

Abstract
Pyranone natural products have attracted great attention in recent years from chemists and biologists due to their fascinating stereoisomeric structural features and impressive bioactivities. A large number of stereoselective total syntheses of these compounds have been described in the literature. The natural pyranones with long side chains have recently received significant importance in the synthetic field. In the present article, we aim to review the modern progress of the stereoselective total syntheses of these natural pyranones containing long-chain substituents.
Keywords: pyranone, side chain, natural product, total synthesis, stereoselectivity
1. Introduction
Pyranones are an important class of natural products [1]. Several natural pyranones have been found to contain long side chains. The side chain is generally present at the C-6 portion of the pyranone ring. These naturally occurring pyranones with long-chain substituents have recently drawn considerable attention from the scientific community due to their impressive structural characteristics as well as interesting biological activities. Structurally, they contain a lactone ring, which is usually within the framework of a 5,6-dihydropyran-2-one (α,β-unsaturated δ-lactone). The H-6 in these molecules may be with the relative stereoposition of α or β. For example, in dodoneine [2] and rugulactone [3], the stereoposition of H-6 is the opposite (Figure 1).
Figure 1.

Structures of natural pyranone having long side chains.
The side chains of the pyranones may be of various lengths; they may even contain more than 20 carbons. Thus, the side chain of passifloricin A consists of 21 carbons [4]. These side chains possess several stereogenic centers with various functionalities. Generally hydroxyl, acetoxy, and carbonyl groups are located at different positions of the side chains. However, natural pyranones (such as rugulactone) having no chiral center in the side chains are also observed. Some compounds are found to possess a double bond [(E) or (Z) stereostructure] in their side chains. Rugulactone contains an (E)-double bond while spicigerolide has a (Z)-double bond in their respective side chains [3,5].
The bioactivity of this class of compounds is promising. They are found to exhibit manifold biological properties including anticancer [3,6], antiviral [7], antifungal [8], antituberculosis [9], and antimicrobial [10] activities. The α,β-unsaturated lactone moiety plays an important role in the bioactivity as it can act as a Micheal acceptor in the presence of protein functional groups [11].
The interesting structural features and important bioactivities of these molecules have inspired synthetic chemists to explore their total syntheses [12,13,14] and biologists to discover novel therapeutics [15,16,17]. In several cases, the pyranones obtained from natural sources are insufficient to conduct further experiments. The total synthesis can generate the compounds in larger amounts required to explore their new medicinal values. Total syntheses are also useful to verify the established structures of the molecules.
Various modern synthetic approaches have now been applied to construct the natural pyranone molecules with proper stereostructures. Efficient diastereoselective and enantioselective synthetic protocols have been employed to introduce the required chirality in their side chains; ring-closing metathesis (RCM) and cross-metathesis (CM) reactions [18] have frequently been applied to construct the lactone rings and side chains, respectively. Various improved approaches have currently been utilized by different synthetic chemists to accomplish successfully stereoselective syntheses of natural pyranones. In this review, we have described the recent progress of the total syntheses of these compounds having long side chains. We have focused our discussion on the total syntheses of the molecules, which have repeatedly been constructed by applying various modern synthetic protocols.
2. Stereoselective Total Syntheses
In recent years, different research groups utilized various efficient procedures for the stereoselective syntheses of naturally occurring pyranones having long-chain substituents. The total syntheses of the following molecules will highlight the current advances in the field.
2.1. Dodoneine
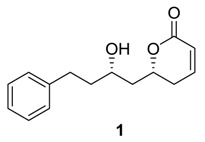
Dodoneine (1), a naturally occurring 5,6-dihydropyran-2-one, was isolated from a medicinal parasitic plant, Tapinunthus dodoneifolius that grows on the sheanut tree in West Africa [2]. The structure of the compound was derived from its spectroscopic data and X-ray crystallographic analysis of its camphorsulfonate derivative. Dodoneine (1) was found to possess relaxation effects on preconstricted rate aortic rings. The compound was also evaluated as a hypotensive agent and as an inhibitor of human carbonic anhydrases [19,20].
Dodoneine (1) has recently been synthesized by several research groups [21,22,23,24,25,26,27,28,29]. The synthetic methods generally involve the asymmetric allylation of an aldehyde for introducing the stereogenic centers and the formation of the pyranone ring by ring-closing metathesis (RCM) or intramolecular transesterification. The first total synthesis of dodoneine (1) was reported independently by Falomir et al. [21] and Srihari et al. [22]. Falomir et al. used commercially available dihydro p-coumaric acid (2) as the starting material (Scheme 1). The acid was converted to silylated dihydro p-coumaraldehyde (3), which underwent asymmetric Keck allylation [30] to generate the homoallylic alcohol 4 (ee ca. 95%). The silylation of 4 and ozonolysis of the product afforded aldehyde 5. Asymmetric allylboration of 5 using (+)-Ipc2BCl/allylmagnesiumbromide (Ipc = isopinocamphenyl) yielded the homoallylic alcohol 6, which was obtained as a single diastereomer after purification. The subsequent acrylation of 6 with acryloyl chloride followed by ring-closing metathesis of the acrylate (7) applying a Grubbs first-generation catalyst provided the pyranone 8. Finally, the cleavage of the two silyl groups of 8 using aqueous HF in MeOH furnished dodoneine (1).
Scheme 1.

Reagents and conditions: (a) allyltri-n-butyltin, (R)-BINOL, Ti(iPrO)4, (approximately 60% overall); (b) TBSOTf, CH2Cl2, 2,6-lutidine, r.t., 2 h, 85%; (c) O3, −78 °C to r.t., PPh3, 2 h; (d) (+)-Ipc2BCl, Et2O,allylMgBr, −90 °C, 2 h, (approximately 60% overall); (e) CH2=CHCOCl, CH2Cl2, iPr2Net, −78 °C, 62%; (f) 10% Grubbs first-generation catalyst, CH2Cl2, Δ, 4 h, 84%; (g) aq HF, MeCN, r.t., 16 h, 89%.
Srihari et al. applied the aldehyde 3 prepared from 4-hydrobenzaldehyde (9) (Scheme 2) [22]. The aldehyde 3 was reacted with (S)-1-(4-benzyl-2-thioxothiazolidin-3-yl) ethanone in the presence of TiCl4 following the Crimmins protocol [31]. The major syn product 10 was converted to TBS ether 11, which upon treatment with DIBAL-H yielded the aldehyde 12. The aldehyde 12 underwent the Crimmins aldol reaction with (S)-1-(4-benzyl-2-thioxothiazolidin-3-yl) ethanone and TiCl4 to produce the 1,3-syn compound 13 as the major product. The hydroxyl group of 13 was protected with MOMCl, and product was treated with DIBAL-H to form the aldehyde 14. The latter was treated with bis-(2,2,2-trifluoromethyl) (methoxycarbonylmethyl) phosphonate following the Horner–Wadsworth–Emmons olefination reaction [32] to produce the cis-olefinic ester 15. At the final stage, it was observed that 3 mol% HCl solution afforded the best result for the simultaneous deprotection and cyclization of the ester 15 to generate dodoneine 1.
Scheme 2.

Reagents and conditions: (a) TiCl4, DIPEA, CH2Cl2, −78 °C, 85%; (b) TiCl4, DIPEA, CH2Cl2, −78 °C, 81%; (c) (CF3CH2O)2P(O)CH2COOCH3, NaH, THF, −78 °C, 82%; (d) PTSA or PPTS or 3 mol% HCl.
A short and efficient synthesis of dodoneine (1) was reported by Cossy et al. [23]. They prepared the aldehyde 3 from the ester 16 (Scheme 3). This aldehyde (3) was treated with allyl titanium complex (S,S)-Ti-I to produce the homoallylic alcohol 4 (ee 96%). A cross-metathesis reaction [33] of 4 with ethyl acrylate in the presence of a Grubbs–Hoveyda second-generation catalyst afforded the unsaturated ester 17. On treatment with benzaldehyde using tert-BuOK, the ester 17 furnished the protected 1,3-diol 18 (syn:anti; 98:2). Reduction of the compound 18 with DIBAL-H afforded the aldehyde 19, which was subjected to Horner–Wadsworth–Emmons reaction [34] using bis (2,2,2-trifluoromethyl) (methoxycarbonylmethyl) phosphonate to produce the unsaturated ester 20 (Z:E = 90:10). Finally, the treatment of 20 with 80% aq. AcOH afforded dodoneine (1).
Scheme 3.

Reagents and conditions: (a) (S,S)-Ti-I, Et2O, −78 °C, 3 h, 97%; (b) GH-II (5 mol%), CH2Cl2, r.t., 2 d, 80%; (c) PhCHO, t-BuOK, THF, 0 °C, 2 h, 68%; (d) DIBAL-H, toluene, −78 °C, 94%; (e) (CF3CH2O)2P(O)CH2COOEt, KHMDS, 18-C-6, THF, −78 °C, 2 h, 70%; (f) aq AcOH, 60 °C, 24 h, 70%.
Das et al. utilized 4-hydroxy benzaldehyde as the starting material and applied Sharpless asymmetric epoxidation, 1,3-syn diastereoselective reduction, and Grubbs ring-closing metathesis in their synthetic sequence for the stereoselective construction of dodoneine (1) (Scheme 4) [24].
Scheme 4.

Reagents and conditions: (a) Ti(iPrO)4, (1.0 equiv), (+)-DIPT (1.1 equiv), TBHP (2.5 equiv), CH2Cl2, −20 °C, 12 h, 92%; (b) Red-Al (3.0 equiv), THF, 0 °C, 0.5 h, 82%; (c) CH2=CHCH2MgBr, Et2O, 0 °C, 1 h, 74%; (d) DMP, NaHCO3, CH2Cl2, 0 °C to r.t., 1 h, 88%; (e) LiAlH4–LiI, Et2O, −100 °C, 30 min, 94%; (f) acryloyl chloride, Et3N, 0 °C to rt, 30 min, 96%; (g). Grubbs first-generation catalyst, CH2Cl2, 50 °C, 24 h, 85%; (h) TiCl4, DCM, 0 °C, 89%.
Sharpless asymmetric epoxidation [35] of 22 was carried out with (+)-DIPT and the diastereoselective reduction of the ketone 27 with LiAlH4-LiI at −100 °C (syn:anti = 94:6). The intramolecular metathesis reaction of 29 was conducted using a Grubbs catalyst of the first generation.
In another synthesis, Sharpless asymmetric dihydroxylation [36] and the regioselective nucleophile opening of cyclic sulfate formed from the resulting diol were used to generate the required chiral center (Scheme 5).
Scheme 5.

Reagents and conditions: (a) AD-mix-α, MeSO2NH2, t-BuOH-H2O (1:1), 0 °C, overnight, 85%; (b) SOCl2, Et3N, CH2Cl2, 0 °C, 30 min, 94%; (c) RuCl3, NaIO4, CCl4-MeCN-H2O: 2:2:3, 0 °C, 30 min, 92%; (d) NaBH4, DMF, 15 min, then THF, cat. Conc. H2SO4, cat. H2O, 20 min, 90%; (e) TBSCl, imidazole, CH2Cl2, 0–25 °C, overnight, 95%; (f) DIBAL-H, CH2Cl2, −78 °C, 10 min, 88%.
Sabitha et al. completed the synthesis of dodoneine (1) starting from the known chiral alcohol 35 (Scheme 6) [26]. The latter was oxidized with IBX to the corresponding aldehyde, which was treated with trimethylsulfoxiumiodide using NaH in DMSO-THF to afford a racemic epoxide. Jacobson’s hydrolytic kinetic resolution (HKR) of this epoxide by applying (S,S)-Salen-Co-OAc catalyst yielded the chiral epoxide 36 (ee 95%) [37]. The epoxide 36 was converted into the homoallylic alcohol 37 by treatment with vinyl magnesium bromide and CuI. The compound is structurally related to 6. It was subsequently transformed into dodoneine (1) following a similar reaction sequence as shown earlier in Scheme 1.
Scheme 6.

