

Abstract
Background
The microbiome is an integral component of many animal species, potentially affecting behavior, physiology, and other biological properties. Despite this importance, bacterial communities remain vastly understudied in many groups of invertebrates, including mites. Quill mites (Acariformes: Syringophilidae) are a poorly known group of permanent bird ectoparasites that occupy quills of feathers and feed on bird subcutaneous tissue and fluids. Most of the known species have strongly female‐biased sex ratio, and it was hypothesized that this is caused by endosymbiotic bacteria. Previously, Anaplasma phagocytophilum (Foggie) and a high diversity of Wolbachia strains were detected in quill mites via targeted PCR screens. Here, we use an unbiased 16S rRNA gene amplicon sequencing approach to determine other bacteria that potentially impact quill mite biology.
Results
We performed 16S rRNA gene amplicon sequencing of 126 quill mite individuals from eleven species parasitizing twelve species (four families) of passeriform birds. In addition to Wolbachia, we found Spiroplasma as potential symbiont of quill mites. Consistently, high Spiroplasma titers were only found in individuals of two mite species associated with finches of the genus Carduelis, suggesting a history of horizontal transfers of Spiroplasma via the bird host. Furthermore, there was evidence for Spiroplasma negatively affecting Wolbachia titers. We found no evidence for the previously reported Anaplasma in quill mites, but detected sequences of high similarity to the potential pathogens Brucella and Bartonella at low abundances. Other amplicon sequence variants (ASVs) could be assigned to a diverse number of bacterial taxa, including several that were previously isolated from bird skin. Further, many frequently found ASVs were assigned to taxa that show a very broad distribution with no strong prior evidence for symbiotic association with animals. We interpret these findings as evidence for a scarcity of resident microbial associates (other than inherited symbionts) in quill mites.
Keywords: 16S rRNA gene amplicon sequencing, Bartonella, birds, Brucella, ectoparasites, quill mites
We found a diverse, but relatively uniform set of bacterial taxa within quill mites that include arthropod endosymbionts, potential pathogens, and bird associated bacteria. The importance of most of these microbes for quill mite biology is unclear, but abundances and distribution patterns suggest that Spiroplasma and Wolbachia are the most important quill mite associates.

1. INTRODUCTION
There is abundant evidence that microbial taxa are an essential component of many animal species (McFall‐Ngai et al., 2013). Bacteria‐encoded traits may significantly impact host phenotypes, for example, through providing essential nutrients (Duron et al., 2018; Hosokawa, Koga, Kikuchi, Meng, & Fukatsu, 2010), defending against pathogens (Ballinger & Perlman, 2017; King et al., 2016), but also affecting ecological features of their hosts, such as mate choice (Sharon et al., 2010) and life history traits (Laughton, Fan, & Gerardo, 2013). Because of their potential importance in understanding the biology of many organisms, the number of microbiome studies has been soaring (Hird, 2017). This popularity is owed to methodological advances (high‐throughput sequencing technologies) allowing comprehensive investigation of the microbial communities (Ji & Nielsen, 2015), but also to the decreasing costs of these approaches (Sboner, Mu, Greenbaum, Auerbach, & Gerstein, 2011). However, the main focus on microbiome studies so far has been vertebrates (Colston & Jackson, 2016) and invertebrates of medical, veterinary, or economical importance. For example, in mites, microbiome studies have been conducted on the stored product pests (Erban et al., 2016; Hubert, Kopecky, Nesvorna, Perotti, & Erban, 2016; Hubert et al., 2019), house dust mites (Chan et al., 2015; Oh, Ishii, Tongu, & Itano, 1986; Valerio, Murray, Arlian, & Slater, 2005), and mites transmitting pathogens, such as sheep scab mites (Hogg & Lehane, 1999), red poultry mites (Hubert et al., 2017; Moro, Thioulouse, Chauve, & Zenner, 2011), and the honey bee parasite Varroa (Hubert, Kamler, et al., 2016).
In the present study, we focus on quill mites (Acariformes: Syringophilidae). These obligatory bird ectoparasites live and reproduce inside the quills of feathers where they feed on subcutaneous fluids (lymph, blood). Quill mite dispersion has been observed on the same individual (from infected to uninfected feathers), between individuals of the same species (e.g., from parents to hatchings) and occasionally by transfer between gregarious bird species (Casto, 1974a,1974b; Kethley, 1970, 1971). This mode of feeding and dispersion makes quill mites potential vectors for bacterial pathogens, similar to ticks or lice (Azad & Beard, 1998). However, only two bacterial taxa were recorded in quill mites so far: (a) Anaplasma phagocytophilum (Foggie) (Alphaproteobacteria, Rickettsiales) was detected in two quill mite species from three bird species (Skoracki et al., 2006); (b) multiple genetically distinct lineages of Wolbachia (Alphaproteobacteria, Rickettsiales) were found in five species of quill mites (Glowska, Dragun‐Damian, Dabert, & Gerth, 2015). As these studies were targeted PCR screens, it remains unclear what other bacteria populate quill mites. Furthermore, the importance of quill mites for bird pathogen dynamics is not known.
To address these questions, we here assess the bacterial composition of 126 quill mite individuals encompassing eleven species with a more unbiased 16S rRNA gene amplicon sequencing approach. We find that the symbionts Wolbachia and Spiroplasma are among the most commonly associated taxa with quill mites. Other taxa include bacteria that were previously found in association with arthropods and bacteria with a very broad distribution. Strikingly, neither quill mite taxonomy nor bird host taxonomy significantly influences bacterial composition in quill mites. Furthermore, we find that despite the detection of Bartonella and Brucella, quill mites do not seem to be major pathogen vectors in birds.
2. MATERIAL AND METHODS
2.1. Animal collection and DNA extraction
A summary of collected quill mite species and their bird hosts can be found in Table 1. All quill mites used in this study were collected in Kopan, Poland, during spring migration of birds monitored by the Bird Migration Research Station, University of Gdansk, April 2009. One secondary flight feather was analyzed from each bird specimen and dissected under a stereo microscope (Olympus ZS30). Individual mites were washed twice and preserved in 96% ethanol, and total genomic DNA was extracted from single specimens using DNeasy Blood & Tissue Kit (Qiagen GmbH), as described previously (Dabert, Ehrnsberger, & Dabert, 2008). This procedure left the exoskeletons intact, and the specimens were subsequently mounted on microscopic slides in Faure medium and determined using the key from Skoracki, Spicer, and Oconnor (2016). All morphological observations were carried out with an Olympus BH2 microscope with differential interference contrast (DIC) optics and a camera lucida. All DNA samples and corresponding voucher specimens are deposited in the collection of the Department of Animal Morphology, Faculty of Biology, Adam Mickiewicz University in Poznan, Poland. To identify potential contaminants, in addition to sequencing a negative control alongside all samples, we further extracted DNA from reagents and materials commonly used in the laboratory this work was carried out in. One library each was created from extraction buffer (ALT), millipore water, microscope swabs, pipette swabs, and swabs of other equipment (pincettes, scalpels, benches, etc). These five libraries were processed and sequenced separately from the other samples, but by using identical procedures.
Table 1.
