

SUMMARY
Pain is an integrated sensory and affective experience. Cortical mechanisms of sensory and affective integration, however, remain poorly defined. Here we investigate the projection from the primary somatosensory cortex (S1), which encodes the sensory pain information, to the anterior cingulate cortex (ACC), a key area for processing pain affect, in freely behaving rats. Using a combination of optogenetics, in vivo electrophysiology and machine learning analysis, we find that a subset of neurons in the ACC receive S1 inputs, and activation of S1 axon terminals increases the response to noxious stimuli in ACC neurons. Chronic pain enhances this cortico-cortical connection, as manifested by an increased number of ACC neurons that respond to S1 inputs and the magnified contribution of these neurons to the nociceptive response in the ACC. Furthermore, modulation of this S1→ACC projection regulates aversive responses to pain. Our results thus define a cortical circuit that plays a potentially important role in integrating sensory and affective pain signals.
Keywords: somatosensory cortex, anterior cingulate cortex, pain, chronic pain
eTOC Blurb
Singh et al. reveal that neurons from the anterior cingulate cortex (ACC) receive inputs from the primary somatosensory cortex (S1), and that activation of these inputs increases the nociceptive response in the ACC and pain-aversive behaviors. Chronic pain, meanwhile, enhances the nociceptive connection between the S1 and the ACC.
INTRODUCTION
Sensory processing requires the transmission of information from sensory cortices into higher order areas in the neocortex that mediate affective and cognitive responses. Such sensory and affective integration is particularly important for the experience of pain, as tissue injury or trauma elicits specific nociceptive signals, which must trigger affective responses to defend against physical harm [1]. On the other hand, unlike acute pain that constitutes a normal response to physical harm, chronic pain represents a maladaptive experience in sensory and affective processing [2, 3]. However, mechanisms of sensory and affective integration of nociception remain incompletely understood, and even less is known about how such mechanisms contribute to chronic pain.
While sensory and affective integration can occur at multiple neural levels, the anterior cingulate cortex (ACC) and the primary somatosensory cortex (S1) are attractive candidates. Canonical ascending nociceptive pathways terminate in the S1 and the ACC to process the sensory and affective aspects of pain, respectively [4–16]. The S1 carries important sensory information of pain; however, it remains unclear to which higher cortical areas the S1 projects its sensory signals. Meanwhile, the ACC has been shown to play a key role in the aversive reactions to pain in studies of animal models as well as human subjects [4–14, 17–20]. It can integrate nociceptive information from the medial thalamus and project to subcortical regions such as the amygdala and nucleus accumbens [21–23]. However, it is not well understood whether the ACC also receives nociceptive information from sensory cortices. A direct projection from the S1 to the ACC would provide a potential mechanism for sensory and affective integration at the cortical level. Furthermore, both the S1 and ACC are known to undergo plasticity in the chronic pain state, raising the question if alterations in the S1→ACC circuit can contribute to chronic pain behaviors.
Here we identified a subset of ACC neurons that received direct inputs from the S1, which were highly responsive to nociceptive stimuli. Unbiased machine learning analysis further supports the specificity of this circuit for nociceptive processing. Chronic pain, meanwhile, strengthens this connection between the S1 and the ACC. At the behavioral level, we found that this S1→ACC projection could regulate the aversive response to pain. Together, these results demonstrate a potential cortical mechanism for the integration of sensory and affective pain signals.
RESULTS
Nociceptive information flow from the S1 to the ACC
We first examined the anatomic link between the S1 and the ACC. We focused on the hind limb region of the S1, to be compatible with our electrophysiological and behavior studies in acute and chronic pain models. Retrograde beads injected in the ACC were found in the cell bodies of neurons in the S1 hind limb region (Figures S1A and S1B). Likewise, anterograde expression of yellow fluorescent protein (YFP) injected in the S1 was found in the ACC (Figures S1C–S1E). These results confirm findings from previous anatomic studies [24, 25].
Having established the direct anatomic projection from the S1 to the ACC, we investigated its functional role in nociception. First, we inserted recording electrodes into the ACC to measure in vivo neural activities in response to either a noxious (pin prick, or PP) or a non-noxious mechanical stimulus (von Frey filaments, or vF) (Figure S2A–S2D) in freely behaving rats.
Approximately 16% of neurons recorded in the ACC responded to the noxious stimulus, whereas only 7% of neurons responded to the non-noxious stimulus, using the same threshold criteria (Figures S2E–S2G; see Methods). When we examined the neurons that responded to both PP and vF, PP triggered a higher firing rate in these neurons (Figure S2H). In order to study whether population responses could differentiate between noxious and non-noxious stimuli, we applied discriminative statistical machine learning analysis (using a support vector machine or SVM; see Methods) based on the firing rates of individual ACC neurons. A peak decoding accuracy of 75% validated the specificity of the nociceptive response in the ACC (Figure S2I and S2J).
Next, we used an optrode approach to investigate if direct S1 inputs to the ACC could alter the nociceptive response of ACC neurons in freely behaving animals. We used a CaMKII promoter to express channelrhodopsin (ChR2) in pyramidal neurons in the S1 hind limb region, and optogenetically activated the axon terminals of S1 neurons that projected to the ACC while simultaneously recording these ACC neurons (Figure 1A). Activation of presynaptic S1 inputs increased the overall spiking rates of ACC neurons evoked by PP, but not vF (Figures 1B–1F). These results suggest that the S1 projection can directly modulate the nociceptive response of ACC neurons. While these results do not completely rule out the effect of S1 inputs on other non-noxious stimuli in general, they demonstrate the significant impact of S1 activation on the nociceptive response in the ACC. To validate the role of S1 inputs for the ACC nociceptive response, we compared unbiased SVM decoding analysis in the presence and absence of S1 activation. Our analysis demonstrates that activation of the S1→ACC projection enriched the specificity of the nociceptive neural codes in the ACC for distinguishing between noxious and non-noxious stimuli (Figures 1G and 1H). These results from unbiased machine learning analysis support the finding that S1 inputs elevate the ensemble nociceptive response in the ACC.
Figure 1. S1 inputs enhance the nociceptive response in the ACC.
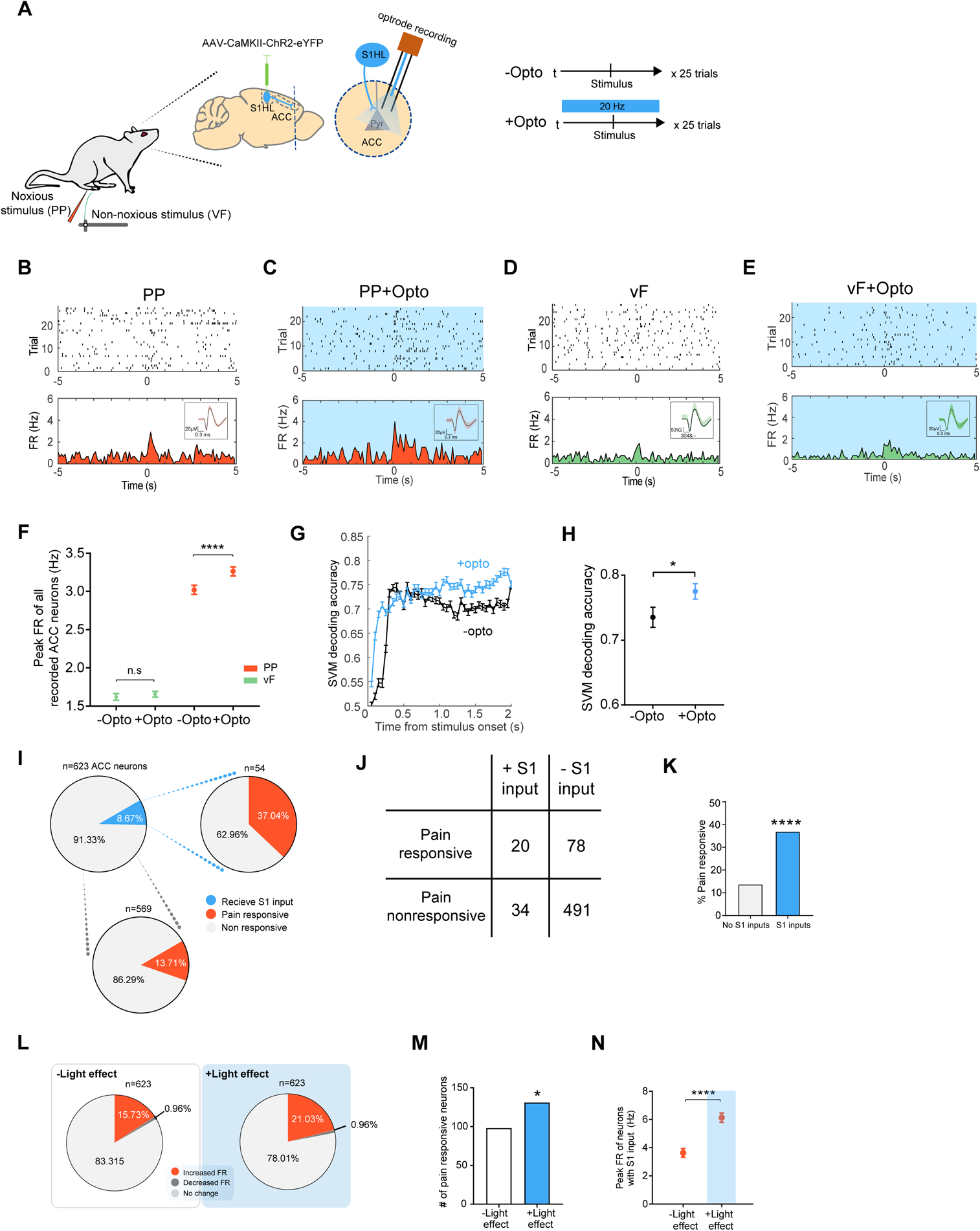
(A) Schematic for in vivo optrode recording experiments. (B) Raster plots and peri-stimulus time histograms (PSTHs) of a representative ACC neuron without activation of S1 inputs. Time 0 indicates the onset of noxious pin prick (PP) stimulation. FR: firing rates. Inset shows representative single cell recordings. (C) Representative recording trace shows that optogenetic activation of the presynaptic S1 inputs increased the firing rates of a pyramidal neuron in the ACC, in response to PP. (D) Raster plots and PSTHs of a representative ACC neuron without activation of S1 inputs. Time 0 indicates the onset of non-noxious von Frey filament (vF) stimulation. (E) Representative recording trace shows that optogenetic activation of the S1 inputs did not change the firing rate response to vF in an ACC neuron. (F) Activation of the presynaptic S1 inputs increased the firing rates of ACC neurons, in response to PP. n = 623 from 5 rats; p < 0.0001, Wilcoxon matched-pairs signed rank test. In contrast, activation of the presynaptic S1 inputs had no impact on the firing rates of ACC neurons, in response to vF. n = 567 from 5 rats; p = 0.3299. (G) A representative session of SVM-based population-decoding analysis to distinguish between PP and vF in the presence of S1 activation, compared to a session without S1 activation. Time 0 denotes the onset of stimulus (PP or vF). The blue curve denotes the decoding accuracy in the presence of S1 activation, (n1 = 25 trials for PP, n2 = 25 trials for vF; C = 8 ACC neurons) derived from the data with true labels; the error bar denotes the SEM from 50 Monte Carlo simulations based on 2-fold cross-validation. (H) S1 activation increases the decoding accuracy to distinguish between noxious and non-noxious stimulation. n1 = 40, n2 = 48; p = 0.0406, Wilcoxon matched-pairs signed rank test. (I) Proportion of ACC neurons that received S1 inputs and their responsiveness to noxious stimulations. See Methods for criteria of responsiveness to S1 inputs. (J) A table illustrating the number of ACC neurons that respond to noxious inputs and number of neurons that respond to S1 activation. (K) ACC neurons that received S1 inputs (20 out of 54 total) were more likely to respond to noxious stimulations that neurons that did not receive S1 inputs (78 out of 569). p < 0.0001, Fisher’s exact test. (L) ACC response to noxious stimulations in the presence of S1 activation. (M) Activation of the S1 inputs increase the ACC response to PP. n = 98 vs 131 out of 623 neurons from 5 rats. p = 0.0191, Fisher’s exact test. (N) S1 inputs increased pain-evoked firing rates of ACC neurons. n = 54; p < 0.0001, Wilcoxon matched-pairs signed rank test. Data represented as mean ± SEM.
