

Abstract
Regulatory T cells (Tregs) are abundant ion human and mouse pancreatic cancer. To understand the contribution to the immunosuppressive microenvironment, we depleted Tregs in a mouse model of pancreatic cancer. Contrary to our expectations, Treg depletion failed to relieve immunosuppression, and led to accelerated tumor progression. We show that Tregs are a key source of TGFβ ligands and, accordingly, their depletion reprogramed the fibroblast population, with loss of tumor-restraining, smooth muscle actin-expressing fibroblasts (myCAFs). Conversely, we observed an increase in chemokines Ccl3, Ccl6, and Ccl8 leading to increased myeloid cell recruitment, restoration of immune suppression and promotion of carcinogenesis, an effect that was inhibited by blockade of the common CCL3/6/8 receptor CCR1. Further, Treg depletion unleashed pathological CD4+ Tcell responses. Our data points to new mechanisms regulating fibroblast differentiation in pancreatic cancer and supports the notion that fibroblasts are a heterogeneous population with different and opposing functions in pancreatic carcinogenesis.
Keywords: Pancreatic Cancer, Pancreatic Intraepithelial Neoplasia, Regulatory T cells, Immunosuppression, Fibroblast, Myeloid cell, Tumor Associated Macrophage, C-C motif Chemokine Ligands, C-C Motif Chemokine Receptor 1, CyTOF
Introduction
Pancreatic ductal adenocarcinoma (PDA) is one of the deadliest human malignancies with a 5-year overall survival rate of ~9% (1). PDA and its most common precursor lesions, Pancreatic Intraepithelial Neoplasia (PanIN), are characterized by an extensively fibrotic microenvironment, which includes abundant infiltrating immune cells. Success in targeting the pancreatic cancer microenvironment has been inconsistent, and immunotherapy approaches such as immune checkpoint inhibition have largely failed (2,3).
Oncogenic mutations are prevalent in pancreatic cancer (4–6) and are present with high frequency in precursor lesions (7). Mouse models genetically engineered to express oncogenic Kras in the pancreas –known as KC mice– develop PanIN lesions that progress to invasive cancer over time (8,9). The formation of PanIN is accompanied by infiltration of suppressive immune cells (10), while cytotoxic CD8+ T cells are rare. Among these, myeloid cells are present in high abundance in the microenvironment, including both macrophages and myeloid derived suppressor cells/immature myeloid cells (10–16).
This immune suppression is functionally important, as activation of a CD8+ T cell mediated immune response is sufficient to block the onset of carcinogenesis (17–19) or induce tumor regression (11,18,20–22). Genetic elimination of CD4+ T cells results in the activation of CD8+ T cells and consequently prevents PanIN progression (17). Similarly, depletion of Th17 cells, a CD4+ T cell subset, inhibits pancreatic carcinogenesis (23). Further, analysis of long-term pancreatic cancer survivors reveal that anti-tumor T cells persist in their peripheral blood years after the initial diagnosis, further supporting the notion that activation of an anti-tumor immune response might be effective (24). However, our limited understanding of the mechanisms underlying immune suppression in pancreatic cancer limits our ability to translate these findings to the clinic.
Regulatory T cells (Tregs), defined as CD4+CD25+Foxp3+ T cells, accumulate in mouse and human PanIN and pancreatic cancer (10,17,25). Moreover, Treg frequency positively correlates with tumor metastasis and poor prognosis in human PDA patients (26,27). Depletion of Tregs has been tested in a model of transplanted pancreatic cancer in mice, where it led to a CD8+ T-cell mediated anti-tumor immune response (21). However, the role of Tregs during the onset and progression of pancreatic cancer, as well as in spontaneous invasive tumors, had not been investigated, and is the focus of our study.
Results
Regulatory T cells are abundant in human PanIN and pancreatic cancer
To study the relative abundance of Tregs, myeloid cells and CD8+ T cells in human pancreatic cancer, we performed Cytometry by Time-of-Flight (CyTOF) analysis. We included 3 surgical samples from Whipple resections and 2 endoscopic ultrasound-guided core needle biopsy samples. High-dimensional analysis using FlowSOM-viSNE (28,29) revealed the presence of a CD45+CD3+CD4+CD25+ Treg population and several subsets of myeloid cells in each sample (Fig 1A and S1A) while CD8+ T cells were rare (Fig. 1A and S1A). To determine the localization of immune cells within the tissue, we have recently optimized the use of fluorescent multiplex immunohistochemistry system (Opal Multiplex IHC, PerkinElmer) for tumor immunophenotyping (30). Using this platform, we stained 39 human pancreatic cancer samples, 37 PanIN samples, and 52 adjacent normal or chronic pancreatitis samples −a non-cancerous inflammatory condition of the pancreas. Out of 52 chronic pancreatitis samples, only 14 had measurable Tregs (defined as CD3+ CD8− FoxP3+). In contrast, Tregs were present in most of the tumor samples and often observed in close proximity of the tumor cells (Fig. 1B and S1B); their abundance positively correlated with macrophages in the same tissue (Fig. S1C) and −with two exceptions– with CD8+ T cells (Fig. S1C). Interestingly, Tregs were abundantly present in the PanIN samples as well, possibly indicating a role during the early stages of carcinogenesis (Fig. 1B, S1D and S1E). Similarly, in PanIN-bearing Kras+/LSL-G12D;Ptf1a+/Cre (KC) mice (8) we observed abundant Treg infiltration by Foxp3 immunostaining (Fig. 1C). To further characterize the Treg population, we performed single cell sequencing analysis of spontaneous PanIN lesions in iKras* mice (31), and iKras*;p53* (32) pancreatic cancer cells orthotopically transplanted in syngeneic mice. In both experimental systems, we detected a cell population co-expressing Cd4, Cytotoxic T-lymphocyte associated protein 4 (Ctla4), Foxp3 and Interleukin-2 receptor alpha (Il2ra), (Fig. 1D and Fig. S1F). Similarly, in human samples (data not shown), Foxp3 was co-expressed with CTLA4 and IL2RA, thus in bona fide Tregs [for a review of Treg classification, see (33)],
Figure 1. Regulatory T cells are prevalent in human PDA and PanINs.
