

Abstract
MicroRNA (miRNA)-binding site variants in 3′ untranslated regions (3′UTRs) are a novel class of germ-line, functional mutations, which are now recognized as powerful biomarkers of human cancer risk and biology. The first mutation discovered in this class is the KRAS-variant, a let-7-binding site mutation in the 3′UTR of the KRAS oncogene. The KRAS-variant predicts increased cancer risk for certain populations, is a predictive biomarker of cancer treatment response across cancer types, leads to conserved tumor biology and elevated AKT signaling in KRAS-variant patient tumors, and was recently found to predict elevated TGF-β and immunosuppression in cancer patients. Based on the functional biology of the KRAS-variant in cancer patients, here we chose to investigate altered normal cellular biology in the presence of the KRAS-variant, through interrogation of an isogenic normal breast epithelial cell line model with and without the KRAS-variant. We find that KRAS-variant normal breast epithelial cells exhibit a mesenchymal phenotype, which appears to be due to numerous molecular changes, including miRNA dysregulation and autocrine pathway alterations, including elevated TGF-β, resulting in ZEB and SNAIL upregulation. Our findings support the hypothesis that the KRAS-variant has a fundamental biological impact on normal cellular biology, that is conserved in these patients when they develop cancer.
Subject terms: Epigenetics, Cell biology
Introduction
The discovery of microRNAs (miRNAs) was a critical step forward in our understanding of human development and biology [1]. Subsequently, variants within the binding sites of miRNAs, which tend to be located in the 3′untranslated region (3′UTR) of important growth and survival genes, were recently discovered and identified as a novel class of germ-line mutations, which are powerful biomarkers of cancer risk and treatment response [2]. The first mutation discovered of this class in cancer is the KRAS-variant, a let-7-binding site mutation in the 3′UTR of the KRAS oncogene [3]. This mutation predicts an increased risk of several cancers, including non-small cell lung cancer [3], triple negative breast cancer in premenopausal women [4] and ovarian cancer [5]. In addition, studies have shown that women with the KRAS-variant are at a significantly increased risk of developing multiple primary cancers, including breast and ovarian cancer, as well as third independent cancers [6]. Due to the higher risk of cancer, especially for KRAS-variant women, we and others investigated the impact of estrogen and estrogen withdrawal on women with the KRAS-variant [7]. We found that estrogen withdrawal was associated with a higher risk of breast cancer development, and that women with the KRAS-variant had up to an 11 times higher risk of developing a second, independent breast cancer compared to women without the KRAS-variant, perhaps due to the strategy of anti-estrogen treatment for the management of breast cancer.
In addition, we and others have found that both breast and ovarian tumors in patients with the KRAS-variant have conserved gene expression, with a KRAS-addicted signature, significantly elevated AKT overexpression, as well as an estrogen negative, basal-like gene expression pattern and altered let-7 miRNAs [4, 8]. These biological differences are clearly significant, as the KRAS-variant has been shown to also be a strong predictive biomarker of response to cancer therapy across numerous cancer types, including ovarian cancer [8], lung cancer [9], colon cancer [10], and head and neck cancer [11, 12]. Furthermore, cancer patients with the KRAS-variant appear to have altered immunity, with elevated TGF-β and immunosuppression [12].
The findings from prior work suggest that the KRAS-variant is a functional mutation, which predicts unique baseline biology in those that harbor it, predicting cancer risk as well as the response to cancer treatment. However, gene expression differences and cytokine signaling has only been studied in KRAS-variant patients with cancer. Thus, to better define the impact of the KRAS-variant on normal cell biology, we created matched isogenic normal breast epithelial cell lines with (MCF10AKRAS+/−, MT1 and MT2) versus without (MCF10AKRAS−/−, WT) the KRAS-variant, which we have previously described [7]. Although these KRAS-variant normal epithelial cell lines are not transformed, here we demonstrate that they have fundamentally different baseline biology, with an epithelial to mesenchymal transition, which is mediated through altered miRNA, gene, and autocrine signaling.
Results and discussion
As KRAS-variant individuals have been shown to have conserved, altered tumor miRNA expression [3, 4, 8, 13], we began by evaluating global miRNA expression in our WT versus MT normal breast epithelial cell lines. We found that miRNA expression was significantly altered in the MT versus the WT line (Supplementary Fig. 1). Our analysis in MT versus WT cells revealed the most significantly altered miRNAs were often involved in the epithelial-to-mesenchymal transition (EMT), including mir-200c, miR-32, miR301b, and miR-205 (Fig. 1a) [14]. In fact there was 1000-fold suppression of mir-200c, which is known to regulate EMT in normal breast stem cells and breast cancer stem cells [15–18] as measured by qRT-PCR in MT cells (Fig. 1b). Of note, many of the most differentially expressed miRNAs also have documented roles in driving EMT and inflammation via the TGF-β pathway, including miR-217, miR-23a, miR215, and miR-376 [19–21]. We additionally found suppression of let-7 family members consistent with previous findings in KRAS-variant tumors (Supplementary Fig. 2).
Fig. 1.

