

Abstract
Objective.
Sickle cell anemia causes chronic inflammation and multi-organ damage. Less understood are the arterial complications, most evident by increased strokes among children. Proteolytic mechanisms, biomechanical consequences, and pharmaceutical inhibitory strategies were studied in a mouse model to provide a platform for mechanistic and intervention studies of large artery damage due to sickle cell disease.
Approach and Results.
Townes humanized transgenic mouse model of sickle cell anemia were used to test the hypothesis that elastic lamina and structural damage in carotid arteries increased with age and was accelerated in mice homozygous for sickle cell anemia (SS) due to of inflammatory signaling pathways activating proteolytic enzymes. Elastic lamina fragmentation observed by 1 month in SS mice compared to heterozygous littermate controls (AS). Positive immunostaining for cathepsin K, a powerful collagenase and elastase, confirmed accelerated proteolytic activity in SS carotids. Larger cross-sectional areas were quantified by magnetic resonance angiography, and increased arterial compliance in SS carotids were also measured. Inhibiting c-jun N-terminal kinase (JNK) signaling with SP600125 significantly reduced cathepsin K expression, elastin fragmentation, and carotid artery perimeters in SS mice. By 5 months of age, continued medial thinning and collagen degradation was mitigated by treatment of SS mice with JNK inhibitor.
Conclusions.
Arterial remodeling due to sickle cell anemia is mediated by JNK signaling, cathepsin proteolytic upregulation and degradation of elastin and collagen. Demonstration in Townes mice establishes their utility for mechanistic studies of arterial vasculopathy, related complications, and therapeutic interventions for large artery damage due to sickle cell anemia.
Keywords: elastin, collagen, hematology, proteases, remodeling, biomechanics
Subject codes: Pathophysiology, Animal Models of Human Disease, Cerebrovascular Disease/Stroke, Vascular Disease
Graphcial abstract
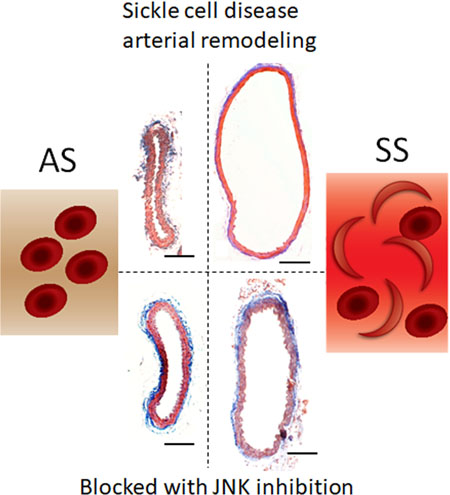
Introduction
Sickle cell anemia (SCA) is a homozygous recessive genetic disorder characterized by hemoglobin polymerization under deoxygenated conditions, damage to red blood cells, and multi-factorial complications in various organ systems and is one of a larger class of hemoglobinopathies known as sickle cell disease (SCD). Though considered a hematological disease, since the mutation causes hemoglobin polymerization and subsequent sickling and deformation of red blood cells (RBCs) after deoxygenation, there are significant vascular complications of SCA because of RBC hemolysis, which induces systemic, chronic inflammation. Mechanisms by which arteries are damaged due to SCA are not well-defined, especially since research is dominant in the deoxygenated microvasculature where sickling of red blood cells occurs, and less so in the arteries containing oxygenated blood. Arterial complications of SCA include aseptic bone necrosis, priapism, and moya moya1. Strokes, however, are one of the most devastating; 11% of children with SCA suffer a major stroke, with the highest risk between 2 and 5 years old2,3 indicating accelerated and early artery damage. Once a patient has one stroke, the likelihood of having another is significantly elevated, indicating a need to protect the integrity and biomechanical properties of the larger arteries.
Post-mortem examination of children with SCA who suffered strokes revealed significant fragmentation of the elastic lamina of the carotid and cerebral arteries4,5. Elastic lamina fragmentation is a well-established marker of arterial remodeling and precursor to cardiovascular disease and ischemic strokes6–8. Specific elastinolytic enzymes involved in this fragmentation, stimulated by the chronic inflammation characteristic of SCA9 are only beginning to be described. Cathepsins K and V (catK, catV) are powerful elastases and collagenase, implicated in internal elastic lamina (IEL) degradation and arterial remodeling in atherosclerosis9. Previously, we identified active catK expression in arterial endothelial cells co-cultured with monocytes isolated from people with SCA was mediated via JNK/c-jun signaling12, leading to the hypothesis that catK may be involved in accelerated elastinolytic remodeling of SCA.
Arterial stiffening is a key and early predictive marker of cardiovascular events to come and well recognized in the atherosclerosis literature10–12, but less so described in sickle cell disease. Changes in arterial stiffness in SCA have reported contradictory results. Lemogoum et al. reported lower blood pressure, increased vasodilation and lower arterial stiffness in 20 SS patients compared to 20 AA subjects13, but in other studies, arterial stiffening in carotid arteries of adults with SCA has been associated with strokes14. This group found that SCA individuals in a cohort with mean age of 27 years that had suffered strokes, had significantly higher arterial stiffness than those with SCA but no history of stroke, confirming the vasculature of people with SCA are aging or advancing through cardiovascular disease at an accelerated rate. Stenoses, occlusions, and arteriopathy have also been quantified in the extracranial carotid artery of children with SCA that were stroke-free at the time of measurement15. These data form the scientific premise of this study to investigate carotid artery remodeling and arterial stiffness as a consequence of SCA.
In this study, humanized knock-out and knock-in model of SCA, Townes sickle transgenic mice, were used which were generated by knocking in the human hemoglobin genes (α, β, and γ), including β-globin S mutation, into mice null for the murine α and β-globin genes16. Townes SS mice have a markedly shorter life span, renal and splenic SCA pathologies at relatively younger ages, and are vulnerable to asymptomatic, idiopathic mortality16,17. Here, we establish its utility as a model of arterial pathological remodeling complications of SCA, and to test pharmacological interventions to prevent this accelerated arterial damage.
