

Abstract
Cardiac myosin binding protein-C (cMyBP-C) phosphorylation is essential for normal heart function and protects the heart from ischemia-reperfusion (I/R) injury. It is known that protein kinase-A (PKA)-mediated phosphorylation of cMyBP-C prevents I/R-dependent proteolysis, whereas dephosphorylation of cMyBP-C at PKA sites correlates with its degradation. While sites on cMyBP-C associated with phosphorylation and proteolysis co-localize, the mechanisms that link cMyBP-C phosphorylation and proteolysis during cardioprotection are not well understood. Therefore, we aimed to determine if abrogation of cMyBP-C proteolysis in association with calpain, a calcium-activated protease, confers cardioprotection during I/R injury. Calpain is activated in both human ischemic heart samples and ischemic mouse myocardium where cMyBP-C is dephosphorylated and undergoes proteolysis. Moreover, cMyBP-C is a substrate for calpain proteolysis and cleaved by calpain at residues 272-TSLAGAGRR-280, a domain termed as the calpain-target site (CTS). Cardiac-specific transgenic (Tg) mice in which the CTS motif was ablated were bred into a cMyBP-C null background. These Tg mice were conclusively shown to possess a normal basal structure and function by analysis of histology, electron microscopy, immunofluorescence microscopy, Q-space MRI of tissue architecture, echocardiography, and hemodynamics. However, the genetic ablation of the CTS motif conferred resistance to calpain-mediated proteolysis of cMyBP-C. Following I/R injury, the loss of the CTS reduced infarct size compared to non-transgenic controls. Collectively, these findings demonstrate the physiological significance of calpain-targeted cMyBP-C proteolysis and provide a rationale for studying inhibition of calpain-mediated proteolysis of cMyBP-C as a therapeutic target for cardioprotection.
Keywords: MYBPC3, cMyBP-C, calpain, cardioprotection, ischemia-reperfusion injury
1. Introduction
Ischemic heart disease is a leading global health concern that affects 5 million people annually [1–3]. Experimental models featuring cardiac ischemia-reperfusion (I/R) injury have been used to study a variety of detrimental effects, ranging from cell necrosis to fibrotic scarring [4–7], hypoxia [8, 9], apoptosis [10–12], and protein degradation. Cardiac sarcomere proteins have been identified as specific substrates for post-ischemic proteolysis [13]. Following ischemia, multiple proteins and pathways associated with the cardiac sarcomere, including cardiac myosin binding protein-C (cMyBP-C), cardiac troponin I, and myosin light chain [13–15], are subject to post-translational modification and degradation. In particular, proteolysis of cMyBP-C during ischemia leads to impaired function in cells that survive I/R injury [15, 16].
Cardiac MyBP-C is a thick and thin myofilament-interacting protein involved in regulating sarcomere structure and function in the heart [17]. Mutations that impair this protein’s function have been linked to familial hypertrophic cardiomyopathy in more than 60 million people worldwide [18–20], highlighting the clinical urgency of fully elucidating the function of cMyBP-C [17]. Previously, we showed that phosphorylation of cMyBP-C by protein kinase A (PKA) regulates myocardial function [21, 22] and confers resistance to proteolysis and protection of cardiac tissue against ischemia-reperfusion (I/R) injury [22, 23]. Cardiac MyBP-C is phosphorylated under physiological conditions by an array of signaling kinases, including PKA, protein kinase C, protein kinase D, ribosomal S6 kinase, glycogen synthase kinase 3β [24], and calcium-calmodulin-activated kinase II (CaMKII) [25]. Calcium activation of CaMKII can influence contractility by phosphorylation of cMyBP-C serine (S) 282 [25, 26]. We have shown that phosphorylation of S282 is required for the subsequent phosphorylation of cMyBP-C and that these signaling events are required for many of the regulatory functions of cMyBP-C [21, 27, 28].
Reduced phosphorylation of cMyBP-C has been observed in mouse models of I/R injury [15], pathological hypertrophy [29], heart failure [30], cytoskeletal muscle LIM protein-knockout mice [21], myocardial stunning [15], as well as human cases of atrial fibrillation [31] and heart failure [32, 33]. Such dephosphorylation is accompanied by proteolysis of cMyBP-C in association with thick filament disruption, loss of contractile regulation, and contractile dysfunction [15, 22]. Transgenic (Tg) mice expressing cMyBP-C with phosphorylatable serine residues mutated to phosphomimetic aspartic acids have reduced cMyBP-C proteolysis following I/R injury [22]. We previously reported that cMyBP-C proteolysis occurs between residues 271 and 272, located in the phosphorylatable M-domain, and results in the generation of a 40kDa N’-terminal fragment (residues 1–271) that includes key regulatory sites [34, 35]. In addition to impaired regulatory function following proteolysis, the cMyBP-C 40kDa fragment has been shown to directly impair contractility in adult rat [14] and human [36] cardiomyocytes. Furthermore, the expression of this fragment leads to heart failure in vivo [34].
We established herein the role of calpain proteases in degrading cMyBP-C and generating the 40kDa fragment in human and mouse ischemic myocardium. Moreover, we specifically characterized a mouse model that expresses a mutant cMyBP-C with the calpain target site (CTS) removed [37]. While this model showed no evidence of structural or functional impairment under normal conditions, we demonstrated that specific abrogation of calpain-mediated proteolysis of cMyBP-C in the ΔCTS model can offer a significant degree of cardioprotection during I/R injury in vivo. Under physiological conditions, these data suggest that preventing proteolysis of full-length cMyBP-C may result in the preservation of myocardial tissue and provide cardioprotection during I/R injury.
2. Methods
2.1. Human heart samples.
LV tissue samples were collected from nine non-failing human hearts that were not used for transplantation (5 males, 4 females, mean age 45 ± 3 years) and eight patients who had a myocardial infarction (7 males, 1 female, mean age 52 ± 3 years). The Institutional Review Boards at the University of Szeged, Hungary, Ohio State University, Columbus, OH, and Loyola University Chicago, Maywood, IL, USA, approved this study. Human hearts were obtained immediately (within seconds) post-explanation and flushed with a cardioplegic solution, as described previously [38, 39]. The explanted hearts were quickly (within 10–30 minutes) transferred to the laboratory in a cold (~4 °C) cardioplegic solution containing (in mM) 110 NaCl, 16 KCL, 16 MgCl2, 10 NaHCO3, and 0.5 CaCl2. Upon arrival in the laboratory, samples were flash-frozen and stored in −80 °C freezers. All hearts were procured and treated with identical protocols, solutions, and timing, irrespective of source. Human tissue experimentation conformed to the Declaration of Helsinki. Informed consent was acquired from cardiac transplant patients, while non-transplantable donor hearts were acquired in identical fashion in the operating room in collaboration with Lifeline of Ohio Organ Procurement.
2.2. Transgenic mouse models
All mouse models were maintained on the FvB/N background. The transgenic lines expressing either the cMyBP-CWT [21] or cMyBP-CΔCTS [37] transgenes were crossed to the cMyBP-C(t/t) mouse line (cMyBP-C(t/t)) [40–42] to study the effect of the transgenic protein without endogenous cMyBP-C (see discussion for further details). All procedures followed the protocol approved by the Institutional Animal Care and Use Committee of Loyola University Chicago and the University of Cincinnati and complied with the Guide for the Use and Care of Laboratory Animals published by the National Institutes of Health. All biochemical and functional assays were performed on 8- to 12-week-old mice of mixed sex with age- and sex-matched controls for each assay after preliminary experiments showed no gender differences.
2.3. Assessment of in vivo cardiac function
Echocardiography measurements were made on 8- to 10-week-old mice using a VisualSonics Vevo 2100 with an MS400 18–38 MHz transducer (FujiFilm, Toronto, Ontario) under 1 % isoflurane anesthesia. Cardiac function was measured from parasternal long axis M-mode recordings as previously described [40, 43]. Pressure-volume catheterization was performed on anesthetized mice to determine cardiac hemodynamics using a pressure-volume catheter (SPR-839; Millar Instruments Inc., Houston, TX) on 8- to 12-week-old mice, as previously described [44].
2.4. Induction of I/R injury
For mouse ischemia/reperfusion studies shown, the mice were anesthetized with a 90 mg/kg pentobarbital injection delivered I.P. and the chest was open through a left thoracotomy as previously described [45]. Cardiac I/R injury was induced by ligation of the left anterior descending coronary artery for 60 minutes of ischemia, followed by 24 hours of reperfusion in 8- to 10-week-old mice [22, 45]. As a terminal procedure, the mice were anesthetized with a second 90 mg/kg pentobarbital injection delivered I.P., followed by removal of the heart. The heart was cannulated through the aorta and the heart was perfused with 1 % triphenyl tetrazolium chloride (TTC), as previously described [45, 46]. This was followed by the re-occlusion of the coronary artery (suture was left in place) and perfusion of the heart through the aorta of a 5 % Phthalo Blue Pigment solution (Heucotech); the occlusion was released and hearts were rinsed and cut into cross-sectional slices, weighed and photographed. The total area of the section, risk and non-risk regions as delineated by TTC and blue stains were quantified for each section using ImageJ software.
2.5. Cellular and molecular analyses
Histological analysis, electron microscopy, and immunofluorescence microscopy were performed, as described previously [40, 43]. Analysis of cMyBP-C and the phosphorylation status of S273, S282, and S302 were performed by Western blot using custom-made antibodies that recognize each residue only when phosphorylated. All buffers used for isolating protein samples contained protease inhibitor (Roche, 4693159001) and phosphatase inhibitor cocktails (Sigma P5726, P0044). TaqMan primers were used to determine gene expression with probes recognizing Myh7 and Nppa normalized to Gapdh (Applied Biosystems, Mm01319006g1; Mm01255748g1; 4352339E, respectively). Probes and templates were used with iTaq Probes Master Mix (BioRad 172–5131) and quantified using a BioRad CFX96 thermocycler. Mybpc3 expression was measured using SybrGreen intercalating dye with forward 5’-ATATAGGCCGGGTCCACAA-3’ and reverse 5’-GCAACAACCACAATGGTGTC-3’ primers (208 bp amplicon) normalized to Actb amplified with forward 5’-GGCTGTATTCCCCTCCATCG-3’ and reverse 5’-CCAGTTGGTAACAATGCCATGT-3’ (154 bp amplicon) primers. All qPCR data were analyzed using the 2−ΔΔCq method.
