

Abstract
Background and objective
Acetyl CoA carboxylase (ACC) regulates the differentiation of Th1, Th2, Th17 cells and Treg cells, which play a critical role in airway inflammation of asthma. Here we investigated the role of ACC in the pathogenesis of asthma.
Methods
Chicken Ovalbumin-sensitized and -challenged mice were divided into three groups, PBS group, DMSO (solvent of TOFA) group and ACC inhibitor 5-tetradecyloxy-2-furoic acid (TOFA) + DMSO group. Airway inflammation was assessed with histology, percentages of CD4+T cell subsets in lung and spleen was assessed with flow cytometry, and airway responsiveness was assessed with FinePointe RC system. The expression of characteristic transcription factors of CD4+T cell subsets was evaluated with real-time PCR. Cytokine levels in bronchoalveolar lavage fluid (BALF) and serum was determined with ELISA.
Results
In asthma mice, the expression of ACC increased, while the expression of phosphorylated ACC (pACC) decreased. TOFA had no significant effect on pACC expression. TOFA reduced serum IgE, airway inflammatory cells infiltration and goblet cell hyperplasia, but dramatically increased airway responsiveness. TOFA significantly reduced the percentages of Th1, Th2, Th17 cells in lung and spleen, the expression of GATA3 and RORγt in lung, and IFN-γ, IL-4, IL-17A levels in BALF and serum. TOFA had no significant effect on the percentage of Treg cells, IL-10 level and the expression of T-bet and Foxp3.
Conclusion
Acetyl-CoA carboxylase inhibitor TOFA might have a distinct effect on asthmatic airway inflammation and airway hyperresponsiveness.
Keywords: Asthma, Acetyl-CoA carboxylase, CD4+T cells, TOFA, Airway inflammation
Introduction
Fatty acids are not only essential constituents of cell membranes, but also important signaling molecules and bioenergetic substrates. Fatty acids needed for cells can be obtained by ligating multiple acetyl units from acetyl-CoA (de novo fatty acid synthesis) or by uptaking exogenous fatty acids [1].
Acetyl-CoA carboxylase (ACC) is a rate-limiting enzyme in fatty acid metabolism that catalyzes the carboxylation of Acetyl-CoA to malonyl-CoA in an ATP-dependent manner [2]. Two isoforms of ACC were identified, ACC1 and ACC2 [3]. Malonyl-CoA synthesized by ACC1 serves as a carbon donor for de novo fatty acid synthesis, whereas malonyl-CoA produced by ACC2 works as an inhibitor of fatty acid oxidation [4]. ACC is rapidly inactivated upon phosphorylation by AMP-activated protein kinase (AMPK) or cAMP-dependent protein kinase A (PKA), whereas dephosphorylation activates the enzyme [5]. Besides, ACC can also be regulated promptly by allosteric agents, such as Soraphen A, which more likely targets ACC1, and 5-(tetradecyloxy)-2-furancarboxylic acid (TOFA), which more likely targets both ACC1 and ACC2 [6, 7].
Fatty acids are essential constituents of cell membranes, important signaling molecules and bioenergetic substrates, which can be obtained by ligating multiple acetyl units from acetyl-CoA (de novo fatty acid synthesis) or by uptaking exogennous fatty acids [1]. Recent studies found that inhibition of ACC activity downregulates the differentiation of Th1, Th2 and Th17 cells, while upregulates the differentiation of Regulatory CD4+ T cells (Treg) [8]. It is found that Th17 cells depend on ACC1-mediated de novo fatty acid synthesis and the underlying glycolytic–lipogenic metabolic pathway for their development, while Treg cells take up exogenous fatty acids for this purpose [8]. In addition, T cell-specific deletion of ACC1 demonstrated that ACC1-mediated de novo lipogenesis is essential for CD8+ T cell expansion [9].
Asthma is a chronic airway inflammatory disease. The prevalence of asthma and its economic and social burden are still increasing [10]. Some patients are not well controlled despite therapy with inhaled glucocorticoids and long-acting β2 agonists [11].
The underlying mechanisms of airway inflammation and remodeling are still not well understood [12], but Th1/Th2 immunity imbalance with Th2 cell bias has been established as the major cause of eosinophilic airway inflammation [13]. In addition, recent studies also demonstrated a role of Th17 immunity in the pathogenesis of asthma, especially neutrophil-dominant airway inflammation, which is associated with severe asthma [14]. Treg cells are responsible for sustaining immune tolerance to allergen, decreased or defected suppressing effects of Treg cells also contribute to the development of allergic asthma [15]. Therefore, imbalance of Th17/Treg is an additional mechanism of asthma and a new target for asthma therapy. Our previous study has shown that abnormal Th17 immunity is also involved in the pathogenesis of allergic asthma [16].