Materials and Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Animals
Townes sickle transgenic mice breeding pairs (B6; 129-Hbatm1(HBA) Tow Hbbtm2 (HBG1, HBB*) Tow/Hbbtm3 (HBG1, HBB) Tow/J) were obtained from The Jackson Laboratory. Data from male and female mice were pooled for analysis because of the low yield per litter and probability of obtaining SS genotype (sickle cell disease). The animals were kept in climate-controlled rooms with a 12 h light-dark cycle and are allowed access to food and water ad libitum. Mice received Lab Diet 5001 after weaning, and for breeding, Lab Diet 5015. Bed-o’Cobs ¼” cage bedding from The Andersons lab bedding was used. Newborn mice were evaluated for sickle status at weaning (21 days old) then drug or vehicle treatment began or in some studies, the drug treatment began at 3 months of age. SP600125 (Invitrogen) was diluted in 10% DMSO and injected daily, intraperitoneally at 50 mg/kg from 1 to 3 months of age or 10 mg/kg doses from 3 to 5 months of age for 8 weeks or with vehicle only. All experiments were approved by Georgia Institute of Technology’s Institutional Animal Care and Use Committee.
Tissue Isolation and Histology
After euthanization, mice were perfusion fixed with 10% neutral buffered formalin. Carotid arteries were carefully isolated, cleaned of outer fat layer, and then embedded in HistoGel (Richard-Allen Scientific) prior to paraffin embedding and sectioning. Thoracic aortas were cleaned of adventitia, and homogenized in zymography lysis buffer18. Modified Verhoeff-van Gieson stain (Electron Microscopy Sciences) was used for elastin visualization and fragment quantification, imaged with Eclipse Ti-E microscope (Nikon Instruments). Carotid artery sections were blinded of sickle status and imaged under 40x magnification. Elastin fraying, fragmentation, and delamination were identified and recorded noting nicks and discontinuities. Mason’s Trichrome stain (Abcam) was used for collagen (blue) stain. Sections were immunostained with rabbit polyclonal anti-catK (Santa Cruz Biotechnology), followed by fluorescent secondary antibody (Invitrogen), and mounted with Prolong Gold anti-fade containing DAPI (Invitrogen).
Magnetic Resonance Angiography
Magnetic resonance angiography was performed with a 7-T MRI system (PharmaScan, BRUKER). Mice were anesthetized by isoflurane. MRA was performed with a 3-dimensional TOF sequence of a repetition time of 14 ms, echo time of 2.6 ms, a flip angle of 30º, field of view of 20 × 20 × 18.562 mm with 256×256×128 resolution. The acquisition time was 17 min 53 s. Angiograms were obtained by generating maximal intensity projection 3D reconstruction of the raw data.
Image Processing and Morphological Analysis
The reconstructive software, Mimics (Materialise, Leuven, Belgium) was used to manually segment and reconstruct the arteries of interest. A minimum threshold was optimized for each specimen, which produced the largest vessel diameter without creating irregularities on the surface. After a threshold was chosen, a preliminary mesh of the vascular network was created, containing arterial segments of the common carotid artery. Irregular artifacts created from the reconstructive process were removed using re-meshing and smoothing algorithms from Vascular Modeling Toolkit (http://www.vmtk.org)19. After the reconstruction process was completed, Mimics was used to calculate several parameters related to the morphology and area along the common carotid artery was graphed with the carotid bifurcation set as the origin.
Mechanical Testing
Carotid arteries were isolated from 1 month or 3-month-old AS or SS mice. The arteries were carefully cleaned of all perivascular fat and stored in a freezing media consisting of 10% DMSO, 50% fetal bovine serum (FBS), and 40% DMEM before biaxial mechanical testing was performed, up to 2 weeks later. Right before the biaxial testing, frozen arteries were subjected to a sequential thawing procedure, then, the artery was examined under a dissecting microscope to ensure it was intact; any remaining perivascular fat was removed. Previous studies have demonstrated that cryopreservation in media and subsequent thawing does not significantly alter arterial mechanics 20,21. The artery was then mounted onto glass cannula and sealed using an 8–0 silk suture tied into a surgeon’s knot. Before pressurizing, video software was used to record the unloaded diameter and length of the artery. Artery was then preconditioned by increasing luminal pressure from 0 to 140 mmHg at increasing axial strains (ratio representing the increase in length from the initial, unloaded length) of 1.3 to 2.0. To estimate in vivo axial strain, data from biaxial mechanical testing of carotid arteries at 5 axial strains within this range were collected for isobaric curves corresponding to a single pressure value. Second-order polynomials were fit to all isobaric curves, and intersection points of all curves were calculated and averaged in order to estimate the in vivo axial strain. As in our previous paper 22, pressure-diameter (p-d) curves were plotted and local compliance was calculated from its p–d response at increments of 10 mmHg using the equation:
where C is compliance (MPa−1), is the difference in outer diameter between two pressures, is the outer diameter at the mean of those two pressures, and ΔP is the difference between pressures.
Multiplex Cathepsin Zymography
Multiplex cathepsin zymography was used to quantify amounts of active cathepsins as described previously18.
Western blotting
Western blotting was used to determine phosphorylated c-jun with primary antibodies from Cell Signaling and secondary antibodies from Li-Cor (Rockland) to be visualized with a LI-COR Odyssey scanner. Densitometry of labeled nitrocellulose membranes was performed using NIH ImageJ.
