

Abstract
With the emergence of large‐scale epidemiologic human microbiome studies, there is a need to understand the reproducibility of microbial DNA sequencing and the impact of specimen collection and processing methods on measures of microbial community composition and structure, with reproducibility studies in infants and young children particularly lacking. Here, we examined batch‐to‐batch variability and reliability of collection, handling, and processing protocols, testing replicate stool samples from infants and young children using Illumina MiSeq sequencing of the bacterial 16S rRNA gene V4‐V5 hypervariable region, evaluating 33 conditions with different protocols and extraction methods. We detected no evidence of batch effects in replicate DNA samples or extractions from the same stool sample. Variability in DNA yield and alpha diversity was observed between the different collection, handling, and processing protocols. However, across all protocols, subject variability was the dominant contributor to microbiome structure, with comparatively little impact of the protocol used. While collection method and DNA extraction kit may affect DNA yield, and correspondingly alpha diversity, our findings suggest that characterization of the structure and composition of the fecal microbiome of infants and young children are reliably measurable by standardized collection, handling, and processing protocols and DNA extraction methods within an individual longitudinal study.
Keywords: cohort studies, gut microbiome, infants, reproducibility
Study question: What is the intralaboratory reliability of microbiome sequencing of infant stool samples and how do results differ with the various collection, handling, and processing extraction protocols? What is already known: Various protocols are used in human microbiome research. Studies of the reliability of these methods have been conducted largely among adults in whom the microbiome is relatively stable. What this study adds: Large‐scaled epidemiologic studies of the human microbiome are beginning to emerge. This study adds to evidence of the reliability of methods used to assess the early developing microbiome during the time of the greatest interperson variability.
1. BACKGROUND
There is a rapidly expanding body of evidence that the gut microbiome profoundly influences multiple aspects of health such as immune function and related disorders (Hooper, Littman, & Macpherson, 2012; Shanahan, 2010; Sjogren et al., 2009), risk of obesity (Hooper et al., 2012; Turta & Rautava, 2016), heart disease, altered drug metabolism (Shanahan, 2010), and neurodevelopment and neurobehavioral disorders such as autism spectrum disorder, anxiety, and cognitive development (Carlson et al., 2018; Kelly, Minuto, Cryan, Clarke, & Dinan, 2017; Sharon, Sampson, Geschwind, & Mazmanian, 2016; Sherwin, Rea, Dinan, & Cryan, 2016; Tognini, 2017). Along with the accessibility of high‐throughput sequencing technologies for human microbiome studies is the need to develop robust standardized methods for the collection, handling, storage, and processing of fecal samples that are feasible in large‐scale, longitudinal epidemiologic studies, and to understand the sources of variability in assay results. As large‐scale compilation studies of infant and child microbiome investigations are beginning, such as the NIH's Environmental influences on Child Health Outcomes study which is combining data from over 50,000 mother–infant pairs with longitudinal microbiome studies of differing protocols, it is critical that protocols be evaluated to determine whether existing data can be combined or compared. Current methods used in human microbiome research vary in substantial ways, making it difficult to perform interstudy comparisons and combined analyses. Data on the reproducibility of the most commonly applied collection, processing, storage, and extraction methods in human fecal microbiome studies are beginning to emerge. Specifically, preservation media, freezing conditions, DNA extraction methods, and sequencing approaches have been identified as potential sources of variation in the microbial composition of samples in previous studies (Ariefdjohan, Savaiano, & Nakatsu, 2010; Cardona et al., 2012; Carroll, Ringel‐Kulka, Siddle, Klaenhammer, & Ringel, 2012; Choo, Leong, & Rogers, 2015; Dominianni, Wu, Hayes, & Ahn, 2014; Flores, Shi, Gail, & Ravel, 2012; Flores et al., 2015; Fu et al., 2016; Hang et al., 2014; Kennedy et al., 2014; Lauber, Zhou, Gordon, Knight, & Fierer, 2010; Maukonen, Simoes, & Saarela, 2012; McOrist, Jackson, & Bird, 2002; Nechvatal et al., 2008; Ott et al., 2004; Rintala et al., 2017; Roesch, Casella, et al., 2009; Sinha, Abnet, White, Knight, & Huttenhower, 2015; Smith, Li, Andersen, Slotved, & Krogfelt, 2011; Tedjo et al., 2015; Walker et al., 2015; Wesolowska‐Andersen et al., 2014; Wu et al., 2010; Yuan, Cohen, Ravel, Abdo, & Forney, 2012). As yet, very few studies have evaluated protocol reliability during the developmental period of intestinal microbiome acquisition in infancy and early childhood, when intersubject variability is pronounced (Walker et al., 2015). Further, the current literature lacks data on the potential for batch effects during microbial DNA sequencing. Here, we examine batch‐to‐batch variation and 11 different collection, handling, and processing protocols and 3 DNA extraction kits applied to stool samples collected from infants and young children.
2. METHODS
2.1. Study population and ethics approval
Eight children, 1–3 years of age, provided stool samples for the evaluation of sequencing batch effects. An additional four children ages 1–4 provided stool samples for the evaluation of sample‐handling protocols and DNA extraction methods. The study was approved by the Committee for the Protection of Human Subjects at Dartmouth College, and all study participants provided written informed consent.
2.2. Stool processing to assess batch effects
To assess batch‐to‐batch variability, diapers containing stool were collected and stored at −80°C until processing, then thawed overnight at 4°C. The stool was aliquoted to cryotubes with RNAlater solution (Corning® 430662) then stored at −80°C or colder. Following centrifugation, samples were processed using a BeadBashing Lysis Tube and extracted using the ZR Fecal DNA MiniPrep™ (Zymo Research D6010) kit.