Overview of quill mites sampled for the study with average abundance of Spiroplasma and Wolbachia
| Quill mite species | Bird host species (common name) | Number of bird individuals | Number of mite individuals |
Average Spiroplasma abundance % |
Average Wolbachia abundance % |
|---|---|---|---|---|---|
| Syringophilopsis kirgizorum | Carduelis carduelis (European goldfinch) | 2 | 9 | 0.43 | 2.60 |
| Torotrogla cardueli | Carduelis carduelis (European goldfinch) | 1 | 6 | 55.10 | 0. |
| Syringophilopsis kirgizorum | Carduelis chloris (European greenfinch) | 1 | 2 | 62.00 | 1.45 |
| Aulobia cardueli | Carduelis flammea (Common redpoll) | 1 | 2 | 0.00 | 0.06 |
| Aulobia cardueli | Carduelis spinus (Eurasian siskin) | 1 | 4 | 0.34 | 1.53 |
| Torotrogla cardueli | Carduelis spinus (Eurasian siskin) | 1 | 13 | 13.70 | 2.41 |
| Torotrogla rubeculi | Erithacus rubecula (European robin) | 3 | 12 | 0.14 | 5.92 |
| Syringophilopsis fringillae | Fringilla coelebs (Common chaffinch) | 1 | 6 | 0.00 | 6.59 |
| Torotrogla gaudi | Fringilla coelebs (Common chaffinch) | 2 | 16 | 0.55 | 0.01 |
| Torotrogla lusciniae | Luscinia luscinia (Thrush nightingale) | 1 | 7 | 0.35 | 0.51 |
| Torotrogla lusciniae | Luscinia svecica (Bluethroat) | 1 | 1 | 1.07 | 0.61 |
| Torotrogla modularis | Prunella modularis (Dunnock) | 1 | 4 | 0.00 | 0.10 |
| Syringophiloidus parapresentalis | Turdus iliacus (Redwing) | 1 | 3 | 0.00 | 0.21 |
| Syringophilopsis turdi | Turdus iliacus (Redwing) | 3 | 15 | 0.00 | 5.92 |
| Torotrogla merulae | Turdus merula (Common blackbird) | 3 | 13 | 0.00 | 4.19 |
| Syringophilopsis turdi | Turdus philomelos (Song thrush) | 1 | 4 | 0.00 | 12.10 |
| Torotrogla merulae | Turdus philomelos (Song thrush) | 2 | 9 | 0.00 | 3.80 |
2.2. Library preparation and sequencing
The V4 hypervariable region of 16S rRNA gene was amplified using PCR primers V4F (GATCAGCAGCCGCGGTAATA) (developed in this study) and V4R (GGACTACCAGGGTATCTAA) (Therese, Anand, & Madhavan, 1998) fused with indexes and Ion Torrent adapters (Table 2). For the PCRs, each 10 µl sample was prepared in two technical replicates containing 2 µl HOT FIREPol Blend Master Mix (Solis BioDyne), 0.25 µM of each double‐indexed fusion primer, and about 1 ng of template DNA. The fusion PCR regime used was 12 min at 95°C, 40 cycles of 15 s at 95°C, 30s at 58°C, 30s at 72°C, and a final 7 min at 72°C. After PCR, all samples were pooled, size‐selected on a 3% agarose gel, purified using the QIAquick Gel Extraction Kit (Qiagen), and quantified on a 2,200 TapeStation (Agilent Technologies, Inc.). Clonal template amplification on Ion Sphere Particles (ISPs) was performed using the Ion Torrent One Touch System II and the Ion PGM™ Hi‐Q™ View OT2 Kit with regard to manufacturer's instructions. Sequencing of the templated ISPs was conducted on the Ion 318™ Chip with the use of Ion PGM™ Hi‐Q™ View Sequencing Kit and Ion PGM system (Ion Torrent, Thermo Fisher Scientific, Inc.) at Molecular Biology Techniques Laboratory, Faculty of Biology, AMU. All reads resulting from the sequencing are available under NCBI BioProject accession PRJNA482380.
Table 2.
Fusion PCR primers sequences used in this study. Unique random barcode sequences are highlighted in bold
| Primer name | Primer sequence |
|---|---|
| V4FA49 | CCATCTCATCCCTGCGTGTCTCCGACTCAGTCCTAACATAACGATCAGCAGCCGCGGTAATA |
| V4FA50 | CCATCTCATCCCTGCGTGTCTCCGACTCAGCGGACAATGGCGATCAGCAGCCGCGGTAATA |
| V4FA51 | CCATCTCATCCCTGCGTGTCTCCGACTCAGTTGAGCCTATTCGATCAGCAGCCGCGGTAATA |
| V4FA52 | CCATCTCATCCCTGCGTGTCTCCGACTCAGCCGCATGGAACGATCAGCAGCCGCGGTAATA |
| V4FA53 | CCATCTCATCCCTGCGTGTCTCCGACTCAGCTGGCAATCCTCGATCAGCAGCCGCGGTAATA |
| V4FA54 | CCATCTCATCCCTGCGTGTCTCCGACTCAGCCGGAGAATCGCGATCAGCAGCCGCGGTAATA |
| V4FA55 | CCATCTCATCCCTGCGTGTCTCCGACTCAGTCCACCTCCTCGATCAGCAGCCGCGGTAATA |
| V4FA56 | CCATCTCATCCCTGCGTGTCTCCGACTCAGCAGCATTAATTCGATCAGCAGCCGCGGTAATA |
| V4FA57 | CCATCTCATCCCTGCGTGTCTCCGACTCAGTCTGGCAACGGCGATCAGCAGCCGCGGTAATA |
| V4FA58 | CCATCTCATCCCTGCGTGTCTCCGACTCAGTCCTAGAACACGATCAGCAGCCGCGGTAATA |
| V4FA59 | CCATCTCATCCCTGCGTGTCTCCGACTCAGTCCTTGATGTTCGATCAGCAGCCGCGGTAATA |
| V4FA60 | CCATCTCATCCCTGCGTGTCTCCGACTCAGTCTAGCTCTTCGATCAGCAGCCGCGGTAATA |
| V4FA61 | CCATCTCATCCCTGCGTGTCTCCGACTCAGTCACTCGGATCGATCAGCAGCCGCGGTAATA |
| V4FA62 | CCATCTCATCCCTGCGTGTCTCCGACTCAGTTCCTGCTTCACGATCAGCAGCCGCGGTAATA |
| V4FA63 | CCATCTCATCCCTGCGTGTCTCCGACTCAGCCTTAGAGTTCGATCAGCAGCCGCGGTAATA |
| V4FA64 | CCATCTCATCCCTGCGTGTCTCCGACTCAGCTGAGTTCCGACGATCAGCAGCCGCGGTAATA |
| V4RP165 | CCTCTCTATGGGCAGTCGGTGATGTCGCTCCAATGGACTACCAGGGTATCTAA |
| V4RP786 | CCTCTCTATGGGCAGTCGGTGATGAGGAACTGGTGGACTACCAGGGTATCTAA |
| V4RP555 | CCTCTCTATGGGCAGTCGGTGATGAAGTTGTAGTGGACTACCAGGGTATCTAA |
| V4RP333 | CCTCTCTATGGGCAGTCGGTGATGATCCAGGCATGGACTACCAGGGTATCTAA |
| V4RP734 | CCTCTCTATGGGCAGTCGGTGATGCGGTTGGCTTGGACTACCAGGGTATCTAA |
| V4RP299 | CCTCTCTATGGGCAGTCGGTGATGCCAGAAGAATGGACTACCAGGGTATCTAA |
| V4RP564 | CCTCTCTATGGGCAGTCGGTGATGACGACAAGGTGGACTACCAGGGTATCTAA |
| V4RP280 | CCTCTCTATGGGCAGTCGGTGATGACCATTAGATGGACTACCAGGGTATCTAA |
| V4RP684 | CCTCTCTATGGGCAGTCGGTGATGAAGAATTCATGGACTACCAGGGTATCTAA |
| V4RP290 | CCTCTCTATGGGCAGTCGGTGATGACCACTCGGTGGACTACCAGGGTATCTAA |
| V4RP178 | CCTCTCTATGGGCAGTCGGTGATGCCGGTAGAATGGACTACCAGGGTATCTAA |
| V4RP322 | CCTCTCTATGGGCAGTCGGTGATGTAGCTTAGGTGGACTACCAGGGTATCTAA |
| V4RP266 | CCTCTCTATGGGCAGTCGGTGATGAATTACAGATGGACTACCAGGGTATCTAA |
| V4RP388 | CCTCTCTATGGGCAGTCGGTGATGTATGGCCGATGGACTACCAGGGTATCTAA |
| V4RP591 | CCTCTCTATGGGCAGTCGGTGATGATCGACTTATGGACTACCAGGGTATCTAA |
| V4RP357 | CCTCTCTATGGGCAGTCGGTGATGTTCATCTCGTGGACTACCAGGGTATCTAA |
2.3. Read processing and statistical analyses
Reads were trimmed of adaptors and primer sites by using cutadapt version 1.16 (Martin, 2011). The remaining reads were dereplicated, denoised, and chimeras eliminated using the DADA2 package version 1.8 (Callahan et al., 2016) within the R statistical programming environment (R Core Team 2015). Taxonomic assignment of the ASVs (amplicon sequence variants), to species level where possible, was also performed within DADA2 using the SILVA database version 132 (Quast et al., 2013). Next, contaminant taxa were identified from the sequenced extraction control using the “prevalence” algorithm implemented in the R package decontam (Davis, Proctor, Holmes, Relman, & Callahan, 2018). Further potential contaminants were identified by processing the five libraries derived from reagents and materials as described, and then excluding all ASVs that were found in any of these control libraries from subsequent analyses.
