

Abstract
The Amyloid Precursor Protein (APP) is infamous for its proposed pivotal role in the pathogenesis of Alzheimer’s disease (AD). Much research on APP focusses on potential contributions to neurodegeneration, mostly based on mouse models with altered expression or mutated forms of APP. However, cumulative evidence from recent years indicates the indispensability of APP and its metabolites for normal brain physiology. APP contributes to the regulation of synaptic transmission, plasticity, and calcium homeostasis. It plays an important role during development and it exerts neuroprotective effects. Of particular importance is the soluble secreted fragment APPsα which mediates many of its physiological actions, often counteracting the effects of the small APP-derived peptide Aβ. Understanding the contribution of APP for normal functions of the nervous system is of high importance, both from a basic science perspective and also as a basis for generating new pathophysiological concepts and therapeutic approaches in AD. In this article, we review the physiological functions of APP and its metabolites, focusing on synaptic transmission, plasticity, calcium signaling, and neuronal network activity.
Keywords: APP, amyloid, Alzheimer’s disease, synaptic transmission, plasticity, oscillations
Introduction
The Amyloid Precursor Protein (APP) is a ubiquitously expressed transmembrane protein with a long extracellular and a short intracellular domain. The predominant isoform in the central nervous system (CNS) consists of 695 amino acids (aa; Gralle and Ferreira 2007; Müller and Zheng 2012; Müller and others 2017). APP forms a protein family together with the homologous APP-like proteins 1 and 2 (APLP1 and APLP2, respectively). APP is highly conserved throughout the animal kingdom, beginning in nematodes, emphasizing its indispensable role for neuronal function. Indeed, triple knock-out (KO) mice lacking all three proteins of the APP/APLP family, as well as APP/APLP2 double knock-outs (DKO) are not viable. They show severe cortical malformations (Herms and others 2004), demonstrating a crucial role of APP/APLP during development. In fact, APP has been shown to promote synapse formation, dendritic sprouting, and neuronal migration (Müller and others 2017). Single APP-KO mice are viable due to compensation by the homologues, but show deficits in long-term potentiation (LTP) at old age (Dawson and others 1999; Ring and others 2007), learning and memory formation as well as higher susceptibility to seizures and hypoxia-ischemia (Hefter and others 2016; Koike and others 2012; Steinbach and others 1998). These findings point to several physiological functions of APP which are addressed in more detail below.
APP is cleaved by the α- or β-secretase (also known as beta-site amyloid precursor protein cleaving enzyme [BACE]) and consecutively by the γ-secretase (Haass and others 2012; Fig. 1A-C). Recently, a third, novel, η-secretase pathway with yet unclear function has been discovered (Willem and others 2015; Fig. 1D). Cleavage by the α-secretase initiates the so-called non-amyloidogenic pathway, which results in the APP-intracellular domain (AICD) and the soluble extracellularly secreted APPsα fragment. APPsα was shown to mediate most of the known neuroprotective and neurotrophic effects of APP (Hefter and others 2016; Mockett and others 2017). In contrast, β-secretase cleavage is the starting point for the amyloidogenic pathway. Besides, the intracellular fragment AICD it produces a secreted APPsβ fragment and, importantly, amyloid β, a small peptide varying from 38 to 43 aa in length. Aβ exists in different monomeric or multimeric soluble forms and can aggregate to fibrils and plaques. Such aggregates are most easily formed by Aβ42, which is less common than the Aβ40 isoform and is more prone to precipitate. The extracellular accumulation of amyloid plaques, along with the intracellular deposition of tau fibrils, is the histopathological hallmark of Alzheimer’s disease (AD) and has been replicated in various mouse models of the disease (Sasaguri and others 2017). Amyloid deposition may be one of the initial steps of the pathophysiological cascade of AD as it usually precedes tau pathology as well as pathophysiological alterations and clinical symptoms. Thus, it is widely being considered crucial for AD pathogenesis, and enormous efforts have been put into development of strategies targeting amyloid (Selkoe and others 2016). However, the correlation between amyloid burden and clinical symptoms is quite weak. In line with this fact, clinical trials based on anti-amyloid treatment strategies for AD received heavy setbacks in recent years (van Dyck 2018). This therapeutic failure makes it even more important to understand the normal functions of APP family proteins and their metabolites in animal models and in humans.
Figure 1.
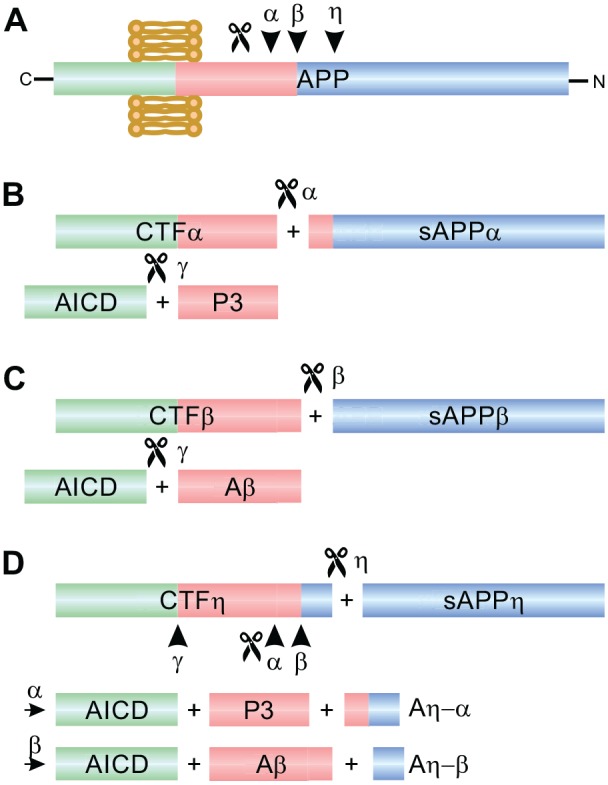
Proteolytic processing of APP by the secretases. (A) Schematic structure of the membrane-bound, full-length Amyloid Precursor Protein (APP). On the left side the shorter C-terminal intracellular domain is depicted in green, on the right side the longer N-terminal extracellular domain is in blue. The amyloid sequence is shown in red. Arrowheads point to the secretase cleavage sites. The length of the fragments is not proportional to the length of the respective amino acid sequence. (B) Cleavage by α-secretase and consecutively by γ-secretase. CTFα = C-terminal fragment alpha; APPsα = extracellularly secreted soluble APP alpha fragment; AICD = APP intracellular domain. (C) Cleavage by β-secretase and consecutively by γ-secretase. CTFβ = C-terminal fragment beta; APPsβ = extracellularly secreted soluble APP beta fragment; Aβ = amyloid beta. (D) Cleavage by η-secretase and consecutively by α, β, and γ-secretases. CTFη = C-terminal fragment eta; APPsη = extracellularly secreted soluble APP eta fragment; Aη-α, Aη-β = amyloid eta-alpha and eta-beta, respectively.
APP metabolites affect synaptic transmission and network function in health and disease, partially counteracting each other. Of special interest are neuroprotective and neurotrophic functions of APP—or its fragment APPsα—which may be essential for normal neuronal function and for resilience toward neurodegenerative diseases. In the following sections we give an overview of physiological functions of APP at the synaptic and network levels.
APP and Synaptic Transmission
APP and its metabolites affect both excitatory and inhibitory synaptic transmission in health and disease. At the postsynaptic site APP interacts with glutamatergic (Hoe and others 2009) and cholinergic (Richter and others 2018) receptors. At the presynaptic site it regulates the function of GABAB receptors (GABAB-R; Dinamarca and others 2019; Fig. 2B), synaptic vesicle release (Klevanski and others 2015), and the abundance of presynaptic proteins (see section “Short-Term Plasticity” for more details; Fig. 2A). Aβ oligomers disrupt synaptic activity and shift the excitation/inhibition ratio toward excitation, altering paired-pulse plasticity and abolishing LTP (Puzzo and others 2008). Application of synthetic Aβ on cultured hippocampal neurons leads to downregulation of dynamin, a protein that is essential for synaptic vesicle endocytosis and interrupts synaptic vesicle recycling (Kelly and others 2005; Kelly and Ferreira 2007). Various direct and indirect interactions with receptors have been proposed to mediate the toxic activity of Aβ (Strooper and Karran 2016). APP and APLP molecules can form cis- and trans-dimers. Cis-homodimerized APP has been proposed to function as a cell-surface G-protein coupled receptor with Aβ and APPsα as putative ligands (Ludewig and Korte 2017). Thus, a recent study showed that synaptic effects of amyloid require the presence of full-length APP (Wang and others 2017). In their trans-forms, transcellular dimers of APP family proteins mediate cell-to-cell adhesion and support synaptic connectivity (Schilling and others 2017). Aη-α, one of the products of the recently discovered η-secretase pathway, was shown to inhibit neuronal activity and LTP in hippocampal slices (Willem and others 2015). While APP and its fragments affect synaptic activity in multiple ways, APP cleavage and amyloid levels are themselves dynamically regulated by neuronal activity (Bero and others 2011; Cirrito and others 2005). Intriguingly, even the relative abundance of amyloid isoforms is regulated by patterns of neuronal firing. Thus, firing of single spikes was shown to favor the formation of the supposedly more toxic Aβ42 peptide, while burst firing facilitated the formation of Aβ40. These effects are mediated by an activity-dependent conformational change of presenilin1, the catalytic partner of γ-secretase (Dolev and others 2013). In the following section we will focus on the interplay of APP and three major neurotransmitter systems which are known to be altered in AD.
Figure 2.

Synaptic actions of APP and its metabolites. (A) Putative and conformed actions of APP and AICD on presynaptic vesicle release. (B) Putative actions of APP dimers at the synapse. APP form dimers with a second APP molecule in the trans- (1) or cis-formation (2). Cis-dimeric APP may act as a receptor for APPsα and Aβ. (3) APP colocalizes in transport vesicles for GABAB-R and is part of the GABAB-R complex at the presynaptic site where it decreases the releasable vesicle pool and increases paired pulse facilitation. (C) Interactions between APP and cholinergic receptors. APP, APPsα, AICD, and picomolar concentrations of Aβ. Present evidence indicates a positive modulation of cholinergic transmission via interactions with α7-nAchR, whereas nanomolar and higher concentrations of Aβ block these receptors. M1-AchR activation promotes cleavage of APP by α-secretase and APPsα secretion; nAchR promotes amyloidogenic processing. Ach = acetylcholine; M1-AchR = M1-type muscarinic acetylcholine receptors; nAchR = nicotinic acetylcholine receptors; Gq = G-Protein q. (D) Interactions between APP and NMDAR. Left: synaptic NMDAR signaling, predominantly GluN2A. Right: extrasynaptic NMDAR signaling, predominantly GluN2B. Aβ inhibits GluN2A-NMDAR-mediated survival pathways. Aβ inhibits GluN2A-NMDAR signaling. Full-length APP positively modulates membrane trafficking and AICD increases expression of GluN2B-NMDAR. GluN2B-NMDAR signaling reduces abundance of APP at the membrane and promotes Aβ secretion. Glu = glutamate.
Glutamatergic Transmission and NMDA Receptor Signaling
Ionotropic glutamate-receptors comprise α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptors (AMPAR) and N-methyl-D-aspartate receptors (NMDAR). The characteristic voltage-dependent gating of NMDAR plays a pivotal role in synaptic plasticity, learning, and memory as well as in pathophysiological cascades in AD and hypoxia-ischemia (Paoletti and others 2013). The NMDAR family comprises a heterogeneous group of tetramers consisting of a combination of the obligatory GluN1 subunit and the facultative GluN2-A, B, C, or D or GluN3 subunits. During development, the expression pattern in the forebrain shifts from the predominant GluN2B to GluN2A subunits (Monyer and others 1994). In the adult brain, GluN2B is commonly associated with excitotoxicity and neuronal degeneration while GluN2A is linked to plasticity and pro-survival pathways (Bading 2017; Hardingham and Bading 2010). Chronic overactivation of NMDAR has been hypothesized as a pathological mechanism in AD, in line with the use of the noncompetitive NMDAR blocker memantine as one of the few approved drugs for treatment of AD (Liu and others 2019). APP as well as APLP1 and APLP2 were found to affect NMDAR trafficking and to enhance its surface expression (Cousins and others 2009; Cousins and others 2015; Fig. 2D). The effects on expression are mutual: APP increases membrane levels and function of GluN2B-containing NMDAR while NMDAR activation decreases surface expression of APP and promotes amyloidogenesis (Hoe and others 2009). Furthermore, APP was found to regulate the homeostasis of D-serine, a potent endogenous co-agonist of NMDAR. APP-deficient transgenic mice show D-serine-dependent impairments in structural plasticity of dendritic spines (Zou and others 2016).