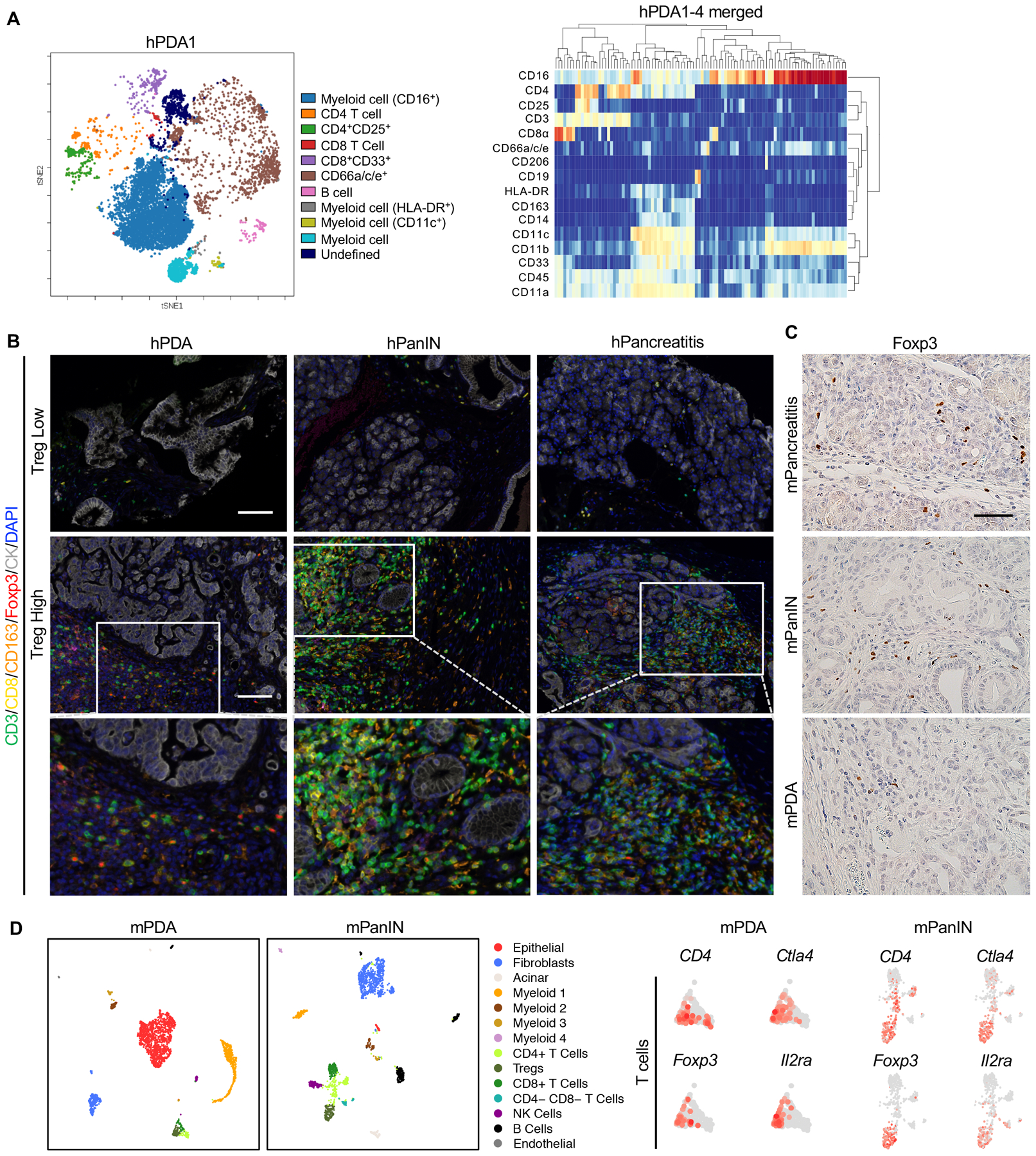
(A) CyTOF immune profiling by FlowSOM-viSNE of human pancreatic tumors demonstrating the presence of CD4+CD25+ Tregs. (B) Representative images of Opal staining on human pancreatic lesion samples. Scale bar 100 μm. (C) Immunohistochemistry staining for Foxp3 staining in mouse pancreatic tissues. Scale bar 50 μm. (D) UMAP plots of single cell RNA sequencing analysis with mouse orthotopic pancreatic cancer samples or PanIN lesions, color-coded by their associated cluster (left) or color-coded for expression (gray to red) of Cd4, Ctla4, Foxp3 and Il2ra.
Treg depletion accelerated pancreatic carcinogenesis.
To study the effect of Treg depletion on the formation of PanIN, we generated Kras+/LSL-G12D; Ptf1a+/Cre;Foxp3tm3(DTR/GFP)Ayr (KC;Foxp3DTR) mice (Fig. 2A) (8,34). In these animals, Tregs can be depleted at will by administering diphtheria toxin (DT). During tissue homeostasis, Tregs play an important role in regulating the activity of the immune system and preventing autoimmune disease (34). Accordingly, depletion of Tregs for 3 weeks, starting at 4–5 weeks of age (Fig. 2B) in Foxp3DTR mice led to systemic inflammation shown by hypertrophy of the lymphoid organs, such as increased spleen to body weight ratio (Fig. 2C, Foxp3DTR mice). In Foxp3DTR pancreata, Treg depletion resulted in acinar cell loss (Fig. 2D) and pancreatitis with distinct histologic changes such as acinar-ductal metaplasia (ADM) –de-differentiation of acinar cells to duct-like cells (Fig. 2D, E)– and elevated immune infiltration (see immunostaining for CD45 in Fig. S2A) with characteristic presence of CD138+ plasma cells (Fig. S2B). In KC;Foxp3DTR mice, Treg depletion led to increased spleen to body weight ratio, as well as increased pancreas to body weight ratio, a measure of tumor burden (Fig. 2C). As expected, KC pancreata had sparse lesions, consistent with their young age (Fig. 2D histopathological quantification and Fig. 2E). In contrast, Treg depletion in KC;Foxp3DTR mice presented with extensive ADM and PanIN (Fig. 2D–E and S2A, PAS staining), as well as abundant immune cell infiltration (Fig. S2A).
Figure 2. Treg depletion results in pancreatitis and promotes PanIN formation and progression.
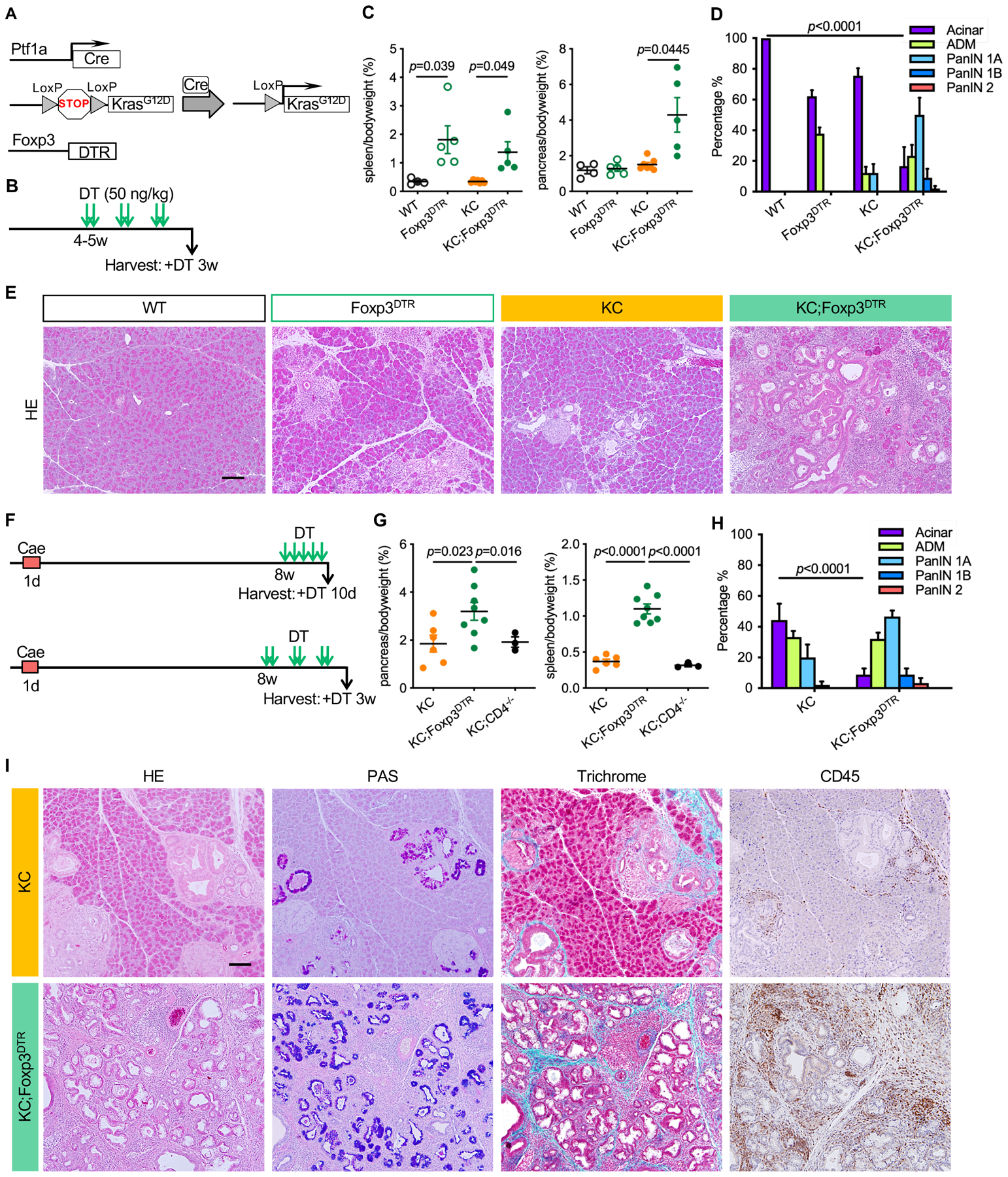
(A) Genetic makeup of the KC;Foxp3DTR mouse model. (B) Experimental design, n=4–7mice/cohort. (C) Pancreas to body weight ratio and spleen to body weight ratio of WT, Foxp3DTR, KC and KC;Foxp3DTR mice after 3 weeks of DT treatment. Data represent mean ± SEM, n=4–7mice/cohort. The statistical difference was determined by two-tailed t-tests. (D) Histopathologic quantification of WT, Foxp3DTR, KC and KC;Foxp3DTR mice after 3 weeks of DT treatment. Data represent mean ± SEM, n = 3–4 mice/cohort. The statistical difference was determined by two-way ANOVA. (E) H&E staining of WT, Foxp3DTR, KC and KC;Foxp3DTR pancreata after 3 weeks of DT treatment. Scale bar 100 μm. (F) Experimental design, n=3–8 mice/cohort. (G) Pancreas to body weight ratio and spleen to body weight ratio of KC, KC;Foxp3DTR and KC;CD4−/− mice that received 3 weeks of DT treatment starting 8 weeks post caerulein. Data represent mean ± SEM, n=3–8 mice/cohort. The statistical difference was determined by two-tailed t-tests. (H) Histopathologic quantification of KC and KC;Foxp3DTR mice received 3 weeks of DT treatment starting 8 weeks post caerulein.. Data represent mean ± SEM, n=3–4mice/cohort. The statistical difference was determined by two-way ANOVA. (I) H&E staining, Periodic acid–Schiff (PAS) staining, Gomori trichrome staining and immunohistochemistry staining for CD45 in KC and KC;Foxp3DTR mice that received 3 weeks of DT treatment starting 8 weeks post caerulein. Scale bar 100 μm.