Statistical Analysis
Experimental conditions were repeated with a minimum of three biological replicates, and data is presented as mean value and standard error of the mean. Representative images are shown. Unpaired student t-tests were used to determine statistical significance (*p<0.05) between experimental groups. Perimeter of internal elastic lamina (IEL) were determined by tracing Van Gieson stained images and measured with ImageJ, and normalized to AS control to compare between sets of experiments. Statistical significance determined with two-way ANOVA for three data sets for AS and SS (column factor) and control and JNK inhibitor (row factor) (Prism 7). we used Anderson-Darling, D’Agostino & Pearson, Shapiro-Wilk, Kolmogorov-Smimorv tests for normality in PRISM 7. Not all data passed normality test, therefore, we used one-way ANOVA followed by multiple comparison tests (Tukey’s multiple comparison or uncorrected Fisher’s LSD test as it is indicated), and for ANOVA, we performed equal variance test using Brown-Forsythe test and variance was not different.
Results
Mice with sickle cell anemia had increased elastic lamina breaks in carotid arteries at 1 month of age.
It was first investigated if sickle cell anemia induced increased elastic lamina fragmentation in mice at age one month to look for early damage to arteries. Transgenic mice homozygous for human beta-globin S mutation (SS) had 8-times more elastin breaks in their carotid arteries compared to littermate heterozygous (AS) mice (Fig 1A and 1B, orange arrowheads, p<0.05, n=4). Immunohistochemistry for cathepsin K in carotid arteries indicated stronger staining in SS mice compared to AS (Fig 1C). These data suggest that accelerated elastin degradation was occurring in SS mice by one month without surgical intervention, and was associated with increased cathepsin K expression.
Figure 1. Sickle cell disease promotes elastin fragmentation and cathepsin K expression in mice by 3 weeks of age.

(A) Perfusion fixed carotid arteries from 3-week AS and SS mice were isolated, sectioned, and stained for elastin morphology using modified Verhoeff elastic-van Gieson stain. (B) elastin fragmentation was quantified along with vessel area with significant increases in the SS mice compared to littermate AS controls. (* denotes p<0.05, n=4) (C) Cathepsin K expression was upregulated in carotid arteries from SS mice.
Longitudinal analysis of carotid arteries with magnetic resonance angiography suggests expansive remodeling in SS carotid arteries compared to AS carotids.
To examine consequences of early elastic lamina degradation and fragmentation, carotid arteries from SS and AS mice were imaged at age 1 month and 3 months for longitudinal study using magnetic resonance angiography (MRA) without contrast agents. MRA images were then reconstructed into three-dimensions, and measurements of cross-sectional area were made along the length of the common carotid arteries from their branch off of aortic arch up to the bifurcation into internal and external carotid artery (Fig 2A). Cross-sectional area differences were detected at 3 months of age from MRA images (Fig 2B). Data from three mice each of genotype AS or SS are shown in figure 2C represented by a unique color to plot data points from the reconstructed carotid artery cross-sectional area measurement from the same mouse when measured at 1 month of age (open) or at 3 months of age (closed). The mean area along the length of the carotid artery from 3-month SS mice was larger than that of AS mice (SS: 0.574 mm2, AS: 0.464 mm2). This average area measurement did not reach statistical significance, as the variability in luminal expansion due to sickle cell anemia in the SS carotid arteries is visible in the graphs compared to that of the AS control mice. Regardless, the change in luminal area from one month to 3 months of mice with SS was larger. Abrupt changes in the cross-sectional area over short distances such as that found in SS#2 can also have an effect on hemodynamics and relevance for stroke stenoses. The body weights of AS vs SS animals were not statistically different (data not shown), therefore differences in cross-sectional area are not assumed to be due to animal size.
Figure 2. Longitudinal analysis of carotid arteries with magnetic resonance also suggests expansive remodeling in SS carotid arteries compared to AS carotids, but with variability.

(A) Reconstruction diagram. (B) Longitudinal magnetic resonance angiography was used on AS, or SS mice beginning at one month of age and those same mice were again imaged at three months of age to compare animal specific growth and remodeling. White arrows are shown to indicate diameter differences between AS and SS arteries in the representative images shown. C) Reconstructed images were used to measure luminal areas of the carotid arteries and to compare both age-related and genotype-related differences. While the one month carotid artery areas were similar between AS and SS, there was greater variability in the SS diameters by 3 months of age. Numbers indicate the mouse label, and the age to show specific change per animal. Area along the length of the artery are presented. Labeling: AS#1 – 1 month indicates sickle cell genotype (AS), animal replicate number of 3 (#1), and age at which MR imaging was conducted (1 mo = 1 month). The same mouse imaged at 1 month was again imaged at 3 months of age (i.e. AS#1 – 1 mo and AS#1 – 3 mo are images from the same mouse). Circle, square, and triangle represent each mouse, open indicates measurements made at 1 mo. and closed indicates measurements made of that mouse at 3 mo.
Carotid arteries from mice with sickle cell disease are more expanded than heterozygous littermate controls
Next, we examined the mechanical properties of carotid arteries from AS or SS mice at ages 1 and 3 months. Diameter-pressure graphs revealed that at 1 month and 3 months of age, SS mice carotid arteries, when inflated with the same pressure value, achieved higher diameters in these vessels compared to AS controls, and even at physiological luminal pressures (n=6, **p<0.05) (Fig 3A). Over the murine blood pressure range of 120/100 mmHg, the average diameter of carotid arteries from SS mice at 1 month was 0.73±0.07 (average ± standard deviation) mm and at 3 months, 0.85±0.05 mm, and AS control arteries were 0.64± 0.05mm and 0.78± 0.04 mm at 1 month and 3 months, respectively. Compliance was calculated from the pressure-diameter curves in windows of 10 mmHg; and at sub-physiological pressures, carotid arteries from SS mice had increased compliance compared to AS; however, at physiological systolic pressures (120 mmHg), there was no significant difference (Fig 3B). These mechanical testing data confirm that carotid arteries from SS mice have larger lumens and are more compliant at subphysiological pressures.
Figure 3. Carotid arteries from mice with sickle cell disease are more expanded than heterozygous littermate controls.

