via assignment of partial charges.68 The approximate nature of these FFs has limited their performance in systems where polarization is important, such as GQs.69 Inaccuracies have been noted in modeling core hydrogen bonding,56,57 cation binding,56,58,64 and flexible linker regions.55,59,63,65 While a careful combination of ion parameters and water models56,70 or system-specific reparameterization71 can diminish these artifacts, the explicit representation of electronic polarization is crucial to modeling GQs.58,67,72 Previously, we compared properties of the c-kit1 GQ using the CHARMM36 (C36) additive73 and Drude-2017 polarizable74,75 FFs and found issues with core-cation binding and preservation of loop structure using C36.67 The Drude polarizable FF, however, provided improved descriptions of K+ coordination in the tetrad core, reversible bulk K+ binding, and structurally important loop interactions.67 These improvements suggest that the inclusion of explicit electronic polarization is essential for investigating the dynamics and energetics of GQs and enable us to provide new insights on GQ conformational sampling as well as critical core-cation interactions.
In the present work, we build upon our previous study and employ the C3673 and Drude-201774,75 FFs to systemically study the c-kit2 and c-kit* GQs. We probe the effects of base substitutions and cation type (K+, Na+, and Li+) on the conformational sampling of the c-kit2 GQ and study the c-kit* GQ in the context of isolated (Figure 2B) and linked structures (Figure 1B). These simulations allow for comparison of the atomistic details of secondary structure, bulk cation binding, local cation alignment, and interaction energies that can be leveraged in the future for the design of selective, anticancer therapeutics.
METHODS
This work comprises MD simulations of isolated c-kit2 and c-kit* GQs and a linked c-kit2/c-kit* oligonucleotide containing both of these GQs (Table 1). The C36 additive73 and Drude-2017 polarizable74,75 FFs were used to simulate each system in ~150 mM KCl. The T12/T21 GQ was simulated in separate solutions of ~150 mM KCl, ~150 mM NaCl, and ~150 mM LiCl for reasons described below. In total, 45 independent simulations were performed, resulting in cumulative time of 43.5 μs (Table 1).
Table 1.
List of the contents and sizes of all simulation systems.
| Box Size (Å) | |||||
|---|---|---|---|---|---|
| c-kit2 WT | N/A† | 3 × 1 μs | 3 × 1 μs | ~150 mM KCl | 61 |
| T21 | 2KQH | 3 × 1 μs | 3 × 1 μs | ~150 mM KCl | 61 |
| A10/T21 | 2KQG | 3 × 1 μs | 3 × 1 μs | ~150 mM KCl | 53 |
| T12/T21 | 2KYP | 3 × 1 μs | 3 × 1 μs | ~150 mM KCl | 54 |
| T12/T21 | 2KYP | 3 × 1 μs | 3 × 1 μs | ~150 mM NaCl | 54 |
| T12/T21 | 2KYP | 3 × 1 μs | 3 × 1 μs | ~150 mM LiCl | 54 |
| c-kit* | 6GH0 | 3 × 1 μs | 2 × 1 μs 1 × 1.5 μs |
~150 mM KCl | 54 |
| Linked c-kit2/c-kit* | N/A† | N/A | 3 × 500 ns | ~150 mM KCl | 113 |
These systems were constructed as described in the Methods.
Isolated GQ structures.
The starting structures for the c-kit2 GQs were taken from the first structure in each deposited NMR ensemble in PDB entries 2KQH, 2KQG, and 2KYP.43,44 The sequence in PDB entry 2KQH differs from the c-kit2 WT only by the substitution of Thy for Gua at position 21. To generate the c-kit2 WT structure, we reverted Thy21 to Gua21 by deleting the Thy base and building the Gua base using the internal coordinate builder in CHARMM.76 For the c-kit* GQ, the starting structure was taken from the first structure in the NMR ensemble in PDB entry 6GH0.45 The deposited GQ structures lack bound ions expected in the tetrad core, so K+ ions were added to each GQ structure using the CHARMM program.76 In T12/T21 NaCl and LiCl simulations, Na+ or Li+ respectively were added to the core. These two ions were positioned to be coincident with the average coordinates of guanine base carbonyl oxygen (O6) atoms in consecutive tetrads, yielding symmetric ion coordination. For c-kit*, a two-tetrad GQ, one K+ ion was positioned at the average coordinates of guanine base O6 atoms in tetrads 1 and 2. Coordinates of all GQ systems with the manually positioned ions are available via the Open Science Framework at https://osf.io/2pfj8.
Linked c-kit2/c-kit*.
Since the folding and stability of the c-kit2 and c-kit* GQs are proposed to be interdependent, we wanted to better understand their dynamics in the context of an extended GQ-forming sequence. To do so, we prepared a linked c-kit2/c-kit* system (Figure 1B). The 10-nucleotide (nt) sequence upstream of c-kit2 was built in ideal B-DNA geometry77,78 (α=298°, β=168°, γ=51°, ε=−164.3°, and ζ=−89.7°) on the 5’-end of the c-kit2 WT structure. One Ade nucleotide separates the c-kit2 and c-kit* GQs and was built on the 3’-end of c-kit2 WT GQ to connect to the 5’-end of c-kit* GQ. The coordinates of c-kit* were translated to position its 5’-hydroxyl group near the 3’-hydroxyl group of this constructed Ade linker to allow for the insertion of the intervening phosphate group using the internal coordinate builder in CHARMM. The 10-nt sequence downstream of c-kit* was then built in ideal B-DNA geometry on the 3’-end of the WT c-kit* structure. K+ ions were placed in the core of the GQs (two ions in WT c-kit2; one ion in c-kit*) as described above in the preparation of the isolated GQ systems. Since an experimental structure of the intact c-kit2/c-kit* GQ complex is unknown, our goal in carrying out the above steps was simply to generate a plausible linked c-kit2/c-kit* structure, free of steric clashes, while acknowledging that multiple initial starting structures are possible given the flexibility of a 1-nt DNA linker. The conformational dependence of this initial structure will be evaluated in future work.
System Construction.
Each simulated system was first prepared with the additive C36 nucleic acid FF73 by centering the GQ in a cubic unit cell (minimum box-solute distance of 10 Å), which was filled with CHARMM-modified TIP3P water79–81 and ~150 mM KCl, including neutralizing K+ counterions. To systematically evaluate the effects of cation type and charge density, the T12/T21 c-kit2 GQ taken from PDB entry 2KYP was also prepared with ~150 mM NaCl and ~150 mM LiCl. Having previously observed a central role for Thy in coordinating ions in the c-kit1 GQ,67 we sought to examine the role of this nucleobase in ligating different ion types. Therefore, we chose to simulate the c-kit2 double Thy mutant in this context. Standard CHARMM ion parameters82 were applied to KCl, NaCl, and LiCl. To relax possible steric clashes within these systems, energy minimization was carried out in CHARMM by performing 500 steps of steepest descent minimization and 500 steps of adopted-basis Newton-Raphson minimization.
After energy minimization, equilibration was carried out in NAMD83 for 1 ns under an NPT ensemble with position restraints applied to all heavy atoms of each structure (GQ and bound ions). Water and mobile ions were unrestrained and therefore free to move during equilibration. The NPT ensemble was maintained at 298 K using the Langevin thermostat84 (friction coefficient = 5 ps−1), and a constant pressure of 1 atm using the Langevin piston method85 (oscillation period = 200 fs, decay time = 100 fs) and isotropic box scaling. Periodic boundary conditions were applied in all spatial dimensions. The short-range van der Waals forces were smoothly switched to zero from 10 – 12 Å. Electrostatic interactions were calculated with the particle mesh Ewald method86,87, a real-space cutoff of 12 Å, and a Fourier grid spacing of 1 Å. All bonds to hydrogen atoms were constrained using the SHAKE88 algorithm. The water molecules were kept rigid with SETTLE89, allowing an integration time step of 2 fs.
Drude systems were prepared from the final coordinates of the additive equilibration simulations by adding Drude oscillators and lone pairs to all heavy atoms in the system with the CHARMM program. The TIP3P water molecules used in the additive simulations were converted to the polarizable SWM4-NDP90 model and the Drude-2017 FF23,24 was applied to the GQ and ions, using the ion parameters of Yu et al.91 and the specific ion-nucleic acid nonbonded parameter refinements described by Savelyev and MacKerell.92 Energy minimization was performed to relax the Drude oscillators using steepest descent minimization and adopted-basis Newton-Raphson energy minimization. NPT equilibration was then carried out in NAMD at 298 K and 1 atm pressure using extended Lagrangian integration, implemented in NAMD as Langevin dynamics.93,94 A “physical” thermostat was coupled to all the real atoms in the system at 298 K with a friction coefficient of 5 ps−1 and Drude oscillators were coupled to a low-temperature relative thermostat at 1 K with a friction coefficient of 20 ps−1. The nonbonded treatment was the same as in the additive equilibration simulations, except that the van der Waals potential, not the force, was switched from 10 – 12 Å, which is standard practice for the Drude FF.95 As in the additive equilibration, heavy atoms of the structure were restrained during equilibration. The time step for polarizable simulations was 1 fs. A “hard wall” constraint96 was also applied to allow a maximum Drude-atom bond length of 0.2 Å and avoid polarization catastrophe. Equilibration of the Drude systems was carried out for 1 ns. For both C36 and Drude systems, three independent replicates were produced for each system by generating different random velocities at the outset of equilibration.
Production MD Simulations.
All unrestrained simulations were performed using OpenMM.97,98 Simulations of isolated GQs were carried out for 1 μs each, resulting in a total sampling time of 3 μs for each system with both FFs. For C36 simulations, temperature was maintained using the Andersen thermostat99 with a collision frequency of 1 ps−1. For Drude simulations, the NPT ensemble was maintained as described above. In both C36 and Drude simulations, the Monte Carlo barostat in OpenMM was used to isotropically regulate pressure, with box scaling attempted every 25 integration steps.
Ion Sampling around GQs.
Ion occupancy maps were generated as described in our previous work.67 Briefly, the system volume was divided into discrete, 1-Å3 volume elements (voxels). All snapshots from each system (replicates 1–3) were analyzed and the location of each ion was assigned to the nearest voxel. An isosurface cutoff of ≥1% was chosen for visualization, meaning a voxel must be occupied by an ion for at least 1% of the frames. This permissive cutoff enables a general description of ion sampling in flexible systems and is sufficient to resolve discrete volumes of preferential ion binding.
Bulk Ion Alignment.
A bulk ion was considered aligned along the GQ tetrad axis if its distance was ≤ 3.5 Å from the nearest tetrad center (above tetrad 1 or below tetrad 3 in c-kit2, above tetrad 1 or below tetrad 2 in c-kit*, respectively; see Figure 2). The tetrad center was defined by the center of mass of guanine base O6 atoms in tetrad 1 and 2 (top alignment) or tetrad 2 and 3 (bottom alignment). A distance analysis was used to determine an appropriate cutoff for ion alignment by tracking the average distance between bound ions and the tetrad center. Between the six isolated GQ systems, the average ion distances were 3.2 ± 0.2 Å for K+, 2.8 ± 0.7 Å for Na+, and 2.7 ± 0.7 Å for Li+. A 3.5-Å cutoff was thus chosen for this analysis as it is approximately one standard deviation above the average for the three cation types, allowing us to account for variability in ion alignment using a uniform criterion.
RMSD-Based Clustering.
The CHARMM clustering function100–102 was employed to generate clusters of the c-kit2 and c-kit* loop regions in isolated and linked systems as well as core-ion clusters in the T12/T21 simulations. Clusters were generated by pooling all replicates within a simulation set and separating frames with a self-defined maximum radius (1 Å for core-ion clusters and 3 Å for loop clusters). The cluster radius set in the core-ion clusters was more stringent than the value in the loop clusters because the overall structure was very rigid, thus requiring a smaller value to discriminate between different conformations.
Interaction Energy.
To determine the impact of water on the interaction energy between core ions and tetrad guanine bases in the various GQs, we performed interaction energy analysis as described in our previous work.67 “With solvent” interaction energies were calculated through direct analysis of the MD trajectories, considering the core ions and guanine tetrad bases with water in the C36 and Drude systems. To calculate the “no solvent” interaction energies, we removed water from the trajectory files and allowed the dipoles to relax by reoptimizing the Drude oscillators with all real atoms fixed and recalculating the interaction energy. The difference between the “with solvent” and “no solvent” interaction energies represents the multi-body contribution of water to the total interaction energy.
RESULTS AND DISCUSSION
Force Field Assessment and Analysis Strategy.
