

Abstract
The Cancer Genome Atlas (TCGA) and other large-scale genomic data pipelines have been integral to the current understanding of the molecular events underlying renal cell carcinoma (RCC). These data networks have focused mostly on primary RCC which often demonstrates indolent behavior. However, metastatic disease is the major cause of mortality associated with RCC and data sets examining metastatic tumors are sparse. Therefore, a more comprehensive analysis of gene expression and DNA methylome profiling of metastatic RCC in addition to primary RCC and normal kidney was performed. Integrative analysis of the methylome and transcriptome identified over 30 RCC specific genes whose mRNA expression inversely correlated with promoter methylation, including several known targets of hypoxia inducible factors (HIFs). Notably, genes encoding several metabolism-related proteins were identified as differentially regulated via methylation including hexokinase 2 (HK2), aldolase C (ALDOC), stearoyl-CoA desaturase (SCD), and estrogen-related receptor-γ(ESRRG) which has a known role in the regulation of nuclear-encoded mitochondrial metabolism genes. Several gene expression changes could portend prognosis in the TCGA cohort. Mechanistically, ESRRG loss occurs via DNA methylation and histone repressive silencing mediated by the polycomb repressor complex 2 (PRC2). Restoration of ESRRG in RCC lines suppresses migratory and invasive phenotypes independently of its canonical role in mitochondrial metabolism.
Keywords: Renal cancer, epigenetics, HIF, ESRRG, methylation
Introduction
Insight into the biologic basis of renal cancer has greatly benefitted from large scale data sets such as The Cancer Genome Atlas (TCGA). These data have confirmed the high prevalence of tumor initiating events such as alterations in the VHL tumor suppressor gene which lead to stabilization of hypoxia inducible factors (HIF-1α and HIF-2α) (1–3). Analyses of these datasets have also indicated the importance of epigenetics to RCC malignancy, including the alteration of PBRM1, which encodes a component of the SWI/SNF chromatin remodeling complex as well as BAP1, a gene encoding a known histone deubiquitinase (1, 4). While TCGA analysis has been invaluable to our understanding of primary ccRCC, far less is known about metastatic ccRCC.
Development of metastasis is the major cause of mortality associated with ccRCC. Patients with metastatic ccRCC have a poor prognosis, and remain largely incurable despite recent advances with combined T-cell checkpoint blockade (5–8). To date, studies of metastatic ccRCC using patient-derived samples has been limited. One recent pan-cancer analysis of metastasis included RCC; however, only 8 of the 500 tumor samples were RCC (9). Two recent studies focused on pharmacogenes and kinases with altered expression in metastatic RCC (10, 11). However, integrative studies performed in an unbiased manner are lacking.
DNA methylation plays a significant role in the regulation of gene expression. Probably the most well characterized alteration is gene promoter methylation that silences the corresponding gene expression. In RCC, VHL inactivation through promoter methylation has been identified (12). While other genes have been described as methylated in RCC through cell line based studies, a recent analysis indicated that the methylomes of cultured RCC cells vastly differs from that of patient samples (11). The TCGA analysis of ccRCC (referred to as KIRC) demonstrated that increased promoter methylation of gene correlated with higher stage and higher grade tumors (1). Recent studies by Sato et al. performed an analysis of over 100 primary ccRCC and identified that a DNA hypermethylator phenotype defined a subgroup of patients with worsened overall survival (13). Although the TCGA analysis provides large-scale data sets, data do not include metastatic tumor tissues. Since gene signatures related to DNA methylation might differ between primary and metastatic RCC, we undertook a combined analysis of the transcriptome and methylome of metastatic as well as primary ccRCC and normal kidney to gain insight into the biological basis of aggressive ccRCC.
We performed concurrent transcriptomic and methylome analyses via array-based methodologies followed by an integrative analysis of these two data sets. These data represent the first such combined analysis for ccRCC that includes normal kidney, primary RCC and metastatic RCC tissues. Our analysis indicates a role for promoter methylation, both hyper- and hypomethylation, in the regulation of genes involved in cellular metabolism. We identified several genes silenced via promoter methylation and whose loss was correlated with worsened prognosis through subsequent analysis of the TCGA cohort. Notably, our data demonstrate a role for promoter hypomethylation in the amplification of HIF signaling in aggressive RCC. Our data also implicate novel genes whose deregulation is associated with tumor progression and invasive phenotypes.
Materials and Methods
Patient Samples
Samples for array (normal=9, primary=9, metastasis n=26) based studies were acquired from the NCI Cooperative Human Tissue Network (CHTN) and were not patient-matched. Patient-matched samples (n=11/group) used for validation RT-qPCR studies were obtained from patients undergoing clinically indicated procedures at UAB Hospital and have been previously described (14). Further detailed information of these patient-matched samples is provided in Supplemental Table 4. All studies were performed in accord with the institutional IRB.
Cell Culture
RCC cell lines were purchased from ATCC except RXF-393 (NCI) and RCC4 (P. Ratcliffe, Oxford). RCC cells RCC4, RCC10, A498, 786–0, and HK2 proximal tubular epithelial cells were grown in DMEM with 10% fetal bovine serum with penicillin-streptomycin (100 U/ml) in 5% CO2 at 37˚C. CAKI-1 cells were cultured in MEM medium containing 10% FBS with antibiotics. Cells were used within 10 passages of the initial stock with periodic testing for Mycoplasma infection. No further characterization was performed.
GSK126 treatment
CAKI-1 cells were seeded on 6 well plates and treated with GSK126 (ThermoFisher Scientific; Waltham, MA) at the indicated dose for 72 hr.
Plasmids and virus infections
Sequence-verified shRNA lentiviral plasmid vectors for the human EZH2 gene and a nonspecific shRNA control (shCTRL) constructs have been previously described (15). Lentiviral transduction was performed as previously described (16). PPARGC1A and ESRRG cDNAs were transduced in RCC cells using adenovirus mediated gene transfer (Vector Biolabs, Malvern PA) for 48 hrs.
Gene Expression Profiling
Gene expression analysis was performed using the human HT-12 BeadChip and iScan system from Illumina, Inc. (San Diego, CA). Total RNA was converted to cDNA by reverse transcription, followed by second-strand synthesis to generate double-stranded cDNA. After purification, the cDNA was converted to biotin-labeled cRNA, hybridized to a human HT-12 BeadChip and stained with strepavidin-Cy3 for visualization. The human HT-12 BeadChips contain sequences representing approximately 46,000 curated and putative genes and ESTs. Quality standards for hybridization, labeling, staining, background signal, and basal level of housekeeping gene expression for each chip were verified. After scanning the probe array, the resulting image was analyzed using the GenomeStudio software (Illumina, Inc., San Diego, CA). Raw data obtained from Illumina’s beadstudio were preprocessed using a bioconductor package “beadarray” (17) in the open source software R v3.2.2 [https://cran.r-project.org/]. Raw signal values were quantile normalized and log2 transformed. Probes with no-match or of poor quality were then filtered out, leaving 34,476 probes for further analysis. Differential expression analysis was performed using the limma package (18). Only probes with an absolute fold change of two or more and a p-value of 0.05 or less were considered differentially expressed. Probes were annotated to their respective genes using the Bioconductor annotation package “illuminaHumanv4.db” (19). The gene expression profiling data has been deposited at Gene Expression Omnibus (GEO, #GSE105288).