MicroRNA and phenotypic differences in KRAS-variant MT cells. a The KRAS-variant alters global microRNA expression. Representation of miRNA expression levels in MCF10A cells with (MCF10aKRAS+/−; MT1 and MT2) versus without (MCF10aKRAS−/−; WT) the KRAS-variant. b Relative mRNA expression of miR-200c in MCF10A cells with (MCF10aKRAS+/−; MT1 and MT2) versus without (MCF10aKRAS−/−; WT) the KRAS-variant at baseline as evaluated by qPCR. c Representative of phase contrast images (top; ×10 objective) and AlexaFluor 488-conjugated phalloidin staining of F-actin (bottom; ×40 objective) in MCF10A cells with (MT1 and MT2) versus without (WT) KRAS-variant grown as a monolayer. The size bars depict 100 and 50 µm length, respectively, in the images. Results of three independent experiments are shown (mean ± SEM). A Student’s t-test (two-tailed) was used to compare two groups and p < 0.05 was considered significant
We next evaluated MT cells to see if the miRNA changes found do indeed predict phenotypic changes and found that MT cells in fact exhibited mesenchymal characteristics. Phenotypically, we found that MT cell lines had taken on a spindle, fibroblast-like morphology (Fig. 1c; top). Phalloidin staining of F-actin confirmed that the MT lines were indeed mesenchymal compared to WT cells, as the actin stress fibers were thicker and more numerous (Fig. 1c; bottom). Also, consistent with a mesenchymal phenotype, we found that cell proliferation in MT cell lines was significantly lower than the WT cells (Supplemental Fig. 3). However, in contrast to tumor cells that have undergone EMT, we did not observe increased migration in MT cells compared to WT parental cells (Supplemental Fig. 4), suggesting that the EMT taking place in the in non-transformed normal epithelial cells harboring the KRAS-variant may be distinct from tumor cells undergoing EMT.
We next wanted to confirm EMT at the molecular level in the KRAS-variant MT cells, and therefore examined known epithelial and mesenchymal markers [19, 22]. Immunofluorescence staining demonstrated surface expression of E-cadherin in WT cells and its absence in MT cells (Fig. 2a; left panels, red). In addition, fibronectin was clearly visible in MT cells and not found in the WT cells (Fig. 2a; right panels, green). Furthermore, by gene expression analysis as measured by qRT-PCR, MT cells expressed significantly lower E-cadherin and Occludin, and significantly higher fibronectin, vimentin, and N-cadherin compared to WT cells (Supplemental Fig. 5). The differences in gene expression levels were also reflected in protein levels as MT cells had significantly lower levels of E-Cadherin, elevated fibronectin, vimentin, and N-cadherin, and modestly reduced β-catenin levels (Fig. 2b), consistent with an EMT phenotype. Next, we investigated the expression of transcription factors SNAIL1, Slug, Twist1 and Twist2, ZEB1 and ZEB2, as repression of E-cadherin is regulated by these factors, which are involved in most physiological EMT processes [19, 22]. We observed significant increases in gene expression levels of SNAIL1, ZEB1, ZEB2, TWIST1 and TWIST2 in MT cells (Supplemental Fig. 6), as well as significantly higher protein levels of SNAIL and ZEB (Fig. 2c), however, reduced protein levels of TWIST, again consistent with an EMT. We hypothesize that differences found between gene expression and protein levels for TWIST are likely due to miRNA regulation. These findings confirm a molecular EMT in KRAS-variant breast epithelial cells.
Fig. 2.

EMT in KRAS-variant epithelial cells. a The representative picture of immunofluorescence images (×40 objective) of MCF10a cells with (MCF10aKRAS+/−; MT1 and MT2) versus without (MCF10aKRAS−/−; WT) the KRAS-variant grown as a monolayer. Cells were stained to detect the expression of the epithelial marker, E-cadherin (red) and the mesenchymal marker, Fibronectin (green). Nuclei were stained with DAPI (blue). The expression of epithelial marker, E-cadherin (Red) is shown in MCF10A WT cells. Upregulation of the mesenchymal marker, Fibronectin (green) is visible in MCF10A MT1 and MT2. The size bars in yellow depict 50 µm length in the images. b Western blot analysis of E-cadherin and occludin (epithelial markers) and Fibronectin, vimentin and N-cadherin (mesenchymal markers) in MCF10A line with (MCF10AKRAS+/−; MT1 and MT2) and without (MCF10AKRAS−/−; WT) the KRAS-variant are shown. The expression of GAPDH was used as an internal control. Results of three independent experiments are shown (mean) and compared quantitatively with GAPDH as baseline using ImageJ software. c Western blot analysis of SNAIL, SLUG, ZEB1 TWIST1 and 2 (EMT transcription factors) in MCF10A line with (MCF10AKRAS+/−; MT1 and MT2) and without (MCF10AKRAS−/−; WT) KRAS-variant are shown. The expression of GAPDH was used as an internal control. Results of three independent experiments are shown (mean) and compared quantitatively using ImageJ software with GAPDH as baseline
We next investigated the potential mechanisms contributing to EMT in KRAS-variant MT cells, starting with the role of miR-200c, as low levels have been shown to lead to an EMT due to ZEB overexpression. Therefore, we restored mir-200c in MT cells by miR200c mimic lentiviral transduction (Supplemental Fig. 7). We found that restoration of miR-200c in KRAS-variant cells resulted in a significant decrease in ZEB1, as well as a modest decrease in SNAIL (Fig. 3a) and decreased mRNA expression of both (Supplemental Figs. 8 and 9). However, there was no visible or molecular reversal of EMT proteins from this single pronged approach (Supplemental Fig. 10). While these findings confirm that mir-200c loss in KRAS-variant MT cells does lead to ZEB1 overexpression and modest SNAIL overexpression, it supports the hypothesis that this pathway is not solely responsible for the observed EMT in these cells.