See also Figures S1 and S2.
We further investigated the mechanisms by which S1 activation could augment the nociceptive response in the ACC. We calculated the proportion of ACC neurons that received S1 inputs based on an increase in firing rate in response to optogenetic stimulation (Figure 1A–1C, see Methods). Less than 9% (54/623) of ACC neurons responded to S1 activation (Figure 1I and 1J). However, 37% (20/54) of these neurons that received S1 inputs were capable of responding to noxious stimulations (Figure 1I–1K). In contrast, among ACC neurons that did not receive S1 inputs, only 14% (78/569) of them demonstrated pain-responsiveness. These results show that while the S1 is not a dominant source of nociceptive input to the ACC, it nevertheless makes an important contribution. Next, we found that optogenetic activation of the presynaptic S1 inputs was able to recruit additional ACC neurons to respond to the pain stimulus (increase from 98 to 131; Figures 1L, M). In addition to this population response, we examined the firing rates of individual neurons. Among ACC neurons that received S1 inputs, S1 activation resulted in a further 69% increase in their firing rate responses to PP (Figures 1N). Together, these findings indicate that S1 inputs could amplify the ACC nociceptive response at both population and single-cell levels, thus providing evidence for a potential pathway to integrate sensory and affective pain circuits.
S1 projection to the ACC regulates pain aversive responses
The ACC is known to process the aversive component of pain. Pain aversion in rodents can be assessed by well-established conditioned place aversion (CPA) assays [12, 17–20, 26]. Here, we used the CPA assay to show that rats could demonstrate aversion to repeated stimulation by a noxious mechanical stimulus (PP). During the preconditioning phase, rats were allowed free access to both chambers. During conditioning, one of the chambers was paired with repeated PP stimulation of the hind paw, whereas the opposite chamber was not paired with noxious stimulations (NS). During the test phase, rats were given free access to both chambers again without peripheral stimulations (Figure 2A). As expected, rats preferred the NS chamber during the test phase, demonstrating their aversive response to acute pain (Figure 2B). We did not observe such aversive response when the rats received a non-noxious (vF) stimulus (Figure S3A and S3B). Having established rats’ aversive reaction to PPs, we examined the effect of the S1→ACC projection on pain aversion [19, 20, 27, 28]. We injected ChR2 into the pyramidal neurons in the S1 hind limb region, and inserted optic fibers bilaterally into the ACC to directly activate the axonal projection of S1 neurons to the ACC (Figures 2C, S3C and S3D). To understand the role of the S1→ACC projection in the aversive response to PP, we paired one chamber with PP, and another chamber with PP as well as optogenetic activation of the S1→ACC projection (Figures 2C and 2D). Rats avoided the chamber associated with S1→ACC activation when presented with PP, suggesting that activation of this cortical circuit enhanced the aversive response to noxious stimuli (Figure 2E). In contrast, rats with YFP control did not avoid the chamber associated with light treatment (Figure S3E and S3F). This enhanced aversive response to PP in the presence of S1→ACC activation could be quantified by a CPA score, which was calculated by subtracting the time rats stayed in the PP chamber during the test phase from the preconditioning phase [19, 20, 27, 28]. Compared with control (YFP) rats, rats that expressed ChR2 showed greater CPA scores, further indicating that S1→ACC activation enhances the aversive response to noxious stimuli (Figure 2F). Activation of the direct S1→ACC projection, however, did not have intrinsic aversive value, indicating that brief activation of this projection specifically enhances the aversive experience of noxious inputs (Figure S3G–J). In contrast, when we selectively inhibited this cortico-cortical projection using halorhodopsin (NpHR) (Figures 2C, 2G and S4), rats preferred the chamber associated with S1→ACC inhibition (Figures 2H and 2I). Together, these data demonstrate the S1 projection plays an important role in the aversive response mediated by the ACC, and this cortico-cortical connection likely confers additional specificity for the affective response to noxious inputs.
Figure 2. S1→ACC projection regulates aversive pain behaviors.
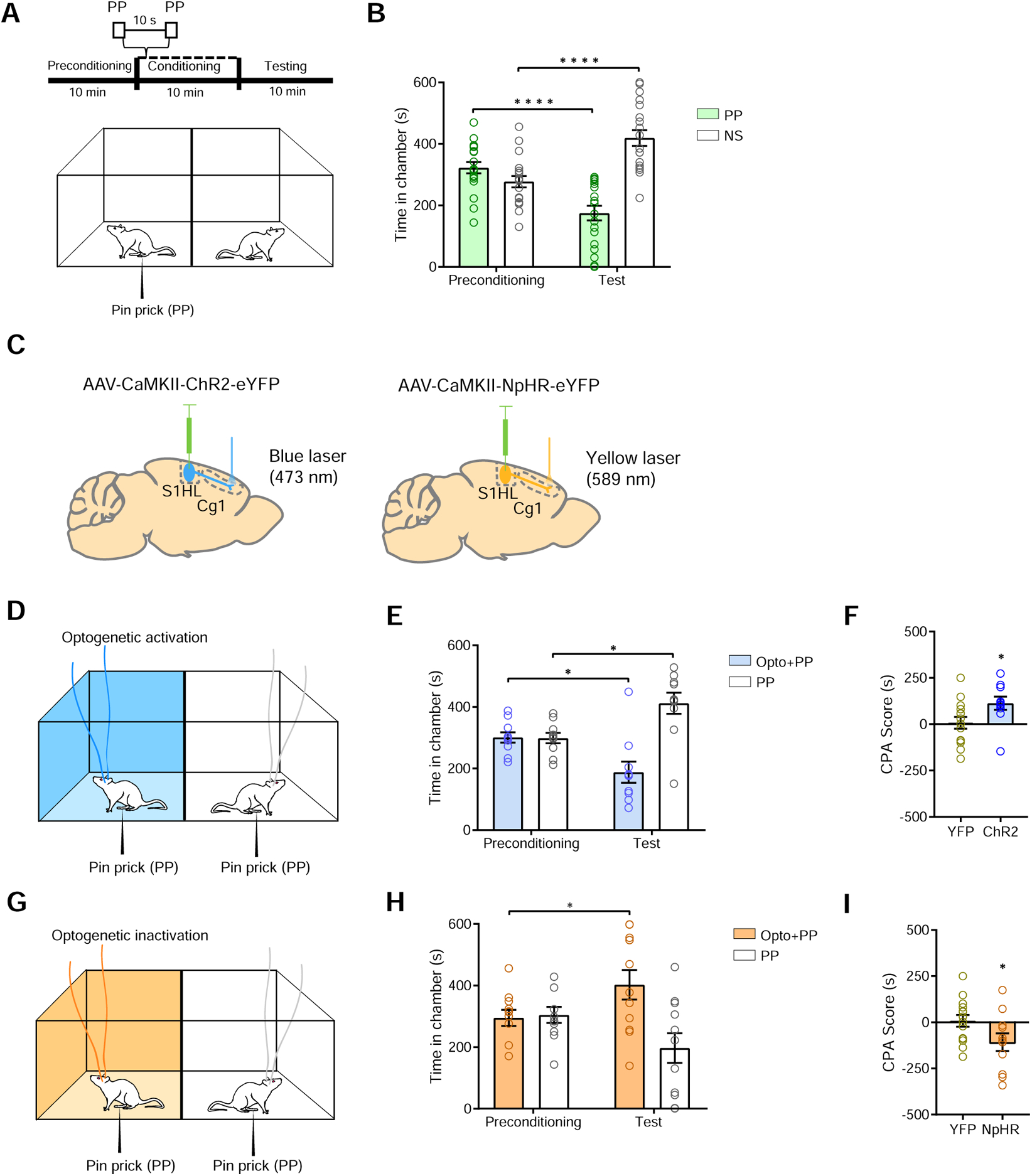
(A) Schematic of the conditioned place preference (CPA) assay. (B) Rats display aversive response to acute mechanical pain. One of the chambers was paired with PP, the other chamber was not paired with a noxious stimulus (NS). n = 19; p < 0.0001, paired Student’s t test. (C) Schematic of injection of channelrhodopsin (ChR2) and halorhodopsin (NpHR) into the S1 hind limb (S1-HL), and insertion optic fibers into the ACC. (D) Schematic of CPA assay with optogenetic activation of the S1→ACC projection in the presence of PP. One of the chambers was paired with optogenetic activation of the S1→ACC projection and PP; the other chamber was paired with PP alone. (E) Rats avoided the chamber associated with S1→ACC activation, when presented with PP. n = 10; p = 0.0114, paired Student’s t test. (F) CPA score for S1→ACC activation in the presence of mechanical pain. n = 10–14; p = 0.0415, unpaired Student’s t test. (G) Schematic of CPA assay with inhibition of S1→ACC circuit. One of the chambers was paired with optogenetic inhibition of the S1→ACC projection and PP; the other chamber was paired with PP alone. (H) Rats preferred the chamber associated with S1→ACC inhibition, when presented with PP. n = 11; p = 0.0486, paired Student’s t test. (I) CPA score for S1→ACC inhibition in the presence of mechanical pain. n = 11–14; p = 0.0495, unpaired Student’s t test. Data represented as mean ± SEM.