Various FFs are currently available for simulating nucleic acids. The ability of these FFs to model diverse noncanonical structures is a frequently discussed topic in the field and an important consideration for evaluating simulation outcomes. Previous MD simulations have made valuable contributions to our understanding of GQs,54–67 however, the use of additive FFs has resulted in incomplete descriptions of the tetrad core,56,57 ion binding,56,58,64 and GQ-linker regions.55,59,63,65 Work by Gkionis et al. showed that explicit electronic polarization is important for modeling ion-ion interactions in the GQ core58 and we previously demonstrated that the Drude polarizable FF models GQ structures with improved structural agreement to experimental data, including proper core ion retention.67,72 The long propeller loop region of the c-kit2 GQ and the expected intrinsic structural plasticity of c-kit* are challenges for any biomolecular FF. To this end, it was important to determine if the C36 or Drude-2017 FFs were able to model the GQs sufficiently, compared to experimental data. Thus, to assess these force fields and further understand the contributions of electronic structure to GQ-ion interactions and conformational sampling, we performed simulations of the c-kit2 and c-kit* GQs with both the C36 nonpolarizable and Drude-2017 polarizable FFs. We characterized the success of these simulations in terms of deviation from experimental structures, including root-mean-square deviation (RMSD) and core hydrogen bond counting. We also examined GQ-ion interactions, paying particular attention to the retention of core ions that should remain coordinated along the tetrad stem of each structure. As part of this investigation, we studied the T12/T21 c-kit2 GQ in the presence of LiCl and NaCl to understand (1) the selectivity of GQs for K+, (2) what role, if any, thymine plays in coordinating monovalent ions, and (3) if there are intrinsic FF limitations across all ion types and if inclusion of electronic polarization leads to altered physical properties given the different polarizabilities of these ions.
Simulations of c-kit2 with the C36 FF produced structures that deviated considerably from the experimental data. Full details of the structural analyses are presented in the Supporting Information and will be summarized here. The c-kit2 structures displayed perturbed tetrad cores, demonstrated by high RMSD of all core nucleotides and tetrad bases (Supporting Information, Table S1 and Figure S1). These perturbations were insensitive to ion type (Supporting Information, Table S1 and Figure S2). Across all c-kit2 C36 simulations, core Hoogsteen hydrogen bonds were lost over time (Supporting Information, Figures S1 and S2), and core ions were expelled (Supporting Information, Figures S3 and S4). The number of Hoogsteen hydrogen bonds oscillated between 5–8 in all tetrads throughout the simulations. These bonds broke and reformed early in the simulations but were lost permanently in many replicates. This disruption occurred in all tetrads of the c-kit2 GQs. Core ion expulsion occurred quickly and consistently in C36 simulations, maintaining the expected coordination for less than 1% of the simulation time (Supporting Information, Figures S3 and S4). We have previously demonstrated that C36 FF simulations led to high RMSD, but now we show that important core interactions (hydrogen bonds and ion occupancy) are disrupted in systems of KCl, NaCl, and LiCl. These issues have long been reported in additive FFs57 and attributed to their nonpolarizable nature;58 however, large perturbation of the tetrad core should be avoidable with proper FF selection or specific combinations of water models and ion parameters.56 Interestingly, simulations of the c-kit2 GQs in LiCl and NaCl produced smaller deviations from the experimental structure and retained a greater number of core Hoogsteen hydrogen bonds. Though all ions were uniformly expelled from the tetrad core, the dominant structures of T12/T21 in LiCl and NaCl may have been trapped in lower-RMSD states, whereas simulations in KCl led to structures that sampled a wider range of conformations. Together, these results highlight fundamental limitations in describing GQs and monovalent ions with nonpolarizable FFs, specifically C36.
So far, the primary shortcomings of the C36 FF seem to arise from core ion expulsion. Since ion expulsion in additive FFs has largely been attributed to inter-cation electrostatic repulsion58,64 and the two-tetrad c-kit* GQ has only one core ion, it was reasonable to believe that its C36 simulations would avoid such deviations. However, the structures produced in c-kit* simulations with the C36 FF also exhibited tetrad core perturbations with high RMSD and disrupted core hydrogen bonding (Supporting Information, Figure S5). Core ion expulsion did not occur, but C36 simulations produced fewer ion-DNA interactions compared to the outcomes of the Drude-2017 simulations (Supporting Information, Figure S6). While we previously believed that ion expulsion disrupted the core and contributed to broad structural deviations in GQs,67,72 the present findings suggest that the observed distortions are not solely attributable to incorrect ion-ion interactions. GQ simulations with the C36 FF may suffer from other force field-specific artifacts in the tetrad core, limiting its relevance in modeling GQs. For these reasons, we will focus on the results of the Drude simulations for the remainder of this work.
Conformational Dynamics of Isolated c-kit2 GQs.
To characterize the conformational dynamics of the c-kit2 GQs with the Drude-2017 FF, we evaluated the RMSD, core hydrogen bonding, root-mean-square-fluctuation (RMSF), and backbone dihedral angle sampling of the WT, T21, A10/T21, and T12/T21 GQs. The tetrad core in each of these GQs was stable, with lower RMSD values, relative to the simulations with C36, and preserved Hoogsteen hydrogen bonding (Supporting Information, Table S2 and Figure S7). Core atoms were more rigid than loop regions in all simulations, demonstrated by lower per-nucleotide RMSD and RMSF values (Supporting Information, Figures S8 and S9). In comparing the simulation outcomes of different structures, we note that the T12/T21 GQ displayed lower RMSD than the WT, T21, and A10/T21 GQs. Though all the c-kit2 GQs simulated here differ in primary structure, the secondary structure (base pairing and stacking) within the WT, T21, and A10/T21 GQs are similar (Figure 2 and Supporting Information, Figure S10). As such, the WT, T21, and A10/T21 GQs displayed similar RMSD and RMSF values for most nucleotides. However, the T12/T21 GQ has a non-canonical base pair (Cyt1:Ade13) stacked above tetrad 1 that helps structure the long propeller loop region and preserve the tetrad core. Due to this base pairing, the flexibility of loop and linker regions in the T12/T21 GQ was lower than that of the WT, T21, and A10/T21 GQs. The 5’-terminal nucleotide (Cyt1) had the lowest fluctuation in the T12/T21 GQ, while the 3’-terminal nucleotide (Gua21) was the least variable in the WT GQ, possibly due to G:G base stacking with tetrad 3 or interactions with bulk K+ ions (discussed later). The Cyt1:Ade13 base pairing is present in all twelve structures of the T12/T21 NMR ensemble, was preserved throughout the simulations, and contributes to the ordered 5-nt loop.43 However, this Cyt1:Ade13 base pairing is not prevalent in the other c-kit2 structures (Supporting Information, Figure S10) and the base pairing of a terminal base may not be possible in the full sequence.
To further evaluate the compatibility of the simulated ensembles with the experimentally derived NMR structures, the backbone and glycosidic dihedral angles and sugar puckering of the T21, A10/T21, and T12/T21 GQs were determined and compared to their respective NMR ensembles (Supporting Information, Figures S11–S13). No comparison could be made in the case of the WT structure, as it was modeled based on the T21 experimental structure. In general, the sampling of these GQs was in good agreement with the experimental ensembles. Gua8, a tetrad guanine, is assigned a North pucker in their respective NMR ensembles, which is uncommon for a base that is also assigned an anti state for its χ dihedral. The A10/T21 and T12/T21 GQs sampled South pucker at this nucleotide in Drude simulations. Even with these few deviations, it is clear that the Drude-2017 FF produced dihedral sampling that is consistent with the respective experimental structural ensembles.
Overall, the Drude FF was better able to model γ sampling than C36, which sampled exclusively gauche+ states as in canonical B-form DNA (Supporting Information, Figures S12 and S13). We note some deviations in γ sampling in propeller loops of the A10/T21 and T12/T21 GQs, whose experimental structures generally contained more trans configurations than predicted by the simulations. Interestingly, studies suggest that the first residue of a propeller loop should adopt γ dihedrals near trans;55 however, Cyt9 γ dihedrals were more predominantly gauche+ in our simulations. Analysis of the entire A10/T21 NMR ensemble yields an average γ value of 155 ± 50°, while the corresponding averages from the C36 and Drude simulations were 65 ± 34° and 90 ± 72°, respectively. Similarly, the average γ value in the T12/T21 NMR ensemble is 216 ± 88°, while C36 produced an average of 63 ± 33° and 62 ± 16° with the Drude FF. In both systems, the Cyt9 γ dihedral sampled the trans state for 10–80 ns before adopting and maintaining a gauche+ geometry. Conversely, the Cyt9 γ dihedral is shifted towards gauche+ in the T21 NMR ensemble, with an average value of 78 ± 73°. Both FFs produced good agreement with the experimental geometry at this position (C36 = 66 ± 20° and Drude = 65 ± 31°). This infrequent Cyt9 γ-trans sampling may indicate a FF limitation in both the C36 and Drude FFs, which has previously been reported in simulations with AMBER FFs.55,103 We note that the Drude FF does not explicitly penalize the γ-trans state and reproduces the QM intrinsic potential energy surface for γ rotation very accurately,74 though these calculations were performed on canonical DNA geometries. Future FF refinement that considers backbone geometries of typical propeller loop dihedrals may be necessary to improve the model.
In short, differences in primary and secondary structure across the c-kit2 GQs resulted in comparable and stable tetrad cores, but varying loop dynamics. Simulations of the isolated structures showed persistent interactions involving 5’-and 3’-terminal residues that influenced structural deviation and base fluctuation. Since the flexibility of c-kit2 terminal regions may differ in the native state, both due to the terminal residue substitutions in the solved structure and interactions of other nucleotides that will be present, further work is needed to understand the c-kit2 GQ in the context of an extended sequence. We discuss such a system later.
Ion Sampling around Isolated c-kit2 GQs in KCl solution.
Nucleic acids contain many ion binding sites with different configurations and affinities; however, ion binding in GQs is particularly important because it governs folding and stability.29,30 Therefore, we evaluated the K+ sampling around the c-kit2 GQs. Core K+ cations were retained in all simulations of the c-kit2 GQs with the Drude-2017 FF and displayed the expected bipyrimidal antiprismatic coordination with guanine O6 atoms. Bulk K+ ions bound to surface-exposed moieties and at the outer faces of tetrads in the GQ stems (above tetrad 1 and below tetrad 3). Such observations emphasize the improvement of the polarizable model over simulations with additive FFs, which generally overestimate inter-cation electrostatic repulsion, resulting in disrupted core ion coordination.56,58,64 These improvements were first demonstrated by our study on the c-kit1 GQ, which showed that loop nucleotides near the core can play an active role in bulk ion alignment.67
Since there are differences in primary and secondary structure in loop regions near the core, analysis of GQ-ion interactions in substituted c-kit2 structures is particularly interesting in determining if these single and double mutations influence K+ binding. The c-kit2 GQs exhibited different ion sampling patterns (Figure 3) that may be due to differences in primary and secondary structure (Supporting Information, Figure S10). Above tetrad 1, the WT, T21, and A10/T21 GQs have a free 5’-cytosine that can interact with bulk ions, whereas the T12/T21 GQ has the Cyt1:Ade13 base-pair that occludes the top of the tetrad. Consequently, we observed similar bulk ion alignment above tetrad 1 in WT, T21, and A10/T21 GQs and no such alignment in T12/T21. Below tetrad 3, the WT GQ has a 3’ guanine, which stacked tightly with the tetrad core during the simulations, while the T21, A10/T21, and T12/T21 GQs have a free 3’-thymine. Bulk ion alignment below tetrad 3 was observed in all four systems with the highest occupancy site in the WT GQ (interacting with the 3’-guanine).
Figure 3.