Whole Genome Methylation Analysis
Genomic DNA was isolated from 25 mg of fresh frozen tissues using the Invitrogen PureLink Genomic DNA kit (ThermoFisher Scientific; Waltham, MA). DNA samples were bisulfite converted with the Zymo EZ DNA Methylation kit (Zymo Research, Irvine, CA, USA). Genome-wide methylation profiling was accomplished with the Illumina Infinium HumanMethylation450 BeadChip kit, which interrogates DNA methylation at 485,755 CpG sites. The BeadChip arrays were processed according to the manufacturer’s instructions and scanned using an Illumina iScan system (Illumina; San Diego, CA). The Illumina GenomeStudio Methylation Module software was used to analyze the BeadChip image data. The raw intensity data were loaded to a biocondutor package “minfi” (20). The raw data were normalized using the subset-quantile within array normalization (SWAN) method and probes with a detection p-value >= 0.01 in at least one sample were excluded from further analysis. Using the getBeta function of minfi, beta values were calculated for each probe and M-values were calculated using the formula: log2 ([methylated_probe+100]/[unmethylated_probe+100]). Differentially methylated probes were identified using M-values determined via the “limma” package (18). Probes with an absolute fold change of 2 or more and p-value of <0.05 were chosen as differentially methylated.
In silico validation
Using the Oncomine database (21), the expression level of 35 RCC associated genes was checked in public RCC microarray data sets (22–24).
In addition, UALCAN (25) was used to study the expression level of these genes in 72 normal kidney and 533 primary RCC samples and their expression effect on patient survival. UALCAN uses “level 3 RNA-seq data” from TCGA [https://tcga-data.nci.nih.gov/].
The promoter CpG methylation status of 35 ccRCC associated genes was validated by KIRC TCGA 450 K methylation data. The raw data for 160 kidney normal and 325 KIRC primary tumors were downloaded from the TCGA portal and processed using the same method as above.
Quantitative RT-PCR
Total RNA from tissues was isolated using the RNeasy Mini Kit (Qiagen). Total RNA was extracted from cultured cells using Trizol reagent (Ambion). cDNA was generated using the high capacity cDNA reverse transcription kit (Applied Biosystems). Real-time PCR (RT-PCR) was performed using TaqMan gene expression reagents mixed with TaqMan primers and indicated cDNAs in QuantStudio™ 6K Flex Real-Time PCR System (Applied Biosystem). Target gene mRNA expression was normalized to either ribosomal protein (RPLPO) or TATA binding protein (TBP). Normalized CT values were calculated via the ΔΔCt analysis. The following probes were used: ALDOA (Hs00605108_g1), ALDOB (Hs01551887_m1), ALDOC(Hs00902799_g1), ESRRA (Hs01067166_g1), ESRRB (Hs01584024_m1),ESRRG (Hs00976243_m1), ACO2(Hs00426616_g1), OGDH (Hs01081865_m1), SUCLG1 (Hs00388749_m1), MDH2 (Hs00938918_m1), TBP (Hs00427620_m1), RPLP0 (Hs99999902_m1).
Immunoblotting analysis
Immunoblotting was performed as described previously (16). Antibodies used included EZH2 (Cell signaling, # 5246), ESRRG (Proteintech, # 14017–1-AP), α-tubulin (Sigma-Aldrich, # T5168), β-actin (Abcam, # Ab8226).
Proteomics analysis
RCC cells and human kidney tissues (n=5/ group) were lysed in T-per lysis buffer with protease inhibitor. Following the BCA protein assay, 40 µg of each lysate was separated onto a 10% SDS-PAGE gels and the band around the 40 kDa was excised and digested with trypsin overnight prior to LC-MS analysis.
Wound healing and migration assay
RCC cells were seeded in 6-well plates in growth medium and transfected with the indicated adenovirus for 48 h. The cells were mechanically disrupted with a pipette tip, and cell migration was observed with an EVOS™ FL imaging system (Invitrogen). For the transwell migration assay, RCC cells were seeded in serum-free medium onto the upper chamber of the transwell (Corning) for 24 h. The migrated cells were fixed and quantified by crystal violet staining.
Matrigel-based invasion assay
Cells were infected with the indicated adenovirus before seeding in the upper chambers of BD BioCoat Matrigel Matrix (Corning). After 16 h, cells on the upper chamber (non-invading cells) were removed with a cotton swab, whereas cells on the lower side of the chamber (invading cells) were stained with Diff-Quik Stain kit (Siemens). Invading cells were observed and counted under a light microscope.
Methylated DNA immunoprecipitation
Genomic DNA was extracted from RCC cells and human kidney tissues using the Wizard genomic DNA isolation kit (Promega). DNA immunoprecipitations were performed with 2 µg of genomic DNA and either 5-methylcytosine (5-mC) rabbit mAb or rabbit mAb IgG using SimpleDIP™ methylated DNA IP (MeDIP) kit (Cell Signaling). The enriched DNA was quantified by real-time PCR (Applied Biosystem). The human ESRRG primers were specifically designed for heavily methylated regions in the ESRRG promoter (5’-GGAGTTGCTGCAGTTTCAAAG-3’; 5’-CGTTCCGAGTCAGCTCCTA-3’). The enrichment of immunoprecipitated DNA was represented as signal relative to the total 10% of input DNA.
Results
Methylome/ transcriptome analysis of normal kidney, primary tumor, and metastasis.
In order to obtain novel insight into the biology of aggressive kidney cancer, we obtained normal kidney, primary ccRCC, and metastatic ccRCC tumor deposits from the Cooperative Human Tissue Network (CHTN). DNA and RNA were extracted from tissue and analyzed via methylome and transcriptomic array analysis, respectively. Sites of metastasis for DNA/RNA array analysis are from sites including adrenal, pancreas, lung, and liver (Supplemental Table 1). Both primary and metastatic tumor were included to assess if alterations in the transcriptome/methylome observed were part of a continuum that paralleled tumor progression. Principal component analysis (PCA) of transcriptomic data demonstrated that metastatic tumor samples clustered together (Supplemental Figure 1S). The pipeline for our integrative analysis is demonstrated in Figure 1. In summary, we first identified genes demonstrating RCC-specific expression alterations. In tandem, we identified CpG sites (i.e., probes) with differential methylation levels in RCC (either primary and/or metastatic tumor). We focused specifically on CpG sites in the expected region of the gene promoter which included probes assigned to the 200 base pairs of the transcription start site (TSS200), within 1500 base of the TSS (TSS1500), 5’ untranslated regions (5’UTR), and the first exon. These analyses identified 35 RCC specific genes changes demonstrating a statistically significant inverse correlation between mRNA expression status and DNA promoter methylation status (Figure 2A). In general, both primary and metastatic tumor demonstrated similar directionality with respect to gene expression changes and methylation. However, metastatic tumor often demonstrated more prominent changes compared with primary tumor (Figure 2A, Supplemental Table 2). For example, HK2 (hexokinase 2) and SLC16A3, which encodes for monocarboxylate transporter 4 (MCT-4), had increased expression in metastasis and a corresponding decrease in promoter methylation.
Figure 1. Analysis pipeline.
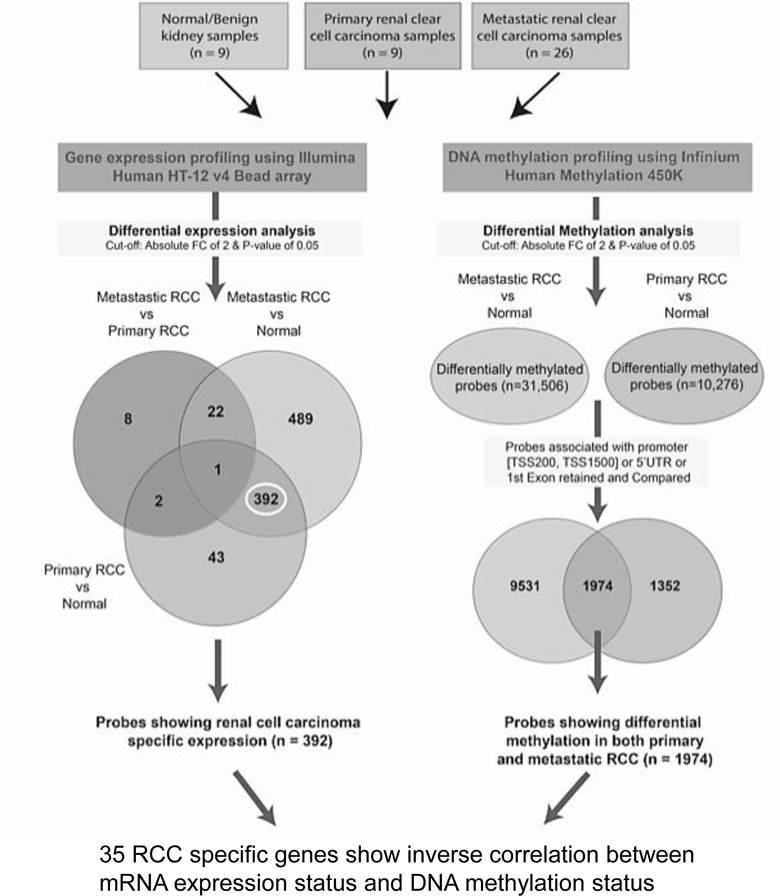
Schematic representation of analysis flow to identify clear cell renal cell carcinoma (RCC) associated genes based on gene expression and DNA methylation profile.