(A) Biomechanical testing was performed on 1 month and 3 months AS and SS mice to generate pressure-diameter curves from pressures 0 to 150 mmHg, (n=5 for 1 month old, n=9 for 3 months old) (* p<0.05, One-way ANOVA, # p<0.05 followed by Tukey’s multiple comparison test between AS and SS at 1 month of age, or $ p<0.05, at 3-month age) (B) Local compliance differences assessed at increments of 20 mmHg (* p<0.05, One-way ANOVA)
Inhibition of JNK signaling reduces arterial cathepsin K expression in SS mice
Since our previous studies using human cells indicated that JNK inhibition could prevent cathepsin K induction23,24, we next wanted to confirm these findings in vivo. To test the hypothesis that inhibition of JNK would reduce cathepsin K expression and elastic lamina breaks in carotid arteries, establishing a causal relationship, AA, AS, and SS mice were treated with daily intraperitoneal injections of the JNK inhibitor SP600125 for 8 weeks, beginning at 1 month of age and sacrificed at 3 months for analysis. Carotid arteries from SS mice had significantly larger internal elastic lamina (IEL) perimeters (Fig 4A; Supp Fig 1A) and higher numbers of elastin breaks over AS mice as well as but treatment of SS mice with SP600125 significantly reduced elastin breaks and artery size (Fig 4B; Supp Fig 1B). Homogenized thoracic aorta from these mice were used for multiplex cathepsin zymography, which qualitatively indicates reduced cathepsin activity with SP600125 treatment, confirm that reduced phosphorylation of JNK’s downstream target c-jun by Western blot, in mice treated with SP600125 (Fig 4B) suggesting that inhibition of JNK/c-Jun signaling cascade may be sufficient to prevent SCA-induced elastin degradation, and the expansive luminal remodeling. Furthermore, arteries from SS mice demonstrated immunopositive catK staining (Fig 4C), that qualitatively appeared to be less in SS mice treated with SP600125 down to levels comparable to untreated AS animals.
Figure 4. JNK inhibition preserves elastic lamina integrity in SS mice.
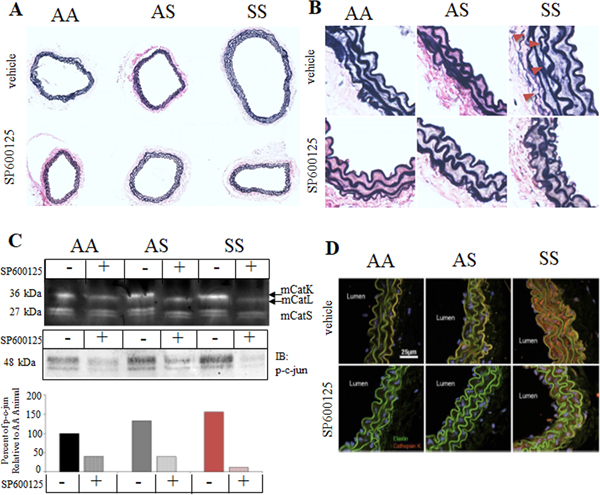
After an 8-week regimen of SP600125 intraperitoneal injections, 3 month old mice were sacrificed and perfusion fixed carotid arteries from AA, AS, and SS mice were isolated, sectioned, and (A) stained for elastin morphology using modified Verhoeff elastic-van Gieson stain. (B) Zoomed in images are shown as well. (C) Thoracic aortas were prepared for multiplex cathepsin zymography and for c-Jun phosphorylation by Western blot to demonstrate efficacy of SP600125 treatment, and confirmed that SS mice had higher baseline cathepsin activity, that was substantially reduced by JNKi, SP600125. (D) Carotid arteries from 3-month old AA, AS and SS mice were immunostained for cathepsin K. Only SS mice showed strong staining for catK (red); elastin fibers are autofluorescent (green).
Carotid arteries from 5-month-old sickle cell disease mouse (SS) were enlarged with decreased medial thickness compared to age-matched AS genotypes.
With expansive remodeling evident from 1 to 3 months, and JNK inhibition in SS mice being able to prevent this by reducing active cathepsins and elastin fragmentation, the next hypothesis tested was if intervention with JNK inhibitor was sufficient to halt arterial remodeling, or if early intervention was required for vascular protection from SCA-mediated remodeling. To test this, JNK treatment was started at mice that were 3 months of age, and after 8 weeks, they reached 5 months of age (Fig 5A). Carotid arteries from 5-month-old, untreated SS mice had IEL perimeters that were significantly larger than AS untreated mice by 124% (AS: 1152 ± 84.5 μm, SS: 1432 ± 86.4 μm). It was also noted that the thickness of the medial layer of SS arteries was significantly decreased in untreated SS mice (AS: 28.68 ± 2.56 μm, SS:.15.44 ± 1.39 μm Fig 5B and C, n=5–7). Administering JNK inhibitor from 3 months to 5 months significantly prevented IEL perimeter expansion and medial thinning compared to SS vehicle control (Fig 5B); however, the late intervention from 3-month-old age could not completely reverse the medial thinning to the thickness of AS control as there was still a significant difference between AS and treated SS (Fig 5B).
Figure 5. Expansive remodeling of carotid arteries continues through 5 months of age for sickle cell disease mouse but can be reduced with JNK inhibition.