Several studies using intrathecal injection of exogenous APPsα in mice suggest that APPsα, but not APPsβ, potently regulates NMDAR function. Indeed, suppression of NMDA currents in hippocampal neurons by APPsα occurs already at picomolar concentrations (Furukawa and Mattson 1998). Another study did not find effects of APPsα on baseline NMDAR currents, but showed its requirement for the induction of LTP (Taylor and others 2008). The effects on NMDAR function appear to be age-dependent. In healthy aged rats APPsα levels were found to be reduced alongside with NMDAR function and LTP. In these animals, exogenous application of APPsα did potentiate LTP while it had no such effect in younger animals (Moreno and others 2015).
The intracellular fragment AICD was shown to promote expression of GluN2B-containing NMDAR and to facilitate GluN2B-mediated synaptic transmission. While APP-knockdown attenuates GluN2B currents, increased AICD production enhances NMDAR function and disrupts LTP (Pousinha and others 2017). On the other hand, Aβ oligomers inhibit NMDAR-activity-dependent trophic cascades leading to synapse loss in the hippocampus (Shankar and others 2007). Vice versa, several studies suggest modulation of APP processing by NMDAR signaling. Activation of synaptic NMDAR stimulates α-secretase processing and inhibits Aβ production (Hoey and others 2009) while activation of extrasynaptic NMDAR promotes amyloidogenesis (Rush and Buisson 2014). In another study, pharmacological block of NMDAR prevented toxic effects of Aβ on dendritic spines (Wei and others 2009). Taken together, an increasing body of evidence indicates that activation of synaptic GluN2A-NMDAR promotes plasticity and survival via facilitation of APPsα production. In contrast, extrasynaptic GluN2B-NMDAR promote cell death by β-secretase activation (Rush and Buisson 2014).
AMPAR are the principal mediators of excitatory glutamatergic transmission in the CNS. Their dysregulation by Aβ is one of the hallmarks of synaptic failure in AD (Paula-Lima and others 2013). Reduced expression and function of AMPAR were found in neurons that overexpress wild-type APP or the APP Swedish double mutation as well as in neurons following exogenous application of (Almeida and others 2005; Chang and others 2006; Hsieh and others 2006). One of the major underlying mechanisms is disrupted AMPAR trafficking mediated by NMDA and metabotropic glutamate receptor signaling (Guntupalli and others 2016). A study on mice overexpressing Aβ showed that its detrimental effect on synapses requires the presence of the AMPAR subunit GluA3 (Reinders and others 2016). On the other hand, AMPAR activation was shown to increase the clearance of Aβ in a dose-dependent manner, thus decreasing its extracellular levels (Hettinger and others 2018). Simultaneously, it promotes non-amyloidogenic cleavage of APP (Hoey and others 2013). These findings demonstrate a complex, reciprocal relationship between APP metabolites and glutamate receptors that depends on receptor composition, location and developmental stage.
GABAergic Transmission
GABA receptors are divided into three main types: the ionotropic GABAA and GABAC (GABAA-ρ) and the metabotropic GABAB-R, all of which are composed from a variety of distinct subunits (Chebib and Johnston 1999). Disrupted GABAergic synaptic transmission, interneuron dysfunction and consecutive aberrant network activity are implicated in the pathophysiology of AD (Verret and others 2012). APP was shown to modulate paired-pulse inhibition and tetanic potentiation in striatal and hippocampal GABAergic interneurons via inhibition of L-type calcium channels (LTCC; Yang and others 2009). Interestingly, APP is highly expressed in GABAergic interneurons, regulating both phasic and tonic inhibitory function. Its selective deletion in GABAergic, but not glutamatergic, neurons disrupts adult hippocampal neurogenesis (Wang and others 2014). APP and APLP2 were found to be part of the GABAB-R complex (Schwenk and others 2016), suggesting a role in presynaptic release dynamics or plasticity (Fig. 2B). Indeed, evidence from two very recent studies supports this hypothesis: Binding of APP to GABAB-R was shown to promote its axonal trafficking, thus facilitating cell-surface expression of these receptors and presynaptic inhibition. At the same time, GABAB-bound APP was less likely to be cleaved by the amyloidogenic β-secretase (Dinamarca and others 2019). These findings link dysfunctional GABAB-R trafficking and deficits in presynaptic inhibition with increased amyloid cleavage in AD. Furthermore, all secreted APP fragments (α, β, and η) were shown act as a GABAB-R ligands. APPsα binding to the sushi domain of GABAB-R inhibited presynaptic vesicle release, thus suppressing synaptic transmission and enhancing short-term facilitation in the hippocampus (Rice and others 2019). Intriguingly, APPsβ binding exerted similar effects. Thus, this study is one of very few to find a physiological role of APPsβ. However, it must be taken into consideration that APP proteins bind to GABAB-R with relatively low affinity (dissociation constants KD of ~200 and ~400 nM, respectively), and 1 µM concentrations have been used in this study, while the physiologically active concentrations of these proteins are in the picomolar to low nanomolar range (Hick and others 2015; Puzzo and others 2008). Therefore, APP’s effects under physiological circumstances rather arise from other interactions than with GABAB-R, while functional implications of these findings require further investigation. Nevertheless, these findings link APP and GABAergic signaling both in health and disease and present an additional regulatory mechanism of presynaptic excitability by APP.
Cholinergic Transmission
Dysfunction of the cholinergic system is strongly implied in the pathogenesis of AD. Several clinically approved antidementives inhibit the acetylcholine-cleaving enzyme acetylcholinesterase, aiming to facilitate cholinergic transmission (Knight and others 2018). Studies in APP-deficient mice show that APP is important for peripheral cholinergic transmission at the neuromuscular junction (Caldwell and others 2013; Weyer and others 2011) as well in the autonomous nervous system (Cai and others 2016; Fig. 2C). Transgenic mouse models of AD confirm that APP overexpression and aberrant amyloid deposition are associated with degeneration of cholinergic neurons beginning with damage of their axons in the cortex (Foidl and others 2016). In support of these findings, inhibition of β-secretase rescued cholinergic dysfunction in a mouse model of AD (Ohno and others 2004). Of special importance for cholinergic function is the intracellular AICD peptide. Mice with knock-in of a specific, dysfunctional motif within AICD show impaired muscular and cognitive performance (Matrone and others 2012). APPsα has been suggested to function as an endogenous positive allosteric modulator of cholinergic signaling by binding to α7-type nicotinic acetylcholine receptors (α7-nAChRs; Richter and others 2018). The aa sequence required for associating APPsα with α7-nAChRs is also present in Aβ. Therefore, it is not surprising that Aβ also potently binds to these receptors, both on presynaptic and postsynaptic sites. The resulting modulation of cholinergic signaling depends on its concentration and the conformation of nAchR (Lasala and others 2019). While physiological concentrations of Aβ in the picomolar to low nanomolar range were shown to activate nAchR and increase synaptic vesicle recycling, higher concentrations inhibit cholinergic currents (Lazarevic and others 2017). This may be of great importance for AD, where levels of Aβ are increased. Similar to GABAergic and glutamatergic transmission, several studies have shown that cholinergic signaling modulates APP processing regulating interstitial amyloid levels. Thus, activation of muscarinic (M1) receptors was shown to inhibit BACE and facilitate non-amyloidogenic cleavage and APPsα secretion (Jiang and others 2014) while activation of nicotinic receptors was found to increase interstitial Aβ levels (Wei and others 2009).
Calcium Signaling
Neuronal activity is accompanied by fluctuations of intracellular calcium concentration. Calcium modulates a plethora of enzymatic cascades, gene transcription, as well as pro-survival and apoptosis pathways; thus, calcium signaling is a major activity-dependent factor affecting the intermediate- and long-term cellular fate (Bading 2013). Furthermore, neuronal excitability and thus network function are heavily influenced by calcium concentrations. Dysregulation of the intracellular calcium homeostasis is considered a common pathomechanism in various neurological conditions, prominently AD and stroke, where cell death and degeneration are involved (Alzheimer’s Association Calcium Hypothesis Workgroup 2017; Green and LaFerla 2008). Cytosolic calcium can derive from extracellular sources such as NMDAR or voltage-gated calcium channels (VGCC) as well as from intracellular stores such as mitochondria and the endoplasmic reticulum (ER; Clapham 2007; Lerdkrai and others 2018). APP and its metabolites regulate calcium homeostasis at multiple levels both in trophic, plasticity-related pathways (via APPsα) and in a destabilizing way in AD (via Aβ; Fig. 3). The interactions between APP and NMDAR have been highlighted above; in the following section we focus on VGCC and internal calcium stores.
Figure 3.
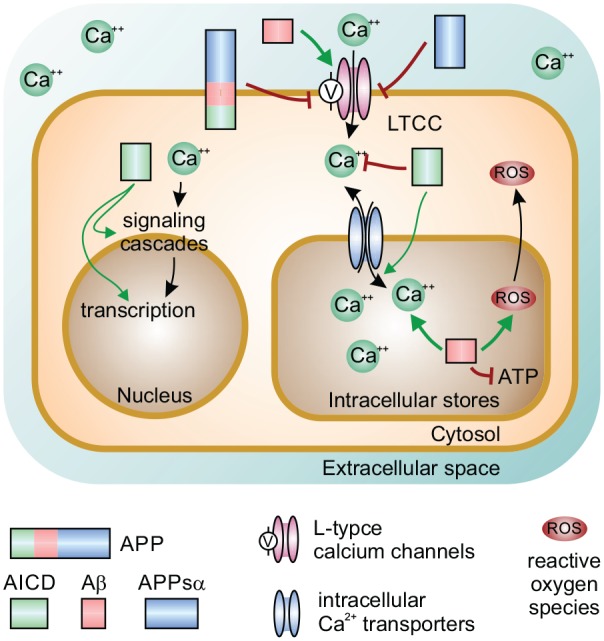
APP and calcium homeostasis. Calcium can enter the cytosol from extracellular space via NMDAR and voltage-gated calcium channels including L-type calcium channels (LTCC) or from intracellular stores such as mitochondria and the endoplasmic reticulum via several calcium transporters. Intracellular calcium triggers various signaling cascades, translocates to the nucleus, and affects gene transcription. APP and APPsα inhibit LTCC. AICD is important for the control of calcium homeostasis by regulating transport to internal stores and transcription of calcium-binding proteins. Aβ oligomers are present in mitochondria where they destabilize mitochondrial membrane potential and function, disturb adenosine triphosphate (ATP) production, and increase reactive oxygen species (ROS) levels.
Voltage-Gated Calcium Channels
VGCC are a family of voltage-gated ion channels with selective permeability for Ca2+. Excessive calcium influx through VGCC is suggested to contribute to calcium overload in degenerative and neurovascular diseases (Cataldi 2013), while VGCC antagonists, on the other hand, show protective effects against cognitive decline (Lovell and others 2015). In several studies amyloid oligomers were shown to activate one subtype of VGCC, Cav1.2 subunit-expressing L-type calcium channels (LTCC). The expression of LTCC is driven by Aβ under hypoxic conditions (Webster and others 2006). Their blockage has been identified as a potential pharmacotherapeutic target for AD (Anekonda and Quinn 2011; Lovell and others 2015). APP regulates LTCC in cultured GABAergic neurons of the hippocampus and striatum, with APP deletion leading to aberrant activity of LTCC and altered short-term plasticity (STP; Yang and others 2009). As expression of APPsα on an APP-KO background was sufficient to rescue deficits linked to enhanced calcium influx through LTCC, these effects are likely mediated by the APPsα fragment (Hefter and others 2016). Expression of human APP in rat primary cortical neurons lead to increased LTCC currents and inhibited calcium oscillations (Santos and others 2009). On the other hand, Aβ appears to inhibit P/Q-type VGCC (Cataldi 2013). The mechanisms of interaction between amyloid and VGCC remain elusive—suggestions include channel phosphorylation by MAP-kinases, free radical formation, and effects on channel trafficking.