In the present study, we used ACC inhibitor TOFA to investigate the role of ACC in the development of allergic airway inflammation in a mouse model of asthma, and the effects of TOFA on CD4+ T cells percentages in asthma mice model.
Materials and methods
Animals
Female C57BL/6 mice (6–8 weeks age) were obtained from Hubei Provincial Center for Disease Control and Prevention and were bred at the Experimental Animal Center of Wuhan University. All procedures were conducted in accordance with the Institution Animal Ethics Committee of Wuhan University.
Animal experimental protocols
Mice were sensitized via intraperitoneal injection on days 0 and 14 with 20 μg chicken ovalbumin (OVA, Sigma-Aldrich, USA) in 148 μl PBS. On day 21, 22, 23, mice were challenged by intranasal administration of 100 μg OVA in PBS in a total volume of 50 μl under narcotization with 1% sodium pentobarbital. Control mice were sensitized and challenged with PBS.
Mice exposed to OVA were divided into three groups and received intraperitoneal administration of 0.5 mM ACC inhibitor TOFA (Sigma-Aldrich, USA), 0.5 mM DMSO, and PBS twice a week, respectively, before OVA challenge. Control mice were intraperitoneally injected with PBS (Fig. 1). The sequence of administration and sensitization to chicken ovalbumin and TOFA was an experimental attempt, for lack of research on the relationship between TOFA and asthma.
Fig. 1.

Protocol of allergic asthma mice model. ip Intraperitoneal, in intranasal, OVA ovalbumin
Determination of airway resistance
After 24 h of last challenge, mice were anesthetized and intubated with a tracheostomy tube, connected to a mechanical ventilator (FinePointe RC system, Wilmington, NC, USA). Aerosolized PBS was used to determine the baseline airway resistance, and aerosolized methacholine (Sigma-Aldrich, USA) was then delivered at increasing concentrations (range from 3.125 to 25 mg/ml), after which the peak airway resistance (RI) was recorded.
Bronchoalveolar lavage
Bronchoalveolar lavage fluid (BALF) was performed 3 times with 0.5 ml PBS via a tracheal cannula. BALF was centrifuged at 1500 r/min for 7 min at 4 °C. Slides for the cellular component were prepared by cytocentrifugation (TXD3 cytocentrifuge, China) and stained with WrightGiemsa Stain kit (Jiancheng Bioengineering Institute, China). Approximately 400 cells were counted in each slide. The number of eosinophils, macrophages, neutrophils, and lymphocytes was expressed as the absolute number from the total cells count. BALF supernatant was stored at − 80 °C until use. After this, the mice were killed with exsanguination and used for further experiments.
Lung histological examination
The left lungs of mice were harvested, dehydrated and embedded in paraffin after complete fixation in 4% paraformaldehyde. Thereafter, paraffin-fixed tissue was cut into sections, deparaffinized, and stained with hematoxylin and eosin (HE) and periodic acid-Schiff (PAS), respectively, for the detection of inflammatory cells infiltration and goblet cell hyperplasia. The severity of inflammatory cells infiltration was classified into 5 classes (0–4). The degree of goblet cell hyperplasia was represented as PAS+ area (A+PAS) per basement membrane perimeter (Pbm).
Analysis of CD4+T cells by flow cytometry
Right lungs and spleens were removed and placed in RPMI1640 media (HyClone™, USA) and penicillin–streptomycin solution (Jinuo, China). The lungs were digested by collagenase I (Biosharp, USA) and the spleens were grinded to obtain mononuclear cells (MNCs). To determine the fraction of Th1, Th2 and Th17 cells by flow cytometry, MNCs were stimulated with 20 ng/ml PMA (Tocris Bioscience, England) and 1 μg/μl Ionomycin (Tocris Bioscience, England) diluted in 10% fetal bovine serum (HyClone™, USA) and 1% penicillin–streptomycin-containing RPMI1640 media for 2 h. Then 5 mg/ml Brefeldin A (eBioscience, USA) was added into the media and cells were cultured further for 4 h. Cells were then surface-stained with PE-Cyanine7-labeled Anti-Mouse CD4 (eBioscience, USA) antibody, fixed and permeabilized with Intracellular Fixation &Permeabilization Buffer Set (eBioscience, USA), and then, respectively, stained with PE-labeled Anti-Mouse IFN-γ, PE-labeled Anti-Mouse IL-4 and PE-labeled Anti-Mouse IL-17A (eBioscience, USA) antibodies. For Treg cells analysis, MNCs were stained directly with PE-Cyanine7-labeled Anti-Mouse CD4 and PE-labeled Anti-Mouse CD25 (eBioscience, USA) without stimulation. Following the fixation and permeabilization by Foxp3/Transcription Factor Fixation/Permeabilization Concentrate and Diluent (eBioscience, USA), cells were stained with FITC-labeled Anti-Mouse Foxp3 (eBioscience, USA). Finally, all stained cells were analyzed on an FACS AriaIII (BD Biosciences, USA).