Reagents and conditions: (a) IBX/DMSO, CH2Cl2, 0 °C to r.t., 5 h; (b) trimethylsulfonium iodide, DMSO, NaH, THF, 0 °C to r.t., 6 h, 60%; (c) (S,S)-Jacobsen catalyst, AcOH, H2O, 12 h, 43%; (d) vinylmagnesium bromide, CuI, THF, 0 °C, 30 min, 85%.
Rauniyar and Hall prepared the chiral alcohol 4 (ee 97%) from the aldehyde 3 by using p-F-Vivol.SnCl4 catalyzed allylboration with allylboron pinacolate (Scheme 7) [27]. In a similar manner, the chiral diol 6 (dr 99:1) was produced from the aldehyde 5. Compound 6 was subsequently converted to dodoneine (1) following a sequence similar to that of Macro et al. [21] (Scheme 1).
Scheme 7.

Reagents and conditions: (a) (R,R)-p-F-vivol 4b (5 mol%), SnCl4 (3.8 mol%), Na2CO3, 4 Å MS, toluene [1.3 M], −78 °C, 4 h, 99%; (b) (R,R)-p-F-vivol 4b (5 mol%), SnCl4 (3.8 mol%), Na2CO3, 4 Å MS, toluene [1.0 M], −78 °C, 4 h, 96%.
In an alternative approach [28], the total synthesis of dodoneine (1) was achieved by applying Keck’s asymmetric allylation, iodine-induced electrophilic cyclization, and ring-closing metathesis (Scheme 8). Compound 40 underwent diastereoselective iodolactoxization with I2 to form the cyclic iodocarbonate 41 as a single diastereoisomer. This iodocarbonate (41) when kept in basic MeOH solution afforded syn-epoxy alcohol 42. The free hydroxyl group of 42 was protected to form TBS-ether 43, which was treated with allylmagnesiumbromide to furnish a diastereoisomeric mixture (syn:anti = 43:57). The desired syn-epoxy alcohol 44 was purified by column chromatography and was converted to dodoneine (1).
Scheme 8.

Reagents and conditions: (a) (R)-BINOL, 4 Å MS, Ti(iPrO)4, allyltributylstannane, CH2Cl2, −78 to −20 °C, 72 h, 79%; (b) (Boc)2O, DMAP, MeCN, r.t., 5 h, 94%; (c) I2, MeCN, −20 °C, 12–15 h, 82%; (d) K2CO3, MeOH, 0 °C to r.t., 2 h, 86%; (e) tBuMe2SiCl, 1H-imidazole, CH2Cl2, r.t., 4.5 h, 88%; (f) CH2=CHMgBr, Et2O, 0 °C, 1.5 h, 76%.
Allais and Ducrot prepared the chiral homoallylic alcohol 4 [29] following the method developed by Rauniyar and Hall (Scheme 9) [27]. This alcohol 4 was treated with OsO4 and NaIO4 to form the corresponding β-hydroxyaldehyde, which was reacted with trimethylallylsilane and SnCl4 to produce the diol 45 (dr > 97:3) favoring the syn-product. The diol 45 was converted to a ketal 46. The latter underwent an oxidative cleavage with OsO4, NaIO4 and the resulting aldehyde was subjected to Horner–Wardsworth–Emmons olefination to furnish the unsaturated ester 47, (Z:E = 90:10). At the final step, the treatment of 47 with 80% aq. AcOH afforded dodoneine (1).
Scheme 9.

Reagents and conditions: (a) OsO4, 2,6-lutidine, NaIO4, dioxane-H2O (3:1), r.t.; (b) allylSiMe3, SnCl4, CH2Cl2, −78 °C, 12 h, 80%; (c) 2,3-DMP, PPTS, CH2Cl2, r.t., 2 h, 96%; (d) OsO4, 2,6-lutidine, NaIO4, dioxane-H2O (3:1); (e) 2-[bis(2,2,2-trifluoroethoxy)phosphoryl]acetate, KHMDS, 18-crown-6, THF, −78 °C, 4 h, 51%; (f) 80% aq AcOH, 60 °C, 1 d, 68%.
2.2. Rugulactone
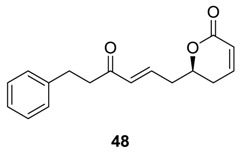
Rugulactone (48) was isolated from Cryptocarya rugulosa [3]. It contains only one chiral center at C-6 with R-stereoconfiguration and an α, β-unsaturated γ-lactone along with an α, β-unsaturated ketone. The compound was found to inhibit constitutive NF-kB activity in human lymphoma cell lines. Several syntheses of rugulactone (48) have recently been reported [38,39,40,41,42,43,44,45,46,47,48]. In these syntheses, the chirality has been introduced by applying different methodologies such as Jacobsen’s hydrolytic kinetic resolution of epoxides, Keck/Maruoka asymmetric allylation, chemoenzymatic process, the chiral pool approach, and allylation with chiral boronic esters.
The first stereoselective total synthesis of rugulactone (48) was reported by Venkateshwarlu et al. [38] as well as by Yadav et al. [39]. The first group used 1,3-propane diol (49) as the starting material (Scheme 10). It was converted to monobenzylether (50), which was oxidized with IBX and the resulting aldehyde underwent Keck allylation to form the homoallylic alcohol 51 (ee 97.5%). Protection of the hydroxyl group and removal of the benzyl group compound 51 yielded the alcohol 52. The latter was oxidized with IBX, and the corresponding aldehyde was converted to the unsaturated ester 53 (Z:E = 95:5) by Still–Gennari modification of the Horner–Emmons olefination reaction. Treatment with 3% HCl in MeOH 53 yielded the pyranone 54.
Scheme 10.

Reagents and conditions: (a) BnBr, NaH, TBAI, THF, 0 °C to r.t., 2 h, 85%; (b) (i) IBX, dry DMSO, CH2Cl2, 5 h, 88%; (ii) (R)-BINOL, 4 Å MS, Ti(iPrO)4, allyltributylstannane, CH2Cl2, –78 °C to –20 °C, 80%; (c) TBDPSCl, imidazole, CH2Cl2, 4 h, 95%; (d) Li in naphthalene, −20 °C, 3 h, 81%; e) (i) IBX, dry DMSO, CH2Cl2, 5 h, 85%; (ii) MeO2CCH2P(O)(OCH2CF3)2, NaH, THF, −78 °C, 2 h, 76%; (f) 3% HCl in MeOH 30 min, 78%; (g) Grubbs second-generation catalyst (5 mol %), CH2Cl2, 40 °C, 12 h, 74%.
The ether part of rugulactone, fragment 56 was prepared from phenyl propanol (55) by treatment with vinyl magnesium bromide followed by oxidation of the generated alcohol with IBX. Finally, the cross-metathesis reaction of 54 and 56 using a second-generation Grubbs catalyst produced the natural rugulactone (48).
Venkateshwarlu et al. in a later publication [41] showed the introduction of chirality through D-proline catalyzed α-aminoxylation of the aldehyde 57 (Scheme 11).
Scheme 11.

Reagents and conditions: (a) i) PhNO, D-proline (25 mol %), CH3CN, −20 °C, 24 h; then, MeOH, NaBH4; ii) CuSO4 (30 mol %), MeOH, 0 °C, 10 h, 85% (over two steps); (b) TsCl, Bu2SnO, Et3N, 0 °C to rt, 4 h, 81%; (c) K2CO3, MeOH, rt, 1 h, 93%; (d) Vinyl magnesium bromide, THF, CuI, −20 °C, 1 h, 88%.
Yadav et al. [39] initiated their synthesis from the chiral epoxide 61 (Scheme 12), which was prepared from the known corresponding racemic compound by Jacobsen’s hydrolytic kinetic resolution using (R,R)-(Salen) CoIII (OAc) catalyst. Epoxide 61 was reacted with vinyl magnesium bromide and CuI to form the homoallyl alcohol 62 (ee 87%). The esterification of 62 with acryloyl chloride, removal of the hydroxyl protection, and subsequently oxidation with DMP yielded the aldehyde 63. This aldehyde 63 was subjected to Horner–Wadsworth–Emmons homologation using dimethyl (2-oxo-4-phenylbutyl) phosphonate to furnish the unsaturated ketone 64. Finally, by treatment of this ketone (64) with Grubb’s first-generation catalyst, rugulactone (48) was formed.
Scheme 12.

Reagents and conditions: (a) Vinylmagnesium bromide, CuI, THF, 0 °C, 2 h, 91%; (b) NaHMDS, dimethyl (2-oxo-4-phenylbutyl) phosphonate, 0 °C, 12 h, 87%.
In a chemoenzymatic synthetic approach, both (R)- and (S)-rugulactone were prepared by applying the Candida rugosa lipase to hydrolyze the butyrate ester of the protected 3-hydroxy homoallylic alcohol 65 (Scheme 13) [42]. The key intermediates (R)-66 (ee > 99%) and (S)-67 (ee > 98%) were obtained with high enantiomeric purity. The ester 66 was hydrolyzed and deprotected to form (R)-68. Both the alcohols (R)-68 and (S)-67 were converted to (R)-48 and (S)-48 respectively following the earlier established method (Scheme 10). (R)-48 is the naturally occurring rugulactone.
Scheme 13.

Reagents and conditions: (a) Tris–HCl buffer, 0.05 M, pH 7.5; Candida rugosa lipase, 48 h.
In another chemoenzymatic synthesis of rugulactone (48), chirality was induced by a stereoselective enzymatic reduction of a ketoester employing NADPH-dependent ketoreductase [40].
A chiral-pool method [43] was developed by Allais et al. for the asymmetric synthesis of rugulactone (48) (Scheme 14). The starting material was commercially available (2S)-glycidyl tosylate (69), which was converted to the olefin 70. The olefin 70 was subjected to a cross-metathesis reaction with 5-phenyl-pent-1-en-3-one using Grubb’s II catalyst to furnish the α,β-unsaturated ketone 71. The thioacetal group of 71 was removed, and the generated aldehyde 72 underwent a Still–Gennari olefination with methyl P,P-bis (2,2,2-trifluoromethyl) phosphonium acetate to form the unsaturated ester 73. The latter on treatment with AcOH yielded natural rugulactone (48).
Scheme 14.

Reagents and conditions: (a) Grubbs second-generation catalyst (5 mol %), CH2Cl2, 40 °C, 12 h, 72%; (b) CaCO3, MeI, MeCN-H2O (9:1), 45 °C to r.t., 2.5 h; (c) MeO2CCH2P(O)(OCH2CF3)2, KHMDS, 18-crown-6, THF, −78 °C, 52%; (d) 80% AcOH, 60 °C, 24 h, 86%.
Das et al. achieved the total synthesis of rugulactone (48) using 3-phenyl propanol (74) as the starting material and applying Maruoka allylation and ring-closing metathesis as the key steps (Scheme 15) [44].
Scheme 15.