Internal Calcium Stores
It has been suggested that intracellular calcium stores play a major role as sources for dysregulation of neuronal calcium levels in aging and AD (Thibault and others 2007). Dysfunction of presynaptic calcium stores leads to neuronal hyperactivity and likely more frequent NMDAR activation in an in vivo mouse model of AD (Lerdkrai and others 2018). Aβ has been suggested to disrupt mitochondrial calcium homeostasis and thus lead to perturbations of cerebral glucose and cholesterol metabolism as well as to accumulation of reactive oxygen species (Barbero-Camps and others 2014; Chen and Yan 2006; Chen and Zhong 2013). In support of this hypothesis, Aβ oligomers were found to promote cell death via endoplasmic reticulum stress, endosomal/lysosomal leakage, and mitochondrial dysfunction (Umeda and others 2011). In contrast to these toxic effects of Aβ, AICD is implicated in regulation of intracellular calcium and energy homeostasis. Possible mechanisms are modulation of calcium transporters between the cytoplasm and intracellular calcium stores (Hamid and others 2007) and transcription of calcium-binding proteins (Cao and Sudhof 2001). Thus, cells lacking AICD exhibit increased calcium levels, hyperpolarized mitochondria, and reduced ATP content (Hamid and others 2007).
In a recent study full-length APP was found to play a critical role in mitochondrial calcium homeostasis in astrocytes. In vivo Ca2+ imaging of astrocytic microdomains revealed a critical role of the amyloid precursor protein for mitochondria (Montagna and others 2019). As astrocytes potently modulate neuronal function by regulating the local supply of ions and energy metabolites, these findings point toward a novel, APP-related mechanism modulating brain function via glial cells.
APP and Synaptic Plasticity
Since many interaction partners of APP mentioned above such as NMDAR and calcium channels are major mediators of short- and long-term synaptic plasticity, it is not surprising that APP intervenes with plasticity-related pathways in various ways (Ludewig and Korte 2017). As synaptic plasticity is the physiological foundation of learning and memory, deficits of which constitute the core symptoms of AD, analysis of APP functions may reveal leverage points to improve these deficits. Therefore, in this section we will highlight the effects of APP on short- and long-term synaptic plasticity, structural plasticity, learning, and memory formation (Fig. 4).
Figure 4.
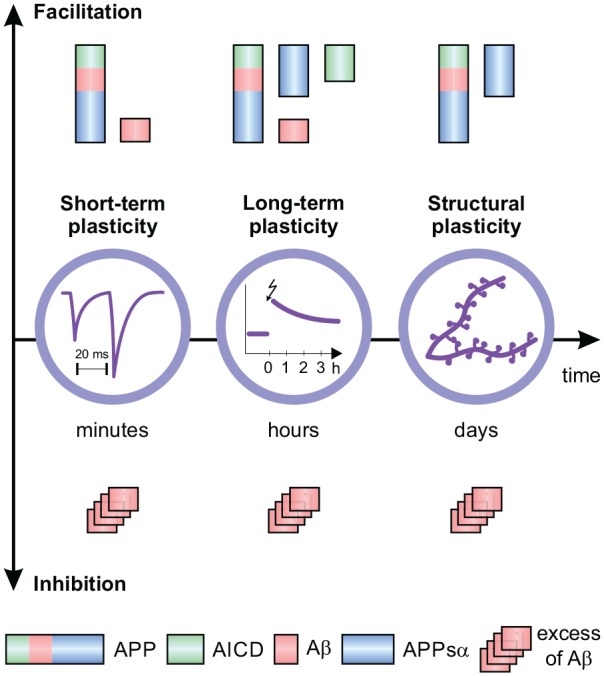
APP and synaptic plasticity. Neuronal plasticity takes place at different time scales: from short-term plasticity (STP) in the order of milliseconds to hundreds of milliseconds to long-term plasticity in the order of hours, days, and years. These functional changes induce structural plasticity such as formation of synapses and dendritic spines. APP and picomolar concentrations of Aβ facilitate STP by interaction with the presynaptic vesicle release machinery, GABAergic and cholinergic receptors, and calcium channels, whereas higher, pathological concentrations of Aβ inhibit STP and long-term potentiation (LTP). APP is necessary for LTP, which is mediated by APPsα and AICD. Physiological concentrations of Aβ promote long-term depression (LTD). APP and APPsα exert trophic effects on spine formation and dendritic arborization promoting structural plasticity, while Aβ oligomers as in AD act toxic and lead to reduced numbers of spines and dendritic branches.
Long-Term Potentiation and Depression
LTP and LTD (long-term depression) are plasticity processes over the time scale of several hours which are mediated by NMDAR and the subsequent intracellular calcium cascades. LTP deficits are a prominent feature of transgenic AD mouse models (Pozueta and others 2013). On the other hand, LTP is also abolished in APP-deficient transgenic mice (Dawson and others 1999; Seabrook and others 1999). Aged APP-KO mice (9 to 12 months) show impaired learning behavior and LTP (Ring and others 2007; Tyan and others 2012). The age-dependent alterations in synaptic plasticity and other biological functions indicate that APLP2—and, possibly, APLP1—may perform redundant functions within the protein family. Interestingly, adult and aged APLP2-KO mice behave like wildtype mice (Midthune and others 2012; Weyer and others 2011), while aged APLP1-KOs have impaired basal synaptic transmission and reduced miniature excitatory-postsynaptic currents frequency, while LTP is normal (Schilling and others 2017; Vnencak and others 2015). It seems likely that endogenous APP is able to compensate for the genetic ablation of APLP2 with age, while vice versa APLP2 is incapable to compensate the loss of APP in aged animals. Furthermore, APLP1 is an essential player in the maintenance of synaptic transmission.
Several studies suggest that the APP intracellular domain (AICD) plays essential roles at synapses. APPΔCT15-DM mice lacking the last 15 aa of APP including the highly conserved YENPTY motif on an APLP2 deficient background show impaired induction and maintenance of LTP. Impaired synaptic plasticity was accompanied by severely altered hippocampus-dependent behavior. Acute hippocampal slices of this mouse strain showed altered postsynaptic properties and a trend toward defective protein synthesis-dependent late-LTP, presumably caused by abolished YENPTY interactions (Klevanski and others 2014). Further known APP interaction partners comprise Dab1, Shc, Grb, and Mint/X11 proteins. Mint/X11 proteins promote clathrin-mediated endocytosis of APP and are involved in the transport of APP to the cell-surface (Aydin and others 2012; van der Kant and Goldstein 2015). Of particular importance might be the interaction with the adapter protein family Fe65. Fe65/Fe65L1 double-deficient mice show a phenotype of cortical dysplasia similar to APP triple-KO animals (Guenette and others 2006). Co-localization of Fe65 with APP in the endoplasmic reticulum (ER) and Golgi apparatus alters the translocation of APP to the cell surface (Sabo and others 1999). As APP family members are enriched in intracellular compartments under resting conditions, this co-localization might be coupled to activity-dependent synaptic plasticity and thus be important for synaptic function (Thinakaran and Koo 2008). The complex of AICD and Fe65 is known to bind Tip60, building a transcription factor and thus regulating gene transcription after shuttling into the nucleus (Cao and Sudhof 2001). This process is essential for protein synthesis-dependent L-LTP. Recently, a study showed that Fe65 proteins bind to all APP family members and might act downstream of cell-surface APP/APLPs at central synapses (Strecker and others 2016). Moreover, a single aa difference between the intracellular domains (ICD) of APP and APLP2 enabled induction of LTD via the AICD, but not the APLP2-ICD (Trillaud-Doppia and others 2016). More recently, the same group reported a role of AICD in metaplasticity (Trillaud-Doppia and Boehm 2018). In this study, neurons expressing full-length chimeric APP (the Aβ domain was exchanged for its homologous domain from APLP2) showed facilitated LTD and completely blocked LTP induction while basic synaptic transmission and plasticity were unaltered. These effects within a very short time frame suggest that AICD might also act independent of gene transcription.
Several studies highlight the LTP-facilitating function of APPsα in chicks and mice (Ishida and others 1997; Meziane and others 1998; Mileusnic and others 2004). Expression of the APPsα fragment on an APP-knockout background (Ring and others 2007) or an APP/APLP2-knockout background (Weyer and others 2014; Zhang and others 2013) was sufficient to rescue LTP deficits. Even acute application of APPsα (10 nΜ), but not APPsβ (50 nΜ), was sufficient to restore impairments in LTP in conditional APP/APLP2 DKO mice (Hick and others 2015). Moreover, in contrast to APPsα the knock-in of APPsβ in mice was unable to rescue the perinatal lethality seen for APP and APLP2 DKO mice (Li and others 2010; Li and others 2011; Weyer and others 2011), confirming functional differences between these two large extracellular domains. Several approaches have been used to reduce the APPsα content, for example, conditional KO of the major α-secretase ADAM-10 (Prox and others 2013) or application of conventional α-secretase inhibitors in vitro and in vivo (Taylor and others 2008). These manipulations resulted in strongly impaired LTP. Recently, it was shown that viral-driven expression of APPsα rescues synaptic plasticity in conditional APP/APLP2 DKO mice (Richter and others 2018; Tan and others 2018). The same approach has restored impaired LTP in the APP/PS1 tg mouse model (Fol and others 2016). In contrast, the modulatory role of APPsβ for synaptic plasticity is less clear (Hick and others 2015; Richter and others 2018; Taylor and others 2008). Of course, the effects of Aβ peptide accumulation and plaque formation during AD progression are relevant and intensely studied (Zott and others 2018). While this disease-related focus points to a negative modulatory function on synaptic plasticity, several studies indicate that picomolar concentrations of monomeric Aβ play an important physiological role. By modulating presynaptic vesicle release, picomolar amounts of Aβ have been proven to be essential for processes of synaptic plasticity like LTP and STP (Abramov and others 2009; Wang and others 2012) via interaction with nicotinic acetylcholine receptors (Lawrence and others 2014). LTP is enhanced following exogenous administration of 200 pM Aβ42 (Puzzo and others 2008). Furthermore, Aβ42 alters the counterpart of LTP, LTD. In vivo injection of 1 to 2 pmol Aβ42 was shown to facilitate LTD as well as reversal of LTD in the CA1 region of the hippocampus (Kim and others 2001). The underlying mechanism is likely related to NMDAR and AMPAR. Synthetic Aβ42 peptides as well as naturally secreted Aβ from APP/PS1 transgenic mice promote the endocytosis of NMDAR from the postsynaptic surface, depressing the NMDAR current in wild-type cortical neurons (Snyder and others 2005). Furthermore, Aβ induced synaptic depression involves enhanced loss of AMPA receptors from dendritic spine surfaces via endocytosis. Interestingly, this is mediated by signaling pathways similar to that occurring during LTD, involving calcineurin and p38 MAP kinase activity (Chen and others 2013; Zhao and others 2004).
Recently an in vivo study on the β-secretase BACE-1 confirmed the essential role of proteolytic peptides generated within the amyloidogenic pathway. Loss of BACE-1 resulted in increased granule cell excitability and prolonged paired-pulse-inhibition as well as altered network gamma oscillations and impaired synaptic plasticity (Vnencak and others 2019).
In contrast, nanomolar concentrations of Aβ42 have deleterious, neurotoxic effects. Especially a ~7 kDa Aβ extract from human AD brains seems to be particularly toxic (Brinkmalm and others 2019). Application of nanomolar amounts of synthetic Aβ42, Aβ40, or Aβ25-35 (200 nM to 1 µM) in acute slices inhibits LTP induction in the hippocampus (Chen and others 2000; Wang and others 2017; Zhao and others 2004). One of the underlying mechanisms is inhibition of AMPAR phosphorylation (Zhao and others 2004). Moreover it has been demonstrated that the intraneuronal injection of 1 to 1000 nM Aβ42 (Ripoli and others 2014) as well as cerebral microinjection of heterogeneous human Aβ (~3 pg) in rats in vivo (Walsh and others 2002) depressed basal synaptic transmission and LTP.
Interestingly, Wang and others (2017) showed that Aβ-mediated impairments of synaptic plasticity depend on the expression of APP. The synaptotoxic effects of Aβ go along with presynaptic alterations disturbing the excitation/inhibition (E/I) balance. The recently discovered peptides Aη-α and Aη-β, both generated via the η-secretase pathway, have been shown to have opposing functions on LTP, similar to the APPsα and Aβ counterbalance (Willem and others 2015). While both peptides had no influence on baseline synaptic transmission, hippocampal LTP was severely impaired by Aη-α but not by Aη-β. Willem and others (2015) used either Aη-α conditioned medium or 100 nM synthetic Aη-α and observed a reduction of LTP. Interestingly, a concentration-dependent action of murine or human recombinant APPsα has been shown (Hick and others 2015; Kuchibhotla and others 2008; Richter and others 2018; Tan and others 2018; Taylor and others 2008). All studies highlight a facilitating action of recombinant APPsα in a range of 1 to 11 nM on LTP, while for Aη-α no other concentrations have been tested so far. Moreover, the application of higher APPsα amounts had no effect or resulted even in reduced LTP when reaching 3300 nM (Taylor and others 2008). Interestingly, by analogy to APPsα and APPsβ, Aη-α and Aη-β differ in their sequence by 16 C-terminal aa, which are only present in APPsα and Aη-α. This peptide sequence contains a predicted neuroprotective domain (Furukawa and others 1996) and was recently shown to be sufficient to facilitate LTP in a manner similar to APPsα (Richter and others 2018).