To reduce the impact of ASVs with very low abundance, we removed all ASVs that were present in only a single sample and also discarded ASVs from bacterial phyla that only occurred once in total. To account for potential biases between samples with uneven sequencing depth, all read counts from the remaining samples were rarefied to the read depth of the sample with the lowest read number. An overview of how our filtering steps affected ASV counts can be found in Table 3. All subsequent statistical analyses were done on log‐transformed read counts. Because the symbionts Wolbachia and Spiroplasma were dominant in some of the samples, we excluded all ASVs corresponding to these taxa prior to statistical comparisons between groups. First, we plotted the abundance of the most frequently found bacterial families using the R packages phyloseq and ggplot2 (McMurdie & Holmes, 2013; Wickham, 2009). Next, ordination analyses were performed with phyloseq using Bray distances and non‐metric multidimensional scaling (NMDS). Differences in abundances of particular taxa between groups (quill mite species, bird host species, developmental stage, Wolbachia positive and negative samples) were determined with Kruskal–Wallis rank sum tests, and p‐values were adjusted to these multiple comparisons to control for the false discovery rate (Benjamini & Hochberg, 1995). These tests were done separately for differences in abundance of bacterial phyla, orders, families, and genera. Furthermore, we calculated Jensen–Shannon distances between the aforementioned groups and used adonis tests (analysis of variance using distance matrices) implemented in the package vegan (Dixon, 2003) to test if they differed significantly. A phyloseq object file containing all data used in the described analyses is available at ://doi.org/10.5281/zenodo.3475151.
Table 3.
Overview on the impact of filtering and decontamination on the number of retained ASVs and samples in this study. For details on each of the steps, please refer to the materials and methods section
| Step | #ASVs | #samples |
|---|---|---|
| Unfiltered | 4,309 | 152 |
| Decontamination with “decontam” prevalence method | 4,275 | 152 |
| Remove phyla with only one representative | 4,248 | 152 |
| Removal of ASVs present in only one sample | 1,016 | 152 |
| Removal of samples with very low read count (<3,500) | 1,016 | 126 |
| Rarefy to even depth | 983 | 126 |
| Remove all taxa present in sequenced laboratory reagents and equipment | 912 | 126 |
3. RESULTS
We have investigated the microbial composition of 126 individuals belonging to eleven quill mite species that parasitize twelve bird host species of passeriform birds. Amplicon sequencing of the v4 region of the 16S rRNA gene on an IonTorrent resulted in 1,582,340 reads, with 9,426 reads/sample on average (4,616–20,231). After processing of reads (quality filtering, denoising, annotation, low abundance filtering, rarefying, decontamination—see methods for details), 912 ASVs were retained. Among the most abundant bacterial genera found in quill mites were Wolbachia and Spiroplasma (Figure 1a). Because these symbionts were not equally abundant across samples and might thus bias estimates of bacterial composition, they were excluded from the subsequent analyses.
Figure 1.

Overview of the bacterial taxa detected in quill mites. (a) Relative abundances for the endosymbionts Spiroplasma and Wolbachia. (b) Relative proportions of the 20 most abundantly found bacterial families in a dataset without the symbionts Spiroplasma and Wolbachia. For (a) and (b), each bar represents the averaged abundances across all samples of a single species. Height of stacks represents relative abundances of each taxon. For abundance plots of all samples, please refer to Appendix Figure A1
Bar plots of ASV abundance and ordination analyses with this filtered dataset revealed that the bacterial composition was relatively uniform across samples, and no clear differentiation between samples extracted from different mite species, or between Wolbachia positive and negative a1 samples could be observed (Figures 1b, 2, see also Appendix Figure A1). However, when trying to identify differential abundance patterns of microbial composition between groups using analysis of variances, we found that bacterial composition was more similar between samples from the same quill mite species or genus and bird host species or genus than expected by chance (p < .01). Furthermore, six bacterial families were found to be differentially abundant between quill mite species with a Kruskal–Wallis test (p < .01, Figure 3), one of which (Xanthobacteraceae) was also found to differ between samples of different bird host genera.
Figure 2.

Similarity of quill mite microbiota without the endosymbionts Spiroplasma and Wolbachia . Ordination analysis is based on non‐metric multidimensional scaling (NMDS) and bray distances. Log‐transformed abundances were analyzed. Colors of the dots represent different quill mite species from which the samples were isolated. Shape of the dots stand for Wolbachia infection status
Figure 3.

Abundance of five bacterial families that were found to be differentially abundant between quill mite species analyzed. Counts for the symbionts Wolbachia and Spiroplasma were excluded
Out of 912 detected ASVs, the 10 most abundantly encountered genera were Micrococcus, Corynebacterium, Acinetobacter, Streptococcus, Burkholderia, Phyllobacterium, Ralstonia, Mycobacterium, Paracoccus, and Sediminibacterium (for a full list of ASVs, see Appendix Table A1 at ://doi.org/10.5281/zenodo.3475151). None of these taxa seemed dominant in any sampled group (based on mite or bird taxonomy), and the 20 most abundant families made up similar proportions of the total ASVs across samples (Figure 1b). Other notable findings were the pathogens Brucella which was detected in 20 samples with an average abundance of 1.3%, and Bartonella which was found in two samples at 1.8% and 0.7% relative abundance, respectively.