Fig. 3.

mir-200c and TGFβ lead to EMT through separate pathways in KRAS-variant cells. a Western blot analysis of EMT transcription factors ZEB1 and SNAIL in MCF10A line with (MT1 and MT2) and without (WT) the KRAS-variant after lentiviral transduction of control (pre-mir-vector) and pre-mir-200c are shown. Results of three independent experiments are shown (mean) and compared quantitatively with GAPDH as baseline using ImageJ software. b TGFβ1, TGFβ2, and TGFβ3 expression levels in MCF10A cells with (MCF10aKRAS+/−; MT1 and MT2) and without (MCF10aKRAS−/−; WT) the KRAS-variant are shown. Levels of TGFβ1 (Abcam), TGFβ2 (Abcam), TGFβ3, and GAPDH (Santa Cruz Biotechnology) were estimated by Western blot analysis. Results of three independent experiments are shown (mean) and compared quantitatively with GAPDH as baseline via ImageJ software. c Western blot analysis for detection of phosphorylation of Smad2, Smad3, ERK, AKT, and GAPDH in WT, MT1, and MT2 cells at baseline. Results of three independent experiments are shown (mean) and compared quantitatively with GAPDH as baseline via ImageJ software. d Impact of TGFβ pathway inhibition on EMT was determined by treating MCF10A WT, MT1 and MT2 cells with 10 µM of Galunisertib for 0, 24, 48, and 72 h. Levels of transcription factors ZEB1 and SNAIL and phosphorylation of AKT were measured by Western blotting. Results of three independent experiments are shown (mean) and compared quantitatively using ImageJ software with GAPDH as baseline. e Following serum starvation, cells were treated with 20 µM of LY294002 (PI3K inhibitor) for 1 h. Detection of phosphorylation of AKT and GSK3β and Snail, and GAPDH in WT, MT1 and MT2 was performed by Western blot analysis. Results of three independent experiments are shown (mean) and compared quantitatively with GAPDH as baseline
We next tested the hypothesis that another pathway that leads to EMT in our MT cells involves elevated TGF-β. This hypothesis is based on the known elevated levels of TGF-β in KRAS-variant cancer patients [12], the finding that TGF-β suppresses miR-200 [23], and the fact that TGF-β signaling is known to be causal for EMT in MCF10A cells through canonical, as well as non-canonical pathways activating PI3K and MAPK [24, 25]. Therefore, we first evaluated secretion of TGF-β into culture-conditioned media by an ELISA assay. We found significantly increased secreted levels of TGF-β1 by MT cells, relative to WT (Supplemental Fig. 11). By immunoblotting, we further confirmed that TGF-β1, TGF-β2, and TGF-β3 protein levels were all significantly higher in MT versus WT cells (Fig. 3b). We next determined if downstream signaling was SMAD-dependent or SMAD-independent in MT cells [26]. At baseline we found similar phosphorylation levels of Smad2 and Smad3 in WT versus MT cells, yet a significant increase in phosphorylation of endogenous ERK (as a readout of MAPK activation) and phosphorylated AKT (as a read out of PI3K activation), (Fig. 3c), consistent with SMAD-independent TGF-β signaling in MT cells. Interestingly, consistent with our cell findings, as previously reported, elevated AKT was found to be one of the most significant elevated proteins in tumors from KRAS-variant patients, including breast and ovarian tumors [4, 8].
Finally, in order to confirm the importance of TGF-β signaling in the EMT of MT cells, we used a TGF-β-specific inhibitor, Galunisertib, and found that this inhibition led to a significant decrease in pAKT, SNAIL, and a modest decrease in ZEB (Fig. 3d), as well as significant decreases in the downstream mesenchymal marker Vimentin (Supplemental Fig. 12). These findings support the hypothesis that TGF-β signaling plays an important role in promoting EMT in KRAS-variant cells, and that this occurs in a SMAD-independent manner and primarily through SNAIL and AKT as has been previously reported [27]. However, it is important to emphasize that TGF-β signaling does not appear to be the only pathway leading to EMT in KRAS-variant cells.