2.3. Batch effect methods
We assessed batch effects in two sets of young children (Tables A1 and A2). For Set 1, one diaper was obtained from five children aged 1 to 3 years. Three aliquots of extracted DNA were taken from each, providing a total of 15 DNA samples for analysis. Analyses of these samples took place over one month . For Set 2, we obtained three additional subjects aged 1–3 years, analyzed two to four aliquots of single DNA extraction for two children and two separate DNA extractions from the same diaper for one child. Set 2 was sequenced in six batches nine months apart (N = 47).
2.4. Collection, processing, and DNA extraction comparisons
To compare the protocols with different collection, handling, processing, and DNA extraction methods, we used four diapers containing freshly collected stool samples, from four subjects aged 1 to 4 years old, that were transported at room temperature to the laboratory within two hours of collection. Comparisons of the various protocols were made to the reference protocol of samples that were immediately −80 C frozen (in trace element‐free and RNAlater‐containing tubes (Corning® 430662)).
A total of 33 protocols were evaluated (Figure 1). Five protocols were tested both with and without RNAlater (Qiagen) storage tubes (Figure 1), totaling 10 protocols, plus an 11th protocol using the Omnigene stool kit. For protocols 1 and 2, the stool was immediately aliquoted and extracted. For protocols 3 and 4, the stool was immediately aliquoted into cryotubes and frozen at −80°C. Frozen tubes later were thawed on ice and extracted. For protocols 5 and 6, diapers were kept at −20°C for 4–12 hr, then thawed for 24 hr in an insulated bag after which stool was frozen at −80°C. Frozen tubes were later thawed on ice and extracted. For protocols 7 and 8, diapers were kept in an insulated bag for 24 hr at room temperature and then transferred into cryotubes and frozen at −80°C. Frozen tubes were thawed on ice and extracted. For protocols 9 and 10, diapers were immediately frozen at −20°C. Frozen diapers were subsequently stored in an insulated transport bag with cold packs for 4 hr and then removed from the bag and stored at −80°C. Diapers were then thawed overnight at 4°C, and stool was aliquoted into cryotubes and frozen at −80°C. Frozen tubes were thawed on ice and extracted. Lastly, protocol 11 involved immediately aliquoting stool into an OMNIgene Gut tube according to the manufacturer's instruction and storing at room temperature prior to extraction.
Figure 1.
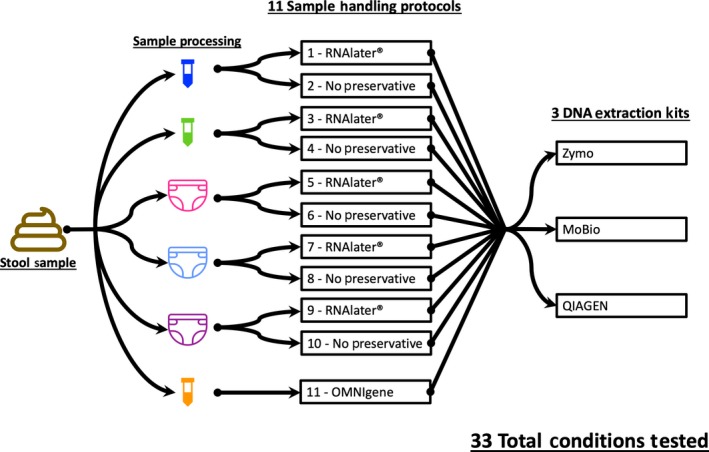
Schema of 33 protocols evaluated. Each stool sample was homogenized then divided and subjected to 6 different sample‐handling conditions. Samples subjected to the first 5 conditions were further tested with and without added RNAlater. The OMNIgene kit includes a preservative and thus was not tested with additional RNAlater. Finally, each of these samples was further divided for extraction with 3 DNA extraction kits
2.5. DNA extraction
Three DNA extraction kits were evaluated: Zymo Research ZR Fecal DNA MiniPrep™ kit, Qiagen QIAamp® Fast Stool Mini Kit, and MO BIO Powersoil®. Tubes containing RNAlater were centrifuged to pellet stool, and RNAlater solution was removed before processing them in the same manner as the nonpreserved stool samples.
For ZR Fecal DNA MiniPrep™ kit extractions, samples were processed following the manufacturer's protocol using the provided 0.5mm glass bead tubes and performing a 6‐min bead beating on a “Disruptor Genie” vortex adapter. DNA was eluted after incubation with Elution buffer and passed through Zymo‐Spin™ IV‐HRC columns.
For the Qiagen QIAamp Fast Stool Mini KitTM extractions, samples were processed following the manufacturer's protocol, using Lysis buffer and a 70°C Lysis step. Samples were strongly vortexed before and after the 70°C incubation. DNA was eluted after incubation with the provided Elution buffer.
For MoBio protocol extractions using the MO BIO Powersoil® kit, C1 solution was added and the sample was bead beaten for 10 min on the MoBio Vortex adapter. The remaining procedures were done exactly per the MoBio protocol instructions. Samples were likewise eluted after 5 min of incubation with the provided Elution buffer.
2.6. Sequencing
Extracted microbial DNA was sent to the Marine Biological Laboratory (MBL) in Woods Hole, MA, an affiliate of the University of Chicago. The V4‐V5 hypervariable regions of bacterial 16S rRNA genes were amplified in 96‐well plate format and sequenced as a multiplexed pool on the Illumina MiSeq. Additional details of sequencing protocols are available in a previous report (Newton et al., 2015). For this study, the average yield per sample after processing, quality control filtering, and chimera exclusion was 117,924 reads and ranged from 528 to 383,728 reads.