Detection of cytokines and serum IgE by ELISA
Serum and BALF were obtained from each group, and the concentrations of IFN-γ, IL-4, IL-17 and IL-10 in BALF and serum, as well as the total level of serum IgE were detected by ELISA kits according to the manufacturer’s protocols (Mouse IFN-γ Instant ELISA, Mouse IL-4 ELISA Ready-SET-Go, Mouse IL-17A ELISA Ready-SET-Go, Mouse IL-10 ELISA Ready-SET-Go, Mouse IgE ELISA Ready-SET-Go, eBioscience, USA).
Real-time quantitative PCR
Total RNA of lungs was extracted with TRIzol reagent (Invitrogen, USA), and the concentration and purity of total RNA was evaluated by spectrophotometer (Thermo Scientific, USA). RNA was reverse-transcribed into cDNA using the ReverTra Ace® qPCR RT Kit (Toyobo, Osaka, Japan) and thereafter cDNA was amplified using an SYBR Premix Ex TaqTM kit (Takara, Tokyo, Japan) according to the manufacturer’s instructions. All Cycle threshold values were normalized to reference gene GAPDH and 2−ΔΔCt method was used to quantify the expression of transcription factors. PCR primers sequences were designed as follows: T-bet, forward 5′-CCATTCCTGTCCTTCACCG-3′ and reverse 5′-CTGCCTTCTGCCTTTCCAC-3′; GATA3, forward 5′-CTGGAGGAGGAACGCTAATG-3′ and reverse 5′-AGATGTGGCTCAGGGATGAC-3′; ROR-γt, forward 5′-TGCGACTGGAGGACCTTCTAC-3′ and reverse 5′-CTCCCACATTGACTTCCTCTGG-3′; Foxp3, forward 5′-ACTCGCATGTTCGCCTACTT-3′ and reverse 5′-AGGGATTGGAGCACTTGTTG-3′; GAPDH, forward 5′-ATGGGTGTGAACCACGAGA-3′ and reverse 5′-CAGGGATGATGTTCTGGGCA-3′.
Measurement of ACC and pACC proteins by western blot
Right lungs were minced in RIPA Lysis Buffer (Beyotime, China) containing protease inhibitors Phenylmethanesulfonyl fluoride (Beyotime, China), and centrifugated at 12,000 rpm for 5 min, supernatants were collected. Protein concentration was determined using the Enhanced BCA Protein Assay Kit (Beyotime, China). The protein was subjected to electrophoresis on SDS–polyacrylamide gels and then transferred onto a polyvinylidene fluoride membrane (Millipore, USA). After blocking in 3% bovine serum albumin (BSA), protein was incubated with Acetyl-CoA Carboxylase Antibody or Phospho-Acetyl-CoA Carboxylase (Ser79) (D7D11) Rabbit mAb (Cell signaling, USA), washed with TBST and then probed with horseradish peroxidase (HRP)-linked secondary antibodies (Boster biological technology, China). Then membranes were imaged with ECL (Thermo Fisher Scientific, USA). The signals were analyzed with Bandscan 5.0.
Statistical analysis
Statistical analyses were performed using SPSS 21.0 software. Data were expressed as mean ± SEM, significance between groups was assessed using one-way ANOVA and Least significant difference (LSD) was used for post hoc test, except those were non-normal distributions or heteroscedasticity, in which Mann–Whitney U tests were used. p < 0.05 was considered statistically significant. Histograms were made using Graphpad Prism 5 software.
Results
TOFA inhibited ACC protein expression in lung of asthmatic mice
We firstly investigated the protein expression of ACC and phosphorylated ACC (pACC) protein, the inactivated form of ACC, in lungs of asthma model group and control mice by western blot. As shown in Fig. 2, the protein expression of ACC was higher in lung of asthma model of mice when compared to that of control mice. By contrast, the protein expression of pACC in lung of asthma model of mice was lower than that in control mice. This protein expression pattern of ACC and pACC suggests a possible role of ACC in asthma. TOFA is a cell-permeable small molecule and also an allosteric inhibitor of ACC. We found that TOFA treatment slightly decreased ACC protein expression in the lung of asthma model of mice. The solvent DMSO had no effects on ACC protein expression (Fig. 2a). However, neither TOFA nor DMSO displayed significant effects on pACC protein expression in lung of asthma model of mice, albeit there was a slight trend of increase in pACC expression in TOFA-treated asthma model of mice (Fig. 2b). Our results suggest that TOFA may reduce the activity of ACC primarily by allosteric effect, but not by phosphorylation of ACC, at least in lung of asthma model of mice.
Fig. 2.