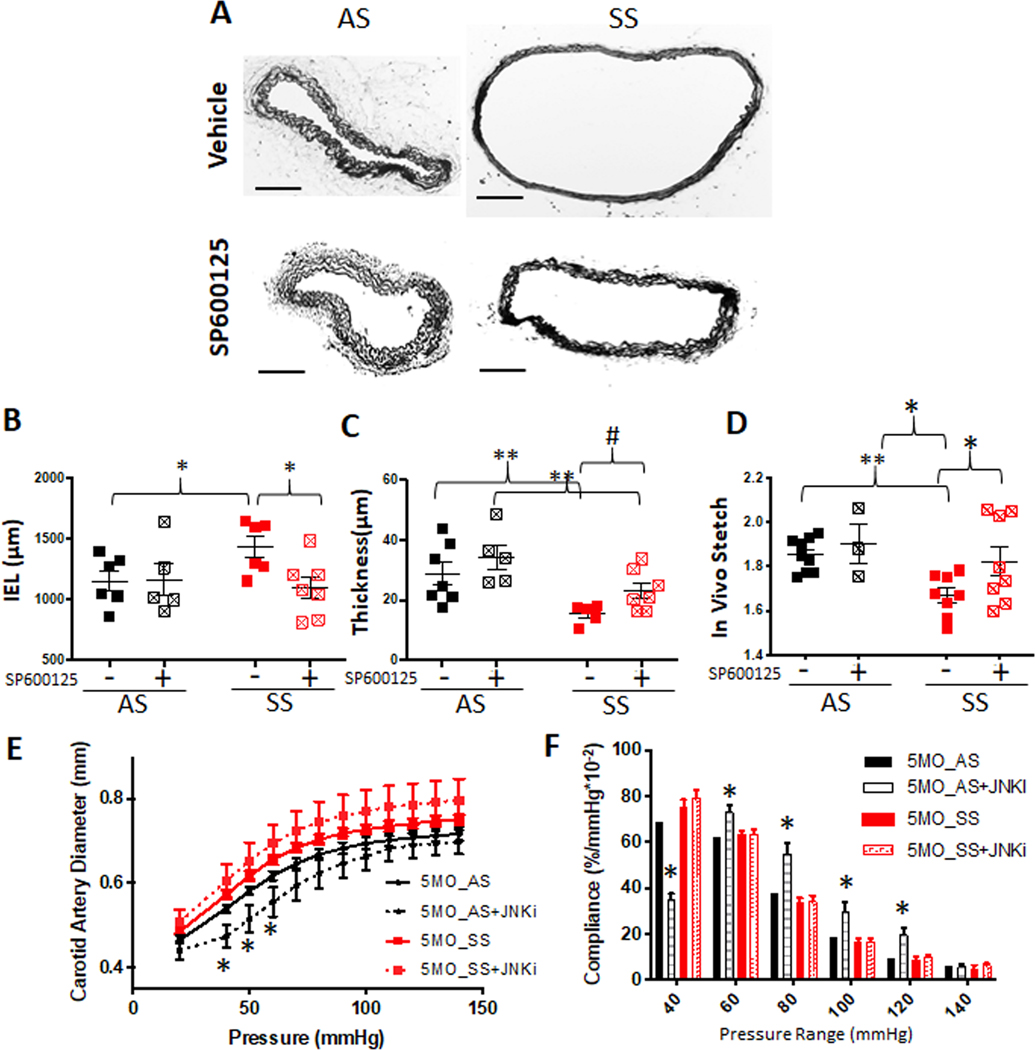
(A) JNK inhibitor (SP600125, 10mg/kg) was injected daily from age 3 months to 5 months. Then, mice were euthanized and perfusion fixed, carotid arteries excised, and processed for sectioning and stained for Van Giesen elastin stain (scale bar= 100 μm). (B) Internal elastin lamina was traced and length was measured using ImageJ. (C) For thickness measurement, medial thickness from four locations were measured and averaged (n of 4–6 animals per genotype and treatment, * p<0.05, ** p<0.01, Two-way ANOVA followed by Fisher’s LSD, # p<0.05, t-test). (D) Biomechanical testing was performed and in vivo stretch was determined and compared between treatments. (E) Pressure-diameter curves from mechanical testing. (F) Local compliance was calculated from P-D curves (One-way ANOVA followed by Tukey’s multiple comparison test. *p<0.05).
Carotid artery mechanics of 5-month old SS mice treated with JNK inhibitor.
In vivo axial strain was measured to quantify artery mechanics in the axial direction. In vivo strain is representative of relative axial elasticity as it is the strain the artery experiences when still attached to the vasculature. At this strain, the artery experiences a nearly constant axial force (the vasculature does no work as blood flows) at all points in the cardiac cycle. Treatment with JNK inhibitor from 3 to 5 months improved in vivo strain in SS mice carotid arteries, though still lower than AS, whether treated or not. However, treatment of mice for two months with JNK inhibitor did not show a significant difference in the pressure-diameter curves except at physiological pressures (Fig 5E), and did not restore compliance of SS mice carotid arteries since the SS + JNKi treatment groups showed no significant difference from the SS groups. Only significance observed in compliance was between AS treated with JNK inhibitor and all other groups. The significant difference for in vivo strain, suggests that axially there may have been structural recovery, but in the radial direction, the material damage persisted.
Axial and radial mechanics were affected differently by JNKi treatment in 5-month-old mice. Carotid arteries were stained with Masson’s Trichrome to provide structural protein information in these expanded, thin carotid arteries. The medial layer of carotid arteries from untreated SS mice at 5 month lost collagen as indicated by the loss of blue staining compared to AS mice (Fig 6A). To quantify this, carotid arteries were homogenized and total collagen was determined indicating that JNK inhibition mitigated this collagen loss in SS mice compared to vehicle control SS mice (Fig 6B).
Figure 6. Carotid arteries from 5 month old sickle cell disease mouse (SS) lost medial collagen and JNKi treatment rescued it.
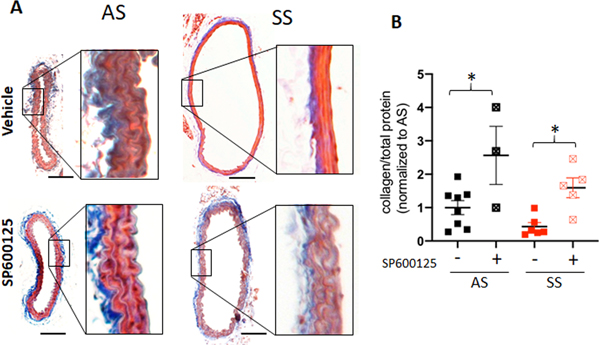
(A) The fixed and processed carotid arteries were stained with Mason’s Trichrome Stain (scale bar= 100 mm). (B) Carotid arteries post mechanical testing were homogenized and hydrolyzed using NaOH at 120’c, and measured total collagen using perchlorate free total collagen assay kit (abcam) was normalized to total protein amount. One way ANOVA was used for statistical analysis.