2.7. Statistical analysis
QIIME (v. 1.9.1) with UCLUST (v. 1.2.22) was used to create OTUs at the 97.5% sequence identity level. Taxonomy was assigned to the OTUs from the Greengenes (v. 13_8) database. To determine batch‐to‐batch variability, we created a heat map to view clustering patterns using the R package pheatmap (v. 1.0.8) with Euclidean clustering (Kolde, 2015). We computed alpha diversity, as a measure of the number of bacterial species present, using the Simpson Index, using the estimate richness function in the phyloseq (v. 1.16.0) package in R (McMurdie & Holmes, 2013). A linear mixed‐effects model was used to compare alpha diversity across batches (Shanahan, 2010) with the Kenward–Roger method to estimate degrees of freedom and approximate p‐value (Halekoh & Højsgaard, 2014). We further calculated the phylogenetic distances, beta diversity, as a measure of the bacterial community structure, between the extracts using generalized UniFrac analysis (GUniFrac R package v. 1.0; Chen et al., 2012), and statistical significance of the clustering using permutational multivariate analysis of variance (PERMANOVA) using the vegan (v. 2.3.5) function Adonis in R (Dixon, 2003). PCoA plots were generated from the GUniFrac distance matrices using the PCoA function in the phyloseq program.
We compared DNA yield, alpha diversity, beta diversity, and the relative abundance of individual taxa across the 33 collection, handling, processing, and DNA extraction protocols. A linear mixed‐effects model was used to compare DNA yield and alpha diversity of each protocol and DNA extraction method (Bates, Mächler, Bolker, & Walker, 2014) again with the Kenward–Roger method to estimate degrees of freedom and approximate p‐values (Halekoh & Højsgaard, 2014). Beta diversity for the samples was, as before, determined using the GUniFrac distances from the GUniFrac R package with PERMANOVA as described above. A generalized linear model was used to determine the statistical significance of clustering for pairwise comparisons of the within versus between‐group GUniFrac distances for the groups defined by protocol, subject, or extraction method (i.e., intragroup minus intergroup distances).
All p‐values were adjusted with Bonferroni correction for multiple testing.
2.8. Sensitivity analyses
All analyses were performed with and without rarefaction to minimum read depth of 3,073 reads based on the rarefaction curve, removing one sample with a depth of 528 reads.
3. RESULTS
To examine batch effects between the sequencing runs, one diaper was collected from each of five children aged 1 to 3 years (Set 1) from which we evaluated three aliquots from one DNA extraction from each diaper (N = 15 samples). These samples were sequenced separately in three different batches over a one‐month period. For three additional children of the same age range (Set 2), two to four aliquots of single DNA extraction for two children and two separate DNA extractions from the same diaper for one child were sequenced in six batches nine months apart (N = 47; Table A1).
3.1. Samples run in separate sequencing batches reliably clustered by subject rather than by sequencing batch
Using Euclidean clustering, we found that the results evaluating batch effects (evaluating the same samples divided and sequenced at different time points) clustered by individual subject and not by sequencing batch in both sets of comparisons (Set 1 vs. Set 2, Figure 2). In generalized UniFrac models, we again found that the results in both sets clustered by subject (PERMANOVA p = .003 and p = .001 for Set 1 and Set 2, respectively) with no evidence of batch effects (PERMANOVA p = .986 and p = .871, respectively, Figure 3a,b). While there was some variance in alpha diversity, these differences were not statistically significant by batch (Figure A1).
Figure 2.
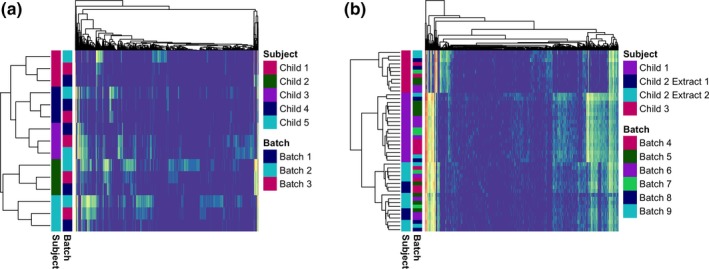
(a) Heat map depicting Set 1 generated with the pheatmap package in R. Samples and OTUs are clustered by Euclidean clustering. Subject and batch are annotated on the y‐axis. (b) Heat map depicting Set 2 generated with the pheatmap package in R. Samples and OTUs are clustered by Euclidean clustering. Subject, extraction, and batch are annotated on the y‐axis
Figure 3.
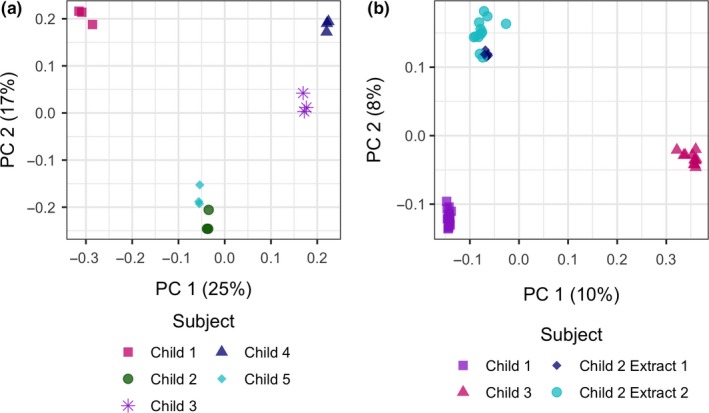
(a) PCoA plot Set 1 generalized UniFrac distances. Samples clustered by subject (PERMANOVA p = .003) and did not cluster by batch (PERMANOVA p = .986). (b) PCoA plot Set 2 generalized UniFrac distances. Samples clustered by subject (PERMANOVA p = .001) and did not cluster by batch (PERMANOVA p = .871)
To compare the 33 protocols with the different collection, handling, processing, and DNA extraction methods, we used four diapers containing freshly collected stool from four subjects aged 1 to 4 years old that were transported at room temperature to the laboratory within two hours of collection. Comparisons of the various protocols were made to the reference protocol of samples that were immediately processed (in trace element‐free and RNAlater‐containing tubes).
