

Abstract
Influenza A virus (IAV) depends on the metabolism of its cellular host to provide energy and essential factors, including lipids, for viral replication. Previous studies have shown that fatty acids (FAs) play an important role in IAV replication and that inhibition of FA biosynthesis can diminish viral replication. However, cellular lipids can either be synthesized intracellularly or be imported from the extracellular environment. Interfering with FA import mechanisms may reduce the cellular lipid content and inhibit IAV replication. To test this hypothesis, MDCK and Detroit 562 cells were infected with IAV followed by exposure to palmitic acid and inhibitors of FA import. Replication of IAV significantly increased when infected cells were supplied with palmitic acid. This enhancement could be reduced by adding an FA import inhibitor. The addition of palmitic acid significantly increased the cellular lipid content, and this increased level was reduced by treatment with an FA import inhibitor. These results show that reducing the cellular lipid level might be an approach for IAV therapy.
Introduction
Influenza A virus (IAV) is an important virus that causes respiratory diseases in humans and many animal species worldwide. The IAV subtypes that have been circulating in humans are H1N1 and H3N2. In the 20th century, there were three major IAV pandemics: Spanish flu in 1918 (H1N1), Asian flu in 1957 (H2N2), Hong Kong flu in 1968 (H3N2). In 2009, WHO declared that a new strain of swine-origin H1N1, known as “swine flu”, was responsible for the first pandemic of the 21st century. The major concept of anti-influenza drugs for humans is targeting conserved viral components that are critical for viral replication. Two types of anti-influenza drugs are commonly used, matrix protein 2 (M2) ion channel blockers and the neuraminidase inhibitors. A new class of cap-dependent endonuclease inhibitor (baloxavir marboxil) has been approved recently for treatment of influenza. However, resistance to the available drugs is a major public-health concern, and development of alternative treatments is required [16, 28].
All viruses depend on cellular factors to complete their replication cycle. Among the host cell factors that are essential for viruses, cellular lipids play a key role in the viral replication cycle. Some viruses can regulate cellular metabolism of infected cells by altering cellular lipid metabolism to support viral replication. Increases in both fatty acid synthesis and lipid beta oxidation have been shown to be induced by various viruses [1, 12, 22, 24, 34]. Like other viruses, IAV has been shown to alter cellular lipid metabolism. Bronchoalveolar lavage fluid of IAV-infected mice has been shown to have significantly increased levels of fatty acids, including palmitic acid, oleic acid, and linoleic acid [6]. Inhibition of fatty acid biosynthesis can inhibit IAV infection. Pharmacological inhibition of fatty acid metabolism pathways can be achieved by treatment with TOFA (5-tetradecyloxy-2-furoic acid), an inhibitor of acetyl-CoA carboxylase (ACC), and C75 (trans-4-carboxy-5-octyl-3-methylene-butyrolactone), an inhibitor of fatty acid synthase (FASN). These enzymes play important roles in the fatty acid biosynthetic pathway. Treatment with both compounds has been shown to cause a significant reduction in IAV replication [26]. Moreover, fatty acids have been shown to play a role in virion assembly and membrane fusion. Palmitoylation (S-acylation) is the post-translational attachment fatty acids (including palmitic acid) with a thioester-type-linkage to cysteine, serine, and threonine residues of proteins, usually peripheral and integral membrane proteins. IAV contains two palmitoylated (S-acylated) proteins: the spike protein HA (haemagglutinin) and the proton channel protein M2. Eliminating the acylation site of viral HA results in defects in genome packaging and irregular morphology. Viral particles possessing HA with deleted palmitoylation sites show defects in viral replication and reduced levels of viral nucleoprotein (NP) and matrix protein 1 (M1) [3, 4, 30, 36]. This indicates that fatty acid metabolism may play an essential role in IAV infection.
Most cells, with the exception of liver and adipose cells, have little capacity for de novo fatty acid synthesis and rely on uptake of fatty acids from extracellular source for their needs [18]. Extracellular fatty acids are taken up through the plasma membrane. Fatty acids can separate from lipoproteins and travel across the plasma membrane by simple passive diffusion [32]. However, there other are two fatty acid import mechanisms that depend on membrane-associated proteins. First, transmembrane protein CD36, originally called fatty acid translocase (FAT), is an 88-kDa transmembrane glycoprotein [35] that can function alone or together with plasma-membrane-associated fatty-acid-binding protein (FABPpm) as an acceptor for fatty acids [9]. Second, fatty acid transport protein 1 (FATP1) is a 71-kDa protein belonging to the FATP/Slc27 protein family that localizes to high-density membranes [38]. This protein enhances cellular uptake of fatty acids and is expressed in several insulin-sensitive tissues [21]. Modulation of the fatty acid import mechanism can affect cellular lipid metabolism. In previous studies, overexpression of murine FATP1 was shown to increase LCFA uptake and triacylglycerol accumulation [13, 20]. Disruption of the FATP1 homolog in yeast was found to significantly impair LCFA uptake, and FATP1 knockout mice showed reduced muscle acyl-CoA levels with increased insulin sensitivity [7, 17]. In animal models, CD36 overexpression in muscle of mice increased fatty acid oxidation and decreased plasma lipids, while deletion of CD36 impaired fatty acid uptake by key metabolic tissues and increased plasma fatty acid and triglyceride (TG) [10].