Discussion
This work, taken together, demonstrates early onset pathological arterial remodeling in a mouse model of sickle cell anemia as early as 1 month old, and without surgical or chemical interventions used to induce strokes in mice25–27 that would not recapitulate the natural, progressive vasculopathy observed in humans with SCA. Proteolytic mechanisms of elastin and collagen degradation were evident and associated with elevated expression of cathepsin K, a powerful elastase and collagenase. Carotid arteries from SS mice underwent 1) elastic lamina fragmentation and breaks, 2) expansive remodeling with increased luminal diameters and internal elastic lamina perimeters, 3) medial thinning with decreased medial collagen content, and 4) altered biomechanical properties with increased compliance in SS carotid arteries. Further, this study demonstrated that inhibiting JNK signaling through the administration of SP600125, was able to mitigate the expansive arterial remodeling and reduced the amount of active cathepsins in the arteries, highlighting mechanistic pathways for future interrogation to protect from SCA-mediated arterial remodeling.
Sickle cell anemia is mainly investigated in the microcirculation and venous side since deoxygenation promotes polymerization of sickle hemoglobin into stiff rods that deform and damage red blood cells. However, there are a number of arterial complications that occur in SCA and that occur in the large arteries that need to be addressed, and this work takes an important step in identifying the structural arteriopathy in carotid arteries while identifying JNK and cysteine cathepsins as targets for intervention. We further demonstrate the utility of Townes mouse model for mechanistic studies and pre-clinical testing of pharmaceutical therapies to preserve the integrity of large arteries by showing that treatment with the JNK inhibitor, SP600125. This corroborates our previous findings that monocyte-endothelial interactions induced cathepsin K via JNK/c-Jun signaling when cultured with monocytes isolated from people with SCA23. Inhibition of JNK signaling reduced the inflammation-induced activation of cathepsins K and V whether due to SS peripheral blood mononuclear cells co-cultured with endothelium, or whether stimulated by TNFα23,24. We also showed that endothelial cell catK expression is regulated by AP-1 and NFκB under disturbed blood flow28. These findings highlight the role of JNK signaling as an integration control point and as a therapeutic target to inhibit the initiation of gene and protein expression in response to inflammatory stimuli resulting in endothelial cell upregulation of catK and V protein and activity. More importantly, the predisposition of SS PBMCs to induce these effects suggests these novel mechanisms may be occurring constantly in the vasculature of individuals with SCA. We hypothesize that cathepsin overexpression in sickle cell arteries is a downstream consequence of the chronic inflammatory state and elevated TNFα consistent with sickle cell disease plasma29. This stimulates a pathway through JNK leading to upregulation of cathepsins, and the consequences of their overexpression in is accelerated degradation of elastin and collagen in the artery wall.
We hypothesized that humanized Townes mouse model would be a useful in vivo model to examine the arteriopathy due to SCA. Carotid arteries from SS mice measured by MRA and histology revealed that they undergo expansive remodeling and medial thinning as they age due to early elastin breaks and loss of collagen. The common carotid is a complex multilayered anisotropic structure. Both elastin and collagen contribute to mechanical properties; however, their organization within the arterial wall affects how and at what pressure thresholds they become fully engaged and contribute. Elastin is responsible for small expansion and collagen is load bearing during large expansion 30. This might contribute to the behavior that we have observed with carotid arteries from SS mice in at subphysiological pressures, arteries from SS shows more compliant due to loss of elastin at 1 and 3-month-old. By 5 months of age, however, when the collagen loss in artery was significant, there was no significant difference in compliance between the genotypes. Taking the material-specific behaviors of elastin and collagen into account, the pressure-diameter curve is linear while collagen is mostly unloaded—once collagen becomes loaded, the curve flattens substantially. This being said, SS arteries with significantly less collagen still exhibited that final plateau at higher pressures suggesting that what little collagen was remaining was sufficient to resist further outward distention, or that elastin had an upper limit of distention. A pressure driven failure test may help to clarify this.
Damage to the carotid arteries has been identified clinically in people with sickle cell disease15,31,32, supporting the findings of this study, but still inconclusive as to the degree of large artery damage causing pediatric strokes in these children33,34. Inflammatory factors such as TNFα are elevated and circulating in blood of people with sickle cell disease, and it has been demonstrated in our work and others to elevate NFKB and JNK signaling pathways in Townes mice35 and human cells23,28 that are linked to collagen and elastin degradation in this study. Regular blood transfusions successfully reduce stroke risk in children with SCA, risks of alloimmunity, iron over load, and infection36 and increased risks during adulthood37,38, all motivate searches for other therapeutic options. By identifying other mechanistic pathways that contribute to vasculopathy, such as proteases and upstream signaling cascades, new therapeutic targets for children with sickle cell anemia can be verified that could be effective in both pediatric strokes and adult strokes, with preclinical testing of mouse models for sickle cell mediated large artery vasculopathy.
Supplementary Material
Highlights.
Mice with sickle cell anemia have arterial damage as early as one month of age with effects on artery biomechanics, and damage accumulates as they age.
Cathepsin K, a potent elastase and collagenase, is elevated in SS Townes mice arteries
JNK inhibition can reduce cathepsin K expression, elastin fragmentation, expansive remodeling, and collagen damage in carotid arteries.
Acknowledgements
PMK, HS, RLG, YH, EAB, and MOP conceived of the experiments. HS, PMK, SA, CPR, YF, VOO, AAC, and SC conducted the experiments, and PMK, HS, YH, WT, VOO, EAB, and MOP analyzed the results. PMK, SA, HS and MOP wrote the manuscript. We also would like to acknowledge George McAlear, Keval Bollavaram, and Oluwasanmi Victor Ariyo for their contributions in artery reconstructions, and Pei Niu, Li Li, and Hao Wu for mice husbandry and preparation at Peking University.