To further confirm the importance of downstream AKT stimulation through TGF-β in SNAIL activation, we next evaluated which AKT isoform was phosphorylated in MT cells, and found that AKT1 was phosphorylated at Serine 473, (Supplemental Fig. 13). This isoform has been specifically implicated in TGF-β-mediated EMT [27]. As the PI3K/AKT pathway has been directly implicated in SNAIL activation and EMT through GSK-3β [25, 28], and studies have shown that phosphorylating GSK3β promotes the stability of SNAIL, leading to its upregulation [28, 29], we next evaluated and found significant increased phosphorylation of GSK-3β (S9) in MT versus WT cells through a phospho-kinase array (Supplemental Fig. 14). To confirm if inhibiting PI3K/AKT would impact SNAIL directly we treated WT and MT cells with the PI3K inhibitor LY294002. We found that phosphorylation of AKT Serine 473 and GSK-3β at Serine 9 were both abrogated by treatment with the PI3K inhibitor, which also significantly reduced the level of SNAIL protein expression without any impact on ZEB (Fig. 3e). These findings confirm the importance of the AKT axis in SNAIL activation in MT cells.
It has been reported that the EMT program in mammary epithelial cells can only be maintained by multiple autocrine signaling pathways, in addition to TGF-β [23, 24, 30]. Therefore, we performed a cytokine array to determine if there were additional cytokines and growth factors differentially released by MT versus WT cells that could be leading to EMT. We found that MT1 and MT2 cells had enhanced secretion of multiple soluble factors (Supplemental Fig. 15), including increased levels of pro-angiogenic factors, such as angiopoietin-1, VEGF, PDGF-AA, FGF-19, and angiogenin, as well as several pro-inflammatory factors, including macrophage inflammatory protein-3α (MIP-3α), IL-6, IL-8, GROα, and macrophage migration inhibitory factor (MIF). As many of these factors are known to increase EMT through AKT stimulation [30–33], we next tested as a proof in principle the hypothesis that blocking the receptors of some of these factors in MT cells would impact AKT activation. We found that inhibiting the CXCR2 receptor with inhibitor SB225002, which is a receptor that binds both IL-8 and GRO, directly decreased AKT phosphorylation (Fig. 4a) above its IC50 of 20 nM.
Fig. 4.

Autocrine signaling in KRAS-variant cells. a The CXCR2 inhibitor SB225002 was used on MT cells at indicated doses for 1 h, and AKT phosphorylation was evaluated by western blot. Results of three independent experiments are shown (mean) and compared quantitatively with tAKT as baseline with the help of ImageJ software. b Conditioned medium (CM) from MCF10A cells with (MCF10KRAS+/−; MT1 and MT2) the KRAS-variant was added to MCF10A WT cells that had been starved for 24 h in serum and growth factor-free medium. Whole cell lysates were prepared after the indicated times and analyzed by Western blotting. Results of three independent experiments are shown (mean) and compared quantitatively with GAPDH as baseline. c MCF10A cells (MCF10KRAS+/−; MT1 and MT2) and (MCF10A KRAS−/−; WT) were grown together in chambered wells with serum starved growth factor-free medium so that MCF10A WT cells had continued access to growth factors exclusively secreted from MT cells for a time duration of 24 and 48 h. Whole cell lysates were prepared after the indicated times and analyzed by Western blotting. Results of three independent experiments are shown (mean) and compared quantitatively with GAPDH as baseline using ImageJ software
Based on our evidence that additional autocrine factors were influencing EMT in MT cells, we next tested the hypothesis that conditioned media from KRAS-variant MT cells would induce AKT activation and downstream pathways in WT cells. We first stimulated WT cells with serum free and growth factor-free medium that had been conditioned by MCF10A MT cells (MT media) using a one-time application. We found that conditioned media from our KRAS-variant MT cells did in fact induce immediate phosphorylation of AKT on Ser473 in WT cells. This effect was over by 16 h, however, when AKT phosphorylation returned to baseline (Fig. 4b). There was a significant immediate increase in ZEB in this time frame, as well as a significant increase in phospho-GSK3β by 2 h, but no impact on SNAIL in this timeframe. To increase time of exposure of the WT cells to the MT-conditioned media we next grew cells in a Transwell chamber. We found that this resulted in a longer period of increased AKT phosphorylation, GSK3β phosphorylation, and a significant increase in SNAIL activation, which were beginning to decrease to baseline at 48 h, when the cells reached full plate capacity (Fig. 4c). We next evaluated EMT protein expression in the WT cells grown in MT-conditioned media, and observed a strong induction of fibronectin but no impact on other proteins (Supplemental Figure 16). These findings support the hypothesis that autocrine factors excreted from KRAS-variant cells can lead to AKT activation and subsequent ZEB and SNAIL overexpression, accompanied by regulation of several proteins important for EMT.
Here we have shown that a 3′UTR mutation, the KRAS-variant, can lead to fundamental biological changes in normal MCF10A epithelial cells, that closely resembles gene expression and cytokine changes in KRAS-variant tumors and cancer patients. The KRAS-variant reprograms the cellular phenotype of mutant cells leading to an EMT. There are several alterations in KRAS-variant cells that appear to influence their EMT. One pathway appears to be through miRNA alterations, with loss of miR200c, accompanied by ZEB elevation. However, the primary mechanism of the KRAS-variant-mediated EMT seems to be through altered secreted angiogenesis factors and pro-inflammatory cytokines, including TGF-β. These secreted factors, in turn signal through an autocrine loop, leading to the activation of the AKT-SNAIL pathway, and could also potentially impact miRNA levels such as miR-200c [23] (Fig. 5).
