

Abstract
The complement system is a critical component of both innate and adaptive immune responses. It has both protective and pathogenic roles in viral infections. There are no studies regarding the role of complement system in Chandipura virus (CHPV) infection. The current study has investigated the role of complement pathways in the in vitro neutralization of CHPV in Vero E6 cells. Using normal human serum (NHS), heat-inactivated serum (HIS), human serum deficient of complement factor, respective reconstituted serum, assays like in vitro neutralization, real-time PCR, and flow cytometry-based tissue culture-based limited dose assay (TC-LDA) were carried out for assessing the activation of different complement pathways. NHS from 9/10 donors showed complement dependent neutralization, reduction in viral load and decrease in percentage of CHPV-positive cells compared to their HIS counterparts. EGTA or EDTA pretreatment experiments indicated that CHPV neutralization proceeds through the alternative pathway of the complement activation. Our data showed a strong dependence on C3 for the in vitro neutralization of CHPV. Disparity in CHPV neutralization levels between factor B-deficient and reconstituted sera could be attributed to amplification loop/“tick-over” mechanism. Assays using C3, C5, and C8 deficient sera indicated that complement-mediated CHPV neutralization and suppression of CHPV infectivity are primarily through C3 and C5, and not dependent on downstream complement factor C8. With no specific anti-viral treatment/vaccine against Chandipura, the current data, elucidating role of human complement system in the neutralization of CHPV, may help in designing effective therapeutics.
Keywords: Alternative pathway, Complement system, Chandipura virus infection
Introduction
Chandipura virus (CHPV), belonging to family Rhabdoviridae, genus vesiculovirus, was discovered in Chandipura village, Nagpur region of India during an outbreak of febrile illness caused by dengue and chikungunya viruses in 1965 [1]. Since then, it has been responsible for several outbreaks in Andhra Pradesh in 2003 [2, 3], Gujarat in 2004 [4], Nagpur in 2007 [5], and Odisha in 2009 [6]. It is one of the major causes of acute encephalitis in pediatric population in India [2]. Chandipura virus has a negative sense, single-strand RNA genome of size 11 kb. Genome comprises of five proteins. Glycoprotein (G) enables virus absorption, assembly, and budding and, therefore, act as major antigenic determinant., matrix protein (M) responsible for cytotoxicity in infected cells, large protein (L) forms the viral RNA-dependent RNA polymerase, thereby acts as a catalytic subunit, Nucleocapsid protein (N) wraps the viral genome, forms template for virus transcription and phospho protein (P) acts as transcriptional activator [7]. Compared to other encephalitic viruses, CHPV-associated fatality occurs within 24 h of onset of illness [2, 6, 8], leading to infrequent appearance of IgM antibodies to CHPV. Thus, the diagnosis is mainly dependent on the detection of the P gene of CHPV in viral RNA in the human clinical samples by RT-PCR [4]. IgG antibodies against G protein to CHPV are a marker of immunity/recovery from CHPV infection [9].
Following the entry of CHPV into the host cells, the innate immune system gets activated involving the activation of complement system, natural killer cells, neutrophils, and other granulocytes and these sentinel cells present antigens to trigger T-cell-mediated immunity [10]. There are very few reports on therapeutic approach against CHPV using si-RNA [11] and targeted peptide [12], and on vaccine development [9, 13, 14]; however, no vaccine or therapy is currently available for use. With no specific anti-viral treatment and vaccine against Chandipura virus, a better understanding of the host–pathogen interactions is needed to identify the key molecules/therapeutic targets that will regulate the immune response towards recovery.
The complement system is a first line of defense against pathogens including viruses. It serves to link innate and adaptive immunity through a large number of activities such as recognition of viruses, viral neutralization, recruitment and stimulation of leukocytes at the sites of infection, opsonization, and activation of T and B cells [15–18]. Complement activation plays an important role in viral pathogenesis [19] and could also serve to improve the effectiveness of vaccines and therapeutic vectors [19]. The complement cascade can be initiated through three main pathways (depending upon virus recognition): the classical pathway, lectin pathway, or alternative pathway [20]. The classical pathway is antibody dependent and is activated upon binding of C1q to antigen antibody complex on the surface of pathogen. The lectin pathway gets activated when carbohydrate patterns on pathogen, attached to serine proteases like Mannan-binding serine proteases (MASPs) associated with mannan-binding lectin (MBL) [21], ficolins [22].
Alternative pathway activation occurs independent of antibodies [18, 23]. These three pathways converge on a central component C3, which is activated by cleavage into C3a and C3b. C3a serves as a potent anaphylatoxin to promote inflammation. C3b can bind covalently to viral components to aid in opsonization and phagocytosis. The association of C3b with components further downstream, such as C5 through C9, can lead to formation of the membrane attack complex (MAC), which is capable of lysing virus particles or infected cells [16, 24]. Progression of the complement cascade depends on the assembly of C3b with other cleavage products from C4, C2, and factor B to form the C3 convertase [25], a protein complex which functions to amplify the signal by further cleavage of C3 molecules in feedback loop. The alternative pathway C3 convertase complex consists of C3b together with a factor B cleavage product to make C3bBb and the classical/lectin pathway C3 convertase consists of C4 and C2 cleavage products to make C4b C2a.
Available literature suggests higher levels of proinflammatory cytokines, virus specific CD4+ T-regulatory cells, and up-regulated expression of TLR 4 in mice experimentally infected with CHPV to be associated with CHPV pathogenesis [26–29]. In a similar line, Roy et al. have reported monocytes, B cells, and enhanced chemokines to support active CHPV replication in human PBMCs [30]. These non-mutually exclusive processes could be responsible towards enhanced viral load and triggered “cytokine storm” resulting in disease severity. Neutralization of vesicular stomatitis virus (VSV), having similarity with CHPV in genetic makeup, follows the classical complement pathway [31]. With this background, the present study investigates (a) the involvement of complement system in Chandipura virus infection and (b) the mechanisms and contributions of complement components in the in vitro neutralization of CHPV.
Materials and methods
Cells and virus
Vero E-6 (African green monkey kidney epithelial cells, ATCC, USA) cells were maintained and grown in Eagles minimum essential medium (Invitrogen, Carlsbad, CA) supplemented with 1.5 g sodium bicarbonate, 200 mM l-glutamine, 10% heat-inactivated fetal bovine serum (Gibco, USA), penicillin (100 U/ml), and streptomycin (100 ug/ml) (Invitrogen, Carlsbad, CA) at 37° with 5% CO2.
CHPV isolate (AP strain 034627) passaged in Vero E6 cell line was used for the present study. Virus titers were determined by TCID50 method [32].
Complement reagents
Normal human serum (NHS) was collected from 10 healthy adult volunteers, to be used as a functional source of complement components in the complement-related assays. Human blood was allowed to clot at 37 °C for 1.5 h, and the serum was separated at 4 °C and then aliquoted and stored at − 80 °C until further use. For inactivation of complement system, NHS was heated at 56 °C for 30 min and used as heat-inactivated serum (HIS) [33].
The involvement of classical, lectin, and alternative pathways in the virus neutralization was determined by treating NHS with 20 mM EDTA (to block all complement pathways) or with 1 mM EGTA and 2 mM MgCl2 (to block classical and MBL pathway) for 30 min at 37 °C. C3, C5, C8, and factor B-deficient human sera (Complement Technology, Tyler, Texas and Quidel, USA) were reconstituted with physiological concentrations of respective purified human proteins [34]; C3 (1.2 mg/ml), C5 (75 µg/ml), C8 (80 µg/ml), and factor B (200 µg/ml) (Complement Technology, Tyler, Texas and Quidel, USA).