3.2. DNA yield differred by DNA extraction kit and by home freezing
With respect to DNA yield (Figure A2), the Qiagen QIAamp Fast Stool Mini KitTM and the MO BIO Powersoil® DNA isolation kit both had lower DNA yield than the ZR Fecal DNA MiniPrep™ kit (Kenward–Roger p < .0001 and p < .03, respectively). A lower yield was observed for protocols involving immediate freezing in a home, −20°C freezer, compared to the reference protocols 1 and 2 of immediate processing of fresh stool (Kenward–Roger p < .03). However, there were no statistically significant differences in DNA yield between protocols involving immediate freezing in −80°C, or transport for up to 24 hr on freezer packs before freezing or processing and the reference protocols. Similarly, no statistically significant differences were detected in DNA yield by whether the collection tube contained the RNA stabilizer RNAlater or not (Figure A3).
3.3. Alpha diversity differred by DNA extraction kit and by the use of OMNIgene Gut collection tubes
Of the three extraction methods, Simpson diversity was higher for the Zymo ZR Fecal DNA MiniPrep™ kit than for the MO BIO Powersoil® DNA isolation kit or the Qiagen QIAamp Fast DNA Stool Mini Kit (Kenward–Roger p < .0001 for both comparisons; Figure 4a). Overall, specimens collected in the OMNIgene Gut collection tubes had lower alpha diversity in comparison with specimens collected using the reference protocol of “trace element‐free” tubes with or without RNAlater that were immediately processed (p = .011) (Figure 4b). These analyses were performed after rarefaction, although models without rarefaction yielded similar results (data not shown).
Figure 4.
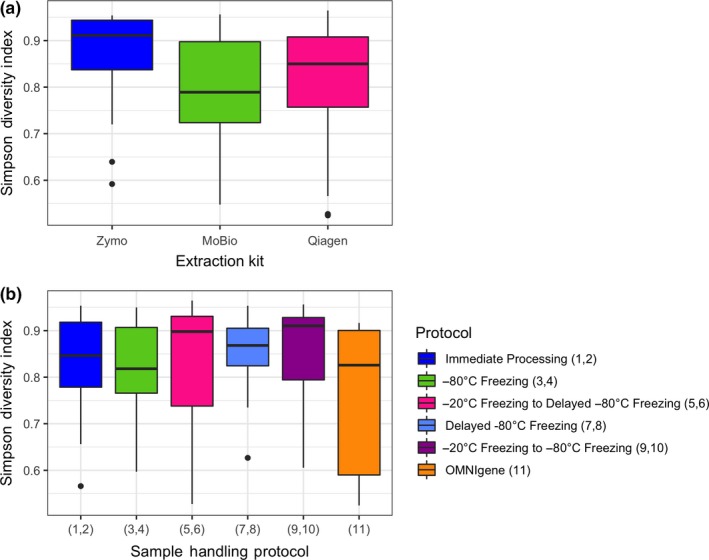
(a) Simpson diversity index by DNA extraction method. The MoBio and Qiagen DNA extraction kits showed a significant reduction in diversity in a linear mixed‐effects model (p < .001). (b) Simpson diversity index by handling protocol. Samples processed with the OMNIgene DNA extraction kit showed a significant reduction in alpha diversity compared to samples immediately processed in a linear mixed‐effects model (p = .011)
3.4. Sample clustered primarily by subject regardless of collection, handling, or processing protocols
Comparing the intra‐ minus intergroup distances from the GUniFrac analyses, we found that samples clustered primarily by subject (Figure 5a), and less so by collection, handling, and processing protocols or DNA extraction kit (Figure 5a). Among the collection, handling, and processing protocols, samples collected using the OMNIgene Gut kit clustered more closely together than those from the other protocols (Figure 5b). At the phylum level for individual taxa, there were no statistically significant differences after Bonferroni correction in the linear mixed‐effects models (data not shown).
Figure 5.
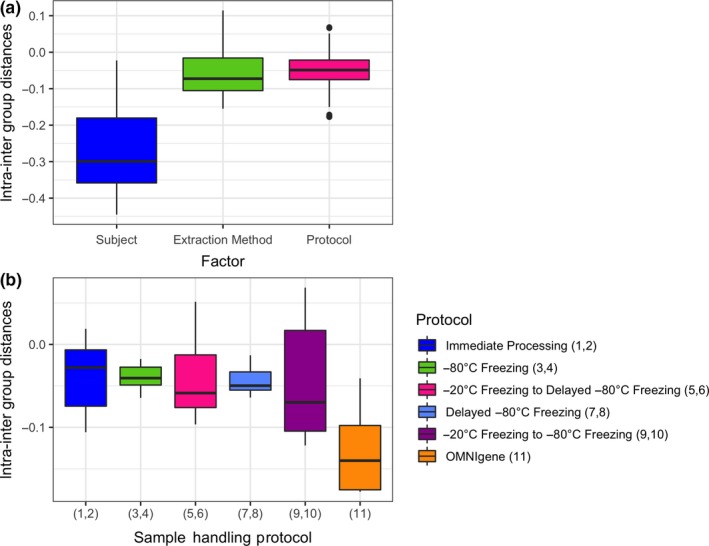
(a) Pairwise generalized UniFrac distances by Child, DNA extraction method, and handling protocol. Samples clustered primarily by subject. (b) Pairwise distance by handling protocol. Samples processed with the OMNIgene kit clustered more closely than samples processed with other sample‐handling methods
4. DISCUSSION
4.1. Principal findings
In samples from our ongoing pregnancy cohort study, we found little to no evidence of batch‐to‐batch variability in DNA extracts from infant stool samples or multiple extracts of the same sample over a period of weeks to years. Immediate freezing or delayed freezing samples that were kept in a home freezer (i.e., −20°C) had somewhat lower DNA yields compared to immediate processing; however, this did not translate to differences in alpha or beta diversity and appeared to have little to no impact on the relative abundance of bacterial phyla. Further, we found both a higher DNA yield and a higher alpha diversity among samples extracted using the Zymo Research ZR Fecal DNA MiniPrep™ kit when compared with the MO BIO Powersoil® DNA isolation kit or the Qiagen QIAamp Fast DNA Stool Mini Kit.™ The OMNIgene Gut collection tube provided sequencing results with lower DNA yield and alpha diversity than the other collection protocols and tended to cluster together more closely than samples collected using the other protocols.