Fig. 5.
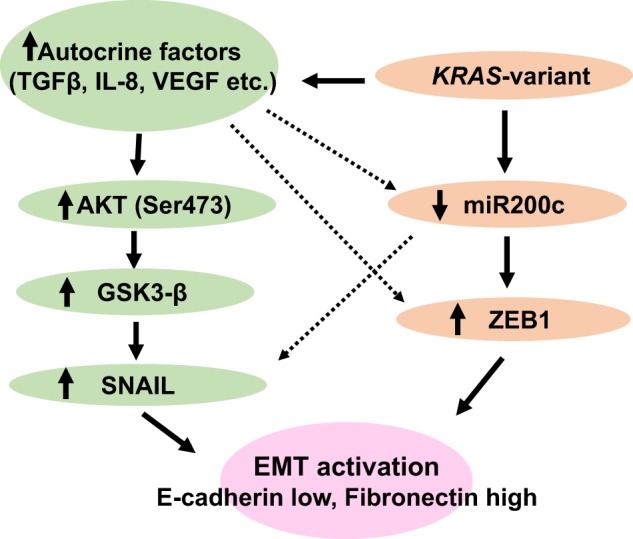
A model of EMT in KRAS-variant MCF10A epithelial cells
Although the KRAS-variant has previously been shown to be functional and lead to altered signaling in tumors, using similar signaling pathways found in this report, such as AKT overexpression and a KRAS “addicted” signature [4, 8], and KRAS tumor mutations have been shown to promote mesenchymal features in breast tumors [34], this is the first evidence of the powerful impact that a 3′UTR germline mutation can have on baseline normal cellular biology. The finding that these KRAS-variant normal epithelial cell lines overexpress TGF-β is especially intriguing, due to the similar finding in KRAS-variant cancer patients [12]. This raises the possibility that immune suppression could be a baseline state for KRAS-variant patients, explaining both their elevated cancer risk, as well as identifying them as a subgroup who would most benefit from both TGF-β inhibitors, as well as immune-stimulating cancer therapy. While understanding how a germline miRNA-binding site mutation leads to altered miRNA and cytokine levels, and which comes first, is work that is still needed, these findings argue for the importance of further elucidating this class of miRNA disrupting normal human variants in biology and clinical applications. It is important to note that better defining baseline biology in such patients is likely the first step towards understanding the triggers for disease in such patients, and could potentially help best direct prevention strategies for such individuals in the near term.
Materials and methods
Cell culture
We previously created perfectly matched isogenic MCF10A cell lines with and without the KRAS-variant, using the CompoZr™ custom designed zinc finger nuclease (ZFN)-targeted genome editing technology, as described [7]. These cells were authenticated by Genetica via bi-allelic short tandem repeat (STR) analysis at 16 different genomic loci, yielding 32 diagnostic markers which confirmed they were the same parental lines, and that the only difference between them was the presence or absence of the KRAS-variant [7]. MCF10A cell lines with (MCF10AKRAS+/−, MT) and without (MCF10AKRAS−/−, WT) the KRAS-variant were maintained in monolayer in Dulbecco’s modified Eagle’s medium-F12 (DMEM/F12) (ThermoFisher Scientific, 10565042) supplemented with 5% horse serum (ThermoFisher Scientific, 26050088), 1% penicillin/streptomycin (Thermo Fisher Scientific, 15140122), 0.5 μg/ml hydrocortisone (Sigma-Aldrich, H-0396), 100 ng/ml cholera toxin (Sigma-Aldrich, C-8052), 10 μg/ml insulin (Sigma-Aldrich, I-0516), and 20 ng/ml recombinant human EGF (Peprotech, 100-15). As previously reported, these cells are not transformed, and do not exhibit anchorage-independent growth [7].
Real-time PCR for epithelial and mesenchymal markers and transcription factors
Total RNA was extracted from two-independent preparations of cells using TRIzol using standard procedures. cDNA libraries were generated (in duplicate) from 1 µg of total RNA using the iScript cDNA Library Synthesis Kit (BioRad). mRNA was analyzed (in triplicate) by qPCR using iQ SYBR Green SuperMix (BioRad) in reactions containing gene-specific primers (listed below). Reactions were amplified in a HT7900 (Applied Biosystems) for 10 min at 95 °C followed by 40 cycles at 95 °C for 15 s and 60 °C for 1 min. mRNA expression was normalized to Beta-Actin and relative expression was calculated using the delta–delta Ct method. Primers were designed to span exon–intron–exon junctions and produced amplicons of ~500 bp. Primers were synthesized at the UCLA Oligonucleotide Synthesis facility. For TaqMan gene expression assays, relative mRNA expression of EMT transcription factor in MCF10A line with (MCF10AKRAS+/−; MT) KRAS-variant cell was compared with MCF10A without (MCF10AKRAS−/−; WT) KRAS-variant. Cells were cultured in regular growth media for MCF10A. Cells were harvested for RNA isolation, which was used in real-time RT-PCR for quantification of SNAI1, SNAI2, ZEB1, ZEB2, TWIST1, TWIST2, and GAPDH mRNA. Relative mRNA expression levels were normalized by GAPDH mRNA expression levels. Total RNA was prepared by using a miRNeasy kit (Qiagen). Primers of SNAI1 (Hs00195591_m1), SNAI2 (Hs00161904_m1), ZEB1 (Hs00232783_m1), ZEB2 (Hs00207691_m1), TWIST1 (Hs01675818_s1), TWIST2 (Hs02379973_s1) SNAI1, SNAI2, ZEB1, ZEB2, TWIST1, TWIST2, and GAPDH (Hs02758991_g1) were purchased from Applied Biosystems. Real-time PCR was done in triplicate with two independent experiments with TaqMan PCR Mastermix (Applied Biosystems).