See also Figures S3 and S4.
Enhanced connection in the S1→ACC circuit in the chronic pain state
Previous studies have shown that chronic pain induces synaptic plasticity in both the S1 and the ACC [19, 29–34]. Thus, we hypothesized that chronic pain could also increase the S1→ACC connectivity to further enhance the integration of sensory and affective nociceptive information. We injected Complete Freund’s Adjuvant (CFA) to induce persistent inflammatory pain. To avoid confounding spinal and peripheral hypersensitivity, we injected CFA in the limb opposite to peripheral stimulation (Figures 3A and 3B). We examined the firing rates of individual neurons in the ACC. In CFA-treated rats, there was an increase in the ACC firing rates in response to noxious stimulations (Figures 3C and 3D), indicating that chronic pain increases the nociceptive response in the ACC, compatible with previous findings [19, 20]. More importantly, activation of the S1 inputs resulted in an additional increase in the firing rates of all ACC neurons after PP (Figures 3E and 3F), indicating increased connection in the S1→ACC circuit.
Figure 3. Persistent pain increases S1-ACC connectivity.
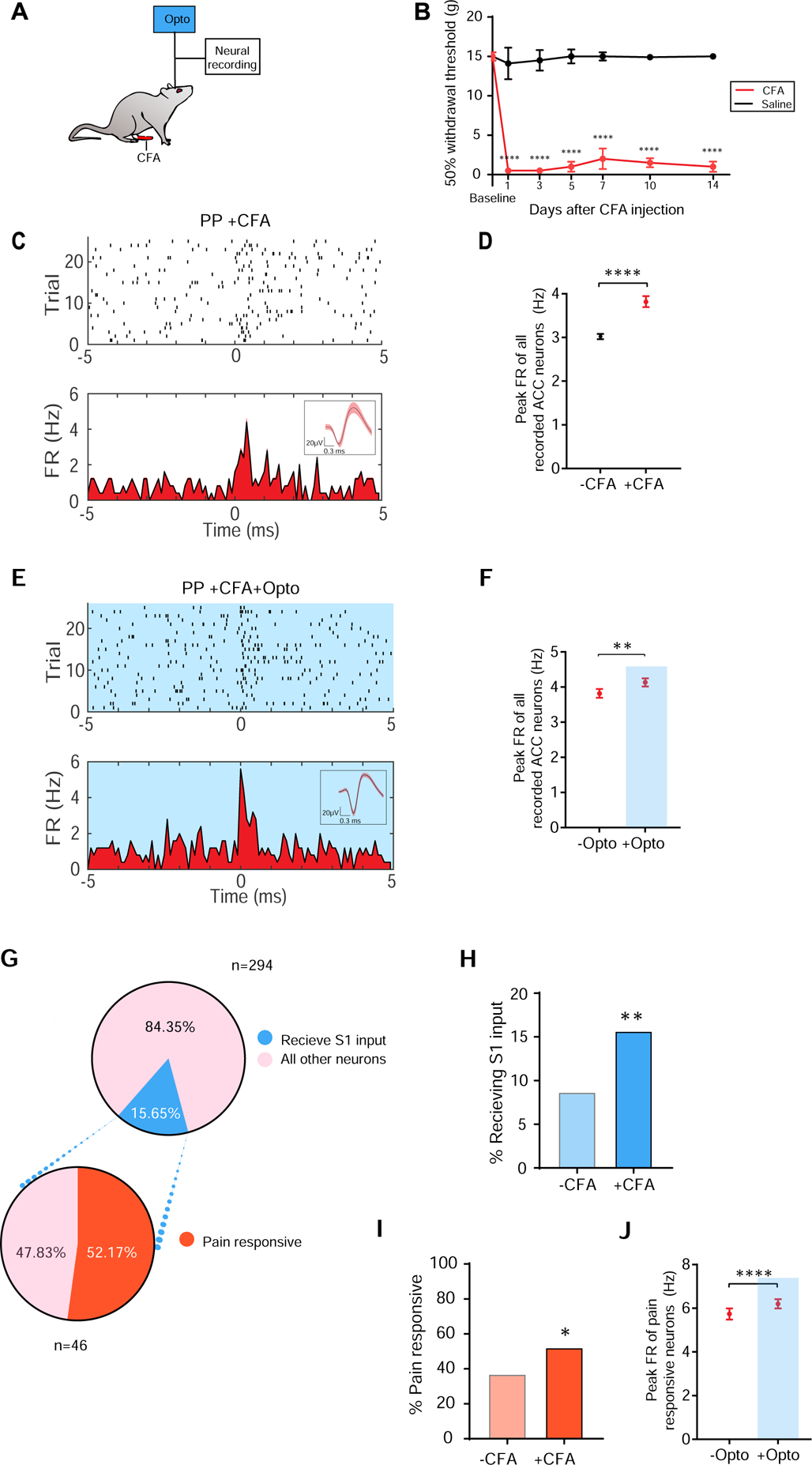
(A) Schematic of the CFA model. (B) CFA treatment induces mechanical allodynia, n = 6 (CFA), n= 6 (Saline). (C) Raster and PSTH of a representative ACC neuron in a CFA-treated rat, in response to PP. (D) Chronic pain increased the peak firing rates of ACC neurons in response to PP. n = 623 (−CFA), n = 294 (+CFA) from 3 rats; p < 0.0001, Mann-Whitney U test. (E) Representative recording trace shows that optogenetic activation of the presynaptic S1 inputs increased the firing rates of a pyramidal neuron in the ACC in response to PP, in a CFA-treated rat. (F) Activation of the S1 inputs increased the firing rates of ACC neurons, in response to PP, in CFA-treated rats. n = 294; p = 0.0083, Wilcoxon matched-pairs signed rank test. (G) Proportions of ACC neurons that receive S1 inputs in the chronic pain condition. (H) Chronic pain increases the proportion of ACC neurons that received S1 inputs. p = 0.0021, Fisher’s exact test. (I) Chronic pain increases the pain-responsiveness of ACC neurons that received S1 inputs. p = 0.0487, Fisher’s exact test. (J) Activation of S1 inputs further enhances the firing rates of pain-responsive ACC neurons. n = 98; p < 0.0001, Wilcoxon matched-pairs signed rank test. Data represented as mean ± SEM.
We examined in detail the mechanisms of this enhanced connection in the S1→ACC circuit in the chronic pain state. We found that the proportion of ACC neurons that received S1 inputs almost doubled in CFA-treated rats (Figure 3G and 3H, see Methods). In addition, in CFA-treated rats, more than 50% of the ACC neurons that receive S1 inputs responded to noxious stimulations, remarkably more than the ratio of pain-responsive neurons without the CFA treatment (Figure 3I), indicating a magnified contribution of the S1 inputs to the overall ACC nociceptive response in the chronic pain state. Together these results suggest an increase in connectivity between S1 and ACC at the level of population response. We also observed that firing rates of pain-responsive ACC neurons, which were already enhanced in the chronic pain state, increased further upon activation of S1 inputs, suggesting that these inputs also contribute to the single neuron response to noxious stimuli (Figure 3J). Therefore, chronic pain likely enhances the S1→ACC connection at both population and single-cell levels.
Enhanced S1→ACC projection contributes to heightened aversive response in the chronic pain state
Finally, we examined the behavioral consequence of enhanced cortico-cortical connectivity in the chronic pain state. We have previously shown that similar to chronic pain patients, rats with persistent pain demonstrate a key phenotype of elevated aversive response to acute noxious stimulations in an anatomically non-specific manner, a phenomenon we termed generalized enhancement of pain aversion [19, 20, 27, 35]. Here, we assessed the impact of CFA injection on the aversive response to noxious PP in the uninjured paw, and found that chronic pain indeed increased the aversive response to noxious stimulations (Figures 4A–4C). We then investigated if activation of the S1→ACC projection could contribute to this enhanced pain aversion. We compared the CPA scores for naïve rats that received S1→ACC activation paired with PP in one chamber, and NS in the opposite chamber (Figures 4D–4F), with CFA-treated rats that received PP in one chamber and NS in the opposite chamber (Figures 4A–4C). The activation of this cortical projection had similar quantitative effect on the CPA scores as chronic pain (Figures 4F), indicating that activation of the S1→ACC circuit when rats were presented with noxious stimulus could mimic the chronic pain phenotype of enhanced aversion to noxious stimulus. To further investigate the role of the S1→ACC projection in the aversive response to chronic pain, we examined how this projection regulates another key feature of chronic pain: aversion related to tonic or spontaneous pain. We adapted a CPA assay that has been used to unmask the tonic-aversive behavior in rodent models of chronic pain [20, 26, 36]. During conditioning, we did not present additional noxious stimulations to CFA-treated rats, but we paired one chamber with S1→ACC activation, and another chamber without S1→ACC activation (Figure 4G). After conditioning to unmask the aversive behavior associated with tonic pain [20, 26, 36], CFA-treated rats avoided the chamber associated with S1→ACC activation (Figures 4H, 4I, and S5). In contrast, we paired one chamber with S1→ACC inhibition and another chamber without S1→ACC inhibition in a conditioned place preference (CPP) test. After conditioning, rats with chronic pain preferred the chamber associated with S1→ACC inhibition (Figures 4J–4L). These data strongly support that the S1→ACC projection can regulate the aversive response associated with tonic pain. Together, results from these CPA/CPP assays indicate that enhanced S1→ACC projection could contribute to elevated evoked and tonic pain responses in the chronic inflammatory pain condition.
Figure 4. Enhanced S1-ACC connectivity contributes to chronic inflammatory pain.