ELISA for detection of IgG anti-CHPV antibodies
G is the spike protein that generates antibody response. Hence, G protein-based ELISA method was used to check CHPV antibodies in serum sample. Serum samples from all donors were checked for IgG anti-CHPV antibodies as described previously [9]. Briefly, infected sf9 supernatants containing (rGp) of CHPV were diluted 1:10 in 50 mM carbonate buffer (pH 9.5) and were coated as 100 µl/well and kept for 2 h at 37 °C. Then, blocking solution was added and kept for 30 min at 37 °C. Following washing with wash buffer, NHS was diluted to 1:25 in blocking solution and 100 µl/well was added to wells. A 1:25 dilution of pre-immune serum served as negative control. Horseradish peroxidase conjugated goat anti-human IgG (Sigma chemicals, St Louis, MO, USA) at 1:10,000 dilution was added to each well and incubated for 30 min at 37 °C. Following the addition of tetramethylbenzidine substrate and incubation in dark for 8–10 min, reaction was stopped and absorbance was measured at 492 nm. A serum sample was considered to be reactive when optimal density (OD) was ≥ cut-off value (mean OD value for three negative controls multiplied by 3). Serum negative for IgG anti-CHPV antibodies was only included in the study.
MTT assay
Cytotoxic nature of complement present in NHS was assessed by MTT assay, a colorimetric test based on reduction of tetrazolium dye by metabolic activity of viable cells. Using three NHS (NHS 7, NHS 8 and NHS 9), MTT assay was performed on Vero E-6 cell line following the protocol followed by [35]. Briefly, 2.4 × 104 Vero E-6 cells were seeded in a 96 well plate at 37 °C for 24 h for monolayer formation. Following this, NHS at a dilution of 1:2 and 1:4 was added to the Vero E-6 monolayers, and cell control wells received only medium. Cells were then kept at 37 °C for 24 h after which they were subjected to MTT assay. As a result, blue formazon crystals formed in viable cells were dissolved in MTT solubilization reagent (DMSO) which were then quantified on ELISA reader (Bio-Rad, USA) at 570 nm after subtracting background reading at 650 nm. The data were represented as percentage of viable cell in the NHS-treated well and cell control wells.
Assessment of anti-CHPV role of complement system by CHPV real-time RT-PCR
Mg and Ca-divalent ions are required for efficient complement activity, i.e., Mg for alternative pathway and Ca-divalent ions for classical and lectin pathway of complement activation [36]. EGTA chelates Ca ions, necessary for the activation of classical and lectin pathways, whereas EDTA chelates both Mg and Ca ions and hence inhibits all the three pathways of complement activation. The addition of Mg divalent ions to EGTA will activate only alternative pathway of complement system. Ten CHPV non-immune NHS and their respective HIS; 20 mM EDTA pre-treated NHS7/NHS8/NHS9; 1 mM EGTA + 2 mM MgCl2 pre-treated NHS7/NHS8/NHS9; C3; C5; C8; and factor B-deficient human serum and deficient serum reconstituted with physiological concentrations of their respective purified human protein C3, C5, C8, and factor B. These sera were twofold diluted and then were incubated with 100TCID50 of CHPV for 1.5 h at 37 °C. Following this, serum + virus mixtures were inoculated on Vero E-6 monolayer grown in 24 well-tissue culture plates (100 µl/well), and control wells received only medium/virus (100 TCID 50 of CHPV). Inoculum was allowed to adsorb for 1 h at 37 °C. Supernatant was then discarded and the cells were washed and further fed with 1 ml of incomplete MEM and were incubated at 37 °C for 24 h. Supernatants and cells were harvested and stored in aliquots at − 80 °C.
A separate Vero E-6 monolayer grown in 96 well-tissue culture plates was also processed in the similar way for the titration of the virus, used in the assay. Briefly, cells were seeded for 24 h and then infected with different dilutions of CHPV. Infected cells were then kept for 24 h at 37 °C. Further supernatant was discarded; cells were washed with incomplete medium and stained with amido black.
Viral RNA was extracted from CHPV stock, 100TCID50 of CHPV, supernatants, and cells harvested after 24 h adsorption, as per the manufacturer’s instructions (Qiagen viral RNA kit). Real-time one-step RT-PCR was performed using 7300 real-time PCR system (Applied Biosystems International, Foster City, CA) [37].
Neutralization assay
Cytopathic effect inhibition-based neutralization assay was performed as described previously with some modifications [33, 38, 39]. In brief, 10 CHPV non-immune NHS and their respective heat-inactivated serum; 20 mM EDTA pre-treated NHS7/NHS8/NHS9; 1 mM EGTA + 2 mM MgCl2 pre-treated NHS7/NHS8/NHS9; C3; C5; C8; and factor B-deficient human serum and the deficient serum reconstituted with physiological concentrations of their respective purified human proteins C3, C5, C8, and factor B were twofold serially diluted and then incubated with 100TCID50 of CHPV for 1.5 h at 37 °C. Serum + virus mixtures were inoculated on Vero E-6 monolayer grown in 96 well plate, control wells received only medium/virus (100 TCID 50 of CHPV). After incubation at 37 °C for 24 h, cells were washed with PBS and stained with amido black. Inhibition to cytotoxicity was checked by counting wells with no cytotoxicity. It was a qualitative analysis. Dilution up to which at least 50% wells showed no cytotoxicity against CHPV was the neutralization titer.
A separate Vero E-6 monolayer grown was also processed in the similar way for the titration of the virus used in the assay.
Tissue-culture-limiting dose assay (TC-LDA) to assess complement response against CHPV
This method directly measures infectivity of CHPV. TC-LDA was used to assess the infectivity of CHPV by incubating with non-immune NHS7/NHS8/NHS 9/HIS7/HIS8/HIS9/NHS7/NHS8/NHS9 pre-treated with 20 mM EDTA/NHS7/NHS8/NHS9 pre-treated with 1 mM EGTA + 2 mM MgCl2/C3-deficient human serum/C3-reconstituted human serum. Briefly, the sera were twofold diluted and then were incubated with 100TCID50 of CHPV for 1.5 h at 37 °C. Following this, serum + CHPV mixtures were inoculated on Vero E-6 monolayer. Control wells received only medium/virus (100 TCID 50 of CHPV). Inoculums were allowed to adsorb on cells for 1 h at 37 °C. After this, supernatants were discarded and cells were fed with 1 ml incomplete MEM and incubated at 37 °C for 24 h. The cells were then harvested and transferred to FACS tubes to perform TC-LDA [40]. Briefly, harvested cells were fixed, and made permeable with cytofix and cytoperm solution (BD Biosciences, USA). Followed by this probing of harvested cells was done with polyclonal purified anti-CHPV IgG antibody raised in mice (1024 reciprocal neutralization titer,) at 4 °C for 1 h. Cells were then washed three times with perm wash buffer followed by addition of donkey anti-mouse IgG Alexa flour 680 (e-Biosciences, USA) and incubated at 4 °C for 30 min. Cells were then washed, resuspended in 2% paraformaldehyde, acquired on FACS Aria II flow cytometer and analyzed using FACS Diva software (BD Biosciences, USA). Cells were gated according to forward and side scatter and at least 10,000 gated events were acquired for assessing cells having antibody conjugated to CHPV-infected cells.
A separate Vero E-6 monolayer grown in 96 well tissue culture plates was also processed in the similar way for the titration of the virus used in the assay in a similar way as that of neutralization assay described previously.
Confirmation of anti-CHPV role of alternative pathway of complement activation by western blot
Activation of factor B by CHPV was assessed by western blot [41]. Briefly, CHPV (7.5 × 103 genome copies) was diluted twofold, and incubated with CHPV non-immune NHS7 (1:10) for 1.5 h at 37 °C. NHS 7 incubated with zymosan (3 µg/µl), was taken as a positive control. The samples were assessed on a 10% SDS-PAGE gel and confirmed by western blotting using mouse monoclonal factor B antibody (Santacruz Biotech, USA) (1:800 dilution), followed by the addition of anti-mouse IgG horseradish peroxidase (Bio-Rad, USA). Blots were visualized by enhanced chemiluminescence (Pierce Chemicals, USA).