Figure 2. Transcriptome and methylome alterations in biologically aggressive kidney cancer.
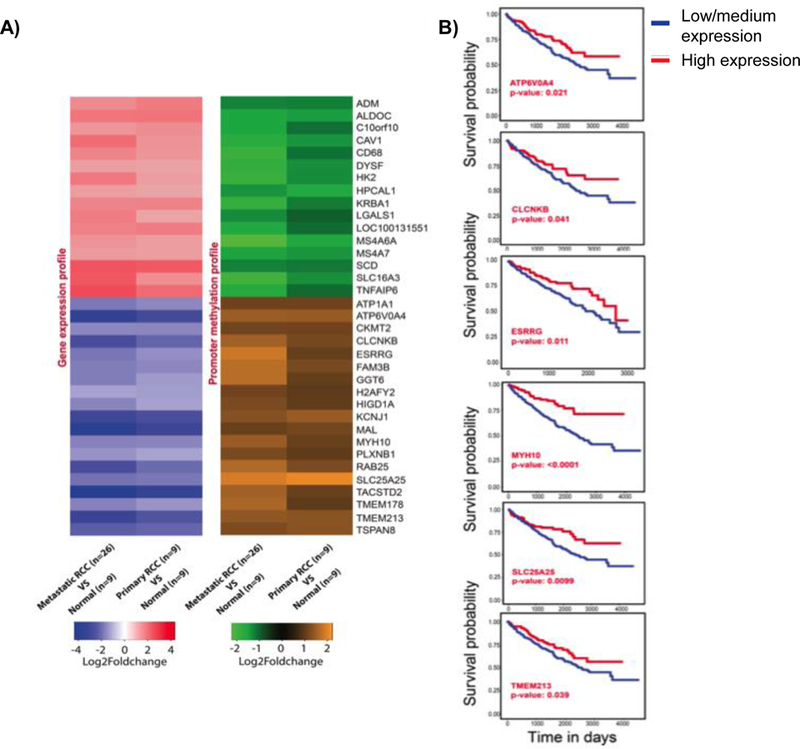
A) Heatmaps showing gene expression and DNA methylation profile of 35 RCC associated genes. Fold change value for each gene is represented by the probe with highest value. Fold change cut-off of 2 and P-value limit of 0.05 were considered to obtain differentially expressed or differentially methylated probes. B) Survival analysis of down-regulated genes SLC25A25, ESRRG, TEMEM213, MYH10, CLCNKB, and ATP6V0A4 using TCGA data. The KM plots were obtained from UALCAN (http://ualcan.path.uab.edu), which uses level 3 RNA-seq data and survival information from TCGA kidney renal clear cell carcinoma (KIRC) patients (n=533). The significance of difference between survival curves was obtained by log rank test.
External validation of transcriptome/ methylome analyses.
We next analyzed gene expression changes in publicly available data sets that mainly analyzed primary ccRCC. Similar gene expression changes were found in the TCGA data set, which performed transcriptomic analysis via RNA sequencing (Supplemental Figures 2S and 3S). The promoter methylation status of the 35 RCC specific genes was also concurrent with KIRC TCGA 450 K methylation data (Supplemental Table 3). Further validation was obtained via analysis of publicly available microarray data sets utilizing the Oncomine analysis platform (Supplemental Figures 4S, 5S, and 6S). Given the more prominent changes in gene expression in metastatic tumors for several genes identified in this analysis, we analyzed TCGA RNAseq data to assess whether gene expression levels in primary tumor had prognostic significance. For several genes downregulated in metastatic RCC, lower expression levels were associated with worsened survival in TCGA analysis (Figure 2B). In contrast, only one of the upregulated genes in metastatic RCC (LGALS1) demonstrated worsened prognosis with higher expression in the TCGA RNAseq data set (Supplemental Figure 7S).
HIF target genes are promoter hypomethylated with increased expression.
Several HIF target genes were hypomethylated with increased expression in metastatic RCC, including ADM (adrenomedullin), TNFAIP6 (TNF alpha induced protein 6), SLC16A3 (monocarboxylate transporter 4), CAV1 (caveolin-1), and LGALS1 (galectin-1) (26–30). A similar pattern was found for the HIF-responsive glycolysis genes HK2 (hexokinase 2) and ALDOC (aldolase C). Our data demonstrate that promoter methylation is an additional layer of regulation in the expression of HIF-regulated genes in RCC. Adolases catalyze the interconversion between fructose 1,6-bisphospate and the intermediates dihydroxyacetone phosphate and glyceraldehyde 3-phosphate. Prior studies have demonstrated alteration in the protein expression of ALDO isoforms in primary RCC (31, 32). In our study, mRNA expression of ALDOA and ALDOC was higher in a panel of RCC lines relative to untransformed HK2 renal epithelial cells (Figures 3A and 3B). ALDOB mRNA levels were extremely low (data not shown). We also examined aldolase isoform expression using a proteomics-based approach as commercial antibodies demonstrated significant cross-reactivity between ALDO isoforms (data not shown). RCC lines demonstrated increased ALDOA and ALDOC protein levels relative to untransformed renal epithelial RPTEC lines (Figure 3C). All cultured lines had very low to undetectable levels of ALDOB relative ALDOA and ALDOC (data not shown). We next utilized proteomics to assess the relative abundance of each ALDO isoform in RCC tissues relative to normal kidney. We identified increased ALDOA and ALDOC protein levels in tumor relative to normal kidney (Figure 3D). ALDOB protein levels were much lower in tumor than normal kidney. We next determined ALDO isoform mRNA expression as a function of tumor progression by assessing transcript levels in normal kidney, primary tumor, and metastatic tumor deposits in patient-matched samples (Figure 3E), identifying a trend toward increased transcript levels of both ALDOA and ALDOC with tumor progression.
Figure 3. The differential distribution of aldolases in RCC cells.