(A) Schematic of the CPA with CFA-treated rats. One of the chambers was paired with PP; the other chamber was paired with NS alone. (B) CFA-treated rats spent significantly less time in the chamber paired with PP. n = 9; p = 0.0007, paired Student’s t test. (C) CFA increases the aversive value of PP, as indicated by a higher CPA score. n = 9–19; p = 0.0460, unpaired Student’s t test. A CPA score was calculated by subtracting the amount of time spent during the test phase from baseline in the chamber paired with PP in CFA-treated and control rats. (D) Schematic of the CPA assay with one chamber paired with optogenetic activation of the S1→ACC projection and PP; the other chamber was paired with NS alone. (E) Rats spent significantly less time during the test phase than at baseline in the chamber paired with S1→ACC activation and PP. n = 15; p < 0.0001, paired Student’s t test. (F) S1→ACC activation caused a similar increase in the aversive response to PP as chronic pain. A CPA score was calculated by subtracting the amount of time spent during the test phase from baseline in the chamber paired with simultaneous S1→ACC activation and PP in (E), compared with the CPA score calculated in (C) n = 9–15; p = 0.4746, unpaired Student’s t test. (G) Schematic of the CPA for tonic-aversive response. One of the chambers was paired with activation of the S1→ACC projection; the other chamber was not. No peripheral stimulus was given. (H) CFA-treated rats avoided the chamber associated with S1→ACC activation. n = 7; p = 0.0017, paired Student’s t test. (I) CPA score for CFA-treated rats which received S1→ACC activation. n = 7; p= 0.0021, unpaired Student’s t test. (J) Schematic of the conditioned place preference (CPP) assay for tonic pain. One of the chambers was paired with inactivation of the S1→ACC projection; the other chamber was not. No peripheral stimulus was given. (K) CFA-treated rats preferred the chamber associated with S1→ACC inhibition. n = 9; p = 0.0128, paired Student’s t test. (L) CPP score for CFA-treated rats which received S1→ACC inhibition. n = 7–9; p = 0.0256, unpaired Student’s t test. Data represented as mean ± SEM.
See also Figure S5.
To validate these findings, we repeated the aversive behavioral tests in the spared-nerve injury (SNI) model of chronic neuropathic pain (Figure 5A). First, we confirmed that rats with chronic neuropathic pain demonstrate elevated aversive response, as manifested by elevated CPA scores to noxious stimulations (Figures 5B–5D). Next, we compared the CPA score for naïve rats that received S1→ACC activation paired with PP in one chamber, and NS in the opposite chamber (Figures 5E–5G), with SNI-treated rats that received PP in one chamber and NS in the opposite chamber (Figures 5B–5D). Similar CPA scores under these conditions indicate that activation of the S1→ACC circuit had the same effects on the aversive phenotype as chronic neuropathic pain (Figure 5G). Finally, we examined the impact of S1→ACC circuit modulation on the tonic pain experience in SNI-treated rats. Whereas activation of the S1→ACC projection enhanced the aversive experience associated with peripheral neuropathy (Figures 5H–5J, and S5), the inhibition of this pathway decreased tonic pain-induced aversion (Figures 5K–5M).
Figure 5. Enhanced S1-ACC connectivity contributes to chronic neuropathic pain.
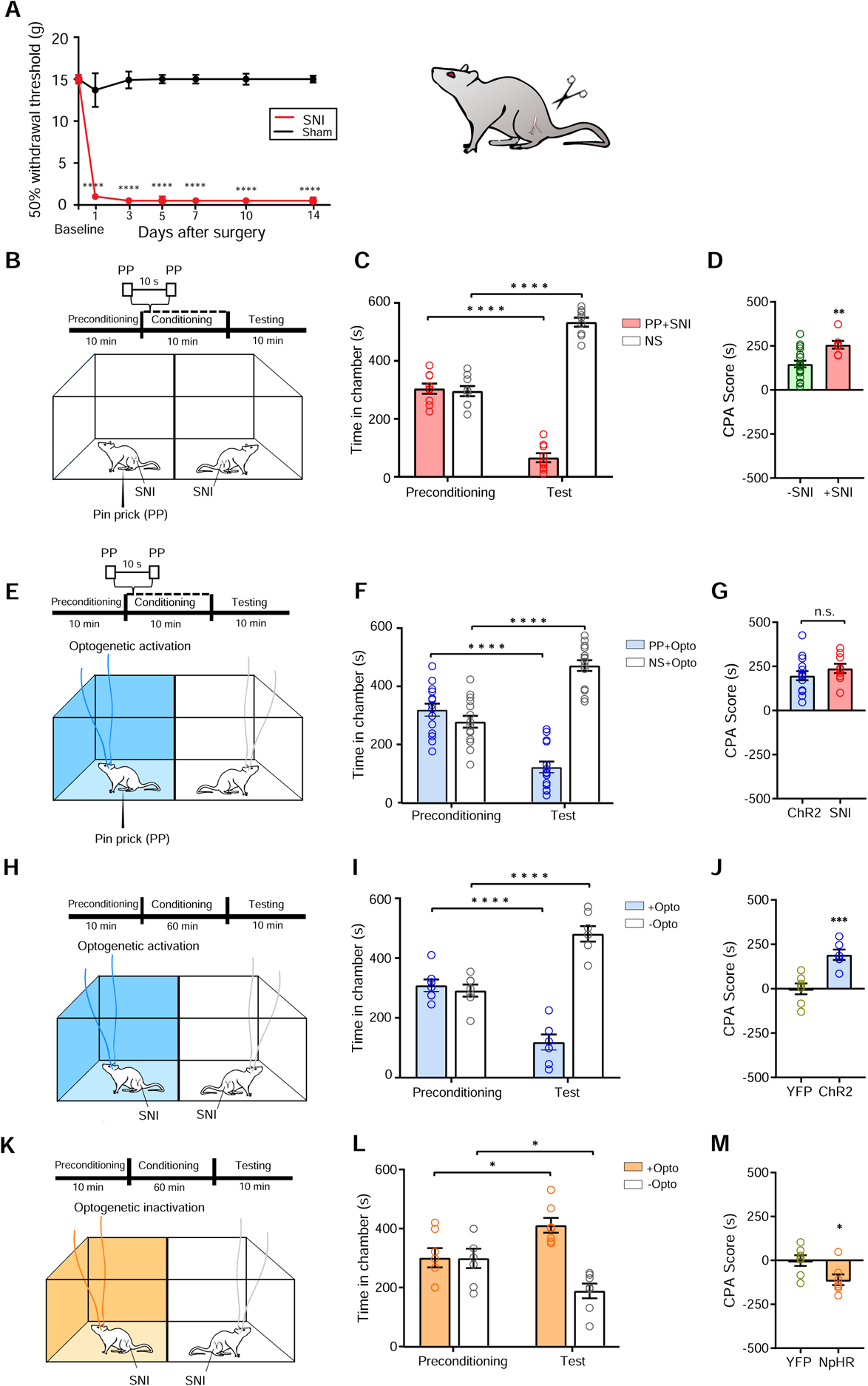
(A) SNI treatment induces mechanical allodynia, n = 6 (SNI), n = 6 (Sham). (B) Schematic of the CPA assay with SNI-treated rats. One of the chambers was paired with PP; the other chamber was paired with NS. (C) SNI treatment gives rise to the aversive value of PP. n = 7; p < 0.0001, paired Student’s t test. (D) SNI increased the aversive value of PP, as indicated by a higher CPA score. n = 7–19; p = 0.0034, unpaired Student’s t test. (E) Schematic of the CPA assay with one chamber paired with optogenetic activation of the S1→ACC projection and PP; the other chamber was paired with NS alone. (F) Rats spent significantly less time during the test phase than at baseline in the chamber paired with S1→ACC activation and PP. n = 15; p < 0.0001, paired Student’s t test. (G) S1→ACC activation caused a similar increase in the aversive response to PP as chronic pain. n = 7–15; p = 0.3012, unpaired Student’s t test. (H) Schematic of the CPA assay for tonic pain in the SNI model. (I) SNI-treated rats avoided the chamber associated with S1→ACC activation. n = 7; p < 0.0001, paired Student’s t test. (J) CPA score for SNI-treated rats which received S1→ACC activation. n = 7 (YFP), n = 6 (ChR2); p = 0.0009, unpaired Student’s t test. (K) Schematic of the CPP assay for tonic pain in the SNI model. One of the chambers was paired with inactivation of the S1→ACC projection; the other chamber was not. No peripheral stimulus was given. (L) SNI-treated rats preferred the chamber associated with S1→ACC inhibition. n = 7; p = 0.0111, paired Student’s t test. (M) CPP score for SNI-treated rats which received S1→ACC inhibition. n = 7; p = 0.0260, unpaired Student’s t test. Data represented as mean ± SEM.
See also Figure S5.
DISCUSSION
Normal physiologic response to acute pain requires the integration of sensory and affective experiences. Whereas the S1 and the ACC are well-known brain regions for processing the sensory and affective components of pain, respectively, a direct functional cortico-cortical circuit linking these two areas has not been previously reported. In this study, we combined in vivo electrophysiology, machine learning, and targeted circuit disruption to demonstrate that a S1→ACC projection allows sensory pain information to be transmitted to a higher order cortical center that regulates the affective experience. Furthermore, we found that enhanced cortico-cortical connectivity contributes to elevated aversive pain behaviors in the chronic pain state
The ACC plays a key role in regulating the affective component of pain [4–14]. Our results show that neurons in the ACC can respond to noxious stimulations by increasing firing rates, consistent with previous findings [19, 20, 37–43]. In addition, our population decoding analysis demonstrates that ensemble activities in the ACC can provide a relatively specific neural code for pain. Previous human functional MRI data have also shown that the ACC encodes pain experience with similarly high specificity [6, 44, 45]. Furthermore, our evidence for the ability of ACC neurons to regulate the aversive response to acute pain is compatible with previous reports [19].
Most importantly, our study provides a simple and direct circuit mechanism for the relay of cortical pain sensory information into higher order cortical centers to drive affective responses. This conclusion is based on several independent lines of evidence. First, our histological findings confirm previous anatomic studies directly linking the S1 with the ACC [24, 25]. Second, even though only a small portion of the ACC neurons receives S1 inputs at baseline, these ACC neurons that receive S1 inputs are highly responsive to noxious inputs. Third, activation of the S1 axons in the ACC further enhances the ACC firing rates in response to noxious stimulations. Fourth, unbiased machine learning analysis confirms that activation of the S1→ACC projection increases the accuracy of pain discriminative power in the ACC. Finally, direct modulation of the S1 input to the ACC regulates the aversive response to pain. Two recent studies further support our findings. Eto et al. found that electrical response of the ACC to peripheral stimulation correlated with S1 neuronal activity [46]. In another study, Tan et al. found that gamma oscillations in parvalbumin-expressing inhibitory interneurons in the S1 led to increased c-fos expression in a number of cortical and subcortical areas including the ACC [47].