Short-Term Plasticity
STP takes place on a timescale of milliseconds to hundreds of milliseconds and is dependent on presynaptic factors such as depletion of the transmitter vesicle pool as well as postsynaptic factors such as feed-forward inhibition. APP was found to affect STP on GABAergic interneurons (Yang and others 2009). These authors reported increased L-type voltage gated Ca2+ channel (LTCC) activity, and Hefter and others (2016) showed that APP, possibly via the APPsα fragment, stabilizes Ca2+ homeostasis by regulating the inhibition of LTCCs. Besides APP, its homologue APLP1 is also implicated in GABAergic transmission. APLP1-KO and aged APP-KO mice exhibit reduced GABAergic mediated paired-pulse depression (PPD; Seabrook and others 1999; Vnencak and others 2015) suggesting that APP/APLP1 interaction with GABA provides a negative-feedback mechanism to preserve homeostatic control of neural circuits. APLP1 localized at the presynaptic active zone, similar to APP and APLP2 (Lassek and others 2016; Rice and others 2019). It contributes to neuronal adhesion (Kaden and others 2009; Mayer and others 2016; Schilling and others 2017) and interacts with signaling molecules of the synaptic vesicle release machinery (Barthet and others 2018; Del Prete and others 2014; Lassek and others 2014; Sullivan and others 2014). Indeed, the intracellular regions of APP, APLP2, and CTF-β interact with several presynaptic proteins involved in vesicle dynamics including Rab, AP-2 subunits, synaptotagmins, clathrin, and complexin (Del Prete and others 2014; Fanutza and others 2015). Depletion of APP causes a reduction in synaptophysin, synaptotagmin-1, and SV2A protein levels pointing toward a role of APP in controlling the synaptic vesicle protein content in the presynaptic active zone. Contrary, an increase in synaptic vesicle proteins is observed, when APLP1 or APLP2 are depleted together with APP (Lassek and others 2014). These results were confirmed in a more recent study by Lassek and others (2016), using the conditional APP/APLP2 DKO mouse model. Moreover, Hick and others (2015) observed altered paired-pulse-facilitation (PPF) behavior at the Schaffer collateral-CA1 pathway for a conditional APP/APLP2 double KO (DKO) mouse model, a deficit that was restored by the viral-mediated re-expression of the APPsα fragment (Richter and others 2018). It seems likely that APPsα interacts with α7-nAChRs at pre-synapses to increase glutamate release via a 16 aa long sequence located within the C-Terminus of APPsα. Consistently as APPsβ does not contain these 16 aa, it fails to enhance α7-nAChRs mediated currents, to restore LTP and PPF (Richter and others 2018). In addition, high amounts of APPsα were found to interact with the GABAB-R, enhancing short-term facilitation via inhibition of synaptic vesicle release in mouse hippocampal synapses (Rice and others 2019). Aβ also regulates STP by modulation of presynaptic nAchR, presynaptic calcium channels, and vesicle proteins (please refer to sections “APP and Synaptic Transmission” and “Calcium Signaling” for more details).
Structural Plasticity-Dendritic Spine Morphology/Arborization
Long-term plasticity over days to years is reflected in structural changes including synaptogenesis, formation of dendritic spines, and dendritic sprouting. These processes are not only important during development but also take place throughout the whole adult life and are indispensable for healthy brain function (Holtmaat and Svoboda 2009). In this respect it is noteworthy that several studies provide evidence that APP is involved in processes of neurite outgrowth and branching (Perez and others 1997; Young-Pearse and others 2008). Whereas spine density is reduced in aged APP-KO animals, it remains unaltered in APLP2-KO mice as well as in organotypic hippocampal cultures (OHCs) of APLP2-KOs in vitro (Lee and others 2010; Midthune and others 2012; Weyer and others 2011). Moreover, the quantitative analysis of adult hippocampal CA1 neurons of conditional APP/APLP2 DKO mice yielded pronounced reductions in total neurite length, dendritic branching, reduced spine density, spine head volume, and proportion of mushroom spines, which are thought to represent mature synapses (Hick and others 2015). Weyer and others (2011) therefore elucidated which secreted APP-fragment (APPsα or APPsβ) contributes to this effect. They observed normal neuronal morphology in organotypic hippocampal cultures and revealed unaltered neurite length and dendritic branching in APPsα DKO mice (no full length APP or APLP2). In addition, this study reported no significant alteration in spine density in the mid-distal portion of apical dendrites, corresponding roughly to the region in which CA3 axons terminate on CA1 dendrites (Weyer and others 2011; Weyer and others 2014). Overall, these experiments suggest that lack of transmembrane APP/APLP2 isoforms in presence of intact APPsα and APLP1 expression does not affect dendritic structure and spine density of CA1 hippocampal neurons of mice. It can be concluded that APPsα is essential for spine density regulation and dynamics, as highlighted by the effects of exogenous application of the extracellular domain (Tyan and others 2012) or by the knock-in of APPsα (Weyer and others 2011). Weyer and others (2014) reported that CA1 neurons from APLP1-KO or APLP2-KO mice showed unaltered neuronal morphology and no changes in spine density, whereas APP-KO mice revealed a highly reduced dendritic complexity in mid-apical dendrites. Despite unaltered morphology of APLP2-KO pyramidal hippocampal neurons, APP/APLP2-DKO showed an additional branching defect in proximal apical dendrites, indicating redundancy and a combined function of APP and APLP2 for dendritic morphology. Hippocampal principal neurons in conventional APP-KO showed a significant reduction in spine density and a deficit in the number of large mushroom spines. No further decrease in spine density, however, was observed in APP/APLP2-DKO mice. Expression of APPsα in these DKO mice, which lack the transmembrane APP and express only the secreted APPsα fragment, was sufficient to rescue the defects in spine density observed in APP-KO mice. Collectively, these studies reveal a combined role of APP and APLP2 for dendritic architecture and a unique function of secreted APPsα for normal synaptic density. These findings are of high relevance for AD, since it has been reported that Aβ release from axons and dendrites reduces local spine number and plasticity (Wei and others 2009).
Authors from the same group reported increased spine numbers in the somatosensory cortex of 4-month-old APP-KO mice (Bittner and others 2009; Zou and others 2016). In a subsequent study, they specified that this finding explicitly included mushroom spines only, while the fraction of thin spines was decreased (Zou and others 2016). Furthermore, they found significantly decreased turnover rates of dendritic spines in APP-KO mice. These alterations of spine morphology might contribute to impaired spine plasticity as thin spines are less stable. This is supported by the finding that 5 weeks exposure to an enriched environment has no effect on spine density in APP-KO mice, while physiologically enhancing it in controls (Montagna and others 2019; Zou and others 2016).
Taken together, APP plays a role in developmental- and plasticity-related changes in spine structure, resulting in a striking decrease in spine density in APP-KO mice.
Learning and Memory
Core symptoms of AD include impaired working, episodic and spatial memory in parallel to hippocampal atrophy, one of the first brain regions affected by the disease (Scheltens and others 2016). These symptoms could be reliably reproduced in several mouse models of AD and show a correlation to the amyloid plaque burden (Sasaguri and others 2017). However, in some models memory deficits arise before there is a detectable amyloid pathology burden (Sasaguri and others 2017). Cognitive deficits in human patients do also lack a strict correlation with detectable amyloid pathology (Jung and others 2017). An important memory-supporting role of APP is suggested by studies in APP-knockout mice, which show spatial memory impairments in the Morris water maze as well as impairments in conditioned avoidance and novel object recognition tests (Dawson and others 1999; Ring and others 2007). Conditional DKO mice lacking both APP and APLP2 exhibit impaired hippocampus-dependent behavior and learning, including deficits in the nesting test, in the Morris water maze and the radial arm maze (Hick and others 2015). Several studies have shown a strong positive correlation between APPsα levels and cognitive performance. Intraventricular application of picomolar concentrations of APPsα enhanced memory both in mice with pharmacologically induced memory deficits as well as in healthy mice (Meziane and others 1998). In aged rats, exogenous intraventricular APPsα injection ameliorated age-dependent LTP and memory deficits, enhancing object location memory (Xiong and others 2017). Remarkably, in an AD mouse model with amyloid plaques and deficient synaptic function and memory, application of APPsα via viral gene transfer could rescue most of these deficits, decreasing plaque load, increasing dendritic spine density, rescuing LTP, and restoring performance in the Morris water maze (Fol and others 2016). In another study of the same group expression of APPsα on the background of conditional knockout of APP and APLP2 also rescued deficits in spine density, LTP, and PPF, as well as spatial reference memory. Notably, APPsβ had no significant effects in these animals (Richter and others 2018). While these results highlight the importance of APPsα, it is important to keep in mind that the AICD has also been shown to harbor important functions in plasticity and memory. Mice lacking the AICD and APLP2 (to prevent compensatory mechanisms) exhibit impairments in LTP, in hippocampus-dependent behavior and malformations of the neuromuscular junction (Klevanski and others 2015). A recently discovered interaction of AICD with heterotrimeric G-protein subunits occurring in lipid raft microdomains was shown to provide a positive, regulatory feedback loop enhancing non-amyloidogenic APP processing, thereby preserving spatial memory impairments in 5xFAD mice (Deyts and others 2019). While having detrimental effects on plasticity and memory in nanomolar concentrations as shown in previous sections, physiological (picomolar) concentrations of Aβ may be involved in memory formation. Thus, enhanced levels of hippocampal Aβ production were quantified during memory induction for contextual fear learning (Puzzo and others 2011).
APP and Network Oscillations
The brain exhibits rhythmic activity over a wide range of frequencies from delta oscillations below 1 Hz to sharp wave-ripple oscillations (SWP-R) of >200 Hz (Buzsáki and Draguhn 2004). These oscillations represent distinct cognitive and behavioral states and entrain neuronal activity within and between brain areas. In the previous sections we outlined multiple functions of APP and its metabolites both at the pre- and postsynaptic sites and in the cytosol. The APP protein family can both negatively and positively modulate main transmission systems including glutamatergic, GABAergic, and cholinergic signaling and tune neuronal excitability by regulating intracellular calcium levels. As these are pivotal factors contributing to network activity, the cellular functions of APP suggest its importance for the rhythmic activity of the brain and cognition. Indeed, AD patients exhibit prominent changes in the function of large brain networks, particularly those involved in memory formation (Sperling and others 2009; Zott and others 2018). These changes include deceleration of the alpha rhythm (8–12 Hz) toward theta frequency (4–8 Hz in humans), aberrant gamma (30–80 Hz) activity (Hamm and others 2015; Kitchigina 2018), and reduction of slow oscillations during sleep (Lucey and Holtzman 2015). Hyperactivity of local networks such as the hippocampus in parallel to dysfunctional connectivity between large brain networks are suggested to underlie cognitive dysfunction in AD (Zott and others 2018). These clinical findings of neuronal hyperactivity are well in line with results from animal studies discussed in previous sections, showing that pathological Aβ deposition along with reduced non-amyloidogenic signaling lead to aberrant neuronal activity. Mouse models lacking APP or expressing mutated forms of the protein exhibit alterations in their network function (Korte and others 2012), demonstrating that APP-dependent changes at synapses and cells cause effects at the network level. In the following section we review the effects of APP on physiological theta/gamma and SPW-R oscillations as well as pathophysiological network states (Fig. 5).
Figure 5.

APP and network activity. Left: Theta-gamma activity in the murine hippocampus in vivo. The local field potential (LFP) trace (bottom) shows ~5 Hz theta activity with superimposed gamma oscillations on each theta peak. These gamma oscillations are clearly distinguishable as peaks in the ~60 Hz range in the time-frequency plot (spectrogram, upper part). Middle column: Sharp wave-ripple activity (SWP-R) in the murine hippocampus in vivo. Sharp waves are of <100 ms duration and superimposed by ~200 Hz ripple activity. Right column: epileptiform activity recorded in an acute hippocampal mouse slice. APP-KO mice exhibit reduced gamma power in slices and reduced gamma-theta coupling in vivo. In AD models gamma power and gamma-theta coupling are reduced. Mice lacking APP or overproducing amyloid show higher susceptibility to seizures. SPW-R oscillations in vitro are, however, unaltered in these mice. APPsα-KI mice on an APP/APLP2-DKO background exhibit unaltered network activity. APPsα-DM mice expressing APPsα on an APP/APLP2-double knockout background also show largely unaltered network activity in vitro, but their network function has not been studied in vivo.