We reasoned that the pancreatitis caused by Treg depletion synergized with oncogenic Kras to promote PanIN formation as shown in other experimental models of pancreatitis (35–40). We then modified the experimental design to address the role of Tregs in pre-existing PanIN. We administered Caerulein (a cholecystokinin agonist) by 8 hourly intraperitoneal injections for one day to induce acute pancreatitis in 6-week old KC and KC;Foxp3DTR mice. KC;CD4−/− mice were included as an additional control, as we have previously shown that lesions in CD4 ablated animals fail to progress because of productive anti-tumor immunity (17). The mice were allowed to develop PanIN lesions for 8 weeks post caerulein treatment and then received DT for 10 days or 3 weeks (Fig. 2F). The two cohorts showed a similar phenotype. Following this treatment, KC;Foxp3DTR mice had increased pancreas/body ratio compared with either KC or KC;CD4−/− mice (Fig. 2G). Histological analysis of the pancreata showed acinar areas with interspersed lesions in KC and KC;CD4−/− mice. In contrast, KC;Foxp3DTR mice had widespread lesions that almost completely replaced the pancreas parenchyma (Fig. 2H–I and S2C).
Treg depletion alters the fibroblast populations in PanIN lesions.
PanIN lesions, like pancreatic cancer, are surrounded by a fibrotic microenvironment. Accordingly, α-smooth muscle actin (SMA) staining of KC pancreata revealed characteristic areas of SMA+ cells surrounding the epithelial lesions (Fig. 3A and B). Intriguingly, lesions in KC;Foxp3DTR pancreata lacked SMA expression, whether carcinogenesis was or was not accelerated by the induction of pancreatitis (Fig. 3A and B), while immunostaining for the fibroblast marker PDGFRβ appeared unchanged (Fig. 3B). We then counted PDGFRβ+ cells and SMA+ cells in KC and KC;Foxp3DTR pancreata, and calculated their ratio. The data revealed no change in total fibroblast numbers, but a reduction in SMA+ fibroblasts, a finding that is consistent with reprogramming of the fibroblast population (Fig. 3B). Consistent with the immunostaining data, α-SMA (Acta2) mRNA expression was lower in KC;Foxp3DTR compared to KC pancreata. Extracellular matrix (ECM) genes such as Collagen Type I (Col1) and Fibronectin 1 (Fn1), as well as genes related to fibroblast activation and ECM synthesis such as Transforming growth factor β1 (Tgfβ1) and Connective tissue growth factor (Ctgf) were also down-regulated in KC;Foxp3DTR compared to KC pancreata (Fig. 3C). We identified the cellular source of Tgfβ1 and putative responsive cells expressing the Tgfβ receptors by single-cell RNA sequencing of mouse PanIN and pancreatic cancer. Tgfb1 was expressed by both epithelial cells and T cells, including Tregs. Conversely, the three Tgfβ receptors were expressed in the majority of fibroblasts, as well as a subset of epithelial cells (Fig. 3D). Supporting these findings, TCGA database (https://www.cbioportal.org/datasets) analysis revealed a positive correlation between a Treg signature and expression of ACTA2 and TGFB signaling genes in human tumors (Fig. S1G–I). SMAhigh fibroblasts define the “myofibroblastic CAFs” (myCAFs) population of cancer-associated fibroblasts, a phenotype that is driven by Tgfβ (41,42). Our data is consistent with the notion that Tregs are a source of Tgfβ and drive myCAF differentiation of fibroblasts.
Figure 3. Treg depletion inactivates stromal fibroblasts.