While previous studies have suggested that alterations of S1 activities could regulate pain behaviors [32, 33], the downstream targets that mediate such nociceptive responses have remained elusive. A recent study suggests the spinal cord to be a target for the S1 regulation of sensory allodynia [48]. Our results here provide a cortical target – the ACC – that is important for processing the aversive component of pain. Interestingly, brief activation of the S1→ACC circuit alone is insufficient to produce CPA in the absence of noxious inputs. Since the S1 encodes specific somatotopic nociceptive information, it is highly likely that the projection from the S1 assigns sensory-specific value to enrich the aversive response in the ACC. This is compatible with findings from human studies showing that whereas the medial pain pathways play an important distinguishing role in the intensity of the unpleasantness for noxious stimuli, the lateral nociceptive pathway has a modulatory role as well [6, 49, 50].
In contrast to cortico-subcortical projections, neural mechanisms for cortico-cortical connectivity in pain processing is not well established. Here, we found that activation of the S1 inputs selectively increased the firing rates of ACC neurons in response to noxious stimuli, without altering their response to non-noxious stimulations. This important finding suggests activity-dependent changes that enables S1 inputs to enhance the ability of pain-responsive ACC neurons to respond more efficiently to noxious signals. At the same time, our results also indicate that activation of the S1 inputs could recruits more ACC neurons to respond to pain stimuli at the population level.
Chronic pain induces synaptic plasticity in a number of cortical and subcortical regions [1, 29, 30, 51]. Such maladaptive plasticity in turn contributes to both sensory hypersensitivity and increased aversion. Pyramidal neurons in the ACC demonstrate increased synaptic plasticity with persistent or chronic pain [19, 20, 29, 30, 52], which could contribute to enhanced S1→ACC projection. It is important to note that in the chronic pain state, we observed an increased number of ACC neurons that receive S1 nociceptive inputs, and at the same time, these neurons that receive S1 inputs also show higher firing rates. These results indicate that enhanced cortical connectivity in the chronic pain state occurs at both population and single-cell levels. Recent studies have suggested that chronic pain can also alter cortio-subcortical projections through similar dual-level mechanisms [53, 54]. Future studies, however, are needed to detail the specific mechanisms of plasticity within the S1-ACC circuit in the chronic pain state, including examinations of dentritic sprouting in the chronic pain state.
Our finding of enhanced cortical integration of sensory and affective nociceptive processing in the chronic pain state is compatible with studies in other sensory systems [55–58]. It should be noted, however, that the S1→ACC projection is likely one of the many circuit mechanisms for the integration of nociceptive signals. It is also possible that nociceptive information flow can also occur in the opposite direction, and the functional significance of a potential ACC→ S1 projection remains to be examined. Meanwhile, S1 projection to the spinal cord and subsequent modulation of ascending nociceptive inputs could provide an additional pathway to integrate sensory pain signals [48]. Other cortical areas, such as the secondary somatosensory cortex (S2), insular, and prefrontal cortex (PFC) can also play key roles in either sensory or affective pain response [59]. The prelimbic region of PFC, in particular, a structure that lies adjacent to the ACC in rodents, can project to a number of subcortical structures to regulate both sensory and affective responses [53, 60, 61]. Recent studies have shown that an interplay of excitatory and inhibitory neurons in the PFC can integrate signals from the amygdala and project in turn to the brain stem to regulate pain aversion [62–64]. Thus, future efforts of circuit dissection will further enhance our understanding of cortical and subcortical connections for the integration of sensory and affective pain information, and how such integration is altered in the chronic pain state.
Our findings may have important translational impact. Optogenetic inhibition of the S1→ACC pathway effectively relieved the aversive component of both acute and chronic pain. In our model, the S1 provides sensory pain information to the ACC to enrich the pain-specific aversive experience. Inhibition of this pathway thus has the potential to specifically reduce pain-associated affective symptoms, and as a result, the S1→ACC projection could be an important target for non-addictive neuromodulation therapy for pain.
In summary, we have discovered a direct projection from the S1 to the ACC that regulates the pain-aversive response. This projection is enhanced in the chronic pain state and thus may form a target for therapeutic neuromodulation.
STAR Methods
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jing Wang (jing.wang2@nyumc.org). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Sprague-Dawley male wild-type rats of the species rattus norvegicus domestica were purchased from Taconic Farms and pair-housed at the vivarium facility in the NYU Langone Science Building in paired housing, with controlled humidity, temperature, and 12-hr (6:30 AM–6:30 PM) light-dark cycle. Vendor health reports indicated that the rats were free of known viral, bacterial, and parasitic pathogens. All rats were purchased at a developmental stage of 7 weeks and given 10 days on average to adjust to the new environment before initiation of experiments. All rats with intracranial implants or injections were naïve to procedures and drugs before surgical procedures. All procedures were performed in accordance with the guidelines of the New York University School of Medicine (NYUSOM) Institutional Animal Care and Use Committee (IACUC) to ensure minimal animal use and discomfort, as consistent with the NIH Guide for the Care and Use of Laboratory Animals.
METHOD DETAILS
Complete Freund’s Adjuvant Administration
0.1 mL of Complete Freund’s Adjuvant (CFA) (Mycobacterium tuberculosis, Sigma-Aldrich) was suspended in an oil:saline (1:1) emulsion and injected subcutaneously into the plantar aspect of the hind paw to induce chronic inflammatory pain. CFA was injected contralateral to the paw that was stimulated by either pin prick (PP) or von Frey filament (vF), in experiments involving peripheral stimulus. In tonic pain experiments, CFA was always injected contralateral to the site of intracranial injection in the S1. Control rats received an equal volume of saline injection.
Spared Nerve Injury
Rats were first anesthetized with isoflurane (1.5 to 2%). An incision was made on the skin on the lateral surface of the thigh of the rat, contralateral to the site of intracranial injection in the S1HL. Next, a section was made through the biceps femoris muscle to expose the sciatic nerve and its three terminal branches: sural, common peroneal, and tibial nerves. The common peroneal and tibial nerves were tied with nonabsorbent 5–0 silk sutures at the proximal point of trifurcation, and cut distal to the knot. To prevent reattachments, approximately 5 mm of the distal ends were removed. Nerves were dissected but not cut in sham surgeries (control group). The muscle layer was then sutured closed, and the skin was stapled. Staples were removed before any behavioral experiments.
Virus Construction and Packaging
Recombinant adeno-associated virus (AAV) vectors were serotyped with AAV1 coat proteins, packaged at Addgene viral vector manufacturing facilities. Viral titers were approximately 5 × 1012 particles per milliliter for AAV1.CaMKII.ChR2-eYFP.WPRE.hGH, AAV1.CaMKII.NpHR-eYFP.WPRE.hGH, and AAV1.CaMKII(1.3).eYFP.WPRE.hGH. Aliquots were stored light protected in a freezer before use.
Intracranial Injection and Fiber Implantation
Rats were anesthetized with 1.5%–2% isoflurane [51]. Virus was delivered to the hind limb region of the primary somatosensory cortex (S1-HL) only in all of the experiments. Rats were unilaterally injected with 0.65 μL of viral particle solution or RetroBeads solution (Lumafluor) at a rate of 0.1 μL/10 s with a 26G 1 μL Hamilton syringe at anteroposterior (AP) −1.5 mm, mediolateral (ML) ± 3.0 mm, and dorsoventral (DV) −1.5 mm. Rats injected with viral particle solution were next implanted bilaterally with 200-μm optic fibers held in 1.25 mm ferrules (Thorlabs) in the ACC (AP +2.6 mm, ML ± 1.2 mm, DV −1.75 mm, angled 28° toward the midline). Fibers with ferrules were held in place by dental acrylic.
Optrode Implant and Intracranial Injections
After exposure of the skull, rats were injected with 0.65 μL of viral particle solution in the at a rate of 0.1 μL/10 s with a 26G 1 μL Hamilton syringe unilaterally in the S1 (AP −1.5 mm, ML ± 3.0 mm, DV −1.5 mm). After the intracranial injection, rats were allowed to recover for a period of three weeks before optrode implantation to allow for adequate viral expression.
Tetrodes were constructed from four twisted 12.7 μm polyimide-coated microwires (Sandvik) and subsequently mounted in an eight-tetrode VersaDrive Optical (NeuraLynx). A 200 μm optic fiber held in 1.25 mm ferrules (Thorlabs) was mounted in the VersaDrive Optical such that the end of the fiber was located approximately 0.5–1 mm above the mounted tetrodes. Electrode tips were gold plated to reduce electrode impedances to 100–500 kΩ at 1 kHz. Rats were anesthetized with 1.5%–2% isoflurane, and the skull was exposed. A 2 mm-diameter hole was drilled above the ACC target region. A durotomy was performed prior to lowering the optrodes slowly unilaterally into the ACC with the stereotaxic apparatus (AP +2.6 mm, ML +0.8 mm, DV −1.75 mm, with tetrode tips angled 15° toward the midline). The drive was secured to the skull screws with dental cement.
After experiments, animals were sacrificed, and 20 μm brain sections were collected using a Leica CM3050S cryostat machine (Leica Biosystems) and analyzed for viral expression, optic fiber localization, and electrode localization with histological staining. Animals with improper fiber or electrode placements, low viral expression, or viral expression in cell bodies outside the S1-HL were excluded from further analysis.
In Vivo Electrophysiological Recordings
Prior to stimulation, animals with chronic optrode implants were allowed 30 min to habituate to a recording chamber over a mesh table, as described previously [19]. Noxious stimulation was applied by pricking the plantar surface of the hind paw contralateral to the brain recording site with pin prick by a 30-gauge needle (PP) in free-moving rats. Noxious stimulation was terminated by withdrawal of the paw. Non-noxious stimulus was applied to the same hind paw using a 2 g vF, continuously for 3 seconds or until paw withdrawal. There were no withdrawal responses to vF in the majority of cases.
All of the recording sessions consisted of approximately 50 trials with variable inter-trial intervals. Half of the recording sessions were conducted using either PP or vF filament peripheral stimulation for the entire session, with the first 25 trials either with or without constant 20 Hz optogenetic stimulation, and the remaining 25 trials under the opposite condition. Trials were also conducted without peripheral stimulus to determine the response of individual neurons to onset of optogenetic stimulus alone. The remaining recording sessions were conducted either with or without constant 20 Hz optogenetic stimulation for the entire session, with PP and vF filament stimulations applied randomly to the rat’s hind paws (equal number of trials for each peripheral stimulation type). Recording conditions were counterbalanced by rats.
A video camera (HC-V550, Panasonic) was used to record the experiments. Long inter-trial intervals of approximately 60 seconds and the breaks between sessions were used to avoid sensitization. No behavioral sensitization or physical damage to the paws was observed. Experiments involving the CFA were performed 7 days after CFA injection. Experiments involving the SNI were performed 14 days after surgery.