4.2. Strengths
Samples and analyses were conducted on a single population in a single laboratory. This limited variation that might arise from comparing samples between laboratories and vastly different populations, thus allowing for a more targeted comparison of the different protocols.
4.3. Limitations
While we examined both batch effects and 33 different protocols, it is important to note that we only studied a small number of subjects limiting our statistical power to detect differences, particularly at the genus level. Further, our study was based on stool samples, and thus, the reliability of other substrates (i.e., skin or saliva) may differ. Additionally, we focused our study on infants and young children, and thus, our findings may not be generalizable to older ages. However, studies have identified that the first 3 years of life represent the most substantial variability in microbiome samples; thus, clarification of protocol‐related impacts on results has potentially the most impact on the reliability of results. While our study based on a single population using a single laboratory was a strength, it also did not enable us to evaluate laboratory‐to‐laboratory variation. We might anticipate that additional statistical considerations will be required to combine or pool data from different studies using disparate protocols. When examining batch effects, we thawed samples overnight at 4°C prior to aliquoting, which produced reproducible DNA sequencing results; however, this approach risks RNA and metabolite degradation and ideally aliquoting would occur immediately to preserve the sample integrity.
4.4. Interpretation
The results of our study are reassuring, as to our knowledge, the issues of batch effects and protocol differences have not been addressed extensively previously in young children. Reliability of microbiome results has been investigated to only a limited extent among adults, with similar results to our study of children (Vogtmann et al., 2017). One study that did assess children's stool samples (Roesch, Lorca, et al., 2009) also noted limited variability in community composition occurring after 72 hr at room temperature. Likewise, several studies in adults have addressed time to extraction and temperature (Choo et al., 2015; Flores et al., 2012, 2015; Hang et al., 2014; Lauber et al., 2010; Tedjo et al., 2015) and found temperature had a minor impact, although rare taxa may be undetected if the sample was not immediately frozen.
Our findings of both a higher DNA yield and a higher alpha diversity among samples extracted using the Zymo Research ZR Fecal DNA MiniPrep™ kit than with the MO BIO Powersoil® DNA isolation kit or Qiagen QIAamp Fast DNA Stool Mini Kit™ are also consistent with prior work. In a study of infants conducted by Walker et al. (2015) mechanical disruption (bead beating) was identified as an important step for the accurate enumeration of Bifidobacteria, which is abundant in the infant gut microbiome. Another study evaluated samples from subjects ages 6 months to 4 years, similar to our study, comparing three DNA extraction methods (Smith et al., 2011) again, results were similar between extraction methods if bead beating was utilized (Walker et al., 2015). Given that we added a bead‐beating step to the MoBio kit, this could not fully explain the higher yield and diversity identified with the Zymo kit.
The OMNIgene Gut collection tube is a simplified, more complete microbiome kit that includes a DNA stabilizer. While the OMNIgene method provided more homogenous, stable results, the loss of diversity could influence a study's statistical power to detect differences, for example, by a predictor of bacterial diversity or the relationship of diversity to a health outcome.
Other studies in adults focusing on extraction (Maukonen et al., 2012; McOrist et al., 2002; Wesolowska‐Andersen et al., 2014; Yuan et al., 2012) and sequencing method reliability (Rintala et al., 2017) have pointed out that DNA extraction method differences are often minor compared to the hypervariable region targeted (Rintala et al., 2017) and that interindividual variation exceeded variation introduced by extraction method (Wesolowska‐Andersen et al., 2014). Overall, most investigations have resulted in using caution when comparing data across different studies employing differing methods. For instance, Sinha et al., for a microbiome quality control project, published their baseline results comparing laboratories in order to address reproducibility among studies (Sinha et al., 2015), concluding that successful reproducibility within laboratories over time and across field sites is possible. Wu et al. (2010) completed a comprehensive evaluation of freezing, storage, and DNA extraction methods and identified individual variation, and purification methods are of most importance in reproducibility of results. Still, others have evaluated home collection in preparation for larger molecular epidemiological studies and have identified reproducibility despite somewhat variable collection and temperature techniques (Nechvatal et al., 2008).
5. CONCLUSIONS
In conclusion, within a single US study of samples from young children and using a single laboratory and technique, we found little evidence of batch effects or between protocol differences in bacterial structure and composition using a standardized collection, handling, processing, and extraction protocols for stool microbiome analysis. These findings have important application for studies that aim to perform large epidemiological investigations, in geographically diverse areas, or attempts to compile extant data to understand potential relationships between the microbiome and disease causation or prevention.
CONFLICT OF INTEREST
None declared.
AUTHORS' CONTRIBUTION
Katherine M. Antosca: Data curation (equal); Formal analysis (lead); Methodology (equal); Software (lead); Visualization (lead); Writing‐original draft (lead); Writing‐review & editing (equal). Anne Hoen: Conceptualization (supporting); Data curation (equal); Methodology (equal); Supervision (equal); Writing‐original draft (equal); Writing‐review & editing (equal). Thomas J. Palys: Data curation (equal); Investigation (equal); Writing‐original draft (supporting); Writing‐review & editing (supporting). Margaret Hilliard: Data curation (equal); Writing‐original draft (supporting); Writing‐review & editing (supporting). Hilary G. Morrison: Data curation (equal); Writing‐original draft (supporting); Writing‐review & editing (supporting). Modupe Coker: Methodology (equal); Writing‐original draft (supporting); Writing‐review & editing (equal). Juliette Madan: Conceptualization (equal); Methodology (equal); Resources (equal); Supervision (equal); Writing‐original draft (equal); Writing‐review & editing (equal). Margaret Karagas: Conceptualization (equal); Funding acquisition (lead); Methodology (equal); Project administration (lead); Resources (equal); Supervision (equal); Writing‐original draft (equal); Writing‐review & editing (equal).