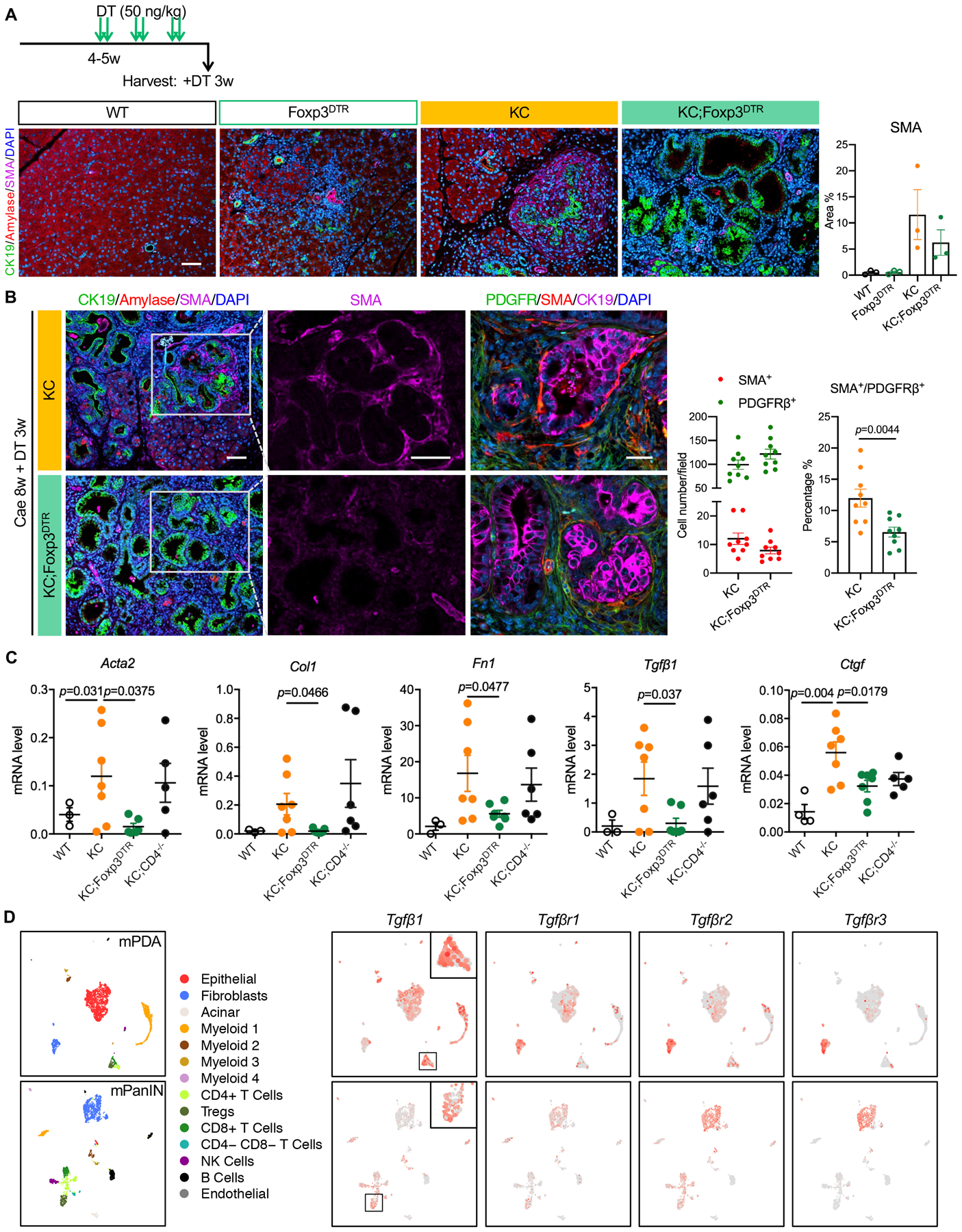
(A) Experimental design (n=4–7 mice/cohort) and co-immunofluorescent staining for CK19 (green), Amylase (red), SMA (magenta) and DAPI (blue) in WT, Foxp3DTR, KC and KC;Foxp3DTR pancreata after 3 weeks of DT treatment. Scale bar 100 μm. Quantification of SMA positive area is shown on the right. Data represent mean ± SEM, n=3 slides/cohort. (B) co-immunofluorescent staining for CK19 (green), Amylase (red), SMA (magenta) and DAPI (blue). Scale bar 100 μm. Quantification of SMA and PDGFRβ positive cells is shown on the right. Data represent mean ± SEM, n=9 images/cohort. The statistical difference was determined by two-tailed t-tests. (C) qRT-PCR for α-SMA (Acta2), Col1, Fn1, Tgfβ1 and Ctgf expression in WT control, KC, KC;Foxp3DTR and KC;CD4−/− pancreata. Mice received DT treatment following 8 weeks post caerulein. Data represent mean ± SEM, n = 3–7 mice/cohort. The statistical difference was determined by two-tailed t-tests. (D) UMAP plots of single cell RNA sequencing analysis with mouse orthotopic pancreatic cancer samples or PanIN lesions, color-coded by their associated cluster (left) or color-coded for expression (gray to red) of Tgfβ1, Tgfβr1, Tgfβr2 and Tgfβr3.
Immunosuppressive myeloid cells increase upon Treg depletion.
We then set out to determine the effect of Treg depletion on the immune microenvironment. To compare the immune infiltration among different groups, we devised a protocol that would result in a similar lesion load in the different genotypes, namely KC, KC;CD4−/− and KC;Foxp3DTR. In brief, adult mice received 8 hourly caerulein injections in a day. Eight weeks later, they were treated with DT for 1 week and then harvested for histology and flow cytometry (Fig. 4A and S2D). PanIN lesions in mice lacking CD4+ T-cell regress over time in a CD8+ T cell-mediated manner (17). Consistently, KC;CD4−/− mice had increased CD8+ T cells and increased Interferon γ (IFNγ) expressing CD8+ T cells compared to KC mice (Fig. 4B). In contrast, in KC;Foxp3DTR pancreata we observed only a modest increase in CD8+ T cells and no change in IFNγ+;CD8+ T cells compared to KC mice. In contrast, we observed an overall increase in immune cells (CD45+) and an increase in CD4+ T cells, as well as the expected loss of FoxP3+ cells. These data indicated that little/no productive immune response was elicited, notwithstanding the depletion of Tregs. By flow cytometry, total myeloid cells (CD11b+) and macrophages (CD11b+F4/80+) (% of total cells) trended towards an increase, while immature myeloid cells/myeloid-derived suppressor cells (MDSCs) (CD11b+Ly6C+Ly6G+F4/80−) were elevated in KC;Foxp3DTR and KC;CD4−/− pancreata compared to KC (Fig. 4C). Immunostaining of KC and KC;Foxp3DTR pancreas tissue showed an increase in CD8+ T cells in proximity of the lesions, but we also observed a comparable increase in F4/80+ macrophages in those areas (Fig. S2E). Myeloid cells have a known immune suppressive function (11,13,18), thus we hypothesized that an increase in their number or suppressive potential might explain the difference in phenotype between Treg depletion and CD4 elimination during pancreatic carcinogenesis.
Figure 4. Characterization of pancreatic immune infiltrates.