As opposed to the general trend in the microbiome composition data, there was strong evidence for differential abundance of the symbionts Wolbachia and Spiroplasma between the bird hosts from which the mites were collected. For example, high Spiroplasma titers were only observed in two mite species collected from the host genus Carduelis (Figures 1a, 4a, Table 1). Further, although Wolbachia was present in mites sampled from all bird hosts, it was especially prevalent in mites collected from birds of the genera Turdus, Erithacus, and Fringilla. On contrast, it was absent or at very low titers in mites parasitizing Luscinia sp. (Figure 4a, Table 1). On average, the abundance of Wolbachia was lower in samples that also contained Spiroplasma (Figure 4b). Notably, this was not an effect of Spiroplasma presence reducing the amount of available reads for Wolbachia (Figure 4b). For mites harboring both symbionts (eleven samples in total), we found that the abundances for Wolbachia and Spiroplasma are positively correlated (Figure 4c).
Figure 4.
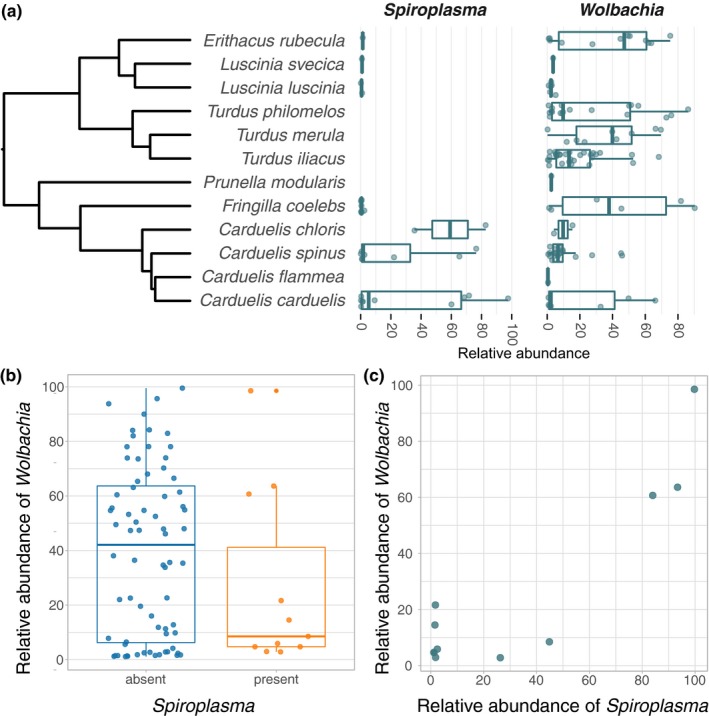
Relative abundances of the endosymbionts Wolbachia and Spiroplasma in quill mite samples. (a) Abundances for all samples that are Spiroplasma and/or Wolbachia positive, sorted by bird host species from which the quill mites were isolated. Bird species phylogeny was taken from Jetz, Thomas, Joy, Hartmann, and Mooers (2012; https://birdtree.org/). (b) Relative Wolbachia abundances in samples with and without Spiroplasma. (c) Correlation of Wolbachia and Spiroplasma abundances for samples in which both symbionts were present. For (b) and (c), only samples with abundances ≥ 1% are shown. Also, to avoid biases of abundance estimates based on a single dominant taxon, Spiroplasma and Wolbachia abundances shown in (b) and (c) were corrected for the presence of the other endosymbiont, that is, Wolbachia and Spiroplasma abundance is plotted relative to the non‐symbiont microbiome. For uncorrected Spiroplasma and Wolbachia abundances for all samples, please refer to Table 1 and Appendix Table A1
4. DISCUSSION
4.1. Origin of microbial DNA in quill mites
We have sequenced microbial taxa from quill mites, an enigmatic group of bird ectoparasites. The taxa detected through 16S rRNA gene sequencing may be (a) resident symbionts of quill mites, (b) environmentally acquired, transient bacteria, or (c) contaminants from reagents and materials. Each of these options comes with a number of assumptions that can be tested with our data.
For “true,” resident symbionts, one would expect high abundances in at least some of the investigated hosts, presence in all individuals of a host species, and specialization of the symbionts, measurable as genetic differentiation between the symbionts of different host taxa. For example, all honey bees (Apis sp.) harbor seven core gut microbial taxa, five of which are present in other corbiculate bees, and two that are not found anywhere else (Kwong, Medina, & Koch, 2017). The composition of these taxa is correlated with phylogenetic distances in this clade of bees, suggesting long‐term association of the microbes with bees. In our dataset, Wolbachia and Spiroplasma are the most likely candidates for true symbiotic associations. Both bacteria are known as endosymbionts from other arthropods and are unable to permanently live outside their hosts (Anbutsu & Fukatsu, 2011; Makepeace & Gill, 2016). Further, we document a very high abundance of these taxa in at least some of the investigated samples (Figure 4), which is in line with the assumptions above. In a previous study, Wolbachia strains of quill mites were investigated with a multi‐locus approach and it has been shown that quill mite associated strains are genetically very different to any other Wolbachia strains described so far (Glowska et al., 2015). Here, we have found eight different ASVs annotated as Wolbachia, each of which is 100% identical to at least one Wolbachia sequence previously isolated from quill mites. For Spiroplasma, we found a single ASV that is only 92% identical to the next closest match in the Silva database. This implies that Spiroplasma in quill mites might be genetically distinct from Spiroplasma of other arthropods, as is the case for Wolbachia. However, sequencing data of more loci are needed to establish the phylogenetic placement of Spiroplasma from quill mites.
For environmentally acquired, transient taxa, the expectation is that the microbial composition detected in the host reflects the microbial composition of its environment stronger than it reflects host‐specific factors. For example, the gut microbiome of some caterpillars is dominated by bacteria that derive from their food, evidenced by similar bacterial composition of leave surfaces and caterpillar feces (Hammer, Janzen, Hallwachs, Jaffe, & Fierer, 2017). Quill mites live permanently within feather quills of their bird hosts; hence, one might expect to find similar taxa in feathers or on bird skin as in quill mites. Unfortunately, none of the bird hosts sampled in our study was investigated previously with regard to resident skin or feather microbes. One of the most comprehensive feather microbiome studies was performed in the Dark‐eyed Juncos Junco hyemalis (L.) and revealed that feathers of these birds harbor bacteria commonly occurring in the soil and phyllosphere (Brevundimonas, Methylobacterium, and Sphingomonas), as well as potential plant pathogens (e.g., Sphingomonas, Microbacterium, Curtobacterium, and Rathayibacter) (Dille, Rogers, & Schneegurt, 2016). All of these taxa were also found in our study, suggesting a potential environmental determinant of the bacterial composition we observed in quill mites. Furthermore, many of the core bacterial families described in bird skin microbiome studies were also found in quill mites (e.g., Pseudomonadaceae, Methylobacteriaceae, Corynebacteriaceae, Moraxellaceae, Mycobacteriaceae, Leuconostocaceae, Staphylococcaceae, Lactobacillaceae, Micrococcaceae, Streptococcaceae, Enterobacteriaceae, Sphingomonadaceae, Neisseriaceae, Xanthomonadaceae, and Weeksellaceae) (Engel et al., 2018; Pearce, Hoover, Jennings, Nevitt, & Docherty, 2017). Despite these similarities, and some statistical support for bird hosts shaping the microbiome community in our study, the lack of clustering in ordination analysis indicates that environment is not the major determining factor of quill mite microbiome composition.