2.6. Assessment of calpain activity
Calpain activity was assessed using the Calpain-Glo Protease Assay (Promega). In 96-well plates, 50 μl of blanks, control samples containing purified μ-calpain (Calbiochem), or test samples were mixed with 50 μl of Calpain-Glo™ reagent with 2 mM CaCl2. Plates were agitated for 30 seconds at 300–500 RPM followed by incubation at room temperature for 5–30 minutes. The plates were read using a luminometer, detecting the luciferase signal generated following cleavage of the Calpain-Glo™ reagent by calpain. To determine the effects of calpain activation on myofilament proteolysis, calpain was inhibited in vitro using 10 nM of the selective inhibitor MDL 28170 (EMD Biosciences, La Jolla, CA) in 200 mM imidazole, 20 mM L-cysteine, and 10 mM CaCl2 at 37 °C. The inhibitor was dissolved in dimethyl sulfoxide (DMSO) and added to the perfusion buffer prior to heart perfusion at a concentration of 10 μM.
2.7. Architectural analysis of the ventricular wall with generalized Q-space MRI
The theory and methods of Q-space MRI (GQI) for imaging myoarchitecture in human (in vivo) and rodent (ex vivo) models were previously described [47, 48]. Employing GQI, multiple gradient pulses were applied with varying orientations and diffusion sensitivities to evaluate signal attenuation in 3D space, employing a 7 Tesla Bruker Biospec magnet equipped with a CryoProbe (Bruker Corp., Billerica, MA). MR scanning was accomplished with a multi-shot EPI pulse sequence employing 512 gradient directions, B value of 750 s/mm2, 8 k-space segments, and a voxel size of 100 × 100 × 300 μm. The resulting GQI image was assessed to discern the orientation and magnitude of the dominant fiber populations with the 3D directionality of the fiber populations encoded to a red-green-blue scale. Intervoxel fiber tracts were constructed by employing streamline methods based on the alignment of the maximal diffusion vectors embodied in the diffusional probability distribution function (pdf). Cardiac fiber orientation was analyzed for helix angle as a function of depth of the myocardium sampled, as previously described (33). In brief, regions of interest (ROI) were placed at various depths across the ventricular wall, and helix-angle at specific locations was quantified and plotted for each heart type. The relative transmural depth (RTD) was calculated for each ROI in a sample using the ratio of the number of ROI positions from the innermost tissue wall divided by the total number of ROIs required to traverse the cardiac wall. The RTD for each ROI in a sample was calculated and displayed as a percentage of the cardiac wall distance.
2.8. Statistics
Statistical analysis was performed using GraphPad Prism 6. Data in Figures 1, 2, 4, 5, and S1 were analyzed by one-way ANOVA to determine group differences. Following ANOVA, group differences were evaluated by a Holm-Sidak post-hoc test. Significance was defined at p < 0.05. Data in Figure 7 were also analyzed with a two-tailed Student’s t-test. Statistical comparisons of transmural helix angles were performed employing one-way ANOVA on a linear regression using least mean squares.
Fig. 1.
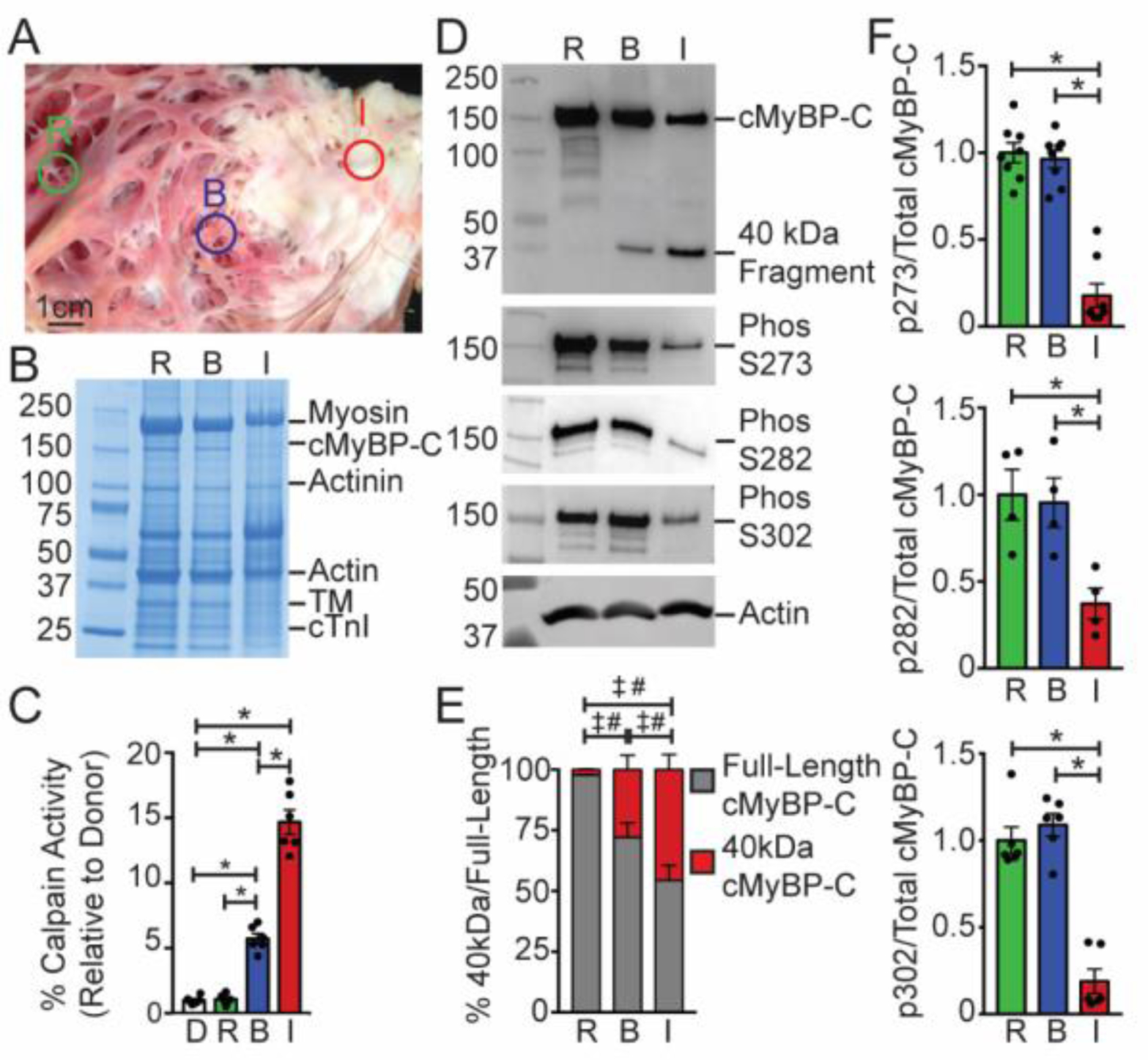
Cardiac MyBP-C is dephosphorylated and degraded in ischemic human myocardium. (A) Representative sample of human infarcted heart tissue with annotated remote [R], border zone [B], and ischemic regions [I]. (B) A representative Coomassie blue-stained SDS-PAGE of myofilament protein preparations from the remote, border, and ischemic regions of an ischemic human heart reveals clear protein degradation in the ischemic region. (C) Calpain protease activity measured in the remote, border, and ischemic regions (n = 6). (D) Western blotting with remote, border, and ischemic protein samples shows full-length and degraded 40kDa cMyBP-C (top panel) and cMyBP-C phosphorylated at residues S273, S282, and S302, with actin shown as a loading control (bottom). (E) The relative percentage of full-length and degraded 40kDa cMyBP-C in the remote, border, and ischemic regions (n = 9; # comparison of full-length cMyBP-C among tissue regions, ‡ comparison of 40kDa cMyBP-C among tissue regions using one-way ANOVA with Holm-Sidak post-hoc test). (F) Quantification of phosphorylated S273, S282, and S302 normalized against total full-length cMyBP-C (n = 8, top; 4, middle; 6, bottom). All data are mean ± SEM (* P<0.05; one-way ANOVA with Holm-Sidak post-hoc test).
Fig. 2.
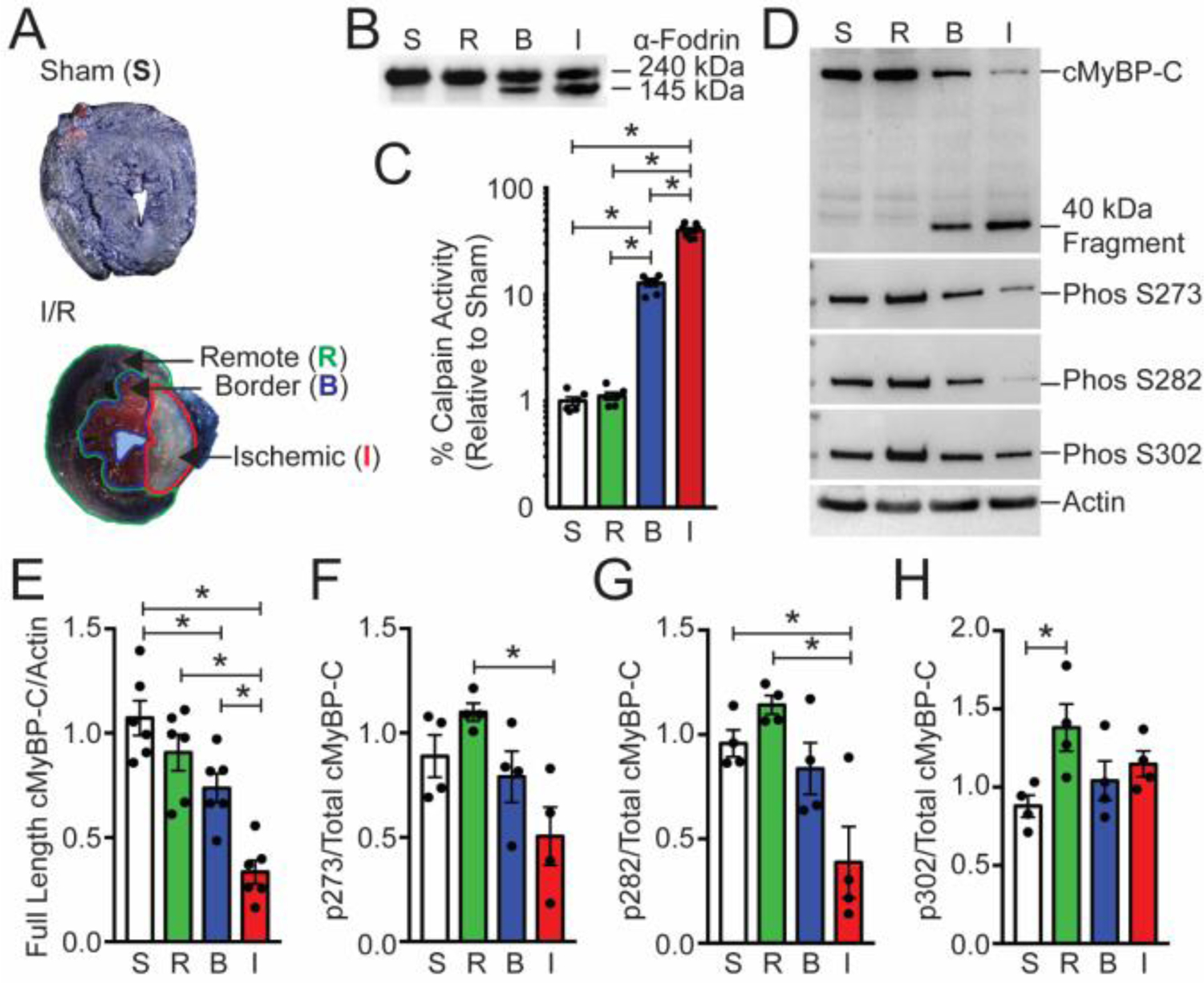
Cardiac MyBP-C is dephosphorylated and degraded in a mouse ischemia/reperfusion model. (A) TTC-staining of I/R-injured mouse hearts showed the remote regions stained blue [R], the border region stained pink [B], and the infarct [I] region in white. The sham heart [S] was stained with TTC and blue dye and shows uniform blue staining. (B) The calpain proteolysis target α-fodrin shows degradation in protein samples from the border and ischemic regions. (C) Calpain activity was determined from sham, remote, border, and infarcted regions (n = 6, S; 6, R; 7, B; 7, I). (D) The levels of cMyBP-C from these regions were determined using Western blot with the anti-cMyBP-C2–14 antibody, and the extent of cMyBP-C degradation was evaluated. The presence of the 40kDa cMyBP-C N’-terminal fragment was identified in the remote and infarct area. Sarcomeric actin was used as a loading control (lower panel). Western blot analysis of cMyBP-C was performed to determine the level of cMyBP-C phosphorylation using phospho-specific antibodies. (E) Quantification of total cMyBP-C and (F–H) phosphorylated cMyBP-C at residues S273, S282, and S302 (n = 6, total protein; 4, phospho-protein). All data are mean ± SEM (* P<0.05; one-way ANOVA with Holm-Sidak post-hoc test).