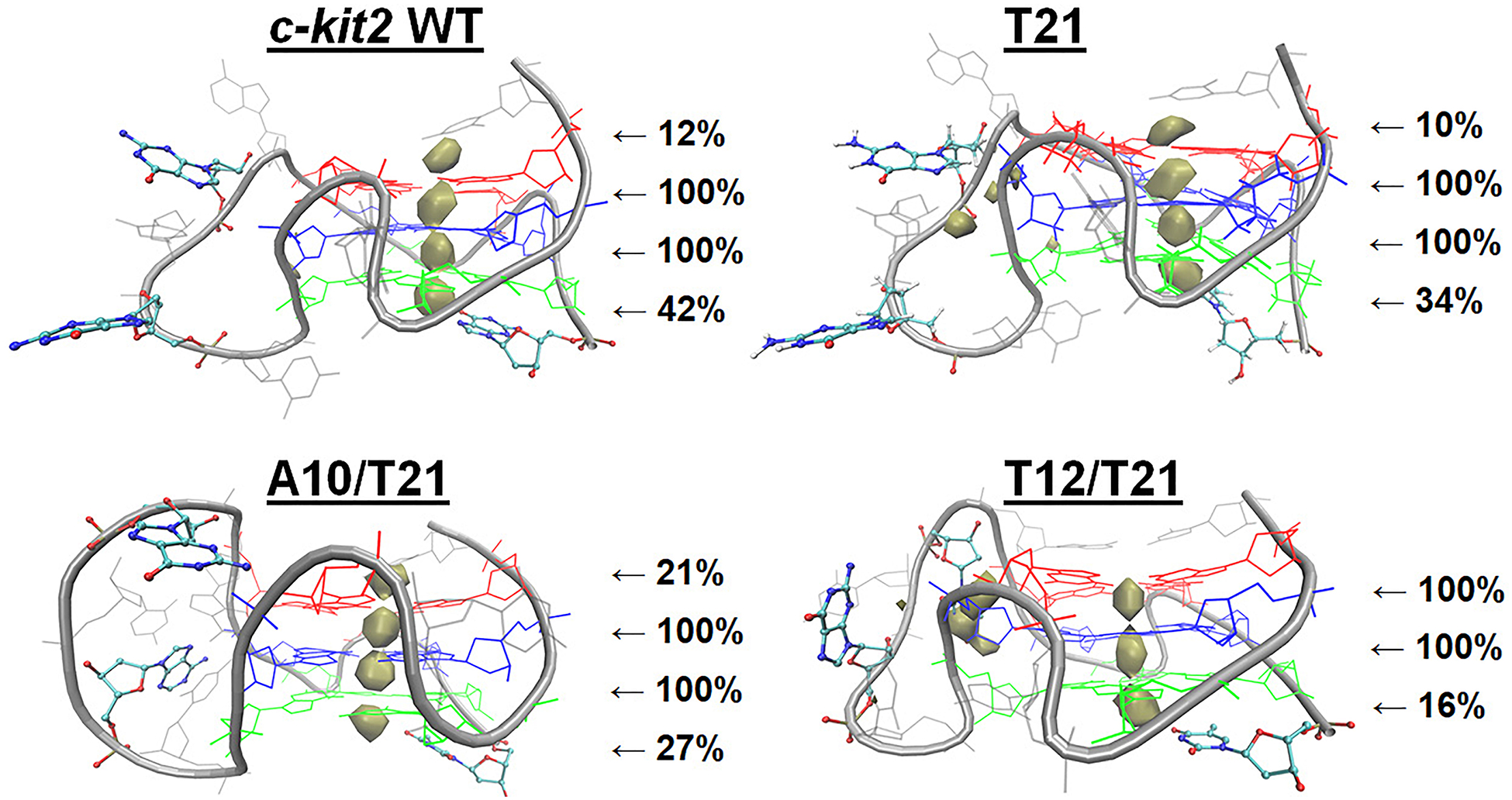
Drude K+ interaction maps for c-kit2 GQs. Ion sampling around c-kit2 GQs is shown at an occupancy threshold of ≥1% and the displayed percentages indicate the persistence of each ion at that location throughout the three replicate simulations.
In previous work with the c-kit1 GQ, we found that an advantageously positioned thymine could participate in bulk K+ alignment in polarizable simulations.67 Here, we report for the first time that other advantageously positioned nucleotides, including cytosine (Cyt1), thymine (Thy21), and guanine (Gua21) can participate in bulk ion alignment. Differences between ion occupancy in these systems suggest that while different bases can help coordinate ion binding, their efficacy may vary. Still, the participation of terminal residues in bulk ion alignment calls for investigations of extended GQ structures, in which their flexibility may differ as a function of other nucleotides present.
Simulations of T12/T21 in KCl, NaCl, and LiCl.
Since we previously observed pronounced thymine-ion interactions in the c-kit1 GQ, we also prepared the T12/T21 systems in KCl, NaCl, and LiCl to systematically compare bulk and bound cation influence on the folded GQ. RMSD and RMSF values were higher in the presence of LiCl and NaCl relative to those obtained in simulations with KCl (Supporting Information, Table S2 and Figures S14–S16). This observation is particularly clear in the case of heavy-atom RMSD (Table S2), where the RMSD trend is LiCl > NaCl > KCl. The RMSD and RMSF of the long propeller loop in c-kit2 GQs were comparable in NaCl and LiCl, both of which were elevated relative to KCl. These results suggest that KCl is better able to stabilize the tetrad core and structure the long propeller loop.
LiCl led to more pronounced Hoogsteen hydrogen bond disruption in the tetrad core that corresponded to increased RMSD (Supporting Information, Figure S14). During GQ folding, cations stabilize the tetrad core by counteracting the repulsion of proximal O6 atoms.29 It is possible that Li+ ions cannot counteract this electronegativity and O6-O6 repulsion leads to increased occurrences of hydrogen bond breaking. In this case, instability in systems with LiCl suggests the Drude FF performs well in modeling ion-DNA interactions, as Li+ is not thought to coordinate GQ folding or promote stability.104–106 Further, the relationship between core hydrogen bond breaking and increasing RMSD values demonstrates that local deviation or destabilization can influence the overall structure of the GQ. In all, these observations agree with the known GQ stabilization preference (K+ > Na+ > Li+)107 and suggests that fundamental differences between cation type affects c-kit2 GQ stability on the microsecond timescale. Future studies will be required to confirm whether this principle applies to GQs with different folds and sequence compositions.
MD simulations have proven to be a robust tool for studying ion binding sites and comparing the arrangements, dynamics, and occupancies of various monovalent ions.92,108–110 To further understand the local differences described above, we examined core ion alignment in systems with KCl, NaCl, and LiCl. Ion-tetrad relative distance distributions and core-ion clusters (Figure 4) show the dominant features of ion coordination in the c-kit2 tetrad core. In KCl, core ions adopted bipyrimidal antiprismatic coordination, whereas core ions sampled both bipyrimidal antiprismatic and planar coordination in NaCl, and planar coordination in LiCl. Further, Li+ ions often interacted closer to two or three guanine bases in each tetrad such that the core ions were not aligned vertically on the tetrad axis. The Na+ ion-tetrad relative distance distributions has two traces between tetrad 1 and 2, as shown in Figure 4. The smaller trace is present because a bulk Na+ ion bound to the tetrad core towards the end of replicate 2, pushing ion 1 out and ion 2 between tetrad 1 and 2. The cation coordination demonstrated in our simulations is supported by a density functional study of guanine tetrads and metal ions, which showed that core K+ ions align symmetrically between stacked tetrads and are too large to adopt planar coordination.111 Conversely, Na+ ions are equally likely to be planar as they are antiprismatic, and Li+ ions have a strong attraction to O6 atoms, resulting in tight, planar coordination with the tetrad.111
Figure 4.
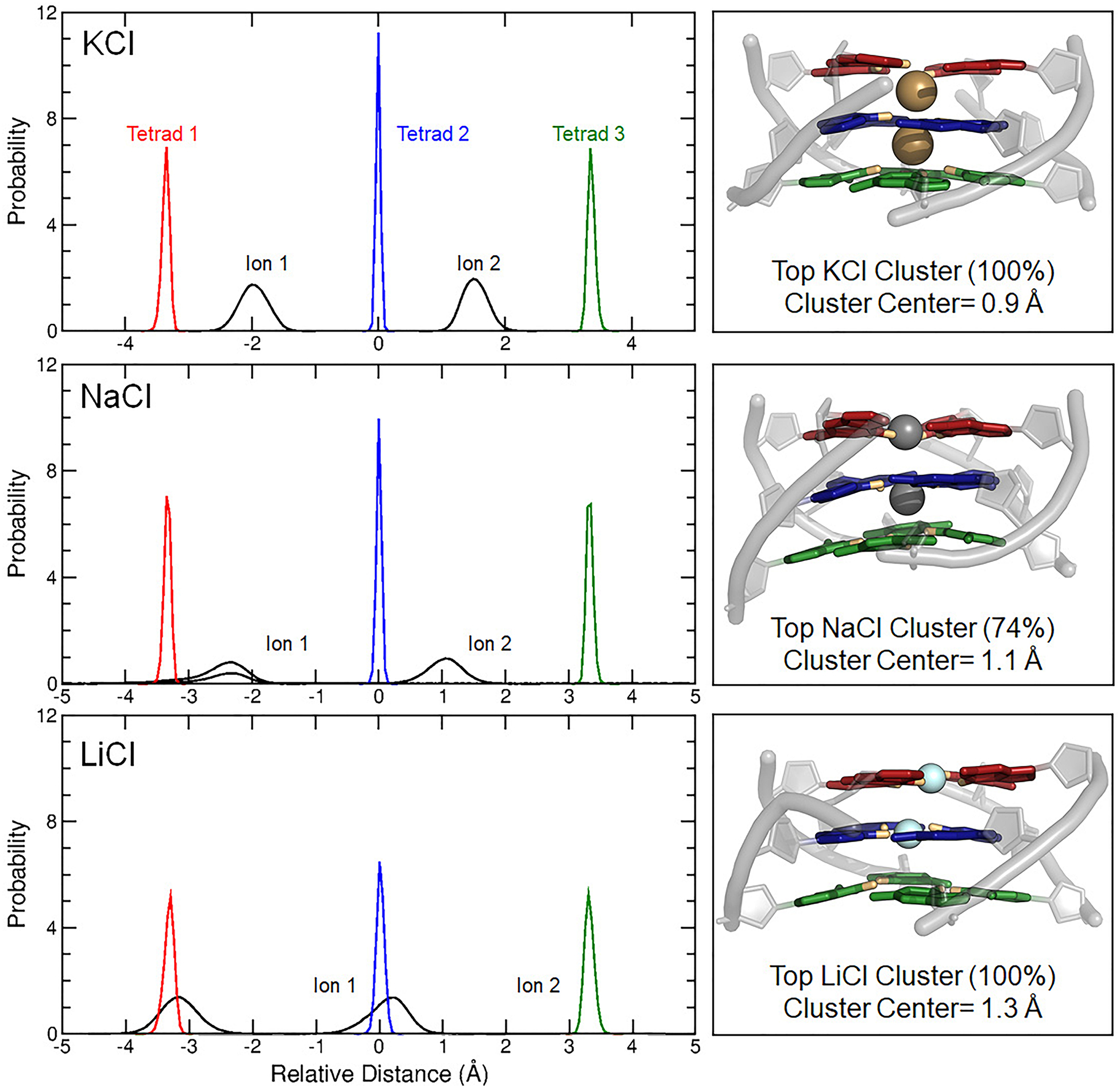
Ion-tetrad relative distance distributions and core-ion clusters show the position of the bound ions in the tetrad core. All distributions are centered on the tetrad core center of mass (relative distance = 0 Å). Ion-tetrad relative distances for all replicates were combined to produce distributions for KCl, NaCl, and LiCl. Cartoon renderings of the top tetrad core-ion cluster in each ion type (right) to illustrate common local alignment in these systems.
We also generated ion interaction maps of the T12/T21 GQ in NaCl and LiCl (Figure 5). In general, cation sampling around the GQ was very similar between KCl and NaCl, though bulk Na+ aligned below tetrad 3 more frequently (45% in NaCl vs 16% in KCl). Systems of LiCl displayed more pronounced sampling around the long propeller loop and the GQ backbone than KCl and NaCl, and bulk Li+ ions aligned below tetrad 3 to K+ ions. Differences in loop and backbone binding are of interest because external (non-core) ion-DNA interactions contribute to GQ folding and stability of the folded state.112,113 Binding of cations to the negatively charged backbone should reduce electrostatic repulsion and stabilize folding,112,113 yet in our simulations, structural deviation was greatest in simulations of LiCl, which had the most extensive ion-backbone interactions. This incongruity, combined with Li+-core coordination disrupting core structure, suggest solutions of LiCl are not suitable for maintaining GQ stability.
Figure 5.