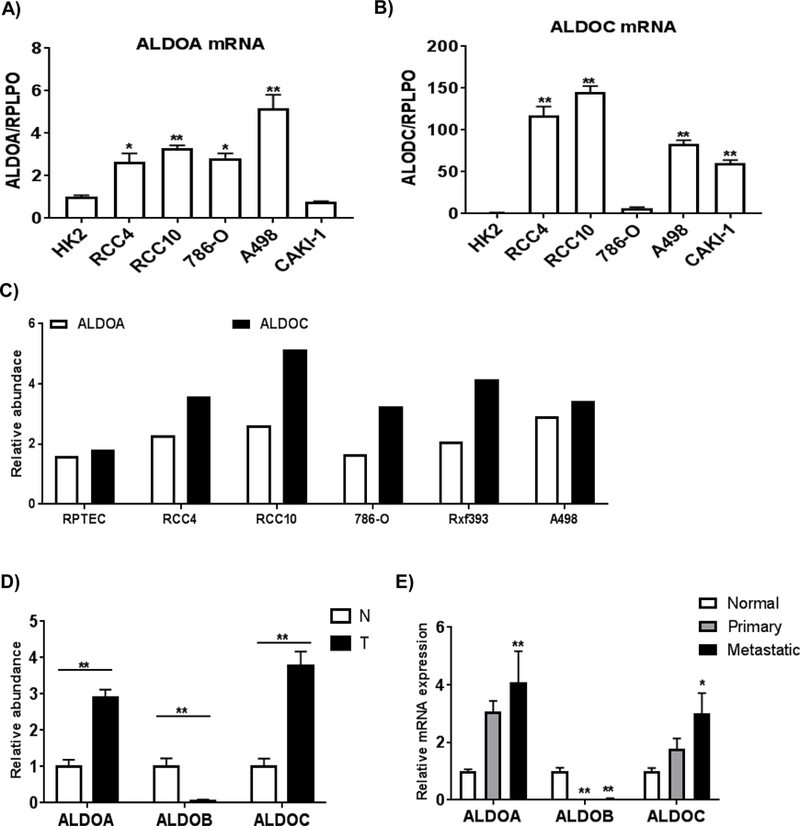
A) and B) Relative mRNA expressions of ALDOA and ALDOC in RCC cells. Total RNA was isolated from RCC cells and renal proximal tubular epithelial cells (HK2) (n=4/group). Transcript levels were normalized to those of RPLPO. Values are means ± SEM and asterisks indicate the differences relative to HK2 cells (*p<0.05, **p<0.01). C) Relative protein levels of aldolases in RCC cells compared to primary renal proximal tubule epithelial cells (RPTEC) by LC-MS proteomic analysis. D) Relative protein levels of aldolases in normal and tumor tissues by LC-MS proteomic analysis (n=5/group). Asterisks indicate difference relative to normal kidney tissue (*p<0.05, **p<0.01). E) Relative mRNA expressions of aldolase genes in patient-matched normal kidney, primary and metastasis were measured by RT-qPCR (n=11/ group). Values are means ± SEM and asterisks indicate the differences relative to normal kidney tissue (*p<0.05, **p<0.01).
Characterization of genes silenced in RCC via promoter methylation.
We identified promoter hypermethylation and silencing of several genes previously reported to be reduced in RCC including ATP1A1 (ATPase Na+/K+ transporting subunit alpha 1), ATP6V0A4 (ATPase H+ transporting V0 subunit A4), KCNJ1 (potassium voltage-gated channel subfamily J member 1), CLCNKB (chloride voltage-gated channel Kb), PLXNB1 (plexin B1), and TMEM213 (transmembrane protein 213) (33–37). A novel finding was reduced expression of the gene encoding the estrogen receptor-related (ERR)-γ (ESRRG). The ERRs consist of 3 members (α, β, and γ) encoded by ESRRA, ESRRB and ESRRG, respectively. The ERRs belong to the superfamily of nuclear hormone receptors.The ERRs most prominent role is the transcriptional regulation of mitochondrial metabolism. We validated the loss of ESRRG expression in patient-matched primary and metastatic tumor relative to normal kidney (Figure 4A). Data on these samples including sites of metastasis and prior systemic therapy is provided in Supplemental Table 4. Although the greatest fold reduction in ESRRG expression was found in primary tumor relative to normal kidney, a further decline in expression was noted in metastatic tumor deposits. Consistent with these data, we demonstrated significantly reduced ESRRG expression in RCC lines relative to normal kidney (Figure 4B). We next performed immunoblotting in another sample cohort and identified reduced ERR-γ protein in primary tumor relative to normal kidney (Figure 4C). We also assessed the mRNA expression of the other ERRs in normal and tumor and identified modest reductions in ESRRA in tumor (Figure 4D). Similar to ESRRG, we identified marked reductions in the expression of ESRRB (Figure 4D). Similar changes in mRNA expression of the ERRs were identified in analyses of TCGA data (data not shown). We next examined the prognostic significance of the various ERR isoforms. As previously noted, low ESRRG expression was associated with worsened overall survival in the TCGA data. In contrast, no significant associations with survival were observed based on expression levels for either ESRRA or ESRRB (Figure 4E).
Figure 4. Suppression of ESRRG in RCC.
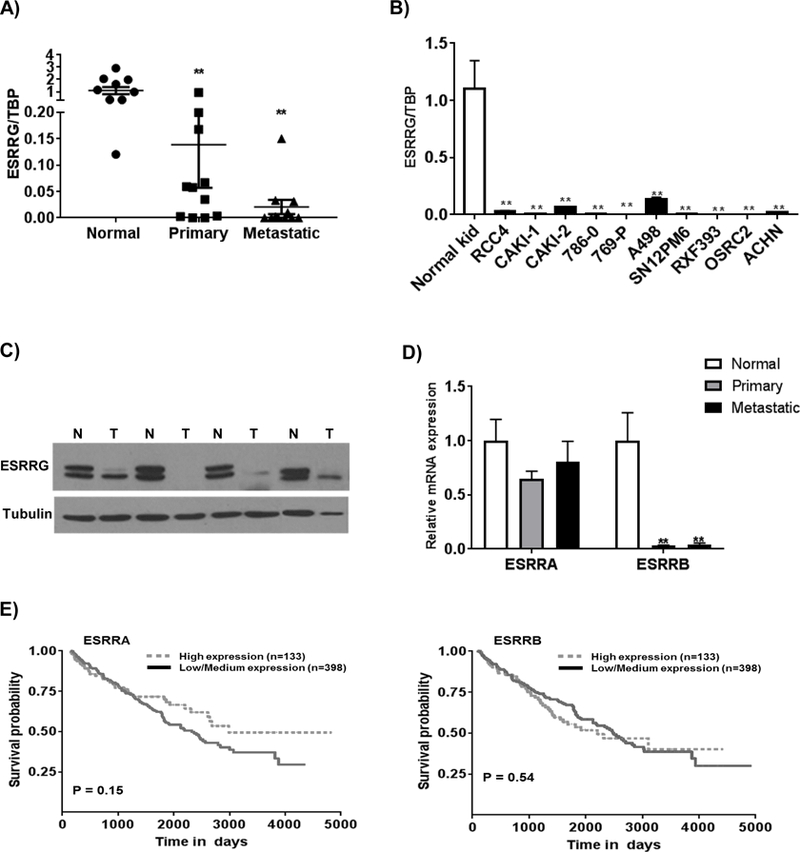
A) Relative mRNA expression of ESRRG in patient-matched normal kidney, primary tumor, and metastatic tumor (n=11/ group). Asterisks indicate differences relative to normal kidney tissue (**p<0.01). B) ESRRG mRNA expression in a panel of RCC cell lines relative to normal kidney. Transcript levels were normalized to those of TATA-binding protein (TBP). C) Western blot analysis of ESRRG in normal (N) and tumor (T). D) Relative mRNA expression of ESRRA and ESRRB in patient-matched normal kidney, primary tumor, and metastatic tumor (n=11/ group). Asterisks indicate differences relative to normal kidney tissue (**p<0.01). E) Kaplan-Meier survival curve analysis of patients from the TCGA data set on ccRCC based on expression of ESRRA and ESRRB.
Mechanism of ESRRG silencing in RCC.