Fig. 4.
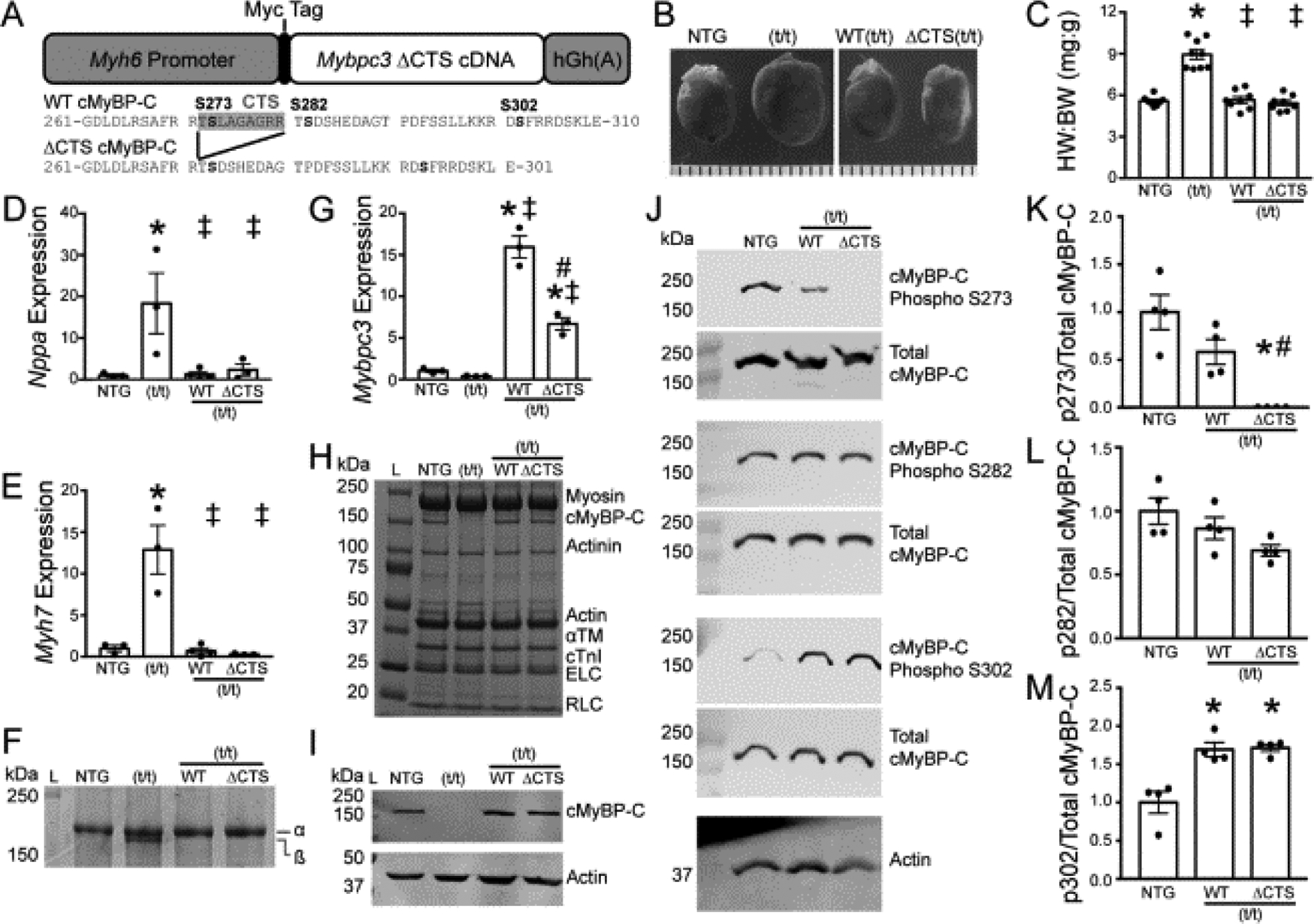
Transgenic expression of cMyBP-C with ablation of the CTS. (A) Schematic representation of the transgenic cMyBP-C construct which includes an α-myosin heavy chain promoter, N’-terminal Myc tag sequence, Mybpc3 cDNA, and an hGh polyadenylation site. The ΔCTS construct removes the region coding for CTS, amino acids 272-TSLAGAGRR-280. (B and C) HW:BW ratios showed cardiac hypertrophy in the t/t hearts, with no changes observed in WT(t/t) or ΔCTS(t/t) compared to NTG (n = 8, 8, 7, 8). (D and E) Expression of the hypertrophic markers Nppa and Myh7 normalized to Gapdh by qPCR showed a significant elevation in the t/t samples only (n = 3). (F) Separation of myosin heavy chain isoforms by SDS-PAGE to identify the hypertrophic marker β-myosin heavy chain showed an increase in the t/t group only. (G) The expression level of total Mybpc3 transcript normalized to Actb by qPCR shows transgenic overexpression in the WT(t/t) and ΔCTS(t/t) hearts. (H) Myofilament protein fractions from NTG, t/t, WT(t/t), and ΔCTS(t/t) hearts resolved by SDS-PAGE and stained with SYPRO-Ruby showed no changes in cMyBP-C stoichiometry. (I) Western blotting of cMyBP-C from whole-heart lysate reveals normal cMyBP-C stoichiometry in the WT and ΔCTS hearts compared to NTG with no detectable cMyBP-C in the t/t hearts. (J) Two-color fluorescent Western blots of total and phospho-serine cMyBP-C. (K–M) Quantification of phosphorylated cMyBP-C at residues 273, 282, and 302 (n = 4). All data are mean ± SEM (* P<0.05 vs. NTG, ‡ P<0.05 vs. t/t, # P<0.05 vs. WT(t/t); one-way ANOVA with Holm-Sidak post-hoc test).
Fig. 5.

Normal cardiac transmural fiber helical progression in CTS(t/t) hearts compared to NTG, WT(t/t), and t/t ascertained by generalized Q-space MRI (GQI) with tractography. (A) Quantification of CTS helical transmural fiber progression with individual readings (points), mean readings at each location (grey line) and the 95% confidence interval (black lines) shown as the function of relative transmural depth (RTD) in the myocardium from endocardium to epicardium (Endo:Epi) (n = 4 hearts). (B) Transmural helix angle fiber progression shown as the linear regression of mean values for the NTG, t/t, WT(t/t), and CTS(t/t) hearts shown as the function of the relative transmural depth in the myocardium. The (t/t) demonstrates significantly reduced helix angle progression compared to all other groups, which indicates a pathological architectural phenotype. (* P<0.05 compared to NTG, ‡ P < 0.05 compared to t/t; one-way ANOVA on linear regression model with least squared means)
Fig. 7.