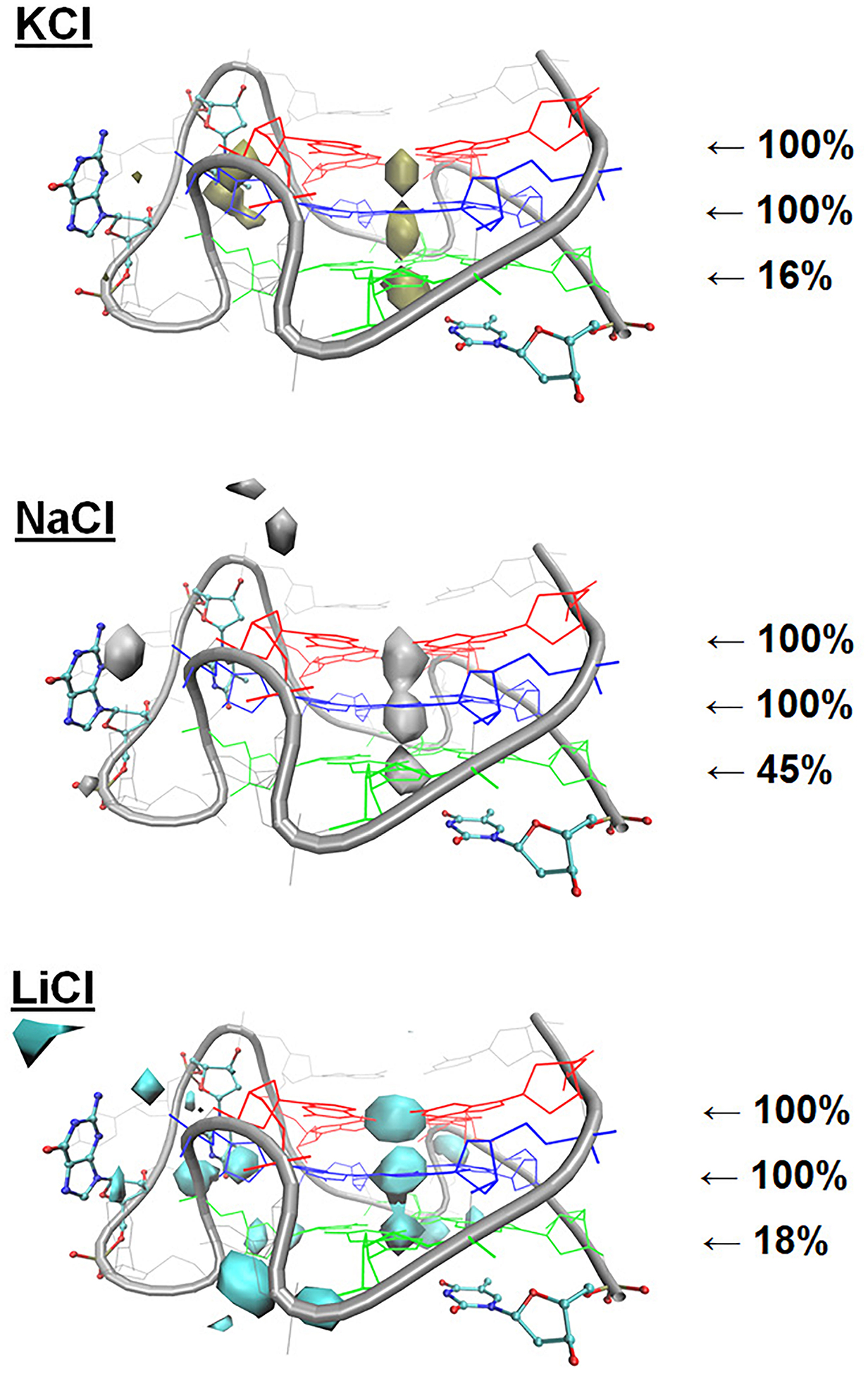
Ion interaction maps around the T12/T21 c-kit2 GQ with the Drude-2017 FF from simulations of the isolated GQs in KCl, NaCl, and LiCl. The isosurface value for ion sampling was set at an occupancy threshold of ≥1%. The displayed percentages reflect the persistence of each ion at the indicated location across the three replicate simulations, expressed as the fraction of snapshots in which an ion was aligned with the tetrad stem (see Methods).
Since base hydration, cation hydration, and ion-DNA interactions are integral to GQ stability and have suggested roles in cation preference,29,113–115 we calculated core-ion interaction energies with and without solvent (see Methods). Evaluating these multi-body effects is possible with polarizable FFs like Drude-2017 and helps clarify how differences in local coordination (described above) and cation charge density influences the tetrad core. The calculated values (Table 2) show that tetrad core-ion interaction energies are strengthened as charge density increases (LiCl < NaCl < KCl). Hybrid density-functional calculations have also reported that tetrad-ion interaction energies are strongest with Li+ ions, followed by Na+ and K+ ions.111 Interaction energies were most variable in NaCl simulations, where ions exchanged between bipyrimidal antiprismatic and planar coordination, as noted above. In general, the core-ion interaction energies in the presence of water (“With Solvent” in Table 2, computed directly from snapshots in the simulation trajectories) were more favorable than those computed after removal of water and dipole relaxation (“No Solvent” in Table 2), indicating that the interaction energies between coordinated ions and the guanine tetrad bases were strengthened by the presence of water. These results demonstrate that water plays an active role in core-ion energetics, and that the strength of this energetic contribution varies as a function of the monovalent cation type.
Table 2.
Drude interaction energies (EINT, kcal/mol) between the two core ions and the guanine bases of each tetrad in the T12/T21 GQ with KCl, NaCl, and LiCl. “With Solvent” are the interaction energies computed directly from the trajectory and “No Solvent” are interaction energies after removal of solvent and reoptimization of the Drude oscillators. ΔEINT is the difference between the “With Solvent” and “No Solvent” interaction energies.
| KCl | ΔEINT | ||
|---|---|---|---|
| Tetrad 1 | −27.5 ± 4.4 | −24.2 ± 4.7 | 3.3 ± 4.6 |
| Tetrad 2 | −66.2 ± 3.9 | −64.3 ± 4.0 | 2.1 ± 4.0 |
| Tetrad 3 | −34.0 ± 3.4 | −31.1 ± 4.4 | 2.9 ± 3.9 |
| NaCl | ΔEINT | ||
| Tetrad 1 | −56.7 ± 6.7 | −49.6 ± 6.6 | 7.1 ± 6.7 |
| Tetrad 2 | −61.0 ± 28.8 | −59.8 ± 27.9 | 1.2 ± 28.4 |
| Tetrad 3 | −24.9 ± 18.6 | −25.1 ± 18.7 | 0.6 ± 18.7 |
| LiCl | ΔEINT | ||
| Tetrad 1 | −102.5 ± 9.9 | −91.7 ± 10.7 | 10.8 ± 10.3 |
| Tetrad 2 | −102.6 ± 9.8 | −98.2 ± 10.7 | 4.4 ± 10.4 |
| Tetrad 3 | −9.6 ± 9.0 | −9.0 ± 9.1 | 0.6 ± 9.0 |
The final analysis we performed on the T12/T21 c-kit2 GQ was to calculate nucleobase dipole moments as a function of ion type. Results for all bases are listed in Supporting Information, Table S3. This analysis revealed that different cations have a small impact on the base dipole moments. Adenine depolarized as a function of decreasing cation size (increasing charge density), and pyrimidine bases (cytosine and thymine) polarized more as a function of increasing charge density. Thymine bases were the most sensitive, increasing from 6.9 ± 0.7 D in KCl to 7.7 ± 0.5 D in NaCl and 8.1 ± 0.4 D in LiCl. The large differences in thymine dipole moments may be due to a combination of advantageous positioning in the c-kit2 GQ (below tetrad 3), increased ion interaction, and the intrinsic flexibility of the thymine base electronic structure. Overall, guanine bases were not generally sensitive to changes in cation type (Supporting Information, Table S3) but when decomposed on a per-tetrad basis, small differences emerged in the guanine bases in different tetrads, though shifts in the base dipole moment were only on the order of 0.1 – 0.2 D. Given the small differences in nucleobase dipole moments, clearly the EINT results shown in Table 2 arise primarily due to changes in ion size, coordination, and charge density.
Conformational Dynamics of the Isolated c-kit* GQ.
Towards a more complete understanding of the dynamics of the promoter c-kit GQs, we performed simulations of the isolated c-kit* GQ, which has been suggested to have structural and functional connections to the c-kit1 and c-kit2 GQs and has a unique, two-tetrad topology.37,45 The heavy-atom RMSD values of the structure were high in some cases and varied across replicates, while the core nucleotide RMSD was ~1.0 Å in each replicate simulation. Hoogsteen hydrogen bonds in the tetrads remained stable throughout the simulations (Supporting Information, Table S2 and Figure S17). The most notable difference in per-nucleotide RMSD and RMSF was in the 3’-tail loop, which includes Cyt18-Cyt19-Gua20-Gua21-Cyt22 (Supporting Information, Figures S18 and S19). The dynamics of this region are of interest because the stability of the c-kit* GQ depends on the network of interactions in the 3’-tail.45 In fact, structural studies show that disruption or truncation of the 3’-tail region inhibits GQ folding.45 In one of our simulations, the 3’-tail region unfolded at ~800 ns, so we decided to extend this simulation to determine if the observed behavior was reversible. After ~300 ns of fluctuation, the 3’-tail region folded back towards the c-kit* GQ structure, adopting a native-like conformation by 1.1 μs and remaining there until the simulation was stopped at 1.5 μs (Supporting Information, Figure S17). The 3’-tail region remained packed against the rest of the c-kit* GQ in the other two replicate simulations, varying only slightly in its position over 1-μs simulations.
To better illustrate these important interactions and the flexibility observed in this region, we performed RMSD-based clustering of the tail nucleotides (Figure 6). Though the predicted fold-back motif was observed in the four most highly populated clusters (Clusters 1–4), secondary structure within the tail was less consistent, demonstrating the variability of the tail region in simulations of the isolated GQ. The important Cyt18:Gua10 WC-base pair remained intact throughout the simulations (100% of the time) and the core structure was preserved. Other studies suggest the c-kit* GQ is in dynamic equilibrium between folded and unfolded states.37 The flexibility of the important 3’-tail supports this possibility, though studying the GQ in the context of an extended sequence is of interest and will be discussed later in the context of the linked c-kit2/c-kit* system.
Figure 6.
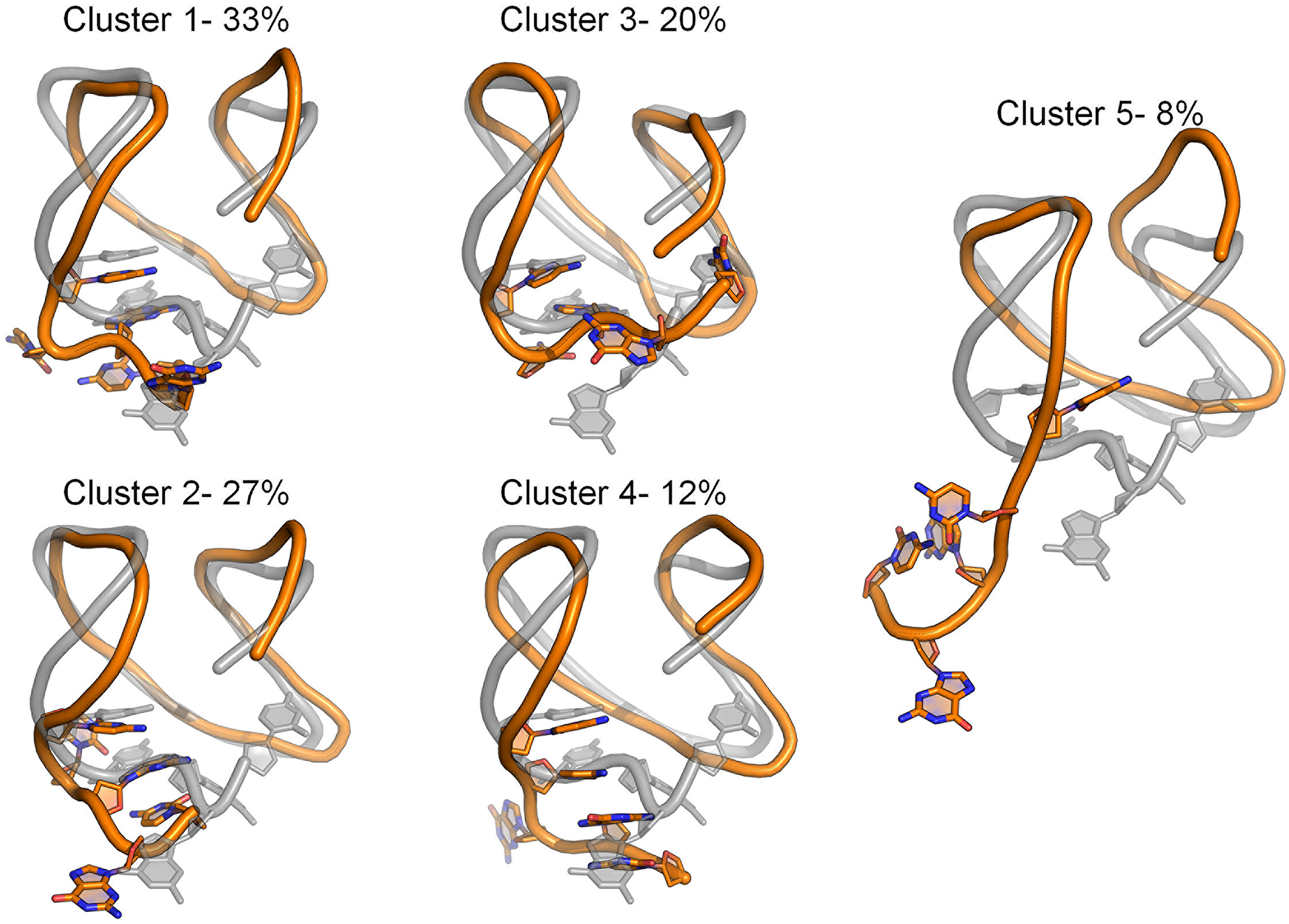
Central structures of each of the five clusters produced by RMSD-based clustering of the c-kit* 3’-tail nucleotides (Cyt18-Cyt19-Gua20-Gua21-Cyt22). Clustering was performed on the pooled Drude simulation trajectories, thus reflecting the entire simulation ensemble. The starting GQ structure (grey) is overlaid with the central structure from each cluster. For reference, the GQ backbone is rendered as a cartoon tube and loop nucleotides are shown in sticks. Percentages denote the occupancy of each cluster.
Ion Sampling around the Isolated c-kit* GQ.