ETHICS STATEMENT
The study was approved by the Committee for the Protection of Human Subjects at Dartmouth College, and all study participants provided written informed consent.
ACKNOWLEDGMENTS
We would like to extend our deepest appreciation of the participants and staff of the New Hampshire Birth Cohort and Center for Molecular Epidemiology. This study was funded in part by the U.S. National Institutes of Environmental Health Sciences grant P01ES022832, U.S. National Institutes of General Medicine grant P20GM104416, and U.S. EPA grant RD 83544201. Katherine Antosca is funded by NIH grant 5T32HL134598. Anne Hoen, Juliette Madan, Modupe Coker, and Margaret Karagas are funded on NIH grant UH3OD023275.
APPENDIX 1.
Sampling Scheme for assessing sequencing batch effects. Table A1 shows samples and batches for set 1. Table A2 shows samples and batches for set 2.
Table A1.
Number of samples sequenced from extractions of each diaper in Set 1 by batch
| Child 1 | Child 2 | Child 3 | Child 4 | Child 5 | |
|---|---|---|---|---|---|
| Batch 1 | 1 | 1 | 1 | 1 | 1 |
| Batch 2 | 1 | 1 | 1 | 1 | 1 |
| Batch 3 | 1 | 1 | 1 | 1 | 1 |
Table A2.
Number of samples sequenced from extractions of each diaper in Set 2 by batch
| Child 1 | Child 2 Extraction 1 | Child 2 Extraction 2 | Child 3 | |
|---|---|---|---|---|
| Batch 4 | 4 | 2 | 2 | 2 |
| Batch 5 | 4 | 2 | 2 | 2 |
| Batch 6 | 4 | 2 | 2 | 2 |
| Batch 7 | 2 | 0 | 2 | 1 |
| Batch 8 | 2 | 0 | 2 | 2 |
| Batch 9 | 2 | 0 | 2 | 2 |
APPENDIX 2.
Figure A1.
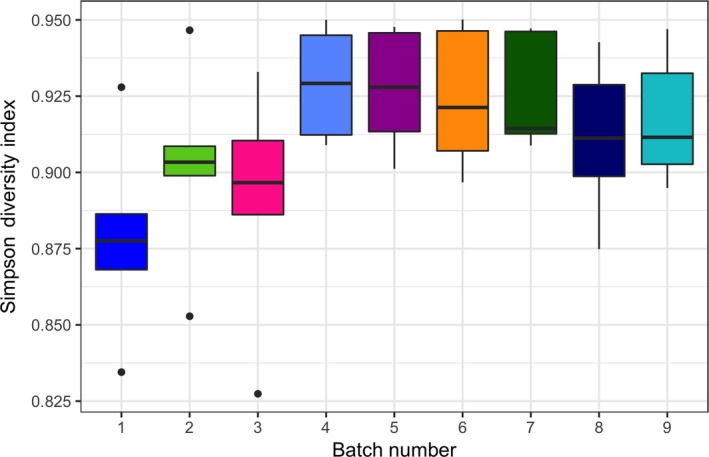
Bar plot of Simpson diversity index for the nine sequencing batches
Figure A2.
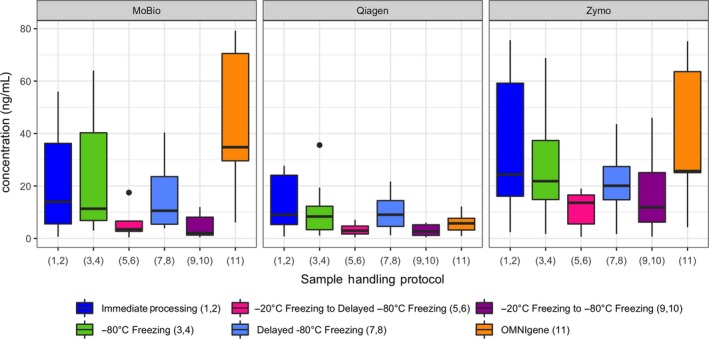
DNA yield for each of the DNA extraction kits by sample‐handling protocol
Figure A3.
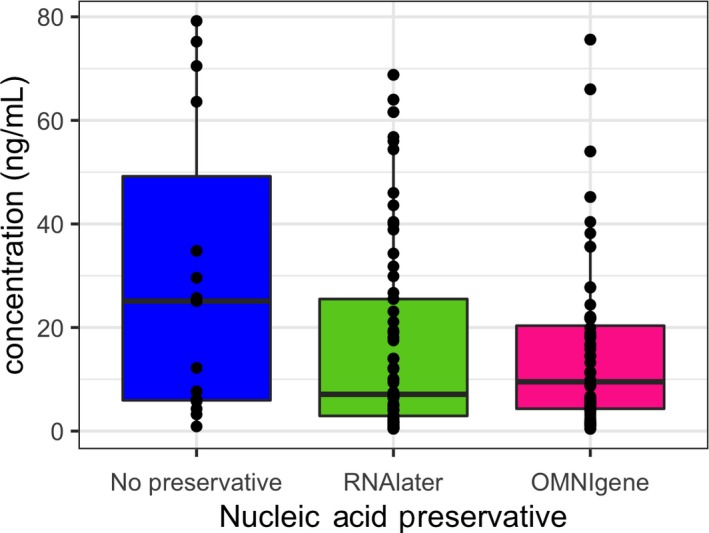
DNA yield by the preservation method
Antosca K, Hoen AG, Palys T, et al. Reliability of stool microbiome methods for DNA yields and sequencing among infants and young children. MicrobiologyOpen. 2020;9:e1018 10.1002/mbo3.1018
DATA AVAILABILITY STATEMENT
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request. Katherine Antosca had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
REFERENCES
- Ariefdjohan, M. W. , Savaiano, D. A. , & Nakatsu, C. H. (2010). Comparison of DNA extraction kits for PCR‐DGGE analysis of human intestinal microbial communities from fecal specimens. Nutrition Journal, 9, 23 10.1186/1475-2891-9-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates, D. , Mächler, M. , Bolker, B. , & Walker, S. (2014). Fitting linear mixed‐effects models using lme4. arXiv preprint arXiv:1406.5823. [Google Scholar]
- Cardona, S. , Eck, A. , Cassellas, M. , Gallart, M. , Alastrue, C. , Dore, J. , … Manichanh, C. (2012). Storage conditions of intestinal microbiota matter in metagenomic analysis. BMC Microbiology, 12, 158 10.1186/1471-2180-12-158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson, A. L. , Xia, K. , Azcarate‐Peril, M. A. , Goldman, B. D. , Ahn, M. , Styner, M. A. , … Knickmeyer, R. C. (2018). Infant gut microbiome associated with cognitive development. Biological Psychiatry, 83, 148–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll, I. M. , Ringel‐Kulka, T. , Siddle, J. P. , Klaenhammer, T. R. , & Ringel, Y. (2012). Characterization of the fecal microbiota using high‐throughput sequencing reveals a stable microbial community during storage. PLoS ONE, 7, e46953 10.1371/journal.pone.0046953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J. , Bittinger, K. , Charlson, E. S. , Hoffmann, C. , Lewis, J. , Wu, G. D. , … Li, H. (2012). Associating microbiome composition with environmental covariates using generalized UniFrac distances. Bioinformatics, 28, 2106–2113. 10.1093/bioinformatics/bts342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo, J. M. , Leong, L. E. , & Rogers, G. B. (2015). Sample storage conditions significantly influence faecal microbiome profiles. Scientific Reports, 5, 16350 10.1038/srep16350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon, P. (2003). VEGAN, a package of R functions for community ecology. Journal of Vegetation Science, 14, 927–930. 10.1111/j.1654-1103.2003.tb02228.x [DOI] [Google Scholar]