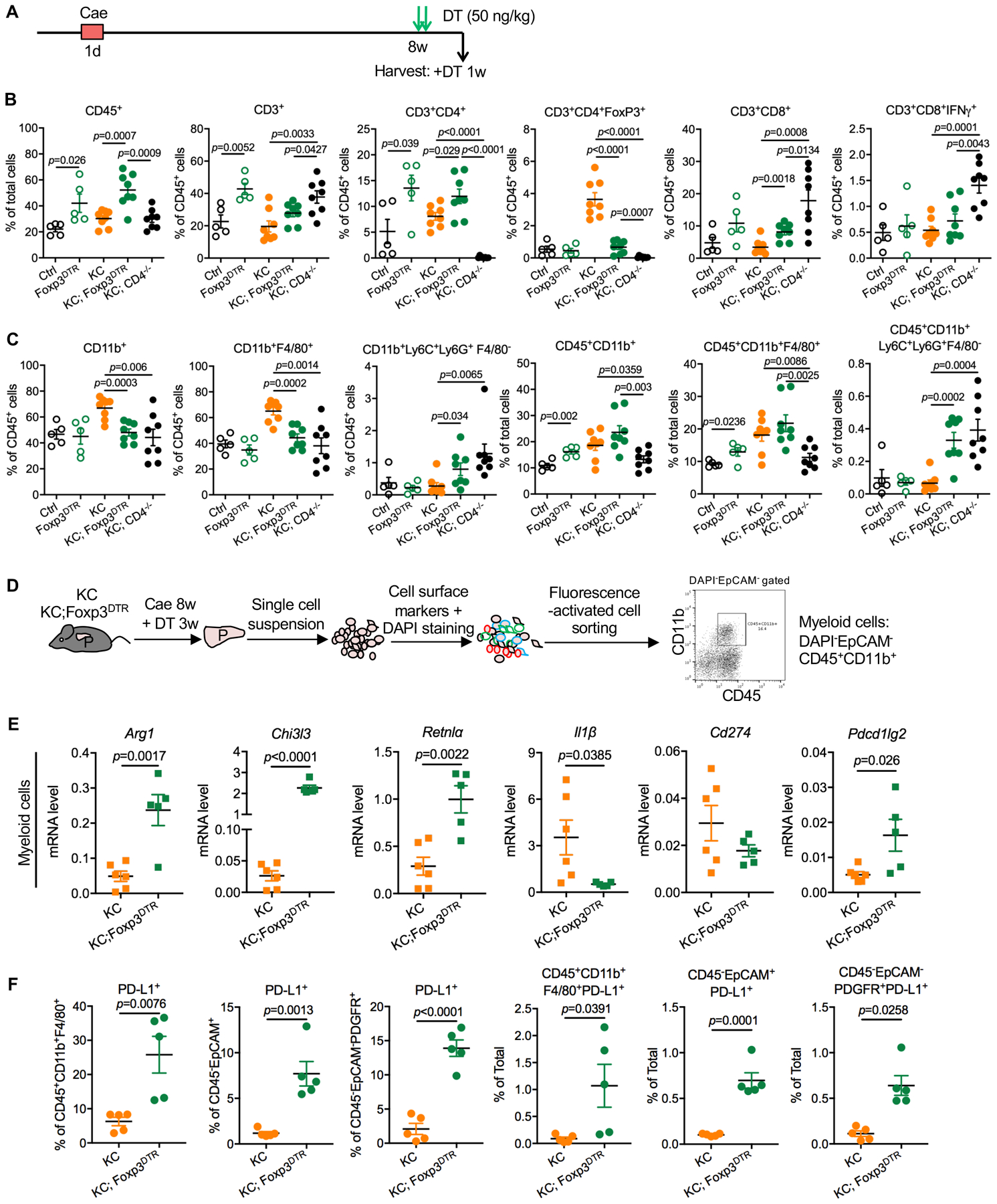
(A) Experimental design, n=4–8 mice/cohort. (B) WT, Foxp3DTR, KC, KC;Foxp3DTR and KC;CD4−/− mice received 1 week DT treatment following 8 weeks post pancreatitis induction. CD45+ leukocytes, CD3+ T cells, CD3+CD4+ T cells, CD3+CD4+FoxP3+ Tregs, CD3+CD8+ T cells, CD3+CD8+IFNγ+ T cells and (C) CD11b+ myeloid cells, CD11b+F4/80+ macrophages and CD11b+Ly6C+Ly6G+F4/80− MDSCs from pancreata were measured by flow cytometry as percentage of total cells or percentage of total leukocytes. Data represent mean ± SEM, the statistical difference between experimental groups was determined by two-tailed t-tests. (D) Schematic illustration of pancreatic infiltrating myeloid cells extraction by fluorescence-activated cell sorting and a representative flow cytometry plot showing the gating strategy. (E) qRT-PCR for Arg1, Chi3l3, Retnl⍺, Il1β, Cd274 and Pdcd1lg2 expression in pancreatic myeloid cells derived from KC and KC;Foxp3DTR mice that received 3-week DT treatment following 8 weeks post caerulein. Data represent mean±SEM, n=5–6. The statistical difference was determined by two-tailed t-tests. (F) The percentage of PD-L1 expressing macrophages, epithelial cells and fibroblasts in KC and KC;Foxp3DTR pancreata were measured by flow cytometry. Data represent mean±SEM, n=5. The statistical difference was determined by two-tailed t-tests.
To assess the changes in the myeloid compartment upon Treg depletion, we performed a comparative transcriptomic analysis of the myeloid cells (DAPI−EpCAM−CD45+CD11b+) sorted from KC and KC;Foxp3DTR tumors (Fig. 4D). The RNAseq results, including Principal Component Analysis (PCA) plot and a volcano plot, illustrating differential gene expression in myeloid cells are shown in Fig. S3A and B. Interestingly, we detected an increase in Arg1, Chi3l3 (also known as Ym1), Retnlα and Programmed cell death 1 ligand 2 (Pdcd1lg2) expression in myeloid cells derived from KC;Foxp3DTR pancreata, while Interleukin-1 (Il1) β was down-regulated (Fig. 4E). Further, we observed an increase in Arginase1 and Chil3l3 expression −markers of tumor associated macrophages (TAM) with immunosuppressive function (43,44)− in both fibroblasts and macrophages (Fig. S2E). By flow cytometry, we observed that cell surface Programmed death-ligand 1 (PD-L1) protein expression was elevated in TAMs (CD45+CD11b+F4/80+), epithelial cells (CD45−EpCAM+) and fibroblasts (CD45−EpCAM−PDGFRα+) in KC;Foxp3DTR compared to KC pancreata (Fig. 4F). Taken together, our data indicates that a compensatory immunosuppressive program is activated upon Treg depletion in mice bearing PanIN lesions.
The changes in expression of individual genes could be explained either by gene expression changes across the entire myeloid population, or by changes in the composition of cells types within the myeloid population. To evaluate these possibilities, we proceeded to immunophenotype our samples by CyTOF with 16 validated antibodies (Table S1). viSNE analysis suggested there were at least 11 different populations in our dataset (Fig. 5A, B and Fig. S3C) and FlowSOM analysis with supervised hierarchical clustering identified 21 separate populations (Fig. 5C). Among these, we identified multiple myeloid subpopulations, including monocytic myeloid-derived suppressor cells (Ly6C+), granulocytic MDSCs (Ly6G+), infiltrating monocytes (CD11b+), and macrophage/dendritic cell subsets (F4/80dim/+ and CD11c+, respectively). Interestingly, we found that both the monocyte/MDSC and macrophage subpopulations could be further divided based on the intracellular expression of arginase 1 (population 5 versus 6 and populations 15–18 versus 19–21). When we analyzed the frequency of each subpopulation in the KC versus KC;FoxP3DTR mice, we found a consistent increase in the frequency of Arg1+ myeloid subsets after Treg depletion (Fig. S3D). By flow cytometry, we found that the percentage of CD206−iNOS+ classically activated macrophages remained unchanged, while, CD206+Arg1+ macrophages increased more than 8-fold upon Treg depletion (Fig. 5D), compared to KC control. More interestingly, we found a non-immune cell population (CD45−CD11b+)−possibly a subset of stromal fibroblasts– also expressed higher level of Arg1 in KC;Foxp3DTR pancreata upon Treg depletion (Fig. 5E). Therefore, Treg depletion resulted in compensatory increase in a prevalently myeloid-driven immune suppression program.
Figure 5. Tumor associated macrophages exhibit high immunosuppressive capacity upon Treg depletion.