It is important to consider that contaminants from reagents and kits may significantly impact microbiome composition estimates, especially when using low biomass samples such as quill mites (de Goffau et al., 2018; Łukasik et al., 2017; Salter et al., 2014). This is problematic in any microbiome study and is very difficult to exclude with certainty. Here, we removed contaminants statistically in silico based on the microbial composition of the sequenced extraction control (Davis et al., 2018). Further, we removed all ASVs present in independently sequenced controls derived from reagents and equipment commonly used in the laboratory where this study was performed (see methods for details). However, a number of ASVs we recovered correspond to common kit contaminants in 16S rRNA microbiome studies (e.g., Ralstonia, Kocuria), human skin bacteria (Corynebacterium), or ubiquitous taxa with no strong evidence for symbiotic associations with arthropods (Pseudomonas, Acinetobacter). These taxa might constitute true associates, but we cannot exclude the possibility that they originate from contaminating sources.
In summary, we found a diverse range of bacteria associated with quill mites. The lack of differentiation between different mite species or between species collected from different bird hosts leads us to conclude that there are no strong associations with typical gut bacteria as observed in other arthropods. However, we cannot exclude that we missed such potential associates due to the limited amount of DNA that can be extracted from the minute hosts.
4.2. Exchange of bacteria via bird hosts
Due to their ectoparasitic life style with occasional host switching, quill mites have the potential to transmit bacteria between their hosts. Here, we detected two pathogenic microbes that might be important in that respect: Brucella and Bartonella. Brucella is the agent of brucellosis, which is considered to be the most widespread zoonotic infection (Pappas, Papadimitriou, Akritidis, Christou, & Tsianos, 2006). Several Brucella species are a human health threat, and people typically become infected through contact with domesticated Brucella infected animals, such as goats, sheep, or swine (Young, 2006). However, several blood‐sucking arthropods, such as ticks and lice, are regarded as possible vectors for Brucella (Neglia et al., 2013; Pritulin, 1954; Rementzova, 1966). To our knowledge, there is no data indicating that Acari other than ticks are natural Brucella carriers. It was hypothesized that birds and other wild animals act as natural reservoirs for Brucella (Zheludkov & Tsirelson, 2010), which is in line with our finding of this bacterium in bird ectoparasites. The importance of quill mites in spreading Brucella between bird species remains to be assessed, but its prevalence (21/126 investigated individuals, 8 different mite species) suggests its finding is of potential importance in understanding this pathogen's dynamics.
Bartonella are gram‐negative bacteria that are typically transmitted by blood‐sucking arthropods and are infectious in mammalian hosts (Billeter, Levy, Chomel, & Breitschwerdt, 2008; Klangthong et al., 2015; Reeves, Nelder, Cobb, & Dasch, 2006). There are also reports on Bartonella incidence in birds (Ebani, Bertelloni, & Mani, 2016; Mascarelli, McQuillan, Harms, Harms, & Breitschwerdt, 2014), and it is conceivable that the bacteria originate from the birds, rather than from the mites. That would suggest that the host range for Bartonella spp. is broader than previously reported and here we expand the list of potential sources for this zoonotic infection. However, Bartonellaceae can be symbiotic in other hosts, such as honey bees and ants (Segers, Kešnerová, Kosoy, & Engel, 2017, Bisch et al. 2018). Further, Bartonella‐like symbionts were recently found in a number astigmatid mites (Kopecký, Nesvorná, & Hubert, 2014), indicating that the Bartonella detected here might be quill mite symbionts, rather than pathogens. With our data, it is not possible to rule out either possibility.
Finally, we found the symbionts Spiroplasma and Wolbachia in quill mites. Both of these are common across a range of arthropod species (Anbutsu & Fukatsu, 2011; Zug & Hammerstein, 2012), are typically transmitted intraovarially, and may cause sex ratio distorting phenotypes (Haselkorn, 2010; Werren, 1997). Whereas Spiroplasma was so far not reported from quill mites, Wolbachia was previously detected and our findings confirm that this is a common symbiont of quill mites (Glowska et al., 2015). The observed presence and abundance of both taxa are not uniform across the sampled taxa (Figures 1, 4a). For example, Wolbachia is most abundant in mites parasitizing birds of the genera Turdus, Erithacus, and Fringilla, whereas Spiroplasma is most strongly associated with mites parasitizing Carduelis. One reason for this may be that some taxa are more susceptible than others for endosymbiosis with certain bacteria, and this phylogenetic effect has been reported for other host taxa as well (Gerth, Saeed, White, & Bleidorn, 2015; Russell, 2012). Strikingly, very high Spiroplasma abundances were only found in two investigated mite species that are specialized parasites of two bird species of the genus Carduelis (Figure 4a., Table 1). A number of samples showed very low Spiroplasma titers, which may be a result of genuine low titer infections or stem from contamination via simultaneously processed libraries (e.g., through index hopping, Kircher, Sawyer, & Meyer, 2012). For the samples with unambiguously high Spiroplasma titers, the bird host phylogeny seems to be the best predictor for a Spiroplasma infection. One interpretation of this pattern is a history of horizontal transmission of the symbiont via the bird hosts. Horizontal transfers have been inferred from phylogenetic data for Wolbachia and Spiroplasma previously (Gerth, Röthe, & Bleidorn, 2013; Haselkorn, Markow, & Moran, 2009), and for both symbionts, horizontal transmissions were also demonstrated experimentally (Huigens et al., 2000; Jaenike, Polak, Fiskin, Helou, & Minhas, 2007). Although the potential mechanism of horizontal symbiont transmission via feather quills is unclear, our data suggest that the bird–parasite interactions may be important for endosymbiont transmission dynamics in quill mites.
Interestingly, we found that Spiroplasma presence leads to reduced Wolbachia titers, although this is based on a small sample size for samples that are both Wolbachia and Spiroplasma positive (N = 11, Figure 4b). Furthermore, in these eleven samples, Spiroplasma and Wolbachia titers seem to be positively correlated (Figure 4c). It is conceivable that sharing of hosts leads to competition for finite resources the host can provide (Vautrin & Vavre, 2009), and thus the growth of one symbiont might limit that of another. In Drosophila, for example, Spiroplasma seem to limit the proliferation of Wolbachia (Goto, Anbutsu, & Fukatsu, 2006) and in aphids, competition between co‐occurring secondary symbionts appears to be harmful to the host (Oliver et al. 2006). Such negative fitness impacts can also be expected when both symbiont titers are very high, as found here in quill mites. Although purely speculative, this may be the reason why we only observed simultaneously high Spiroplasma and Wolbachia titers in very few of the 126 investigated quill mites (Figure 4c).
5. SUMMARY
We found a diverse, but relatively uniform set of bacterial taxa within quill mites that includes arthropod endosymbionts, pathogens, and bird‐associated bacteria. The importance of most of these microbes for quill mite biology is unclear, but abundances and distribution patterns suggest that Spiroplasma and Wolbachia are the most important quill mite associates.
CONFLICT OF INTEREST
None declared.
AUTHOR CONTRIBUTIONS
EG, ZF, and MG involved in study conception and design. EG involved in collecting and identification of the mite material, funding acquisition, project administration, supervision, writing review and editing and contributed resources. ZF and MD involved in acquisition of data. MG analyzed the data. EG and MG interpreted the data and wrote the paper.
ETHICAL STATEMENT
None required.
ACKNOWLEDGMENTS
We thank Dr. J. K. Nowakowski, Bird Migration Research Station, University of Gdansk for the permission to collect the material. We further thank Dr. Piotr Łukasik for comments on an earlier version of this manuscript. This study was supported by the National Science Centre of Poland grant 2015/19/D/NZ8/00191 (EG) and the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska‐Curie grant agreement No. 703379 (MG).
APPENDIX 1.