Because inhibition of intracellular fatty acid synthesis inhibits IAV replication, it may be possible to decrease viral replication by disrupting the action of fatty-acid-importing proteins. In this study, the compounds used to inhibit fatty acid transport were arylpiperazines and sulfo-N-succinimidyl oleate. Arylpiperazines are a class of FATP1 inhibitors. In a previous report, derivatives the arylpiperazine 5k and 12a were identified as potential compounds for in vitro inhibition of human and mouse FATP1s with excellent pharmacokinetic properties [25]. Sulfo-N-succinimidyl oleate (SSO) is an irreversible inhibitor of the fatty acid translocase CD36. It has been shown to reduce the uptake of palmitate by mouse insulinoma MIN6 cells and prevent palmitate-induced changes in insulin secretion [27]. These inhibitors were used to demonstrate the possibility of inhibiting viral replication by interfering with cellular lipid importation, which may be a novel approach for broad-spectrum antiviral therapy.
Materials and methods
Cell culture and viruses
MDCK (Madin-Darby canine kidney) cells were grown in minimum essential medium (MEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS), penicillin, streptomycin, gentamicin and Fungizone. Detroit 562 (human pharynx epithelial cells) were grown in same medium used for MDCK cells supplemented with 1% nonessential amino acids. The viruses used in this study included a seasonal H1N1 influenza virus strain (A/Thailand/Siriraj-12/2006 (A/New Caledonia/20/99-like virus]), a pandemic H1N1 influenza virus strain isolated from a patient with influenza-like illness in 2009 (A/Thailand/104/2009), PR8 H1N1 (A/Puerto Rico/8/1934), and a seasonal H3N2 influenza strain (A/Thailand/Siriraj-04/2003). All of these viruses were propagated in MDCK cells.
Fatty acid and fatty acid import inhibitors
Palmitic acid (Sigma-Aldrich, Missouri, USA) was conjugated to fatty-acid-free (FFA) bovine serum albumin (BSA) (Sigma-Aldrich, Missouri, USA). Palmitic acid (51.2 mg) was dissolved in 100% ethanol (1 ml) to make a 200 mM stock solution. FFA low-endotoxin BSA was prepared in 0.1 M Tris-HCl, pH 8.0, at a concentration of 10%. Forty microliters of 200 mM palmitate and 1.96 ml of 10% FFA BSA were mixed for at least 2 hours to make a 4 mM palmitate stock solution, which was then passed through a 0.22-µm filter and diluted to the working concentrations in VGM (serum-free MEM plus 1 µg of TPCK-treated trypsin per ml). Sulfo-N-succinimidyl oleate (Cayman Chemical, Michigan, USA) and arylpiperazine derivatives 5k and 12a (Daiichi Sankyo, Tokyo, Japan) were diluted to their working concentrations in VGM.
Assessment the cytotoxicity of fatty acids and fatty acid metabolism inhibitors
MDCK and Detroit 562 cells were plated in 96-well culture plates (3.5 × 104 cells/well and 1.2 × 105 cells/well, respectively) and incubated overnight at 37°C in a 5% CO2 atmosphere. The cells were treated with various concentrations of fatty acids and fatty acid import inhibitors for 24 hours at 37 C, 5% CO2, and the cytotoxicity of all compounds was evaluated by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. MTT dye (Invitrogen, California, USA) was diluted 1:10 in sterile PBS. All compounds from the experimental plate were removed, and the plate was washed with 200 μl of PBS per well. One hundred microliters of diluted MTT dye was added to each well, and the plate was incubated for 4 hours at 37° C, 5% CO2. Seventy microliters of diluted MTT dye was removed, and 70 μl of 100% DMSO was added to each well. The optical density (OD) of was then measured using a spectrophotometer at a wavelength of 570 nm.
Plaque assay
The IAV stocks were tested by plaque assay in confluent MDCK cell monolayers. Each IAV stock was serially diluted and inoculated onto an MDCK cell monolayer in a 12-well tissue culture plate. After incubation for 1.3 hours at 37° C with 5% CO2, the infected MDCK cell monolayer was overlaid with a mixture (1:1) of 2X VGM (serum-free MEM plus TPCK-treated trypsin) and 1.6% low-melting-point agarose (Promega, Wisconsin, USA). After 48 h of incubation at 37°C with 5% CO2, the agar overlay was removed. The infected MDCK cell monolayer was incubated with 10% formaldehyde for 30 minutes at room temperature and stained with 1% crystal violet. The IAV titer was determined using a plaque assay and expressed in plaque-forming units (PFU)/ml.
Virus titration by 50% tissue culture infectious dose (TCID50) assay
IAV was titrated on an MDCK cell monolayer, using a viral-nucleoprotein-based ELISA (enzyme-linked immunosorbent assay) as the endpoint measurement for viral infection. The TCID50 titers were calculated by the Reed–Muench method. Each culture supernatant from the experiment was serially diluted and inoculated into an MDCK cell monolayer in a 96-well tissue culture plate. After incubation overnight at 37° C with 5% CO2, the cells were fixed with 80% cold acetone (20 ml of 1X PBS + 80 ml of acetone) at 4° C for 1 hour and blocked with 3% H2O2 at room temperature for 30 minutes. The level of viral infection was measured by ELISA, using a mouse anti-influenza-virus nucleoprotein monoclonal antibody as the primary antibody (Merck Millipore, Massachusetts, USA) diluted 1:1000 in blocking buffer (1% BSA and 0.1% Tween20 in 1X PBS, pH 7.2). The primary antibody was added to each well, and the cells were incubated at 37° C for 1 hour. After incubation, the plate was washed, and horseradish-peroxidase-conjugated goat anti-mouse immunoglobulin (Dako, Glostrup, Denmark), diluted 1:2000 in blocking buffer was added to each well as the secondary antibody. The plate was incubated at 37° C for 1 hour and washed. TMB peroxidase substrate was added to each well, and the plate was incubated in the dark for 10 minutes at room temperature. After the incubation, 1 M H2SO4 was added to stop the reaction, and the optical density at 450/630 nm was measured in an ELISA plate reader.