Sources of funding
This work was funded by NIH New Innovator grant #1DP2OD007433-01 (MOP) from the Office of the Director, National Institutes of Health, NIH grant #1R56HL136210-01 (MOP, RLG, EAB), and American Heart Association Grant-in-Aid 17GRNT33710016 (MOP, RLG, EAB). PMK was supported by an NSF graduate research fellowship.
Abbreviations:
- SS
sickle cell anemia homozygous genotype
- AS
sickle cell trait heterozygous genotype
- AA
homozygous wildtype hemoglobin genotype
- JNK
c-jun N-terminal kinase
- SCA
sickle cell anemia
Footnotes
Disclosures
There are no competing financial interests.
References
- 1.Francis RB. Large-vessel occlusion in sickle cell disease: Pathogenesis, clinical consequences, and therapeutic implications. Med Hypotheses. 1991;35:88–95. [DOI] [PubMed] [Google Scholar]
- 2.Adams R, McKie V, Nichols F, Carl E, Zhang DL, McKie K, Figueroa R, Litaker M, Thompson W, Hess D. The use of transcranial ultrasonography to predict stroke in sickle cell disease. N Engl J Med. 1992;326:605–610. [DOI] [PubMed] [Google Scholar]
- 3.Powars D, Wilson B, Imbus C, Pegelow C, Allen J. The natural history of stroke in sickle cell disease. Am J Med. 1978;65:461–471. [DOI] [PubMed] [Google Scholar]
- 4.Merkel KH, Ginsberg PL, Parker JC, Post MJ. Cerebrovascular disease in sickle cell anemia: A clinical, pathological and radiological correlation. Stroke. 1978;9:45–52. [DOI] [PubMed] [Google Scholar]
- 5.Switzer JA, Hess DC, Nichols FT, Adams RJ. Pathophysiology and treatment of stroke in sickle-cell disease: Present and future. Lancet neurology. 2006;5:501–512. [DOI] [PubMed] [Google Scholar]
- 6.Platt MO, Ankeny RF, Shi GP, Weiss D, Vega JD, Taylor WR, Jo H. Expression of cathepsin K is regulated by shear stress in cultured endothelial cells and is increased in endothelium in human atherosclerosis. Am J Physiol Heart Circ Physiol. 2007;292:H1479–1486. [DOI] [PubMed] [Google Scholar]
- 7.Lutgens E, Lutgens SP, Faber BC, Heeneman S, Gijbels MM, de Winther MP, Frederik P, van der Made I, Daugherty A, Sijbers AM, Fisher A, Long CJ, Saftig P, Black D, Daemen MJ, Cleutjens KB. Disruption of the cathepsin K gene reduces atherosclerosis progression and induces plaque fibrosis but accelerates macrophage foam cell formation. Circulation. 2006;113:98–107. [DOI] [PubMed] [Google Scholar]
- 8.Aikawa E, Aikawa M, Libby P, Figueiredo JL, Rusanescu G, Iwamoto Y, Fukuda D, Kohler RH, Shi GP, Jaffer FA, Weissleder R. Arterial and aortic valve calcification abolished by elastolytic cathepsin S deficiency in chronic renal disease. Circulation. 2009;119:1785–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Platt OS. Sickle cell anemia as an inflammatory disease. J Clin Invest. 2000;106:337–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Laurent S, Boutouyrie P, Asmar R, Gautier I, Laloux B, Guize L, Ducimetiere P, Benetos A. Aortic stiffness is an independent predictor of all-cause and cardiovascular mortality in hypertensive patients. Hypertension. 2001;37:1236–1241. [DOI] [PubMed] [Google Scholar]
- 11.Mattace-Raso FU, van der Cammen TJ, Hofman A, van Popele NM, Bos ML, Schalekamp MA, Asmar R, Reneman RS, Hoeks AP, Breteler MM, Witteman JC. Arterial stiffness and risk of coronary heart disease and stroke: The Rotterdam study. Circulation. 2006;113:657–663. [DOI] [PubMed] [Google Scholar]
- 12.Weber T, Auer J, O’Rourke MF, Kvas E, Lassnig E, Berent R, Eber B. Arterial stiffness, wave reflections, and the risk of coronary artery disease. Circulation. 2004;109:184–189. [DOI] [PubMed] [Google Scholar]
- 13.Lemogoum D, Van Bortel L, Najem B, Dzudie A, Teutcha C, Madu E, Leeman M, Degaute JP, van de Borne P. Arterial stiffness and wave reflections in patients with sickle cell disease. Hypertension. 2004;44:924–929. [DOI] [PubMed] [Google Scholar]
- 14.Belizna C, Loufrani L, Ghali A, Lahary A, Primard E, Louvel JP, Henrion D, Levesque H, Ifrah N. Arterial stiffness and stroke in sickle cell disease. Stroke. 2012;43:1129–1130. [DOI] [PubMed] [Google Scholar]
- 15.Verlhac S, Balandra S, Cussenot I, Kasbi F, Vasile M, Kheniche A, Elmaleh-Berges M, Ithier G, Benkerrou M, Bernaudin F, Sebag G. Extracranial carotid arteriopathy in stroke-free children with sickle cell anemia: Detection by submandibular doppler sonography. Pediatr Radiol. 2014;44:587–596. [DOI] [PubMed] [Google Scholar]
- 16.Ryan TM, Ciavatta DJ, Townes TM. Knockout-transgenic mouse model of sickle cell disease. Science. 1997;278:873–876. [DOI] [PubMed] [Google Scholar]
- 17.Beuzard Y. Mouse models of sickle cell disease. Transfusion clinique et biologique. 2008 [DOI] [PubMed] [Google Scholar]
- 18.Wilder CL, Park K-Y, Keegan PM, Platt MO. Manipulating substrate and pH in zymography protocols selectively distinguishes cathepsins K, L, S, and V activity in cells and tissues. Arch Biochem Biophys. 2011;516:52–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Piccinelli M, Veneziani A, Steinman DA, Remuzzi A, Antiga L. A framework for geometric analysis of vascular structures: Application to cerebral aneurysms. Ieee T Med Imaging. 2009;28:1141–1155. [DOI] [PubMed] [Google Scholar]