Decreased ACC protein expression in TOFA-treated asthma mice. a Western blot analysis of ACC protein expression in mice lung tissue. b Western blot analysis of pACC protein expression in mice lung tissue. All data are represented as mean ± SEM (n = 3 mice per group). *p < 0.05; **p < 0.01; ***p < 0.001; NS, p > 0.05
TOFA reduced airway inflammation and serum IgE level in asthma mice
We then determined the effects of ACC inhibitor TOFA on airway inflammation in mice of asthma model. As shown in Fig. 3a–c, OVA-exposed mice manifested typical asthma features such as increased lung inflammatory cells infiltration and goblet cell hyperplasia. DMSO treatment had no significant effects on airway inflammation and goblet cell proliferation (Fig. 3b, c), while treatment with TOFA significantly alleviated lung inflammatory cells infiltration (Fig. 3b) and goblet cell hyperplasia (Fig. 3c). We tried to observe cellular component in BALF, unfortunately most of cells membrane were ruptured and unable to be recognized in TOFA-treated mice. This may be due to the influence of TOFA on cell membrane in view of the role of TOFA on fatty acid metabolism.
Fig. 3.

Decreased airway inflammation and serum IgE level in TOFA-treated asthma mice. a Lung sections stained with HE and PAS (× 200). b Inflammatory score and c PAS-stained areas per unit length were determined. d Serum IgE level was analyzed by ELISA. All data are represented as mean ± SEM (n = 5–6 mice per group). *p < 0.05; ***p < 0.001; NS, p > 0.05
In accordance with lung inflammation, serum IgE level was significantly higher in OVA-exposed mice than that in control mice. Treatment of OVA-exposed mice with TOFA, but not DMSO, significantly reduced serum IgE level (Fig. 3d). Taken together, these results demonstrated that ACC inhibition with TOFA reduced OVA-induced airway inflammation and serum IgE level in a mice model of asthma.
Effects of TOFA on airway resistance in asthma mice
Next, we assessed the effect of ACC inhibitor TOFA on airway resistance in asthma model of mice. As shown in Fig. 4, both the baseline airway resistance and airway resistance induced by increasing concentration of methacholine were higher in asthma model of mice than that in control mice. Treatment with DMSO had no significant effect on airway resistance in asthma model of mice. Unexpectedly, TOFA treatment slightly increased baseline airway resistance and also airway resistance induced by methacholine in asthma model of mice. The resistance to methacholine peaked at 12.5 mg/ml, and then decreased even though with increased concentration of methacholine. These data suggested that inhibition of ACC activity might result in increased airway resistance.
Fig. 4.
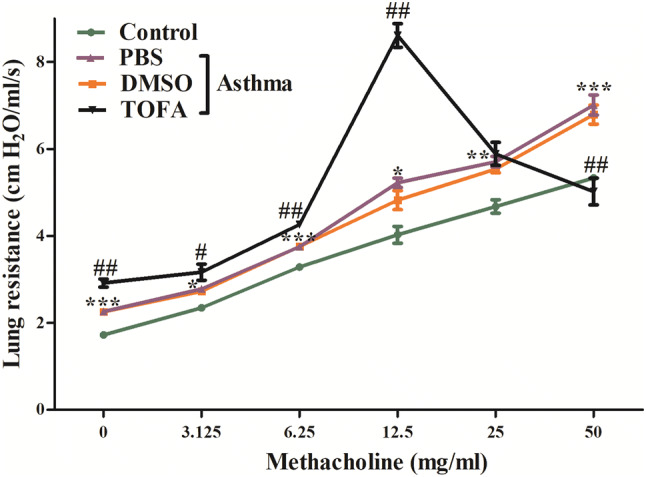
Increased airway resistance in TOFA-treated asthma mice. *Control vs PBS/Asthma, #TOFA/Asthma vs PBS/Asthma. All data are represented as mean ± SEM (n = 5 mice per group). *p < 0.05, **p < 0.01, ***p < 0.001, #p < 0.05, ##p < 0.001
TOFA regulated CD4+ T cells differentiation in asthma mice
To further explore the mechanism underlying the effect of ACC in asthma model of mice, we investigated the differentiation of CD4+ T cells, which highly related to the pathogenesis of asthma. Flow cytometric analysis demonstrated that TOFA significantly reduced the percentage of Th1, Th2 and Th17 cells in mononuclear cells (MNC) of lung in asthma model of mice (Fig. 5a, b). Similar results were found in spleen MNC (Fig. 5c, d). In consistent with these results, a decrease in the level of IFN-γ, IL-4 and IL-17A in BALF and serum (Fig. 6a) was found in TOFA-treated asthma model of mice. However, no significance difference was detected in the percentage of Treg cells and the level of IL-10 among different treatment groups (Figs. 5, 6a).
Fig. 5.