Lipid measurement by the sulfo-phospho-vanillin (SPV) method
MDCK and Detroit 562 cells were plated in 12-well culture plates and incubated overnight at 37° C, 5% CO2. The cells were treated with various concentrations of fatty acids and fatty acid metabolism inhibitors for 24 hours at 37° C in a 5% CO2 atmosphere. The supernatant of the treated cells was removed, and the cells were trypsinized, collected by sedimentation, and suspended in 20 μl of deionized water. This was followed by addition of 180 μl of concentrated sulfuric acid and heating at 100° C for 10 min. The cell sediment was cooled to room temperature, and 500 μl of phosphovanillin (PV) reagent (a solution of 6 mg of vanillin dissolved in 100 ml of water and diluted to 500 ml with 85% phosphoric acid) (Sigma-Aldrich, Missouri, USA) was added to each sediment sample for color development. The sediment mixture was incubated at 37° C for 15 min, cooled to room temperature, and transferred to a 96-well microtiter plate. The plate was stored in a dark box for 45 min, and the optical density was measured at 530 nm [2]. For the standard curve, commercial soybean oil was used as the standard for the SPV method. Soybean oil was dissolved in chloroform in different concentrations (0 - 1200 μg/ml).
Measurement of CD36 and FATP1 expression in cell culture
Expression of CD36 and FATP1 in MDCK cells was measured by reverse transcription polymerase chain reaction (RT-PCR). The oligonucleotide primers used for amplification of the canine CD36 gene were forward, 5’-AAGGAAGTTGTACTTGAAGAAGGT-3’, and reverse, 5’-TCCTGTGCATTTTGCACGTC-3’, and the product size was 106 bp [36]. The primers used for amplification of both the human and canine FATP1 gene were forward, 5’- GTGAAGGTCAATGAGGACAC-3’, and reverse, 5’-CCCTGCTTCAGGTCCAGGAA-3’, and the product size was 613 bp. Total RNA was extracted from MDCK, A549 and RD cells using a PARIS™ Kit (Thermo Fisher Scientific, Massachusetts, USA). Complementary DNA (cDNA) was synthesized using AMV reverse transcriptase (Promega, Wisconsin, USA) for later use as a template to detect mRNA. The PCR mixture contained 4 μl of cDNA and 10 µM each of forward and reverse primers in Phusion DNA polymerase master mix (New England Biolabs, Massachusetts, USA). The reaction was run in a Bio-Rad T100™ Thermal Cycler (California, USA) for 30 cycles. PCR products were separated by electrophoresis in a 2% agarose gel and stained with SYBR™ Safe DNA Gel Stain (Thermo Fisher Scientific, Massachusetts, USA).
Western blot
Detroit 562 cells were trypsinized, and the cell pellets were washed twice with 1X PBS. Proteins from the cells were extracted by adding ice-cold RIPA buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 0.1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM sodium orthovanadate, and 1 mM sodium fluoride). The cell lysates were centrifuged at 14,000 × g for 10 minutes, and the supernatants were transferred to a fresh tube. The protein concentration was measured using the Bradford protein assay (Bio-Rad, California, USA) according to the manufacturer’s instructions. Thirty micrograms of each protein sample was mixed with 4x loading buffer and boiled at 70°C for 10 minutes. The protein samples were loaded into a 10% SDS-polyacrylamide gel (Invitrogen, California, USA) along with the molecular weight marker and separated by electrophoresis at 110 V for 90 minutes. The separated protein bands were then blotted onto a nitrocellulose membrane by placing the gel into 1X transfer buffer for 10 minutes and then placing the gel on the membrane and assembling a filter paper stack as a sandwich (avoiding air bubbles). The proteins were transferred to membrane using electroblotting (Amersham Biosciences, Little Chalfont, UK) at 40 mA for 75 minutes. The blotted membrane was briefly rinsed in distilled water and stained with Ponceau S solution (Cell Signaling Technology, Massachusetts, USA) to check the transfer quality. After washing five times with washing buffer (0.1% Tween 20 in TBS) to remove the Ponceau S solution, the approximate sizes of protein bands of CD36 (75-100 kDa) and GAPDH (35 kDa) were marked. The membrane was cut for detecting CD36 and GAPDH (housekeeping protein) separately according to the size of protein bands. The blot was blocked then with blocking buffer (3% skim milk and 0.2% Tween 20 in TBS) at room temperature for 1 hour. After washing three times with washing buffer, the blot was probed with a 1:1000 dilution of anti-CD36 rabbit monoclonal antibody (Abcam, Cambridge, UK) or a 1:5000 dilution of anti-GAPDH mouse monoclonal antibody (Santa Cruz Biotechnology, Texas, USA) diluted in the blocking buffer as the primary antibody and incubated overnight with gentle agitation on a rocker at 4° C. The blot was extensively washed with washing buffer (3 × 10 minutes) with gentle agitation. CD36 protein was detected by incubating for 1 hour with a 1:1000 dilution of HRP-conjugated goat anti-rabbit IgG (Agilent Technologies, California, USA), and GAPDH protein was detected by incubating for 1 hour with a 1:1000 dilution of HRP-conjugated rabbit anti-mouse IgG (Agilent Technologies, California, USA) as the secondary antibody. The blot was extensively washed with washing buffer (3 × 10 minutes) with gentle agitation. Protein bands were detected by adding ECL western blotting substrate (Thermo Fisher Scientific, Massachusetts, USA) and visualized by the chemiluminescent method using an ImageQuant™ LAS 4000 biomolecular imager (GE Healthcare, Illinois, USA).