Statistical analysis
Viral loads were log-transformed for improvement of normality. Statistics for viral copies from real-time PCR data were done on logarithmic scale and represented the same on Y-axis in graphs. Viral copies obtained from real-time RT-PCR and percentage of population obtained from TC-LDA method were expressed as mean ± standard deviation. Statistics were performed on three independent experiments (n = 3) for all assays. The statistical significance was determined by unpaired student t test for comparison was among two groups as well as by ANOVA with Tukey’s post hoc test when comparison was among multiple groups, using Graphpad prism 5 software. A p ≤ 0.05 was considered to be significant.
Results
Based on the consensus results obtained using different dilutions of 10 CHPV non-immune NHS and respective HIS, 1:4 dilution was decided to be the optimum dilution for both NHS and HIS to be used in all assays. 100 TCID50 of CHPV used for infection worked well at the above mentioned sera dilution. NHS 7 was used as a reference in all assays done with deficient/reconstituted human serum. EGTA/EDTA experiments were done with individual use of NHS7, NHS8, and NHS9.
Effect of treatment of NHS on the proliferation of Vero E-6 cells (MTT assay)
Three NHS at dilutions 1:2 and 1:4 did not exhibit any significant change in the proliferation of Vero E-6 cells (Fig. 1) (p = 0.42).
Fig. 1.

Cytotoxicity assay of NHS Effect of normal human sera treatment on survival of Vero E-6 cells was assessed by MTT cytotoxicity assay. NHS was used at dilutions 1:2 and 1:4. For each serum, 3 independent experiments were performed
Putative role of complement in Chandipura virus infection
Complement-mediated in vitro neutralization of CHPV
Human sera from 10 donors, negative for anti-CHPV antibodies, were assessed for the ability to neutralize CHPV. NHS from donor no. 8 showed the highest reciprocal neutralization titer (reciprocal neutralization titre, 16), whereas donor no 4 serum was ineffective to neutralize CHPV. Sera 1, 3, 5, 6,9,10 showed average neutralization activity. HIS from none of 10 donors were able to neutralize CHPV (Table 1i, ii). Neutralization activity of CHPV naïve human serum could be attributed to complement activation, as their counterpart HIS were not able to neutralize CHPV.
Table 1.
Complement mediated in vitro neutralization of CHPV
| (i) Sera | Reciprocal neutralization titer | TCID50 of CHPV used in assay |
|---|---|---|
| NHS1 | 8 | 7 |
| NHS2 | 4 | 7 |
| NHS3 | 8 | 7 |
| NHS4 | Not able to neutralize | 7 |
| NHS5 | 8 | 7 |
| NHS6 | 8 | 7 |
| NHS7 | 4 | 7 |
| NHS8 | 16 | 7 |
| NHS9 | 8 | 7 |
| NHS10 | 8 | 7 |
| (ii) Sera | Reciprocal neutralization titer | TCID50 of CHPV used in assay |
|---|---|---|
| HIS1 | Not able to neutralize | 7 |
| HIS2 | Not able to neutralize | 7 |
| HIS3 | Not able to neutralize | 7 |
| HIS4 | Not able to neutralize | 7 |
| HIS5 | Not able to neutralize | 7 |
| HIS6 | Not able to neutralize | 7 |
| HIS7 | Not able to neutralize | 7 |
| HIS8 | Not able to neutralize | 7 |
| HIS9 | Not able to neutralize | 7 |
| HIS10 | Not able to neutralize | 7 |
Efficiency of NHS in reducing CHPV replication in vitro
Infectivity of CHPV post-incubation with CHPV non-immune NHS/HIS was assessed by real-time RT-PCR. Overall, (p < 0.0001), NHS could high significantly suppress CHPV copies by 5 log10 (p = ***) in “NHS + CHPV” group compared to CHPV only group, whereas virus copies were comparable in “HIS + CHPV” and CHPV only groups (p = NS). The reduction in the CHPV copies was by 3 log10 (p = ***) when “NHS + CHPV” group was compared with “HIS + CHPV” group (Fig. 2a). At individual serum level (except for one serum), NHS could effectively suppress CHPV copies by 1–6 log10 in “NHS + CHPV” group compared to CHPV only group and by 1–6 log10 in “HIS + CHPV” group (Fig. 2b). CHPV viral load per well was determined by absolute quantification using standard curve method. In brief, a standard curve was generated by the amplification of serial dilutions of in vitro transcribed CHPV RNA (108 to 102 serial dilutions), followed by comparison of test sample with the standard curve and subsequent extrapolation. Viral loads were expressed as RNA copies per well. Detection limit of real-time PCR was 10 copies per reaction.
Fig. 2.



a, b Efficiency of normal human sera (NHS) in reducing CHPV replication in vitro: individual serumwise representation: Vero E-6 cells were seeded in 6 well plate and infected with non-immune NHS/heat-inactivated human sera “(HIS) + CHPV” or “CHPV” only. Total RNA was extracted after 24 h PI. CHPV RNA was quantitated by real-time RT-PCR. “NHS + CHPV” group shows significant inhibition in CHPV replication compared to mentioned groups. Results represent mean of three independent experiments with error bar representing standard deviations. Values are given as mean log 10 CHPV RNA copies/well. Statistical significance was determined by ANOVA Tukeys post hoc test in a and by unpaired student t test in b. Standard bars represent standard deviation. c Measurement of infectious CHPV by TC-LDA. (i) gating strategy Vero E-6 cells were gated based on forward, side scattering. A minimum of 10,000 gated events was acquired for assessing cells having antibody conjugated to CHPV-infected cells. (A) Unstained uninfected Vero E-6 cells for setting, (B) only secondary antibody stained vero E-6 cells. (ii) Representative flowcytometry histogram showing results of stained uninfected Vero E-6; CHPV-infected Vero E-6; NHS7 + CHPV; HIS7 + CHPV (100 TCID 50, 24 hpi). Vero E-6 cells were infected with NHS7 + CHPV; HIS7 + CHPV; CHPV only (100 TCID 50, 24 hpi). Cells were then fixed, permeabilized, and probedwith mouse polyclonal purified anti-CHPV antibody followed by anti-mouse Alexa Flour 680 and acquired in the APC channel of flow cytometer. CHPV-positive cells were gated in the P3 region, set on the basis of CHPV and cell controls in the assay. d Measurement of infectious CHPV by TC-LDA –NHS group shows substantial decrease in CHPV-positive cells percentage compared to other groups. Results represent mean of three independent experiments with error bar representing standard deviations. Values are given as mean ± SD of CHPV-positive cell percent of parent population. Statistical significance was determined by student unpaired t test
Measurement of infectious CHPV by TC-LDA
A decrease of 18.35% in CHPV-positive cell percentage in “NHS7/NHS8/NHS9 +CHPV” group compared to CHPV only group (p = 0.05) was observed. A decrease of 23.08% in CHPV-positive cell percentage in “NHS7/NHS8/NHS9 +CHPV” group compared to “HIS7/HIS8/HIS9 +CHPV” group (p = 0.0103) was observed. However, the percentages of CHPV-positive infected cell in “HIS7/HIS8/HIS9 +CHPV” and CHPV only groups were comparable (p = NS) (Fig. 2c, d).
C3 plays a significant role in CHPV neutralization, replication and infectivity
Neutralization assay of C3 deficient and reconstituted human serum
C3-deficient serum was not able to neutralize CHPV, whereas C3 reconstituted serum showed a reciprocal neutralization titer of 4 (Table 2). Complement 3 deficient human serum upon reconstitution gives reciprocal neutralization titer against CHPV the same as NHS.