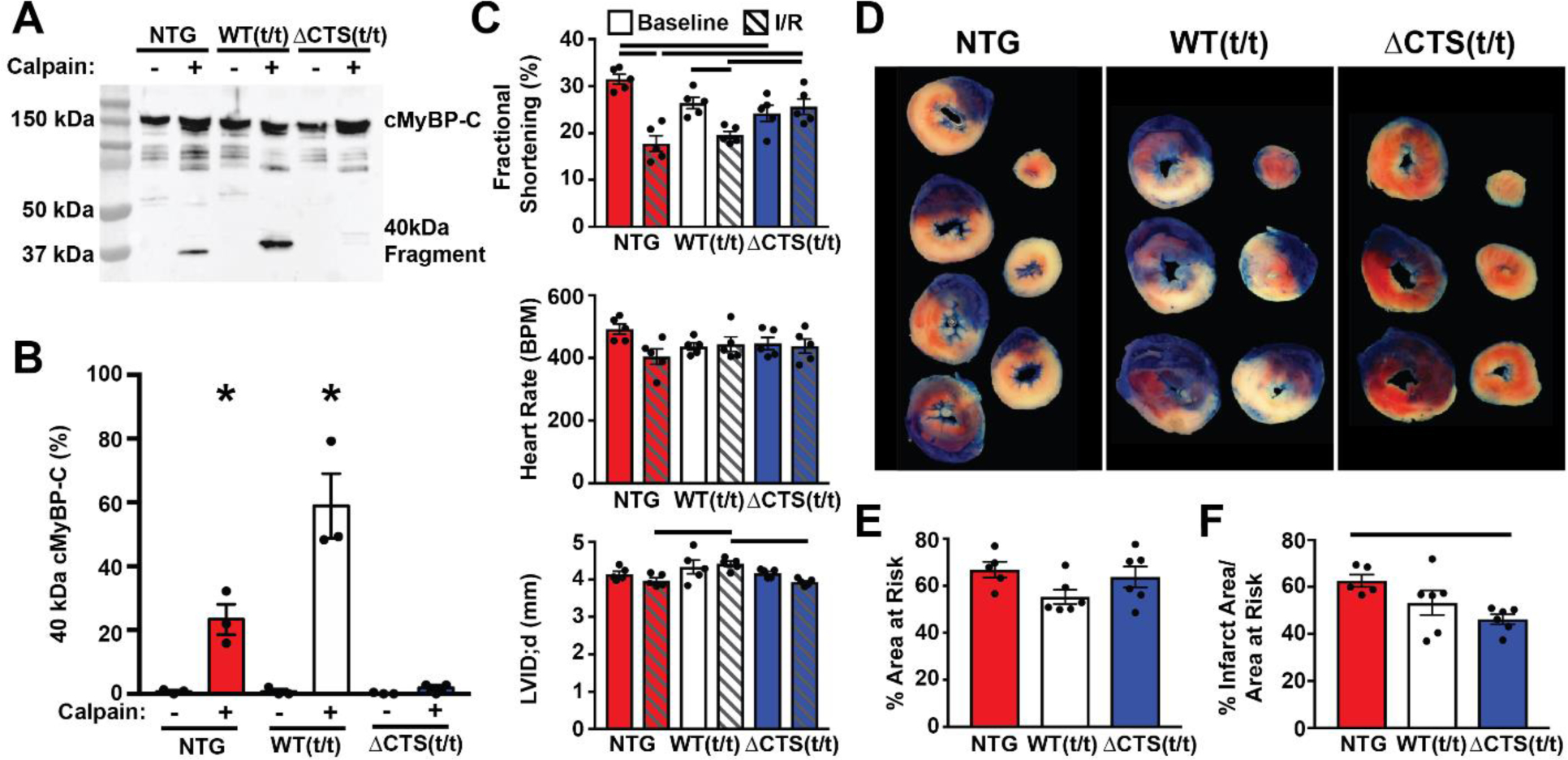
Prevention of calpain proteolysis of cMyBP-C reduces infarct size following ischemia-reperfusion injury. (A) Western blot of cMyBP-C from NTG, WT(t/t), and ΔCTS(t/t) myofilaments incubated with and without calpain in 1.25 mM calcium shows the generation of the N’-terminal 40 kDa cMyBP-C proteolysis fragment. The fragment appears at a higher weight in the WT(t/t) sample due to the presence of a Myc tag. (B) Quantification of the percent of 40kDa cMyBP-C fragment to total cMyBP-C (n = 3) (* P<0.05; two-way ANOVA). (C) Echocardiography results from NTG, WT(t/t), and ΔCTS(t/t) mice pre- and post-I/R show preserved systolic function in ΔCTS(t/t) mice but not in NTG or WT(t/t) controls. Additionally, NTG and WT(t/t) hearts showed ventricular wall thinning following I/R, whereas this was not apparent in ΔCTS(t/t) hearts (n=5) (horizontal black bars represent p<0.05 between indicated groups; two-way ANOVA). (D) Example tissue sections of NTG, WT(t/t), and ΔCTS(t/t) hearts stained with TTC and Phthalo Blue pigment solution following I/R injury. After cutting into cross sections, tissue mass, area at risk, and infarct areas were measured. Blue myocardium represents the remote area, red stained myocardium represents the area-at-risk, and the white regions are infarcted tissue. (E) Quantification of these areas show the area at risk was similar between the three groups. (F) The % infarct area was significantly reduced in ΔCTS(t/t) hearts. (n = 5, NTG; 6, WT(t/t); 6, ΔCTS(t/t)). Horizontal black bar represents p<0.05 between indicated groups; one-way ANOVA). All data are mean ± SEM.
3. Results
3.1. Calpain proteases are activated and cMyBP-C is degraded in infarcted human hearts.
To investigate the role of cMyBP-C proteolysis following ischemia, we first evaluated the active state of calpain and whether cMyBP-C is degraded in human heart samples following MI. Using samples of infarcted human hearts, we isolated ischemic tissue, border zone tissue adjacent to the infarct area, and tissue from an area remote from the ischemic site (Fig. 1A). We also used non-ischemic human heart tissue from unused donor hearts as controls. Myofilament protein extractions from the ischemic region exhibited general protein degradation compared to remote and border tissues (Fig. 1B). Using a calpain activity assay, we observed significantly higher calpain activity in the border zone and ischemic region compared to the remote area and control donor hearts (Fig. 1C). Western blotting revealed a reduction in full-length cMyBP-C and a concomitant increase in 40kDa N’-terminal cMyBP-C proteolysis in the border and infarct tissues compared to the remote area (Fig. 1D and E). Dephosphorylation of residues S273, S282, and S302 is a required step that precedes cMyBP-C proteolysis and generation of the 40kDa fragment, as previously reported [14, 33]. Using antibodies that detect the phosphorylated epitope of these residues, we observed decreased phosphorylation of cMyBP-C in human ischemic tissue compared to remote or border tissues (Fig. 1F).
3.2. I/R Injury is associated with increased calpain activity and cMyBP-C proteolysis in mice.
I/R injury or sham surgery was performed on non-transgenic (NTG) wild-type mice, and tissue from ischemic, border zone, and remote myocardium was identified with Phthalo Blue Pigment and TTC staining (Fig. 2A). Total protein lysates from these regions were assessed for full-length and degraded α-fodrin, a known substrate of calpain proteases [49]. An increase of the 145 kDa α-fodrin proteolysis product was identified in border zone and ischemic tissues compared to sham and remote samples (Fig. 2B). Calpain activity was significantly elevated in border and infarcted tissues compared to remote and sham tissue controls (Fig. 2C). Levels of total cMyBP-C, 40kDa cMyBP-C, and phosphorylated cMyBP-C were assessed by Western blot (Fig. 2 D). Full-length cMyBP-C was reduced in border and ischemic tissues compared to sham and remote samples (Fig. 2E). Levels of phosphorylated cMyBP-C residues normalized to total cMyBP-C revealed a significant reduction of phosphorylated S273 and S282 in ischemic tissue only (Fig. 2F–H).
3.3. Calpain proteases degrade cMyBP-C and generate a 40kDa fragment.
In silico analysis [50] of the cMyBP-C amino acid sequence identified a candidate CTS with proteolysis expected between residue R271 and T272 (Fig. 3A). The CTS includes residues 272-TSLAGAGRR-280 and is consistent with the previously determined sequence of the 40kDa N’-terminal fragment that contains cMyBP-C residues 1–271 [50]. To establish cMyBP-C as a calpain substrate, we incubated 20 μg of myofilament protein from wild-type mouse hearts with increasing concentrations of μ-calpain at 37 °C for 1 hour with 10 mM calcium. The results demonstrated a dose-dependent increase in the proteolysis of full-length cMyBP-C and generation of the 40kDa fragment (Fig. 3B). To further demonstrate the specific calpain-mediated degradation of cMyBP-C, we incubated myofilament proteins with 1 μg calpain in the presence and absence of 10 mM calcium and with 10 nM of the calpain inhibitor MDL 28170. In the presence of calcium, calpain could degrade cMyBP-C, whereas removal of calcium from the buffer or inclusion of MDL 28170 prevented the generation of the 40kDa cMyBP-C proteolytic fragment (Fig. 3C). In order to compare calpain-mediated proteolysis of cMyBP-C and other known myofilament calpain substrates, 1 U of μ-calpain was incubated with 20 μg of myofilament protein for different durations with 10 mM calcium. Digested proteins were resolved using 4–15 % SDS-PAGE that revealed protein degradation after 12 hours of calpain treatment (Fig. 3D). Western blot analysis using anti-cMyBP-C2–14 antibodies showed a reduction in full-length cMyBP-C at 12 hours of incubation with a corresponding rise in the 40kDa N’-terminal fragment (Fig. 3E). Cardiac troponin T and troponin I are known targets of calpain proteolysis [13, 51]. Western blotting for these troponins revealed similar proteolytic activity in both troponins and an increased amount of troponin proteolytic fragments with increasing calpain incubation time (Fig. 3F and G). These results confirm that calpain cleaves cMyBP-C into multiple fragments, principally including a 40kDa fragment.
Fig. 3.
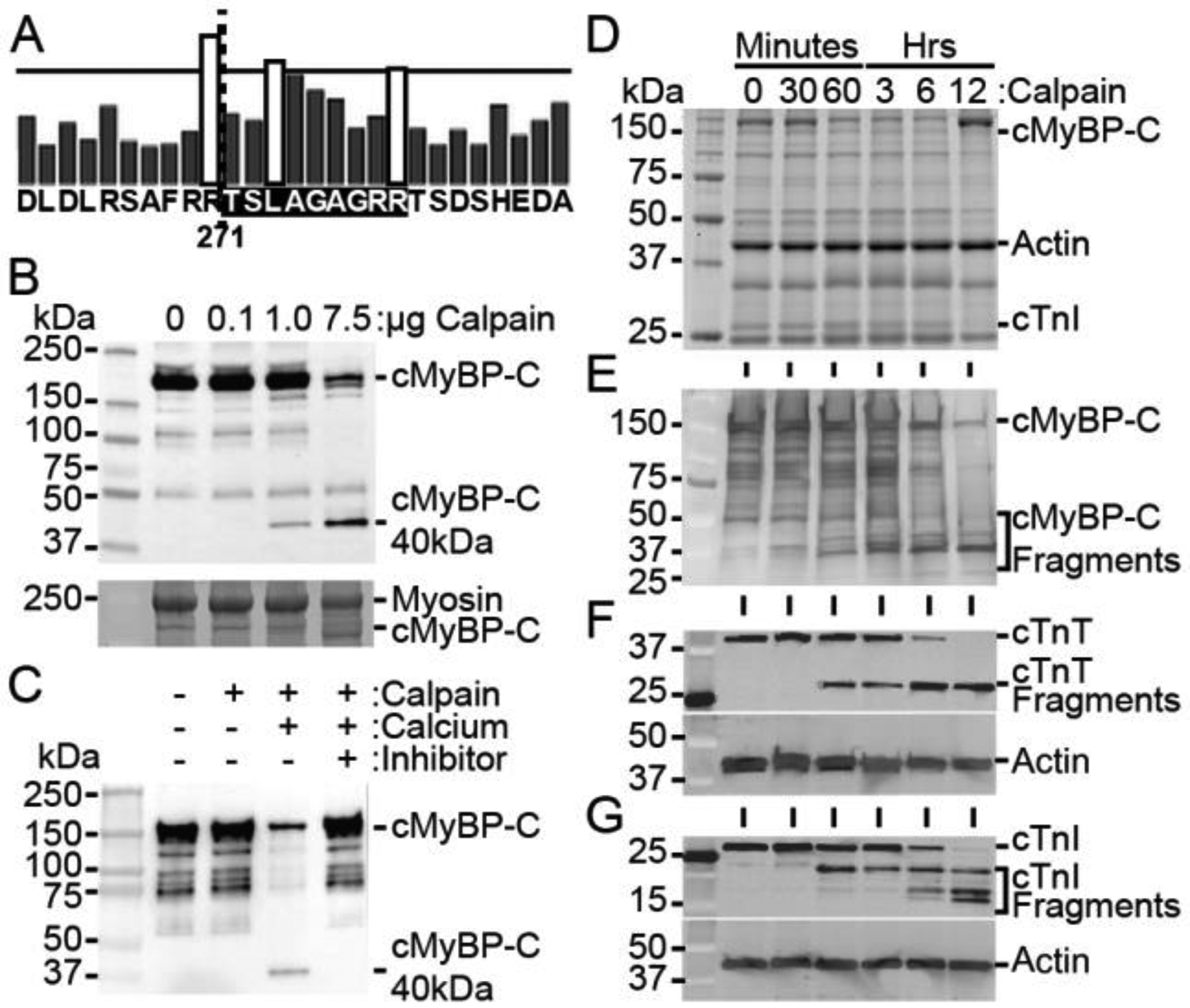
Calpain proteases degrade cMyBP-C and generate the 40kDa fragment. (A) In silico analysis of calpain proteolysis sites on cMyBP-C, with bar height representing the likelihood of cleavage, indicates R271 as a potential location of cleavage (dotted line), corresponding to the known sequence of the 40kDa fragment. The calpain-target site is highlighted in black. (B) Incubation of myofilaments with increasing concentration of calpain with 10 mM calcium for 1 hour at 37 °C shows a dose-dependent increase in cMyBP-C proteolysis and the generation of the 40kDa fragment with a SYPRO Ruby-stained SDS-PAGE loading control (bottom). (C) Proteolysis of cMyBP-C in myofilament fractions incubated with 1 μg calpain for 1 hour at 37 °C is prevented in the absence of calcium or in the presence of 10 nM of the calpain inhibitor MDL 28170. (D) Myofilament protein fractions demonstrate proteolysis at time points following incubation of 20 μg of total myofilament protein with 1 U μ-calpain with 10 mM calcium. (E) Western blotting for cMyBP-C shows reduction in full-length cMyBP-C and an increase in 40kDa cMyBP-C with longer calpain incubation. (F and G) The known calpain targets cTnT and cTnI show a reduction in full-length protein and an increase in degraded protein with increasing incubation time with calpain.