Statistical analysis
A t-test was used for comparing the differences in the mean values for IAV replication and levels of cellular lipid among the groups of tested subjects. A p-value of <0.05 was considered statistically significant.
Results
The effect of palmitic acid to IAV replication
As cell culture medium does not usually contain fatty acids, in order to investigate effect of inhibition of fatty acid import on viral infection, infected cells were exposed to fatty acid import inhibitors along with fatty acid. Palmitic acid (CH3(CH2)14COOH) was used in this study because this fatty acid is the most common saturated fatty acid found in animals, plants, and microorganisms. It is the first fatty acid produced during fatty acid synthesis and is the precursor of longer fatty acids [31]. In addition, some proteins are modified by the addition of a palmitoyl group in a process known as palmitoylation. Palmitoylation is important for membrane localization of many proteins, including IAV HA and M2 [3, 4, 30, 37].
To investigate the role of fatty acids in IAV replication, MDCK and Detroit 562 cells were inoculated with four IAV strains, seasonal H1N1 (H1N1 12/06), pandemic H1N1 (pH1N1 TH104), PR8 H1N1, and seasonal H3N2, for 1 hour at a multiplicity of infection (MOI) of 0.1 and 5, respectively. The viral suspension was removed, and infected cells were washed with VGM and incubated for 24 hours with palmitic acid at various concentrations. A palmitic acid concentration of 100 µM was the highest concentration that was well tolerated by both cell types, with ≥90% cellular metabolic activity. The cytotoxicity of palmitic acid was measured by MTT assay (Fig. 1A and Fig. 3A). Palmitic acid supplementation enhanced viral replication of all IAV subtypes (Fig. 1B-E and Fig. 3B-C). Replication of all IAV subtypes increased significantly when the cells were treated with palmitic acid (t-test, P < 0.05) at a concentration of 100 µM. The fatty acid treatment increased viral replication in a dose-dependent manner. In MDCK cells, the increase in viral titer due to supplementation with 100 µM palmitic acid was 41%, 69%, 38% and 64% for H1N1, pH1N1, PR8 H1N1, and H3N2, respectively. In Detroit 562 cells, the increase in viral titer due to supplementation with 100 µM palmitic acid was 19.8% and 16.8% for pH1N1 and PR8 H1N1, respectively.
Fig. 1.

The mean and standard deviation values for (A) the percent cellular activity of MDCK cells as measured by MTT assay, the viral titers of (B) seasonal H1N1 (12/06 H1N1), (C) pandemic 2009 H1N1 (TH104 pH1N1), (D) seasonal H3N2, and (E) PR8 H1N1, and (F) the cellular lipid content in MDCK cells after an incubation for 24 hours with various concentrations of palmitic acid. All experiments were performed in triplicate. The p-values were determined by t-test. *, P < 0.05 relative to the cell control (viral infection without palmitic acid treatment). **, P < 0.05 relative to the cell control (MDCK cells without palmitic acid treatment)
Fig. 3.

The mean and standard deviation values for (A) the percent cellular metabolic activity of Detroit 562 cells as measured by MTT assay, the viral titers of (B) pandemic 2009 H1N1 (TH104 pH1N1) and (C) PR8 H1N1, and (D) the cellular lipid content in Detroit 562 cells after an incubation for 24 hours with various concentrations of palmitic acid. All experiments were performed in triplicate. The p-values were determined by t-test. *, P < 0.05 relative to the cell control (viral infection without palmitic acid treatment). **, P < 0.05 relative to the cell control (Detroit 562 cells without palmitic acid treatment)
The effect of fatty acid import inhibitors on IAV replication
Under normal cell culture conditions cells receive only a small amount of lipid from FBS. We therefore investigated the effect of fatty acid import inhibitors in cell culture medium supplemented with fatty acids, which enhanced IAV replication. Palmitic acid at 100 µM and fatty acid import inhibitors at 150 µM for MDCK and 125 µM for Detroit 562 were the highest concentrations in the mixture of compounds that were well tolerated by both cell types, with ≥ 90% cellular metabolic activity. MDCK and Detroit 562 cells were infected with IAV and incubated for 24 hours with a mixture of 100 µM palmitic acid and fatty acid import inhibitors (Fig. 2A-C and Fig. 4A). Cytotoxicity was measured by MTT assay. The level of viral output was reduced significantly by treatment with SSO in both MDCK and Detroit 562 cells (t-test, P < 0.05) (Fig. 2D and Fig. 4B-C). Treatment with arylpiperazines slightly reduced IAV replication in MDCK but this did not reach statistical significance (t-test, P > 0.05) (Fig. 2E-F). However, SSO was unable to completely abolish the enhancement of viral replication by palmitic acid.
Fig. 2.