Reduced percentages of Th1, Th2, Th17 cells in TOFA-treated asthma mice. IFN-γ+, IL-4+, IL-17+ Th cells from CD4+ cells, Foxp3+ Th cells from CD4+CD25+ Th cells in lung tissue (a) and in spleen (c) were examined by flow cytometric analysis. The percentage of CD4+ cells expressing IFN-γ+, IL-4+, IL-17+ and the percentage of CD4+CD25+ cells expressing Foxp3+ in lung tissue (b) and in spleen (d). All data are represented as mean ± SEM (n = 5–6 mice per group). *p < 0.05; **p < 0.01; ***p < 0.001; NS, p > 0.05
Fig. 6.

Regulated cytokines level and characteristic transcriptional factor expression of CD4+T cells in TOFA-treated asthma mice. a Cytokines IFN-γ, IL-4, IL-17A and IL-10 in BALF and in serum were examined by ELISA. b The transcription factors T-bet, GATA3, RORγt and Foxp3 were measured in lung tissue by Real-time quantitative PCR. All data are represented as mean ± SEM (n = 5–6 mice per group). *p < 0.05; **p < 0.01; ***p < 0.001; NSp > 0.05
We also checked the expression of characteristic transcription factors of each CD4+T cell subset in lung tissue. Figure 6b showed that in asthma model of mice, administration of TOFA effectively downregulated the expression of GATA3 and RORγt, the characteristic transcription factor of Th2 and Th17, respectively, but had no significant effects on the expression of T-bet and Foxp3, the characteristic transcriptional factor of Th1 and Treg, respectively. These results indicated that ACC inhibition with TOFA will inhibit the differentiation of Th2 and Th17 cells, but not Th1 and Treg cells in lung of asthma model of mice.
Discussion
In the present study, we demonstrated a possible role of ACC in the pathogenesis of asthma airway inflammation using ACC inhibitor TOFA. Our results showed that TOFA intraperitonially administration in asthma mice could significantly reduce the protein expression of ACC and airway inflammation, and the effect of TOFA may be due to its regulation on the differentiation of Th2 and Th17 cells, but not Th1 and Treg cells.
ACC is a rate-limiting enzyme that catalyzes the carboxylation of acetyl-CoA to form malonyl-CoA and plays a key role in the regulation of fatty acid metabolism. The two isoforms of ACC, ACC1 and ACC2, have distinct tissue and cellular distribution. ACC is inactivated while phosphorylated to pACC. Our results demonstrated the protein expression of ACC in lung of mice, and this expression may be slightly inhibited by ACC allosteric inhibitor TOFA. Indeed, a recent study using another ACC inhibitor Soraphen A showed that it will increase pACC protein levels. Therefore, we also checked the expression of pACC after treatment with TOFA [8]. We showed that TOFA had no significant effect on the protein expression of pACC, the inactivate form of ACC, although a slight trend of increase of pACC. Therefore, phosphorylation of ACC plays no significant role in the inhibiting effect of TOFA on ACC.
AMPK–ACC signaling has been demonstrated in different biochemical and physiological processes, including platelet function and thrombus formation [17], fasting- and cold- induced appetite [18], in human skeletal muscle contraction [19] and in metformin-induced anti-fibrotic effect in renal fibrosis [20]. AMPK activation leads to phosphorylation and inactivation of ACC. Role of AMPK in airway inflammation of asthma have been demonstrated in toluene diisocyanate-induced occupational asthma model [21] and in OVA-induced allergic asthma model [22]. In addition, AMPK signaling is also involved in airway smooth muscle cell proliferation [23, 24], suggesting a possible role of AMPK in airway remodeling of asthma. However, as the downstream signaling molecule of AMPK, the role of ACC in airway inflammation and remodeling is still unknown. Our results showed that ACC is involved in airway inflammation of OVA-induced asthma mice model for the first time, so far as we know.
Resveratrol is a polyphenolic phytoalexin with anti-inflammatory, antioxidant, and anticancer effects. Recent studies have proved it can ameliorate airway inflammation induced by OVA [25] or HDM [26]. In addition, resveratrol could inhibit mucus overproduction [27] and airway remodeling [25, 26, 28] in asthma mice models. Resveratrol could inhibit ACC1 expression and increase ACC1 phosphorylation via AMPK signaling [29], suggesting a role of ACC in asthma. As expected, inhibition ACC directly with TOFA also attenuated OVA-induced airway inflammation, as shown in our present study.
Interestingly, TOFA increased, but not reduced airway resistance of asthma mice, as shown in Fig. 4. Not only the baseline airway resistance, but also the resistance induced by increasing concentration of methacholine was increased after treatment of TOFA. There is no report on the effects of acetyl-CoA carboxylase and its inhibitor TOFA on functional regulation of airway smooth muscle such as proliferation, apoptosis, contraction, migration and airway remodeling. The role of upstream regulating molecule of ACC, AMPK, has been described above [29]. Instead, the downstream molecule of ACC, PI3K were shown to be important for the IL-13-induced airway hyperreactivity [30]. According to these studies, TOFA induces PI3K inhibition will result in decreased airway hyperreactivity, this is contrast to our results.