Table 2.
Reciprocal neutralization titer for deficient/reconstituted human serum
| Deficient/reconstituted human serum | Deficient reciprocal neutralization titer | Reconstituted reciprocal neutralization titer | TCID 50 |
|---|---|---|---|
| C3 | Not able to neutralize | 4 | 7 |
| Factor B | 4 | 8 | 7 |
| C5 | Not able to neutralize | 4 | 7 |
| C8 | 4 | 8 | 7 |
Effect of C3 deficient and reconstituted human serum on CHPV replication
Infectivity of CHPV incubated with C3 deficient human serum/C3 reconstituted human serum was assessed by infection followed by real-time PCR. C3-reconstituted human serum incubated with CHPV suppressed CHPV copies by 2 log10 (p value = 0.005) compared to C3 deficient human serum (Fig. 3a).
Fig. 3.

a Effect of C3 deficient and reconstituted human serum on CHPV replication Vero E-6 cells were infected with “C3 deficient/reconstituted human serum/NHS7 + CHPV” (100 TCID 50). Viral RNA was extracted 24 h PI and was detected by measuring P gene RNA copies by real-time RT-PCR. “C3 reconstituted + CHPV” shows drastic inhibition in CHPV replication compared to other groups. Statistics were performed on 3 independent experiments. Values are given as mean log 10 CHPV RNA copies/well. Standard bars represent standard deviation. Statistical significance was determined by unpaired student t test. b Effect of C3 deficient and reconstituted human serum on CHPV infectivity by TC-LDA. Vero E-6 cells were infected with “C3 deficient/C3 reconstituted serum +CHPV” or “NHS7 + CHPV” (100 TCID 50, 24 h post-infection). Cells were then fixed, permeabilized, and stained with mouse polyclonal purified anti-CHPV antibody followed by the addition of anti-mouse Alexa Flour 680. C3 reconstituted serum shows a significant decrease in CHPV-positive cells percentage compared to other groups. Statistics were performed on three independent experiments. Values are given as mean ± SD of CHPV-positive cell percent of parent population. Statistical significance was determined by student unpaired t test
Effect of C3 deficient and reconstituted human serum on CHPV infectivity by TC-LDA
A significant decrease of 16.8% (p = 0.009) in CHPV-positive cell percentage in “C3 reconstituted human serum +CHPV” group compared to “C3 deficient human serum + CHPV” group was observed (Fig. 3b).
Alternative pathway of complement activation is involved in CHPV neutralization
To assess which pathway was involved in CHPV neutralization, non-immune NHS was needed to be pre-treated with EGTA + MgCl2 and EDTA. For this concentration of EGTA, MgCl2 and EDTA were optimized to get the concentrations at which they had no effect on CHPV activity. It was found that the treatment of 1 mM EGTA + 2 mM MgCl2 and 20 mM EDTA with CHPV showed clear cytopathic effect (CPE) in accordance with R.M. Dez Prez et al. [36]. Henceforth, NHS7/NHS8/NHS9 were pre-treated with 1 mM EGTA + 2 mM MgCl2/20 mM EDTA and incubated with 100 TCID 50 CHPV. Infectivity was assessed by neutralization assay, real-time RT-PCR, and TC-LDA methods.
Neutralizing ability of EDTA/EGTA pre-treated NHS
NHS with 20 mM EDTA failed to show protection, whereas NHS with 1 mM EGTA + 2 mM MgCl2 showed a reciprocal neutralization titers of 4–8 when done with 3 individual NHS (NHS7, NHS8, and NHS9) (p = 0.035) suggesting possible involvement of alternative pathway in CHPV neutralization (Fig. 4a).
Fig. 4.

a Neutralizing ability of EDTA/EGTA on pre-treated NHS. 100 TCID 50 of CHPV was incubated with 3 individual CHPV non-immune NHS7,8,9 either pre-treated with (a) EGTA and MgCl2 to inactivate classical and lectin pathways or with (b) EDTA to inactivate all the three complement pathways. Viral infectivity was measured by neutralization test on Vero E-6 cells. Results represent mean of three independent experiments with error bar representing standard deviations. Statistical significance was determined by student unpaired t test. b Effect of EGTA/EDTA pretreatment on CHPV copies by real-time RT-PCR. Vero E-6 cells were infected with CHPV non-immune NHS7/NHS8/NHS9 pre-treated either with EGTA and MgCl2 or with EDTA then incubated with CHPV (100 TCID 50). Viral RNA was extracted 24 h post-infection. CHPV RNA was detected by measuring P gene RNA copies by real-time PCR. “NHS7/NHS8/NHS9 pre-treated with EGTA and MgCl2 + CHPV” shows significant decrease in CHPV copies compared to other groups. Statistics were performed on three independent experiments. Values are given as mean log 10CHPV RNA copies/well. Standard bars represent standard deviation. Statistical significance was determined by ANOVA withTukeys post hoc testt. c Effect of EGTA/EDTA pretreatment on CHPV infectivity by TC-LDA. Vero E-6 cells were infected with CHPV non-immune NHS7/NHS8/NHS9 alone/pre-treated either with “EGTA and MgCl2” or with “EDTA then incubated with CHPV” (100 TCID 50). Cells were harvested 24 h post-infection, fixed, permeabilized, and probedwith mouse polyclonal purified anti-CHPV antibody followed by anti-mouse Alexa Flour 680. NHS7/NHS8/NHS9 pre-treated with EGTA and MgCl2 shows significant decrease in CHPV-positive cells percentage compared to other groups. Statistics were performed on three independent experiments. Values are given as mean ± SD of CHPV-positive cell percent of parent population. Statistical significance was determined by student unpaired t test
Effect of EGTA/EDTA pretreatment on CHPV copies by real-time RT-PCR
Overall (p = 0.0017) CHPV copies decreased by 2 log 10 (p = *7) in “NHS7/NHS8/NHS9 + 1 mM EGTA + 2 mM MgCl2 + CHPV” group compared to “NHS7/NS8/NHS9 + 20 mM EDTA + CHPV” group. Three log 10 (p = **) decrease in CHPV copies in “NHS7/NHS8/NHS9 pre-treated with 1 mM EGTA + 2 mM MgCl2 + CHPV” group was observed compared to CHPV only group. CHPV copies were comparable in “NHS7/NHS8/NHS9 pre-treated with 20 mM EDTA + CHPV” and CHPV only groups (Fig. 4b).
Effect of EGTA/EDTA pretreatment on CHPV infectivity by TC-LDA
A decrease of 27.93% (p = 0.0017) of CHPV-positive cell percentage in “NHS7/NHS8/NHS9 pre-treated with 1 mM EGTA + 2 mM MgCl2 incubated with CHPV” group compared to CHPV only group was observed. A decrease of 11.97% (p = 0.0442) of CHPV-positive cell percentage in “NHS7/NHS8/NHS9 pre-treated with 1 mM EGTA + 2 mM MgCl2 incubated with CHPV” group compared to “NHS7/NHS8/NHS9 pre-treated with 20 mM EDTA incubated with CHPV” group was observed. Difference (15.97%) in CHPV-positive cell percentage in “NHS7/NHS8/NHS9 pre-treated with 20 mM EDTA incubated with CHPV” group and CHPV only group was non-significant (p = 0.08) (Fig. 4c).
Confirmation of alternative pathway involvement in CHPV neutralization
To confirm the alternative complement activation pathway participation in CHPV, infectivity of CHPV incubated with human factor B-deficient serum was compared with CHPV incubated with human reconstituted factor B serum. Infectivity was assessed by neutralization assay, real-time PCR, and western blot.