As with the isolated c-kit2 GQs, we performed ion interaction analysis on the isolated c-kit* GQ. Drude simulations showed two distinct binding sites above and below the tetrad core with 25% and 70% occupancy, respectively (Supporting Information, Figure S6). The binding site above tetrad 1 was only occupied in one replicate because an Ade5-Gua14 base stacking interaction typically occluded tetrad 1. In replicate 2, this stacking interaction was disrupted and an ion was bound for 88% of the simulation. Ion alignment below the tetrad core strengthened the interaction energies between the core K+ and tetrad 2 (Supporting Information, Table S5), again demonstrating that ion alignment trends can influence the energetics of the GQ. In simulations of the c-kit* GQ, it is also important to note that the variability of the loop and tail structures influenced ion binding patterns, as some conformations occluded the tetrad core more than others (Supporting Information, Figure S17). Once again, the sampling of these regions could change in the context of an extended sequence, possibly affecting ion binding patterns and core energetics. For GQ secondary structure, there is limited structural data available in biologically relevant conditions, leaving interpretation of these interactions open for discussion. These possibilities, as well as others noted throughout the Discussion above, prompted us to study the linked c-kit2/c-kit* GQs with nucleotides upstream and downstream from these structures.
Linked c-kit2/c-kit* GQ Introduction.
The outcomes of our isolated systems, and several findings in previous studies, motivated us to perform simulations of the c-kit2 and c-kit* GQs as a linked structure. Previous work has shown that the stability of these GQs is dependent upon one another and that the c-kit* GQ may only be able to stably fold in the presence of the folded c-kit2 GQ.37 Further, isolated simulations of c-kit2 and c-kit* GQs exhibited some interesting behavior in the 5’-and 3’-terminal regions. In simulations of the isolated c-kit2 GQs, both the 5’-and 3’-residues participated in bulk ion alignment and did so with different affinity. We also observed that base pairing and base stacking near the tetrad core influences ion sampling. Lastly, sampling of the 3’ tail in the c-kit* GQs was very flexible and in some cases, occluded a potential binding site. These properties might be impacted by the presence of downstream nucleotides in the promoter region.
Conformational Dynamics of the Linked c-kit2/c-kit* GQ.
The RMSD of all nucleotides, core nucleotides, and core bases were low and comparable to values calculated from the isolated simulations, indicating that these systems were in similar agreement to the experimental structures (Supporting Information, Table S2). However, differences between the isolated and linked systems emerged in per-nucleotide RMSD and RMSF, specifically in loop and terminal region sampling. The c-kit2 long propeller loop (nucleotides 9–13) had lower RMSD in the linked system (Figure 7A) but similar RMSF (Figure 7B), meaning the structure of the loop deviated less from the experimental structure but had similar flexibility. These results demonstrate that the flexibility of the loop region is independent of upstream and downstream flanking nucleotides. The c-kit2 nucleotides had systematically lower RMSF values in the linked c-kit2/c-kit* system, apart from the long loop and the termini, which suggests that overall, the c-kit2 GQ is more rigid in the presence of c-kit* and flanking nucleotides. The c-kit2 terminal residues had both high RMSD and RMSF values, showing that these residues can sample different states and adopt a different conformation than the experimental structure. The dominant conformations produced by RMSD-based clustering (Figure 7C) help illustrate that the overall fold of the c-kit2 GQ was preserved in simulations of the linked system but differences emerged in the interactions of loop and terminal nucleotides.
Figure 7.
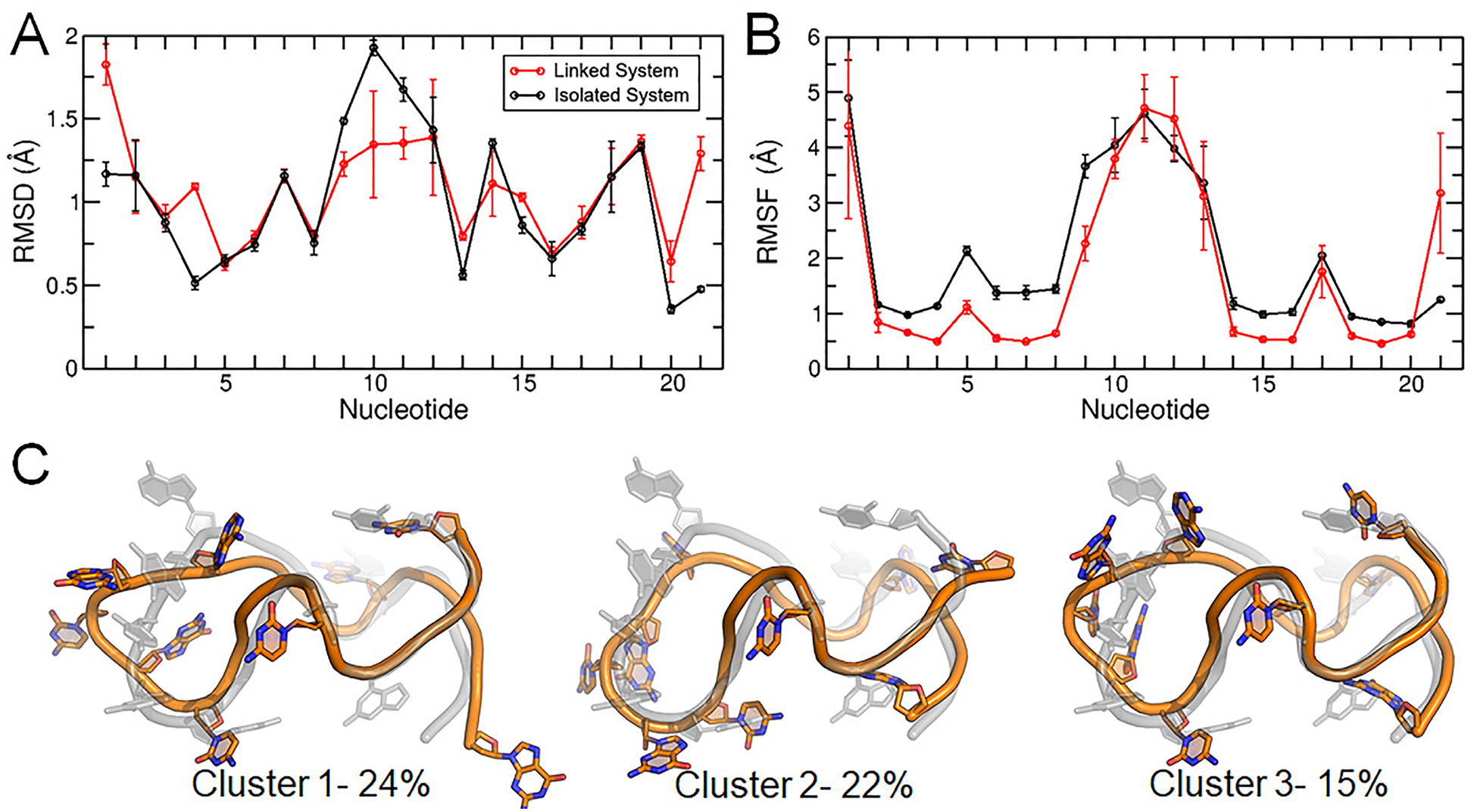
Structural characterization of the c-kit2 WT GQ in the linked c-kit2/c-kit* system. (A) Per-nucleotide RMSD, (B) per-nucleotide RMSF, and (C) central structures of the top three clusters of the c-kit2 GQ from RMSD-based clustering with associated occupancy percentages. Error bars in panels (A) and (B) represent the standard deviation of the averages of three replicate simulations.
The 3’-tail behaved differently in the c-kit* isolated and linked structures. The RMSD values (Figure 8A) of this region in the linked c-kit2/c-kit* simulations were higher, while the RMSF values (Figure 8B) were lower. This outcome suggests that the 3’-tail may adopt different, and less flexible, conformations than what was observed in the case of the isolated structure. RMSD-based clustering analysis of loop nucleotides (Figure 8C) illustrates differences in the loop and terminal regions. The fold back motif of the c-kit* 3’-tail persisted in simulations of the linked c-kit2/c-kit* GQs, while secondary interactions differed. In replicate 1, the fold back motif was maintained in part by Gua10-Gua20 base stacking interactions. In replicate 2, the motif was maintained by Ade8-Cyt22 base stacking interactions. In replicate 3, the tail was structured by both Gua9-Gua21 and Gua10-Gua20 base stacking interactions. The Cyt18:Gua10 WC-base pair remained intact throughout the simulations (100% of the time).
Figure 8.
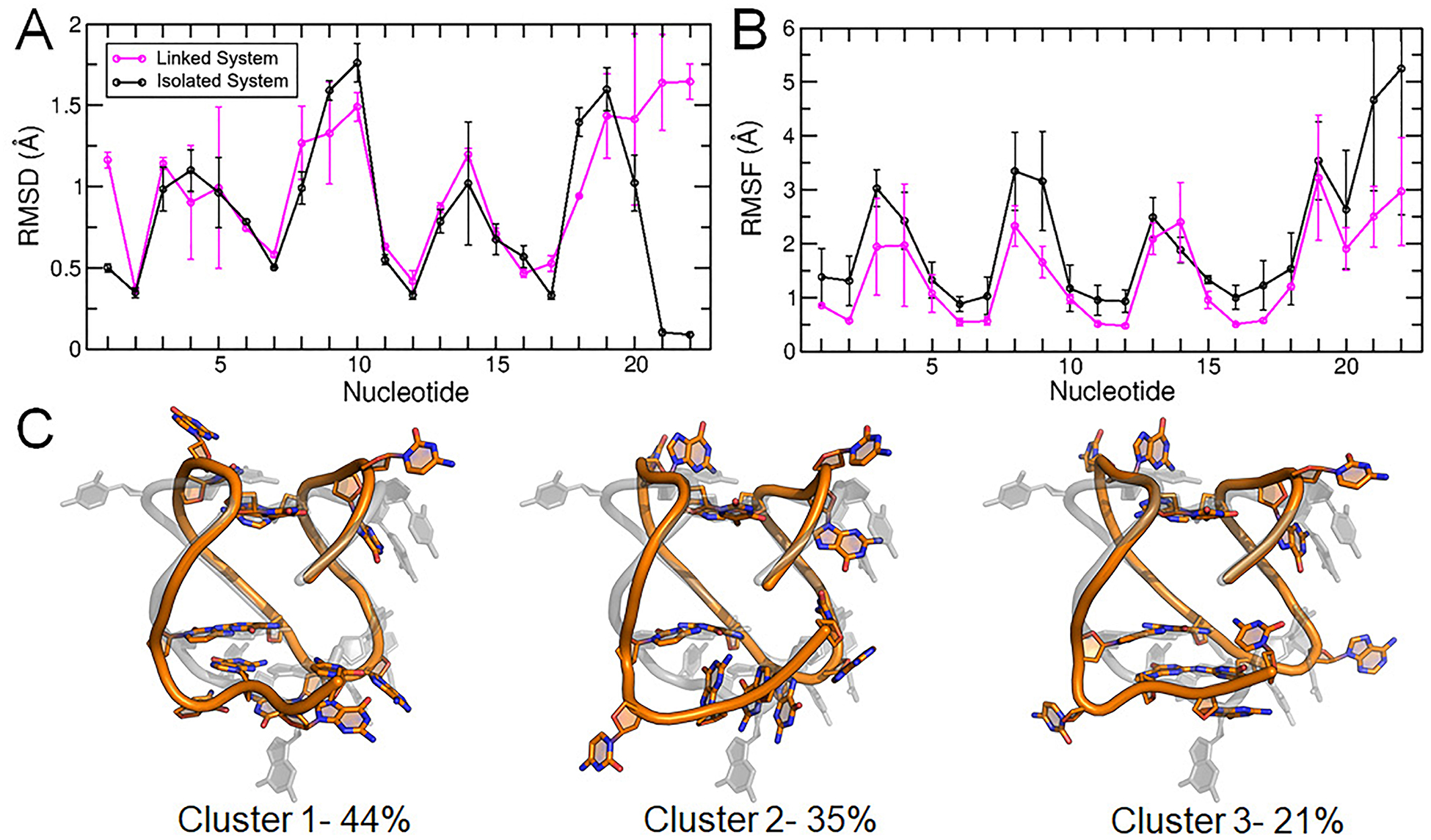
Structural characterization of the c-kit* GQ in the linked c-kit2/c-kit* system. (A) Per-nucleotide RMSD, (B) per-nucleotide RMSF, and (C) central structures of the top three clusters of the c-kit* GQ from RMSD-based clustering with associated occupancy percentages. Error bars in panels (A) and (B) represent the standard deviation of the averages of three replicate simulations.
Ion Sampling around the Linked c-kit2/c-kit* GQ.
Differences in loop conformation and sampling influenced ion-DNA interactions in the c-kit2 and c-kit* GQs. For the c-kit2 GQ, there was ion sampling above and below the tetrad core as in the isolated system. Ion interaction percentages above tetrad 1 were similar to the isolated system (12% in the isolated GQ and 19% in the linked system), though the 5’-terminal Cyt1 did not always interact with the bulk ion that bound. Ion interactions below tetrad 3 were slightly less prevalent in the linked system (42% in the isolated GQ and 30% in the linked system), likely because tetrad 3 was less solvent-exposed as a consequence of the proximity of the c-kit* GQ. While the ion occupancy map shows four binding sites in the tetrad core (Figure 9), four ions were not generally bound at the same time. When a fourth ion bound to the core, the opposing bulk ion left. Still, the highest ion occupancy sites were found proximal to the tetrad core in each simulation. These ions were always coordinated in bipyrimidal antiprismatic coordination with guanine O6 atoms. This consistency, and the ability to bind with and without interacting with an advantageously positioned nucleotide, suggests the high occupancy regions (above tetrad 1, between tetrads 1 and 2, between tetrads 2 and 3, and below tetrad 3) are relevant ion binding sites and are intrinsically favorable in the c-kit2 GQ structure.