Although the dissociation between airway inflammation and airway resistance has been revealed in asthma [31–33], and also airway inflammation-dependent and -independent airway remodeling [34], this cannot clearly explain our results and also the inconsistency with previous studies. Therefore, further studies with more selective ACC inhibitors or with ACC knock-out mice are necessary. In addition, the ex vivo effect of TOFA on contraction of airway smooth muscle strips, which minimize the impact of airway inflammation and other cytokines, will give deep insights into the underlying mechanisms of increased AHR induced by TOFA.
In the present study, we used chicken OVA as allergen to induce an acute allergic bronchial inflammation, which is characterized by Th2 type inflammation and eosinophil infiltration with elevated serum IgE level. The clinically relevant allergen house dust mite (HDM) induces airway inflammation by proteinase activation receptor (PAR)-dependent mechanisms [35], and innate lymphoid cells type 2 (ILC2) are critical player in Type 2 inflammation of this kind of asthma model [36]. As shown in our results, TOFA decreased Th2 inflammation in OVA-exposed mice. The effects of TOFA on airway inflammation in an HDM-induced mice model of asthma will deepen our understanding of the role of ACC in airway inflammation of asthma.
Besides, ACC has also been shown to regulate the balance of Th17 and Treg. Our data also confirmed that ACC inhibitor TOFA inhibited Th17 differentiation in OVA-exposed asthma model of mice. Since Th17 induces neutrophil-dominant airway inflammation, a mice model of asthma characterized by neutrophil-dominant inflammation, e.g., mice exposed to ovalbumin and polyinosinic:polycytidylic acid (Poly I:C), will give more information about the effect of TOFA. In fact, recent study showed that HDM induced both Th2 and Th17 cytokine profile and mixture of eosinophilic and neutrophilic airway inflammation [37]; therefore, effects of TOFA should be conducted with HDM-induced airway inflammation.
There are other limitations of our study. First, TOFA is a non-selective ACC inhibitor. Therefore, we could not distinguish the respective effects of ACC1 and ACC2 in the development of allergic asthma. It is necessary to verify our results in an OVA- or HDM-induced asthma model with ACC1 and/or ACC2 knock-out mice. Since deficiency of ACC1 in mice is embryonic lethal [38], mice with heterozygous genotype of ACC1 may be used to elucidate this question. In addition, it is better to confirm the role of ACC in asthma and in CD4+T cells differentiation with CD4+T cell-specific ACC1-deficient (TACC1) and/or ACC2-deficient (TACC2) mice.
In conclusion, our results demonstrated that ACC inhibitor TOFA have distinct effects on asthmatic airway inflammation and airway hyperresponsiveness.
Acknowledgements
This study was supported by academic grant of Health Commission of Hubei Province (WJ2017M031).
Compliance with ethical standards
Conflict of interest
The authors declared that they have no conflicts of interest to this work.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Lochner M, Berod L, Sparwasser T. Fatty acid metabolism in the regulation of T cell function. Trends Immunol. 2015;36:81–91. doi: 10.1016/j.it.2014.12.005. [DOI] [PubMed] [Google Scholar]
- 2.Kim KH, Lopez-Casillas F, Bai DH, Luo X, Pape ME. Role of reversible phosphorylation of acetyl-CoA carboxylase in long-chain fatty acid synthesis. Faseb J. 1989;3:2250–2256. doi: 10.1096/fasebj.3.11.2570725. [DOI] [PubMed] [Google Scholar]
- 3.Abu-Elheiga L, Jayakumar A, Baldini A, Chirala SS, Wakil SJ. Human acetyl-CoA carboxylase: characterization, molecular cloning, and evidence for two isoforms. Proc Natl Acad Sci USA. 1995;92:4011–4015. doi: 10.1073/pnas.92.9.4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harriman G, Greenwood J, Bhat S, Huang X, Wang R, Paul D, et al. Acetyl-CoA carboxylase inhibition by ND-630 reduces hepatic steatosis, improves insulin sensitivity, and modulates dyslipidemia in rats. Proc Natl Acad Sci USA. 2016;113:E1796–E1805. doi: 10.1073/pnas.1520686113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hardie DG, Pan DA. Regulation of fatty acid synthesis and oxidation by the AMP-activated protein kinase. Biochem Soc Trans. 2002;30:1064–1070. doi: 10.1042/bst0301064. [DOI] [PubMed] [Google Scholar]