Reagents and conditions: (a) i) Oxalyl chloride, CH2Cl2, DMSO, Et3N, −78 °C, 2.5 h, 82%; ii) BuLi, dry THF, THP-protected homopropargyl alcohol, −78 °C, 3 h; 87%; (b) LiAlH4, dry THF, 0 °C to reflux, 3 h; 78%; (c) i) IBX, DMSO, CH2Cl2, 0 °C to r.t., 2 h; 76%; ii) (S,S)-I, allyl(tributyl)tin, −15 °C to 0 °C, 16 h, 74%; (d) Acryloyl chloride, Et3N, DMAP, CH2Cl2, 0 °C to r.t., 4 h, 78%.
Compound 75 was prepared from 74 by oxidation of the latter under Swern conditions and treatment of the corresponding aldehyde with THP protected homopropargyl alcohol. The alkenol 76 was obtained by reduction of 75 with LiAlH4 and subsequently, it was converted to 77. This alcohol (77) was oxidized with IBX, and the resulting aldehyde was subjected to Maruoka allylation [49] to form the homoallylic alcohol 78 (ee 97%). The latter was esterified with acryloyl chloride and the ester 79 was then converted to rugulactone (48).
Das et al. also synthesized rugulactone (48) through an alternative route (Scheme 16) [47]. They prepared the chiral aldehyde 81 from propane 1,3-diol applying Maruoka allylation and ring-closing metathesis. This aldehyde 81 underwent Wittig olefination with the phosphorane, PhCH2CH2COCH=PPh3 to yield rugulactone (48).
Scheme 16.

Reagents and conditions: (a) prop-2-enoyl chloride, CH2Cl2, Et3N, DMAP, 0 °C to r.t., 30 min, 96%; (b) PhCH2CH2COCH=PPh3, C6H6, reflux, 14 h, 72%.
The intermediate 81 was prepared by Barua et al. [45] from the chiral epoxide 82 (Scheme 17) and by Pietruszka et al. [46] from the allylic boronic ester 85 (Scheme 18).
Scheme 17.

Reagents and conditions: (a) Ethyl propionate, BF3.Et2O, n-BuLi, –78 °C, THF, 93%; (b) H2, Lindlar’s catalyst, quinoline, EtOAc, 91%.
Scheme 18.

Reagents and conditions: (a) CH2Cl2, 0 °C to r.t., 98.4% ee, 92%; (b) PhI(OAc)2, TEMPO, CH2Cl2, r.t., 4 h, 92%.
In a synthetic approach, Reddy and Singh [48] applied Sharpless asymmetric epoxidation of an allyl alcohol to generate a chiral alcohol, which was applied as a key intermediate. The total synthesis of racemic rugulactone has also recently been reported [50].
2.3. Synargentolide A
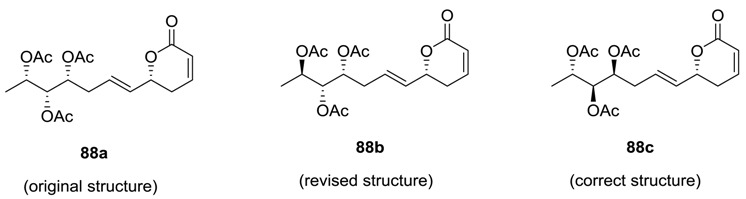
Synargentolide A was isolated from the South African plant, Syncolostemon argenteus [51]. The compound is a member of 6-substituted α, β-unsaturated δ-lactones, which are well known for their cytotoxic and antitumor properties [3,6]. The structure of the compound was originally proposed as 88a on the basis of spectroscopic studies, Mosher ester analysis, and acetonide formation [51]. Macro et al. synthesized the structure 88a and observed that the synthetic compound was not identical to natural product [52]. Sabitha et al. also synthesized 88a and its one stereoisomer 88b (Scheme 19) [53]. Their synthesis was initiated with the known (R)-benzyl glycidyl ether 89, which was converted to allyl alcohol 90. The latter was subjected to Sharpless asymmetric epoxidation to form the single isomer 91. This epoxide (91) was transformed to the alcohol 93, which on epoxidation generated the epoxide 94. Later, this epoxide (94) was converted to acetonides 95a and 95b.
Scheme 19.

Reagents and conditions: (a) l-(+) DIPT, Ti(iPrO)4, cumenehydroperoxide, CH2Cl2, −24 °C, 4 h, 98%; (b) TPP, I2, imidazole, Et2O/CH3CN (3:1), 0 °C to rt, 20 min, 92%; (c) Zn dust, EtOH, reflux, 2 h, 92%; (d) i) m-CPBA, NaHCO3, CH2Cl2, −10 °C, 10 h, 92%, dr (1:1); ii) BnBr, NaH, THF, 0 °C to rt, 3 h, 95%; (e) Grubbs second-generation catalyst, refluxing CH2Cl2, 2 h, 65%.
Next, these acetonides 95a and 95b furnished separately the triacetates 96a and 96b respectively by deprotection, acetylation, and partial reduction. Finally, the cross-metathesis reaction between 95a/95b and vinyl lactone (97) using Grubbs second-generation catalyst produced 88a/88b. After inspection of the NMR spectra of 88a and 88b, the authors revised the structure of synargentolide A as 88b.
Several other syntheses of 88b have recently been reported [54,55,56,57,58,59]. Das et al. developed an efficient synthesis of both 88a and 88b starting from D-tartaric acid (Scheme 20) [56]. The compound was converted to the alcohol 98, which was subjected to Swern oxidation, and the corresponding aldehyde was treated with methyl magnesium bromide to produce the second alcohol 99. The deprotection and acetylation of this compound (99) yields 96a and 96b, which were then converted to 88a and 88b, respectively (following the method shown earlier in Scheme 19).
Scheme 20.

Reagents and conditions: (a) i) (COCl)2, DMSO, Et3N, CH2Cl2, 1 h, 89%; ii) MeMgBr, Et2O, −50 °C, 2 h, 62%; (b) i) MeOH, 2N HCl, r.t., 1 h, ii) Ac2O, Et3N, DMAP, CH2Cl2, 0 °C to r.t., 1 h, 92% (2 steps); (c) Grubbs second-generation catalyst, refluxing CH2Cl2, 2 h, 67% for 88a and 66% for 88b.
It is interesting to mention here that the calculation of density functional theory (DFT) NMR parameters has recently suggested that both the structures 88a and 88b are incorrect and 88c is the correct structure of the natural synargentolide A [60].
2.4. Synargentolide B
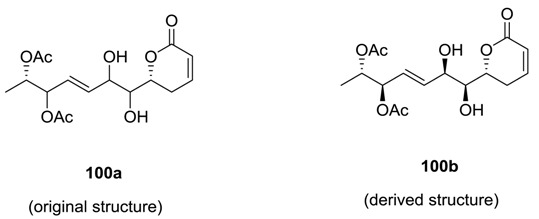
Synargentolide B was isolated from the South Africa species Syncolostemon argenteus [51]. Its structure was tentatively proposed as 100a. Prasad and Gutala [61] carried out the total synthesis of possible diastereoisomers of the compound (Scheme 21) and derived the structure of the natural product as 100b, which was earlier reported for a constituent of Hyptis oblongifolia [62].
Scheme 21.

Reagents and conditions: (a) allylmagnesiumbromide, THF, 0 °C, 1 h, 40%; (b) NaBH4/CeCl3, MeOH, −78 °C, 1 h, 94%.
Compound 100b was synthesized [61] from the aldehyde 101, which was converted to the major allyl alcohol 102. This allyl alcohol 102 was elaborated to the ester 103. On the reduction of 103 with NaBH4/CeCl3, two diastereoisomeric compounds 104 and 105 were obtained. Compound 104 was transformed to 105 by Mitsunobu inversion. Subsequently, 105 was converted to synargentolide B (100b) by reaction sequences involving deprotection, acetylation, and cross-metathesis.
In a tandem ring-closing/cross-metathesis approach for the synthesis of synargentolide B (100b), d-(-)-diethyl tartarate and d-ribose were used as starting materials (Scheme 22) [63].
Scheme 22.

Reagents and conditions: (a) Grubbs second-generation catalyst, benzene, r.t. to reflux, 1.5 h, 83%; (b) TFA, CH2Cl2, 12 h, 87%.
Akkewar et al. obtained the diacetyl compound 107 from l-ascorbic acid, and they prepared the other part of 100b from d-ribose employing the Bestmann–Ohira reaction, zinc allylation, and ring-closing metathesis (Scheme 23) [64].
Scheme 23.

Reagents and conditions: (a) Bestmaan–Ohira reaction, reflux, 8 h, 65%; (b) i) Grubbs second-generation catalyst, CH2Cl2, reflux, 6 h, 67%; ii) PTSA, MeOH, reflux, 12 h, 78%.
Liu et al. prepared the intermediates 107 and 110 from L-ethyl lactate and D-mannitol respectively [65]. They also synthesized the enantiomer of natural synargentolide B [66]. A diastereoselective synthesis of 5′-epi- synargentolide B has also been reported [67].
Suresh Babu et al. followed a different strategy for the stereoselective synthesis of synargentolide B (Scheme 24) [68]. They started their synthesis with ethyl (S)-2-hydroxypropanoate (111), which was protected to form 112. The latter on reduction with DIBAL-H followed by treatment with ethyl propiolate and LiHMDS furnished the hydroxyl ester 113. This ester (113) was subsequently converted to the protected allyl alcohol 114 following a reaction sequence involving protection, reduction, and Wittig olefination. Compound 114 underwent Sharpless asymmetric dihydroxylation using an AD-mix-β to form the diol 115 (dr 97.5:2.5). Next, this diol (115) was used to produce the allyl alcohol 116, which was converted to two isomeric acryloylesters, and the major isomer was subsequently transformed to natural synargentolide B (100b).
Scheme 24.

Reagents and conditions: (a) TBSCl, CH2Cl2, imidazole, r.t., 8 h, 93%; (b) (i) DIBAL-H, CH2Cl2, −78 °C, 0.5 h, 84%; (ii) ethyl propiolate, LIHMDS, THF, −78 °C to r.t., 3 h, 76%.
2.5. Synrotolide
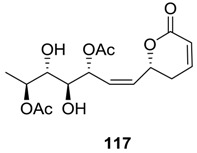
Synrotolide (117) was isolated from the leaves of Syncolostemon ratundifolius [69]. Its structure was determined from spectroscopic analysis and X-ray crystallographic studies. The structure of synrotolide (117) is interesting, as it contains a five-chiral center and a cis-double bond. Initially, the synthesis of its diacetate was reported [70,71], and later, the total synthesis of the natural product was published [72,73].
The total synthesis of synrotolide (117) was started from (S)-ethyl lactate, which was converted to the allyl alcohol 118 (Scheme 25) [72]. The Sharpless epoxidation of this allyl alcohol (118) using l-(+)-DIPT and TBHP yielded the epoxy alcohol 119 (dr 97:3). The ring opening of the epoxide 119 with 0.5 N NaOH in t-BuOH:H2O (1:5) afforded the alcohol 120, which was converted to aldehyde 121. Treatment of 121 with the protected hydroxyl propyne generated the compound 122 (dr 97:3), which by following protection/deprotection methodologies produced the alcohol 123. Oxidation of the alcohol 123 with IBX and stereoselective allylation of the aldehyde with (+)-(IPC)2 Ballyl furnished the homoallyl alcohol 124 (dr 97:3). This homoallyl alcohol with different protecting groups was also prepared from (S)-ethyl lactate through an alternative route. Next, the alcohol 124 was converted to the pyranone derivative 125, which on partial hydrogenation with Lindlar’s catalyst followed by treatment with H2SiF6 provided the natural synrotolide (117).
Scheme 25.