The percent cellular metabolic activity of MDCK cells as measured by MTT assay at 24 hours after exposure to a mixture of 100 µM palmitic acid and various concentrations of (A) SSO, (B) arylpiperazine 5k, and (C) arylpiperazine 12a. The levels of IAV replication in cells at 24 hours after exposure to a mixture of 100 µM palmitic acid and 150 µM (D) SSO, (E) arylpiperazine 5k and (F) arylpiperazine 12a compared with 100 µM of palmitic acid treatment alone and virus control without treatment were determined. (G) Cellular lipid content of cells at 24 hours after exposed to a mixture of 100 µM palmitic acid and various concentrations of fatty-acid-import inhibitors. All experiments were performed in triplicate. The p-values were determined by t-test (p-value < 0.05). *, P < 0.05 relative to treatment with 100 µM palmitic acid alone (without treatment with the fatty-acid-import inhibitor)
Fig. 4.

The percent cellular metabolic activity of Detroit 562 cells as measured by MTT assay at 24 hours after exposure to a mixture of 100 µM palmitic acid and various concentrations of SSO (A). The levels of IAV replication in cells at 24 hours after exposed to a mixture of 100 µM palmitic acid and 125 µM SSO, compared with 100 µM palmitic acid treatment alone and virus control without treatment. (B and C) Viral titers of pandemic 2009 H1N1 (TH104 pH1N1) replication (B) and PR8 H1N1 replication (C). (D) Cellular lipid content of cells at 24 hours after treatment with a mixture of 100 µM palmitic acid and various concentrations of SSO. All experiments were performed in triplicate. The p-values were evaluated by t-test (p < 0.05). *, P < 0.05 relative to treatment with 100 µM palmitic acid alone (without SSO treatment)
The effect of palmitic acid and fatty acid import inhibitors on cellular lipid levels
To confirm that palmitic acid and the fatty acid uptake inhibitors affected cellular lipid content, we measured cellular lipids in treated MDCK and Detroit 562 cells by the sulfo-phospho-vanillin (SPV) method. Both cell types were treated with palmitic acid and fatty acid import inhibitors at the same concentration and duration as in the above experiments on IAV infection. Palmitic acid at the same concentrations that increased viral replication significantly increased lipid levels in MDCK and Detroit 562 cells (t-test, P < 0.05) (Fig. 1F and Fig. 3D). The increase in cellular lipid content by palmitic acid showed a dose response at a concentration of 100 μM that was similar to the dose response curve for enhancement of viral replication. The lipid content in each test was calculated based on a standard curve made using soybean oil. The standard curve measured by the SPV method had a correlation coefficient of 0.968 and a regression equation of y = 0.0016x - 0.1345. Each point in the regression represents the mean of three experiments (Fig. 4). SSO at 150 μM and 125 μM significantly reduced the cellular lipid content (t-test, P < 0.05) of MDCK and Detroit 562 cells, respectively (Fig. 2G and Fig. 5D), but SSO treatment did not reduce the cellular lipid content to the original level without palmitic acid supplementation.
Fig. 5.
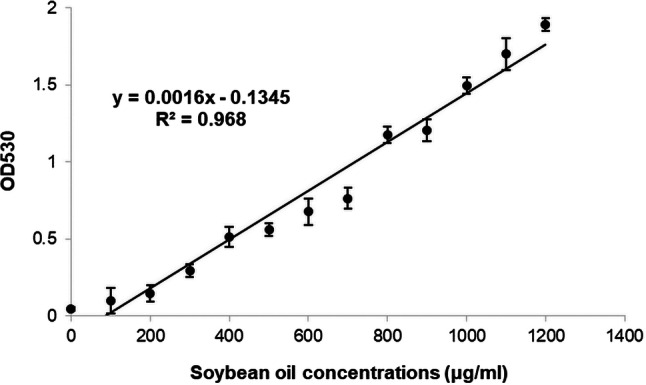
The standard curve obtained by plotting lipid content of various soybean oil concentrations (0-1200 μg/ml). The correlation coefficient is 0.968, and the regression equation is y = 0.0016x - 0.1345. Each point is the average of three experiments
Expression of CD36 and FATP1 in cell culture
To measure the expression of CD36 and FATP1 in MDCK cells, we measured cellular mRNA expression of both genes by RT-PCR using specific primers for the canine CD36 and FATP1 genes. CD36 expression was detected in MDCK cells as shown in Fig. 6A, but no expression of FATP1 was observed in MDCK cells (Fig. 6B). The lack of expression of FATP1 was consistent with the finding that the arylpiperazine FATP1 inhibitors did not reduce IAV replication in this cell line. In Detroit 562 cells, CD36 expression was detected by western blot using a specific antibody against the human CD36 protein, as shown in Fig. 6C.
Fig. 6.