Neural Data Collection and Preprocessing
The neuronal activity and the onset of stimulation were simultaneously recorded with acquisition equipment (Open Ephys) via an RHD2132 amplifier board (Intan Technologies). Signals were monitored and recorded from 32 low-noise amplifier channels at 30 kHz, and band-pass filtered (0.3–7.5 kHz). In order to identify spike activity, the raw data were high-pass filtered at 300 Hz with subsequent thresholding and offline sorting by commercial software (Offline Sorter, Plexon). The threshold was below the 3-sigma peak heights line and was manually optimized based on the signal-to-noise ratio. The features of three valley electrodes were used for spike sorting. Trials were aligned to the initiation of the peripheral or optogenetic stimulus to compute the peri-stimulus time histogram (PSTH) for each single unit using MATLAB (MathWorks).
Immunohistochemistry
Rats were deeply anesthetized with isoflurane and transcardially perfused with ice-cold PBS and paraformaldehyde (PFA). After extraction, brains were fixed in PFA overnight and then cryoprotected in 30% sucrose in PBS for 48 hours or until sinking [60]. 20 μm coronal sections were washed in PBS and coverslipped with Vectashield mounting medium. Sections from brains containing tetrodes were stained with cresyl violet and imaged at 10x magnification with an Axio Zoom widefield microscope (Carl Zeiss). Sections also were made after viral transfer for opsin verification, and these sections were stained with anti-rabbit GFP (1:500, #AB290, Abcam), CaMKII-α (6G9) mouse monoclonal antibody (mAb) (1:200, #50049, Cell Signaling Technology) antibodies, and cover-slipped with DAPI (1:200, Vector Laboratories). Secondary antibodies were anti-rabbit immunoglobulin G (IgG) conjugated to Alexa Fluor 488, and anti-mouse IgG conjugated to Alexa Fluor 647 (1:200, Life Technologies). Sections with RetroBeads solution (Lumafluor) injection were cover-slipped with DAPI (1:200, Vector Laboratories). Images were acquired with a Zeiss LSM 700 confocal microscope (Carl Zeiss).
Animal Behavioral Tests
Behavioral tests involving optogenetic stimulation were conducted approximately 2–4 weeks after viral injection of channelrhodopsin-2 (ChR2), and approximately 3–5 weeks after viral injection of halorhodopsin (NpHR). Prior to each experiment, optic fibers were connected to a laser diode (Shanghai Dream Lasers Technology) through a mating sleeve, as described previously [60]. Pulsed laser light was delivered at a frequency of 20 Hz using a transistor-transistor logic (TTL) pulse generator (Doric Lenses), and intensity was measured with a power meter (Thorlabs, Newton, NJ, USA). Laser diodes of wavelength 473 nm were used for channelrhodopsin-2, and laser diodes of wavelength 589 nm were used for halorhodopsin. Experiments involving CFA were performed 7 days after CFA injection. Experiments involving the SNI were performed 14 days after surgery.
Conditioned Place Aversion Assay
Conditioned Place Aversion (CPA) experiments were conducted similarly to those described previously [19], in a standard two-compartment apparatus (Stoelting Co., Wood Dale, IL, USA) consisting of two large compartments of equal size connected with an opening large enough for a rat to travel through freely. The CPA protocol included preconditioning (baseline), conditioning, and testing phases. The preconditioning phase was 10 min, and animals spending >500 s or <100 s of the total time in either chamber during the preconditioning phase were eliminated from further analysis. Immediately following the preconditioning phase, the rats underwent conditioning. During conditioning, at least one of the two chambers was paired with either peripheral stimulation (PP or vF), or 20 Hz optogenetic stimulation, or both. The peripheral stimulus was repeated every 10 s. The order of peripheral stimulation and optogenetic activation was counterbalanced, e.g. half of the rats received optogenetic activation first, whereas the other half received control treatment first during conditioning. Likewise, chamber pairings were counterbalanced. For experiments involving peripheral stimulation, the conditioning phase was 10 minutes. For experiments without peripheral stimulation, the conditioning phase was 60 min, with the rats spending 30 min in each of the two treatment (optogenetic and control) chambers. During the test phase, the animals did not receive any treatment and had free access to both compartments for 10 min. The time between each experimental phase was on average 5–10 min. Movements of the rats in each chamber were recorded by a camera and analyzed with ANY-maze software. Decreased time spent in a chamber during the test phase as compared with the baseline indicated avoidance (aversion) of that chamber, whereas increased time spent in a chamber during the test phase as compared with the baseline indicated preference for that chamber.
Mechanical Allodynia Test
Mechanical allodynia was measured using a Dixon up-down method with vF filaments. Rats were placed individually into plexiglass chambers over a mesh table and allowed to acclimate for 20 min prior to testing. vF filaments were applied to the lateral one-third of the hind paw in the distribution of the sural nerve with logarithmically incremental stiffness (0.45, 0.75, 1.20, 2.55, 4.40, 6.10, 10.50, and 15.10 g), beginning with 2.55 g, as described previously [60]. 50% withdrawal thresholds were calculated.
Statistical Analysis
Behavioral results were given as mean ± SEM. To compare mechanical allodynia withdrawal thresholds for CFA-treated, SNI-treated, and control rats, a two-way ANOVA with repeated-measures and post hoc multiple pairwise comparison Bonferroni tests or unpaired t tests were used whenever appropriate. For the CPA assay, a paired Student’s t test was used to compare the time spent in each treatment chamber before and after conditioning (i.e., preconditioning versus test phase for each chamber). CPA scores were computed by subtracting the time spent in the more noxious chamber during the test phase from the time spent in that chamber at baseline [19]. A two-tailed unpaired Student’s t test was used to compare differences in CPA scores under various testing conditions.
To determine whether the firing rate of a particular neuron was altered in response to a peripheral stimulus, we used the method described previously [27]. We calculated PSTHs using a 5 second range before and after peripheral stimulus (i.e., PP or vF) with a bin size of 100 ms. We calculated the basal spontaneous firing rate of each neuron by averaging the PSTH over the pre-stimulus bins. The peak evoked firing rate was then calculated as the maximum value of the PSTH after stimulus onset (within 5 s from the stimulus). The baseline mean is the average of the PSTH bins before stimulus onset, and the SD is the standard deviation of the PSTH bins before stimulus onset. We calculated a Z scored firing rate using the following equation: Z = (FR – mean of FRb)/SD of FRb, where FR indicates the firing rate for each bin and FRb indicates the baseline firing rate before the stimulus onset. To define a positive responding neuron, we used the following criteria: (1) the absolute value of the Z scored firing rate of least one time bin after stimulation must be ≥2.5, and (2) if the first criterion is passed, at least the next two bins must be >1.645. These criteria must be fulfilled within 3 s after the peripheral stimulus.
To determine whether the firing rate of a particular ACC neuron was altered in response to optogenetic activation of the S1 axon terminals, we used the method described previously [27]. We calculated PSTHs using a 5 second range before and after onset of optogenetic stimulus with a bin size of 100 ms. We calculated the basal spontaneous firing rate of each neuron by averaging the PSTH over the pre-stimulus bins. The peak optogenetically evoked firing rate was then calculated as the maximum value of the PSTH after stimulus onset (within 5 s from the stimulus). The baseline mean is the average of the PSTH bins before stimulus onset, and the SD is the standard deviation of the PSTH bins before stimulus onset. We calculated a Z scored firing rate using the following equation: Z = (FR – mean of FRb)/SD of FRb, where FR indicates the firing rate for each bin and FRb indicates the baseline firing rate before the stimulus onset. To define a positive responding neuron, we used the following criteria: (1) the absolute value of the Z scored firing rate of least one time bin after stimulation must be ≥2.5, and (2) if the first criterion is passed, at least the next two bins must be >1.645. These criteria must be fulfilled within 3 s after the onset of optogenetic stimulus.
When comparing groups of neurons, nonparametric tests were performed as neuronal FRs had non-Gaussian distributions, compatible with a previous report [65]. For unpaired data, a Mann-Whitney U test was performed to test the equivalence of distributions. For paired data, the Wilcoxon matched-pairs signed-rank test was used to test the equivalence of distributions. Fisher’s exact test was used to analyze the population changes of positive responding neurons. Due to the negligible number of neurons that decreased their FRs in response to stimulation (PP or vF filament), we assigned those neurons to the category of non-responders.
Support Vector Machine Population Decoding
After spike sorting, we obtained population spike trains from simultaneously recorded ACC neurons. For each single neuronal recording, we binned spikes into 50 ms to obtain spike count data in time. To simulate the online decoding, we used a 50-ms moving window to accumulate spike count statistics from the onset of the peripheral stimulus (time 0) up to 3 s (i.e., 60 bins). We assessed the decoding accuracy at each time bin based on the cumulative spike count statistics. Therefore, for a total of C neurons, the input dimensionality ranged from C (the first bin) to 60C (all bins). In these experiments in which we randomly mixed different stimulations (PP and vF filament), we assumed that we had n1 trials of PP and n2 trials of vF filament. We split the total (n1 + n2) trials into two groups: 50% used for training and 50% used for testing. The goal of population-decoding analysis was to classify the trial labels of different stimulations (PP versus vF filament) based on population spike data. We used a support vector machine (SVM) classifier. The SVM is a discriminative supervised learning model that constructs the classification boundary by a separating hyperplane with maximum margin. Specifically, the SVM can map the input x into high-dimensional feature spaces, which allows nonlinear classification, as follows:
where yi denotes the class label for the training sample xi (some of which associated with nonzero αi are called support vectors), b denotes the bias, and K (x, xi) denotes the kernel function. We used a polynomial kernel and trained the nonlinear SVM with a sequential minimal optimization algorithm (MATLAB Machine Learning Toolbox “fitcsvm” function). Finally, the decoding accuracy was assessed by 2-fold cross-validation from 50 Monte Carlo simulations. We reported the means ± SEMs. In all of the population-decoding analyses, we used only the recording sessions with ≥5 simultaneously recorded ACC units, independent of the cell-firing properties.
QUANTIFICATION AND STATISTICAL ANALYSIS
Significance was defined at a level of 0.05 for all the statistical tests used in the study.