Reagents and conditions: (a) Ti(iPrO)4, (+)-DIPT, TBHP, CH2Cl2, −20 °C, 24 h, 91%; (b) 0.5 N NaOH, t-BuOH:H2O (1:5), 75 °C, 15 h, 72%; (c) n-BuLi, THF, −78 °C, 2 h, 91%; (d) i) IBX, DMSO, CH2Cl2, 0 °C to r.t., 2 h; ii) (+)IPC2Ballyl, Et2O, −100 °C, 81%, over two steps.
In another approach of the synthesis of synrotolide (117), the intermediates 128 (related to 124 with different protecting groups) was prepared from d-(-)-ribose (Scheme 26) [73]. This intermediate (128) was subsequently transformed to synrotolide (117).
Scheme 26.

Reagents and conditions: (a) EtMgBr, THF, 0 °C to r.t., 3 h, 92%.
The synthesis synrotolide (117) prepared by this method (Scheme 26) was evaluated for cyclotoxic activity. The compound was found to inhibit the growth of PANC1 cell lines [73].
2.6. Lippialactone
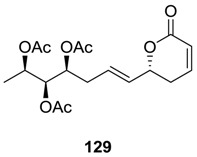
Lippialactone (129) was isolated from the acrial parts of Lippia javanica [74]. The compound is structurally related to synargentolide A (88c), but its stereoconfiguration is different. Lippialactone (129) was found to be active against the chloroquine-sensitive D10 strain of Plasmodium falciparum.
The total synthesis of 129 was initiated from l (−)-threonine, which was converted to the alcohol 130 (Scheme 27) [75]. This alcohol (130) was oxidized, and the corresponding aldehyde was subjected to Keck allylation to produce the allyl alcohol 131 (dr = 84:16). Then, this allyl alcohol (131) was transformed to the required triacetate 132. Finally, the cross-metathesis reaction between the triacetate 132 and vinyl lactone (97) using Grubbs second-generation catalyst furnished lippialactone (129).
Scheme 27.

Reagents and conditions: (a) i) DMP, CH2Cl2, 0 °C to r.t., 1 h; ii) CH2=CHCH2SnBu3, MgBr2·OEt2, CH2Cl2, –78 °C, 2 h, 66% (over two steps); (b) Grubbs second-generation catalyst, CH2Cl2, reflux, 4 h, 68%.
In another total synthesis of lippialactone (129), D-mannitol was used as the starting material (Scheme 28) [76]. It was converted to the diol (133), which was esterified with PivCl to form the ester 134. Mesylation of the ester 134 and then treatment with anhydrous K2CO3 yielded the epoxide 135. Ring opening of the epoxide (135) with vinyl magnesium bromide and CuI furnished a homoallyl alcohol, which on acetylation afforded the monoacetate 136. The latter was converted to lippialactone (129) by a cross-metathesis reaction with vinyl lactone (97) followed by deprotection and acetylation.
Scheme 28.

Reagents and conditions: (a) PivCl, Et3N, DMAP, CH2Cl2, 0 °C to r.t., 4 h, 87%; (b) i) MsCl, Et3N, DMAP, CH2Cl2, −80 °C to −20 °C, 12 h, ii) K2CO3, MeOH, rt, 2 h, 85% for two steps.
2.7. Spicigerolide
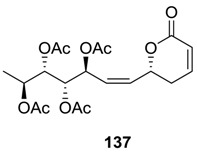
Spicigerolide (137) was isolated from the Mexican traditional medicinal plant, Hyptis spicigera [5]. The compound showed cytotoxic activity is some cell tumoral lines. The first synthesis of the compound [77,78] was described by Marco et al. [12]. Later, some other total syntheses of 137 were reported [79,80].
Garcia et al. used protected (S)-lactaldehyde as the starting material (Scheme 29) [79]. This was treated with (R)-1-phenylprop-2-ynyl acetate (137) under Carreira’s condition [81] to form the anti-syn alcohol 138, which was converted to the olefin 139. This olefin (139) was subjected to a Pd-catalyzed [3,3]-sigmatropic rearrangement to form the triacetate 140. The latter was transformed to the aldehyde 141, which on treatment with 2-tert-butyldiphenylsilyloxy-1-propyne followed by acetylation afforded the tetraacetyl compound 142 as a single diastereoisomer. The partial hydrogenation of 142 using Lindlars’ catalyst and removal of the silicon-protecting group furnished the allyl alcohol 143. Next, the later was oxidized to the aldehyde 143 under Swern condition, and this aldehyde (144) was allylated using Duthaler’s Ti-TADDOL-mediated allylation [82] to form the alcohol 145 (dr 87:13). Compound 145 with proper stereoconfiguration was subsequently converted to spicigerolide (137).
Scheme 29.

Reagents and conditions: (a) Zn(OTf)2, Et3N, (-)-NME, toluene, 4 h, 95%; (b) PdCl2(NCPh)2, CH2Cl2, r.t., 24 h, 70%; (c) Swern oxidation conditions; (d) CpTiCl-(S,S)-TADDOL, Et2O, CH2=CHCH2MgBr, –78 °C, 2.5 h, 84%.
In a recent synthetic approach, spicigerolide (137) was prepared from l-(+)-DET, which was transformed to the aldehyde (146) (Scheme 30) [80]. The Grignard reagent prepared from this aldehyde and ethyl bromide and Mg was treated with the alkyne 147 to produce the alcohol 148 (1;1 mixture of diastereoisomers). The alcohol (148) was oxidized, and the corresponding ketone was reduced using (S)-CBS catalyst [83] to furnish the chiral propargyl alcohol 149 (dr 9:1). Next, the aldehyde 150 generated from this alcohol (149) was used for chain elongation using a Still–Gennari reagent to form the ester 151, which was subsequently converted to spicigerolide (137).
Scheme 30.

Reagents and conditions: (a) EtMgBr, THF, reflux, 2 h, 80%, (1:1 dr); (b) NaH, (F3CCH2O)2POCH2COOCH3, THF, −78 °C, 1 h, 80%.
Recently, a total synthesis of an epimer of spicigerolide has also been reported. d-xylose was employed as a chiral source to generate the four stereogenic centers in the side chain [84].
2.8. Cryptofolione
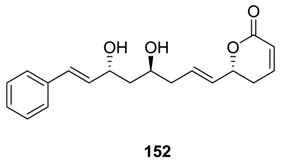
Cryptofolione (152) was isolated from Cryptocarya myrtrifolia and C. moschata, which are indigenous to South Africa and Brazil, respectively [85,86]. The compound was evaluated to be active against the trypomastigots of Trypanosoma eruzi, reducing their number by 77% at 250 µg/mL. The first synthesis of 152 [87] was mentioned by Macro et al. [12]. In recent years, several other syntheses of the molecule have been reported [88,89,90,91,92].
In a recent synthetic approach, the required intermediate was prepared from a chiral allyl epoxide (153) (Scheme 31) [88]. This epoxide (153) was treated with acyl anion equivalent 154, and the product was converted to the ketone 155. A diastereoselective reaction of 155 with borane–dimethyl sulfide adduct using (S)-CBS catalyst yielded the chiral alcohol 156 (de > 95%). The removal of the MOM protecting group of the latter afforded the required diol-intermediate, which was transformed to the acetonide 157. The same intermediate was also prepared by Prins cyclization of a chiral homoallylic alcohol with trans-cinnamaldehyde. Finally, the cross-metathesis reaction between 157 and the vinyl lactone (97) in the presence of Grubbs second-generation catalyst followed by treatment of the product with aq 4% HCl furnished the natural cryptofolione (152).
Scheme 31.

Reagents and conditions: (a) S-CBS catalyst, toluene, BH3.DMS, 0 °C, 0.5 h, 78%, 98% de.
Das et al. initiated the synthesis of cryptofolione (152) starting from propane-1,3-diol, which was transformed to the alcohol 158 (Scheme 32) [89]. Oxidation of this alcohol (158) with IBX followed by reduction with BH3-Me2S using the catalyst (R)-2-methyl-CBS-oxazaborolidine furnished the chiral alcohol 159 (ee 97%). Acetylation of the free –OH group, removal of the PMB group, and oxidation of the generated alcohol with IBX formed an aldehyde. This aldehyde underwent Maruoka allylation to produce the allyl alcohol 160, which was subsequently converted to cryptofolione (152).
Scheme 32.

Reagents and conditions: (a) IBX, DMSO, CH2Cl2, 0 °C to r.t., 4 h, 88%; (b) (R)-(Me)-CBS (1.0 M in toluene), THF, BH3.Me2S, −20 °C, 2 h, 70%; (c) (S,S)-I, allyl tributyl stannane, CH2Cl2, 0 °C, 18 h, 74%,.
In some current synthesis of natural cryptofolione, an asymmetric aldol reaction has been applied to generate the required chirality of the molecule [90,91,92]. Das et al. also completed the stereoselective synthesis of the non-lactonic portion of (Z)- cryptofolione [93].
2.9. Passifloricin A
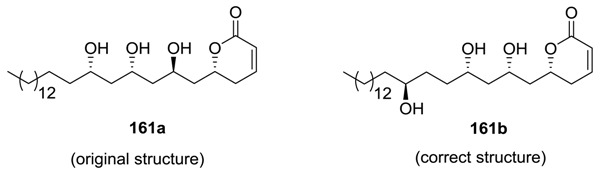
Passifloricin A was isolated from Passiflora foctida var. hispida [4]. Its structure was originally proposed to be 161a from its spectroscopic data, and its correct structure was settled as 161b on the basis of the syntheses of its different isomers [94,95,96,97]. The compound was found to inhibit impressive leishmanicidal and antiprotozoal properties [4].
In recent years, several new syntheses of passifloricin (161b) have been reported. Chandrasekar et al. prepared the compound starting from the olefin (162) (Scheme 33) [98]. This was converted to the chiral epoxide 163 by epoxidation followed by resolution of the racemic form using Jacobson’s catalyst. The epoxide 163 was transformed to the allyl alcohol 164 following a series of known reactions. This allyl alcohol 164 was subjected to Sharpless asymmetric epoxidation using (+)-DET to produce the chiral epoxy alcohol 165. The latter was converted to the α,β-unsaturated ester 166, which was treated with benzaldehyde and potassium tert-butoxide to form the acetal 167. The ester group of 167 was reduced to aldehyde, and the product underwent Maruoka allylation to furnish the major syn-isomer 168. This allyl alcohol (168) was subjected to its conversions to passifloricin A (161b) with proper stereo configuration.
Scheme 33.

Reagents and conditions: (a) Ti(iPrO)4, (+)-DET, tBuOOH, CH2Cl2, 4 Å MS, −20 °C, 8 h, 85%; (b) PhCHO, KOtBu, THF, 0 °C, 45 min, 72%.
A similar intermediate as 168 with different protecting groups was also prepared by a different research group [99]. They employed Prins cyclization [100] as the key step.
Das et al. accomplished the total synthesis of passifloricin A (161b) starting from protected glyceraldehyde and employing Maruoka allylation, iodo-carbonate cyclization, and olefin metathesis as the key reactions in their synthesis sequence (Scheme 34) [101].
Scheme 34.