3.4. Transgenic mouse model with ablation of the CTS in cMyBP-C has normal levels and preserved phosphorylation of cMyBP-C.
Previous investigations using transgenic mice with residues S273, S282, and S302 mutated to phosphomimetic aspartic acids were resistant to proteolysis of cMyBP-C and had significant improvement of cardiac function following ischemia [22]. Conversely, dephosphorylation has been associated with increased cMyBP-C proteolysis following ischemic injury [35]. These data led us to investigate whether abrogation of calpain-mediated proteolysis of full-length cMyBP-C would be sufficient to confer the previously observed phosphorylation-mediated cardioprotection. To test this question, we used a transgenic mouse with a deletion of the cMyBP-C CTS (residues 272-TSLAGAGRR-280) [35], termed as ΔCTS [37]. This transgenic cMyBP-C ΔCTS construct includes an N’-terminal Myc tag and a Myh6 promoter for cardiac-specific expression (Fig. 4A). This construct was expressed in the cMyBP-C(t/t) mouse line that does not express endogenous cMyBP-C and normally has profound dilation and systolic dysfunction [41–43]. In order to control for transgenic overexpression of cMyBP-C, an additional transgenic mouse line was used that expresses full-length wild-type cMyBP-C with an N’-terminal Myc tag driven by the Myh6 promoter [21]. This, too, was expressed in the cMyBP-C(t/t) background and is referred to as WT(t/t). A further description of the cMyBP-C(t/t) and transgenic mouse models of cMyBP-C is included in the discussion.
Deletion of the CTS residues 272-TSLAGAGRR-280 removes S273, although the amino acid sequence immediately preceding S282 remains largely unaltered (Fig. 4A). These phosphorylation sites are critical for regulating cardiac function [27]. To assess potential cardiac hypertrophy following CTS deletion, we evaluated the heart weight-to-body weight ratio, which showed no differences among non-transgenic (NTG), WT(t/t), and ΔCTS(t/t), but with a significant increase in the t/t group (Fig. 4B and C). Expression of the hypertrophic markers Nppa and Myh7 was significantly elevated in t/t samples compared to NTG, but with no elevation observed in WT(t/t) or ΔCTS(t/t) (Fig. 4D and E). Separation of α- and β-myosin heavy chain proteins using 6 % glycerol SDS-PAGE and visualized with SYPRO Ruby staining showed an increase of the hypertrophic marker β-myosin heavy chain in the t/t group, again with no differences among NTG, WT(t/t), and ΔCTS(t/t) (Fig. 4F).
Expression of the Mybpc3 transcript shows transgenic overexpression in both WT(t/t) and ΔCTS(t/t) groups (Fig. 4G). Myofilament protein isolation from NTG, t/t, WT(t/t), and ΔCTS(t/t) hearts resolved with SDS-PAGE and visualized with SYPRO Ruby showed normal stoichiometry of cMyBP-C and other myofilament proteins between mouse lines, excepting the t/t group that does not express detectable levels of cMyBP-C (Fig. 4H). Western blotting of whole-heart protein lysates for cMyBP-C showed normal protein levels between the NTG, WT(t/t), and ΔCTS(t/t) groups, confirming that cMyBP-C stoichiometry is not altered by transgenic overexpression, whereas no cMyBP-C was detected in the (t/t) group (Fig. 4I). To evaluate how the loss of CTS affects the phosphorylation of cMyBP-C, two-color fluorescent Western blots were performed to detect total and phosphorylated cMyBP-C and S273, S282, and S302 (Fig. 4J). Quantification of phosphorylated cMyBP-C showed that the phosphorylated S273 epitope was lost in the ΔCTS(t/t) group, but with no significant differences between NTG and WT(t/t) (Fig. 4K). Phosphorylation of S282 was not different between groups, whereas S302 phosphorylation showed a significant increase in the WT(t/t) and ΔCTS(t/t) groups compared to NTG (Fig. 4L, M). Taken together, these data demonstrate that the loss of cMyBP-C CTS does not result in overt cardiac pathology or alter normal cMyBP-C protein levels. Importantly, the deletion of CTS does not alter phosphorylation of the functionally critical S282 residue [27].
3.5. Loss of the calpain target site does not alter gross cardiac morphology.
Morphological analysis of the ΔCTS(t/t) model was performed to assess the consequences of the change in the protein sequence. Myocardial sections were stained with hematoxylin & eosin or Masson’s trichrome to detect fibrosis (Fig. S1A). Staining revealed increased fibrosis in the t/t group, but no abnormalities among NTG, WT(t/t), and ΔCTS(t/t) hearts. Electron microscopy images of cardiac sarcomeres revealed normal M-line structure and proper orientation in NTG, WT(t/t), and ΔCTS(t/t) samples, while t/t sarcomeres showed disarray and improper M-line arrangement (Fig. S1B). Immunofluorescence microscopy performed on isolated cardiomyocytes showed typical cMyBP-C (green) C-zone doublet patterning between the α-actinin (red)-stained Z-disks (left panel) (Fig. S2A); no cMyBP-C was observed in the t/t samples. Anti-Myc antibodies were used to detect Myc-tagged WT and CTS transgenic protein constructs (green) (Fig. S2B). NTG and t/t groups showed no Myc staining, but WT(t/t) and ΔCTS(t/t) showed Myc doublet staining (green) between the α-actinin (red)-stained Z-disks, which is consistent with proper cMyBP-C localization. The myoarchitecture of the left ventricular wall, as represented by the transmural helix angle transition (Fig. S3), was well preserved in the ΔCTS(t/t) mouse hearts (Fig. 5A and B) compared with WT(t/t) mouse hearts and previously published myoarchitectural studies [47, 48].
3.6. ΔCTS(t/t) mice show normal cardiac function.
M-mode echocardiography did not show changes in wall thickness or left ventricular diameter in the ΔCTS(t/t) mice compared to NTG or WT(t/t) (Fig. 6A and B, Table S1). No significant change in % fractional shortening was measured among NTG, WT(t/t), and ΔCTS(t/t) in contrast to the significantly reduced systolic function and chamber dilation in the t/t group (Fig. 6B–D). Ejection fraction measured directly by left ventricular pressure-volume catheterization revealed a similar pattern with a significant reduction in systolic function only in the t/t group (Fig. 6I). The left ventricular volumes at end systole and end diastole were significantly increased only in the t/t hearts, which is consistent with the dilated phenotype associated with this mouse model (Fig. 6H). Preload recruitable stroke work was significantly decreased only in the t/t group, indicating impaired contractility (Fig. 6G). Diastolic function measured as the time to relaxation (τ-Weiss) was also significantly slower only in the t/t mice (Fig. 6J). These data, combined with normal protein levels, protein localization, and cardiac morphology, suggest that the ΔCTS(t/t) mouse is functionally indistinguishable from the NTG and WT(t/t) controls under normal conditions.
Fig. 6.

Transgenic expression of ΔCTS in the t/t background shows normal cardiac structure and function. (A–C) Representative parasternal long axis M-mode echocardiography images showed dilation and reduced contractility only in the t/t hearts (n = 6, 6, 4, 7). (D) Representative pressure-volume loops. (E) Pressure-volume catheterization derived ejection fraction, (F) end diastolic volumes, (G) contractility shown by preload recruitable stroke work (PRSW), and (H) relaxation (Tau) were not significantly different in WT(t/t) and ΔCTS(t/t) compared to NTG, whereas deficits were observed in t/t hearts across all parameters (n = 6, 6, 6, 7). All data are mean ± SEM (* P<0.05 vs. NTG, ‡ P<0.05 vs. t/t; one-way ANOVA with Holm-Sidak post-hoc test).
3.7. Ablation of the CTS of cMyBP-C prevents calpain proteolysis and is cardioprotective following I/R injury.
To directly assess whether the ΔCTS cMyBP-C is, in fact, resistant to calpain proteolysis, we incubated myofilament protein extracted from NTG, WT(t/t), and ΔCTS(t/t) hearts with μ-calpain. NTG myofilament samples treated with μ-calpain showed proteolysis of cMyBP-C with the appearance of the N’-terminal 40 kDa fragment, whereas calpain treatment of ΔCTS(t/t) samples did not produce any detectable cMyBP-C proteolysis (Fig. 7A and B). To determine if ablation of the CTS is protective following ischemic injury, NTG, WT(t/t), and ΔCTS(t/t) mice were subjected to 60 minutes of ischemia followed by 24 hours of reperfusion. Echocardiography was performed pre- and post-ischemia to assess changes in cardiac function. ΔCTS(t/t) animals showed preserved systolic function, with no significant reduction in % fractional shortening following ischemia (Fig. 7C, Table S2). Following ischemia, heart samples were stained with TTC and Phthalo Blue Pigment solution to identify the infarct area and the area at risk (Fig. 7D). Quantification of the area at risk showed that the extent of ischemia was not significantly different between NTG, WT(t/t), and ΔCTS(t/t) hearts, indicating that all groups received a similar surgical insult (Fig. 7E). However, the infarct area was significantly reduced in the ΔCTS(t/t) hearts compared to NTG, whereas WT(t/t) infarct area was not significantly reduced compared to NTG (Fig. 7F). This result was recapitulated using a separate set of I/R surgeries on NTG and ΔCTS(t/t) mice under the same conditions at a separate institution (Fig. S4).