- 6.Jump DB, Torres-Gonzalez M, Olson LK. Soraphen A, an inhibitor of acetyl CoA carboxylase activity, interferes with fatty acid elongation. Biochem Pharmacol. 2011;81:649–660. doi: 10.1016/j.bcp.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parker RA, Kariya T, Grisar JM, Petrow V. 5-(Tetradecyloxy)-2-furancarboxylic acid and related hypolipidemic fatty acid-like alkyloxyarylcarboxylic acids. J Med Chem. 1977;20:781–791. doi: 10.1021/jm00216a009. [DOI] [PubMed] [Google Scholar]
- 8.Berod L, Friedrich C, Nandan A, Freitag J, Hagemann S, Harmrolfs K, et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat Med. 2014;20:1327–1333. doi: 10.1038/nm.3704. [DOI] [PubMed] [Google Scholar]
- 9.Lee J, Walsh MC, Hoehn KL, James DE, Wherry EJ, Choi Y. Regulator of fatty acid metabolism, acetyl coenzyme a carboxylase 1, controls T cell immunity. J Immunol. 2014;192:3190–3199. doi: 10.4049/jimmunol.1302985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ding B, Small M. Disease burden of mild asthma in China. Respirology. 2018;23:369–377. doi: 10.1111/resp.13189. [DOI] [PubMed] [Google Scholar]
- 11.Loftus PA, Wise SK. Epidemiology of asthma. Curr Opin Otolaryngol Head Neck Surg. 2016;24:245–249. doi: 10.1097/MOO.0000000000000262. [DOI] [PubMed] [Google Scholar]
- 12.King GG, James A, Harkness L, Wark P. Pathophysiology of severe asthma: we’ve only just started. Respirology. 2018;23:262–271. doi: 10.1111/resp.13251. [DOI] [PubMed] [Google Scholar]
- 13.Lambrecht BN, Hammad H. The immunology of asthma. Nat Immunol. 2015;16:45–56. doi: 10.1038/ni.3049. [DOI] [PubMed] [Google Scholar]
- 14.Hirose K, Iwata A, Tamachi T, Nakajima H. Allergic airway inflammation: key players beyond the Th2 cell pathway. Immunol Rev. 2017;278:145–161. doi: 10.1111/imr.12540. [DOI] [PubMed] [Google Scholar]
- 15.Chen CC, Kobayashi T, Iijima K, Hsu FC, Kita H. IL-33 dysregulates regulatory T cells and impairs established immunologic tolerance in the lungs. J Allergy Clin Immunol. 2017;140:1351–1363. doi: 10.1016/j.jaci.2017.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao Y, Yang J, Gao YD, Guo W. Th17 immunity in patients with allergic asthma. Int Arch Allergy Immunol. 2010;151:297–307. doi: 10.1159/000250438. [DOI] [PubMed] [Google Scholar]
- 17.Lepropre S, Kautbally S, Octave M, Ginion A, Onselaer MB, Steinberg GR, et al. AMPK-ACC signaling modulates platelet phospholipids and potentiates thrombus formation. Blood. 2018;132:1180–1192. doi: 10.1182/blood-2018-02-831503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Galic S, Loh K, Murray-Segal L, Steinberg GR, Andrews ZB, Kemp BE. AMPK signaling to acetyl-CoA carboxylase is required for fasting- and cold-induced appetite but not thermogenesis. Elife. 2018;7:e32656. doi: 10.7554/eLife.32656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen ZP, McConell GK, Michell BJ, Snow RJ, Canny BJ, Kemp BE. AMPK signaling in contracting human skeletal muscle: acetyl-CoA carboxylase and NO synthase phosphorylation. Am J Physiol Endocrinol Metab. 2000;279:E1202–E1206. doi: 10.1152/ajpendo.2000.279.5.E1202. [DOI] [PubMed] [Google Scholar]
- 20.Lee M, Katerelos M, Gleich K, Galic S, Kemp BE, Mount PF, et al. Phosphorylation of acetyl-CoA carboxylase by AMPK reduces renal fibrosis and is essential for the anti-fibrotic effect of metformin. J Am Soc Nephrol. 2018;29:2326–2336. doi: 10.1681/ASN.2018010050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park SJ, Lee KS, Kim SR, Chae HJ, Yoo WH, Kim DI, et al. AMPK activation reduces vascular permeability and airway inflammation by regulating HIF/VEGFA pathway in a murine model of toluene diisocyanate-induced asthma. Inflamm Res. 2012;61:1069–1083. doi: 10.1007/s00011-012-0499-6. [DOI] [PubMed] [Google Scholar]
- 22.Andre DM, Calixto MC, Sollon C, Alexandre EC, Leiria LO, Tobar N, et al. Therapy with resveratrol attenuates obesity-associated allergic airway inflammation in mice. Int Immunopharmacol. 2016;38:298–305. doi: 10.1016/j.intimp.2016.06.017. [DOI] [PubMed] [Google Scholar]