Theta/Gamma Oscillations
Theta-nested gamma oscillations are associated with active waking and exploratory behavior in rodents and with cognitive activity in humans (Tamura and others 2017). Compelling evidence from both in vivo and in vitro studies shows reductions in the power of gamma oscillations in mouse models of AD (Korte and others 2012; Verret and others 2012). For example, mice expressing human APP with an AD-associated mutation show impaired gamma activity in vivo. This deficit is associated with reduced expression of the voltage-dependent sodium channel subunit Nav1.1 in parvalbumin-positive interneurons and can be rescued by increasing expression of this subunit (Verret and others 2012). Due to compensation by the homologues, gamma activity is only slightly affected in mice lacking either APP or APLP2 (Zhang and others 2013). In this ex vivo study, gamma frequency in the hippocampal CA3 region in acute slices from APLP2-KO mice was significantly reduced as compared to WT or mice expressing APPsα on the otherwise lethal APP/APLP2-KO background. These data suggest that APP and APLP2 are not essential for gamma oscillations, but rather fine-tune this network activity, for example, modulating its frequency.
The rescue of baseline synaptic transmission, network function, and short-term plasticity by APPsα once again emphasizes the pivotal role of this fragment. While the power of theta and gamma oscillations remained unaltered in APP-deficient mice in vivo, the coupling between these oscillations was strongly diminished specifically in the parietal cortex and the hippocampus, suggesting perturbed communication between large brain networks in these animals (Zhang and others 2016). The role of APLP2 in theta/gamma oscillations in vivo has not been studied yet.
Sharp Wave-Ripple Oscillations (SPW-R)
SPW-R represent a form of network activity that is prominent in the hippocampal formation during sleep and awake immobility. During ripples (~200 Hz oscillations) distinct populations of neurons (ensembles) fire with sub-millisecond precision, possibly strengthening connections between co-active neurons and their targets. In line with this notion, SPW-R are crucial for memory consolidation (Buzsáki 2015). Similar to gamma, SPW-R oscillations prevail in APP-deficient mice in vitro (Hefter and others 2016; Zhang and others 2013). In the Swedish mutant mouse model of AD SPW-R activity was also unaltered in comparison to WT despite reduced synaptic excitation in the hippocampal CA1 (Hermann and others 2009). Whether APP affects SPW-R in vivo remains elusive. Further in vivo studies on the influence of APP on SPW-R as a sleep-related activity highly relevant for memory formation might yield promising results as reciprocal connections between sleep disturbances, memory deficits, and amyloid levels have been established in AD patients (Zott and others 2018).
Neuronal Hyperactivity and Epilepsy
Epilepsy is a pathological condition of recurrent hypersynchronized network activity. AD patients are prone to develop epileptic seizures (Born 2015; Kitchigina 2018), similar to mouse models of AD (Born 2015; Verret and others 2012). On the other hand, mice deficient of APP are also more susceptible to seizures, as shown in the kainate model of epilepsy (Steinbach and others 1998). Neurons in vicinity of amyloid plaques exhibit aberrant activity (Busche and others 2008), which might lead to the development of an epileptic focus. This hyperactivity may, in turn, favor amyloid deposition and compromise the metabolic homeostasis of neurons (Bero and others 2011). In a recent study on another transgenic AD mouse model hyperactivity was found in several brain regions including the hippocampus, beginning at a relatively early stage of the disease. This was associated with the extent of the subsequent tau pathology (Liu and others 2018). Reduction of Aβ levels by BACE inhibition could rescue circuit function and memory by ameliorating neuronal hyperactivity in a mouse model of AD (Keskin and others 2017). However, the use of BACE inhibitors as potential therapeutic tools for AD is restricted by the importance of its cleavage products in physiological concentrations for synaptic plasticity and network function (as described in section “Long-Term Potentiation and Depression”). In line with this finding, decreasing interstitial amyloid levels is not sufficient to normalize the neuronal hyperactivity typical for AD. Thus, application of amyloid antibodies in a mouse model of AD successfully decreased Aβ levels, but even exacerbated neuronal dysfunction by facilitating hyperactivity (Busche and others 2015).
An Integrative Approach to APP Functions in the CNS
In the sections above, we highlighted various functions of APP family proteins at the synaptic, cellular, network, and systemic levels. The current body of knowledge on APP derives to a large extent from transgenic mouse lines with full or partial depletion of APP/APLP or with APP overexpression and increased amyloidogenesis. While this approach has significantly advanced our understanding of particular APP functions, our knowledge about the physiological role of APP in the nervous system is still far from complete. Moreover, the relationship between altered protein levels and disturbed functions may be indirect and nonlinear. Indeed, a large body of literature suggests a U-shaped relationship between APP levels and various observables. For instance, both mice lacking as well as overexpressing APP exhibit age-dependent deficits in LTP and learning, disturbed calcium homeostasis, altered oscillatory activity, and increased vulnerability to seizures and metabolic stress (for details, see sections above). Further complicating factors are the mutual compensation of deficient functions by the homologous proteins APP, APLP1, and APLP2 (Herms and others 2004), differences between functions of full-length APP or its fragments, and the complex regulation of APP processing by secretases. The unexpected discovery of the η-secretase pathway (Willem and others 2015) after three decades of research on APP demonstrates how much is still to be explored. Importantly, APP is not confined to neurons, but is ubiquitously expressed und thus is also suggested to affect the function of astrocytes, microglia, other immune and endothelial cells (Montagna and others 2019). It is likely that APP does also influence brain function via its effects in non-neuronal cells. The diverse expression of α- versus β-secretases may heavily influence effects of APP in different neuronal subtypes, together with the different availability of downstream interaction partners (e.g., the predominance of GluN2A vs. GluN2B NMDAR). Finally, research on cognitive performance in rodents may not be fully applicable to the situation in humans, despite the high conversation of APP and its isoforms (Sasaguri and others 2017).
In face of all these obstacles, it is not surprising that many questions remain open. Nevertheless, some functions and mechanisms have been firmly established: Expression and processing of APP is controlled by neuronal activity; APP and its metabolites potently modulate synaptic function in various neurotransmitter systems; APP regulates intracellular calcium levels via interaction with calcium channels as well as intracellular stores in an activity-dependent manner; it affects neuronal excitability, gene expression, cell proliferation, and pro-apoptotic pathways. It is known that APP family proteins or their fragments unfold their action in the nucleus (AICD), mitochondria, ER, and lysosomes (Aβ), in the cytosol (AICD and Aβ), at the cell membrane (full-length APP), or in the extracellular space (APPsα). The effects of the APP metabolites APPsα and Aβ mostly antagonize each other (inhibition vs. excitation of synaptic transmission, increasing vs. decreasing calcium levels and cellular excitability, pro-survival vs. pro-apoptotic pathways). In line with this functional antagonism, APPsα lowers Aβ levels by direct inhibition of BACE (Obregon and others 2012). Therefore, regulation of APP is a key mechanism for development and function of the nervous system, and possibly a key target for therapeutic interventions.
Its multiple functions make APP a perfect candidate for acting as an acute-phase protein under conditions of cellular stress, for example, in hypoxia-ischemia (HI) or traumatic brain injury (TBI). Indeed, APP is rapidly upregulated in response to metabolic stress and following TBI (Van Den Heuvel and others 2007), and APP-knockout mice experience increased mortality in response to HI and TBI (Koike and others 2012; Plummer and others 2016). These protective effects were shown to be once again carried out by APPsα (Hefter and others 2016; Plummer and others 2016) via inhibition of LTCC (Hefter and others 2016). However, presence of membrane-bound APP (but not APLP1 or APLP2) was required to mediate neuroprotective effects of APPsα by activation of the Akt survival pathway via its C-terminal domain (Milosch and others 2014).
Several lines of evidence indicate a relationship between AD and HI conditions: stroke, heart failure, and metabolic diseases comprise risk factors for AD. Vice versa, AD patients are at a higher risk to suffer a stroke (Tolppanen and others 2013). TBI is also a well-known risk factor for AD. We hypothesize that APP exerts protective effects mostly via APPsα in various acute conditions of brain injury. However, chronic and severe injuries may lead to a maladaptation of APP expression and processing, followed by diminished secretion of APPsα, excessive production of Aβ, and development of AD (Fig. 6). Restoring the balance between APP metabolites toward APPsα is being increasingly discussed as a potential therapeutic strategy in AD (Haass and Willem 2019; Habib and others 2017; Hefter and Draguhn 2017; Mockett and others 2017).
Figure 6.

Summary of APP functions on multiple biological scales. APP expression and processing are tightly regulated by neuronal activity. Under physiological conditions (e.g., during learning) and compensable stress, upregulation of APP expression and α-secretase cleavage leads to increased secretion of APPsα and can promote synaptic transmission and plasticity, regulate calcium signaling and homeostasis, thus influencing network activity and promoting learning and memory formation. Furthermore, AICD, which is a product of all APP processing pathways, also modulates these processes. Αt physiological concentrations, Aβ also appears to harbor important functions in regulation of synaptic transmission and plasticity. Chronic and severe insults such as hypoxia-ischemia, on the other hand, might lead to overexpression of APP and/or shift the balance to amyloidogenic signaling. Aβ at high concentrations forms oligomers and plaques that act toxic at multiple scales, inhibiting synaptic transmission and plasticity, leading to chronically increased cellular calcium levels, impairments in network function, and ultimately to degeneration and dementia.
Currently available treatment options for AD are purely symptomatic and can only slightly delay the progression of dementia (Knight and others 2018). The recent failure of large clinical anti-amyloid trials (van Dyck 2018) is a major challenge for presently developed therapeutic approaches. While AD is likely diagnosed far too late for any complete remission, there are no means of prediction or prevention of the disease. We are in urgent need of new interventions at the synaptic, cellular, and network levels, especially at early stages of the disease. Therefore, it seems imperative to tackle the issues described above and advance our understanding of function and malfunction of APP family proteins.
Acknowledgments
We thank Dr. Martin Both and Dr. Yevgenij Yanovsky for providing the representative recording traces for Figure 5A and B, respectively.
Footnotes
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work has been supported by a grant from the Deutsche Forschungsgemeinschaft (DFG, DR 326/10-1 and KO 1674/11-1). DH is supported by the Physician Scientist Program of the Medical Faculty of Heidelberg University. MK is supported by the Helmholtz-Gemeinschaft, Zukunftsthema “Immunology and Inflammation” (ZT-0027).