4. Discussion
Myocardial dysfunction is a clinically important consequence of myocardial ischemia or infarction [1]. Impairments in calcium handling, contractile protein phosphorylation, and sarcomere integrity often accompany contractile dysfunction, but the subcellular mechanisms responsible for these deficits remain incompletely defined [13, 51]. Phosphorylation of cMyBP-C has a direct effect on the heart’s contractile properties, as well as sarcomere organization, and contributes to cardioprotection during I/R injury [15, 17, 22]. Cardiac MyBP-C degradation during I/R injury may contribute to sarcomere disorganization and contractile dysfunction in the recovering myocardium [14, 34].
4.1. Animal models of cMyBP-C
Many studies have used mouse models of cMyBP-C to establish the involvement of cMyBP-C in cardioprotection during ischemic injury. As in previous studies, we have used transgenic models of cMyBP-C expressed on the cMyBP-C t/t background to evaluate the function of transgenic cMyBP-C constructs without confounding effects from endogenous cMyBP-C. The cMyBP-C t/t model was engineered to have a truncating stop site inserted in exon 30 to model a human mutation with a similar truncation [42]. When the cMyBP-C t/t model was originally reported by McConnell et al. (1999), Western blot data showed a substantial amount of cMyBP-C in the t/t hearts [42]. However, in numerous subsequent reports by several research groups using this mouse line, no cMyBP-C protein has been detected by Western blot or immunofluorescence microscopy, using several high-quality antibodies [21, 43, 52]. While this discrepancy has never been explained, the possibility remains that undetectable levels of mutant cMyBP-C could cause alterations in cardiac function. This possibility appears unlikely based on the evidence that models expressing various transgenic cMyBP-C constructs on the t/t background do not share the t/t model’s pathology, despite rigorous cardiac phenotyping [21, 22, 40].
Transgenic cMyBP-C constructs have been used to elucidate the functions of phosphorylatable residues in the cMyBP-C M-domain [21, 22, 27, 53], and in these studies, transgenic overexpression of WT cMyBP-C on the t/t background has never been shown to be significantly different from NTG controls at baseline or after I/R injury. Differential phosphorylation of the three major serines, S273, S282, and S302, alter the ability of cMyBP-C to regulate cross-bridge cycling [17]. The deletion of CTS results in loss of the S273 site and alteration of residues near S282. Phosphorylation of S282 is critical for subsequent S302 phosphorylation and regulation of myofilament function [27]; therefore, evidence that S282 can still be phosphorylated after the loss of CTS is likely critical for the absence of any functional deficits in this model.
4.2. Calpain proteolysis of myofilament proteins
During cardiac ischemia, increased intracellular calcium activates calpain proteases [51, 54, 55], resulting in the degradation of myofilament proteins [56, 57]. This proteolysis regulates physiological myofilament turnover [58] and hypertrophic remodeling during cardiac stress [59]. Inactive calpains localize to the myofilament Z-disk under normal physiological conditions, emphasizing their importance in myofilament proteolysis [60]. Calpain proteolysis precedes the release of cardiac troponin fragments into the circulation following ischemic injury, and measurement of these fragments is the current gold standard for clinical diagnosis of myocardial ischemia [61, 62]. Experimental methods have long used calpains to isolate specific myofilament components and have shown that these calcium-dependent, nonlysosomal cysteine proteases can degrade cMyBP-C [28]. Observations of cMyBP-C proteolysis in ischemic tissue have suggested calpain as the protease principally responsible for generating the 40kDa cMyBP-C fragment [14, 35]. We have now identified the exact location of calpain cleavage that contributes to cMyBP-C proteolysis and that it generates the previously identified 40kDa N’-terminal fragment. Future studies will evaluate the effects of calpain proteolysis in vivo in the ΔCTS(t/t) during ischemia.
4.3. Regulation of cMyBP-C by proteolysis
Identification of this 40kDa N’-terminal fragment of cMyBP-C in ischemic myocardium was performed by using cMyBP-C N’-terminal antibodies [34, 35]. Viral expression of the 40kDa cMyBP-C fragment in adult rat cardiomyocytes and addition of the recombinant 40kDa fragment into permeabilized human cardiomyocytes have both resulted in impaired myofilament contractile function [14, 36]. In addition, the proteolysis of cMyBP-C removes the N’-terminal domains from their functional location in the myofilament where these domains normally regulate actomyosin interactions [28]. Systemic release of cMyBP-C fragments causes an acute inflammatory response that may provide further insult to the heart during reperfusion [63]. The known cytotoxic effects of cMyBP-C proteolysis and the observed reduction in infarct size in ΔCTS(t/t) mouse hearts following ischemic injury suggest that degradation of cMyBP-C may not only occur as a result of ischemic signaling events but can potentially exacerbate the progression of cell death within the ischemic myocardium [34].
The calpain proteolysis site is located within the phosphorylatable M-domain of cMyBP-C. Colocalization of these sites suggests a regulatory interaction between the phosphorylation status of cMyBP-C and its degradation. A mouse line expressing a phospho-mimetic cMyBP-C transgene in which phosphorylatable sites S273, S282, and S302 were mutated to aspartic acid mitigates cMyBP-C degradation [22]. These mice exhibit cardioprotection during I/R injury similar to that observed from ischemic preconditioning and to an extent similar to that observed in the current study. This stands in contrast to cMyBP-C proteolysis that occurs with reduced cMyBP-C phosphorylation levels following hypoxia [35]. Accordingly, we have hypothesized that phosphorylation of cMyBP-C inhibits the ability of calpain to degrade cMyBP-C within the phosphorylatable M-domain. This could explain how cMyBP-C phosphorylation prevents cMyBP-C proteolysis to confer cardioprotection following ischemia [21, 22]; although whether cMyBP-C phosphorylation prevents calpain proteolysis by altering the recognition site or sterically inhibiting the enzyme remain to be determined. While the myocardial tissue response to ischemia is multi-faceted, embodying spatially distributed patterns of apoptosis [10–12], hypoxia [8, 9], and fibrogenesis [4–7], our current findings provide evidence that calpain-mediated cMyBP-C proteolysis is a pivotal event in the injury sequence occurring in vivo.
Conclusions
We assessed whether the abrogation of calpain-directed degradation of cMyBP-C is responsible for cardioprotection by ablating the calpain-target sequence in cMyBP-C and assessing the effect on infarct size. Ablation of the CTS site in the phosphorylatable M-domain of cMyBP-C was associated with significantly reduced infarct size following I/R injury. These findings support the beneficial effect of stabilizing cMyBP-C during ischemia to preserve myocardial function in reperfused tissue. Previous efforts to explain cMyBP-C phosphorylation-mediated cardioprotection have focused on the importance of preserving cMyBP-C phosphorylation and the role of phosphorylation in altering myofilament contractility as the mechanism of cardioprotection. By demonstrating that cardioprotection occurs through the prevention of calpain-mediated cMyBP-C proteolysis, even in the absence of direct manipulation of cMyBP-C phosphorylation, we provide a rationale for a renewed investigation into calpain and the CTS as therapeutic targets and the resultant regulation of calpain proteolysis during and after ischemic injury.
Supplementary Material
Highlights.
Calpain-mediated proteolysis of cMyBP-C occurs in human and mouse ischemic injury.
In silico analysis shows that calpain targets cMyBP-C residues 272-TSLAGAGRR-280.
Loss of the calpain target site (CTS) prevents calpain-mediated cMyBP-C proteolysis.
Mice expressing cMyBP-C with no CTS have normal cardiac function.
Loss of the CTS is sufficient for cardioprotection in I/R injury.
Acknowledgements
The cMyBP-C ΔCTS mouse model was provided by Jeffrey Robbins, PhD, Cincinnati Children’s Hospital.
Source of Funding
The authors were supported by NIH National Heart, Lung, and Blood grants, including R01HL130356, R01HL105826, and K02HL114749 (to S.S.); R01DC011528 (to R.J.G.); and F32HL131304 (to D.Y.B), R01-HL131517 and R01-HL136389 (to D.D.), the American Heart Association Midwest Affiliate Research Programs, including Cardiovascular Genome-Phenome Study 15CVGPSD27020012 and Catalyst 17CCRG33671128 (to S.S.), Predoctoral Fellowship 11PRE7240022 (to D.Y.B.), Postdoctoral Fellowship 13POST14720024 (to S.G.), Postdoctoral Fellowship 13POST17220009 (to D.W.D.K.), Predoctoral Fellowship 15PRE22430028 (to T.L.L.IV.), Postdoctoral Fellowship 17POST33630095 (to J.W.M), and a British Heart Foundation Programme Grant RG/11/21/29335 (to P.K.L.).
Nonstandard Abbreviations and Acronyms:
- CaMKII
Calcium-calmodulin-activated kinase II
- CTS
Calpain-targeted site
- ΔCTS
Ablation of calpain-targeted site
- cMyBP-C
Cardiac myosin binding protein-C
- GQI
Generalized Q-space MRI
- I/R
Ischemia-reperfusion
- MHC
Myosin heavy chain
- Mybpc3
Cardiac myosin binding protein-C gene
- Myh7
Cardiac β-myosin heavy chain gene
- NTG
Non-transgenic mice
- Nppa
Atrial natriuretic factor gene
- PKA
Protein kinase A
- ROI
Region of interest
- RTD
Relative transmural depth
- TTC
Triphenyl tetrazolium chloride
- t/t
cMyBP-C null mice
- WT
Wild-type mice
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
Dr. Sadayappan provides consulting services to AstraZeneca, Merck, and Amgen unrelated to the content of this manuscript. No other disclosures are reported.