Immunoblotting and antibody phosphokinase array
Cells were lysed in RIPA buffer with a phosphatase and protease inhibitor (Thermofisher). Protein concentration was determined using BCA assay (Thermo Fisher). Thirty micrograms of protein was separated by SDS–PAGE and transferred to nitrocellulose membranes which were blocked with 5% non-fat milk TBST for 1 h at room temperature before overnight incubation with primary antibody diluted in 5% non-fat milk TBST. HRP-conjugated secondary antibody (Cell Signaling Technology) was applied and detected by ECL Western Blotting Detection Reagent (GE Healthcare Life Sciences) and X-ray film (Bioland). Serum starved cell were harvested, washed with PBS, lysed with RIPA lysis buffer with a phosphatase and protease inhibitor (Thermofisher). Antibody array used was the human phosphor-kinase (ARY003B) array from R&D Systems. Each membrane was applied with 200 µg cell lysate according to manufacturer’s instructions.
Immunofluorescence
MCF10A WT and MCF10A MT were seeded on a four-well Lab-Tek Slide (Thermo Scientific). After 5 days, cells were washed with phosphate-buffered saline (PBS) twice and fixed with 4% paraformaldehyde in PBS for 20 min at room temperature. The cells were then washed three times with PBS and incubated with the blocking solution, which contains 1% bovine serum albumin (BSA), 10% normal donkey serum, and 0.3% Triton X 100 in PBS for 1 h at room temperature. Cells were then incubated with the primary antibodies (anti-E-cadherin (NL557 polyclonal Ab, goat IgG [NL648R] and human anti-Fibronectin MAB, Mouse IgG1 [MAB1918]; R&D Systems) in the blocking solution for overnight at 4 °C. Cells were washed three times with 0.1% BSA in PBS for 5 min each wash and incubated with NL 493 and NL 557-conjugated secondary antibodies (R&D Systems) in 1% BSA in PBS for 2 h at room temperature in the dark. The slides were washed with PBS and mounted with Antifade Mountant with DAPI (Invitrogen) and were cured for 24 h at room temperature in the dark.
miRNA microarray analysis
Total RNA was extracted from cells using Trizol using standard procedures. miRNA cDNA libraries were generated from total RNA (1 µg) using Megaplex RT Primers (Human Pool A, Applied Biosystems). MicroRNA expression was analyzed using a TaqMan Array Human miRNA Card A v2.0 (Applied Biosystems). Reactions were cycled in a HT7900 (Applied Biosystems) for 10 min at 95 °C followed by 40 cycles at 95 oC for 15 s and 60 oC for 1 min. miRNAs that had a Ct value of >350,000 were excluded from further analysis. Relative expression was calculated using the delta–delta Ct method.
Transduction of miRNA mimics
Transduction of MCF10A with KRAS-variant cells with either hsa-mir-200c lentivirus (Biosettia) or mir-control lentivirus (Biosettia) was performed using manufacturer’ protocols. Briefly, 5 × 104 cells per well were infected with 1 × 107 lentiviral transduction units (TU) (multiplicity of infection [MOI] = 1.5) with the addition of hexadimethrine bromide (8 µg/ml; Sigma-Aldrich) as adjuvants. After 24 h of incubation (at 37 °C and 5% CO2), the medium was changed, and cells were incubated for an additional 48 h in adequate medium with the inclusion of puromycin (1 µg/ml) before further analysis.
ELISA
Cells were starved overnight in no phenol red DMEM/F12 medium (ThermoFisher Scientific). Starvation medium was removed, and 4 ml of fresh assay medium containing 1% charcoal stripped horse serum in no phenol red DMEM/F12 medium was added to cells in 10-cm culture dish. After 24 h, culture medium was collected by centrifugation. All ELISAs were performed using commercially available kits according the manufacturer’s instructions. For TGFβ-1 and TGFβ-2 ELISA kits were purchased from R&D systems. All ELISA readouts were normalized by cell number.
Statistical analysis
Data are presented as mean ± SEM when appropriate. A Student’s t-test (two-tailed) was used to compare two groups (p < 0.05 was considered significant) unless otherwise indicated.
Supplementary information
Acknowledgements
JBW was supported by a grant from the NIH (CA157749) and DA was supported by NIH grant (R01CA220693-01) for this study. The funding body had no role in the design of the study, the collection, analysis, and interpretation of the data, or in writing of the manuscript.