Figure 9.
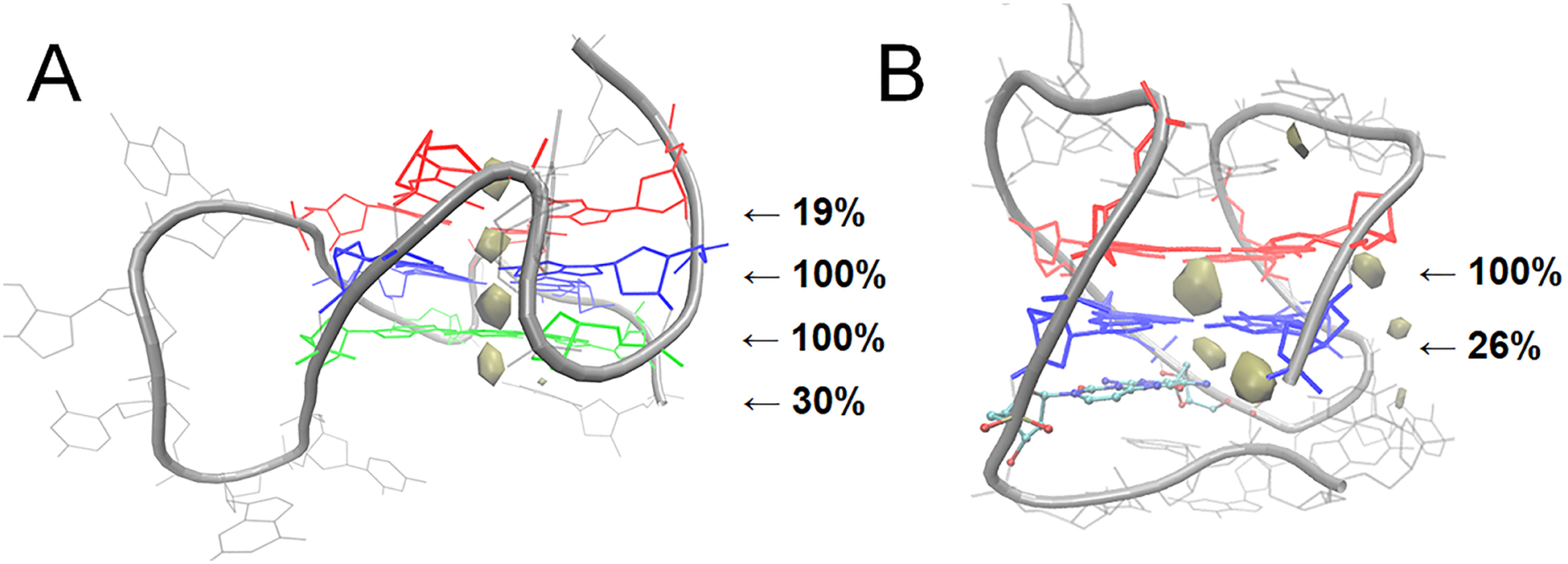
Ion interaction maps in the linked c-kit2/c-kit* GQ system. Ion sampling around the (A) c-kit2 and (B) c-kit* GQs are shown with an occupancy threshold of ≥1%.
Ion sampling around the c-kit* GQ greatly decreased in the linked structure. Tetrad 1 was blocked by base stacking interactions, preventing ion binding above tetrad 1. Similarly, tetrad 2 was less solvent exposed in the linked systems because the fold back motif of the 3’-tail largely occluded the core from solution. These findings suggest that the ion binding site above tetrad 1 identified in the isolated system is not relevant in an extended system, while the ion binding site below tetrad 2 remains accessible, even when single-stranded downstream nucleotides are present. For the c-kit* GQ, we predict it to have two ion binding sites in the core, between tetrads 1 and 2 and below tetrad 2.
The differences in ion sampling are also reflected in tetrad core-ion interaction energies (Table 3), especially for the c-kit* systems which had the greatest difference in ion sampling. With more sampling above tetrad 1 in the linked c-kit2 GQ, the tetrad 1 core-ion interaction energy decreased, indicating more favorable interaction energy. Conversely, there was less sampling below tetrad 3 in the linked system and the tetrad 3 core-ion interaction energy increased, indicating weakened interaction energy. With respect to the c-kit* GQ in the linked system, we observed no ion alignment above tetrad 1 and the interaction energy was slightly weakened. We observed reduced ion alignment below tetrad 2 as well and the interaction energy was consequently weakened. These results show that the core-ion interaction energies are strengthened by bulk ion alignment. In comparing the isolated and linked simulations, a connection can be made between secondary structure, loop sampling, bulk ion binding, and core-interaction energy. This interrelatedness is evidence that several factors, including primary structure, secondary structure, and ion binding, play an important role in the dynamics of GQ structures, even on the microsecond timescale.
Table 3.
Interaction energies (EINT, kcal/mol) between the core K+ ions and the guanine bases of each tetrad in the c-kit2 and c-kit* GQs as isolated and linked structures. Interaction energies computed directly from the trajectory without any reoptimization of the Drude oscillators.
| c-kit2 | Isolated | Linked |
|---|---|---|
| Tetrad 1 | −30.4 ± 5.0 | −31.8 ± 5.2 |
| Tetrad 2 | −67.0 ± 3.9 | −65.9 ± 4.1 |
| Tetrad 3 | −35.1 ± 3.3 | −32.1 ± 4.8 |
| c-kit* | Isolated | Linked |
| Tetrad 1 | −27.1 ± 4.8 | −25.6 ± 3.4 |
| Tetrad 2 | −42.4 ± 6.6 | −27.1 ± 4.1 |
CONCLUSIONS
In this work, we systemically studied the c-kit2 and c-kit* GQs, evaluating nonpolarizable and polarizable FFs, sequence mutations in the c-kit2 structure, different cation solutions, and isolated versus linked GQs. Our results suggest that GQ simulations with the C36 FF suffer from inadequate ion interactions and local instability in the core of two-and three-tetrad GQs and in KCl, NaCl, and LiCl solutions. In the same systems, the Drude FF produced good agreement with experimental structures and yielded an improved representation of ion interactions. We also showed that differences in primary structure, and resulting secondary structure interactions, influence loop sampling, ion binding, and core-ion energetics of the c-kit2 GQ. Moreover, local coordination of different cations in the c-kit2 core varied. Whereas K+ ions displayed antiprismatic coordination, Na+ ions exchanged between antiprismatic and planar coordination, and Li+ had a preference for planar coordination. The core-ion interaction energies of these systems were strongest in LiCl solution and weakened with increasing cation size (LiCl > NaCl > KCl). In simulations of isolated c-kit*, we highlighted deviations in the structurally important 3’-tail that may support the hypothesis that this GQ is in dynamic equilibrium between folded and unfolded states.
Simulations of the linked c-kit2/c-kit* GQs emphasized that differences in primary structure and resulting secondary interactions influence GQ dynamics. In the context of an extended sequence, the c-kit2 terminal regions adopted different conformations than the experimental structure and were less advantageously positioned for recruiting bulk ions. Still, four high-affinity ion occupancy sites persisted in the c-kit2 tetrad core, so we predicted two new ion binding sites, above tetrad 1 and below tetrad 3. The important c-kit* 3’-tail differed from the experimental structure but more consistently displayed the predicted fold back motif in the linked system. The secondary structure of the tail and loop regions above tetrad 1 occluded the core more than in the isolated systems. We predicted one new binding site in the c-kit* GQ, below tetrad 2. Based on these differences in loop secondary structure, ion sampling and ultimately core-ion interaction energies varied between isolated and linked structures. Overall, this work draws connections between primary structure, secondary structure, loop sampling, bulk ion binding, and core-ion interaction energy on the microsecond timescale. The interrelatedness of these properties calls for future simulation studies aimed at deconstructing the forces governing GQ dynamics.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Dr. Yue Yu, Brian D. Ratnasinghe, and Jesse R. Janoski for contributions at the initial stage of this project and Virginia Tech Advanced Research Computing for providing computing time and resources.
FUNDING
This work was supported by the National Institutes of Health (grant R35GM133754), the Thomas F. and Kate Miller Jeffress Memorial Trust (Bank of America, Trustee), USDA-NIFA (project number VA-160092), and startup funding from the Virginia Tech Office of the Provost, College of Agriculture and Life Sciences, and Department of Biochemistry.
Footnotes
Publisher's Disclaimer: This document is confidential and is proprietary to the American Chemical Society and its authors. Do not copy or disclose without written permission. If you have received this item in error, notify the sender and delete all copies.
SUPPORTING INFORMATION
The Supporting Information is available free of charge on the ACS Publications website and details the following analysis:
For C36 simulations of c-kit2 WT, T21, A10/T21, T12/T21, and c-kit*: RMSD over time, core hydrogen bonding over time, ion interaction maps, and dihedral angles. For Drude simulations of c-kit2 WT, T21, A10/T21, T12/T21, c-kit*, and linked c-kit2/c-kit*: RMSD over time, core hydrogen bonding over time, base dipole moments, tetrad-ion interaction energies, ion interaction maps, per-nucleotide RMSD, per-nucleotide RMSF, central cluster snapshots, and dihedral angles.