References
- [1].Finegold JA, Asaria P, Francis DP, Mortality from ischaemic heart disease by country, region, and age: statistics from World Health Organisation and United Nations, Int J Cardiol 168(2) (2013) 934–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Thygesen K, Alpert JS, Jaffe AS, Simoons ML, Chaitman BR, White HD, E.S.C.A.A.H.A.W.H.F.T.F.f.U.D.o.M.I. Joint, C. Authors/Task Force Members, Thygesen K, Alpert JS, White HD, Biomarker S, Jaffe AS, Katus HA, Apple FS, Lindahl B, Morrow DA, E.C.G. Subcommittee, Chaitman BR, Clemmensen PM, Johanson P, Hod H, Imaging S, Underwood R, Bax JJ, Bonow JJ, Pinto F, Gibbons RJ, Classification S, Fox KA, Atar D, Newby LK, Galvani M, Hamm CW, Intervention S, Uretsky BF, Steg PG, Wijns W, Bassand JP, Menasche P, Ravkilde J, Trials, S. Registries, Ohman EM, Antman EM, Wallentin LC, Armstrong PW, Simoons ML, Trials, S. Registries, Januzzi JL, Nieminen MS, Gheorghiade M, Filippatos G, Trials, S. Registries, Luepker RV, Fortmann SP, Rosamond WD, Levy D, Wood D, Trials, S. Registries, Smith SC, Hu D, Lopez-Sendon JL, Robertson RM, Weaver D, Tendera M, Bove AA, Parkhomenko AN, Vasilieva EJ, Mendis S, E.S.C.C.f.P. Guidelines, Bax JJ, Baumgartner H, Ceconi C, Dean V, Deaton C, Fagard R, Funck-Brentano C, Hasdai D, Hoes A, Kirchhof P, Knuuti J, Kolh P, McDonagh T, Moulin C, Popescu BA, Reiner Z, Sechtem U, Sirnes PA, Tendera M, Torbicki A, Vahanian A, Windecker S, Document R, Morais J, Aguiar C, Almahmeed W, Arnar DO, Barili F, Bloch KD, Bolger AF, Botker HE, Bozkurt B, Bugiardini R, Cannon C, Lemos J. de, Eberli FR, Escobar E, Hlatky M, James S, Kern KB, Moliterno DJ, Mueller C, Neskovic AN, Pieske BM, Schulman SP, Storey RF, Taubert KA, Vranckx P, Wagner DR , Third universal definition of myocardial infarction, J Am Coll Cardiol 60(16) (2012) 1581–98. [DOI] [PubMed] [Google Scholar]
- [3].Benjamin EJ, Virani SS, Callaway CW, Chamberlain AM, Chang AR, Cheng S, Chiuve SE, Cushman M, Delling FN, Deo R, de Ferranti SD, Ferguson JF, Fornage M, Gillespie C, Isasi CR, Jimenez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Lutsey PL, Mackey JS, Matchar DB, Matsushita K, Mussolino ME, Nasir K, O’Flaherty M, Palaniappan LP, Pandey A, Pandey DK, Reeves MJ, Ritchey MD, Rodriguez CJ, Roth GA, Rosamond WD, Sampson UKA, Satou GM, Shah SH, Spartano NL, Tirschwell DL, Tsao CW, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P, American E Heart Association Council on, C. Prevention Statistics, S. Stroke Statistics, Heart Disease and Stroke Statistics-2018 Update: A Report From the American Heart Association, Circulation 137(12) (2018) e67–e492. [DOI] [PubMed] [Google Scholar]
- [4].van den Borne SW, Diez J, Blankesteijn WM, Verjans J, Hofstra L, Narula J, Myocardial remodeling after infarction: the role of myofibroblasts, Nat Rev Cardiol 7(1) (2010) 30–7. [DOI] [PubMed] [Google Scholar]
- [5].Daskalopoulos EP, Janssen BJ, Blankesteijn WM, Myofibroblasts in the infarct area: concepts and challenges, Microsc Microanal 18(1) (2012) 35–49. [DOI] [PubMed] [Google Scholar]
- [6].Li AH, Liu PP, Villarreal FJ, Garcia RA, Dynamic changes in myocardial matrix and relevance to disease: translational perspectives, Circ Res 114(5) (2014) 916–27. [DOI] [PubMed] [Google Scholar]
- [7].Quinn KP, Sullivan KE, Liu Z, Ballard Z, Siokatas C, Georgakoudi I, Black LD, Optical metrics of the extracellular matrix predict compositional and mechanical changes after myocardial infarction, Sci Rep 6 (2016) 35823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Sabbah HN, Sharov VG, Goldstein S, Cell death, tissue hypoxia and the progression of heart failure, Heart Fail Rev 5(2) (2000) 131–8. [DOI] [PubMed] [Google Scholar]
- [9].Tekin D, Dursun AD, Xi L, Hypoxia inducible factor 1 (HIF-1) and cardioprotection, Acta Pharmacol Sin 31(9) (2010) 1085–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Saraste A, Pulkki K, Kallajoki M, Henriksen K, Parvinen M, Voipio-Pulkki LM, Apoptosis in human acute myocardial infarction, Circulation 95(2) (1997) 320–3. [DOI] [PubMed] [Google Scholar]
- [11].Olivetti G, Quaini F, Sala R, Lagrasta C, Corradi D, Bonacina E, Gambert SR, Cigola E, Anversa P, Acute myocardial infarction in humans is associated with activation of programmed myocyte cell death in the surviving portion of the heart, J Mol Cell Cardiol 28(9) (1996) 2005–16. [DOI] [PubMed] [Google Scholar]
- [12].Teringova E, Tousek P, Apoptosis in ischemic heart disease, J Transl Med 15(1) (2017) 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Van Eyk JE, Powers F, Law W, Larue C, Hodges RS, Solaro RJ, Breakdown and release of myofilament proteins during ischemia and ischemia/reperfusion in rat hearts: identification of degradation products and effects on the pCa-force relation, Circ Res 82(2) (1998) 261–71. [DOI] [PubMed] [Google Scholar]
- [14].Govindan S, Sarkey J, Ji X, Sundaresan NR, Gupta MP, de Tombe PP, Sadayappan S, Pathogenic properties of the N-terminal region of cardiac myosin binding protein-C in vitro, J Muscle Res Cell Motil 33(1) (2012) 17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Decker RS, Decker ML, Kulikovskaya I, Nakamura S, Lee DC, Harris K, Klocke FJ, Winegrad S, Myosin binding protein C phosphorylation, myofibril structure, and contractile function during low-flow ischemia, Circulation 111(7) (2005) 906–12. [DOI] [PubMed] [Google Scholar]
- [16].Lynch TL, Sadayappan S, Surviving the infarct: A profile of cardiac myosin binding protein-C pathogenicity, diagnostic utility, and proteomics in the ischemic myocardium, Proteomics Clin Appl 8(7–8) (2014) 569–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Barefield D, Sadayappan S, Phosphorylation and function of cardiac myosin binding protein-C in health and disease, J Mol Cell Cardiol 48(5) (2010) 866–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Watkins H, Conner D, Thierfelder L, Jarcho JA, MacRae C, McKenna WJ, Maron BJ, Seidman JG, Seidman CE, Mutations in the cardiac myosin binding protein-C gene on chromosome 11 cause familial hypertrophic cardiomyopathy, Nat Genet 11(4) (1995) 434–7. [DOI] [PubMed] [Google Scholar]
- [19].Bonne G, Carrier L, Bercovici J, Cruaud C, Richard P, Hainque B, Gautel M, Labeit S, James M, Beckmann J, Weissenbach J, Vosberg HP, Fiszman M, Komajda M, Schwartz K, Cardiac myosin binding protein-C gene splice acceptor site mutation is associated with familial hypertrophic cardiomyopathy, Nat Genet 11(4) (1995) 438–40. [DOI] [PubMed] [Google Scholar]
- [20].Dhandapany PS, Sadayappan S, Xue Y, Powell GT, Rani DS, Nallari P, Rai TS, Khullar M, Soares P, Bahl A, Tharkan JM, Vaideeswar P, Rathinavel A, Narasimhan C, Ayapati DR, Ayub Q, Mehdi SQ, Oppenheimer S, Richards MB, Price AL, Patterson N, Reich D, Singh L, Tyler-Smith C, Thangaraj K, A common MYBPC3 (cardiac myosin binding protein C) variant associated with cardiomyopathies in South Asia, Nat Genet 41(2) (2009) 187–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Sadayappan S, Gulick J, Osinska H, Martin LA, Hahn HS, Dorn GW 2nd, Klevitsky R, Seidman CE, Seidman JG, Robbins J, Cardiac myosin binding protein-C phosphorylation and cardiac function, Circ Res 97(11) (2005) 1156–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Sadayappan S, Osinska H, Klevitsky R, Lorenz JN, Sargent M, Molkentin JD, Seidman CE, Seidman JG, Robbins J, Cardiac myosin binding protein-C phosphorylation is cardioprotective, Proc Natl Acad Sci U S A 103(45) (2006) 16918–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Sanada S, Asanuma H, Tsukamoto O, Minamino T, Node K, Takashima S, Fukushima T, Ogai A, Shinozaki Y, Fujita M, Hirata A, Okuda H, Shimokawa H, Tomoike H, Hori M, Kitakaze M, Protein kinase A as another mediator of ischemic preconditioning independent of protein kinase C, Circulation 110(1) (2004) 51–7. [DOI] [PubMed] [Google Scholar]
- [24].Kuster DW, Sequeira V, Najafi A, Boontje NM, Wijnker PJ, Witjas-Paalberends ER, Marston SB, Dos Remedios CG, Carrier L, Demmers JA, Redwood C, Sadayappan S, van der Velden J, GSK3beta phosphorylates newly identified site in the proline-alanine-rich region of cardiac myosin-binding protein C and alters cross-bridge cycling kinetics in human: short communication, Circ Res 112(4) (2013) 633–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Gautel M, Zuffardi O, Freiburg A, Labeit S, Phosphorylation switches specific for the cardiac isoform of myosin binding protein-C: a modulator of cardiac contraction?, Embo J 14(9) (1995) 1952–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Hartzell HC, Glass DB, Phosphorylation of purified cardiac muscle C-protein by purified cAMP-dependent and endogenous Ca2+-calmodulin-dependent protein kinases, J Biol Chem 259(24) (1984) 15587–96. [PubMed] [Google Scholar]
- [27].Sadayappan S, Gulick J, Osinska H, Barefield D, Cuello F, Avkiran M, Lasko VM, Lorenz JN, Maillet M, Martin JL, Brown JH, Bers DM, Molkentin JD, James J, Robbins J, A critical function for Ser-282 in cardiac myosin binding protein-C phosphorylation and cardiac function, Circ Res 109(2) (2011) 141–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Previs MJ, Beck Previs S, Gulick J, Robbins J, Warshaw DM, Molecular mechanics of cardiac myosin-binding protein C in native thick filaments, Science 337(6099) (2012) 1215–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].van Dijk SJ, Dooijes D, dos Remedios C, Michels M, Lamers JM, Winegrad S, Schlossarek S, Carrier L, ten Cate FJ, Stienen GJ, van der Velden J, Cardiac myosin-binding protein C mutations and hypertrophic cardiomyopathy: haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction, Circulation 119(11) (2009) 1473–83. [DOI] [PubMed] [Google Scholar]