CD36 and FATP1 expression in cell lines. (A) Expression of mRNA CD36 in MDCK cells. (B) Expression of mRNA FATP1 in MDCK cells. Expression of both genes was analyzed by RT-PCR using specific primers for the canine CD36 and FATP1 genes. (C) Protein expression of CD36 in the Detroit 562 cell line, analyzed by western blot using a specific antibody to the human CD36 protein
Discussion
In this study, addition of palmitic acid significantly increased the replication of all IAV strains in both cell lines. Supplementing infected cells with palmitic acid might enhance viral replication by increasing lipid availability in the infected cells. The same palmitic acid concentration (100 µM) that significantly increased the cellular lipid content also increased IAV replication. Previous studies have demonstrated alterations in lipid metabolism upon viral infection [1, 12]. Some viruses take control of cellular metabolism by promoting synthesis of fatty acids to enhance energy and facilitate the formation of the viral lipid membrane [22, 24]. Lipids are an energy source that is utilized during viral replication. Some viruses such as hepatitis C virus can induce lipid metabolism at both the transcriptional and post-transcriptional steps, which can lead to increased lipid oxidation and ATP generation. Dengue virus infection can induce the process of autophagy, which leads to degradation of lipid droplets. Lipids released from these droplets are oxidized in mitochondria to generate ATP [24, 34]. Moreover, several reports have shown that fatty acids play a critical role in IAV replication. Bronchoalveolar lavage samples from IAV-infected mice have been shown to contain increased levels of fatty acids, including palmitic acid. Palmitic acid plays a role as a component of the viral spike protein HA (haemagglutinin) and the proton-channel protein M2 via palmitoylation, which has been shown to affect the association of the virus with membrane rafts, the entry of IAV into target cells by HA-mediated membrane fusion, and the release of newly assembled virions from infected cells [3, 4, 30, 37]. In addition, inhibition of fatty acid biosynthesis by TOFA (an inhibitor of acetyl-CoA carboxylase) and C75 (an inhibitor of fatty acid synthase) has been shown to reduce IAV replication [26]. However, fatty acid synthesis mainly occurs in liver and adipose cells [18], while the major targets of human influenza A virus infection are ciliated epithelial cells in the upper respiratory tract [15].
We hypothesized that it may be possible to decrease viral replication by disrupting the action of the fatty-acid-importing proteins CD36 and FATP1. This study was conducted in both MDCK and Detroit 562 cells. MDCK cells are the most commonly used cell line for IAV culture, and Detroit 562 cells are human pharynx epithelial cells originating from target area of human influenza A viruses infection. Although we did not know whether the inhibitors would work with the canine fatty acid transporters, both target proteins (CD36 and FATP1) are highly conserved in mammals. It has been reported previously, that SSO inhibits CD36 in both humans and rats. This compound inhibits oxidized low-density lipoprotein (oxLDL) uptake by human macrophages (THP-1) by about 50% [19] and modulates metabolism in diabetic heart tissue of Wistar rats by reducing fatty acid uptake [23]. The amino acid sequences of CD36 of humans and rats (Rattus norvegicus) are 86.9% identical, while those of humans and dogs (Canis lupus) are 83.2% identical. Between dogs and rats, percent identity in amino acid sequence of CD36 is 87.4%. FATP1 is a very-long-chain acyl-CoA synthetase that facilitates fatty acid uptake via esterification-coupled influx [5, 11]. The arylpiperazines inhibit the acyl-CoA synthetase activity of human and mouse FATP1 [25]. Previously, those compounds were tested by oral administration to mice, but a reduction in tissue triglyceride (TG) accumulation was not observed [25]. The FATP1 amino acid sequences, of humans and mice (Mus musculus) are 89.5% identical. The percent identity between human and dog (Canis lupus) FATP1 is 92.7% and between dogs and mice, the percent identity is 88.1%.
In this study, we demonstrate that SSO treatment at 150 µM (MDCK) and 125 µM (Detroit 562) significantly reduces IAV replication and cellular lipid content. Our results suggest a relationship between IAV replication and cellular lipid content. Disruption of fatty acid uptake can cause a reduction in cellular lipid content. Reducing cellular lipid levels might have an effect on IAV replication. The effect of the inhibitors on lipid uptake and IAV replication might depend on both the efficiency of the inhibitor itself and the level of expression of the target proteins.
The target of SSO, CD36, is found on renal tubular epithelial cells in mammals and plays an important role in lipid metabolism [8, 14, 29, 39]. The inability of arylpiperazines to inhibit IAV replication and cellular lipid synthesis might be due to the lack of FATP1 expression in MDCK cells, since FATP1 is not the major FATP in renal tubular epithelial cells [33].
Further studies are needed to examine the effect of inhibitors on various cell lines with different levels of target proteins to further develop this approach into a new therapeutic strategy. Although the ability of SSO to inhibit IAV replication is limited and probably insufficient for its use as an antiviral drug, our results showed that inhibition of fatty-acid-importing proteins can reduce cellular lipid levels and viral replication, and that this approach might therefore be a new therapeutic strategy that should be explored further.
Acknowledgements
We thank Dr. Tsuyoshi Shinozuka from End-organ Disease Laboratories, Daiichi Sankyo Co., Ltd for generously providing the arylpiperazines. All virus isolates used in this research were kindly provided by Professor Pilaipan Puthavathana and the Department of Microbiology, Faculty of Medicine, Siriraj Hospital. This work was financial supported by the Thailand Research Fund (Grant no. IRN60W0002).
Compliance with ethical standards
Conflict of interest
The authors declare no conflicts of interest.