REFERENCES
- (1).Onel B; Clement L; DanZhou Y DNA G-Quadruplex and Its Potential as Anticancer Drug Target. Sci. China Chem 2016, 57 (12), 1605–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Mulholland K; Wu C Binding of Telomestatin to a Telomeric G-Quadruplex DNA Probed by All-Atom Molecular Dynamics Simulations with Explicit Solvent. J. Chem. Inf. Model 2016, 56 (10), 2093–2102. [DOI] [PubMed] [Google Scholar]
- (3).Balasubramanian S; Hurley LH; Neidle S Targeting G-Quadruplexes in Gene Promoters: A Novel Anticancer Strategy? Nat. Rev. Drug Discov 2011, 10 (4), 261–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Phan AT; Kuryavyi V; Darnell JC; Serganov A; Majumdar A; Ilin S; Raslin T; Polonskaia A; Chen C; Clain D; et al. Structure-Function Studies of FMRP RGG Peptide Recognition of an RNA Duplex-Quadruplex Junction. Nat. Struct. Mol. Biol 2011, 18 (7), 796–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Armas P; David A; Calcaterra NB Transcriptional Control by G-Quadruplexes: In Vivo Roles and Perspectives for Specific Intervention. Transcription 2017, 8 (1), 21–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).David AP; Margarit E; Domizi P; Banchio C; Armas P; Calcaterra NB G-Quadruplexes as Novel Cis-Elements Controlling Transcription during Embryonic Development. Nucleic Acids Res. 2016, 44 (9), 4163–4173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Fleming AM; Zhu J; Ding Y; Burrows CJ 8-Oxo-7,8-Dihydroguanine in the Context of a Gene Promoter G-Quadruplex Is an On–Off Switch for Transcription. ACS Chem. Biol 2017, 12 (9), 2417–2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Maizels N Genomic Stability: FANCJ-Dependent G4 DNA Repair. Curr. Biol 2008, 18 (14), R613–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Simone R; Fratta P; Neidle S; Parkinson GN; Isaacs AM G-Quadruplexes: Emerging Roles in Neurodegenerative Diseases and the Non-Coding Transcriptome. FEBS Lett. 2015, 589 (14), 1653–1668. [DOI] [PubMed] [Google Scholar]
- (10).Ozdilek BA; Thompson VF; Ahmed NS; White CI; Batey RT; Schwartz JC Intrinsically Disordered RGG/RG Domains Mediate Degenerate Specificity in RNA Binding. Nucleic Acids Res. 2017, 45 (13), 7984–7996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Johnson JE; Cao K; Ryvkin P; Wang L-S; Johnson FB Altered Gene Expression in the Werner and Bloom Syndromes Is Associated with Sequences Having G-Quadruplex Forming Potential. Nucleic Acids Res. 2010, 38 (4), 1114–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Wu Y; Brosh RM G-Quadruplex Nucleic Acids and Human Disease. FEBS J. 2010, 277 (17), 3470–3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Cimino-Reale G; Zaffaroni N; Folini M Emerging Role of G-Quadruplex DNA as Target in Anticancer Therapy. Curr. Pharm. Des 2017, 22 (44), 6612–6624. [DOI] [PubMed] [Google Scholar]
- (14).Chambers VS; Marsico G; Boutell JM; Di Antonio M; Smith GP; Balasubramanian S High-Throughput Sequencing of DNA G-Quadruplex Structures in the Human Genome. Nat. Biotechnol 2015, 33 (8), 877–881. [DOI] [PubMed] [Google Scholar]
- (15).Huppert JL; Balasubramanian S Prevalence of Quadruplexes in the Human Genome. Nucleic Acids Res. 2005, 33 (9), 2908–2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Eddy J; Maizels N Gene Function Correlates with Potential for G4 DNA Formation in the Human Genome. Nucleic Acids Res. 2006, 34 (14), 3887–3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Kikin O; D’Antonio L; Bagga PS QGRS Mapper: A Web-Based Server for Predicting G-Quadruplexes in Nucleotide Sequences. Nucleic Acids Res. 2006, 34 (Web Server), W676–W682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Huppert JL; Balasubramanian S G-Quadruplexes in Promoters throughout the Human Genome. Nucleic Acids Res. 2007, 35 (2), 406–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Dexheimer TS; Sun D; Hurley LH Deconvoluting the Structural and Drug-Recognition Complexity of the G-Quadruplex-Forming Region Upstream of the Bcl-2 P1 Promoter. J. Am. Chem. Soc 2006, 128 (16), 5404–5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Siddiqui-Jain A; Grand CL; Bearss DJ; Hurley LH Direct Evidence for a G-Quadruplex in a Promoter Region and Its Targeting with a Small Molecule to Repress c-MYC Transcription. Proc. Natl. Acad. Sci. U. S. A 2002, 99 (18), 11593–11598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Yamamoto K; Tojo A; Aoki N; Shibuya M Characterization of the Promoter Region of the Human C-Kit Proto-Oncogene. Jpn. J. Cancer Res 1993, 84 (11), 1136–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Rankin S; Reszka AP; Huppert J; Zloh M; Parkinson GN; Todd AK; Ladame S; Balasubramanian S; Neidle S Putative DNA Quadruplex Formation within the Human c-Kit Oncogene. J. Am. Chem. Soc 2005, 127 (30), 10584–10589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Raiber E-A; Kranaster R; Lam E; Nikan M; Balasubramanian S A Non-Canonical DNA Structure Is a Binding Motif for the Transcription Factor SP1 in Vitro. Nucleic Acids Res. 2012, 40 (4), 1499–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Saini N; Zhang Y; Usdin K; Lobachev KS When Secondary Comes First--the Importance of Non-Canonical DNA Structures. Biochimie 2013, 95 (2), 117–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Wang J-M; Huang F-C; Kuo MH-J; Wang Z-F; Tseng T-Y; Chang L-C; Yen S-J; Chang T-C; Lin J-J Inhibition of Cancer Cell Migration and Invasion through Suppressing the Wnt1-Mediating Signal Pathway by G-Quadruplex Structure Stabilizers. J. Biol. Chem 2014, 289 (21), 14612–14623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Cogoi S; Xodo LE G-Quadruplex Formation within the Promoter of the KRAS Proto-Oncogene and Its Effect on Transcription. Nucleic Acids Res. 2006, 34 (9), 2536–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Burge S; Parkinson GN; Hazel P; Todd AK; Neidle S Quadruplex DNA: Sequence, Topology and Structure. Nucleic Acids Res. 2006, 34 (19), 5402–5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Gellert M; Lipsett MN; Davies DR Helix Formation by Guanylic Acid. Proc. Natl. Acad. Sci. U. S. A 1962, 48 (12), 2013–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Williamson JR; Raghuraman MK; Cech TR Monovalent Cation-Induced Structure of Telomeric DNA: The G-Quartet Model. Cell 1989, 59 (5), 871–880. [DOI] [PubMed] [Google Scholar]
- (30).Gu J; Leszczynski J; Bansal M A New Insight into the Structure and Stability of Hoogsteen Hydrogen-Bonded G-Tetrad: An Ab Initio SCF Study. Chem. Phys. Lett 1999, 311 (3–4), 209–214. [Google Scholar]
- (31).Lee JY; Okumus B; Kim DS; Ha T Extreme Conformational Diversity in Human Telomeric DNA. Proc. Natl. Acad. Sci 2005, 102 (52), 18938–18943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Le HT; Dean WL; Buscaglia R; Chaires JB; Trent JO An Investigation of G-Quadruplex Structural Polymorphism in the Human Telomere Using a Combined Approach of Hydrodynamic Bead Modeling and Molecular Dynamics Simulation. J. Phys. Chem. B 2014, 118 (20), 5390–5405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Yang D; Okamoto K Structural Insights into G-Quadruplexes: Towards New Anticancer Drugs. Future Med. Chem 2010, 2 (4), 619–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Nambiar M; Goldsmith G; Moorthy BT; Lieber MR; Joshi MV; Choudhary B; Hosur RV; Raghavan SC Formation of a G-Quadruplex at the BCL2 Major Breakpoint Region of the t(14;18) Translocation in Follicular Lymphoma. Nucleic Acids Res. 2011, 39 (3), 936–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Agrawal P; Lin C; Mathad RI; Carver M; Yang D The Major G-Quadruplex Formed in the Human BCL-2 Proximal Promoter Adopts a Parallel Structure with a 13-Nt Loop in K + Solution. J. Am. Chem. Soc 2014, 136 (5), 1750–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Onel B; Carver M; Wu G; Timonina D; Kalarn S; Larriva M; Yang D A New G-Quadruplex with Hairpin Loop Immediately Upstream of the Human BCL2 P1 Promoter Modulates Transcription. J. Am. Chem. Soc 2016, 138 (8), 2563–2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Ducani C; Bernardinelli G; Högberg B; Keppler BK; Terenzi A Interplay of Three G-Quadruplex Units in the KIT Promoter. J. Am. Chem. Soc 2019, 141 (26), 10205–10213. [DOI] [PubMed] [Google Scholar]
- (38).Heinrich MC; Corless CL; Demetri GD; Blanke CD; Von Mehren M; Joensuu H; McGreevey LS; Chen CJ; Van Den Abbeele AD; Druker BJ; et al. Kinase Mutations and Imatinib Response in Patients with Metastatic Gastrointestinal Stromal Tumor. J. Clin. Oncol 2003, 21 (23), 4342–4349. [DOI] [PubMed] [Google Scholar]
- (39).Diveshkumar KV; Sakrikar S; Rosu F; Harikrishna S; Gabelica V; Pradeepkumar PI Specific Stabilization of C-MYC and c-KIT G-Quadruplex DNA Structures by Indolylmethyleneindanone Scaffolds. Biochemistry 2016, 55 (25), 3571–3585. [DOI] [PubMed] [Google Scholar]
- (40).Abbaspour Babaei M; Kamalidehghan B; Saleem M; Huri HZ; Ahmadipour F Receptor Tyrosine Kinase (c-Kit) Inhibitors: A Potential Therapeutic Target in Cancer Cells. Drug Des. Devel. Ther 2016, 10, 2443–2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Wei D; Husby J; Neidle S Flexibility and Structural Conservation in a C-KIT G-Quadruplex. Nucleic Acids Res. 2015, 43 (1), 629–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Phan AT; Kuryavyi V; Burge S; Neidle S; Patel DJ Structure of an Unprecedented G-Quadruplex Scaffold in the Human c-Kit Promoter. J. Am. Chem. Soc 2007, 129 (14), 4386–4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Kuryavyi V; Phan AT; Patel DJ Solution Structures of All Parallel-Stranded Monomeric and Dimeric G-Quadruplex Scaffolds of the Human c-Kit2 Promoter. Nucleic Acids Res. 2010, 38 (19), 6757–6773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Hsu S-TD; Varnai P; Bugaut A; Reszka AP; Neidle S; Balasubramanian S A G-Rich Sequence within the c-Kit Oncogene Promoter Forms a Parallel G-Quadruplex Having Asymmetric G-Tetrad Dynamics. J. Am. Chem. Soc 2009, 131 (37), 13399–13409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Kotar A; Rigo R; Sissi C; Plavec J Two-Quartet Kit* G-Quadruplex Is Formed via Double-Stranded Pre-Folded Structure. Nucleic Acids Res. 2019, 47 (5), 2641–2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Gunaratnam M; Swank S; Haider SM; Galesa K; Reszka AP; Beltran M; Cuenca F; Fletcher JA; Neidle S Targeting Human Gastrointestinal Stromal Tumor Cells with a Quadruplex-Binding Small Molecule. J. Med. Chem 2009, 52 (12), 3774–3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Waller ZAE; Sewitz SA; Hsu S-TD; Balasubramanian S A Small Molecule That Disrupts G-Quadruplex DNA Structure and Enhances Gene Expression. J. Am. Chem. Soc 2009, 131 (35), 12628–12633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).McLuckie KIE; Waller ZAE; Sanders DA; Alves D; Rodriguez R; Dash J; McKenzie GJ; Venkitaraman AR; Balasubramanian S G-Quadruplex-Binding Benzo[a]Phenoxazines Down-Regulate c-KIT Expression in Human Gastric Carcinoma Cells. J. Am. Chem. Soc 2011, 133 (8), 2658–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Bejugam M; Gunaratnam M; Müller S; Sanders DA; Sewitz S; Fletcher JA; Neidle S; Balasubramanian S Targeting the c-Kit Promoter G-Quadruplexes with 6-Substituted Indenoisoquinolines. ACS Med. Chem. Lett 2010, 1 (7), 306–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Zorzan E; Da Ros S; Musetti C; Shahidian LZ; Coelho NFR; Bonsembiante F; Létard S; Gelain ME; Palumbo M; Dubreuil P; et al. Screening of Candidate G-Quadruplex Ligands for the Human c-KIT Promotorial Region and Their Effects in Multiple in-Vitro Models. Oncotarget 2016, 7 (16), 21658–21675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Wei D; Parkinson GN; Reszka AP; Neidle S Crystal Structure of a C-Kit Promoter Quadruplex Reveals the Structural Role of Metal Ions and Water Molecules in Maintaining Loop Conformation. Nucleic Acids Res. 2012, 40 (10), 4691–4700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Park GH; Plummer HK; Krystal GW Selective Sp1 Binding Is Critical for Maximal Activity of the Human C-Kit Promoter. Blood 1998, 92 (11), 4138–4149. [PubMed] [Google Scholar]
- (53).Rigo R; Sissi C Characterization of G4–G4 Crosstalk in the c-KIT Promoter Region. Biochemistry 2017, 56 (33), 4309–4312. [DOI] [PubMed] [Google Scholar]
- (54).Islam B; Sgobba M; Laughton C; Orozco M; Sponer J; Neidle S; Haider S Conformational Dynamics of the Human Propeller Telomeric DNA Quadruplex on a Microsecond Time Scale. Nucleic Acids Res. 2013, 41 (4), 2723–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Islam B; Stadlbauer P; Gil-Ley A; Pérez-Hernández G; Haider S; Neidle S; Bussi G; Banas P; Otyepka M; Sponer J Exploring the Dynamics of Propeller Loops in Human Telomeric DNA Quadruplexes Using Atomistic Simulations. J. Chem. Theory Comput 2017, 13 (6), 2458–2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Havrila M; Stadlbauer P; Islam B; Otyepka M; Šponer J Effect of Monovalent Ion Parameters on Molecular Dynamics Simulations of G-Quadruplexes. J. Chem. Theory Comput 2017, 13 (8), 3911–3926. [DOI] [PubMed] [Google Scholar]
- (57).Špačková N; Imre Berger; Šponer J Nanosecond Molecular Dynamics Simulations of Parallel and Antiparallel Guanine Quadruplex DNA Molecules. 1999.