Neutralization ability of factor B-deficient and reconstituted human serum with CHPV
Vero E-6 cells infected with “factor B-deficient serum + CHPV” group showed a reciprocal neutralization titer of 4, whereas “factor B reconstituted serum + CHPV” group showed a reciprocal neutralization titer of 8 (Table 2).
Effect of factor B-deficient and reconstituted human serum on CHPV replication
Difference was 1 log 10 (p = 0.0458) when “factor B reconstituted human serum + CHPV” group was compared with “factor B-deficient +CHPV” group (Fig. 5a).
Fig. 5.
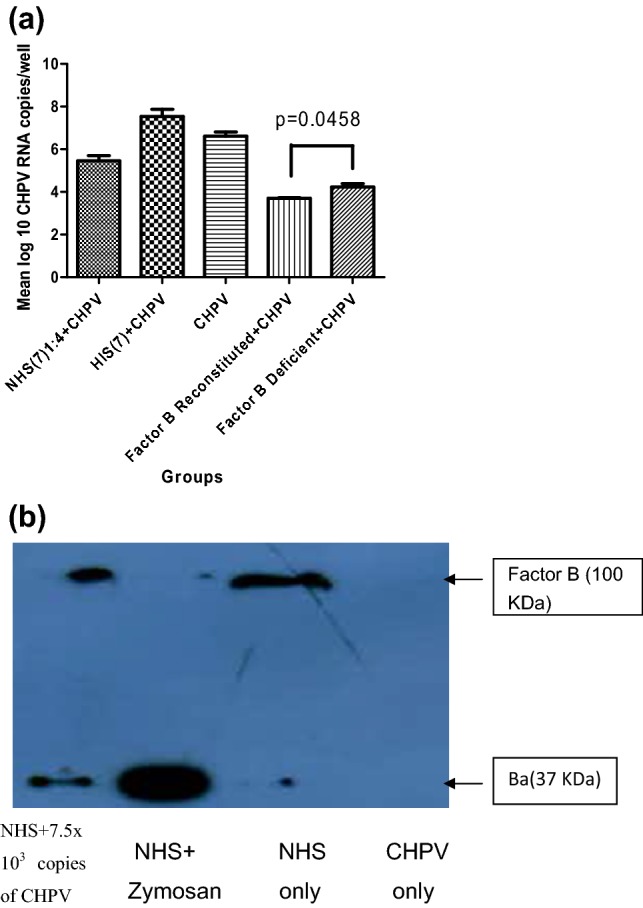
a Effect of factor B-deficient/reconstituted human serum on CHPV replication. Vero E-6 cells were infected with factor B-deficient/reconstituted human serum/NHS7 + CHPV (100 TCID 50), viral RNA was extracted 24 h post-infection. CHPV RNA was detected by measuring P gene RNA copies by real-time RT-PCR. Factor B reconstituted + CHPV shows suppression in CHPV replication compared to factor B-deficient + CHPV group. Values given in figure are mean log 10 CHPV RNA copies/well. Statistics were performed on three independent experiments. Standard bars represent standard deviation. Statistical significance was determined by unpaired student t test. b Western blot confirms that complement activation proceeds through alternative pathway via factor B. NHS7 (1:10 dilution) was incubated for 1.5 h with medium only/zymosan (positive control)/with 7.5 × 103 copies of CHPV. Samples were then assayed for the presence of the factor B (100 KDa) and its cleavage product Bb (37 KDa) by performing western blot
Confirmation of alternative pathway via factor B by western blot
Expression of factor B cleaved product Ba (37 kDa) was observed in NHS7 incubated with 7.5 × 103 copies of CHPV for 1.5 h, and this showed alternative pathway of complement activation. In NHS 7, only, i.e., in the absence of incubation with CHPV, factor B cleaved product (37 kDa) was absent, while the highest expression of cleaved factor B (37 kDa) was observed in the positive control (Fig. 5b).
CHPV in vitro neutralization is C5 dependent and C8 is partly involved
Role of C5 and C8, key molecules for MAC formation, was examined using C5, C8-deficient human serum and C5, C8-deficient human serum reconstituted with their respective proteins, and incubated with 100 TCID 50 of CHPV. The infectivity was assessed by neutralization assay and real-time RT-PCR assay.
Neutralizing role of C5 and C8
C5-deficient serum failed to neutralize CHPV, whereas C5 reconstituted human serum showed a reciprocal neutralization titer of 4. C8 deficient serum showed a reciprocal neutralization titer of 4, whereas C8 reconstituted human serum showed a reciprocal neutralization titer of 8 (Table 2).
Effect of C5 and C8 deficient/reconstituted sera on CHPV replication
C5 deficient/reconstituted sera
CHPV copies decreased by 1 log 10 (p = 0.0065) in “C5 reconstituted human serum + CHPV” group compared to “C5 deficient human serum + CHPV” group (Fig. 6a).
Fig. 6.

a Effect of C5 deficient/reconstituted sera on CHPV replication. Vero E-6 cells were infected with C5 deficient/reconstituted human serum/NHS7 + CHPV (100 TCID 50). Viral RNA was extracted 24 h post-infection. CHPV RNA was detected by measuring P gene RNA copies by real-time RT-PCR. “C5 reconstituted + CHPV” shows drastic inhibition in CHPV replication compared to “C5 deficient + CHPV” group. Values are given as mean log 10 CHPV RNA copies/well. Statistics were performed on three independent experiments. Standard bars represent standard deviation. Statistical significance was determined by unpaired student t test. b Effect of C8 deficient/reconstituted sera on CHPV replication. Vero E-6 cells were infected with C8 deficient/reconstituted human serum/NHS7 + CHPV (100 TCID 50). Viral RNA was extracted 24 h post-infection. CHPV RNA was detected by measuring P gene RNA copies by real-time RT-PCR. No significant difference was observed among all the groups studied. Statistics were performed on three independent experiments. Values are given as mean log 10 CHPV RNA copies/well. Standard bars represent standard deviation. Statistical significance was determined by unpaired student t test
C8 deficient/reconstituted sera
CHPV copies decreased by < 1 log 10 and was statistically non-significant (p = NS) in “C8 reconstituted human serum +CHPV” group compared to “C8 deficient human serum + CHPV” group (Fig. 6b).
Discussion
Complement system plays an important role in the neutralization of a large number of RNA/DNA viruses. Although complement activation partly inhibits infection of vesicular stomatitis virus (VSV) a virus having similarity in genetical make up with CHPV [42–44], the protective/pathogenic role of complement system in CHPV infection remains unexplored. Here, we have determined the extent and mechanisms by which complement system contributes to the neutralization of CHPV in vitro.
Shuang Hu group, while exploring alternative options for vectors for gene delivery, reported that a pseudotype lentiviral vector containing plasmid of amplified CHPV G gene showed some level of neutralization by human sera and compared the complement sensitivity of the VSV G protein to the glycoproteins of Chandipura virus and Piry virus [45]. Here, our data indicated that 9 out of 10 NHS (at 1:4 dilution) showed neutralizing activity against CHPV with reciprocal neutralization titres ranging from 2 to 16 and 7 out of 10 counterparts HIS did not neutralize CHPV at any dilutions (Table 1), suggesting that the observed neutralization could be complement mediated as reported in VSV infection [46]. The observation was further supported by (1) real-time RT-PCR assay showing suppression of CHPV copies by 1 log10 in “NHS + CHPV” group compared to both CHPV only and “HIS + CHPV” groups (Fig. 2a, b). However, factors like antimicrobial peptides present in the serum could be attributed to the observed neutralization of CHPV for HIS1, HIS3 and to some extent for HIS10 (Fig. 2b) compared to their respective virus controls. Besides, in a biological system, all individuals may not behave in a similar manner in response to a pathogen. (2) Flow cytometry-based TC-LDA assay showed decrease of 18.25% and 23.08% in “NHS7/NHS8/NHS9 + CHPV”-positive cells compared to CHPV only and “HIS7/HIS8/HIS9 + CHPV” groups (Fig. 2d), thus establishing the activation of complement system.