Figure A1.

Overview of the bacterial taxa detected in quill mites. Bar plots show the 20 most abundantly found bacterial families. Each stacked bar represents one sample, and the samples are ordered by quill mite species. Height of stacks represents relative abundances of each taxon. Note that all Anaplasmataceae ASVs are Wolbachia, and all Spiroplasmataceae ASVs are Spiroplasma
Table A1 List of all ASVs detected in this study, ordered by abundance (relative abundances summed over all samples) available at http://doi.org/10.5281/zenodo.3475151
Glowska E, Karolina Filutowska Z, Dabert M, Gerth M. Microbial composition of enigmatic bird parasites: Wolbachia and Spiroplasma are the most important bacterial associates of quill mites (Acariformes: Syringophilidae). MicrobiologyOpen. 2020;9:e964 10.1002/mbo3.964
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are openly available under NCBI BioProject accession number PRJNA482380 and in Zenodo repository at ://doi.org/10.5281/zenodo.3475151.
REFERENCES
- Anbutsu, H. , & Fukatsu, T. (2011). Spiroplasma as a model insect endosymbiont. Environmental Microbiology Reports, 3, 144–153. 10.1111/j.1758-2229.2010.00240.x [DOI] [PubMed] [Google Scholar]
- Azad, A. F. , & Beard, C. B. (1998). Rickettsial pathogens and their arthropod vectors. Emerging Infectious Diseases, 4, 179 10.3201/eid0402.980205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballinger, M. , & Perlman, S. (2017). Generality of toxins in defensive symbiosis: Ribosome‐inactivating proteins and defense against parasitic wasps in Drosophila . PLoS Path, 13, e1006431 10.1371/journal.ppat.1006431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini, Y. , & Hochberg, Y. (1995). Controlling the false discovery rate: A practical and powerful approach to multiple testing. Journal of the Royal Statistical Society Series B, 57, 289–300. [Google Scholar]
- Billeter, S. , Levy, M. , Chomel, B. , & Breitschwerdt, E. (2008). Vector transmission of Bartonella species with emphasis on the potential for tick transmission. Medical and Veterinary Entomology, 22, 1–15. 10.1111/j.1365-2915.2008.00713.x [DOI] [PubMed] [Google Scholar]
- Bisch, G. , Neuvonen, M. M. , Pierce, N. E. , Russell, J. A. , Koga, R. , … Andersson, S. G. E. (2018). Genome evolution of bartonellaceae symbionts of ants at the opposite ends of the trophic scale. Genome Biology and Evolution, 10(7), 1687–1704. 10.1093/gbe/evy126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan, B. J. , McMurdie, P. J. , Rosen, M. J. , Han, A. W. , Johnson, A. J. A. , & Holmes, S. P. (2016). DADA2: High‐resolution sample inference from Illumina amplicon data. Nature Methods, 13, 581–583. 10.1038/nmeth.3869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casto, S. D. (1974a). A nocturnal dispersal rhythm in the quill mite, Syringophiloidus minor (Berlese) (Prostigmata: Syringophilidae). Journal of Medical Entomology, 11, 113–114. 10.1093/jmedent/11.1.113 [DOI] [PubMed] [Google Scholar]
- Casto, S. D. (1974b). Entry and exit of syringophilid mites (Acarina: Syringophilidae) from the lumen of the quill. The Wilson Bulletin, 86, 272–278. [Google Scholar]
- Chan, T. F. , Ji, K. M. , Yim, A. K. , Liu, X. Y. , Zhou, J. W. ,… Tsui, S. K. W. (2015). The draft genome, transcriptome, and microbiome of Dermatophagoides farinae reveal a broad spectrum of dust mite allergens. Journal of Allergy and Clinical Immunology, 135, 539–548. 10.1016/j.jaci.2014.09.031 [DOI] [PubMed] [Google Scholar]
- Colston, T. J. , & Jackson, C. R. (2016). Microbiome evolution along divergent branches of the vertebrate tree of life: What is known and unknown. Molecular Ecology, 25, 3776–3800. 10.1111/mec.13730 [DOI] [PubMed] [Google Scholar]
- R Core Team (2015). A language and environment for statistical computing. Vienna, Austria: (R Foundation for Statistical Computing. [Google Scholar]
- Dabert, J. , Ehrnsberger, R. , & Dabert, M. (2008). Glaucalges tytonis sp. n. (Analgoidea, Xolalgidae) from the barn owl Tyto alba (Strigiformes, Tytonidae): Compiling morphology with DNA barcode data for taxon descriptions in mites (Acari). Zootaxa, 1719, 41–52. [Google Scholar]
- Davis, N. M. , Proctor, D. M. , Holmes, S. P. , Relman, D. A. & Callahan B. J. (2018). Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome, 6, 226 10.1186/s40168-018-0605-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Goffau, M. C. , Lager, S. , Salter, S. J. , Wagner, J. , Kronbichler, A. , Charnock‐Jones, D. S. , … Parkhill, J. (2018). Recognizing the reagent microbiome. Nature Microbiology, 3, 851–853. 10.1038/s41564-018-0202-y [DOI] [PubMed] [Google Scholar]
- Dille, J. W. , Rogers, C. M. , & Schneegurt, M. A. (2016). Isolation and characterization of bacteria from the feathers of wild Dark‐eyed Juncos (Junco hyemalis). The Auk, 133, 155–167. [Google Scholar]
- Dixon, P. (2003). VEGAN, a package of R functions for community ecology. Journal of Vegetation Science, 14, 927–930. 10.1111/j.1654-1103.2003.tb02228.x [DOI] [Google Scholar]
- Duron, O. , Morel, O. , Noël, V. , Buysse, M. , Binetruy, F. , Lancelot, R. , … Vial, L. (2018). Tick‐bacteria mutualism depends on B vitamin synthesis pathways. Current Biology, 28, 1896–1902.e5. 10.1016/j.cub.2018.04.038 [DOI] [PubMed] [Google Scholar]
- Ebani, V. V. , Bertelloni, F. , & Mani, P. (2016). Molecular survey on zoonotic tick‐borne bacteria and chlamydiae in feral pigeons (Columba livia domestica). Asian Pacific Journal of Tropical Medicine, 9, 324–327. 10.1016/j.apjtm.2016.03.005 [DOI] [PubMed] [Google Scholar]
- Engel, K. , Sauer, J. , Jünemann, S. , Winkler, A. , Wibberg, D. , Kalinowski, J. , … Caspers, B. A. (2018). Individual‐ and species‐specific skin microbiomes in three different estrildid finch species revealed by 16S amplicon sequencing. Microbial Ecology, 76, 518–529. 10.1007/s00248-017-1130-8 [DOI] [PubMed] [Google Scholar]
- Erban, T. , Klimov, P. B. , Smrz, J. , Phillips, T. W. , Nesvorna, M. , Kopecky, J. , & Hubert, J. (2016). Populations of stored product mite tyrophagus putrescentiae differ in their bacterial communities. Frontiers in Microbiology, 7, 1046 10.3389/fmicb.2016.01046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerth, M. , Röthe, J. , & Bleidorn, C. (2013). Tracing horizontal Wolbachia movements among bees (Anthophila): A combined approach using MLST data and host phylogeny. Molecular Ecology, 22, 6149–6162. [DOI] [PubMed] [Google Scholar]
- Gerth, M. , Saeed, A. , White, J. A. , & Bleidorn, C. (2015). Extensive screen for bacterial endosymbionts reveals taxon‐specific distribution patterns among bees (Hymenoptera, Anthophila). FEMS Microbiology Ecology, 91, fiv047 10.1093/femsec/fiv047 [DOI] [PubMed] [Google Scholar]