We sought further insight into the silencing of ESRRG in renal cancer given that its expression had prognostic significance. Analysis using the UCSC genome browser identified to two CpG islands near the 5’ region of the ESRRG gene (Figure 5A). We identified multiple CpG sites within a CpG island situated right at the transcription start site whose methylation levels inversely correlated with ESRRG mRNA expression (Figure 5A-CpG island #1). We therefore performed ChIP studies using an antibody specific to 5-methylcytosine (5mC). ChIP qPCR utilizing primers to this CpG island demonstrated strong enrichment of 5mC in this region in three RCC cell lines as well as tumors from patients (Figures 5B, 5C and 5D). Recent studies of the methylation landscape of ccRCC indicate that many DNA hypermethylated sites are enriched for targets of the polycomb repressor complex 2 (PRC2) (13). PRC2 lays down the repressive histone mark H3K27me3. Prior studies have demonstrated coordination between DNA methylation and histone repressive marks in the silencing of gene expression. The catalytic subunit, EZH2 (enhancer of zeste homolog 2) is required for PRC2 activity. We therefore assessed the role of PRC2 in ESRRG silencing in RCC cells via a knockdown approach. We confirmed EZH2 knockdown via stable transduction with shRNA (Figure 5E). Knockdown of EZH2 with two nonoverlapping shRNAs led to increased ESRRG expression in CAKI-1 cells (Figure 5F). We further evaluated the effect of EZH2 inhibition on ESRRG expression using a pharmacologic inhibitor (GSK126). GSK126 reduced H3K27me3 levels consistent with EZH2/PRC2 inhibition (Figure 5G). Similar to EZH2 knockdown, GSK126 treatment resulted in the increased expression of ESRRG in CAKI-1 cells (Figure 5H). Collectively, these data demonstrate coordination between DNA and histone methylation in the silencing of ESRRG in RCC.
Figure 5. Silencing of ESRRG gene by CpG methylation in ccRCC.
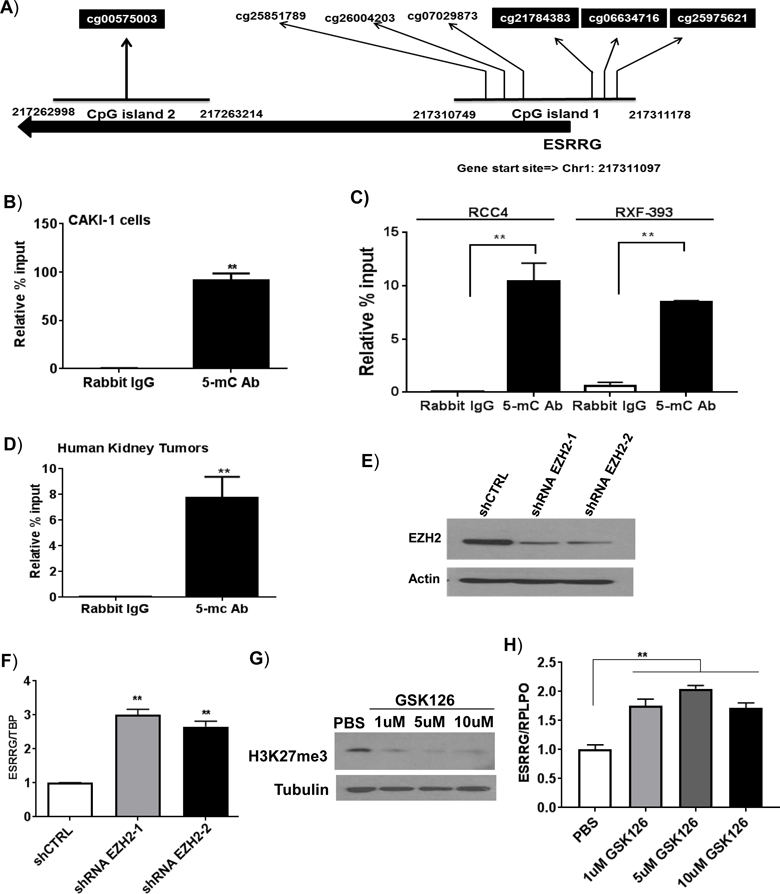
A) A cluster of CpG loci in the 5’ region of the ESRRG gene. The marked CpG loci are heavily methylated in kidney tumors. B and C) Analysis of methylated DNA enrichment by ChIP-qPCR. Genomic DNA was extracted with either B) CAKI-1 cells, or C) RCC4 and RXF-393 cells (n=3/group). D) ChIP was performed on kidney tumor samples with either rabbit IgG or 5-methylcystoine (5-mC) antibodies (n=3/group). The enriched DNA was quantified by real-time PCR with primer sets targeting highly methylated regions of the ESRRG promoter. E) Immunoblot analysis of EZH2 knockdown in CAKI-1 cells transduced with shCTRL or two non-overlapping EZH2-targeting shRNAs (shRNA EZH2–1 and shRNA2–2). F) CAKI-1 cells were stably transduced with shEZH2–1, shEZH2–2 or control shRNA (ShCTRL). The mRNA levels of ESRRG were analyzed by qRT-PCR (n=3 per group). G) Immunoblot analysis of H3K27me3 in CAKI-1 cells treated with GSK126 at indicated doses for 72 hr. H) Relative mRNA expression of ESRRG in CAKI-1 cells treated with GSK126 at indicated doses for 72 hr. (n=3/group, 3 independent experiments).
ESRRG suppresses migratory and invasive phenotypes in RCC.
Based on these data, we next assessed the biologic significance of ESRRG silencing in RCC. We transiently expressed ERR-γ via adenovirus. Adenovirus with a GFP construct was used as a control. We first assessed ESRRG mRNA levels in transduced cells. RT-qPCR analysis demonstrated that ESRRG expression was comparable to ESRRG transcript levels in normal kidney (Figure 6A). We confirmed ERR-γ expression at the protein level via immunoblotting (Figure 6B). We next assessed for effects on in vitro tumor phenotypes. In both CAKI-1 and RCC4 cells, ERR-γ suppressed wound healing as determined by scratch assay (Figure 6C and 6D). RCC cell migration as determined by a transwell assay was significantly reduced by the expression of ERR-γ (Figure 6E and 6F). We next assessed cell invasiveness using a Matrigel Boyden chamber assay. In both RCC lines tested, ERR-γ expression led to a significant reduction in cell invasiveness (Figure 6G and 6H). As noted previously, ERRs including ERR-γ have well-established roles in mitochondrial metabolism. ERR-γ is known to promote the transcription of nuclear encoded genes related to mitochondrial metabolism including genes encoded tricarboxylic acid (TCA) cycle enzymes. ERR-γ has also been demonstrated to have a role in mesenchymal-to epithelial transition (MET) in line with our invasion assay results (38). We next wanted to assess if ERR-γ’s effects on cell invasiveness were related to its role in mitochondrial metabolism. Notably, we did not observe significant effects on the expression of TCA cycle enzymes, known targets of ERRs (Figure 6I). In contrast, transduction with peroxisome proliferator-activated receptor gamma coactivator 1-α (PGC-1α), a known transcriptional activator of nuclear encoded mitochondrial metabolism genes, induced mRNA expression of several TCA cycle enzymes (Figure 6I). Collectively, these data demonstrate that ERR-γ suppresses invasive phenotypes independent of its canonical role in mitochondrial metabolism.
Figure 6. ESRRG reverts migratory and invasive behaviors in RCC cells.