Since all the three complement pathways converge to activate C3, the central molecule of the complement cascade [47, 48], we assessed the activation of C3 by real-time PCR, neutralization assay, and flow cytometry-based TC-LDA. Our data showed a strong C3 dependence for the CHPV neutralization, with C3-deficient human serum having no effect on virus infectivity, while reconstitution with physiological concentrations of C3 restored neutralizing capacity with a reciprocal titre of 4 (Table 2). Real-time PCR supported this with a suppression of CHPV copies by 2 log10 in “C3 reconstituted human serum + CHPV” group compared to “C3 deficient human serum + CHPV” group (Fig. 3a), and TC-LDA assay yielded parallel observations showing decrease of 16.8% CHPV-positive cells in “C3 reconstituted human serum + CHPV” compared to “C3 deficient human serum + CHPV” (Fig. 3b). In a similar fashion, Johnson et al. have reported a strong C3 dependence for the inactivation of Simian Virus 5 and Mumps virus showing no effect of C3-depleted serum on virus infectivity, while reconstitution with physiological concentrations of C3 fully restored the neutralizing capacity to C3-depleted serum [33].
Ca2+ is required for the activation of the classical and lectin pathways and Mg2+ is required for the activation of alternative pathway. Since EDTA chelates both Ca2+ and Mg2+ ions, hence, it inhibits all the three pathways [25], whereas EGTA chelates Ca2+ ions, has lower affinity for Mg2+ ions, and thus inhibits classical and lectin pathways, but does not affect the alternative pathway. The pleiotropic effects of EGTA, MgCl2, and EDTA on the function of various pathways/proteins were ruled out using optimized concentrations, where by themselves they had no effect on the virus. Moreover, the concentrations of the chelators and Mg2+ ions used in the current study were much lower than the related published report [36], where the authors had checked the pleiotropic effects of chelators on the integrity of host cells as well as the complement system.
Pretreatment of NHS7/8/9 with 20 mM EDTA failed to show complement-mediated CHPV neutralization, whereas NHS7/8/9 pre-treated with 1 mM EGTA + 2 mM MgCl2 showed a reciprocal neutralization titre of 4 (Fig. 4a), hereby suggesting the participation of alternative pathway. Similar results showing decrease in CHPV copies by 2 log10 in “NHS7/8/9 pre-treated with EGTA + MgCl2” group compared to “NHS7/8/9 pre-treated with EDTA” group as assessed by real-time PCR further confirmed that the observed neutralization is through the alternative pathway of the complement system (Fig. 4b). Flow cytometry-based TC-LDA also supported this observation (Fig. 4c).).
The alternative pathway is activated by recognizing the foreign pathogens with factor B, factor D, and properdin to form C3 convertase (C3BbP) which cleaves C3 to form C3b [49]. The binding of factor B to C3b is Mg2+ dependent [50] and results in the cleavage of factor B into Ba and Bb. Bb combine to the hydrolyzed form of C3 to form C3 (H2O) Bb. Therefore, a constant level of C3b remains in human plasma during this process of self-defense and no pathogen recognition molecule participates in this pathway, and this phenomenon is known as “tick-over” [51–53]. The role and mechanisms of alternative pathway involvement were further explored using in vitro studies, where factor B-deficient human serum showed reciprocal neutralization titre of 4 that was amplified to 8 upon reconstitution with factor B. Reciprocal neutralization titre of 4 in factor B-deficient human serum could very well be attributed to an amplification loop/“tick-over” mechanism. Through the “Tick-over “mechanism, a steady level of C3b remains in human plasma even when pathogen is absent [51–53] and this leads to vigorous activation of alternative pathway upon pathogen recognition [54, 55]. Similar disparities have been reported in collagen antibody-induced arthritis involving in vivo and in vitro studies [56].
Binding of C3b-to-C3 convertase generates C5 convertase, which cleaves C5 into C5a and C5b. C5b initiates the assembly of the MAC by interacting with the downstream molecules on the membrane of the pathogen [57]. C5 can also directly contribute to virus neutralization [44, 58]. Since C8 is also one of the major constituents of the MAC, the effect of C5 and C8 depletion on CHPV neutralization and infectivity was examined. C5-depleted serum failed to exhibit any neutralizing activity against CHPV, but the same was restored when the physiological concentration of C5 was the same as NHS. Parallel evidence of decrease in CHPV infectivity (by real-time PCR) in reconstituted C5 suggested C5 as an essential factor for the in vitro neutralization of CHPV. C8-depleted serum by itself was capable of neutralizing CHPV (4 reciprocal neutralization titer), reconstituting C8 resulted in more effective in vitro neutralization of CHPV (8 reciprocal neutralizing titer) indicating that C8 is not essential for in vitro neutralization of CHPV. However, C8 enhanced neutralization of CHPV at low concentrations of serum (8 reciprocal neutralizing titer).
In a pilot experiment in human-derived cells line involving NHS and HIS, CHPV displayed similar results in neutralization and in virus replication, indicating that complement activation is dependent on the viral component rather than on the producer cell line.
Several studies have reported that C3 activation results in virion lysis through MAC formation [59–61], virion aggregation, opsonization and through activation product deposition on virus particle [62]. Initial studies on VSV have shown that C3b deposition on VSV hinders its attachment to host cell, thereby leading to virus neutralization [63]. Johnson et al. have reported that C3 upon activation by paramyxoviruses, mumps virus and simian virus gets aggravated and deposited on virion which leads to viral lysis [33]. Though C5 is the initiator of MAC formation, it can also directly participate in virus neutralization [44, 58]. In a similar line, C5 involvement but non-involvement of C8 and C9 in the CHPV neutralization of the current study probably denies MAC formation.
In our studies, C5 had a major effect and was found to be essential towards in vitro neutralization of CHPV, whereas C8 had a minor effect and was not an essential factor as evident from the real-time PCR data.
Taken together, our results indicate that the protective involvement of alternative pathway of complement activation towards in vitro neutralization and suppression of infectivity in Chandipura virus infection is primarily C3 and C5 mediated.
Acknowledgements
We thank Director, ICMR-National Institute of Virology for the kind support. Ms Pooja Gupta would like to thank the University Grant Commission, New Delhi, India for providing the Senior Research Fellowship. This research did not receive any specific grant from any funding agency.
Funding
None.
Data availability
All data generated or analyzed during this study are included in this published article.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics statement
The study was approved by the Institutional Ethical Committee for Research on Humans as per the guidelines of the Indian Council of Medical Research.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Pooja Gupta, Email: pooja7gupta@gmail.com.