- Glowska, E. , Dragun‐Damian, A. , Dabert, M. , & Gerth, M. (2015). New Wolbachia supergroups detected in quill mites (Acari: Syringophilidae). Infection, Genetics and Evolution, 30, 140–146. 10.1016/j.meegid.2014.12.019 [DOI] [PubMed] [Google Scholar]
- Goto, S. , Anbutsu, H. , & Fukatsu, T. (2006). Asymmetrical interactions between Wolbachia and Spiroplasma endosymbionts coexisting in the same insect host. Applied and Environmental Microbiology, 72, 4805–4810. 10.1128/AEM.00416-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer, T. J. , Janzen, D. H. , Hallwachs, W. , Jaffe, S. P. , & Fierer, N. (2017). Caterpillars lack a resident gut microbiome. Proceedings of the National Academy of Sciences of the United States of America, 114, 9641–9646. 10.1073/pnas.1707186114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haselkorn, T. S. (2010). The Spiroplasma heritable bacterial endosymbiont of Drosophila . Fly, 4, 80–87. 10.4161/fly.4.1.10883 [DOI] [PubMed] [Google Scholar]
- Haselkorn, T. S. , Markow, T. A. , & Moran, N. A. (2009). Multiple introductions of the Spiroplasma bacterial endosymbiont into Drosophila . Molecular Ecology, 18, 1294–1305. [DOI] [PubMed] [Google Scholar]
- Hird, S. M. (2017). Evolutionary biology needs wild microbiomes. Frontiers in Microbiology, 8, 725 10.3389/fmicb.2017.00725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogg, J. , & Lehane, M. (1999). Identification of bacterial species associated with the sheep scab mite (Psoroptes ovis) by using amplified genes coding for 16S rRNA. Applied and Environmental Microbiology, 65, 4227–4229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa, T. , Koga, R. , Kikuchi, Y. , Meng, X.‐Y. , & Fukatsu, T. (2010). Wolbachia as a bacteriocyte‐associated nutritional mutualist. Proceedings of the National Academy of Sciences of the United States of America, 107, 769–774. 10.1073/pnas.0911476107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubert, J. , Erban, T. , Kopecky, J. , Sopko, B. , Nesvorna, M. , Lichovnikova, M. , … Sparagano, O. (2017). Comparison of microbiomes between red poultry mite populations (Dermanyssus gallinae): Predominance of Bartonella‐like bacteria. Microbial Ecology, 74, 947–960. 10.1007/s00248-017-0993-z [DOI] [PubMed] [Google Scholar]
- Hubert, J. , Kamler, M. , Nesvorna, M. , Ledvinka, O. , Kopecky, J. , & Erban, T. (2016). Comparison of Varroa destructor and worker honeybee microbiota within hives indicates shared bacteria. Microbial Ecology, 72, 448–459. 10.1007/s00248-016-0776-y [DOI] [PubMed] [Google Scholar]
- Hubert, J. , Kopecky, J. , Nesvorna, M. , Perotti, M. A. , & Erban, T. (2016). Detection and localization of Solitalea‐like and Cardinium bacteria in three Acarus siro populations (Astigmata: Acaridae). Experimental and Applied Acarology, 70, 309–327. 10.1007/s10493-016-0080-z [DOI] [PubMed] [Google Scholar]
- Hubert, J. , Nesvorna, M. , Klimov, P. , Dowd, S. E. , Sopko, B. , & Erban, T. (2019). Differential allergen expression in three Tyrophagus putrescentiae strains inhabited by distinct microbiome. Allergy, 10.1111/all.13921. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Huigens, M. E. , Luck, R. F. , Klaassen, R. H. G. , Maas, M. , Timmermans, M. , & Stouthamer, R. (2000). Infectious parthenogenesis. Nature, 405, 178–179. 10.1038/35012066 [DOI] [PubMed] [Google Scholar]
- Jaenike, J. , Polak, M. , Fiskin, A. , Helou, M. , & Minhas, M. (2007). Interspecific transmission of endosymbiotic Spiroplasma by mites. Biology Letters, 3, 23–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jetz, W. , Thomas, G. H. , Joy, J. B. , Hartmann, K. , & Mooers, A. O. (2012). The global diversity of birds in space and time. Nature, 491, 444–448. 10.1038/nature11631 [DOI] [PubMed] [Google Scholar]
- Ji, B. , & Nielsen, J. (2015). From next‐generation sequencing to systematic modeling of the gut microbiome. Frontiers in Genetics, 6, 219 10.3389/fgene.2015.00219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kethley, J. (1970). A revision of the family Syringophilidae (Prostigmata: Acarina). Contributions of the American Entomological Institute, 5, 1–76. [Google Scholar]
- Kethley, J. (1971). Population regulation in quill mites (Acarina: Syringophilidae). Ecology, 52, 1113–1118. 10.2307/1933821 [DOI] [Google Scholar]
- King, K. C. , Brockhurst, M. A. , Vasieva, O. , Paterson, S. , Betts, A. , Ford, S. A. , … Hurst, D. D. G. (2016). Rapid evolution of microbe‐mediated protection against pathogens in a worm host. The ISME Journal, 10, 1915–1924. 10.1038/ismej.2015.259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher, M. , Sawyer, S. , & Meyer, M. (2012). Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform. Nucleic Acids Research, 40, e3 10.1093/nar/gkr771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klangthong, K. , Promsthaporn, S. , Leepitakrat, S. , Schuster, A. L. , McCardle, P. W. , Kosoy, M. , & Takhampunya, R. (2015). The distribution and diversity of Bartonella species in rodents and their ectoparasites across Thailand. PLoS One, 10, e0140856 10.1371/journal.pone.0140856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopecký, J. , Nesvorná, M. , & Hubert, J. (2014). Bartonella‐like bacteria carried by domestic mite species. Experimental and Applied Acarology, 64, 21–32. 10.1007/s10493-014-9811-1 [DOI] [PubMed] [Google Scholar]
- Kwong, W. K. , Medina, L. A. , Koch, H. et al. (2017). Dynamic microbiome evolution in social bees. Science Advances, 3, e1600513 10.1126/sciadv.1600513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laughton, A. M. , Fan, M. H. , & Gerardo, N. M. (2013). The combined effects of bacterial symbionts and aging on life history traits in the pea aphid, Acyrthosiphon pisum . Applied and Environmental Microbiology, 80, 470–477. 10.1128/AEM.02657-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Łukasik, P. , Newton, J. A. , Sanders, J. G. , Hu, Y. , Moreau, C. S. , Kronauer, D. J. C. , … Russell, J. A. (2017). The structured diversity of specialized gut symbionts of the New World army ants. Molecular Ecology, 26, 3808–3825. 10.1111/mec.14140 [DOI] [PubMed] [Google Scholar]
- Makepeace, B.L. , Gill, A.C. (2016) Wolbachia In: Thomas S., (Eds). Rickettsiales. Cham, Switzerland: Springer. [Google Scholar]
- Martin, M. (2011). Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnet.journal, 17, 10–12. [Google Scholar]