- Dominianni, C. , Wu, J. , Hayes, R. B. , & Ahn, J. (2014). Comparison of methods for fecal microbiome biospecimen collection. BMC Microbiology, 14, 103 10.1186/1471-2180-14-103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores, R. , Shi, J. , Gail, M. H. , & Ravel, J. (2012). Goedert JJ. Assessment of the human faecal microbiota: I. Measurement and reproducibility of selected enzymatic activities. European Journal of Clinical Investigation, 42, 848–854. 10.1111/j.1365-2362.2012.02660.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores, R. , Shi, J. , Yu, G. , Ma, B. , Ravel, J. , Goedert, J. J. , & Sinha, R. (2015). Collection media and delayed freezing effects on microbial composition of human stool. Microbiome, 3, 33 10.1186/s40168-015-0092-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, B. C. , Randolph, T. W. , Lim, U. , Monroe, K. R. , Cheng, I. , Wilkens, L. R. , … Lampe, J. W. (2016). Characterization of the gut microbiome in epidemiologic studies: The multiethnic cohort experience. Annals of Epidemiology, 26, 373–379. 10.1016/j.annepidem.2016.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halekoh, U. , & Højsgaard, S. (2014). A kenward‐roger approximation and parametric bootstrap methods for tests in linear mixed models–the R package pbkrtest. Journal of Statistical Software, 59, 1–30.26917999 [Google Scholar]
- Hang, J. , Desai, V. , Zavaljevski, N. , Yang, Y. , Lin, X. , Satya, R. V. , … Kuschner, R. A. (2014). 16S rRNA gene pyrosequencing of reference and clinical samples and investigation of the temperature stability of microbiome profiles. Microbiome, 2, 31 10.1186/2049-2618-2-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper, L. V. , Littman, D. R. , & Macpherson, A. J. (2012). Interactions between the microbiota and the immune system. Science, 336, 1268–1273. 10.1126/science.1223490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly, J. R. , Minuto, C. , Cryan, J. F. , Clarke, G. , & Dinan, T. G. (2017). Cross talk: The microbiota and neurodevelopmental disorders. Frontiers in Neuroscience, 11, 490 10.3389/fnins.2017.00490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy, N. A. , Walker, A. W. , Berry, S. H. , Duncan, S. H. , Farquarson, F. M. , Louis, P. , … Hold, G. L. (2014). The impact of different DNA extraction kits and laboratories upon the assessment of human gut microbiota composition by 16S rRNA gene sequencing. PLoS ONE, 9, e88982 10.1371/journal.pone.0088982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolde, R. (2015). R pheatmap: Pretty Heatmaps. R package version 1.0.8. Retrieved from http://CRAN.R-project.org/package=pheatmap [Google Scholar]
- Lauber, C. L. , Zhou, N. , Gordon, J. I. , Knight, R. , & Fierer, N. (2010). Effect of storage conditions on the assessment of bacterial community structure in soil and human‐associated samples. FEMS Microbiology Letters, 307, 80–86. 10.1111/j.1574-6968.2010.01965.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maukonen, J. , Simoes, C. , & Saarela, M. (2012). The currently used commercial DNA‐extraction methods give different results of clostridial and actinobacterial populations derived from human fecal samples. FEMS Microbiology Ecology, 79, 697–708. 10.1111/j.1574-6941.2011.01257.x [DOI] [PubMed] [Google Scholar]
- McMurdie, P. J. , & Holmes, S. (2013). phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE, 8, e61217 10.1371/journal.pone.0061217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McOrist, A. L. , Jackson, M. , & Bird, A. R. (2002). A comparison of five methods for extraction of bacterial DNA from human faecal samples. Journal of Microbiol Methods, 50, 131–139. 10.1016/S0167-7012(02)00018-0 [DOI] [PubMed] [Google Scholar]
- Nechvatal, J. M. , Ram, J. L. , Basson, M. D. , Namprachan, P. , Niec, S. R. , Badsha, K. Z. , … Kato, I. (2008). Fecal collection, ambient preservation, and DNA extraction for PCR amplification of bacterial and human markers from human feces. Journal of Microbiol Methods, 72, 124–132. 10.1016/j.mimet.2007.11.007 [DOI] [PubMed] [Google Scholar]
- Newton, R. J. , McLellan, S. L. , Dila, D. K. , Vineis, J. H. , Morrison, H. G. , Eren, A. M. , & Sogin, M. L. (2015). Sewage reflects the microbiomes of human populations. mBio, 6, e02574 10.1128/mBio.02574-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott, S. J. , Musfeldt, M. , Timmis, K. N. , Hampe, J. , Wenderoth, D. F. , & Schreiber, S. (2004). In vitro alterations of intestinal bacterial microbiota in fecal samples during storage. Diagnostic Microbiology and Infectious Disease, 50, 237–245. 10.1016/j.diagmicrobio.2004.08.012 [DOI] [PubMed] [Google Scholar]