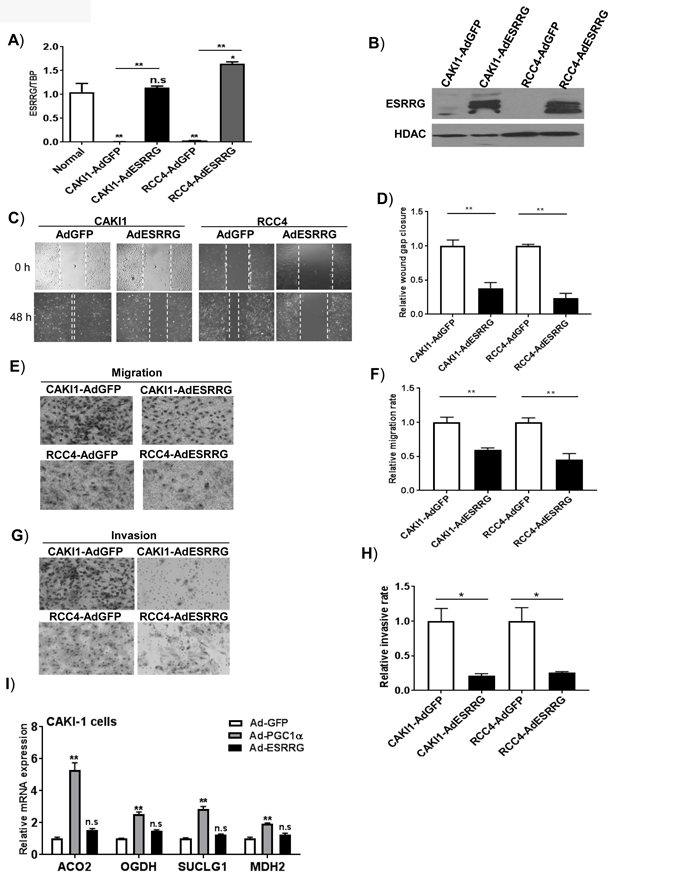
A) RCC cells were transduced with either Ad-GFP or Ad-ESRRG for 48 hr. Relative mRNA quantification of ESRRG was analyzed by qRT-PCR (n=3, 2 independent experiments). B) Western blot analysis of ESRRG in nuclear lysates from RCC cells transduced with either Ad-GFP or Ad-ESRRG. C) and D) Representative images of wound healing over time and the measurement of cap distance in RCC cells following ESRRG expression (n=3/ group, 3 independent experiments). E) and F) Representative images and quantification of Transwell migratory assay in RCC cells (n=3/group, 3 independent experiments). G) and H) Boyden invasion chamber with Matrigel insert was used to assess invasiveness of RCC cells following ESRRG expression (n=3/ group, 3 independent experiments). All data are presented as mean±SEM. I) Relative mRNA expression of TCA cycle enzymes in RCC cells transduced with the indicated adenovirus. After forty-eight hours, mRNA was extracted from RCC cells and analyzed by RT-qPCR for expression of TCA cycle enzymes (n=3/ group).
Discussion
Similar to many other malignancies, RCC has excellent outcomes when organ confined. In contrast, patients with metastatic disease face a poor prognosis. Despite the difference in prognosis between localized and metastatic disease, datasets examining metastatic tumor deposits in the context of RCC are generally lacking. While prior studies from our group and others have performed transcriptomic analyses of primary and metastatic sites, molecular analyses using epigenetic profiling platforms were not performed (14, 39, 40). A recent study analyzed the transcriptomes and methylomes of both primary and metastatic RCC samples with a focus on pharmacogenes; however, normal samples were not included in the analysis (11). Thus, the current study presents a unique data set with insight into the biology of aggressive renal cancer performed in an unbiased manner. The integration with methyl array data provides additional insight into the contribution of epigenetics to observed changes in gene expression.
A notable finding from the current study is the presence of multiple alterations in metabolism related genes in metastatic RCC. These include the glycolysis genes HK2 and ALDOC. However, several other HIF responsive genes were identified as hypomethylated in RCC, including ADM, TNFAIP6, and SLC16A3. HIF signaling is activated in the setting of VHL alterations, the most common tumor initiating event in ccRCC. Hence the increased expression in primary RCC is consistent with prior studies. However, further increases within metastatic tumor deposits with corresponding promoter hypomethylation indicate an epigenetic basis for the amplification of HIF signaling with tumor progression. To the best of our knowledge, these are the first data from patient-derived samples to implicate a role for methylation in the regulation of HIF target genes in the context of ccRCC. These data are consistent with recent experimental studies demonstrating further increases in HIF signaling beyond the tumor initiating event of VHL loss. Studies by Gao et al. demonstrate that inactivation of PBRM1, a gene commonly mutated in ccRCC, promotes HIF signaling in VHL null ccRCC (41). Experimental studies of RCC by Vanharanta et al. demonstrate that DNA demethylation promotes the expression of the HIF target gene CYTIP in an in vivo model of ccRCC metastasis (42). Given the large number of HIF target genes, a major challenge in the field has been to identify which of these genes are contributory to the malignant phenotype. Adding another level of complexity in renal cancer is the findings that HIF-1α is tumor suppressive whereas HIF-2α is tumor promoting (43, 44). Hence, the identification of HIF targets genes that are epigenetically amplified through mechanisms such as DNA promoter hypomethylation or chromatin remodeling (e.g. PBRM1 mutation) may reveal those HIF target genes critical to tumor development and/or progression.
We also demonstrate clear evidence for cooperative effects between both DNA and histone repressive methylation in the silencing of ESRRG, which encodes for ERR-γ. The ERRs are known to physically interact with PGC-1α in the promotion on mitochondrial metabolism related genes (45, 46). Our RT-qPCR studies identified markedly reduced expression of ESRRB and ESRRG in metastatic RCC with modest reductions in ESRRA. However, these are the first data to identify the role for histone and DNA repressive methylation of ESRRG in cancer. Our data are in line with prior studies in ccRCC demonstrating that DNA hypermethylated regions are enriched for targets of the PRC2 complex which lays down the repressive H3K27me3 mark (13, 47). Treatment of RCC cells with azacytidine did not result in increased ESRRG expression (data not shown). However, our studies with EZH2 knockdown and EZH2 inhibition suggest that hypermethylated/silenced genes may be reactivated via PRC2 inhibition. EZH2 has been shown to be overexpressed in RCC and other cancer such as prostate, bladder, and breast cancer among others (48–51). Our data are relevant as EZH2 inhibitors are currently in clinical trials.
Loss of ESRRG has previously been implicated in other malignancies including prostate cancer where it impacts proliferation (52, 53). ESRRG has also been implicated in breast cancer. For example, high ESRRG expression is associated with a better prognosis in breast cancer patients (54). However, data on its role on tumor biology are conflicting. Expression of ERR-γ in MCF-7 breast cancer cells has been shown to promote proliferation(55). In contrast, ERR-γ expression in MDA-MB-231 breast carcinoma cells suppresses invasion(38). We would like to point out that ERR-γ is highly expressed in the kidney in contrast to prostate and breast tissues. Moreover, we expressed ERR-γ in RCC cells at levels comparable to normal kidney to maintain physiologic relevance. Our data demonstrating that ERR-γ re-expression in RCC cells suppresses invasive phenotypes is consistent with a role for this transcription factor in mesenchymal to epithelial transition as previously reported. Further supportive evidence is provided by the fact that lower ESRRG expression correlates with worsened survival. No correlation with survival was found with the genes encoding the other ERR isoforms, ESRRA or ESRRB. Furthermore, despite the strong effects on invasive phenotypes in our studies, we did not see effects on the expression on mitochondrial metabolism genes under our experimental conditions. Hence, our data would support a role for ERR-γ independent of activities known to be redundant among the ERR isoforms (e.g. transcriptional regulation of nuclear encoded mitochondrial metabolism genes).
In addition, we identified several other genes that were hypermethylated and repressed that have been shown to suppress in vitro tumor phenotypes in RCC cells including PLXNB1, ATP1A, and KCNJ1 (33, 35, 37). Our study provides mechanistic insight into the silencing of these genes in kidney cancer. Moreover, they provide further rationale to assess the role of these genes in renal carcinogenesis.
We would like to acknowledge the limitations of the current study. Our initial transcriptomic analysis did not utilize patient-matched samples. Moreover, our initial analysis used relatively few normal tissues and primary RCCs. Nevertheless, we were able to validated expression changes of ESRRG in a patient-matched set of tissues. Moreover, many of the changes observed in both our transcriptomic and methylome analyses were consistent with publically available data sets including TCGA data. In addition, there were some discordancy between expression changes and correlation with survival. TMEM178, PLXNB1, TSPAN8 demonstrated reduced expression in RCC but lower expression correlated with improved survival in the TCGA cohort. In contrast, DYSF expression was increased in tumor but higher expression correlated with improved outcomes in the TCGA cohort. One potential explanation is that these factors may have different roles in primary RCC as compared with metastasis.