- Mascarelli, P. E. , McQuillan, M. , Harms, C. A. , Harms, R. V. , & Breitschwerdt, E. B. (2014). Bartonella henselae and B. koehlerae DNA in birds. Emerging Infectious Diseases, 20, 490–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFall‐Ngai, M. , Hadfield, M. G. , Bosch, T. C. , Carey, H. V. , Domazet‐Lošo, T. , Douglas, A. E. et al. (2013). Animals in a bacterial world, a new imperative for the life sciences. Proceedings of the National Academy of Sciences of the United States of America, 110, 3229–3236. 10.1073/pnas.1218525110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurdie, P. J. , & Holmes, S. (2013). Phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE, 8, e61217 10.1371/journal.pone.0061217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moro, C. V. , Thioulouse, J. , Chauve, C. , & Zenner, L. (2011). Diversity, geographic distribution, and habitat‐specific variations of microbiota in natural populations of the chicken mite, Dermanyssus gallinae . Journal of Medical Entomology, 48, 788–796. 10.1603/ME10113 [DOI] [PubMed] [Google Scholar]
- Neglia, G. , Veneziano, V. , De Carlo, E. , Galiero, G. , Borriello, G. , Francillo, M. , … Manna, L. (2013). Detection of Brucella abortus DNA and RNA in different stages of development of the sucking louse Haematopinus tuberculatus . BMC Veterinary Research, 9, 236 10.1186/1746-6148-9-236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh, H. , Ishii, A. , Tongu, Y. , & Itano, K. (1986). Microorganisms associated with the house‐dust mite Dermatophagoides. Medical Entomology and Zoology, 37, 229–235. 10.7601/mez.37.229 [DOI] [Google Scholar]
- Oliver, K. M. , Moran, N. A. , & Hunter, M. S. (2006). Costs and benefits of a superinfection of facultative symbionts in aphids. Proceedings of the Royal Society B: Biological Sciences, 273(1591), 1273–1280. 10.1098/rspb.2005.3436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappas, G. , Papadimitriou, P. , Akritidis, N. , Christou, L. , & Tsianos, E. V. (2006). The new global map of human brucellosis. The Lancet Infectious Diseases, 6, 91–99. 10.1016/S1473-3099(06)70382-6 [DOI] [PubMed] [Google Scholar]
- Pearce, D. S. , Hoover, B. A. , Jennings, S. , Nevitt, G. A. , & Docherty, K. M. (2017). Morphological and genetic factors shape the microbiome of a seabird species (Oceanodroma leucorhoa) more than environmental and social factors. Microbiome, 5, 146 10.1186/s40168-017-0365-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritulin, P. (1954). On the transmission of brucellosis by the pasture ticks Dermacentor nuttallia and Hyalomma marginatum . Veterinariya, 31, 31–33. [Google Scholar]
- Quast, C. , Pruesse, E. , Yilmaz, P. , Gerken, J. , Schweer, T. , Yarza, P. , … Glöckner, F. O. (2013). The SILVA ribosomal RNA gene database project: Improved data processing and web‐based tools. Nucleic Acids Research, 41, D590–D596. 10.1093/nar/gks1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves, W. K. , Nelder, M. P. , Cobb, K. D. , & Dasch, G. A. (2006). Bartonella spp. in deer keds, Lipoptena mazamae (Diptera: Hippoboscidae), from Georgia and South Carolina, USA. Journal of Wildlife Diseases, 42, 391–396. 10.7589/0090-3558-42.2.391 [DOI] [PubMed] [Google Scholar]
- Rementzova, M. (1966).Parasitical arthropods vectors of the brucellic infectionIn Corradetti A. (Eds.), Proceedings of the first international congress of parasitology (pp. 147–150). Oxford, UK: Pergamon. [Google Scholar]
- Russell, J. A. (2012). The ants (Hymenoptera: Formicidae) are unique and enigmatic hosts of prevalent Wolbachia (Alphaproteobacteria) symbionts. Myrmecological News, 16, 7–23. [Google Scholar]
- Salter, S. J. , Cox, M. J. , Turek, E. M. , Calus, S. T. , Cookson, W. O. , Moffatt, M. F. , … Walker, A. W. (2014). Reagent and laboratory contamination can critically impact sequence‐based microbiome analyses. BMC Biology, 12, 87 10.1186/s12915-014-0087-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sboner, A. , Mu, X. , Greenbaum, D. , Auerbach, R. K. , & Gerstein, M. B. (2011). The real cost of sequencing: Higher than you think!. Genome Biology, 12, 125 10.1186/gb-2011-12-8-125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segers, F. H. I. D. , Kešnerová, L. , Kosoy, M. , & Engel, P. (2017). Genomic changes associated with the evolutionary transition of an insect gut symbiont into a blood‐borne pathogen. The ISME Journal, 11, 1232–1244. 10.1038/ismej.2016.201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon, G. , Segal, D. , Ringo, J. , Hefetz, A. , Zilber‐Rosenberg, I. , & Rosenberg, E. (2010). Commensal bacteria play a role in mating preference of Drosophila melanogaster . Proceedings of the National Academy of Sciences of the United States of America, 107, 20051–20056. 10.1073/pnas.1009906107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoracki, M. , Michalik, J. , Skotarczak, B. , Rymaszewska, A. , Sikora, B. , Hofman, T. , … Sawczuk, M. (2006). First detection of Anaplasma phagocytophilum in quill mites (Acari: Syringophilidae) parasitizing passerine birds. Microbes and Infection, 8, 303–307. 10.1016/j.micinf.2005.06.029 [DOI] [PubMed] [Google Scholar]
- Skoracki, M. , Spicer, G. S. , & Oconnor, B. M. (2016). A systematic review of the subfamily Syringophilinae (Acari: Syringophilidae) of the Nearctic region. Part 1: Quill mites associated with passerines (Aves: Passeriformes). Zootaxa, 4084, 451. [DOI] [PubMed] [Google Scholar]
- Therese, K. L. , Anand, A. R. , & Madhavan, H. N. (1998). Polymerase chain reaction in the diagnosis of uveitis. British Journal of Ophthalmology, 82, 1078–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valerio, C. , Murray, P. , Arlian, L. , & Slater, J. (2005). Bacterial 16S ribosomal DNA in house dust mite cultures. Journal of Allergy and Clinical Immunology, 116, 1296–1300. 10.1016/j.jaci.2005.09.046 [DOI] [PubMed] [Google Scholar]
- Vautrin, E. , & Vavre, F. (2009). Interactions between vertically transmitted symbionts: Cooperation or conflict? Trends in Microbiology, 17, 95–99. [DOI] [PubMed] [Google Scholar]
- Werren, J. H. (1997). Biology of Wolbachia . Annual Review of Entomology, 42, 587–609. [DOI] [PubMed] [Google Scholar]
- Wickham, H. (2009). ggplot2: Elegant graphics for data analysis. New York, NY: Springer‐Verlag. [Google Scholar]
- Young, E. J. (2006). Brucella sppIn Gillespie S. H., & Hawkey P. M. (Eds.), Principles and practice of clinical bacteriology (pp. 265–271). Chichester, UK: John Wiley and Sons Ltd. [Google Scholar]
- Zheludkov, M. , & Tsirelson, L. (2010). Reservoirs of Brucella infection in nature. Biology Bulletin, 37, 709–715. 10.1134/S106235901007006X [DOI] [Google Scholar]
- Zug, R. , & Hammerstein, P. (2012). Still a host of hosts for Wolbachia: Analysis of recent data suggests that 40% of terrestrial arthropod species are infected. PLoS One, 7, e38544 10.1371/journal.pone.0038544 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are openly available under NCBI BioProject accession number PRJNA482380 and in Zenodo repository at ://doi.org/10.5281/zenodo.3475151.