Research involving human participants and/or animals
No part of this study was performed with human participants or animals.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Alvisi G, Madan V, Bartenschlager R. Hepatitis c virus and host cell lipids: An intimate connection. RNA Biol. 2011;8(2):258–269. doi: 10.4161/rna.8.2.15011. [DOI] [PubMed] [Google Scholar]
- 2.Anschau A, Caruso CS, Kuhn RC, Franco TT. Validation of the sulfo-phospho-vanillin (SPV) method for the determination of lipid content in oleaginous microorganisms. Braz J Chem Eng. 2017;34(1):19–27. doi: 10.1590/0104-6632.20170341s20140222. [DOI] [Google Scholar]
- 3.Basu J. Protein palmitoylation and dynamic modulation of protein function. Curr Sci. 2004;87(2):12–17. [Google Scholar]
- 4.Blanc M, Blaskovic S, van der Goot FG. Palmitoylation pathogens and their host. Biochem Soc Trans. 2013;41(1):84–88. doi: 10.1042/BST20120337. [DOI] [PubMed] [Google Scholar]
- 5.Coe NR, Smith AJ, Frohnert BI, Watkins PA, Bernlohr DA. The fatty acid transport protein (FATP1) is a very long chain acyl-CoA synthetase. J Biol Chem. 1999;274(51):36300–36304. doi: 10.1074/jbc.274.51.36300. [DOI] [PubMed] [Google Scholar]
- 6.Cui L, Zheng D, Lee YH, Chan TK, Kumar Y, Ho WE, et al. Metabolomics investigation reveals metabolite mediators associated with acute lung injury and repair in a murine model of influenza pneumonia. Sci Rep. 2016;6:26076. doi: 10.1038/srep26076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Færgeman NJ, Dirusso CC, Elberger A, Knudsen J, Black PN. Disruption of the Saccharomyces cerevisiae homologue to the murine fatty acid transport protein impairs uptake and growth on long-chain fatty acids. J Biol Chem. 1997;272(13):8531–8538. doi: 10.1074/jbc.272.13.8531. [DOI] [PubMed] [Google Scholar]
- 8.Gao X, Wu J, Qian Y, Fu L, Wu G, Xu C, et al. Oxidized high-density lipoprotein impairs the function of human renal proximal tubule epithelial cells through CD36. Int J Mol Med. 2014;34(2):564–572. doi: 10.3892/ijmm.2014.1799. [DOI] [PubMed] [Google Scholar]
- 9.Glatz JFC, Luiken JJFP. Dynamic role of the transmembrane glycoprotein CD36 (SR-B2) in cellular fatty acid uptake and utilization. J Lipid Res. 2018;59(7):1084–1093. doi: 10.1194/jlr.R082933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goldberg IJ, Eckel RH, Abumrad NA. Regulation of fatty acid uptake into tissues: lipoprotein lipase- and CD36-mediated pathways. J Lipid Res. 2008;50(Suppl):S86–S90. doi: 10.1194/jlr.R800085-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hall AM, Smith AJ, Bernlohr DA. Characterization of the acyl-CoA synthetase activity of purified murine fatty acid transport protein 1. J Biol Chem. 2003;278(44):43008–43013. doi: 10.1074/jbc.M306575200. [DOI] [PubMed] [Google Scholar]
- 12.Heaton NS, Randall G. Multifaceted roles for lipids in viral infection. Trends Microbiol. 2011;19(7):368–375. doi: 10.1016/j.tim.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holloway GP, Chou CJ, Lally J, Stellingwerff T, Maher AC, Gavrilova O, et al. Increasing skeletal muscle fatty acid transport protein 1 (FATP1) targets fatty acids to oxidation and does not predispose mice to diet-induced insulin resistance. Diabetologia. 2011;54(6):1457–1467. doi: 10.1007/s00125-011-2114-8. [DOI] [PubMed] [Google Scholar]
- 14.Iwao Y, Nakajou K, Nagai R, Kitamura K, Anraku M, Maruyama T, et al. CD36 is one of important receptors promoting renal tubular injury by advanced oxidation protein products. Am J Physiol Ren Physiol. 2008;295(6):F1871–F1880. doi: 10.1152/ajprenal.00013.2008. [DOI] [PubMed] [Google Scholar]
- 15.Ibricevic A, Pekosz A, Walter MJ, Newby C, Battaile JT, Brown EG, et al. Influenza virus receptor specificity and cell tropism in mouse and human airway epithelial cells. J Virol. 2006;80(15):7469–7480. doi: 10.1128/JVI.02677-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kilbourne ED. Influenza pandemics of the 20th century. Emerg Infect Dis. 2006;12:9–14. doi: 10.3201/eid1201.051254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim JK, Gimeno RE, Higashimori T, Kim H-J, Choi H, Punreddy S, et al. Inactivation of fatty acid transport protein 1 prevents fat-induced insulin resistance in skeletal muscle. J Clin Investig. 2004;113(5):756–763. doi: 10.1172/JCI200418917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bensaad K, Harris AL (2016) Lipid metabolism. In: Koumenis C, Hammond E, Giaccia A (eds) Tumor microenvironment and cellular stress: signaling, metabolism, imaging, and therapeutic targets. Springer, New York, p 18 [DOI] [PubMed]
- 19.Kuda O, Pietka TA, Demianova Z, Kudova E, Cvacka J, Kopecky J, et al. Sulfo-N-succinimidyl oleate (SSO) inhibits fatty acid uptake and signaling for intracellular calcium via binding CD36 lysine 164: SSO also inhibits oxidized low density lipoprotein uptake by macrophages. J Biol Chem. 2013;288(22):15547–15555. doi: 10.1074/jbc.M113.473298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lobo S, Wiczer BM, Smith AJ, Hall AM, Bernlohr DA. Fatty acid metabolism in adipocytes: functional analysis of fatty acid transport proteins 1 and 4. J Lipid Res. 2006;48(3):609–620. doi: 10.1194/jlr.M600441-JLR200. [DOI] [PubMed] [Google Scholar]