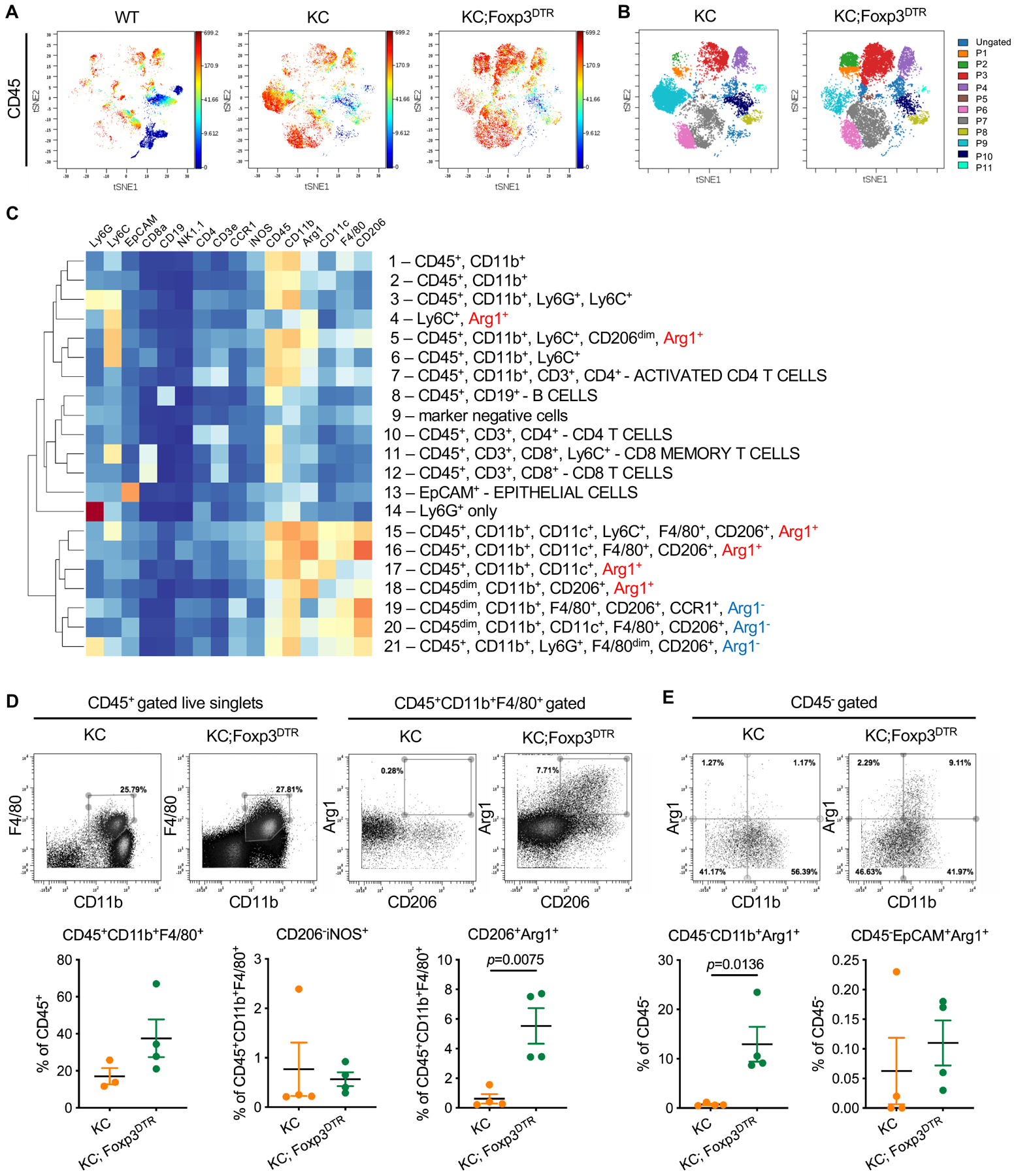
(A) Representative viSNE plots (dot plots colored by CD45 channel) of WT, KC and KC;Foxp3DTR pancreata that received 1-week DT treatment following 8 weeks post caerulein. (B) Immune cell populations identified by manual gating in viSNE maps, and (C) by supervised clustering of SPADE-identified subpopulations. (D) Top: representative CyTOF dot plots showing gating strategy defining tumor associated macrophages and CD206+Arg1+ M2-like macrophage subset. Bottom: quantification of the total macrophage and CD206−iNOS+ M1-like and CD206+Arg1+ M2-like macrophage subsets. (E) Representative CyTOF dot plots showing gating strategy defining CD11b+Arg1+ non-immune cell population, and quantification of the Arg1+ non-immune cell populations. Data represent mean±SEM, n=3–4. The statistical difference between KC and KC;Foxp3DTR pancreata was determined by two-tailed t-tests.
Treg depletion results in compensatory immune suppression in late-stage disease.
Treg depletion was recently reported to induce anti-tumor immune responses and inhibit tumor growth in a transplantation model of pancreatic cancer (21). We thus reasoned that either the stage of disease (onset vs advanced disease) or different models (spontaneous vs transplanted tumors) explained the divergent findings. To investigate this apparent inconsistency, we implanted a KPC (Ptf1aCre; LSL-Kras; P53R172H/+) (45) pancreatic cancer cell line (7940B) (46) orthotopically into syngeneic C57BL/6 Foxp3DTR mice and depleted Tregs 11 days later (Fig. S4A). At harvest (day 20) we observed smaller tumors in the Foxp3DTR cohort (Fig. S4B), and an increase in the expression of CD8+ T cell activation markers Ifnγ, Granzyme B (Gzmb) and Perforin (Prf1) (Fig. S4C). Consistent with activation of anti-tumor immunity, concurrent depletion of CD8+ T cells partially rescued tumor growth (Fig. S4B). Flow cytometry analysis revealed increased CD8+ T cells and myeloid cells, including macrophages, upon Treg depletion (Fig. S4D). Immunostaining confirmed increased immune infiltration and apoptosis in Treg depleted tumors (Fig. S4E). At the same time, we observed fewer SMA positive fibroblasts and more expression of immunosuppressive factors Chi3l3 (notably in tumor epithelial cells) and Arg1 in Foxp3DTR tumors (Fig. S4E and F), which were consistent with our findings in the KC;Foxp3DTR model. We then repeated a similar set of experiments in KPC;Foxp3DTR (Ptf1aCre; LSL-Kras; P53R172H/+; Foxp3DTR) mice that develop spontaneous invasive pancreatic cancer. Upon detection of a tumor by ultrasound imaging, we administered DT to deplete Tregs (or used vehicle control) and followed tumor growth (Fig. S4G). In this setting, we observed continued tumor growth even following Treg depletion. Upon harvesting and immunostaining, we observed both an increase in CD8+ T cells and an increase in myeloid cells (Fig. S4G). Our data are consistent with Treg depletion failing to elicit a productive anti-tumor immune response at late stage disease, likely due to compensatory myeloid-driven immune suppression.
Multiple CCR1 ligands are upregulated in epithelial cells and fibroblasts upon Treg depletion.
To elucidate a potential causal link between the changes in the fibroblast and epithelial compartments and the shift in immune composition upon Treg depletion, we flow sorted epithelial cells (CD45−EpCAM+) and fibroblasts (CD45−EpCAM−PDGFRα+) from KC and KC;Foxp3DTR pancreata (n=3 for each group) (Fig. 6A, B) and performed RNA sequencing. We then analyzed the secretome transcripts in both compartments. Interestingly, overall gene expression, as well as expression of secreted factors, showed a distinct pattern in both epithelial cells and fibroblasts upon Treg depletion (Fig. 6C and Fig. S5A, B). Among more than 400 differentially expressed secreted chemokines, cytokines and immunosuppressive factors, we observed that the C-C motif chemokine ligands Ccl3, Ccl6, Ccl8, (Fig. 6D) were among the top upregulated secreted factors in fibroblasts and/or epithelial cells upon Treg depletion, while interleukins such as Il1β were downregulated but to a lesser extent compared to what we saw earlier in the myeloid cell compartment (Fig. 4E). We validated these changes by qRT-PCR in 4–8 samples/genotype (Fig. 6E and S5C). Finally, immunostaining confirmed upregulated expression of CCL8 in KC;Foxp3DTR pancreata (Fig. 6F) compared to KC. Other notable changes in epithelial cells and fibroblasts were an increase in immunosuppressive factors such as Arg1 and Chi3l3, as well as immune check point molecules Cd274 (also known as PD-L1) and Pdcd1lg2 (Fig. 6E and S5C).