Author contributions
JBW contributed to the conception and design of the study; SJ, PM, KCN, DS, YQ, EHP, JK, and EV contributed to data acquisition, SJ, KCN, EV, and DA contributed to data analysis; all authors contributed to the interpretation of the results; SJ, PM, and JBW drafted the manuscript and all authors contributed to the intellectual content of the revisions; all authors approved the final version of the manuscript.
Compliance with ethical standards
Conflict of interest
JBW has founded a company that has licensed IP around the KRAS-variant from Yale University. No funds were supplied from the company for this work, nor do the findings in this manuscript impact the value of the IP owned by Yale University. The other authors declare that they have no conflict of interest.
Footnotes
Edited by G. Del Sal
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version of this article (10.1038/s41418-019-0320-y) contains supplementary material, which is available to authorized users.
References
- 1.Reinhart B, Slack F, Basson M, Pasquinelli A, Bettinger J, Rougvie A, et al. The 21 nucleotide let-7 RNA regulates C. elegans developmental timing. Nature. 2000;403:901–6. doi: 10.1038/35002607. [DOI] [PubMed] [Google Scholar]
- 2.Cipollini M, Landi S, Gemignani F. MicroRNA binding site polymorphisms as biomarkers in cancer management and research. Pharmgenomics Pers Med. 2014;7:173–91. doi: 10.2147/PGPM.S61693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chin LJ, Ratner E, Leng S, Zhai R, Nallur S, Babar I, et al. A SNP in a let-7 microRNA complementary site in the KRAS 3’ untranslated region increases non-small cell lung cancer risk. Cancer Res. 2008;68:8535–40. doi: 10.1158/0008-5472.CAN-08-2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paranjape T, Heneghan H, Lindner R, Keane F, Hoffman A, Hollestelle A, et al. A 3’-untranslated region KRAS variant and triple-negative breast cancer: a case-control and genetic analysis. Lancet Oncol. 2011;12:377–86. doi: 10.1016/S1470-2045(11)70044-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ratner E, Lu L, Boeke M, Barnett R, Nallur S, Chin LJ, et al. A KRAS-variant in ovarian cancer acts as a genetic marker of cancer risk. Cancer Res. 2010;70:6509–15. doi: 10.1158/0008-5472.CAN-10-0689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pilarski R, Patel DA, Weitzel J, McVeigh T, Dorairaj JJ, Heneghan HM, et al. The KRAS-variant is associated with risk of developing double primary breast and ovarian cancer. PLoS One. 2012;7:e37891. doi: 10.1371/journal.pone.0037891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McVeigh TP, Jung SY, Kerin MJ, Salzman DW, Nallur S, Nemec AA, et al. Estrogen withdrawal, increased breast cancer risk and the KRAS-variant. Cell Cycle. 2015;14:2091–9. doi: 10.1080/15384101.2015.1041694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ratner ES, Keane FK, Lindner R, Tassi RA, Paranjape T, Glasgow M, et al. A KRAS variant is a biomarker of poor outcome, platinum chemotherapy resistance and a potential target for therapy in ovarian cancer. Oncogene. 2012;31:4559–66. doi: 10.1038/onc.2011.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weidhaas Joanne B., Kim Edward S., Herbst Roy S., Yu Jun, Slack Frank, Blumenschein George R., Tsao Anne S., Wistuba Ignacio Ivan, Lee J. Jack, Papadimitrakopoulou Vassiliki, Hong Waun Ki, Heymach John, Tran Hai T. The KRAS-variant and treatment response in BATTLE-1. Journal of Clinical Oncology. 2014;32(15_suppl):8135–8135. doi: 10.1200/jco.2014.32.15_suppl.8135. [DOI] [Google Scholar]
- 10.Saridaki Z, Weidhaas JB, Lenz HJ, Laurent-Puig P, Jacobs B, De Schutter J, et al. A let-7 microRNA-binding site polymorphism in KRAS predicts improved outcome in patients with metastatic colorectal cancer treated with salvage cetuximab/panitumumab monotherapy. Clin Cancer Res. 2014;20:4499–510. doi: 10.1158/1078-0432.CCR-14-0348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chung CH, Lee JW, Slebos RJ, Howard JD, Perez J, Kang H, et al. A 3’-UTR KRAS-variant is associated with cisplatin resistance in patients with recurrent and/or metastatic head and neck squamous cell carcinoma. Ann Oncol. 2014;25:2230–6. doi: 10.1093/annonc/mdu367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weidhaas JB, Harris J, Schaue D, Chen A, Chin R, Axelrod R, et al. The KRAS-variant and cetuximab response in head and neck squamous cell cancer, a secondary analysis of a randomized cinical trial. JAMA Oncology. 2017;3:483–91. doi: 10.1001/jamaoncol.2016.5478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Christensen B, Moyer B, Avissar M, Ouellet L, Plaza S, McClean M, et al. A let-7 microRNA binding site polymorphism in the KRAS 3’UTR is associatied with reduced survival in oral cancers. Carcinogenesis. 