We also note that the methyl array analysis was performed with standard bisulfite sequencing, which cannot resolve between 5-mC and 5-hydroxymethylcytosine (5-hmC). The conversion of 5-mC to 5-hmC is catalyzed by the ten-eleven translocation enzymes (TETs 1–3). This has traditionally been thought of as the initial step of converting of 5-mC to unmethylated cytosine. However, recent studies indicate that 5-hmC may actually be a stable mark that can accumulate in various genomic regions including enhancer elements and promote gene transcription (56, 57). In contrast to 5-mC, 5-hmC levels tend to have lower levels in promoter regions (58). Based on these data, we therefore focused on methylation in gene promoters that anticorrelated with gene expression. Studies from our lab and others have demonstrated loss of 5-hmC in ccRCC (16, 59). Hence, this may represent an alternate mechanism for gene repression. Future studies should consider these alternate methyl markers and their contribution to gene expression.
In summary, our studies provide critical insight into the biology of aggressive kidney cancers and provide valuable global profiling data of metastatic RCC. We demonstrate an epigenetic basis for the propagation of HIF-related pathways, such as glycolysis, beyond tumor initiating events such as VHL loss. Furthermore, our study is the first demonstrate a role for DNA/repressive methylation in the silencing of ESRRG in cancer and that this process promotes invasive phenotypes in RCC cells. Taken together, our study provides compelling evidence that ccRCC is an epigenetically–driven malignancy. Given the emergence of pharmacologic agents that can target the epigenetic modifiers, our data provide rationale for the consideration of epigenetic–based approaches to advanced kidney cancer.
Supplementary Material
Implications:
Collectively, these data provide significant insight into the biology of aggressive RCC and demonstrate a novel role for DNA methylation in the promotion of HIF signaling and invasive phenotypes in renal cancer.
Acknowledgments
Array data were generated by the UT Health San Antonio Cancer Center Genomics Shared Resource which is supported by NIH-NCI P30 CA054174 and UT Health San Antonio. Proteomics studies were formed at the UAB Cancer Center Mass Spectrometry/Proteomics (MSP) Shared Facility. We thank Dr. Gregory M. Pavela (University of Alabama, Birmingham) for his editing of the manuscript.
Financial Support: Support for this study was provided by Department of Veterans Affairs Merit Award BX002930 (SS), NIH R01CA200653 (SS) and NIH R01CA157845 (SV). The research described was supported in part by the UAB Comprehensive Cancer Center (P30CA013148).
Footnotes
Conflicts of Interest: None to Declare
Footnote: Supplementary data for this article are available at Molecular Cancer Research Online (http://mcr.aacrjournals.org/).
References
- 1.Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013;499:43–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999;399:271–5. [DOI] [PubMed] [Google Scholar]
- 3.Iliopoulos O, Levy AP, Jiang C, Kaelin WG Jr., Goldberg MA. Negative regulation of hypoxia-inducible genes by the von Hippel-Lindau protein. Proceedings of the National Academy of Sciences of the United States of America 1996;93:10595–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pena-Llopis S, Vega-Rubin-de-Celis S, Liao A, Leng N, Pavia-Jimenez A, Wang S, et al. BAP1 loss defines a new class of renal cell carcinoma. Nature genetics 2012;44:751–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Rixe O, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med 2007;356:115–24. [DOI] [PubMed] [Google Scholar]
- 6.Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med 2015;373:1803–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choueiri TK, Escudier B, Powles T, Mainwaring PN, Rini BI, Donskov F, et al. Cabozantinib versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med 2015;373:1814–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Escudier B ea. CheckMate 214: Efficacy and safety of nivolumab + ipilimumab (N+I) v sunitinib (S) for treatment-naïve advanced or metastatic renal cell carcinoma (mRCC), including IMDC risk and PD-L1 expression subgroups. ESMO Congress, Madrid, Spain, October 2017; LBA 5. [Google Scholar]
- 9.Robinson DR, Wu YM, Lonigro RJ, Vats P, Cobain E, Everett J, et al. Integrative clinical genomics of metastatic cancer. Nature 2017;548:297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghosh AP, Willey CD, Anderson JC, Welaya K, Chen D, Mehta A, et al. Kinomic profiling identifies focal adhesion kinase 1 as a therapeutic target in advanced clear cell renal cell carcinoma. Oncotarget 2017;8:29220–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Winter S, Fisel P, Buttner F, Rausch S, D’Amico D, Hennenlotter J, et al. Methylomes of renal cell lines and tumors or metastases differ significantly with impact on pharmacogenes. Scientific reports 2016;6:29930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Herman JG, Latif F, Weng Y, Lerman MI, Zbar B, Liu S, et al. Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma. Proceedings of the National Academy of Sciences of the United States of America 1994;91:9700–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sato Y, Yoshizato T, Shiraishi Y, Maekawa S, Okuno Y, Kamura T, et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nature genetics 2013;45:860–7. [DOI] [PubMed] [Google Scholar]
- 14.Ghatalia P, Yang ES, Lasseigne BN, Ramaker RC, Cooper SJ, Chen D, et al. Kinase Gene Expression Profiling of Metastatic Clear Cell Renal Cell Carcinoma Tissue Identifies Potential New Therapeutic Targets. PLoS One 2016;11:e0160924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu J, Cao Q, Mehra R, Laxman B, Yu J, Tomlins SA, et al. Integrative genomics analysis reveals silencing of beta-adrenergic signaling by polycomb in prostate cancer. Cancer Cell 2007;12:419–31. [DOI] [PubMed] [Google Scholar]
- 16.Shim EH, Livi CB, Rakheja D, Tan J, Benson D, Parekh V, et al. L-2-Hydroxyglutarate: an epigenetic modifier and putative oncometabolite in renal cancer. Cancer Discov 2014;4:1290–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dunning MJ, Smith ML, Ritchie ME, beadarray Tavare S.. R classes and methods for Illumina bead-based data. Bioinformatics 2007;23:2183–4. [DOI] [PubMed] [Google Scholar]
- 18.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015;43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dunning M, Lynch A, Eldridge M. illuminaHumanv4.db: Illumina HumanHT12v4 annotation data (chip illuminaHumanv4). R package version 1.26.0. 2015.