- Rintala, A. , Pietila, S. , Munukka, E. , Eerola, E. , Pursiheimo, J. P. , Laiho, A. , … Huovinen, P. (2017). Gut microbiota analysis results are highly dependent on the 16S rRNA gene target region, whereas the impact of DNA extraction is minor. Journal of Biomolecular Techniques, 28, 19–30. 10.7171/jbt.17-2801-003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roesch, L. F. , Casella, G. , Simell, O. , Krischer, J. , Wasserfall, C. H. , Schatz, D. , … Triplett, E. W. (2009). Influence of fecal sample storage on bacterial community diversity. Open Microbiology Journal, 3, 40–46. 10.2174/1874285800903010040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roesch, L. F. , Lorca, G. L. , Casella, G. , Giongo, A. , Naranjo, A. , Pionzio, A. M. , … Triplett, E. W. (2009). Culture‐independent identification of gut bacteria correlated with the onset of diabetes in a rat model. ISME Journal, 3, 536–548. 10.1038/ismej.2009.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanahan, F. (2010). 99th Dahlem conference on infection, inflammation and chronic inflammatory disorders: Host‐microbe interactions in the gut: Target for drug therapy, opportunity for drug discovery. Clinical and Experimental Immunology, 160, 92–97. 10.1111/j.1365-2249.2010.04135.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon, G. , Sampson, T. R. , Geschwind, D. H. , & Mazmanian, S. K. (2016). The central nervous system and the gut microbiome. Cell, 167, 915–932. 10.1016/j.cell.2016.10.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherwin, E. , Rea, K. , Dinan, T. G. , & Cryan, J. F. (2016). A gut (microbiome) feeling about the brain. Current Opinion in Gastroenterology, 32, 96–102. 10.1097/MOG.0000000000000244 [DOI] [PubMed] [Google Scholar]
- Sinha, R. , Abnet, C. C. , White, O. , Knight, R. , & Huttenhower, C. (2015). The microbiome quality control project: Baseline study design and future directions. Genome Biology, 16, 276 10.1186/s13059-015-0841-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjogren, Y. M. , Tomicic, S. , Lundberg, A. , Bottcher, M. F. , Bjorksten, B. , Sverremark‐Ekstrom, E. , & Jenmalm, M. C. (2009). Influence of early gut microbiota on the maturation of childhood mucosal and systemic immune responses. Clinical and Experimental Allergy, 39, 1842–1851. 10.1111/j.1365-2222.2009.03326.x [DOI] [PubMed] [Google Scholar]
- Smith, B. , Li, N. , Andersen, A. S. , Slotved, H. C. , & Krogfelt, K. A. (2011). Optimising bacterial DNA extraction from faecal samples: Comparison of three methods. Open Microbiology Journal, 5, 14–17. 10.2174/1874285801105010014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedjo, D. I. , Jonkers, D. M. , Savelkoul, P. H. , Masclee, A. A. , van Best, N. , Pierik, M. J. , & Penders, J. (2015). The effect of sampling and storage on the fecal microbiota composition in healthy and diseased subjects. PLoS ONE, 10, e0126685 10.1371/journal.pone.0126685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tognini, P. (2017). Gut microbiota: A potential regulator of neurodevelopment. Frontiers in Cellular Neuroscience, 11, 25 10.3389/fncel.2017.00025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turta, O. , & Rautava, S. (2016). Antibiotics, obesity and the link to microbes ‐ what are we doing to our children? BMC Medicine, 14, 57 10.1186/s12916-016-0605-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogtmann, E. , Chen, J. , Amir, A. , Shi, J. , Abnet, C. C. , Nelson, H. , … Sinha, R. (2017). Comparison of collection methods for fecal samples in microbiome studies. American Journal of Epidemiology, 185, 115–123. 10.1093/aje/kww177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker, A. W. , Martin, J. C. , Scott, P. , Parkhill, J. , Flint, H. J. , & Scott, K. P. (2015). 16S rRNA gene‐based profiling of the human infant gut microbiota is strongly influenced by sample processing and PCR primer choice. Microbiome, 3, 26 10.1186/s40168-015-0087-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesolowska‐Andersen, A. , Bahl, M. I. , Carvalho, V. , Kristiansen, K. , Sicheritz‐Ponten, T. , Gupta, R. , & Licht, T. R. (2014). Choice of bacterial DNA extraction method from fecal material influences community structure as evaluated by metagenomic analysis. Microbiome, 2, 19 10.1186/2049-2618-2-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, G. D. , Lewis, J. D. , Hoffmann, C. , Chen, Y. Y. , Knight, R. , Bittinger, K. , … Bushman, F. D. (2010). Sampling and pyrosequencing methods for characterizing bacterial communities in the human gut using 16S sequence tags. BMC Microbiology, 10, 206 10.1186/1471-2180-10-206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan, S. , Cohen, D. B. , Ravel, J. , Abdo, Z. , & Forney, L. J. (2012). Evaluation of methods for the extraction and purification of DNA from the human microbiome. PLoS ONE, 7, e33865 10.1371/journal.pone.0033865 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request. Katherine Antosca had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.