2009;30:1003–7. doi: 10.1093/carcin/bgp099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abba Mohammed, Patil Nitin, Leupold Jörg, Allgayer Heike. MicroRNA Regulation of Epithelial to Mesenchymal Transition. Journal of Clinical Medicine. 2016;5(1):8. doi: 10.3390/jcm5010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shimono Y, Zabala M, Cho RW, Lobo N, Dalerba P, Qian D, et al. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell. 2009;138:592–603. doi: 10.1016/j.cell.2009.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dou J, He XF, Cao WH, Zhao FS, Wang XY, Liu YR, et al. Overexpression of microRna-200c in CD44+CD133+ CSCS inhibits the cellular migratory and invasion as well as tumorigenicity in mice. Cell Mol Biol. 2013;59(Suppl):OL1861–8. [PubMed] [Google Scholar]
- 17.Park SM, Gaur AB, Lengyel E, Peter ME. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008;22:894–907. doi: 10.1101/gad.1640608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, Sonntag A, et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. 2009;11:1487–95. doi: 10.1038/ncb1998. [DOI] [PubMed] [Google Scholar]
- 19.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial–mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15:178–96. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Butz Henriett, Rácz Károly, Hunyady László, Patócs Attila. Crosstalk between TGF-β signaling and the microRNA machinery. Trends in Pharmacological Sciences. 2012;33(7):382–393. doi: 10.1016/j.tips.2012.04.003. [DOI] [PubMed] [Google Scholar]
- 21.Guo L, Zhang Y, Zhang L, Huang F, Li J, Wang S. MicroRNAs, TGF-beta signaling, and the inflammatory microenvironment in cancer. Tumour Biol. 2016;37:115–25. doi: 10.1007/s13277-015-4374-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131–42. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- 23.Gregory P, Bracken C, Smith E, Bert A, Wright J, Roslan S, et al. A TGF-B/ZEB/miR-200 network controls EMT. Mol Biol Cell. 2011;22:1686–98. doi: 10.1091/mbc.e11-02-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scheel C, Eaton E, Hsin-Jung LIS, Chaffer C, Reinhardt F, Kah KJ, et al. Paracrine and autocrine signals induce and maintain mesenchymal stem cell states in the breast. Cell. 2011;145:926–40. doi: 10.1016/j.cell.2011.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bellacosa A., Larue L. Cancer Genome and Tumor Microenvironment. New York, NY: Springer New York; 2009. PI3K/AKT Pathway and the Epithelial-Mesenchymal Transition; pp. 11–32. [Google Scholar]
- 26.Massague J. TGFbeta in cancer. Cell. 2008;134:215–30. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang L, Zhou F, Dijke P. Signaling interplay between transforming growth factor-B receptor and PI3K/AKT pathways in cancer. Trends Biochem Sci. 2013;38:612–20. doi: 10.1016/j.tibs.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 28.Zhou BP, Deng J, Xia W, Xu J, Li YM, Gunduz M, et al. Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat Cell Biol. 2004;6:931–40. doi: 10.1038/ncb1173. [DOI] [PubMed] [Google Scholar]
- 29.Wu Y, Deng J, Rychahou PG, Qiu S, Evers BM, Zhou BP. Stabilization of snail by NF-kappaB is required for inflammation-induced cell migration and invasion. Cancer Cell. 2009;15:416–28. doi: 10.1016/j.ccr.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Long X, Ye Y, Zhang L, Liu P, Yu W, Wei F, et al. IL-8, a novel messenger to cross-link inflammation and tumor EMT via autocrine and paracrine pathways. Int J Oncol. 2016;48:5–12. doi: 10.3892/ijo.2015.3234. [DOI] [PubMed] [Google Scholar]
- 31.Wegiel B, Bjartell A, Culig Z, Persson J. Interleukin-6 activated PI3K/Akt pathway and regulates cyclin A1 to promote prostate cancer cell survival. Int J Cancer. 2008;122:1521–9. doi: 10.1002/ijc.23261. [DOI] [PubMed] [Google Scholar]
- 32.Wang L, Tang C, Cao H, Kuangfa L, Pang X, Zhong L, et al. Activation of IL-8 via PI3K/Akt-dependent pathway is involved in leptin-mediated epithelial-mesenchymal transition in human breast cancer cells. Cancer Biol Ther. 2015;16:1220–30. doi: 10.1080/15384047.2015.1056409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Papapetropoulos Andreas, Fulton David, Mahboubi Keyvan, Kalb Robert G., O'Connor Daniel S., Li Fengzhi, Altieri Dario C., Sessa William C. Angiopoietin-1 Inhibits Endothelial Cell Apoptosis via the Akt/Survivin Pathway. Journal of Biological Chemistry. 2000;275(13):9102–9105. doi: 10.1074/jbc.275.13.9102. [DOI] [PubMed] [Google Scholar]
- 34.Kim RK, Suh Y, Yoo KC, Cui YH, H K, Kim MJ, et al. Activation of KRAS promotes the mesenchymal features of basal-type breast cancer. Exp Mol Med. 2015;47:e137. doi: 10.1038/emm.2014.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.