Behavioral results were given as mean ± SEM, with n as number of animals (exact values of n are noted in the Figure Legends). To compare mechanical allodynia withdrawal thresholds for CFA-treated, SNI-treated, and control rats, a two-way ANOVA with repeated-measures and post hoc multiple pairwise comparison Bonferroni tests or unpaired t tests were used whenever appropriate. For the CPA assay, a paired Student’s t test was used to compare the time spent in each treatment chamber before and after conditioning (i.e., preconditioning versus test phase for each chamber). A two-tailed unpaired Student’s t test was used to compare differences in CPA scores under various testing conditions. When comparing groups of neurons, nonparametric tests were performed as neuronal FRs had non-Gaussian distributions. N was number of neurons (exact values of n are noted in the Figure Legends). For unpaired data, a Mann-Whitney U test was performed to test the equivalence of distributions. For paired data, the Wilcoxon matched-pairs signed-rank test was used to test the equivalence of distributions. Fisher’s exact test was used to analyze the population changes of positive responding neurons. P values are noted in the Figure Legends. Sample sizes were determined so as to be comparable with previous studies.
For behavioral experiments, the order of peripheral stimulation and optogenetic activation was counterbalanced, e.g. half of the rats received optogenetic activation first, whereas the other half received control treatment first during conditioning. Likewise, chamber pairings were counterbalanced. In electrophysiological experiments, the order of conditions with or without optogenetic activation for each peripheral stimulus was also counterbalanced by rats.
Animals with improper fiber or electrode placements, low viral expression, or viral expression in cell bodies outside the S1-HL were excluded from further analysis.
GraphPad Prism 7 (GraphPad Software) and MATLAB (MathWorks) were used to calculate statistical significance and GraphPad Prism 7 software was used to plot all the graphs.
Further information of quantification and statistical tests used are described in the figure legends.
DATA CODE AND AVAILABILITY
Software used for analysis are listed in the Key Resources Table. All data and code supporting the findings of this study are available at https://github.com/aks2213/Mapping-Cortical-Integration-of-Sensory-and-Affective-Pain-Pathways or from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-GFP (polyclonal ChIP-grade) | Abcam | Cat# ab290; RRID: AB_303395 |
| CaMKII-a (6G9) mouse monoclonal antibody (mAb) | Cell Signaling Technology | Cat# 50049; RRID: AB_2721906 |
| VECTASHIELD Antifade Mounting Medium with DAPI | Vector Laboratories | Cat# H-1200; RRID: AB_2336790 |
| Alexa Fluor 488-conjugated anti-rabbit IgG | Life Technologies | Cat# A-11034; RRID: AB_2576217 |
| Alexa Fluor 647-conjugated anti-mouse IgG | Life Technologies | Cat# A-21236; RRID: AB_2535805 |
| Bacterial and Virus Strains | ||
| AAV1.CaMKII.ChR2-eYFP.WPRE.hGH | Addgene | Cat# 26969-AAV1; RRID: Addgene_26969 |
| AAV1.CaMKII.NpHR-eYFP.WPRE.hGH | Addgene | Cat# 26971-AAV1; RRID: Addgene_26971 |
| AAV1.CaMKII(1.3).eYFP.WPRE.hGH | Addgene | Cat# 105622-AAV1; RRID: Addgene_105622 |
| Mycobacterium tuberculosis: Complete Freund’s Adjuvant | Sigma-Aldrich | Cat# F5881–10ML |
| Experimental Models: Organisms/Strains | ||
| Rat: Sprague-Dawley | Taconic | Model SD |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Red RetroBeads (100 uL) | Lumafluor | https://www.lumafluor.com |
| Software and Algorithms | ||
| MATLAB R2019a | MathWorks | https://www.mathworks.com/products/matlab.html |
| GraphPad Prism 7 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Offline Sorter 4.4.0 | Plexon | https://plexon.com/products/offline-sorter/ |
| ANY-maze Behavioral Tracking Software 5.11 | ANY-maze | http://www.anymaze.co.uk |
| Other | ||
| Electrophysiological data, behavioral data, analyses, histological imaging files | This paper | https://github.com/aks2213/Mapping-Cortical-Integration-of-Sensory-and-Affective-Pain-Pathways |
| Ceramic ferrules | Thorlabs | Cat# CFLC126–10 |
| Compact Power and Energy Meter Console, Digital “4 LCD | Thorlabs | Cat# PM100D |
| 12.7 μm polyimide-coated microwires | Sandvik | Item No. PX000003 |
| VersaDrive-8 Optical | NeuraLynx | Item Code VersaDrive-8 Optical |
| Acquisition equipment | Open Ephys | https://open-ephys.org/ |
| RHD2132 amplifier board | Intan Technologies | Part #D8214 |
| Transistor-transistor logic pulse generator | Doric Lenses | Code OTPG_4 |
Supplementary Material
Highlights.
Anterior cingulate cortex (ACC) receives inputs from the somatosensory cortex (S1)
Activation of the S1 inputs increases the nociceptive response in the ACC
This cortico-cortical projection regulates pain-aversive behaviors
Chronic pain enhances the connection between the S1 and the ACC
ACKNOWLEDGMENTS
This work was supported by the NIH grants GM115384 (J.W.) and the NSF grant CBET-1835000 (Z.C., J.W.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Basbaum AI, Bautista DM, Scherrer G, and Julius D (2009). Cellular and molecular mechanisms of pain. Cell 139, 267–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kuner R, and Flor H (2016). Structural plasticity and reorganisation in chronic pain. Nature reviews. Neuroscience 18, 20–30. [DOI] [PubMed] [Google Scholar]
- 3.Vardeh D, Mannion RJ, and Woolf CJ (2016). Toward a Mechanism-Based Approach to Pain Diagnosis. The journal of pain : official journal of the American Pain Society 17, T50–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Turnbull IM (1972). Bilateral cingulumotomy combined with thalamotomy or mesencephalic tractotomy for pain. Surgery, gynecology & obstetrics 134, 958–962. [PubMed] [Google Scholar]
- 5.Craig AD, Reiman EM, Evans A, and Bushnell MC (1996). Functional imaging of an illusion of pain. Nature 384, 258–260. [DOI] [PubMed] [Google Scholar]
- 6.Rainville P, Duncan GH, Price DD, Carrier B, and Bushnell MC (1997). Pain affect encoded in human anterior cingulate but not somatosensory cortex. Science 277, 968–971. [DOI] [PubMed] [Google Scholar]
- 7.Foltz EL, and White LE (1968). The role of rostral cingulumotomy in “pain” relief. International journal of neurology 6, 353–373. [PubMed] [Google Scholar]
- 8.Talbot JD, Villemure JG, Bushnell MC, and Duncan GH (1995). Evaluation of pain perception after anterior capsulotomy: a case report. Somatosensory & motor research 12, 115–126. [DOI] [PubMed] [Google Scholar]
- 9.Koyama T, Kato K, and Mikami A (2000). During pain-avoidance neurons activated in the macaque anterior cingulate and caudate. Neuroscience letters 283, 17–20. [DOI] [PubMed] [Google Scholar]
- 10.Koyama T, Kato K, Tanaka YZ, and Mikami A (2001). Anterior cingulate activity during pain-avoidance and reward tasks in monkeys. Neuroscience research 39, 421–430. [DOI] [PubMed] [Google Scholar]
- 11.Qu C, King T, Okun A, Lai J, Fields HL, and Porreca F (2011). Lesion of the rostral anterior cingulate cortex eliminates the aversiveness of spontaneous neuropathic pain following partial or complete axotomy. Pain 152, 1641–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johansen JP, Fields HL, and Manning BH (2001). The affective component of pain in rodents: direct evidence for a contribution of the anterior cingulate cortex. Proceedings of the National Academy of Sciences of the United States of America 98, 8077–8082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.LaGraize SC, Borzan J, Peng YB, and Fuchs PN (2006). Selective regulation of pain affect following activation of the opioid anterior cingulate cortex system. Experimental neurology 197, 22–30. [DOI] [PubMed] [Google Scholar]
- 14.Lubar JF (1964). Effect of Medial Cortical Lesions on the Avoidance Behavior of the Cat. Journal of comparative and physiological psychology 58, 38–46. [DOI] [PubMed] [Google Scholar]
- 15.Melzack R, and Wall PD (1965). Pain mechanisms: a new theory. Science 150, 971–979. [DOI] [PubMed] [Google Scholar]
- 16.Melzack R.a.C., K.L. (1968). Sensory, motivational, and central control determinants of pain: a new conceptual model.. The Skin Senses, 423–443. [Google Scholar]
- 17.Navratilova E, Xie JY, Okun A, Qu C, Eyde N, Ci S, Ossipov MH, King T, Fields HL, and Porreca F (2012). Pain relief produces negative reinforcement through activation of mesolimbic reward-valuation circuitry. Proceedings of the National Academy of Sciences of the United States of America 109, 20709–20713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johansen JP, and Fields HL (2004). Glutamatergic activation of anterior cingulate cortex produces an aversive teaching signal. Nature neuroscience 7, 398–403. [DOI] [PubMed] [Google Scholar]
- 19.Zhang Q, Manders T, Tong AP, Yang R, Garg A, Martinez E, Zhou H, Dale J, Goyal A, Urien L, et al. (2017). Chronic pain induces generalized enhancement of aversion. eLife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou H, Zhang Q, Martinez E, Dale J, Hu S, Zhang E, Liu K, Huang D, Yang G, Chen Z, et al. (2018). Ketamine reduces aversion in rodent pain models by suppressing hyperactivity of the anterior cingulate cortex. Nature communications 9, 3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vogt BA, and Sikes RW (2000). The medial pain system, cingulate cortex, and parallel processing of nociceptive information. Progress in brain research 122, 223–235. [DOI] [PubMed] [Google Scholar]
- 22.Shyu BC, Sikes RW, Vogt LJ, and Vogt BA (2010). Nociceptive processing by anterior cingulate pyramidal neurons. Journal of neurophysiology 103, 3287–3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meda KS, Patel T, Braz JM, Malik R, Turner ML, Seifikar H, Basbaum AI, and Sohal VS (2019). Microcircuit Mechanisms through which Mediodorsal Thalamic Input to Anterior Cingulate Cortex Exacerbates Pain-Related Aversion. Neuron 102, 944–959 e943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sesack SR, and Pickel VM (1992). Prefrontal cortical efferents in the rat synapse on unlabeled neuronal targets of catecholamine terminals in the nucleus accumbens septi and on dopamine neurons in the ventral tegmental area. J Comp Neurol 320, 145–160. [DOI] [PubMed] [Google Scholar]
- 25.Sesack SR, Deutch AY, Roth RH, and Bunney BS (1989). Topographical organization of the efferent projections of the medial prefrontal cortex in the rat: an anterograde tract-tracing study with Phaseolus vulgaris leucoagglutinin. J Comp Neurol 290, 213–242. [DOI] [PubMed] [Google Scholar]