- (58).Gkionis K; Kruse H; Platts JA; Mládek A; Koča J; Šponer J Ion Binding to Quadruplex DNA Stems. Comparison of MM and QM Descriptions Reveals Sizable Polarization Effects Not Included in Contemporary Simulations. J. Chem. Theory Comput 2014, 10 (3), 1326–1340. [DOI] [PubMed] [Google Scholar]
- (59).Islam B; Stadlbauer P; Krepl M; Havrila M; Haider S; Sponer J Structural Dynamics of Lateral and Diagonal Loops of Human Telomeric G-Quadruplexes in Extended MD Simulations. J. Chem. Theory Comput 2018, 14 (10), 5011–5026. [DOI] [PubMed] [Google Scholar]
- (60).Rueda M; Luque FJ; Orozco M G-Quadruplexes Can Maintain Their Structure in the Gas Phase. J. Am. Chem. Soc 2006, 128 (11), 3608–3619. [DOI] [PubMed] [Google Scholar]
- (61).Haider S; Parkinson GN; Neidle S Molecular Dynamics and Principal Components Analysis of Human Telomeric Quadruplex Multimers. Biophys. J 2008, 95 (1), 296–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Li H; Cao E; Gisler T Force-Induced Unfolding of Human Telomeric G-Quadruplex: A Steered Molecular Dynamics Simulation Study. Biochem. Biophys. Res. Commun 2009, 379 (1), 70–75. [DOI] [PubMed] [Google Scholar]
- (63).Fadrná E; Špačková N; Sarzyñska J; Koča J; Orozco M; Cheatham TE; Kulinski T; Šponer J Single Stranded Loops of Quadruplex DNA As Key Benchmark for Testing Nucleic Acids Force Fields. J. Chem. Theory Comput 2009, 5 (9), 2514–2530. [DOI] [PubMed] [Google Scholar]
- (64).Šponer J; Bussi G; Stadlbauer P; Kührová P; Banáš P; Islam B; Haider S; Neidle S; Otyepka M Folding of Guanine Quadruplex Molecules–Funnel-like Mechanism or Kinetic Partitioning? An Overview from MD Simulation Studies. Biochim. Biophys. Acta - Gen. Subj 2017, 1861 (5), 1246–1263. [DOI] [PubMed] [Google Scholar]
- (65).Fadrná E; Špačková N; Štefl R; Koča J; Cheatham TE; Šponer J Molecular Dynamics Simulations of Guanine Quadruplex Loops: Advances and Force Field Limitations. Biophys. J 2004, 87 (1), 227–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Islam B; Stadlbauer P; Krepl M; Koca J; Neidle S; Haider S; Sponer J Extended Molecular Dynamics of a C-Kit Promoter Quadruplex. Nucleic Acids Res. 2015, 43 (18), 8673–8693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Salsbury AM; Lemkul JA Molecular Dynamics Simulations of the c-Kit1 Promoter G-Quadruplex: Importance of Electronic Polarization on Stability and Cooperative Ion Binding. J. Phys. Chem. B 2019, 123 (1), 148–159. [DOI] [PubMed] [Google Scholar]
- (68).Mackerell AD Empirical Force Fields for Biological Macromolecules: Overview and Issues. J. Comput. Chem 2004, 25 (13), 1584–1604. [DOI] [PubMed] [Google Scholar]
- (69).MacKerell AD; Bashford D; Bellott M; Dunbrack RL; Evanseck JD; Field MJ; Fischer S; Gao J; Guo H; Ha S; et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102 (18), 3586–3616. [DOI] [PubMed] [Google Scholar]
- (70).Rebič M; Laaksonen A; Šponer J; Uličný J; Mocci F Molecular Dynamics Simulation Study of Parallel Telomeric DNA Quadruplexes at Different Ionic Strengths: Evaluation of Water and Ion Models. J. Phys. Chem. B 2016, 120 (30), 7380–7391. [DOI] [PubMed] [Google Scholar]
- (71).Song J; Ji C; Zhang JZH The Critical Effect of Polarization on the Dynamical Structure of Guanine Quadruplex DNA. Phys. Chem. Chem. Phys 2013, 15 (11), 3846. [DOI] [PubMed] [Google Scholar]
- (72).Lemkul JA Same Fold, Different Properties: Polarizable Molecular Dynamics Simulations of Telomeric and TERRA G-Quadruplexes. Nucleic Acids Res. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Hart K; Foloppe N; Baker CM; Denning EJ; Nilsson L; MacKerell AD Optimization of the CHARMM Additive Force Field for DNA: Improved Treatment of the BI/BII Conformational Equilibrium. J. Chem. Theory Comput 2012, 8 (1), 348–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Lemkul JA; MacKerell AD Polarizable Force Field for DNA Based on the Classical Drude Oscillator: I. Refinement Using Quantum Mechanical Base Stacking and Conformational Energetics. J. Chem. Theory Comput 2017, 13 (5), 2053–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Lemkul JA; MacKerell AD Polarizable Force Field for DNA Based on the Classical Drude Oscillator: II. Microsecond Molecular Dynamics Simulations of Duplex DNA. J. Chem. Theory Comput 2017, 13 (5), 2072–2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Brooks BR; Brooks CL; Mackerell AD; Nilsson L; Petrella RJ; Roux B; Won Y; Archontis G; Bartels C; Boresch S; et al. CHARMM: The Biomolecular Simulation Program. J. Comput. Chem 2009, 30 (10), 1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Foloppe N; Hartmann B; Nilsson L; MacKerell AD Intrinsic Conformational Energetics Associated with the Glycosyl Torsion in DNA: A Quantum Mechanical Study. Biophys. J 2002, 82 (3), 1554–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).MacKerell AD; Jr. Contribution of the Intrinsic Mechanical Energy of the Phosphodiester Linkage to the Relative Stability of the A, BI and BII Forms of Duplex DNA. J. Phys. Chem. B 2009, 113 (10), 3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys 1983, 79 (2), 926–935. [Google Scholar]
- (80).Durell SR; Brooks BR; Ben-Naim A Solvent-Induced Forces between Two Hydrophilic Groups. J. Phys. Chem 1994, 98 (8), 2198–2202. [Google Scholar]
- (81).Neria E; Fischer S; Karplus M Simulation of Activation Free Energies in Molecular Systems. J. Chem. Phys 1998, 105 (5), 1902. [Google Scholar]
- (82).Beglov D; Roux B Finite Representation of an Infinite Bulk System: Solvent Boundary Potential for Computer Simulations. J. Chem. Phys 1994, 100 (12), 9050–9063. [Google Scholar]
- (83).Phillips JC; Braun R; Wang W; Gumbart J; Tajkhorshid E; Villa E; Chipot C; Skeel RD; Kalé L; Schulten K Scalable Molecular Dynamics with NAMD. J. Comput. Chem 2005, 26 (16), 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Izaguirre JA; Catarello DP; Wozniak JM; Skeel RD Langevin Stabilization of Molecular Dynamics. J. Chem. Phys 2001, 114 (5), 2090–2098. [Google Scholar]
- (85).Feller SE; Zhang Y; Pastor RW; Brooks BR Constant Pressure Molecular Dynamics Simulation: The Langevin Piston Method. J. Chem. Phys 1995, 103 (11), 4613–4621. [Google Scholar]
- (86).Darden T; York D; Pedersen L Particle Mesh Ewald: An N log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys 1993, 98 (12), 10089–10092. [Google Scholar]
- (87).Essmann U; Perera L; Berkowitz ML; Darden T; Lee H; Pedersen LG A Smooth Particle Mesh Ewald Method. J. Chem. Phys 1995, 103 (19), 8577–8593. [Google Scholar]
- (88).Ryckaert J-P; Ciccotti G; Berendsen HJ. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys 1977, 23 (3), 327–341. [Google Scholar]
- (89).Miyamoto S; Kollman PA Settle: An Analytical Version of the SHAKE and RATTLE Algorithm for Rigid Water Models. J. Comput. Chem 1992, 13 (8), 952–962. [Google Scholar]
- (90).Lamoureux G; Harder E; Vorobyov IV; Roux B; MacKerell AD A Polarizable Model of Water for Molecular Dynamics Simulations of Biomolecules. Chem. Phys. Lett 2006, 418 (1–3), 245–249. [Google Scholar]
- (91).Yu H; Whitfield TW; Harder E; Lamoureux G; Vorobyov I; Anisimov VM; MacKerell AD; Roux B Simulating Monovalent and Divalent Ions in Aqueous Solution Using a Drude Polarizable Force Field. J. Chem. Theory Comput 2010, 6 (3), 774–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Savelyev A; MacKerell AD Competition among Li+, Na+, K+, and Rb+ Monovalent Ions for DNA in Molecular Dynamics Simulations Using the Additive CHARMM36 and Drude Polarizable Force Fields. J. Phys. Chem. B 2015, 119 (12), 4428–4440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Jiang W; Hardy DJ; Phillips JC; MacKerell AD; Schulten K; Roux B High-Performance Scalable Molecular Dynamics Simulations of a Polarizable Force Field Based on Classical Drude Oscillators in NAMD. J. Phys. Chem. Lett 2011, 2 (2), 87–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Lamoureux G; Roux B Modeling Induced Polarization with Classical Drude Oscillators: Theory and Molecular Dynamics Simulation Algorithm. J. Chem. Phys 2003, 119 (6), 3025–3039. [Google Scholar]
- (95).Lemkul JA; Huang J; Roux B; Mackerell AD An Empirical Polarizable Force Field Based on the Classical Drude Oscillator Model: Development History and Recent Applications. Chem. Rev 2016, 116 (9), 4983–5013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Chowdhary J; Harder E; Lopes PEM; Huang L; MacKerell AD; Roux B A Polarizable Force Field of Dipalmitoylphosphatidylcholine Based on the Classical Drude Model for Molecular Dynamics Simulations of Lipids. J. Phys. Chem. B 2013, 117 (31), 9142–9160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Eastman P; Swails J; Chodera JD; McGibbon RT; Zhao Y; Beauchamp KA; Wang L-P; Simmonett AC; Harrigan MP; Stern CD; et al. OpenMM 7: Rapid Development of High Performance Algorithms for Molecular Dynamics. PLOS Comput. Biol 2017, 13 (7), e1005659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Huang J; Lemkul JA; Eastman PK; MacKerell AD Molecular Dynamics Simulations Using the Drude Polarizable Force Field on GPUs with OpenMM: Implementation, Validation, and Benchmarks. J. Comput. Chem 2018, 39 (21), 1682–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Andersen HC; C. H Molecular Dynamics Simulations at Constant Pressure and/or Temperature. J. Chem. Phys 1980, 72 (4), 2384–2393. [Google Scholar]
- (100).Carpenter GA; Grossberg S ART 2: Self-Organization of Stable Category Recognition Codes for Analog Input Patterns. Appl. Opt 1987, 26 (23), 4919. [DOI] [PubMed] [Google Scholar]
- (101).Carpenter GA; Grossberg S The ART of Adaptive Pattern Recognition by a Self-Organizing Neural Network. Computer (Long. Beach. Calif) 1988, 21 (3), 77–88. [Google Scholar]
- (102).Karpen ME; Tobias DJ; Brooks CL Statistical Clustering Techniques for the Analysis of Long Molecular Dynamics Trajectories: Analysis of 2.2-Ns Trajectories of YPGDV. Biochemistry 1993, 32 (2), 412–420. [DOI] [PubMed] [Google Scholar]
- (103).Havrila M; Stadlbauer P; Kührová P; Banáš P; Mergny J-L; Otyepka M; Šponer J Structural Dynamics of Propeller Loop: Towards Folding of RNA G-Quadruplex. Nucleic Acids Res. 2018, 46 (17), 8754–8771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Venczel EA; Sen D Parallel and Antiparallel G-DNA Structures from a Complex Telomeric Sequence. Biochemistry 1993, 32 (24), 6220–6228. [DOI] [PubMed] [Google Scholar]
- (105).Włodarczyk A; Grzybowski P; Patkowski A; Dobek A Effect of Ions on the Polymorphism, Effective Charge, and Stability of Human Telomeric DNA. Photon Correlation Spectroscopy and Circular Dichroism Studies. J. Phys. Chem. B 2005, 109 (8), 3594–3605. [DOI] [PubMed] [Google Scholar]
- (106).Todd AK; Haider SM; Parkinson GN; Neidle S Sequence Occurrence and Structural Uniqueness of a G-Quadruplex in the Human c-Kit Promoter. Nucleic Acids Res. 2007, 35 (17), 5799–5808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Hardin CC; Watson T; Corregan M; Bailey C Cation-Dependent Transition between the Quadruplex and Watson-Crick Hairpin Forms of d(CGCG3GCG). Biochemistry 1992, 31 (3), 833–841. [DOI] [PubMed] [Google Scholar]
- (108).Savelyev A; MacKerell AD Differential Impact of the Monovalent Ions Li+, Na+, K+, and Rb+ on DNA Conformational Properties. J. Phys. Chem. Lett 2015, 6 (1), 212–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Noy A; Soteras I; Javier Luque F; Orozco M The Impact of Monovalent Ion Force Field Model in Nucleic Acids Simulations. Phys. Chem. Chem. Phys 2009, 11 (45), 10596. [DOI] [PubMed] [Google Scholar]
- (110).Lavery R; Maddocks JH; Pasi M; Zakrzewska K Analyzing Ion Distributions around DNA. Nucleic Acids Res. 2014, 42 (12), 8138–8149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Meyer M; Steinke T; Brandl M; Suhnel J Density Functional Study of Guanine and Uracil Quartets and of Guanine Quartet/Metal Ion Complexes. J. Comput. Chem 2001, 22 (1), 109–124. [Google Scholar]
- (112).Gray RD; Chaires JB Linkage of Cation Binding and Folding in Human Telomeric Quadruplex DNA. Biophys. Chem 2011, 159 (1), 205–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Bhattacharyya D; Mirihana Arachchilage G; Basu S Metal Cations in G-Quadruplex Folding and Stability. Front. Chem 2016, 4, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Hud NV; Smith FW; Anet FAL; Feigon J The Selectivity for K+ versus Na+ in DNA Quadruplexes Is Dominated by Relative Free Energies of Hydration: A Thermodynamic Analysis by 1 H NMR. Biochemistry 1996, 35, 15383–15390. [DOI] [PubMed] [Google Scholar]
- (115).Miller MC; Buscaglia R; Chaires JB; Lane AN; Trent JO Hydration Is a Major Determinant of the G-Quadruplex Stability and Conformation of the Human Telomere 3′ Sequence of d(AG 3 (TTAG 3) 3). J. Am. Chem. Soc 2010, 132 (48), 17105–17107. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.