Reagents and conditions: (a) Me(CH2)13Br, Mg, THF, 70 °C, 8 h, 94%; (b) i) DIBAL-H, CH2Cl2, −78 °C, 2 h, 79%; ii) (R,R)-BINOL (10 mol%), allyl(tributyl)-stannane, 4 Å MS, CH2Cl2, 0 °C, 25 h, 81%; (c) (Boc)2O, CH2Cl2, Et3N, DMAP, 0 °C to r.t., 4 h, 93%; (d) N-Iodosuccinimide, MeCN, −20 °C, 2 h, 81%.
Protected glyceraldehyde was treated with 1-bromotetradecane and the major product, anti-isomer 169, was purified by chromatography. This compound (169) was converted to the ester 170, which was reduced with DIBAL-H, and the resulting aldehyde was subjected to Maruoka allylation to give the allyl alcohol 171 (ee 97%). The free hydroxyl group of 171 was protected with Boc2O, and the product underwent iodo-carbonate cyclization with NIS to furnish the major syn-isomer 173 (>95%). The purified 173 was reacted with K2CO3 in MeOH, and the resulting epoxide was treated with vinyl Grignard reagent and CuI to produce the allyl 1,3-diol 174. The protection of two hydroxyl groups of 174, conversion of the olefin moiety to aldehyde, and again Maruoka allylation produced the required intermediate 175, which generated passifloricin A in a stereoselective manner.
2.10. Strictifolione
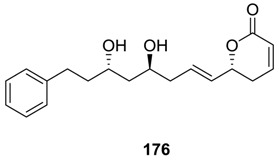
Strictifolione (176) was isolated from the stem bark of Cryptocarya strictifolia that grows in Indonesia [102]. Its structure was deduced from spectroscopic analysis. The compound was found to display antifungal property. Its earlier syntheses were mentioned by Macro et al. [12]. Recently, a large number of new syntheses of strictifolione (176) have been reported [92,103,104,105,106,107,108,109].
In a recent synthesis, known chiral allyl alcohol 177 was used as a starting material (Scheme 35) [103]. The Prins reaction between 177 and benzaldehyde followed by hydrolysis of the generated trifluoroacetate and protection of the hydroxyl group afforded 178. The tetrahydropyran ring of 178 was opened with Li in liquid NH3 to form the open chain compound 179. The protection of the primary hydroxyl group of 179 as a tosyl derivative and treatment of the product with NaH yielded the epoxide 180. The opening of this epoxide (180) with Li acetylide afforded the homopropargyl alcohol 181. The partial reduction of 181 with Lindlar’s catalyst and deprotection of the MOM group furnished the required intermediate 182. Finally, the cross-metathesis reaction between 182 and vinyl lactone 97 using Grubbs second-generation catalyst afforded natural strictifolione (176).
Scheme 35.

Reagents and conditions: (a) i) PhCHO, TFA, CH2Cl2, K2CO3, MeOH, r.t., 0.5 h, 59%; ii) MOMCl, Hunig’s base, 0 °C to r.t., 2 h, 90%; (b) Li-liq NH3, dry THF, −78 °C, 60%; (c) LiC≡CH, DMSO, 0 °C to r.t., 4 h, 80%.
During the studies [104,110,111,112,113,114] on the syntheses of natural pyranones, Das et al. accomplished the stereoselective total synthesis of strictifolione (Scheme 36) [104]. The starting material, phenyl propanal, was subjected to 2C-Wittig homologation with (carboethylmethylene) triphenylα phosphorane to produce the α,β-unsaturated ester 183. Reduction of the ester 183 with DIBAL-H and allylation of the resulting aldehyde afforded the racemic alcohol 184. The Sharpless kinetic resolution of 184 by applying (+)-DIPT yielded the chiral epoxy alcohol 185 (ee 97%). The epoxide ring of 185 was opened with Red-Al to give the intermediate 182, which was transformed to strictifolione (176).
Scheme 36.

Reagents and conditions: (a) Ph3PCHCOOEt, CH2Cl2, r.t., 8 h, 84%; (b) Ti(iPrO)4, (+)-DIPT, TBHP, CH2Cl2, 4 Å MS, −20 °C, 5 h, 45%; (c) Red-Al, THF, 0 °C to r.t., 3 h, 77%.
The same intermediate 182 or its protected form was also prepared by different other research groups by utilizing various synthetic methodologies, such as hydrolytic kinetic resolution [105,106], chemoenzymatic means [107], and asymmetric aldol reaction [92]. In addition, one modular approach that utilized phosphate tether mediate protocol was also developed for the synthesis of 182 [108].
An efficient synthesis of strictifolione (176) was achieved by She et al. by employing one-pot double allyl boration and ring-closing metathesis (Scheme 37) [109]. The required intermediate 188 was prepared by the treatment of 3-butenal with boryl-substituted allylborane 186 and then with the known aldehyde 187 utilizing a double allylboration methodology. Compound 188 was obtained with high diastereoselectivity and enanteioselectivity (dr ≥ 20:1, ee 92%). The esterification of this compound with acryloyl chloride and ring-closing metathesis of the resulting diester followed by deprotection of the product 189 furnished the ketone 190. Finally, the reduction of this ketone (190) with Me4NBH (OAc)3 yielded strictifolione (176).
Scheme 37.