ORCID iD: Dimitri Hefter  https://orcid.org/0000-0003-2861-7865
https://orcid.org/0000-0003-2861-7865
References
- Abramov E, Dolev I, Fogel H, Ciccotosto GD, Ruff E, Slutsky I. 2009. Amyloid-beta as a positive endogenous regulator of release probability at hippocampal synapses. Nat Neurosci 12(12):1567–76. [DOI] [PubMed] [Google Scholar]
- Almeida CG, Tampellini D, Takahashi RH, Greengard P, Lin MT, Snyder EM, and others. 2005. Beta-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiol Dis 20(2):187–98. [DOI] [PubMed] [Google Scholar]
- Alzheimer’s Association Calcium Hypothesis Workgroup. 2017. Calcium hypothesis of Alzheimer’s disease and brain aging: a framework for integrating new evidence into a comprehensive theory of pathogenesis. Alzheimer’s Dement 13(2):178–82. [DOI] [PubMed] [Google Scholar]
- Anekonda TS, Quinn JF. 2011. Calcium channel blocking as a therapeutic strategy for Alzheimer’s disease: the case for isradipine. Biochim Biophys 1812(12):1584–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aydin D, Weyer SW, Muller UC. 2012. Functions of the APP gene family in the nervous system: insights from mouse models. Exp Brain Res 217(3–4):423–34. [DOI] [PubMed] [Google Scholar]
- Bading H. 2013. Nuclear calcium signalling in the regulation of brain function. Nat Rev Neurosci 14(9):593–608. [DOI] [PubMed] [Google Scholar]
- Bading H. 2017. Therapeutic targeting of the pathological triad of extrasynaptic NMDA receptor signaling in neurodegenerations. J Exp Med 214(3):569–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbero-Camps E, Fernandez A, Baulies A, Martinez L, Fernandez-Checa JC, Colell A. 2014. Endoplasmic reticulum stress mediates amyloid beta neurotoxicity via mitochondrial cholesterol trafficking. Am J Pathol 184(7):2066–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthet G, Jorda-Siquier T, Rumi-Masante J, Bernadou F, Muller U, Mulle C. 2018. Presenilin-mediated cleavage of APP regulates synaptotagmin-7 and presynaptic plasticity. Nat Commun 9(1):4780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bero AW, Yan P, Hon Roh J, Cirrito JR, Stewart FR, Raichle ME, and others. 2011. Neuronal activity regulates the regional vulnerability to amyloid-β deposition. Nat Neurosci 14(6):750–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittner T, Fuhrmann M, Burgold S, Jung CKE, Volbracht C, Steiner H, and others. 2009. Gamma-secretase inhibition reduces spine density in vivo via an amyloid precursor protein-dependent pathway. J Neurosci 29:10405–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Born HA. 2015. Seizures in Alzheimer’s disease. Neuroscience 286:251–63. [DOI] [PubMed] [Google Scholar]
- Brinkmalm G, Hong W, Wang Z, Liu W, O’Malley TT, Sun X, and others. 2019. Identification of neurotoxic cross-linked amyloid-beta dimers in the Alzheimer’s brain. Brain 142(5):1441–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busche MA, Eichhoff G, Adelsberger H, Abramowski D, Wiederhold K, Haass C, and others. 2008. Clusters of hyperactive neurons near amyloid plaques in a mouse model. Science 321(5896):1686–90. [DOI] [PubMed] [Google Scholar]
- Busche MA, Grienberger C, Keskin AD, Song B, Neumann U, Staufenbiel M, and others. 2015. Decreased amyloid-β and increased neuronal hyperactivity by immunotherapy in Alzheimer’s models. Nat Neurosci 18(12):1725–7. [DOI] [PubMed] [Google Scholar]
- Buzsáki G. 2015. Hippocampal sharp wave-ripple: a cognitive biomarker for episodic memory and planning. Hippocampus 25(10):1073–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsáki G, Draguhn A. 2004. Neuronal oscillations in cortical networks. Science 304(5679):1926–9. [DOI] [PubMed] [Google Scholar]
- Cai Z, Zhang J, Chen M, Wang J, Xiao P, Yang L, and others. 2016. Both pre- and post-synaptic alterations contribute to aberrant cholinergic transmission in superior cervical ganglia of APP-/- mice. Neuropharmacology 110(pt A):493–502. [DOI] [PubMed] [Google Scholar]
- Caldwell JH, Klevanski M, Saar M, Müller UC. 2013. Roles of the amyloid precursor protein family in the peripheral nervous system. Mech Dev 130(6–8):433–46. [DOI] [PubMed] [Google Scholar]
- Cao X, Sudhof TC. 2001. A transcriptionally [correction of transcriptively] active complex of APP with Fe65 and histone acetyltransferase Tip60. Science 293(5527):115–20. [DOI] [PubMed] [Google Scholar]
- Cataldi M. 2013. The changing landscape of voltage-gated calcium channels in neurovascular disorders and in neurodegenerative diseases. Curr Neuropharmacol 11(3):276–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang EH, Savage MJ, Flood DG, Thomas JM, Levy RB, Mahadomrongkul V, and others. 2006. AMPA receptor downscaling at the onset of Alzheimer’s disease pathology in double knockin mice. Proc Natl Acad Sci U S A 103(9):3410–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chebib M, Johnston GAR. 1999. The “ABC” of GABA receptors: a brief review. Clin Exp Pharmacol Physiol 26(11):937–40. [DOI] [PubMed] [Google Scholar]
- Chen QS, Kagan BL, Hirakura Y, Xie CW. 2000. Impairment of hippocampal long-term potentiation by Alzheimer amyloid beta-peptides. J Neurosci Res 60(1):65–72. [DOI] [PubMed] [Google Scholar]
- Chen X, Lin R, Chang L, Xu S, Wei X, Zhang J, and others. 2013. Enhancement of long-term depression by soluble amyloid beta protein in rat hippocampus is mediated by metabotropic glutamate receptor and involves activation of p38MAPK, STEP and caspase-3. Neuroscience 253:435–43. [DOI] [PubMed] [Google Scholar]
- Chen X, Yan S Du. 2006. Mitochondrial Aβ: a potential cause of metabolic dysfunction in Alzheimer’s disease. IUBMB Life. 58(12):686–94. [DOI] [PubMed] [Google Scholar]
- Chen Z, Zhong C. 2013. Decoding Alzheimer’s disease from perturbed cerebral glucose metabolism: implications for diagnostic and therapeutic strategies. Prog Neurobiol 108:21–43. [DOI] [PubMed] [Google Scholar]
- Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, and others. 2005. Synaptic activity regulates interstitial fluid amyloid-β levels in vivo. Neuron 48(6):913–22. [DOI] [PubMed] [Google Scholar]
- Clapham DE. 2007. Calcium signaling. Cell 131(6):1047–58. [DOI] [PubMed] [Google Scholar]
- Cousins SL, Dai W, Stephenson FA. 2015. APLP1 and APLP2, members of the APP family of proteins, behave similarly to APP in that they associate with NMDA receptors and enhance NMDA receptor surface expression. J Neurochem 133(6):879–85. [DOI] [PubMed] [Google Scholar]
- Cousins SL, Hoey SEA, Anne Stephenson F, Perkinton MS. 2009. Amyloid precursor protein 695 associates with assembled NR2A- and NR2B-containing NMDA receptors to result in the enhancement of their cell surface delivery. J Neurochem 111(6):1501–13. [DOI] [PubMed] [Google Scholar]
- Dawson GR, Seabrook GR, Zheng H, Smith DW, Graham S, O’Dowd G, and others. 1999. Age-related cognitive deficits, impaired long-term potentiation and reduction in synaptic marker density in mice lacking the beta-amyloid precursor protein. Neuroscience 90(1):1–13. [DOI] [PubMed] [Google Scholar]
- Del Prete D, Lombino F, Liu X, D’Adamio L. 2014. APP is cleaved by Bace1 in pre-synaptic vesicles and establishes a pre-synaptic interactome, via its intracellular domain, with molecular complexes that regulate pre-synaptic vesicles functions. PLoS One 9(9):e108576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deyts C, Clutter M, Pierce N, Chakrabarty P, Ladd TB, Goddi A, and others. 2019. APP-mediated signaling prevents memory decline in Alzheimer’s disease mouse model. Cell Rep 27(5):1345–55.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinamarca MC, Raveh A, Schneider A, Fritzius T, Fruh S, Rem PD, and others. 2019. Complex formation of APP with GABAB receptors links axonal trafficking to amyloidogenic processing. Nat Commun 10(1):1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolev I, Fogel H, Milshtein H, Berdichevsky Y, Lipstein N, Brose N, and others. 2013. Spike bursts increase amyloid-b 40/42 ratio by inducing a presenilin-1 conformational change. Nat Neurosci 16:587–95. [DOI] [PubMed] [Google Scholar]
- Fanutza T, Del Prete D, Ford MJ, Castillo PE, D’Adamio L. 2015. APP and APLP2 interact with the synaptic release machinery and facilitate transmitter release at hippocampal synapses. Elife 4:e09743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foidl BM, Do-Dinh P, Hutter-Schmid B, Bliem HR, Humpel C. 2016. Cholinergic neurodegeneration in an Alzheimer mouse model overexpressing amyloid-precursor protein with the Swedish-Dutch-Iowa mutations. Neurobiol Learn Mem 136:86–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fol R, Braudeau J, Ludewig S, Abel T, Weyer S, Roederer JP, and others. 2016. Viral gene transfer of APPsα rescues synaptic failure in an Alzheimer’s disease mouse model. Acta Neuropathol 131(2):247–66. [DOI] [PubMed] [Google Scholar]
- Furukawa K, Mattson M. 1998. Secreted amyloid precursor protein α selectively suppresses N-methyl-D-aspartate currents in hippocampal neurons: involvement of cyclic GMP. Neuroscience 83(2):429–38. [DOI] [PubMed] [Google Scholar]
- Furukawa K, Sopher BL, Rydel RE, Begley JG, Pham DG, Martin GM, and others. 1996. Increased activity-regulating and neuroprotective efficacy of alpha-secretase-derived secreted amyloid precursor protein conferred by a C-terminal heparin-binding domain. J Neurochem 67(5):1882–96. [DOI] [PubMed] [Google Scholar]
- Gralle M, Ferreira ST. 2007. Structure and functions of the human amyloid precursor protein: the whole is more than the sum of its parts. Prog Neurobiol 82(1):11–32. [DOI] [PubMed] [Google Scholar]
- Green KN, LaFerla FM. 2008. Linking calcium to Abeta and Alzheimer’s disease. Neuron. 59(2):190–4. [DOI] [PubMed] [Google Scholar]
- Guenette S, Chang Y, Hiesberger T, Richardson JA, Eckman CB, Eckman EA, and others. 2006. Essential roles for the FE65 amyloid precursor protein-interacting proteins in brain development. EMBO J 25(2):420–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guntupalli S, Widagdo J, Anggono V. 2016. Amyloid-beta-induced dysregulation of AMPA receptor trafficking. Neural Plast 2016:3204519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C, Kaether C, Thinakaran G, Sisodia S. 2012. Trafficking and proteolytic processing of APP. Cold Spring Harb Perspect Med 2(5):a006270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C, Willem M. 2019. Spotlight secreted APP modulates synaptic activity: a novel target for therapeutic intervention ? Neuron 101(4):557–9. [DOI] [PubMed] [Google Scholar]
- Habib A, Sawmiller D, Tan J. 2017. Restoring soluble amyloid precursor protein alpha functions as a potential treatment for Alzheimer’s disease. J Neurosci Res 95(4):973–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamid R, Kilger E, Willem M, Vassallo N, Kostka M, Bornhövd C, and others. 2007. Amyloid precursor protein intracellular domain modulates cellular calcium homeostasis and ATP content. J Neurochem 102(4):1264–75. [DOI] [PubMed] [Google Scholar]
- Hamm V, Heraud C, Cassel JC, Mathis C, Goutagny R. 2015. Precocious alterations of brain oscillatory activity in Alzheimer’s disease: a window of opportunity for early diagnosis and treatment. Front Cell Neurosci 9:491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. 2010. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci 11(10):682–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefter D, Draguhn A. 2017. APP as a protective factor in acute neuronal insults. Front Mol Neurosci 10:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefter D, Kaiser M, Weyer SW, Papageorgiou IE, Both M, Kann O, and others. 2016. Amyloid precursor protein protects neuronal network function after hypoxia via control of voltage-gated calcium channels. J Neurosci 36(32):8356–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann D, Both M, Ebert U, Gross G, Schoemaker H, Draguhn A, and others. 2009. Synaptic transmission is impaired prior to plaque formation in amyloid precursor protein-overexpressing mice without altering behaviorally-correlated sharp wave-ripple complexes. Neuroscience 162(4):1081–90. [DOI] [PubMed] [Google Scholar]
- Herms J, Anliker B, Heber S, Ring S, Fuhrmann M, Kretzschmar H, and others. 2004. Cortical dysplasia resembling human type 2 lissencephaly in mice lacking all three APP family members. EMBO J 23(20):4106–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hettinger JC, Lee H, Bu G, Holtzman DM, Cirrito JR. 2018. AMPA-ergic regulation of amyloid-beta levels in an Alzheimer’s disease mouse model. Mol Neurodegener 13(1):22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hick M, Herrmann U, Weyer SW, Mallm JP, Tschäpe JA, Borgers M, and others. 2015. Acute function of secreted amyloid precursor protein fragment APPsα in synaptic plasticity. Acta Neuropathol 129(1):21–37. [DOI] [PubMed] [Google Scholar]