Figure 6. Gene expression profiling for pancreatic epithelial cells and fibroblasts upon Treg depletion.
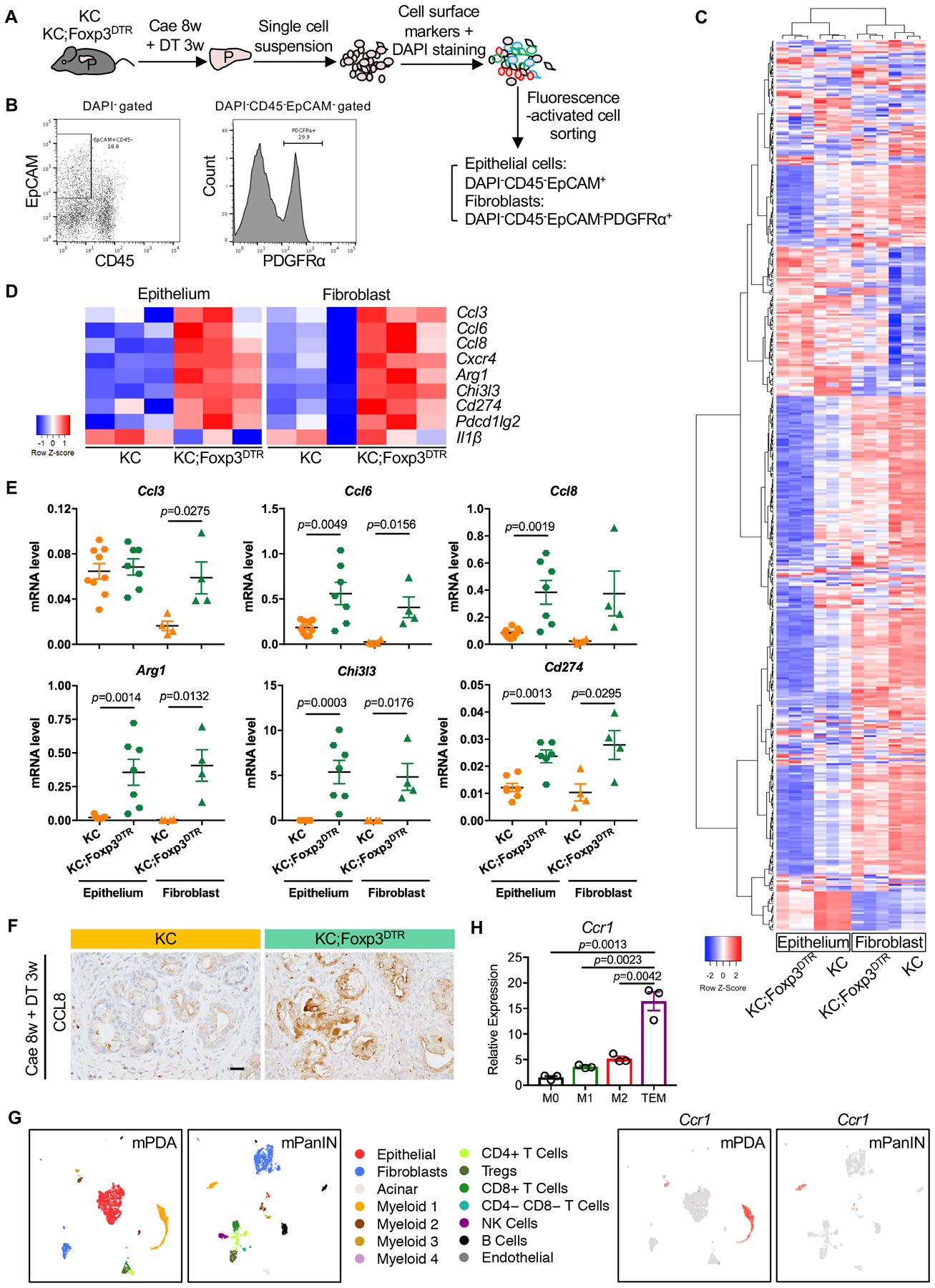
(A) Schematic illustration of pancreatic epithelial cells and fibroblasts extraction by fluorescence-activated cell sorting. (B) Representative flow cytometry plots showing gating strategy to identify epithelial cells and fibroblasts. (C) Heat map showing differentially expressed secretome genes and (D) Top differentially expressed secretome genes in epithelial cells or fibroblasts between KC and KC;Foxp3DTR pancreata. (E) qRT-PCR for Ccl3, Ccl6, Ccl8, Arg1, Chi3l3 and Cd274 expression in pancreatic epithelial cells or fibroblasts derived from KC and KC;Foxp3DTR mice that received 3 weeks of DT treatment following 8 weeks post caerulein. Data represent mean ± SEM, n=4–9. (F) Immunohistochemistry staining for CCL8 in KC and KC;Foxp3DTR pancreata. Scale bar 50 μm. (G) UMAP plots of single cell RNA sequencing analysis with mouse orthotopic pancreatic cancer samples or PanIN lesions, color-coded by their associated cluster (left) or color-coded for expression (gray to red) of Ccr1. (H) qRT-PCR for Ccr1 expression in bone marrow derived macrophages under different polarization status. Data represent mean ± SEM, n=3. The statistical difference was determined by two-tailed t-tests.
CCL3, CCL6, and CCL8 bind a common receptor, CCR1, and they are known chemo-attractants for myeloid cells (47). We identified 4 subsets of myeloid cells in PanINs and 3 in PDA by Single-cell sequencing analysis (Fig. S1F). Myeloid 1 = MDSCs (high expression of Itgam, Cd14, Fcgr3, S100a8, Ly6g); myeloid 2 = macrophages (high expression of Itgam, Cd68, Adgre1, Mrc1); myeloid 3 = dendritic cells (high expression of Itgax, H2-Eb1, Batf3, Itgae, Clec9a) and myeloid 4 = monocytes (high expression of Itgax, Ly6c2). Of note, myeloid 4 was not detected in tumors, but only in PanIN samples. Single-cell sequencing analysis of mouse PDA or PanINs confirmed that expression of Ccr1 was restricted to the myeloid compartment and included MDSCs and macrophages (Fig. 6G). We validated these findings in vitro by qRT-PCR from bone-marrow-derived macrophages (M0) polarized to M1 with lipopolysaccharide (LPS), to M2 with Interleukin-4 (IL4) and to Tumor Educated Macrophages (TEMs) with pancreatic cancer cell conditioned media. Ccr1 expression was lowest in M1 macrophages, and highest in TEMs (Fig. 6H). Our single cell sequencing analysis also revealed that myeloid cells are the main source of Il1α both in PanINs and in pancreatic cancer, while the receptor Il1r is expressed in a subset of epithelial cells and in the vast majority of fibroblasts (Fig S5D). The expression of Il1α was not different in KC and KC;FoxP3DTR myeloid cells, epithelial cells and fibroblasts (Fig. S5E). IL1α drives differentiation of “inflammatory CAFs” (iCAFs), a pro-inflammatory subset of fibroblasts which also expresses elevated IL6 (41), and Il6 was similarly unchanged in our model (data not shown). Thus, while fibroblasts in KC;FoxP3DTR become more inflammatory at least pertaining recruitment of myeloid cells, they might not fit exactly in the iCAF subtype, consistent with the complexity of this cell type (42,48).