Reagents and conditions: (a) 186, Et2O, −78 °C, 2 h, then 187, r.t., 24 h, 55%, 92% ee; (b) Me4NBH(OAc)3, AcOH/CH3CN (1:1), −20 °C, 10 h, 91%.
3. Conclusions
In the present article, we have described briefly the recent progress in the stereoselective total syntheses of natural pyranones having long-chain substituents. A large number of molecules have currently been synthesized by different workers following various synthetic approaches. As for examples, nine syntheses of dodoneine (from 2008) and 12 syntheses of rugulactone (from 2009) have been reported. The interesting structural features as well as promising biological activities of natural pyranones stimulated the research groups to develop new methodologies for their total syntheses. We have considered some important bioactive natural pyranones having long side chains and discussed the different modern approaches for their stereoselective syntheses. From this review, it is apparent that the rapid achievement in the diastereoselective and enantioselective synthetic protocols have made it possible to introduce proper chirality in the pyranone molecules. The ring-closing metathesis and cross-metathesis reactions have been largely utilized for the construction of their lactone rings and side chains, respectively. It is expected that the knowledge generated from the modern synthetic endeavors of the described natural pyranones in this article will enable the further development of more concise, efficient, and practical syntheses of this class of compounds.
Acknowledgments
The authors would like to thank the University of Nizwa for the generous support of this project.
Abbreviations
| HF | Hydrofluoric acid | TsCl | Para-toluene sulfonyl chloride |
| TBS | tert-Butyldimethylsilyl | TESCl | Triethylsilyl Chloride |
| DIBAL-H | Diisobutylaluminium hydride | DET | N,N-Diethyltryptamine |
| DIPEA | N,N-Diisopropylethylamine | (S)-CBS | (S)-5,5-Diphenyl-2-methyl-3,4-propano-1,3,2-oxazaborolidine |
| PTSA | para-toluenesulfonic acid | NIS | N-Bromo scuccinimide |
| PPTS | Pyridinium p-toluenesulfonate | TBSOTf | Trimethylsilyl trifluoromethanesulfonate |
| DIPT | Diisopropyltartrate | t-BuOK | Potassium tert-butoxide |
| TBHP | tert-Butylhydroperoxide | AcOH | Acetic acid |
| DMP | 2,2-Dimethoxypropane | LiAlH4 | Lithium aluminium hydride |
| TBSCl | tert-Butyldimethylsilyl chloride | Et3N | Triethyl amine |
| IBX | 2-Iodoxybenzoic acid | t-BuOH | tert-Butyl alcohol |
| BINOL | 1,1′-Bi-2-naphthol | NaBH4 | Sodium borohydride |
| Ti(iPrO)4 | Titanium isopropoxide | BnBr | Benzyl Bromide |
| PMB | 4-Methoxybenzyl | THF | Tetrahydrofuran |
| KHMDS | Potassium hexamethyldisilazide | CHCl3 | Chloroform |
| NF-kB | Nuclear factor kappa-light-chain-enhancer of activated B cells | DMAP | 4-Dimethylaminopyridine |
| TBAI | tert-Butylammonium iodide | NaH | Sodium hydride |
| TBDPSCl | tert-Butyldiphenylsilyl chloride | NH4Cl | Ammonium chloride |
| NaHMDS | Sodium bis (trimethylsilyl) amide | NaIO4 | Sodium periodate |
| NADPH | Nicotinamide adenine dinucleotide phosphate | Ac | Acetyl |
| THP | Tetrahydropyran | TFA | Trifluoroacetic acid |
| TEMPO | (2,2,6,6-Tetramethylpiperidin-1-yl)oxyl | MOMCl | Methoxymethyl chloride |
| TPP | Thiamine pyrophosphate |
Funding
This research received no external funding.
Conflicts of Interest
All authors confirm that this article content has no conflict of interest.
References
- 1.McGlacken G.P., Fairlamb I.J.S. 2-Pyrone natural products and mimetics: Isolation, characterisation and biological activity. Nat. Prod. Rep. 2005;22:369–385. doi: 10.1039/b416651p. [DOI] [PubMed] [Google Scholar]
- 2.Ouedraogo M., Carreyre H., Vandebrouck C., Bescond J., Raymond G., Guissou I.-P., Cognard C., Becq F., Potreau D., Cousson A., et al. Structure Elucidation of a Dihydropyranone from Tapinanthus dodoneifolius. J. Nat. Prod. 2007;70:2006–2009. doi: 10.1021/np070355x. [DOI] [PubMed] [Google Scholar]
- 3.Meragelman T.L., Scudiero D.A., Davis R.E., Staudt L.M., McCloud T.G., Cardellina J.H., Shoemaker R.H. Inhibitors of the NF-κB Activation Pathway from Cryptocarya rugulosa. J. Nat. Prod. 2009;72:336–339. doi: 10.1021/np800350x. [DOI] [PubMed] [Google Scholar]
- 4.Echeverri F., Arango V., Quiñones W., Torres F., Escobar G., Rosero Y., Archbold R. Passifloricins, polyketides α-pyrones from Passiflora foetida resin. Phytochemistry. 2001;56:881–885. doi: 10.1016/S0031-9422(00)00478-7. [DOI] [PubMed] [Google Scholar]
- 5.Pereda-Miranda R., Fragoso-Serrano M., Cerda-García-Rojas C.M. Application of molecular mechanics in the total stereochemical elucidation of spicigerolide, a cytotoxic 6-tetraacetyloxyheptenyl-5,6-dihydro-α-pyrone from Hyptis spicigera. Tetrahedron. 2001;57:47–53. doi: 10.1016/S0040-4020(00)00987-X. [DOI] [Google Scholar]
- 6.Kasaplar P., Yılmazer Ö., Çağır A. 6-Bicycloaryl substituted (S)- and (R)-5,6-dihydro-2H-pyran-2-ones: Asymmetric synthesis, and anti-proliferative properties. Bioorg. Med. Chem. 2009;17:311–318. doi: 10.1016/j.bmc.2008.10.069. [DOI] [PubMed] [Google Scholar]
- 7.Grover A., Agrawal V., Shandilya A., Bisaria V.S., Sundar D. Non-nucleosidic inhibition of Herpes simplex virus DNA polymerase: Mechanistic insights into the anti-herpetic mode of action of herbal drug withaferin A. BMC Bioinform. 2011;12:S22. doi: 10.1186/1471-2105-12-S13-S22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.USUI T. Actin- and Microtubule-Targeting Bioprobes: Their Binding Sites and Inhibitory Mechanisms. Biosci. Biotechnol. Biochem. 2007;71:300–308. doi: 10.1271/bbb.60516. [DOI] [PubMed] [Google Scholar]
- 9.McCracken S.T., Kaiser M., Boshoff H.I., Boyd P.D.W., Copp B.R. Synthesis and antimalarial and antituberculosis activities of a series of natural and unnatural 4-methoxy-6-styryl-pyran-2-ones, dihydro analogues and photo-dimers. Bioorg. Med. Chem. 2012;20:1482–1493. doi: 10.1016/j.bmc.2011.12.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sassa T., Kato H., Kajiura H. Isolation and structure of pycnophorin, a novel diterpene α-pyrone with antimicrobial activity, produced by phytopathogenic macrophoma kuwatsukai. Tetrahedron Lett. 1986;27:2121–2124. doi: 10.1016/S0040-4039(00)84464-0. [DOI] [Google Scholar]
- 11.Kalesse M., Christmann M., Bhatt U., Quitschalle M., Claus E., Saeed A., Burzlaff A., Kasper C., Haustedt L.O., Hofer E., et al. The Chemistry and Biology of Ratjadone. Chem. Bio. Chem. 2001;2:709–714. doi: 10.1002/1439-7633(20010903)2:9<709::AID-CBIC709>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 12.Marco J.A., Carda M., Murga J., Falomir E. Stereoselective syntheses of naturally occurring 5,6-dihydropyran-2-ones. Tetrahedron. 2007;63:2929–2958. doi: 10.1016/j.tet.2006.12.047. [DOI] [Google Scholar]
- 13.Boucard V., Broustal G., Campagne J.M. Synthetic Approaches to α,β-Unsaturated δ-Lactones and Lactols. Eur. J. Org. Chem. 2007;2007:225–236. doi: 10.1002/ejoc.200600570. [DOI] [Google Scholar]
- 14.Eskandari K., Rafieian-Kopaei M. Synthesis of 5,6-dihydro-2H-pyran-2-ones (microreview) Chem. Heterocycl. Compd. 2016;52:158–160. doi: 10.1007/s10593-016-1853-3. [DOI] [Google Scholar]
- 15.Wilson Cardona G., Winston Quiñones F., Fernando Echeverri L. Leishmanicidal Activity of Passifloricin A and Derivatives. Molecules. 2004;9:666–672. doi: 10.3390/90800666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carreyre H., Coustard J.-M., Carré G., Vandebrouck C., Bescond J., Ouédraogo M., Marrot J., Vullo D., Supuran C.T., Thibaudeau S. Natural product hybrid and its superacid synthesized analogues: Dodoneine and its derivatives show selective inhibition of carbonic anhydrase isoforms I, III, XIII and XIV. Bioorg. Med. Chem. 2013;21:3790–3794. doi: 10.1016/j.bmc.2013.04.041. [DOI] [PubMed] [Google Scholar]
- 17.Cuccarese M.F., Wang Y., Beuning P.J., O’Doherty G.A. Cryptocaryol Structure–Activity Relationship Study of Cancer Cell Cytotoxicity and Ability to Stabilize PDCD4. ACS Med. Chem. Lett. 2014;5:522–526. doi: 10.1021/ml4005039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grubbs R.H. Olefin-Metathesis Catalysts for the Preparation of Molecules and Materials (Nobel Lecture) Angew. Chem. Int. Ed. 2006;45:3760–3765. doi: 10.1002/anie.200600680. [DOI] [PubMed] [Google Scholar]
- 19.Carreyre H., Carré G., Ouedraogo M., Vandebrouck C., Bescond J., Supuran C.T., Thibaudeau S. Bioactive Natural Product and Superacid Chemistry for Lead Compound Identification: A Case Study of Selective hCA III and L-Type Ca2+ Current Inhibitors for Hypotensive Agent Discovery. Molecules. 2017;22:915. doi: 10.3390/molecules22060915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carré G., Carreyre H., Ouedraogo M., Becq F., Bois P., Thibaudeau S., Vandebrouck C., Bescond J. The hypotensive agent dodoneine inhibits L-type Ca2+ current with negative inotropic effect on rat heart. Eur. J. Pharmacol. 2014;728:119–127. doi: 10.1016/j.ejphar.2014.01.059. [DOI] [PubMed] [Google Scholar]
- 21.Álvarez-Bercedo P., Falomir E., Murga J., Carda M., Marco J.A. Stereoselective Synthesis of the Naturally Occurring 2-Pyranone Dodoneine. Eur. J. Org. Chem. 2008;2008:4015–4018. doi: 10.1002/ejoc.200800389. [DOI] [Google Scholar]
- 22.Srihari P., Rajendar G., Srinivasa Rao R., Yadav J.S. First stereoselective total synthesis of (+)-dodoneine. Tetrahedron Lett. 2008;49:5590–5592. doi: 10.1016/j.tetlet.2008.07.027. [DOI] [Google Scholar]
- 23.Dittoo A., Bellosta V., Cossy J. Total synthesis of (+)-dodoneine. Synlett. 2008:2459–2460. doi: 10.1055/s-2008-1078052. [DOI] [Google Scholar]
- 24.Das B., Suneel K., Satyalakshmi G., Kumar D.N. Stereoselective total synthesis of dodoneine. Tetrahedron Asymmetry. 2009;20:1536–1540. doi: 10.1016/j.tetasy.2009.05.036. [DOI] [Google Scholar]
- 25.Vuppalapati S.V.N., Putapatri S.R., Kantevari S. A formal enantioselective synthesis of (+)-dodoneine via cyclic sulfate methodology. Arkivoc. 2009;2009:217–226. [Google Scholar]
- 26.Sabitha G., Bhaskar V., Reddy S.S.S., Yadav J.S. Total synthesis of (+)-dodoneine and its 6-epimer. Synthesis. 2009;2:3285–3292. doi: 10.1055/s-0029-1216953. [DOI] [Google Scholar]
- 27.Rauniyar V., Hall D.G. Rationally Improved Chiral Brønsted Acid for Catalytic Enantioselective Allylboration of Aldehydes with an Expanded Reagent Scope. J. Org. Chem. 2009;74:4236–4241. doi: 10.1021/jo900553f. [DOI] [PubMed] [Google Scholar]