- Hoe HS, Fu Z, Makarova A, Lee JY, Lu C, Feng L, and others. 2009. The effects of amyloid precursor protein on postsynaptic composition and activity. J Biol Chem 284(13):8495–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoey SE, Buonocore F, Cox CJ, Hammond VJ, Perkinton MS, Williams RJ. 2013. AMPA receptor activation promotes non-amyloidogenic amyloid precursor protein processing and suppresses neuronal amyloid-beta production. PLoS One 8(10):e78155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoey SE, Williams RJ, Perkinton MS. 2009. Synaptic NMDA receptor activation stimulates alpha-secretase amyloid precursor protein processing and inhibits amyloid-beta production. J Neurosci 29(14):4442–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtmaat A, Svoboda K. 2009. Experience-dependent structural synaptic plasticity in the mammalian brain. Nat Rev Neurosci 10(9):647–58. [DOI] [PubMed] [Google Scholar]
- Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, and others. 2006. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron 52(5):831–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida A, Furukawa K, Keller JN, Mattson MP. 1997. Secreted form of beta-amyloid precursor protein shifts the frequency dependency for induction of LTD, and enhances LTP in hippocampal slices. Neuroreport 8(9–10):2133–7. [DOI] [PubMed] [Google Scholar]
- Jiang S, Li Y, Zhang C, Zhao Y, Bu G, Xu H, and others. 2014. M1 muscarinic acetylcholine receptor in Alzheimer’s disease. Neurosci Bull 30(2):295–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung Y, Whitwell JL, Duffy JR, Edythe A, Machulda MM, Senjem ML, and others. 2017. Regional β-amyloid burden does not correlate with cognitive or language deficits in Alzheimer’s disease presenting as aphasia. Eur J Neurosci 23(2):313–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaden D, Voigt P, Munter LM, Bobowski KD, Schaefer M, Multhaup G. 2009. Subcellular localization and dimerization of APLP1 are strikingly different from APP and APLP2. J Cell Sci 122(pt 3):368–77. [DOI] [PubMed] [Google Scholar]
- Kelly BL, Ferreira A. 2007. Beta-amyloid disrupted synaptic vesicle endocytosis in cultured hippocampal neurons. Neuroscience 147(1):60–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly BL, Vassar R, Ferreira A. 2005. Beta-amyloid-induced dynamin 1 depletion in hippocampal neurons. A potential mechanism for early cognitive decline in Alzheimer disease. J Biol Chem 280(36):31746–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keskin AD, Kekus M, Adelsberger H, Neumann U, Shimshek DR, Song B, and others. 2017. BACE inhibition-dependent repair of Alzheimer’s pathophysiology. Proc Natl Acad Sci U S A 114(32):8631–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Anwyl R, Suh YH, Djamgoz MB, Rowan MJ. 2001. Use-dependent effects of amyloidogenic fragments of (beta)-amyloid precursor protein on synaptic plasticity in rat hippocampus in vivo. J Neurosci 21(4):1327–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitchigina VF. 2018. Alterations of coherent theta and gamma network oscillations as an early biomarker of temporal lobe epilepsy and Alzheimer’s disease. Front Integr Neurosci 12:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klevanski M, Herrmann U, Weyer SW, Fol R, Cartier N, Wolfer DP, and others. 2015. The APP intracellular domain is required for normal synaptic morphology, synaptic plasticity, and hippocampus-dependent behavior. J Neurosci 35(49):16018–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klevanski M, Saar M, Baumkotter F, Weyer SW, Kins S, Muller UC. 2014. Differential role of APP and APLPs for neuromuscular synaptic morphology and function. Mol Cell Neurosci 61:201–10. [DOI] [PubMed] [Google Scholar]
- Knight R, Khondoker M, Magill N, Stewart R, Landau S. 2018. A systematic review and meta-analysis of the effectiveness of acetylcholinesterase inhibitors and memantine in treating the cognitive symptoms of dementia. Dement Geriatr Cogn Disord 45(3–4):131–51. [DOI] [PubMed] [Google Scholar]
- Koike MA, Lin AJ, Pham J, Nguyen E, Yeh JJ, Rahimian R, and others. 2012. APP knockout mice experience acute mortality as the result of ischemia. PLoS One 7(8):e42665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korte M, Herrmann U, Zhang X, Draguhn A. 2012. The role of APP and APLP for synaptic transmission, plasticity, and network function: lessons from genetic mouse models. Exp Brain Res 217(3–4):435–40. [DOI] [PubMed] [Google Scholar]
- Kuchibhotla KV, Goldman ST, Lattarulo CR, Wu HY, Hyman BT, Bacskai BJ. 2008. Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron 59(2):214–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasala M, Fabiani C, Corradi J, Antollini S, Bouzat C. 2019. Molecular modulation of human α7 nicotinic receptor by amyloid-β peptides. Front Cell Neurosci 13:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassek M, Weingarten J, Acker-Palmer A, Bajjalieh SM, Muller U, Volknandt W. 2014. Amyloid precursor protein knockout diminishes synaptic vesicle proteins at the presynaptic active zone in mouse brain. Curr Alzheimer Res 11(10):971–80. [DOI] [PubMed] [Google Scholar]
- Lassek M, Weingarten J, Wegner M, Mueller BF, Rohmer M, Baeumlisberger D, and others. 2016. APP is a context-sensitive regulator of the hippocampal presynaptic active zone. PLoS Comput Biol 12(4):e1004832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence JL, Tong M, Alfulaij N, Sherrin T, Contarino M, White MM, and others. 2014. Regulation of presynaptic Ca2+, synaptic plasticity and contextual fear conditioning by a N-terminal beta-amyloid fragment. J Neurosci 34(43):14210–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarevic V, Fienko S, Andres-Alonso M, Anni D, Ivanova D, Montenegro-Venegas C, and others. 2017. Physiological concentrations of amyloid beta regulate recycling of synaptic vesicles via alpha7 acetylcholine receptor and CDK5/calcineurin signaling. Front Mol Neurosci 10:221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KJ, Moussa CEH, Lee Y, Sung Y, Howell BW, Turner RS, and others. 2010. Beta amyloid-independent role of amyloid precursor protein in generation and maintenance of dendritic spines. Neuroscience 169(1):344–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerdkrai C, Asavapanumas N, Brawek B, Kovalchuk Y, Mojtahedi N, Olmedillas Del Moral M, and others. 2018. Intracellular Ca(2+) stores control in vivo neuronal hyperactivity in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A 115(6):E1279–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Wang B, Wang Z, Guo Q, Tabuchi K, Hammer RE, and others. 2010. Soluble amyloid precursor protein (APP) regulates transthyretin and Klotho gene expression without rescuing the essential function of APP. Proc Natl Acad Sci U S A 107(40):17362–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Jin M, Koeglsperger T, Shepardson NE, Shankar GM, Selkoe DJ. 2011. Soluble Abeta oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J Neurosci 31(18):6627–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Lu H, Stein E, Zhou Z, Yang Y, Mattson MP. 2018. Brain regional synchronous activity predicts tauopathy in 3xTgAD mice. Neurobiol Aging 70:160–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Chang L, Song Y, Li H, Wu Y, Jackson MF. 2019. The role of NMDA receptors in Alzheimer’s disease. Front Neurosci 13. doi: 10.3389/fnins.2019.00043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell MA, Abner E, Kryscio R, Xu L, Fister SX, Lynn BC. 2015. Calcium channel blockers, progression to dementia, and effects on amyloid beta peptide production. Oxid Med Cell Longev 2015:787–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucey BP, Holtzman DM. 2015. How amyloid, sleep and memory connect. Nat Neurosci 18(7):933–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludewig S, Korte M. 2017. Novel insights into the physiological function of the APP (gene) family and its proteolytic fragments in synaptic plasticity. Front Mol Neurosci 9:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matrone C, Luvisetto S, La Rosa LR, Tamayev R, Pignataro A, Canu N, and others. 2012. Tyr682 in the Abeta-precursor protein intracellular domain regulates synaptic connectivity, cholinergic function, and cognitive performance. Aging Cell 11(6):1084–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer MC, Schauenburg L, Thompson-Steckel G, Dunsing V, Kaden D, Voigt P, and others. 2016. Amyloid precursor-like protein 1 (APLP1) exhibits stronger zinc-dependent neuronal adhesion than amyloid precursor protein and APLP2. J Neurochem 137(2):266–76. [DOI] [PubMed] [Google Scholar]
- Meziane H, Dodart JC, Mathis C, Little S, Clemens J, Paul SM, and others. 1998. Memory-enhancing effects of secreted forms of the beta-amyloid precursor protein in normal and amnestic mice. Proc Natl Acad Sci U S A 95(21):12683–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midthune B, Tyan SH, Walsh JJ, Sarsoza F, Eggert S, Hof PR, and others. 2012. Deletion of the amyloid precursor-like protein 2 (APLP2) does not affect hippocampal neuron morphology or function. Mol Cell Neurosci 49(4):448–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mileusnic R, Lancashire CL, Rose SPR. 2004. The peptide sequence Arg-Glu-Arg, present in the amyloid precursor protein, protects against memory loss caused by Abeta and acts as a cognitive enhancer. Eur J Neurosci 19(7):1933–8. [DOI] [PubMed] [Google Scholar]
- Milosch N, Tanriover G, Kundu A, Rami A, Francois JC, Baumkotter F, and others. 2014. Holo-APP and G-protein-mediated signaling are required for sAPPalpha-induced activation of the Akt survival pathway. Cell Death Dis 5:e1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mockett BG, Richter M, Abraham WC, Müller UC. 2017. Therapeutic potential of secreted amyloid precursor protein APPsα. Front Mol Neurosci 10:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagna E, Crux S, Luckner M, Herber J, Colombo AV, Marinkovic P, and others. 2019. In vivo Ca2+ imaging of astrocytic microdomains reveals a critical role of the amyloid precursor protein for mitochondria. Glia 67(5):985–98. [DOI] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. 1994. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 12(3):529–40. [DOI] [PubMed] [Google Scholar]
- Moreno L, Rose C, Mohanraj A, Allinquant B, Billard JM, Dutar P. 2015. sAbetaPPalpha improves hippocampal NMDA-dependent functional alterations linked to healthy aging. J Alzheimers Dis 48(4):927–35. [DOI] [PubMed] [Google Scholar]
- Müller UC, Deller T, Korte M. 2017. Not just amyloid: physiological functions of the amyloid precursor protein family. Nat Neurosci 18(5):281–98. [DOI] [PubMed] [Google Scholar]
- Müller UC, Zheng H. 2012. Physiological functions of APP family proteins. Cold Spring Harb Perspect Med 2(2):a006288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obregon D, Hou H, Giunta B, Tian J, Darlington D, Shahaduzzaman M, and others. 2012. sAPP-α modulates β-secretase activity and amyloid-β generation. Nat Commun 3:777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno M, Sametsky EA, Younkin LH, Oakley H, Younkin SG, Citron M, and others. 2004. BACE1 deficiency rescues memory deficits and cholinergic dysfunction in a mouse model of Alzheimer’s disease. Neuron 41(1):27–33. [DOI] [PubMed] [Google Scholar]
- Paoletti P, Bellone C, Zhou Q. 2013. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci 14(6):383–400. [DOI] [PubMed] [Google Scholar]
- Paula-Lima AC, Brito-Moreira J, Ferreira ST. 2013. Deregulation of excitatory neurotransmission underlying synapse failure in Alzheimer’s disease. J Neurochem 126(2):191–202. [DOI] [PubMed] [Google Scholar]
- Perez RG, Zheng H, Van der Ploeg LH, Koo EH. 1997. The beta-amyloid precursor protein of Alzheimer’s disease enhances neuron viability and modulates neuronal polarity. J Neurosci 17(24):9407–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plummer S, Van den Heuvel C, Thornton E, Corrigan F, Cappai R. 2016. The neuroprotective properties of the amyloid precursor protein following traumatic brain injury. Aging Dis 7(2):163–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pousinha PA, Mouska X, Raymond EF, Gwizdek C, Dhib G, Poupon G, and others. 2017. Physiological and pathophysiological control of synaptic GluN2B-NMDA receptors by the C-terminal domain of amyloid precursor protein. Elife 6. doi: 10.7554/eLife.25659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozueta J, Lefort R, Shelanski ML. 2013. Synaptic changes in Alzheimer’s disease and its models. Neuroscience 251:51–65. [DOI] [PubMed] [Google Scholar]