- 23.Pan Y, Liu L, Li S, Wang K, Ke R, Shi W, et al. Activation of AMPK inhibits TGF-beta1-induced airway smooth muscle cells proliferation and its potential mechanisms. Sci Rep. 2018;8:3624. doi: 10.1038/s41598-018-21812-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu L, Pan Y, Song Y, Su X, Ke R, Yang L, et al. Activation of AMPK alpha2 inhibits airway smooth muscle cells proliferation. Eur J Pharmacol. 2016;791:235–243. doi: 10.1016/j.ejphar.2016.09.003. [DOI] [PubMed] [Google Scholar]
- 25.Chen G, Tang J, Ni Z, Chen Q, Li Z, Yang W, et al. Antiasthmatic effects of resveratrol in ovalbumin-induced asthma model mice involved in the upregulation of PTEN. Biol Pharm Bull. 2015;38:507–513. doi: 10.1248/bpb.b14-00610. [DOI] [PubMed] [Google Scholar]
- 26.Chen J, Zhou H, Wang J, Zhang B, Liu F, Huang J, et al. Therapeutic effects of resveratrol in a mouse model of HDM-induced allergic asthma. Int Immunopharmacol. 2015;25:43–48. doi: 10.1016/j.intimp.2015.01.013. [DOI] [PubMed] [Google Scholar]
- 27.Ni ZH, Tang JH, Chen G, Lai YM, Chen QG, Li Z, et al. Resveratrol inhibits mucus overproduction and MUC5AC expression in a murine model of asthma. Mol Med Rep. 2016;13:287–294. doi: 10.3892/mmr.2015.4520. [DOI] [PubMed] [Google Scholar]
- 28.Lee HY, Kim IK, Yoon HK, Kwon SS, Rhee CK, Lee SY. Inhibitory effects of resveratrol on airway remodeling by transforming growth factor-beta/smad signaling pathway in chronic asthma model. Allergy Asthma Immunol Res. 2017;9:25–34. doi: 10.4168/aair.2017.9.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park SJ, Ahmad F, Philp A, Baar K, Williams T, Luo H, et al. Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting cAMP phosphodiesterases. Cell. 2012;148:421–433. doi: 10.1016/j.cell.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jiang H, Xie Y, Abel PW, Toews ML, Townley RG, Casale TB, et al. Targeting phosphoinositide 3-kinase gamma in airway smooth muscle cells to suppress interleukin-13-induced mouse airway hyperresponsiveness. J Pharmacol Exp Ther. 2012;342:305–311. doi: 10.1124/jpet.111.189704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leavy O. Asthma and allergy: an IFNgamma bias in severe asthma. Nat Rev Immunol. 2015;15:466–467. doi: 10.1038/nri3891. [DOI] [PubMed] [Google Scholar]
- 32.Singapuri A, McKenna S, Brightling CE, Bradding P. Mannitol and AMP do not induce bronchoconstriction in eosinophilic bronchitis: further evidence for dissociation between airway inflammation and bronchial hyperresponsiveness. Respirology. 2010;15:510–515. doi: 10.1111/j.1440-1843.2010.01712.x. [DOI] [PubMed] [Google Scholar]
- 33.Birrell MA, Battram CH, Woodman P, McCluskie K, Belvisi MG. Dissociation by steroids of eosinophilic inflammation from airway hyperresponsiveness in murine airways. Respir Res. 2003;4:3. doi: 10.1186/rr197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Elliot JG, Noble PB, Mauad T, Bai TR, Abramson MJ, McKay KO, et al. Inflammation-dependent and independent airway remodelling in asthma. Respirology. 2018;23:1138–1145. doi: 10.1111/resp.13360. [DOI] [PubMed] [Google Scholar]
- 35.de Boer JD, Van’T VC, Stroo I, van der Meer AJ, de Vos AF, van der Zee JS, et al. Protease-activated receptor-2 deficient mice have reduced house dust mite-evoked allergic lung inflammation. Innate Immun. 2014;20:618–625. doi: 10.1177/1753425913503387. [DOI] [PubMed] [Google Scholar]
- 36.Li B, Beerens D, Brem MD, Hendriks RW. Characterization of group 2 innate lymphoid cells in allergic airway inflammation models in the mouse. Methods Mol Biol. 2017;1559:169–183. doi: 10.1007/978-1-4939-6786-5_12. [DOI] [PubMed] [Google Scholar]
- 37.Ullah MA, Revez JA, Loh Z, Simpson J, Zhang V, Bain L, et al. Allergen-induced IL-6 trans-signaling activates gammadelta T cells to promote type 2 and type 17 airway inflammation. J Allergy Clin Immunol. 2015;136:1065–1073. doi: 10.1016/j.jaci.2015.02.032. [DOI] [PubMed] [Google Scholar]
- 38.Abu-Elheiga L, Matzuk MM, Kordari P, Oh W, Shaikenov T, Gu Z, et al. Mutant mice lacking acetyl-CoA carboxylase 1 are embryonically lethal. Proc Natl Acad Sci USA. 2005;102:12011–12016. doi: 10.1073/pnas.0505714102. [DOI] [PMC free article] [PubMed] [Google Scholar]