- 20.Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014;30:1363–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rhodes DR, Kalyana-Sundaram S, Mahavisno V, Varambally R, Yu J, Briggs BB, et al. Oncomine 3.0: genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia (New York, NY 2007;9:166–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beroukhim R, Brunet JP, Di Napoli A, Mertz KD, Seeley A, Pires MM, et al. Patterns of gene expression and copy-number alterations in von-hippel lindau disease-associated and sporadic clear cell carcinoma of the kidney. Cancer research 2009;69:4674–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gumz ML, Zou H, Kreinest PA, Childs AC, Belmonte LS, LeGrand SN, et al. Secreted frizzled-related protein 1 loss contributes to tumor phenotype of clear cell renal cell carcinoma. Clin Cancer Res 2007;13:4740–9. [DOI] [PubMed] [Google Scholar]
- 24.Jones J, Otu H, Spentzos D, Kolia S, Inan M, Beecken WD, et al. Gene signatures of progression and metastasis in renal cell cancer. Clin Cancer Res 2005;11:5730–9. [DOI] [PubMed] [Google Scholar]
- 25.Chandrashekar DS, Bashel B, Balasubramanya SAH, Creighton CJ, Ponce-Rodriguez I, Chakravarthi B, et al. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia (New York, NY 2017;19:649–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garayoa M, Martinez A, Lee S, Pio R, An WG, Neckers L, et al. Hypoxia-inducible factor-1 (HIF-1) up-regulates adrenomedullin expression in human tumor cell lines during oxygen deprivation: a possible promotion mechanism of carcinogenesis. Mol Endocrinol 2000;14:848–62. [DOI] [PubMed] [Google Scholar]
- 27.Wan J, Chai H, Yu Z, Ge W, Kang N, Xia W, et al. HIF-1alpha effects on angiogenic potential in human small cell lung carcinoma. J Exp Clin Cancer Res 2011;30:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ullah MS, Davies AJ, Halestrap AP. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. J Biol Chem 2006;281:9030–7. [DOI] [PubMed] [Google Scholar]
- 29.Xie L, Xue X, Taylor M, Ramakrishnan SK, Nagaoka K, Hao C, et al. Hypoxia-inducible factor/MAZ-dependent induction of caveolin-1 regulates colon permeability through suppression of occludin, leading to hypoxia-induced inflammation. Mol Cell Biol 2014;34:3013–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao XY, Chen TT, Xia L, Guo M, Xu Y, Yue F, et al. Hypoxia inducible factor-1 mediates expression of galectin-1: the potential role in migration/invasion of colorectal cancer cells. Carcinogenesis 2010;31:1367–75. [DOI] [PubMed] [Google Scholar]
- 31.Unwin RD, Craven RA, Harnden P, Hanrahan S, Totty N, Knowles M, et al. Proteomic changes in renal cancer and co-ordinate demonstration of both the glycolytic and mitochondrial aspects of the Warburg effect. Proteomics 2003;3:1620–32. [DOI] [PubMed] [Google Scholar]
- 32.Zhu YY, Takashi M, Miyake K, Kato K. An immunochemical and immunohistochemical study of aldolase isozymes in renal cell carcinoma. J Urol 1991;146:469–72. [DOI] [PubMed] [Google Scholar]
- 33.Gomez Roman JJ, Garay GO, Saenz P, Escuredo K, Sanz Ibayondo C, Gutkind S, et al. Plexin B1 is downregulated in renal cell carcinomas and modulates cell growth. Translational research : the journal of laboratory and clinical medicine 2008;151:134–40. [DOI] [PubMed] [Google Scholar]
- 34.Girgis AH, Iakovlev VV, Beheshti B, Bayani J, Squire JA, Bui A, et al. Multilevel whole-genome analysis reveals candidate biomarkers in clear cell renal cell carcinoma. Cancer Res 2012;72:5273–84. [DOI] [PubMed] [Google Scholar]
- 35.Guo Z, Liu J, Zhang L, Su B, Xing Y, He Q, et al. KCNJ1 inhibits tumor proliferation and metastasis and is a prognostic factor in clear cell renal cell carcinoma. Tumour Biol 2015;36:1251–9. [DOI] [PubMed] [Google Scholar]
- 36.Zodro E, Jaroszewski M, Ida A, Wrzesinski T, Kwias Z, Bluyssen H, et al. FUT11 as a potential biomarker of clear cell renal cell carcinoma progression based on meta-analysis of gene expression data. Tumour Biol 2014;35:2607–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang D, Zhang P, Yang P, He Y, Wang X, Yang Y, et al. Downregulation of ATP1A1 promotes cancer development in renal cell carcinoma. Clinical proteomics 2017;14:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tiraby C, Hazen BC, Gantner ML, Kralli A. Estrogen-related receptor gamma promotes mesenchymal-to-epithelial transition and suppresses breast tumor growth. Cancer Res 2011;71:2518–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ho TH, Serie DJ, Parasramka M, Cheville JC, Bot BM, Tan W, et al. Differential gene expression profiling of matched primary renal cell carcinoma and metastases reveals upregulation of extracellular matrix genes. Ann Oncol 2017;28:604–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 366:883–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gao W, Li W, Xiao T, Liu XS, Kaelin WG Jr. Inactivation of the PBRM1 tumor suppressor gene amplifies the HIF-response in VHL-/- clear cell renal carcinoma. Proceedings of the National Academy of Sciences of the United States of America 2017;114:1027–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vanharanta S, Shu W, Brenet F, Hakimi AA, Heguy A, Viale A, et al. Epigenetic expansion of VHL-HIF signal output drives multiorgan metastasis in renal cancer. Nature medicine 2013;19:50–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shen C, Beroukhim R, Schumacher SE, Zhou J, Chang M, Signoretti S, et al. Genetic and Functional Studies Implicate HIF1alpha as a 14q Kidney Cancer Suppressor Gene. Cancer Discov 2011;1:222–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kondo K, Kim WY, Lechpammer M, Kaelin WG Jr. Inhibition of HIF2alpha is sufficient to suppress pVHL-defective tumor growth. PLoS Biol 2003;1:E83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huss JM, Kopp RP, Kelly DP. Peroxisome proliferator-activated receptor coactivator-1alpha (PGC-1alpha) coactivates the cardiac-enriched nuclear receptors estrogen-related receptor-alpha and -gamma. Identification of novel leucine-rich interaction motif within PGC-1alpha. J Biol Chem 2002;277:40265–74. [DOI] [PubMed] [Google Scholar]
- 46.Eichner LJ, Giguere V. Estrogen related receptors (ERRs): a new dawn in transcriptional control of mitochondrial gene networks. Mitochondrion 2011;11:544–52. [DOI] [PubMed] [Google Scholar]
- 47.Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 2002;298:1039–43. [DOI] [PubMed] [Google Scholar]
- 48.Weikert S, Christoph F, Kollermann J, Muller M, Schrader M, Miller K, et al. Expression levels of the EZH2 polycomb transcriptional repressor correlate with aggressiveness and invasive potential of bladder carcinomas. International journal of molecular medicine 2005;16:349–53. [PubMed] [Google Scholar]
- 49.Wang Y, Chen Y, Geng H, Qi C, Liu Y, Yue D. Overexpression of YB1 and EZH2 are associated with cancer metastasis and poor prognosis in renal cell carcinomas. Tumour Biol 2015;36:7159–66. [DOI] [PubMed] [Google Scholar]
- 50.Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002;419:624–9. [DOI] [PubMed] [Google Scholar]
- 51.Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proceedings of the National Academy of Sciences of the United States of America 2003;100:11606–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yu S, Wang X, Ng CF, Chen S, Chan FL. ERRgamma suppresses cell proliferation and tumor growth of androgen-sensitive and androgen-insensitive prostate cancer cells and its implication as a therapeutic target for prostate cancer. Cancer Res 2007;67:4904–14. [DOI] [PubMed] [Google Scholar]
- 53.Audet-Walsh E, Yee T, McGuirk S, Vernier M, Ouellet C, St-Pierre J, et al. Androgen-Dependent Repression of ERRgamma Reprograms Metabolism in Prostate Cancer. Cancer Res 2017;77:378–89. [DOI] [PubMed] [Google Scholar]
- 54.Ariazi EA, Clark GM, Mertz JE. Estrogen-related receptor alpha and estrogen-related receptor gamma associate with unfavorable and favorable biomarkers, respectively, in human breast cancer. Cancer Res 2002;62:6510–8. [PubMed] [Google Scholar]
- 55.Ijichi N, Shigekawa T, Ikeda K, Horie-Inoue K, Fujimura T, Tsuda H, et al. Estrogen-related receptor gamma modulates cell proliferation and estrogen signaling in breast cancer. The Journal of steroid biochemistry and molecular biology 2011;123:1–7. [DOI] [PubMed] [Google Scholar]
- 56.Rampal R, Alkalin A, Madzo J, Vasanthakumar A, Pronier E, Patel J, et al. DNA hydroxymethylation profiling reveals that WT1 mutations result in loss of TET2 function in acute myeloid leukemia. Cell reports 2014;9:1841–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hahn MA, Qiu R, Wu X, Li AX, Zhang H, Wang J, et al. Dynamics of 5-hydroxymethylcytosine and chromatin marks in Mammalian neurogenesis. Cell reports 2013;3:291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Skvortsova K, Zotenko E, Luu PL, Gould CM, Nair SS, Clark SJ, et al. Comprehensive evaluation of genome-wide 5-hydroxymethylcytosine profiling approaches in human DNA. Epigenetics & chromatin 2017;10:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen K, Zhang J, Guo Z, Ma Q, Xu Z, Zhou Y, et al. Loss of 5-hydroxymethylcytosine is linked to gene body hypermethylation in kidney cancer. Cell research 2016;26:103–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.