- Prox J, Bernreuther C, Altmeppen H, Grendel J, Glatzel M, D’Hooge R, and others. 2013. Postnatal disruption of the disintegrin/metalloproteinase ADAM10 in brain causes epileptic seizures, learning deficits, altered spine morphology, and defective synaptic functions. J Neurosci 33(32):12915–28, 12928a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puzzo D, Privitera L, Fa M, Staniszewski A, Hashimoto G, Aziz F, and others. 2011. Endogenous amyloid-beta is necessary for hippocampal synaptic plasticity and memory. Ann Neurol 69(5):819–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puzzo D, Privitera L, Leznik E, Fa M, Staniszewski A, Palmeri A, and others. 2008. Picomolar amyloid-beta positively modulates synaptic plasticity and memory in hippocampus. J Neurosci 28(53):14537–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinders NR, Pao Y, Renner MC, da Silva-Matos CM, Lodder TR, Malinow R, and others. 2016. Amyloid-beta effects on synapses and memory require AMPA receptor subunit GluA3. Proc Natl Acad Sci U S A 113(42):E6526–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice HC, de Malmazet D, Schreurs A, Frere S, van Molle I, Volkov AN, and others. 2019. Secreted amyloid-beta precursor protein functions as a GABABR1a ligand to modulate synaptic transmission. Science 363(6423). doi: 10.1126/science.aao4827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter MC, Ludewig S, Winschel A, Abel T, Bold C, Salzburger LR, and others. 2018. Distinct in vivo roles of secreted APP ectodomain variants APPsα and APPsβ in regulation of spine density, synaptic plasticity, and cognition. EMBO J 37(11). doi: 10.15252/embj.201798335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring S, Weyer SW, Kilian SB, Waldron E, Pietrzik CU, Filippov MA, and others. 2007. The secreted beta-amyloid precursor protein ectodomain APPs alpha is sufficient to rescue the anatomical, behavioral, and electrophysiological abnormalities of APP-deficient mice. J Neurosci 27(29):7817–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripoli C, Cocco S, Li Puma DD, Piacentini R, Mastrodonato A, Scala F, and others. 2014. Intracellular accumulation of amyloid-beta (Abeta) protein plays a major role in Abeta-induced alterations of glutamatergic synaptic transmission and plasticity. J Neurosci 34(38):12893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush T, Buisson A. 2014. Reciprocal disruption of neuronal signaling and Aβ production mediated by extrasynaptic NMDA receptors: a downward spiral. Cell Tissue Res 356(2):279–86. [DOI] [PubMed] [Google Scholar]
- Sabo SL, Lanier LM, Ikin AF, Khorkova O, Sahasrabudhe S, Greengard P, and others. 1999. Regulation of beta-amyloid secretion by FE65, an amyloid protein precursor-binding protein. J Biol Chem 274(12):7952–7. [DOI] [PubMed] [Google Scholar]
- Santos SF, Pierrot N, Morel N, Gailly P, Sindic C, Octave JN. 2009. Expression of human amyloid precursor protein in rat cortical neurons inhibits calcium oscillations. J Neurosci 29(15):4708–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaguri H, Nilsson P, Hashimoto S, Nagata K, Saito T, Strooper B, and others. 2017. APP mouse models for Alzheimer’s disease preclinical studies. EMBO J 36(17):2473–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheltens P, Blennow K, Breteler MMB, Strooper B, Frisoni GB, Salloway S, and others. 2016. Alzheimer’s disease. Lancet 388(10043):505–17. [DOI] [PubMed] [Google Scholar]
- Schilling S, Mehr A, Ludewig S, Stephan XJ, Zimmermann M, August A, and others. 2017. APLP1 is a synaptic cell adhesion molecule, supporting maintenance of dendritic spines and basal synaptic transmission. J Neurosci 37(21):5345–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwenk J, Perez-Garci E, Schneider A, Kollewe A, Gauthier-Kemper A, Fritzius T, and others. 2016. Modular composition and dynamics of native GABAB receptors identified by high-resolution proteomics. Nat Neurosci 19(2):233–42. [DOI] [PubMed] [Google Scholar]
- Seabrook GR, Smith DW, Bowery BJ, Easter A, Reynolds T, Fitzjohn SM, and others. 1999. Mechanisms contributing to the deficits in hippocampal synaptic plasticity in mice lacking amyloid precursor protein. Neuropharmacology 38(3):349–59. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, Hardy J, Selkoe D, Hardy J. 2016. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 8(6):595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. 2007. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci 27(11):2866–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, and others. 2005. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci 8(8):1051–8. [DOI] [PubMed] [Google Scholar]
- Sperling RA, Laviolette PS, Keefe KO, Brien JO, Rentz M, Pihlajamaki M, and others. 2009. Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron 63(2):178–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbach JP, Müller U, Leist M, Li ZW, Nicotera P, Aguzzi A. 1998. Hypersensitivity to seizures in beta-amyloid precursor protein deficient mice. Cell Death Differ 5(10):858–66. [DOI] [PubMed] [Google Scholar]
- Strecker P, Ludewig S, Rust M, Mundinger TA, Gorlich A, Krachan EG, and others. 2016. FE65 and FE65L1 share common synaptic functions and genetically interact with the APP family in neuromuscular junction formation. Sci Rep 6:25652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strooper B, Karran E. 2016. The cellular phase of Alzheimer’s disease. Cell 164:603–15. [DOI] [PubMed] [Google Scholar]
- Sullivan SE, Dillon GM, Sullivan JM, Ho A. 2014. Mint proteins are required for synaptic activity-dependent amyloid precursor protein (APP) trafficking and amyloid beta generation. J Biol Chem 289(22):15374–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura M, Spellman TJ, Rosen AM, Gogos JA, Gordon JA. 2017. Hippocampal-prefrontal theta-gamma coupling during performance of a spatial working memory task. Nat Commun 8(1):2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan VTY, Mockett BG, Ohline SM, Parfitt KD, Wicky HE, Peppercorn K, and others. 2018. Lentivirus-mediated expression of human secreted amyloid precursor protein-alpha prevents development of memory and plasticity deficits in a mouse model of Alzheimer’s disease. Mol Brain 11(1):7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor CJ, Ireland DR, Ballagh I, Bourne K, Marechal NM, Turner PR, and others. 2008. Endogenous secreted amyloid precursor protein-alpha regulates hippocampal NMDA receptor function, long-term potentiation and spatial memory. Neurobiol Dis 31(2):250–60. [DOI] [PubMed] [Google Scholar]
- Thibault O, Gant JC, Landfield PW. 2007. Expansion of the calcium hypothesis of brain aging and Alzheimer’s disease: minding the store. Aging Cell 6(3):307–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thinakaran G, Koo EH. 2008. Amyloid precursor protein trafficking, processing, and function. J Biol Chem 283(44):29615–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolppanen AM, Lavikainen P, Solomon A, Kivipelto M, Soininen H, Hartikainen S. 2013. Incidence of stroke in people with Alzheimer disease: a national register-based approach. Neurology 80(4):353–8. [DOI] [PubMed] [Google Scholar]
- Trillaud-Doppia E, Boehm J. 2018. The amyloid precursor protein intracellular domain is an effector molecule of metaplasticity. Biol Psychiatry 83(5):406–15. [DOI] [PubMed] [Google Scholar]
- Trillaud-Doppia E, Paradis-Isler N, Boehm J. 2016. A single amino acid difference between the intracellular domains of amyloid precursor protein and amyloid-like precursor protein 2 enables induction of synaptic depression and block of long-term potentiation. Neurobiol Dis 91:94–104. [DOI] [PubMed] [Google Scholar]
- Tyan S, Shih A, Walsh JJ, Maruyama H, Sarsoza F, Ku L, and others. 2012. Amyloid precursor protein (APP) regulates synaptic structure and function. Mol Cell Neurosci 51(1–2):43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umeda T, Tomiyama T, Sakama N, Tanaka S, Lambert MP, Klein WL, and others. 2011. Intraneuronal amyloid β oligomers cause cell death via endoplasmic reticulum stress, endosomal/lysosomal leakage, and mitochondrial dysfunction in vivo. J Neurosci Res 89(7):1031–42. [DOI] [PubMed] [Google Scholar]
- Van Den Heuvel C, Thornton E, Vink R. 2007. Traumatic brain injury and Alzheimer’s disease: a review. Prog Brain Res 161:303–16. [DOI] [PubMed] [Google Scholar]
- van der Kant R, Goldstein LS. 2015. Cellular functions of the amyloid precursor protein from development to dementia. Dev Cell 32(4):502–15. [DOI] [PubMed] [Google Scholar]
- van Dyck CH. 2018. Review anti-amyloid-beta monoclonal antibodies for Alzheimer’s disease: pitfalls and promise. Biol Psychiatry 83(4):311–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verret L, Mann EO, Hang GB, Barth AMI, Cobos I, Ho K, and others. 2012. Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell 149(3):708–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vnencak M, Paul MH, Hick M, Schwarzacher SW, Del Turco D, Muller UC, and others. 2015. Deletion of the amyloid precursor-like protein 1 (APLP1) enhances excitatory synaptic transmission, reduces network inhibition but does not impair synaptic plasticity in the mouse dentate gyrus. J Comp Neurol 523(11):1717–29. [DOI] [PubMed] [Google Scholar]
- Vnencak M, Scholvinck ML, Schwarzacher SW, Deller T, Willem M, Jedlicka P. 2019. Lack of beta-amyloid cleaving enzyme-1 (BACE1) impairs long-term synaptic plasticity but enhances granule cell excitability and oscillatory activity in the dentate gyrus in vivo. Brain Struct Funct 224(3):1279–90. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, and others. 2002. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416(6880):535–9. [DOI] [PubMed] [Google Scholar]
- Wang B, Wang Z, Sun L, Yang L, Li H, Cole AL, and others. 2014. The amyloid precursor protein controls adult hippocampal neurogenesis through GABAergic interneurons. J Neurosci 34(40):13314–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XZ, Jackson RJ, Hong W, Taylor WM, Corbett GT, Moreno A, and others. 2017. Human brain-derived A-beta oligomers bind to synapses and disrupt synaptic activity in a manner that requires APP. J Neurosci 37(49):11947–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Yang L, Zheng H. 2012. Role of APP and Abeta in synaptic physiology. Curr Alzheimer Res 9(2):217–26. [DOI] [PubMed] [Google Scholar]
- Webster NJ, Ramsden M, Boyle JP, Pearson HA, Peers C. 2006. Amyloid peptides mediate hypoxic increase of L-type Ca2+ channels in central neurones. Neurobiol Aging 27(3):439–45. [DOI] [PubMed] [Google Scholar]
- Wei W, Nguyen LN, Kessels HW, Hagiwara H, Sisodia S, Malinow R. 2009. Amyloid beta from axons and dendrites reduces local spine number and plasticity. Nat Neurosci 13(2):190–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyer SW, Klevanski M, Delekate A, Voikar V, Aydin D, Hick M, and others. 2011. APP and APLP2 are essential at PNS and CNS synapses for transmission, spatial learning and LTP. EMBO J 30(11):2266–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyer SW, Zagrebelsky M, Herrmann U, Hick M, Ganss L, Gobbert J, and others. 2014. Comparative analysis of single and combined APP/APLP knockouts reveals reduced spine density in APP-KO mice that is prevented by APPsα expression. Acta Neuropathol Commun 2:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willem M, Tahirovic S, Busche MA, Ovsepian SV, Chafai M, Kootar S, and others. 2015. eta-Secretase processing of APP inhibits neuronal activity in the hippocampus. Nature 526(7573):443–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong M, Jones OD, Peppercorn K, Ohline SM, Tate WP, Abraham WC. 2017. Secreted amyloid precursor protein-alpha can restore novel object location memory and hippocampal LTP in aged rats. Neurobiol Learn Mem 138:291–9. [DOI] [PubMed] [Google Scholar]
- Yang L, Wang Z, Wang B, Justice NJ, Zheng H. 2009. Amyloid precursor protein regulates Cav1.2 L-type calcium channel levels and function to influence GABAergic short-term plasticity. J Neurosci 29(50):15660–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young-Pearse TL, Chen AC, Chang R, Marquez C, Selkoe DJ. 2008. Secreted APP regulates the function of full-length APP in neurite outgrowth through interaction with integrin beta1. Neural Dev 3:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Herrmann U, Weyer SW, Both M, Müller UC, Korte M, and others. 2013. Hippocampal network oscillations in APP/APLP2-deficient mice. PLoS One 8(4):e61198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Zhong W, Brankack J, Weyer SW, Muller UC, Tort ABL, and others. 2016. Impaired theta-gamma coupling in APP-deficient mice. Sci Rep 6:21948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao D, Watson JB, Xie CW. 2004. Amyloid beta prevents activation of calcium/calmodulin-dependent protein kinase II and AMPA receptor phosphorylation during hippocampal long-term potentiation. J Neurophysiol 92(5):2853–8. [DOI] [PubMed] [Google Scholar]
- Zott B, Busche MA, Sperling RA, Konnerth A. 2018. What happens with the circuit in Alzheimer’s disease in mice and humans? Annu Rev Neurosci 41:277–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou C, Crux S, Marinesco S, Montagna E, Sgobio C, Shi Y, and others. 2016. Amyloid precursor protein maintains constitutive and adaptive plasticity of dendritic spines in adult brain by regulating D-serine homeostasis. EMBO J 35(20):2213–22. [DOI] [PMC free article] [PubMed] [Google Scholar]