- 28.Chinnababu B., Reddy S.P., Rao C.B., Rajesh K., Venkateswarlu Y. Stereoselective Total Synthesis of Dodoneine. Helv. Chim. Acta. 2010;93:1960–1966. doi: 10.1002/hlca.200900478. [DOI] [Google Scholar]
- 29.Allais F., Ducrot P.H. Stereoselective total synthesis of (+)-dodoneine. Synthesis. 2010:1649–1653. doi: 10.1055/s-0029-1218741. [DOI] [Google Scholar]
- 30.Keck G.E., Covel J.A., Schiff T., Yu T. Pyran Annulation: Asymmetric Synthesis of 2,6-Disubstituted-4-methylene Tetrahydropyrans. Org. Lett. 2002;4:1189–1192. doi: 10.1021/ol025645d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crimmins M.T., Chaudhary K. Titanium Enolates of Thiazolidinethione Chiral Auxiliaries: Versatile Tools for Asymmetric Aldol Additions. Org. Lett. 2000;2:775–777. doi: 10.1021/ol9913901. [DOI] [PubMed] [Google Scholar]
- 32.Ando K. Highly Selective Synthesis of Z-Unsaturated Esters by Using New Horner−Emmons Reagents, Ethyl (Diarylphosphono)acetates. J. Org. Chem. 1997;62:1934–1939. doi: 10.1021/jo970057c. [DOI] [PubMed] [Google Scholar]
- 33.Kingsbury J.S., Harrity J.P.A., Bonitatebus P.J., Hoveyda A.H. A Recyclable Ru-Based Metathesis Catalyst. J. Am. Chem. Soc. 1999;121:791–799. doi: 10.1021/ja983222u. [DOI] [Google Scholar]
- 34.Still W.C., Gennari C. Direct synthesis of Z-unsaturated esters. A useful modification of the horner-emmons olefination. Tetrahedron Lett. 1983;24:4405–4408. doi: 10.1016/S0040-4039(00)85909-2. [DOI] [Google Scholar]
- 35.Katsuki T., Sharpless K.B. The first practical method for asymmetric epoxidation. J. Am. Chem. Soc. 1980;102:5974–5976. doi: 10.1021/ja00538a077. [DOI] [Google Scholar]
- 36.Kolb H.C., VanNieuwenhze M.S., Sharpless K.B. Catalytic Asymmetric Dihydroxylation. Chem. Rev. 1994;94:2483–2547. doi: 10.1021/cr00032a009. [DOI] [Google Scholar]
- 37.Schaus S.E., Brandes B.D., Larrow J.F., Tokunaga M., Hansen K.B., Gould A.E., Furrow M.E., Jacobsen E.N. Highly Selective Hydrolytic Kinetic Resolution of Terminal Epoxides Catalyzed by Chiral (salen)CoIII Complexes. Practical Synthesis of Enantioenriched Terminal Epoxides and 1,2-Diols. J. Am. Chem. Soc. 2002;124:1307–1315. doi: 10.1021/ja016737l. [DOI] [PubMed] [Google Scholar]
- 38.Reddy D.K., Shekhar V., Reddy T.S., Reddy S.P., Venkateswarlu Y. Stereoselective first total synthesis of (R)-rugulactone. Tetrahedron Asymmetry. 2009;20:2315–2319. doi: 10.1016/j.tetasy.2009.09.016. [DOI] [Google Scholar]
- 39.Mohapatra D.K., Das P.P., Sai Reddy D., Yadav J.S. First total syntheses and absolute configuration of rugulactone and 6(R)-(4′-oxopent-2′-enyl)-5,6-dihydro-2H-pyran-2-one. Tetrahedron Lett. 2009;50:5941–5944. doi: 10.1016/j.tetlet.2009.08.028. [DOI] [Google Scholar]
- 40.Tyrikos-Ergas T., Giannopoulos V., Smonou I. An efficient chemoenzymatic approach towards the synthesis of rugulactone. Molecules. 2018;23:640. doi: 10.3390/molecules23030640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reddy D.K., Shekhar V., Prabhakar P., Chinna Babu B., Siddhardha B., Murthy U.S.N., Venkateswarlu Y. Stereoselective synthesis and biological evaluation of (R)-rugulactone, (6R)-((4R)-hydroxy-6-phenyl-hex-2-enyl)-5,6-dihydro-pyran-2-one and its 4S epimer. Eur. J. Med. Chem. 2010;45:4657–4663. doi: 10.1016/j.ejmech.2010.07.036. [DOI] [PubMed] [Google Scholar]
- 42.Reddipalli G., Venkataiah M., Fadnavis N.W. Chemo-enzymatic synthesis of both enantiomers of rugulactone. Tetrahedron Asymmetry. 2010;21:320–324. doi: 10.1016/j.tetasy.2010.01.016. [DOI] [Google Scholar]
- 43.Allais F., Aouhansou M., Majira A., Ducrot P.H. Asymmetric total synthesis of rugulactone, an α-pyrone from Cryptocarya rugulosa. Synthesis. 2010:2787–2793. doi: 10.1055/s-0029-1218836. [DOI] [Google Scholar]
- 44.Das B., Srinivas Y., Sudhakar C., Raghavendar Reddy P. Stereoselective Total Synthesis of Rugulactone. Helv. Chim. Acta. 2011;94:1290–1295. doi: 10.1002/hlca.201000396. [DOI] [Google Scholar]
- 45.Goswami A., Saikia P.P., Saikia B., Chaturvedi D., Barua N.C. An improved stereoselective total synthesis of (R)-rugulactone. Tetrahedron Lett. 2011;52:5133–5135. doi: 10.1016/j.tetlet.2011.07.109. [DOI] [Google Scholar]
- 46.Böse D., Fernández E., Pietruszka J. Stereoselective Synthesis of Both Enantiomers of Rugulactone. J. Org. Chem. 2011;76:3463–3469. doi: 10.1021/jo2004583. [DOI] [PubMed] [Google Scholar]
- 47.Shinde D.B., Kanth B.S., Satyakumar A., Kamble V.T., Das B. Simple Stereoselective Synthesis of Unsaturated Lactone Intermediates and Their Conversion into Natural Dihydropyranones and Their Enantiomers. Lett. Org. Chem. 2013;10:317–323. doi: 10.2174/1570178611310050003. [DOI] [Google Scholar]
- 48.Reddy B., Singh R. A Facile Stereoselective Total Synthesis of (R)-Rugulactone. ISRN Org. Chem. 2014;2014:767954. doi: 10.1155/2014/767954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kii S., Maruoka K. Practical approach for catalytic asymmetric allylation of aldehydes with a chiral bidentate titanium(IV) complex. Tetrahedron Lett. 2001;42:1935–1939. doi: 10.1016/S0040-4039(01)00025-9. [DOI] [Google Scholar]
- 50.Cros F., Pelotier B., Piva O. Regioselective Tandem Ring Closing/Cross Metathesis of 1,5-Hexadien-3-ol Derivatives: Application to the Total Synthesis of Rugulactone. Eur. J. Org. Chem. 2010;2010:5063–5070. doi: 10.1002/ejoc.201000187. [DOI] [Google Scholar]
- 51.Collett L.A., Davies-Coleman M.T., Rivett D.E.A. 5,6-Dihydro-α-pyrones from Syncolostemon argenteus. Phytochemistry. 1998;48:651–656. doi: 10.1016/S0031-9422(97)01075-3. [DOI] [Google Scholar]
- 52.García-Fortanet J., Murga J., Carda M., Marco J.A. Stereoselective synthesis of the published structure of synargentolide A and of one of its stereoisomers. Arkivoc. 2005;2005:175–188. [Google Scholar]
- 53.Sabitha G., Gopal P., Reddy C.N., Yadav J.S. Stereoselective synthesis of the published structure of synargentolide A and of one of its stereoisomers. Tetrahedron Lett. 2009;50:6298–6302. doi: 10.1016/j.tetlet.2009.08.109. [DOI] [Google Scholar]
- 54.Kamal A., Balakrishna M., Reddy P.V., Faazil S. An efficient synthesis of synargentolide A from d-mannitol. Tetrahedron Asymmetry. 2010;21:2517–2523. doi: 10.1016/j.tetasy.2010.10.001. [DOI] [Google Scholar]
- 55.Prasad K.R., Penchalaiah K. Stereoselective total synthesis of (+)-synargentolide A. Tetrahedron Asymmetry. 2010;21:2853–2858. doi: 10.1016/j.tetasy.2010.11.005. [DOI] [Google Scholar]
- 56.Das B., Balasubramanyam P., Veeranjaneyulu B., Chinna Reddy G. An Efficient Stereoselective Total Synthesis of Synargentolide A and Its Epimer. Helv. Chim. Acta. 2011;94:881–884. doi: 10.1002/hlca.201000342. [DOI] [Google Scholar]
- 57.Sabitha G., Reddy S.S.S., Raju A., Yadav J.S. A concise stereoselective total synthesis of synargentolide a from 3,4,6-Tri-o-acetyl-d-glucal. Synthesis. 2011:1279–1282. doi: 10.1055/s-0030-1258458. [DOI] [Google Scholar]
- 58.Yadav J.S., Thirupathaiah B., Singh V.K., Ravishashidhar V. Total synthesis of (+)-synargentolide A. Tetrahedron Asymmetry. 2012;23:931–937. doi: 10.1016/j.tetasy.2012.06.021. [DOI] [Google Scholar]
- 59.Enders D., Reichenbach L.F. Asymmetric synthesis of synargentolide A and its 3-epimer using the RAMP-hydrazone methodology. Synthesis. 2013;45:959–965. doi: 10.1055/s-0032-1316865. [DOI] [Google Scholar]
- 60.Juárez-González F., Suárez-Ortiz G.A., Fragoso-Serrano M., Cerda-García-Rojas C.M., Pereda-Miranda R. DFT 1H–1H coupling constants in the conformational analysis and stereoisomeric differentiation of 6-heptenyl-2H-pyran-2-ones: Configurational reassignment of synargentolide A. Magn. Reson. Chem. 2015;53:203–212. doi: 10.1002/mrc.4178. [DOI] [PubMed] [Google Scholar]
- 61.Prasad K.R., Gutala P. Total Synthesis and Determination of the Absolute Configuration of 5,6-Dihydro-α-pyrone Natural Product Synargentolide B. J. Org. Chem. 2013;78:3313–3322. doi: 10.1021/jo400083v. [DOI] [PubMed] [Google Scholar]
- 62.Pereda-Miranda R., García M., Delgado G. Structure and stereochemistry of four α-pyrones from Hyptis oblongifolia. Phytochemistry. 1990;29:2971–2974. doi: 10.1016/0031-9422(90)87117-D. [DOI] [Google Scholar]
- 63.Sabitha G., Shankaraiah K., Yadav J.S. Tandem Ring-Closing/Cross-Metathesis Approach for the Synthesis of Synargentolide B and Its Stereoisomers. Eur. J. Org. Chem. 2013;2013:4870–4878. doi: 10.1002/ejoc.201300434. [DOI] [Google Scholar]
- 64.Konda S., Bhaskar K., Nagarapu L., Akkewar D.M. A convenient approach to total synthesis of synargentolide-B from l-ascorbic acid and d-ribose. Tetrahedron Lett. 2014;55:3087–3089. doi: 10.1016/j.tetlet.2014.03.133. [DOI] [Google Scholar]
- 65.Liu J., Gao Y., Wang L., Du Y. Chiron approach for the total synthesis of (+)-synargentolide B. Tetrahedron. 2017;73:6443–6447. doi: 10.1016/j.tet.2017.09.041. [DOI] [Google Scholar]
- 66.Gao Y., Liu J., Qiao J., Liu Y., He P., Du Y. Stereoselective synthesis of (−)-synargentolide B. Tetrahedron Lett. 2018;59:291–294. doi: 10.1016/j.tetlet.2017.12.039. [DOI] [Google Scholar]
- 67.Kumaraswamy G., Raghu N., Jayaprakash N., Ankamma K. A concise diastereoselective synthesis of 5′-epi synargentolide-B. Tetrahedron. 2015;71:5472–5477. doi: 10.1016/j.tet.2015.06.080. [DOI] [Google Scholar]
- 68.Ramulu U., Rajaram S., Ramesh D., Suresh Babu K. Total synthesis of synargentolide B. Tetrahedron Asymmetry. 2015;26:928–934. doi: 10.1016/j.tetasy.2015.07.007. [DOI] [Google Scholar]
- 69.Davies Coleman M.T., English R.B., Rivett D.E.A. Synrotolide, an α-pyrone from Syncolostemon rotundifolius. Phytochemistry. 1987;26:1497–1499. doi: 10.1016/S0031-9422(00)81843-9. [DOI] [Google Scholar]
- 70.Srihari P., Prem Kumar B., Subbarayudu K., Yadav J.S. A convergent approach for the total synthesis of (−)-synrotolide diacetate. Tetrahedron Lett. 2007;48:6977–6981. doi: 10.1016/j.tetlet.2007.07.172. [DOI] [Google Scholar]
- 71.Krishna P.R., Reddy P.S. Stereoselective total synthesis of (−)-synrotolide diacetate from d-ribose. Tetrahedron. 2007;63:3995–3999. doi: 10.1016/j.tet.2007.03.011. [DOI] [Google Scholar]
- 72.Sabitha G., Rao A.S., Sandeep A., Yadav J.S. The First Stereoselective Total Synthesis of (–)-Synrotolide. Eur. J. Org. Chem. 2014;2014:455–465. doi: 10.1002/ejoc.201301215. [DOI] [Google Scholar]