- 21.Glatz JFC, Luiken JJFP (2016) Myocardial fatty acid uptake. In: Lopaschuk GD, Dhalla NS (eds) Cardiac energy metabolism in health and disease. Springer, New York, p 54
- 22.Lorizate M, Krausslich HG. Role of lipids in virus replication. Cold Spring Harb Perspect Biol. 2011;3(10):a004820. doi: 10.1101/cshperspect.a004820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mansor LS, Fialho MDLS, Yea G, Coumans WA, West JA, Kerr M, et al. Inhibition of sarcolemmal FAT/CD36 by sulfo-N-succinimidyl oleate rapidly corrects metabolism and restores function in the diabetic heart following hypoxia/reoxygenation. Cardiovasc Res. 2017;113(7):737–748. doi: 10.1093/cvr/cvx045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martín-Acebes Miguel A, Ángela Vázquez-Calvo, Caridi F, Sobrino F, Saiz JC. Lipid involvement in viral infections: present and future perspectives for the design of antiviral strategies. Rijeka: INTECH Open Access Publisher; 2013. [Google Scholar]
- 25.Matsufuji T, Ikeda M, Naito A, Hirouchi M, Kanda S, Izumi M, et al. Arylpiperazines as fatty acid transport protein 1 (FATP1) inhibitors with improved potency and pharmacokinetic properties. Bioorg Med Chem Lett. 2013;23(9):2560–2565. doi: 10.1016/j.bmcl.2013.02.116. [DOI] [PubMed] [Google Scholar]
- 26.Munger J, Bennett BD, Parikh A, Feng XJ, Mcardle J, Rabitz HA, et al. Systems-level metabolic flux profiling identifies fatty acid synthesis as a target for antiviral therapy. Nat Biotechnol. 2008;26(10):1179–1186. doi: 10.1038/nbt.1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Noushmehr H, Damico E, Farilla L, Hui H, Wawrowsky KA, Mlynarski W, et al. Fatty acid translocase (FAT/CD36) is localized on insulin-containing granules in human pancreatic β-cells and mediates fatty acid effects on insulin secretion. Diabetes. 2005;54(2):472–481. doi: 10.2337/diabetes.54.2.472. [DOI] [PubMed] [Google Scholar]
- 28.O’Hanlon R, Shaw ML. Baloxavir marboxil: the new influenza drug on the market. Curr Opin Virol. 2019;35:14–18. doi: 10.1016/j.coviro.2019.01.006. [DOI] [PubMed] [Google Scholar]
- 29.Okamura DM, Pennathur S, Pasichnyk K, López-Guisa JM, Collins S, Febbraio M, et al. CD36 regulates oxidative stress and inflammation in hypercholesterolemic CKD. J Am Soc Nephrol. 2009;20(3):495–505. doi: 10.1681/ASN.2008010009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rocks O, Peyker A, Kahms M, Verveer PJ, Koerner C, Lumbierres M, Kuhlmann J, Waldmann H, Wittinghofer A, Bastiaens PI. An acylation cycle regulates localization and activity of palmitoylated Ras isoforms. Science. 2005;307(5716):1746–1752. doi: 10.1126/science.1105654. [DOI] [PubMed] [Google Scholar]
- 31.Rustan AC, Drevon CA. Fatty acids: structures and properties. eLS. 2001 doi: 10.1038/npg.els.0003894. [DOI] [Google Scholar]
- 32.Schwenk RW, Holloway GP, Luiken JJ, Bonen A, Glatz JF. Fatty acid transport across the cell membrane: regulation by fatty acid transporters. Prostaglandins Leukot Essent Fatty Acids. 2010;82(4–6):149–154. doi: 10.1016/j.plefa.2010.02.029. [DOI] [PubMed] [Google Scholar]
- 33.Schaffer JE, Lodish HF. Expression cloning and characterization of a novel adipocyte long chain fatty acid transport protein. Cell. 1994;79(3):427–436. doi: 10.1016/0092-8674(94)90252-6. [DOI] [PubMed] [Google Scholar]
- 34.Stapleford KA, Miller DJ. Role of cellular lipids in positive-sense RNA virus replication complex assembly and function. Viruses. 2010;2(5):1055–1068. doi: 10.3390/v2051055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tarhda Z, Ibrahimi A. Insight into the mechanism of lipids binding and uptake by CD36 receptor. Bioinformation. 2015;11(6):302–306. doi: 10.6026/97320630011302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tomura S, Uchida M, Yonezawa T, Kobayashi M, Bonkobara M, Arai S, et al. Molecular cloning and gene expression of canine apoptosis inhibitor of macrophage. J Vet Med Sci. 2014;76(12):1641–1645. doi: 10.1292/jvms.14-0166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Veit M, Serebryakova MV, Kordyukova LV. Palmitoylation of influenza virus proteins. Biochem Soc Trans. 2013;41(1):50–55. doi: 10.1042/BST20120210. [DOI] [PubMed] [Google Scholar]
- 38.Wu Q, Ortegon AM, Tsang B, Doege H, Feingold KR, Stahl A. FATP1 is an insulin-sensitive fatty acid transporter involved in diet-Induced obesity. Mol Cell Biol. 2006;26(9):3455–3467. doi: 10.1128/MCB.26.9.3455-3467.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang X, Okamura DM, Lu X, Chen Y, Moorhead J, Varghese Z, et al. CD36 in chronic kidney disease: novel insights and therapeutic opportunities. Nat Rev Nephrol. 2017;13(12):769–781. doi: 10.1038/nrneph.2017.126. [DOI] [PubMed] [Google Scholar]