- 26.King T, Vera-Portocarrero L, Gutierrez T, Vanderah TW, Dussor G, Lai J, Fields HL, and Porreca F (2009). Unmasking the tonic-aversive state in neuropathic pain. Nature neuroscience 12, 1364–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dale J, Zhou H, Zhang Q, Martinez E, Hu S, Liu K, Urien L, Chen Z, and Wang J (2018). Scaling Up Cortical Control Inhibits Pain. Cell reports 23, 1301–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martinez E, Lin HH, Zhou H, Dale J, Liu K, and Wang J (2017). Corticostriatal Regulation of Acute Pain. Frontiers in cellular neuroscience 11, 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li XY, Ko HG, Chen T, Descalzi G, Koga K, Wang H, Kim SS, Shang Y, Kwak C, Park SW, et al. (2010). Alleviating neuropathic pain hypersensitivity by inhibiting PKMzeta in the anterior cingulate cortex. Science 330, 1400–1404. [DOI] [PubMed] [Google Scholar]
- 30.Xu H, Wu LJ, Wang H, Zhang X, Vadakkan KI, Kim SS, Steenland HW, and Zhuo M (2008). Presynaptic and postsynaptic amplifications of neuropathic pain in the anterior cingulate cortex. The Journal of neuroscience : the official journal of the Society for Neuroscience 28, 7445–7453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sellmeijer J, Mathis V, Hugel S, Li XH, Song Q, Chen QY, Barthas F, Lutz PE, Karatas M, Luthi A, et al. (2018). Hyperactivity of Anterior Cingulate Cortex Areas 24a/24b Drives Chronic Pain-Induced Anxiodepressive-like Consequences. The Journal of neuroscience : the official journal of the Society for Neuroscience 38, 3102–3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cichon J, Blanck TJJ, Gan WB, and Yang G (2017). Activation of cortical somatostatin interneurons prevents the development of neuropathic pain. Nature neuroscience 20, 1122–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim SK, and Nabekura J (2011). Rapid synaptic remodeling in the adult somatosensory cortex following peripheral nerve injury and its association with neuropathic pain. The Journal of neuroscience : the official journal of the Society for Neuroscience 31, 5477–5482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blom SM, Pfister JP, Santello M, Senn W, and Nevian T (2014). Nerve injury-induced neuropathic pain causes disinhibition of the anterior cingulate cortex. The Journal of neuroscience : the official journal of the Society for Neuroscience 34, 5754–5764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou H, Martinez E, Lin HH, Yang R, Dale JA, Liu K, Huang D, and Wang J (2018). Inhibition of the Prefrontal Projection to the Nucleus Accumbens Enhances Pain Sensitivity and Affect. Frontiers in cellular neuroscience 12, 240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.De Felice M, Eyde N, Dodick D, Dussor GO, Ossipov MH, Fields HL, and Porreca F (2013). Capturing the aversive state of cephalic pain preclinically. Annals of neurology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hutchison WD, Davis KD, Lozano AM, Tasker RR, and Dostrovsky JO (1999). Pain-related neurons in the human cingulate cortex. Nature neuroscience 2, 403–405. [DOI] [PubMed] [Google Scholar]
- 38.Iwata K, Kamo H, Ogawa A, Tsuboi Y, Noma N, Mitsuhashi Y, Taira M, Koshikawa N, and Kitagawa J (2005). Anterior cingulate cortical neuronal activity during perception of noxious thermal stimuli in monkeys. Journal of neurophysiology 94, 1980–1991. [DOI] [PubMed] [Google Scholar]
- 39.Yamamura H, Iwata K, Tsuboi Y, Toda K, Kitajima K, Shimizu N, Nomura H, Hibiya J, Fujita S, and Sumino R (1996). Morphological and electrophysiological properties of ACCx nociceptive neurons in rats. Brain research 735, 83–92. [DOI] [PubMed] [Google Scholar]
- 40.Kung JC, Su NM, Fan RJ, Chai SC, and Shyu BC (2003). Contribution of the anterior cingulate cortex to laser-pain conditioning in rats. Brain research 970, 58–72. [DOI] [PubMed] [Google Scholar]
- 41.Kuo CC, and Yen CT (2005). Comparison of anterior cingulate and primary somatosensory neuronal responses to noxious laser-heat stimuli in conscious, behaving rats. Journal of neurophysiology 94, 1825–1836. [DOI] [PubMed] [Google Scholar]
- 42.Sikes RW, and Vogt BA (1992). Nociceptive neurons in area 24 of rabbit cingulate cortex. Journal of neurophysiology 68, 1720–1732. [DOI] [PubMed] [Google Scholar]
- 43.Zhang Y, Wang N, Wang JY, Chang JY, Woodward DJ, and Luo F (2011). Ensemble encoding of nociceptive stimulus intensity in the rat medial and lateral pain systems. Molecular pain 7, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Coghill RC, Sang CN, Maisog JM, and Iadarola MJ (1999). Pain intensity processing within the human brain: a bilateral, distributed mechanism. Journal of neurophysiology 82, 1934–1943. [DOI] [PubMed] [Google Scholar]
- 45.Buchel C, Bornhovd K, Quante M, Glauche V, Bromm B, and Weiller C (2002). Dissociable neural responses related to pain intensity, stimulus intensity, and stimulus awareness within the anterior cingulate cortex: a parametric single-trial laser functional magnetic resonance imaging study. The Journal of neuroscience : the official journal of the Society for Neuroscience 22, 970–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eto K, Wake H, Watanabe M, Ishibashi H, Noda M, Yanagawa Y, and Nabekura J (2011). Inter-regional contribution of enhanced activity of the primary somatosensory cortex to the anterior cingulate cortex accelerates chronic pain behavior. The Journal of neuroscience : the official journal of the Society for Neuroscience 31, 7631–7636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tan LL, Oswald MJ, Heinl C, Retana Romero OA, Kaushalya SK, Monyer H, and Kuner R (2019). Gamma oscillations in somatosensory cortex recruit prefrontal and descending serotonergic pathways in aversion and nociception. Nature communications 10, 983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu Y, Latremoliere A, Li X, Zhang Z, Chen M, Wang X, Fang C, Zhu J, Alexandre C, Gao Z, et al. (2018). Touch and tactile neuropathic pain sensitivity are set by corticospinal projections. Nature 561, 547–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bushnell MC, Duncan GH, Hofbauer RK, Ha B, Chen JI, and Carrier B (1999). Pain perception: is there a role for primary somatosensory cortex? Proceedings of the National Academy of Sciences of the United States of America 96, 7705–7709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Price DD (2000). Psychological and neural mechanisms of the affective dimension of pain. Science 288, 1769–1772. [DOI] [PubMed] [Google Scholar]
- 51.Goffer Y, Xu D, Eberle SE, D’Amour J, Lee M, Tukey D, Froemke RC, Ziff EB, and Wang J (2013). Calcium-permeable AMPA receptors in the nucleus accumbens regulate depression-like behaviors in the chronic neuropathic pain state. The Journal of neuroscience : the official journal of the Society for Neuroscience 33, 19034–19044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Koga K, Descalzi G, Chen T, Ko HG, Lu J, Li S, Son J, Kim T, Kwak C, Huganir RL, et al. (2015). Coexistence of two forms of LTP in ACC provides a synaptic mechanism for the interactions between anxiety and chronic pain. Neuron 85, 377–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cheriyan J, and Sheets PL (2018). Altered Excitability and Local Connectivity of mPFC-PAG Neurons in a Mouse Model of Neuropathic Pain. The Journal of neuroscience : the official journal of the Society for Neuroscience 38, 4829–4839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kiritoshi T, Ji G, and Neugebauer V (2016). Rescue of Impaired mGluR5-Driven Endocannabinoid Signaling Restores Prefrontal Cortical Output to Inhibit Pain in Arthritic Rats. The Journal of neuroscience : the official journal of the Society for Neuroscience 36, 837–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rodgers CC, and DeWeese MR (2014). Neural correlates of task switching in prefrontal cortex and primary auditory cortex in a novel stimulus selection task for rodents. Neuron 82, 1157–1170. [DOI] [PubMed] [Google Scholar]
- 56.Fyall AM, El-Shamayleh Y, Choi H, Shea-Brown E, and Pasupathy A (2017). Dynamic representation of partially occluded objects in primate prefrontal and visual cortex. eLife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Loureiro M, Achargui R, Flakowski J, Van Zessen R, Stefanelli T, Pascoli V, and Luscher C (2019). Social transmission of food safety depends on synaptic plasticity in the prefrontal cortex. Science 364, 991–995. [DOI] [PubMed] [Google Scholar]
- 58.Chow SS, Romo R, and Brody CD (2009). Context-dependent modulation of functional connectivity: secondary somatosensory cortex to prefrontal cortex connections in two-stimulus-interval discrimination tasks. The Journal of neuroscience : the official journal of the Society for Neuroscience 29, 7238–7245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Apkarian AV, Bushnell MC, Treede RD, and Zubieta JK (2005). Human brain mechanisms of pain perception and regulation in health and disease. European journal of pain 9, 463–484. [DOI] [PubMed] [Google Scholar]
- 60.Lee M, Manders TR, Eberle SE, Su C, D’Amour J, Yang R, Lin HY, Deisseroth K, Froemke RC, and Wang J (2015). Activation of corticostriatal circuitry relieves chronic neuropathic pain. The Journal of neuroscience : the official journal of the Society for Neuroscience 35, 5247–5259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kiritoshi T, and Neugebauer V (2018). Pathway-Specific Alterations of Cortico-Amygdala Transmission in an Arthritis Pain Model. ACS Chem Neurosci 9, 2252–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huang J, Gadotti VM, Chen L, Souza IA, Huang S, Wang D, Ramakrishnan C, Deisseroth K, Zhang Z, and Zamponi GW (2019). A neuronal circuit for activating descending modulation of neuropathic pain. Nature neuroscience 22, 1659–1668. [DOI] [PubMed] [Google Scholar]
- 63.Zhang Z, Gadotti VM, Chen L, Souza IA, Stemkowski PL, and Zamponi GW (2015). Role of Prelimbic GABAergic Circuits in Sensory and Emotional Aspects of Neuropathic Pain. Cell reports 12, 752–759. [DOI] [PubMed] [Google Scholar]
- 64.Ji G, Sun H, Fu Y, Li Z, Pais-Vieira M, Galhardo V, and Neugebauer V (2010). Cognitive impairment in pain through amygdala-driven prefrontal cortical deactivation. The Journal of neuroscience : the official journal of the Society for Neuroscience 30, 5451–5464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Buzsaki G, and Mizuseki K (2014). The log-dynamic brain: how skewed distributions affect network operations. Nature reviews. Neuroscience 15, 264–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.