Anuradha S. Tripathy, Email: anuradhastripathy@hotmail.com
References
- 1.Bhatt PN, Rodrigues FM. Chandipura: a new arbovirus isolated in India from patients with febrile illness. Indian J Med Res. 1967;55:1295–1305. [PubMed] [Google Scholar]
- 2.Rao BL, Basu A, Wairagkar NS, Gore MM, Arankalle VA, Thakare JP, Jadi RS, Rao KA, Mishra AC. A large outbreak of acute encephalitis with high fatality rate in children in Andhra Pradesh, India, in 2003, associated with Chandipura virus. Lancet. 2004;364:869–874. doi: 10.1016/S0140-6736(04)16982-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tandale BV, Tikute SS, Arankalle VA, Sathe PS, Joshi MV, Ranadive SN, Kanojia PC, Eshwarachary D, Kumarswamy M, Mishra AC. Chandipura virus: a major cause of acute encephalitis in children in north Telangana, Andhra Pradesh, India. J Med Virol. 2004;80:118–124. doi: 10.1002/jmv.21041. [DOI] [PubMed] [Google Scholar]
- 4.Chadha MS, Arankalle VA, Jadi RS, Joshi MV, Thakare JP, Mahadev PV, Mishra AC. An outbreak of Chandipura virus encephalitis in the eastern districts of Gujarat State, India. Am J Trop Med Hyg. 2005;73:566–570. [PubMed] [Google Scholar]
- 5.Gurav YK, Tandale BV, Jadi RS, Gunjikar RS, Tikute SS, Jamgaonkar AV. Chandipura virus encephalitis outbreak among children in Nagpur division, Maharashtra, 2007. Indian J Med Res. 2010;132:395–399. [PubMed] [Google Scholar]
- 6.Dwibedi B, Sabat J, Hazra RK, Kumar A, Dinesh DS, Kar SK. Chandipura virus infection causing encephalitis in a tribal population of Odisha in eastern India. Natl Med J India. 2015;28:185–187. [PubMed] [Google Scholar]
- 7.Basak S, Mondal A, Polley S, Mukhopadhyay S, Chattopadhyay D. Reviewing Chandipura: a vesiculovirus in human epidemics. Biosci Rep. 2007;27:275–298. doi: 10.1007/s10540-007-9054-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rao NS, Wairagkar NS, Mohan MV, Khetan M, Somavathi S. Brain stem encephalitis associated with Chandipura virus in Andhra Pradesh outbreak. J Trop Pediatr. 2008;54:25–30. doi: 10.1093/tropej/fmm078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Venkateswarlu CH, Arankalle VA. Recombinant glycoprotein based vaccine for Chandipura virus infection. Vaccine. 2009;27:2845–2850. doi: 10.1016/j.vaccine.2009.02.089. [DOI] [PubMed] [Google Scholar]
- 10.Ghosh S, Basu A. Neuropathogenesis by Chandipura virus: an acute encephalitis syndrome in India. Natl Med J India. 2017;30:21–25. [PubMed] [Google Scholar]
- 11.Kumar S, Arankalle VA. Intracranial administration of P gene siRNA protects mice from lethal Chandipura virus encephalitis. PLoS One. 2010;5:1–13. doi: 10.1371/journal.pone.0008615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roy A, Chakraborty P, Polley S, Chattopadhyay D, Roy S. A peptide targeted against phosphoprotein and leader RNA interaction inhibits growth of Chandipura virus—an emerging Rhabdovirus. Antivir Res. 2013;100:346–355. doi: 10.1016/j.antiviral.2013.09.003. [DOI] [PubMed] [Google Scholar]
- 13.Venkateswarlu CH, Arankalle VA. Evaluation of the immunogenicity of a recombinant glycoprotein-based Chandipura vaccine in combination with commercially available DPT vaccine. Vaccine. 2010;28:1463–1467. doi: 10.1016/j.vaccine.2009.11.072. [DOI] [PubMed] [Google Scholar]
- 14.Jadi RS, Anakkathil SB, Barde PV, Arankalle VA, Mishra AC. Development of an inactivated candidate vaccine against Chandipura virus (Rhabdoviridae: vesiculovirus) Vaccine. 2011;29:4613–4617. doi: 10.1016/j.vaccine.2011.04.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kemper C, Atkinson JP. T-cell regulation: with complements from innate immunity. Nat Rev Immunol. 2007;7:9–18. doi: 10.1038/nri1994. [DOI] [PubMed] [Google Scholar]
- 16.Carroll MC. The complement system in regulation of adaptive immunity. Nat Immunol. 2004;5:981–986. doi: 10.1038/ni1113. [DOI] [PubMed] [Google Scholar]
- 17.Blue CE, Spiller OB, Blackbourn DJ. The relevance of complement to virus biology. Virology. 2004;319:176–184. doi: 10.1016/j.virol.2003.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gasque P. Complement: a unique innate immune sensor for danger signals. Mol Immunol. 2004;41:1089–1098. doi: 10.1016/j.molimm.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 19.Johnson JB, Grant K, Parks GD. The paramyxoviruses simian virus 5 and mumps virus recruit host cell CD46 To evade complement-mediated neutralization. J Virol. 2009;83:7602–7611. doi: 10.1128/JVI.00713-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walport MJ. Complement. First of two parts. N Engl J Med. 2001;344:1058–1066. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- 21.Stahl PD, Ezekowitz RA. The mannose receptor is a pattern recognition receptor involved in host defense. Curr Opin Immunol. 1998;10:50–55. doi: 10.1016/s0952-7915(98)80031-9. [DOI] [PubMed] [Google Scholar]
- 22.Lu J, Le Y. Ficolins and the fibrinogen like domain. Immunobiology. 1998;199:190–199. doi: 10.1016/S0171-2985(98)80026-0. [DOI] [PubMed] [Google Scholar]
- 23.Pangburn MK, Schreiber RD, Müller-Eberhard HJ. Formation of the initial C3 convertase of the alternative pathway: acquisition of C3b-like activities by spontaneous hydrolysis of the putative thioester in native C3. J Exp Med. 1981;154:856–867. doi: 10.1084/jem.154.3.856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stoermer KA, Morrison TE. Complement and viral pathogenesis. Virology. 2011;411:362–373. doi: 10.1016/j.virol.2010.12.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Biswas M, Johnson JB, Kumar SRP, Parks GD, Subbiaha E. Incorporation of host complement regulatory proteins into newcastle disease virus enhances complement evasion. J Virol. 2012;86:12708. doi: 10.1128/JVI.00886-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Balakrishnan A, Mishra AC. Immune response during acute Chandipura viral infection in experimentally infected susceptible mice. Virol J. 2008;5:121. doi: 10.1186/1743-422X-5-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Balakrishnan A, Shahir P. Immune regulation in Chandipura virus infection: characterization of CD4+ T regulatory cells from infected mice. Virol J. 2011;8:259. doi: 10.1186/1743-422X-8-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Balakrishnan A, Shahir P. Chandipura Virus infection in mice: the role of toll like receptor 4 in pathogenesis. BMC Infect Dis. 2012;12:125. doi: 10.1186/1471-2334-12-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Balakrishnan A, Balasubramaniam GA, Shelke VN, Gunjikar R, Shewale P. Neuro-invasion of Chandipura virus mediates pathogenesis in experimentally infected mice. Int J Clin Exp Pathol. 2013;6:1272–1281. [PMC free article] [PubMed] [Google Scholar]
- 30.Roy S, Pavitrakar D, Gunjikar R, Ayachit VM, Bondre VP, Sapkal GN. Monocytes and B cells support active replication of Chandipura virus. BMC Infect Dis. 2016;16:487. doi: 10.1186/s12879-016-1794-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beebe DP, Cooper NR. Neutralization of vesicular stomatitis virus (VSV) by human complement requires a natural IgM antibody present in human serum. J Immunol. 1981;126:1562–1568. [PubMed] [Google Scholar]
- 32.Reed JL, Muench H. A simple method of estimating fifty percent endpoints. Am J Hyg. 1938;27:493–497. [Google Scholar]
- 33.Johnson JB, Capraro GA, Parks GD. Differential mechanisms of complement-mediated neutralization of the closely related paramyxoviruses Simian virus 5 and Mumps virus. Virology. 2008;376:112–123. doi: 10.1016/j.virol.2008.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shevach EM. Chapter 13: Complement. Curr Protoc Immunol. 2003;58:13.0.1–13.0.4. doi: 10.1002/0471142735.im1300s58. [DOI] [Google Scholar]