- 73.Sabitha G., Rao A.S., Sandeep A., Latha B.M., Reddy D.V. Total synthesis of (−)-synrotolide and the evaluation of its antiproliferative activity. Tetrahedron Asymmetry. 2014;25:856–859. doi: 10.1016/j.tetasy.2014.04.010. [DOI] [Google Scholar]
- 74.Ludere M.T., van Ree T., Vleggaar R. Isolation and relative stereochemistry of lippialactone, a new antimalarial compound from Lippia javanica. Fitoterapia. 2013;86:188–192. doi: 10.1016/j.fitote.2013.03.009. [DOI] [PubMed] [Google Scholar]
- 75.Krishna P.R., Nomula R., Kunde R. First total synthesis of lippialactone and its C9 epimer. Synthesis. 2014;46:307–312. doi: 10.1055/s-0033-1340314. [DOI] [Google Scholar]
- 76.Gowravaram S., Karra P., Sudina P. Stereoselective total synthesis of lippialactone. Reports Org. Chem. 2015;5:57–63. [Google Scholar]
- 77.Falomir E., Murga J., Carda M., Marco J.A. Stereoselective synthesis of spicigerolide. Tetrahedron Lett. 2003;44:539–541. doi: 10.1016/S0040-4039(02)02588-1. [DOI] [Google Scholar]
- 78.Falomir E., Murga J., Ruiz P., Carda M., Marco J.A., Pereda-Miranda R., Fragoso-Serrano M., Cerda-García-Rojas C.M. Stereoselective Synthesis and Determination of the Cytotoxic Properties of Spicigerolide and Three of Its Stereoisomers. J. Org. Chem. 2003;68:5672–5676. doi: 10.1021/jo034470y. [DOI] [PubMed] [Google Scholar]
- 79.Georges Y., Ariza X., Garcia J. Stereoselective Synthesis of (−)-Spicigerolide. J. Org. Chem. 2009;74:2008–2012. doi: 10.1021/jo8025753. [DOI] [PubMed] [Google Scholar]
- 80.Sabitha G., Reddy C.N., Raju A., Yadav J.S. Stereoselective total synthesis of the cytotoxic lactone (−)-spicigerolide. Tetrahedron Asymmetry. 2011;22:493–498. doi: 10.1016/j.tetasy.2011.02.010. [DOI] [Google Scholar]
- 81.Anand N.K., Carreira E.M. A Simple, Mild, Catalytic, Enantioselective Addition of Terminal Acetylenes to Aldehydes. J. Am. Chem. Soc. 2001;123:9687–9688. doi: 10.1021/ja016378u. [DOI] [PubMed] [Google Scholar]
- 82.Hafner A., Duthaler R.O., Marti R., Rihs G., Rothe-Streit P., Schwarzenbach F. Enantioselective syntheses with titanium carbohydrate complexes. Part 7. Enantioselective allyltitanation of aldehydes with cyclopentadienyldialkoxyallyltitanium complexes. J. Am. Chem. Soc. 1992;114:2321–2336. doi: 10.1021/ja00033a005. [DOI] [Google Scholar]
- 83.Corey E.J., Helal C.J. Reduction of Carbonyl Compounds with Chiral Oxazaborolidine Catalysts: A New Paradigm for Enantioselective Catalysis and a Powerful New Synthetic Method. Angew. Chem. Int. Ed. 1998;37:1986–2012. doi: 10.1002/(SICI)1521-3773(19980817)37:15<1986::AID-ANIE1986>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 84.Subba Reddy B.V., Veerabhadra Reddy V., Praneeth K. Stereoselective total synthesis of (+)-boronolide, (+)-anamarine, 8-epi-spicegerolide. Tetrahedron Lett. 2014;55:1398–1401. doi: 10.1016/j.tetlet.2013.12.061. [DOI] [Google Scholar]
- 85.Sehlapelo B.M., Drewes S.E., Scott-Shaw R. A 6-substituted 5,6-dihydro-α-pyrone from two species of Cryptocarya. Phytochemistry. 1994;37:847–849. doi: 10.1016/S0031-9422(00)90368-6. [DOI] [Google Scholar]
- 86.Cavalheiro A.J., Yoshida M. 6-[ω-arylalkenyl]-5,6-dihydro-α-pyrones from Cryptocarya moschata (Lauraceae) Phytochemistry. 2000;53:811–819. doi: 10.1016/S0031-9422(99)00532-4. [DOI] [PubMed] [Google Scholar]
- 87.Matsuoka Y., Aikawa K., Irie R., Katsuki T. Asymmetric synthesis of cryptofolione and determination of its absolute configuration. Heterocycles. 2005;66:187–194. doi: 10.1002/chin.200622209. [DOI] [Google Scholar]
- 88.Sabitha G., Reddy S.S.S., Reddy D.V., Bhaskar V., Yadav J.S. Stereoselective approaches to the total synthesis of (6R,4′S, 6′R)-cryptofolione. Synthesis. 2010:3453–3460. doi: 10.1055/s-0030-1258215. [DOI] [Google Scholar]
- 89.Das B., Nagendra S., Ravindra Reddy C. Stereoselective total synthesis of (+)-cryptofolione and (+)-goniothalamin. Tetrahedron Asymmetry. 2011;22:1249–1254. doi: 10.1016/j.tetasy.2011.06.029. [DOI] [Google Scholar]
- 90.Kumar R.N., Meshram H.M. Total synthesis of (−)-diospongin A and (+)-cryptofolione via asymmetric aldol reaction. Tetrahedron Lett. 2011;52:1003–1007. doi: 10.1016/j.tetlet.2010.12.070. [DOI] [Google Scholar]
- 91.Yadav J., Ganganna B., Bhunia D. First stereoselective total synthesis of cryptomoscatone D2 and syntheses of (5R,7S)-kurzilactone and (+)-cryptofolione by an asymmetric acetate aldol approach. Synthesis. 2012;44:1365–1372. doi: 10.1055/s-0031-1290771. [DOI] [Google Scholar]
- 92.Li X., Wang G., Zhang Z., Wu N., Yang Q., Huang S., Wang X. A concise and straightforward approach to total synthesis of (+)-Strictifolione and formal synthesis of Cryptofolione via a unified strategy. Synth. Commun. 2019;49:1031–1039. doi: 10.1080/00397911.2019.1580373. [DOI] [Google Scholar]
- 93.Nagendra S., Krishna Reddy V., Das B. Stereoselective Synthesis of the Non-Lactonic Portion of (Z)-Cryptofolione and Approaches towards Its Conversion to (Z)-Cryptofolione. Helv. Chim. Acta. 2015;98:520–526. doi: 10.1002/hlca.201400242. [DOI] [Google Scholar]
- 94.García-Fortanet J., Murga J., Carda M., Marco J.A. On the Structure of Passifloricin A: Asymmetric Synthesis of the δ-Lactones of (2Z,5S,7R,9S,11S)- and (2Z,5R,7R,9S,11S)-Tetrahydroxyhexacos-2-enoic Acid. Org. Lett. 2003;5:1447–1449. doi: 10.1021/ol034182o. [DOI] [PubMed] [Google Scholar]
- 95.Murga J., García-Fortanet J., Carda M., Marco J.A. Asymmetric synthesis of passifloricin A: A correction in structure. Tetrahedron Lett. 2003;44:7909–7912. doi: 10.1016/j.tetlet.2003.09.007. [DOI] [Google Scholar]
- 96.Murga J., García-Fortanet J., Carda M., Marco J.A. Stereoselective Synthesis of the Antiprotozoal Lactone Passifloricin A and Seven Isomers Thereof. J. Org. Chem. 2004;69:7277–7283. doi: 10.1021/jo049275d. [DOI] [PubMed] [Google Scholar]
- 97.Curran D.P., Moura-Letts G., Pohlman M. Solution-Phase Mixture Synthesis with Fluorous Tagging En Route: Total Synthesis of an Eight-Member Stereoisomer Library of Passifloricins. Angew. Chem. Int. Ed. 2006;45:2423–2426. doi: 10.1002/anie.200600041. [DOI] [PubMed] [Google Scholar]
- 98.Chandrasekhar S., Rambabu C., Reddy A.S. Asymmetric synthesis of (+)-passifloricin A and its 6-epimer. Tetrahedron Lett. 2008;49:4476–4478. doi: 10.1016/j.tetlet.2008.05.070. [DOI] [Google Scholar]
- 99.Sabitha G., Prasad M.N., Shankaraiah K., Reddy N.M., Yadav J.S. Convergent synthesis of passifloricin a via a prins cyclisation and olefin cross-metathesis approach. Synthesis. 2010:3891–3898. doi: 10.1055/s-0030-1258253. [DOI] [Google Scholar]
- 100.Arundale E., Mikeska L.A. The Olefin-Aldehyde Condensation. The Prins Reaction. Chem. Rev. 1952;51:505–555. doi: 10.1021/cr60160a004. [DOI] [Google Scholar]
- 101.Reddy C.R., Veeranjaneyulu B., Nagendra S., Das B. Stereoselective Total Synthesis of Passifloricin A. Helv. Chim. Acta. 2013;96:505–513. doi: 10.1002/hlca.201200356. [DOI] [Google Scholar]
- 102.Juliawaty L.D., Kitajima M., Takayama H., Achmad S.A., Aimi N. A 6-substituted-5,6-dihydro-2-pyrone from Cryptocarya strictifolia. Phytochemistry. 2000;54:989–993. doi: 10.1016/S0031-9422(00)00077-7. [DOI] [PubMed] [Google Scholar]
- 103.Sabitha G., Fatima N., Gopal P., Reddy C.N., Yadav J.S. Stereoselective total synthesis of (+)-strictifolione and (6R)-6-[(4R,6R)-4,6-dihydroxy-10-phenyldec-1-enyl]-5,6-dihydro-2H-pyran-2-one by Prins reaction and olefin cross-metathesis. Tetrahedron Asymmetry. 2009;20:184–191. doi: 10.1016/j.tetasy.2008.12.004. [DOI] [Google Scholar]
- 104.Das B., Veeranjaneyulu B., Balasubramanyam P., Srilatha M. Stereoselective total synthesis of (+)-strictifolione and (6R)-6[(E,4R,6R)-4,6-dihydro-10-phenyl-1-decenyl)-5,6-dihydro-2H-2-pyrone. Tetrahedron Asymmetry. 2010;21:2762–2767. doi: 10.1016/j.tetasy.2010.11.003. [DOI] [Google Scholar]
- 105.Kumar P., Gupta P. Hydrolytic kinetic resolution as an emerging tool in the synthesis of bioactive molecules. Synlett. 2009:1367–1382. doi: 10.1055/s-0029-1217171. [DOI] [Google Scholar]
- 106.Kumar P., Pandey M., Gupta P., Naidu S.V., Dhavale D.D. Enantio- and Diastereocontrolled Total Synthesis of (+)-Strictifolione. Eur. J. Org. Chem. 2010;2010:6993–7004. doi: 10.1002/ejoc.201001221. [DOI] [Google Scholar]
- 107.Ghadigaonkar S., Koli M.R., Gamre S.S., Choudhary M.K., Chattopadhyay S., Sharma A. A chemoenzymatic asymmetric synthesis of (+)-strictifolione. Tetrahedron Asymmetry. 2012;23:1093–1099. doi: 10.1016/j.tetasy.2012.06.015. [DOI] [Google Scholar]
- 108.Jayasinghe S., Venukadasula P.K.M., Hanson P.R. An Efficient, Modular Approach for the Synthesis of (+)-Strictifolione and a Related Natural Product. Org. Lett. 2014;16:122–125. doi: 10.1021/ol403110p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tang S., Xie X., Wang X., He L., Xu K., She X. Concise Total Syntheses of (+)-Strictifolione and (6R)-6-[(4R,6R)-4,6-Dihydroxy-10-phenyldec-1-enyl]-5,6-dihydro-2H-pyran-2-one. J. Org. Chem. 2010;75:8234–8240. doi: 10.1021/jo101875w. [DOI] [PubMed] [Google Scholar]
- 110.Das B., Laxminarayana K., Krishnaiah M., Kumar D.N. Stereoselective total synthesis of a potent natural antifungal compound (6S)-5,6,dihydro-6-[(2R)-2-hydroxy-6-phenyl hexyl]-2H-pyran-2-one. Bioorg. Med. Chem. Lett. 2009;19:6396–6398. doi: 10.1016/j.bmcl.2009.09.063. [DOI] [PubMed] [Google Scholar]
- 111.Shinde D.B., Kanth B.S., Srilatha M., Das B. First stereoselective total synthesis of naturally occurring (6R)-6-(4-Oxopentyl)-5,6-dihydro-2H-pyran-2-one and Its (6S)-enantiomer. Synthesis. 2012;44:469–473. [Google Scholar]
- 112.Chinna Reddy G., Balasubramanyam P., Salvanna N., Sreenivasulu Reddy T., Das B. The first stereoselective total synthesis of (Z)-cryptomoscatone D2, a natural G2 checkpoint inhibitor. Bioorg. Med. Chem. Lett. 2012;22:2415–2417. doi: 10.1016/j.bmcl.2012.02.025. [DOI] [PubMed] [Google Scholar]
- 113.Reddy P.R., Lingaiah M., Das B. Stereoselective Total Synthesis of psiAβ—A Sporogenic Psi Factor from Aspergillus nidulans 1. Synlett. 2014;25:975–976. [Google Scholar]
- 114.Raghavendar Reddy P., Das B. Stereoselective Total Synthesis of Crassalactone A, a Natural Cytotoxic Styryl Lactone. Helv. Chim. Acta. 2015;98:509–514. doi: 10.1002/hlca.201400237. [DOI] [Google Scholar]