- [30].El-Armouche A, Pohlmann L, Schlossarek S, Starbatty J, Yeh YH, Nattel S, Dobrev D, Eschenhagen T, Carrier L, Decreased phosphorylation levels of cardiac myosin-binding protein-C in human and experimental heart failure, J Mol Cell Cardiol 43(2) (2007) 223–9. [DOI] [PubMed] [Google Scholar]
- [31].El-Armouche A, Boknik P, Eschenhagen T, Carrier L, Knaut M, Ravens U, Dobrev D, Molecular determinants of altered Ca2+ handling in human chronic atrial fibrillation, Circulation 114(7) (2006) 670–80. [DOI] [PubMed] [Google Scholar]
- [32].Jacques AM, Copeland O, Messer AE, Gallon CE, King K, McKenna WJ, Tsang VT, Marston SB, Myosin binding protein C phosphorylation in normal, hypertrophic and failing human heart muscle, J Mol Cell Cardiol 45(2) (2008) 209–16. [DOI] [PubMed] [Google Scholar]
- [33].Copeland O, Sadayappan S, Messer AE, Steinen GJ, van der Velden J, Marston SB, Analysis of cardiac myosin binding protein-C phosphorylation in human heart muscle, J Mol Cell Cardiol 49(6) (2010) 1003–11. [DOI] [PubMed] [Google Scholar]
- [34].Razzaque MA, Gupta M, Osinska H, Gulick J, Blaxall BC, Robbins J, An endogenously produced fragment of cardiac myosin-binding protein C is pathogenic and can lead to heart failure, Circ Res 113(5) (2013) 553–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Govindan S, McElligott A, Muthusamy S, Nair N, Barefield D, Martin JL, Gongora E, Greis KD, Luther PK, Winegrad S, Henderson KK, Sadayappan S, Cardiac myosin binding protein-C is a potential diagnostic biomarker for myocardial infarction, J Mol Cell Cardiol 52(1) (2012) 154–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Witayavanitkul N, Ait Mou Y, Kuster DW, Khairallah RJ, Sarkey J, Govindan S, Chen X, Ge Y, Rajan S, Wieczorek DF, Irving T, Westfall MV, de Tombe PP, Sadayappan S, Myocardial infarction-induced N-terminal fragment of cardiac myosin-binding protein C (cMyBP-C) impairs myofilament function in human myocardium, J Biol Chem 289(13) (2014) 8818–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Yang Q, Hewett TE, Klevitsky R, Sanbe A, Wang X, Robbins J, PKA-dependent phosphorylation of cardiac myosin binding protein C in transgenic mice, Cardiovasc Res 51(1) (2001) 80–8. [DOI] [PubMed] [Google Scholar]
- [38].Milani-Nejad N, Canan BD, Elnakish MT, Davis JP, Chung JH, Fedorov VV, Binkley PF, Higgins RS, Kilic A, Mohler PJ, Janssen PM, The Frank-Starling mechanism involves deceleration of cross-bridge kinetics and is preserved in failing human right ventricular myocardium, Am J Physiol Heart Circ Physiol 309(12) (2015) H2077–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Elnakish MT, Canan BD, Kilic A, Mohler PJ, Janssen PM, Effects of zacopride, a moderate IK1 channel agonist, on triggered arrhythmia and contractility in human ventricular myocardium, Pharmacol Res 115 (2017) 309–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Barefield D, Kumar M, Gorham J, Seidman JG, Seidman CE, de Tombe PP, Sadayappan S, Haploinsufficiency of MYBPC3 exacerbates the development of hypertrophic cardiomyopathy in heterozygous mice, J Mol Cell Cardiol 79 (2015) 234–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].McConnell BK, Fatkin D, Semsarian C, Jones KA, Georgakopoulos D, Maguire CT, Healey MJ, Mudd JO, Moskowitz IP, Conner DA, Giewat M, Wakimoto H, Berul CI, Schoen FJ, Kass DA, Seidman CE, Seidman JG, Comparison of two murine models of familial hypertrophic cardiomyopathy, Circ Res 88(4) (2001) 383–9. [DOI] [PubMed] [Google Scholar]
- [42].McConnell BK, Jones KA, Fatkin D, Arroyo LH, Lee RT, Aristizabal O, Turnbull DH, Georgakopoulos D, Kass D, Bond M, Niimura H, Schoen FJ, Conner D, Fischman DA, Seidman CE, Seidman JG, Dilated cardiomyopathy in homozygous myosin-binding protein-C mutant mice, J Clin Invest 104(12) (1999) 1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Barefield D, Kumar M, de Tombe PP, Sadayappan S, Contractile dysfunction in a mouse model expressing a heterozygous MYBPC3 mutation associated with hypertrophic cardiomyopathy, Am J Physiol Heart Circ Physiol 306(6) (2014) H807–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Nagayama T, Takimoto E, Sadayappan S, Mudd JO, Seidman JG, Robbins J, Kass DA, Control of in vivo left ventricular contraction/relaxation kinetics by myosin binding protein C: protein kinase A phosphorylation dependent and independent regulation, Circulation 116(21) (2007) 2399–408. [DOI] [PubMed] [Google Scholar]
- [45].Ren X, Wang Y, Jones WK, TNF-alpha is required for late ischemic preconditioning but not for remote preconditioning of trauma, J Surg Res 121(1) (2004) 120–9. [DOI] [PubMed] [Google Scholar]
- [46].Fishbein MC, Meerbaum S, Rit J, Lando U, Kanmatsuse K, Mercier JC, Corday E, Ganz W, Early phase acute myocardial infarct size quantification: validation of the triphenyl tetrazolium chloride tissue enzyme staining technique, Am Heart J 101(5) (1981) 593–600. [DOI] [PubMed] [Google Scholar]
- [47].Hoffman MP, Taylor EN, Aninwene GE 2nd, Sadayappan S, Gilbert RJ , Assessing the multiscale architecture of muscular tissue with Q-space magnetic resonance imaging: Review, Microsc Res Tech (2016). [DOI] [PubMed] [Google Scholar]
- [48].Taylor EN, Hoffman MP, Barefield DY, Aninwene GE 2nd, Abrishamchi AD, Lynch T.L.t., Govindan S, Osinska H, Robbins J, Sadayappan S, Gilbert RJ, Alterations in Multi-Scale Cardiac Architecture in Association With Phosphorylation of Myosin Binding Protein-C, J Am Heart Assoc 5(3) (2016) e002836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Fukuda S, Harada K, Kunimatsu M, Sakabe T, Yoshida K, Postischemic reperfusion induces alpha-fodrin proteolysis by m-calpain in the synaptosome and nucleus in rat brain, J Neurochem 70(6) (1998) 2526–32. [DOI] [PubMed] [Google Scholar]
- [50].DuVerle DA, Ono Y, Sorimachi H, Mamitsuka H, Calpain cleavage prediction using multiple kernel learning, PLoS One 6(5) (2011) e19035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Kositprapa C, Zhang B, Berger S, Canty JM Jr., Lee TC, Calpain-mediated proteolytic cleavage of troponin I induced by hypoxia or metabolic inhibition in cultured neonatal cardiomyocytes, Mol Cell Biochem 214(1–2) (2000) 47–55. [DOI] [PubMed] [Google Scholar]
- [52].Palmer BM, McConnell BK, Li GH, Seidman CE, Seidman JG, Irving TC, Alpert NR, Maughan DW, Reduced cross-bridge dependent stiffness of skinned myocardium from mice lacking cardiac myosin binding protein-C, Mol Cell Biochem 263(1) (2004) 73–80. [DOI] [PubMed] [Google Scholar]
- [53].Sadayappan S, Gulick J, Klevitsky R, Lorenz JN, Sargent M, Molkentin JD, Robbins J, Cardiac myosin binding protein-C phosphorylation in a {beta}-myosin heavy chain background, Circulation 119(9) (2009) 1253–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Gao WD, Atar D, Liu Y, Perez NG, Murphy AM, Marban E, Role of troponin I proteolysis in the pathogenesis of stunned myocardium, Circ Res 80(3) (1997) 393–9. [PubMed] [Google Scholar]
- [55].Inserte J, Hernando V, Garcia-Dorado D, Contribution of calpains to myocardial ischaemia/reperfusion injury, Cardiovasc Res 96(1) (2012) 23–31. [DOI] [PubMed] [Google Scholar]
- [56].Neti G, Novak SM, Thompson VF, Goll DE, Properties of easily releasable myofilaments: are they the first step in myofibrillar protein turnover?, Am J Physiol Cell Physiol 296(6) (2009) C1383–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Martin-Garrido A, Biesiadecki BJ, Salhi HE, Shaifta Y, Dos Remedios CG, Ayaz-Guner S, Cai W, Ge Y, Avkiran M, Kentish JC, Monophosphorylation of cardiac troponin-I at Ser-23/24 is sufficient to regulate cardiac myofibrillar Ca(2+) sensitivity and calpain-induced proteolysis, J Biol Chem 293(22) (2018) 8588–8599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Galvez AS, Diwan A, Odley AM, Hahn HS, Osinska H, Melendez JG, Robbins J, Lynch RA, Marreez Y, Dorn GW, 2nd, Cardiomyocyte degeneration with calpain deficiency reveals a critical role in protein homeostasis, Circ Res 100(7) (2007) 1071–8. [DOI] [PubMed] [Google Scholar]
- [59].Patterson C, Portbury AL, Schisler JC, Willis MS, Tear me down: role of calpain in the development of cardiac ventricular hypertrophy, Circ Res 109(4) (2011) 453–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Kumamoto T, Kleese WC, Cong JY, Goll DE, Pierce PR, Allen RE, Localization of the Ca(2+)-dependent proteinases and their inhibitor in normal, fasted, and denervated rat skeletal muscle, Anat Rec 232(1) (1992) 60–77. [DOI] [PubMed] [Google Scholar]
- [61].Katus HA, Remppis A, Looser S, Hallermeier K, Scheffold T, Kubler W, Enzyme linked immuno assay of cardiac troponin T for the detection of acute myocardial infarction in patients, J Mol Cell Cardiol 21(12) (1989) 1349–53. [DOI] [PubMed] [Google Scholar]
- [62].Reichlin T, Hochholzer W, Bassetti S, Steuer S, Stelzig C, Hartwiger S, Biedert S, Schaub N, Buerge C, Potocki M, Noveanu M, Breidthardt T, Twerenbold R, Winkler K, Bingisser R, Mueller C, Early diagnosis of myocardial infarction with sensitive cardiac troponin assays, N Engl J Med 361(9) (2009) 858–67. [DOI] [PubMed] [Google Scholar]
- [63].Lipps C, Nguyen JH, Pyttel L, T.L.t. Lynch C. Liebetrau G. Aleshcheva S. Voss O. Dorr HM. Nef H. Mollmann CW. Hamm S. Sadayappan C. Troidl, N-terminal fragment of cardiac myosin binding protein-C triggers pro-inflammatory responses in vitro, J Mol Cell Cardiol 99 (2016) 47–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.