- 35.Parasha D, Paingankar Mandar S, Kumar Satyendra, Gokhale Mangesh D, Sudeep AB, Shinde Sapana B, Arankalle VA. Administration of E2 and NS1 siRNAs inhibit chikungunya virus replication in vitro and protects mice infected with the virus. PLoS Negl Trop Dis. 2013;7(9):e2405. doi: 10.1371/journal.pntd.0002405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Des Prez RM, Bryan CS, Hawiger J, Colley DG. Function of the classical and alternate pathways of human complement in serum treated with ethylene glycol tetraacetic acid and MgCl2-Ethylene glycol tetraacetic acid. Infect Immun. 1975;11:1235–1243. doi: 10.1128/iai.11.6.1235-1243.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kumar S, Jadi RS, Anakkathil SB, Tandale BV, Mishra AC, Arankalle VA. Development and evaluation of a real-time one step Reverse-Transcriptase PCR for quantitation of Chandipura Virus. BMC Infect Dis. 2008;8:168. doi: 10.1186/1471-2334-8-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tiwari M, Parida M, Santhosh SR, Khan M, Dash PK, Rao PVL. Assessment of immunogenic potential of Vero adapted formalin inactivated vaccine derived from novel ECSA genotype of Chikungunya virus. Vaccine. 2009;27:2513–2522. doi: 10.1016/j.vaccine.2009.02.062. [DOI] [PubMed] [Google Scholar]
- 39.Kumar M, Anakkathil SB, Arankalle VA. Evaluation of recombinant E2 protein-based and whole-virus inactivated candidate vaccines against chikungunya virus. Vaccine. 2012;30:6142–6149. doi: 10.1016/j.vaccine.2012.07.072. [DOI] [PubMed] [Google Scholar]
- 40.Hammarlund E, Amanna IJ, Dubois ME, Barron A, Engelmann F. A flow cytometry-based assay for quantifying non-plaque forming strains of yellow fever virus. PLoS One. 2012;7:e41707. doi: 10.1371/journal.pone.0041707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Johnson JB, Aguilar HC, Lee B, Parks GD. Interactions of human complement with virus particles containing the Nipah virus glycoproteins. J Virol. 2011;85:5940–5948. doi: 10.1128/JVI.00193-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ochsenbein AF, Pinschewer DD, Odermatt B, Carroll MC, Hengartner H, Zinkernagel RM. Protective T cell-independent antiviral antibody responses are dependent on complement. J Exp Med. 1999;190:1165–1174. doi: 10.1084/jem.190.8.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tesfay MZ, Ammayappan A, Federspiel MJ, Barber GN, Stojdl D, Peng KW, Russell SJ. Vesiculovirus neutralization by natural IgM and complement. J Virol. 2014;88:6148–6157. doi: 10.1128/JVI.00074-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hirsch RL, Winkelstein JA, Griffin DE. The role of complement in viral infections III. Activation of the classical and alternative complement pathways by Sindbis virus. J Immunol. 1980;124:2507–2510. [PubMed] [Google Scholar]
- 45.Shuang Hu, Kumar Dipu Mohan, Sax Chelsea, Schuler Clayton, Akkina Ramesh. Pseudotyping of lentiviral vector with novel vesiculovirus envelope glycoproteins derived from Chandipura and Piry viruses. Virology. 2016;488:162–168. doi: 10.1016/j.virol.2015.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mills BJ, Beebe DP, Cooper NR. Antibody-independent neutralization of vesicular stomatitis virus by human complement. II. Formation of VSV lipoprotein complexes in human serum and complement-dependent viral lysis. J Immunol. 1979;121:1549–1557. [PubMed] [Google Scholar]
- 47.Kerr MA. The human complement system: assembly of the classical pathway C3 convertase. Biochem J. 1980;189:173–181. doi: 10.1042/bj1890173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sahu A, Lambris JD. Structure and biology of complement C3, a connecting link between innate and acquired immunity. Immunol Rev. 2001;180:35–48. doi: 10.1034/j.1600-065x.2001.1800103.x. [DOI] [PubMed] [Google Scholar]
- 49.Harrison RA. The properdin pathway: an “alternative activation pathway” or a “critical amplification loop” for C3 and C5 activation? Semin Immunopathol. 2018;40:15–35. doi: 10.1007/s00281-017-0661-x. [DOI] [PubMed] [Google Scholar]
- 50.Pangburn MK, Müller-Eberhard HJ. The C3 convertase of the alternative pathway of human complement. Enzymic properties of the bimolecular proteinase. Biochem J. 1986;235:723–730. doi: 10.1042/bj2350723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bexborn F, Andersson PO, Chen H, Nilsson B, Ekdahl KN. The tick-over theory revisited: formation and regulation of the soluble alternative complement C3 convertase (C3(H2O)Bb) Mol Immunol. 2008;45:2370–2379. doi: 10.1016/j.molimm.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nilsson B, Nilsson Ekdahl K. The tick-over theory revisited: is C3 a contact-activated protein? Immunobiology. 2012;217:1106–1110. doi: 10.1016/j.imbio.2012.07.008. [DOI] [PubMed] [Google Scholar]
- 53.Xue X, Wu J, Ricklin D, Forneris F, Di Crescenzio P, Schmidt CQ, Granneman J, Sharp TH, Lambris JD. Gros P Regulator dependent mechanisms of C3b processing by factor I allow differentiation of immune responses. Nat Struct Mol Biol. 2017;24:643–651. doi: 10.1038/nsmb.3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thurman JM, Holers VM. The central role of the alternative complement pathway in human disease. J Immunol. 2006;176:1305–1310. doi: 10.4049/jimmunol.176.3.1305. [DOI] [PubMed] [Google Scholar]
- 55.Harboe M, Ulvund G, Vien L, Fung M, Mollnes TE. The quantitative role of alternative pathway amplification in classical pathway induced terminal complement activation. Clin Exp Immunol. 2004;138:439–446. doi: 10.1111/j.1365-2249.2004.02627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Banda NK, Takahashi K, Wood AK, Holers VM, Arend WP. Pathogenic complement activation in collagen antibody-induced arthritis in mice requires amplification by the alternative pathway. J Immunol. 2007;179:4101–4109. doi: 10.4049/jimmunol.179.6.4101. [DOI] [PubMed] [Google Scholar]
- 57.Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement system part I—molecular mechanisms of activation and regulation. Front Immunol. 2015;6:262. doi: 10.3389/fimmu.2015.00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Friedman HM, Wang L, Pangburn MK, Lambris JD, Lubinski J. Novel mechanism of antibody independent complement neutralization of Herpes Simplex virus type 1. J Immunol. 2000;165:4528–4536. doi: 10.4049/jimmunol.165.8.4528. [DOI] [PubMed] [Google Scholar]
- 59.Bartholomew RM, Esser AF, Muller-Eberhard HJ. Lysis of oncornaviruses by human serum Isolation of the viral complement (C1) receptor and identification as p15E. J Exp Med. 1978;147:844–853. doi: 10.1084/jem.147.3.844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cooper NR, Jensen FC, Welsh RM, Jr, Oldstone MB. MBA Lysis of RNA tumor viruses by human serum: direct antibody-independent triggering of the classical complement pathway. J Exp Med. 1976;44:970–984. doi: 10.1084/jem.144.4.970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mills BJ, Beebe DP, Cooper NR. Antibody-independent neutralization of vesicular stomatitis virus by human complement II Formation of VSV lipoprotein complexes in human serum and complement-dependent viral lysis. J Immunol. 1979;123:2518. [PubMed] [Google Scholar]
- 62.Oldstone MBA, Cooper NR, Larson DL. Formation and biologic role of polyoma virus-antibody complexes A critical role for complement. J Exp Med. 1974;1974(140):549–565. doi: 10.1084/jem.140.2.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Beebe DP, Cooper NR. Neutralization of vesicular stomatitis virus (VSV) by human complement requires a natural IgM antibody present in human serum. J Immunol. 1981;126:1562–1568. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.