- 20.Venkatasubramanian RT, Grassl ED, Barocas VH, Lafontaine D, Bischof JC. Effects of freezing and cryopreservation on the mechanical properties of arteries. Annals of Biomedical Engineering. 2006;34:823–832. [DOI] [PubMed] [Google Scholar]
- 21.Pukacki F, Jankowski T, Gabriel M, Oszkinis G, Krasinski Z, Zapalski S. The mechanical properties of fresh and cryopreserved arterial homografts. European Journal of Vascular and Endovascular Surgery. 2000;20:21–24. [DOI] [PubMed] [Google Scholar]
- 22.Kim CW, Pokutta-Paskaleva A, Kumar S, Timmins LH, Morris AD, Kang DW, Dalal S, Chadid T, Kuo KM, Raykin J, Li H, Yanagisawa H, Gleason RL Jr., Jo H, Brewster LP. Disturbed flow promotes arterial stiffening through thrombospondin-1. Circulation. 2017;136:1217–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Keegan PM, Surapaneni S, Platt MO. Sickle cell disease activates peripheral blood mononuclear cells to induce cathepsins K and V activity in endothelial cells. Anemia. 2012;2012:201781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keegan PM, Wilder CL, Platt MO. Tumor necrosis factor alpha stimulates cathepsin K and V activity via juxtacrine monocyte-endothelial cell signaling and JNK activation. Mol Cell Biochem. 2012;367:65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chiang T, Messing RO, Chou WH. Mouse model of middle cerebral artery occlusion. J Vis Exp. 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Atochin DN, Murciano JC, Gursoy-Ozdemir Y, Krasik T, Noda F, Ayata C, Dunn AK, Moskowitz MA, Huang PL, Muzykantov VR. Mouse model of microembolic stroke and reperfusion. Stroke. 2004;35:2177–2182. [DOI] [PubMed] [Google Scholar]
- 27.Schaffer CB. Optical tools to produce and study small strokes in animal models. Conf Proc IEEE Eng Med Biol Soc. 2010;2010:3377–3378. [DOI] [PubMed] [Google Scholar]
- 28.Keegan PM, Anbazhakan S, Kang B, Pace BS, Platt MO. Biomechanical and biochemical regulation of cathepsin k expression in endothelial cells converge at AP-1 and NF-KappaB. Biol Chem. 2016;397:459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Belcher JD, Marker PH, Weber JP, Hebbel RP, Vercellotti GM. Activated monocytes in sickle cell disease: Potential role in the activation of vascular endothelium and vaso-occlusion. Blood. 2000;96:2451–2459. [PubMed] [Google Scholar]
- 30.Wang R, Raykin J, Li H, Gleason RL Jr., Brewster LP. Differential mechanical response and microstructural organization between non-human primate femoral and carotid arteries. Biomech Model Mechanobiol. 2014;13:1041–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hadeed K, Hascoet S, Castex MP, Munzer C, Acar P, Dulac Y. Endothelial function and vascular properties in children with sickle cell disease. Echocardiography. 2015;32:1285–1290. [DOI] [PubMed] [Google Scholar]
- 32.Buch K, Arya R, Shah B, Nadgir RN, Saito N, Qureshi MM, Sakai O. Quantitative analysis of extracranial arterial tortuosity in patients with sickle cell disease. J Neuroimaging. 2017;27:421–427. [DOI] [PubMed] [Google Scholar]
- 33.Bernaudin F, Verlhac S, Arnaud C, Kamdem A, Vasile M, Kasbi F, Hau I, Madhi F, Fourmaux C, Biscardi S, Epaud R, Pondarre C. Chronic and acute anemia and extracranial internal carotid stenosis are risk factors for silent cerebral infarcts in sickle cell anemia. Blood. 2015;125:1653–1661. [DOI] [PubMed] [Google Scholar]
- 34.Guilliams KP, Fields ME, Ragan DK, Chen Y, Eldeniz C, Hulbert ML, Binkley MM, Rhodes JN, Shimony JS, McKinstry RC, Vo KD, An H, Lee JM, Ford AL. Large-vessel vasculopathy in children with sickle cell disease: A magnetic resonance imaging study of infarct topography and focal atrophy. Pediatr Neurol. 2017;69:49–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Charrin E, Ofori-Acquah SF, Nader E, Skinner S, Connes P, Pialoux V, Joly P, Martin C. Inflammatory and oxidative stress phenotypes in transgenic sickle cell mice. Blood Cells Mol Dis. 2016;62:13–21. [DOI] [PubMed] [Google Scholar]
- 36.Adams RJ, McKie VC, Vichinsky E, Pegelow C, Abboud M, Gallagher D, Kutlar A, Nichols FT, Bonds DR, Brambilla D. Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial doppler ultrasonography. N Engl J Med. 1998;339:5–11. [DOI] [PubMed] [Google Scholar]
- 37.Gueguen A, Mahevas M, Nzouakou R, Hosseini H, Habibi A, Bachir D, Brugiere P, Lionnet F, Ribei JA, Godeau B, Girot R, Ibrahima V, Calvet D, Galacteros F, Bartolucci P. Sickle-cell disease stroke throughout life: A retrospective study in an adult referral center. Am J Hematol. 2014;89:267–272. [DOI] [PubMed] [Google Scholar]
- 38.DeBaun MR, Kirkham FJ. Central nervous system complications and management in sickle cell disease. Blood. 2016;127:829–838. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.