CCR1 inhibition rescues Treg depletion-induced PanIN progression.
Our data to this point demonstrated that Treg depletion resulted in an increase in several CCR1 ligands, and a corresponding influx of immune-suppressive myeloid cells. To determine whether CCR1 activation is responsible for the increased myeloid infiltration and increased tumorigenesis, we utilized a commercial CCR1 inhibitor, BX471 (hereby termed CCR1i). We treated PanIN-bearing KC;Foxp3DTR mice with CCR1i for a week and at the same time we depleted Tregs (Fig. 7A). At endpoint, KC;Foxp3DTR mice that received DT and vehicle control showed enlarged pancreata and spleens compared to KC mice, an effect that was reversed by CCR1i treatment (Fig. S6A). Further, histopathological analysis revealed more acini and less ADM/PanIN (CK19+ cells) in KC;Foxp3DTR mice treated with CCR1i (Fig. 7B and S6B). Immunostaining for CD45-F4/80 showed decreased total immune infiltration and macrophages (Fig. 7B and S6C), although the level of immune infiltration remained elevated compared to KC mice (data not shown). Thus, CCR1 inhibition bypassed Treg-depletion acceleration of carcinogenesis in KC mice driven by infiltration of immunosuppressive myeloid cells.
Figure 7. CCR1 inhibition and CD4+ T cell depletion abrogate Treg depletion induced PanIN progression.
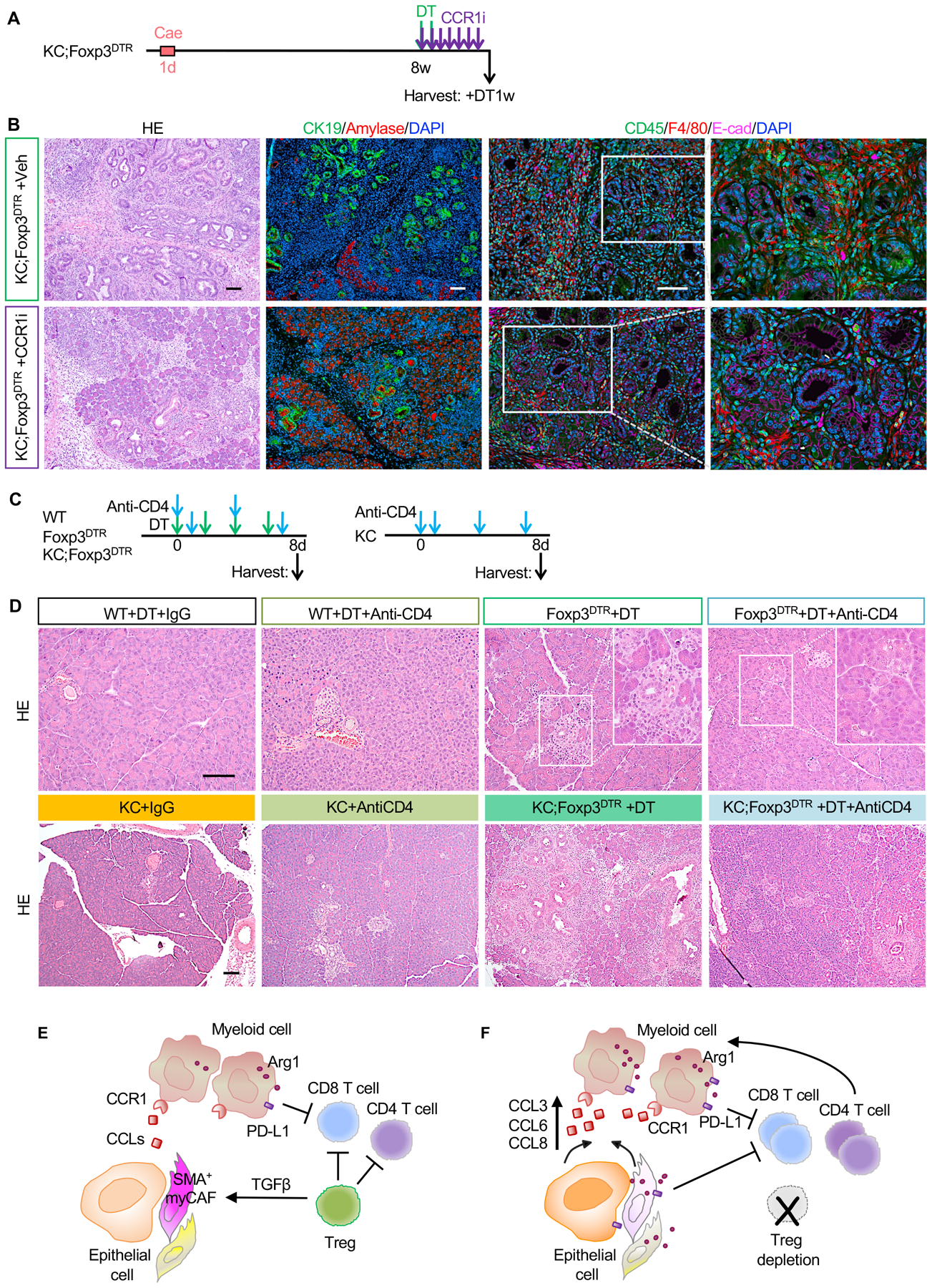
(A) Experimental design, n=5–6 mice/cohort. (B) H&E staining, co-immunofluorescent staining for CK19 (green), Amylase (red) and DAPI (blue), and co-immunofluorescent staining for CD45 (green), F4/80 (red), E-cad (magenta) and DAPI (blue) in control and CCR1 inhibitor treated KC;Foxp3DTR pancreata. Scale bar 100 μm. (C) Experimental design and (D) H&E staining for WT, Foxp3DTR, KC and KC;Foxp3DTR mice that received DT treatment and/or anti-CD4 antibody. n=3–4 mice/cohort. Scale bar 100 μm. (E) Working model. Tregs inhibit CD8+ T cells, but also restrain pathogenic CD4+ T cell responses. Tregs regulate the production of TGFβ thus promoting the differentiation of Smooth Muscle Actin (SMA)+ fibroblasts (myCAFs). (F) Treg depletion results in loss of TGFβ and compensatory changes in the fibroblast population that in turn secrete myeloid-recruiting chemokines, driving increased recruitment of myeloid cells and failing to alleviate immunosuppression. Treg depletion also unleashes pathogenic CD4+ T cell responses that promote pancreatic inflammation and carcinogenesis.
CD4+ T cells promote carcinogenesis upon Treg depletion.
Reprogramming of fibroblasts and consequent increased infiltration of myeloid cells restored immune suppression upon Treg depletion. However, we were intrigued by the increase in total CD4+ T cells upon Treg depletion, and by the acceleration of carcinogenesis in contrast with genetic or antibody-driven CD4+ T cell depletion (17,23). We thus reasoned that Tregs might normally restrain pathogenic Th2 or Th17 responses, both of which have been associated with tumor progression in pancreatic cancer (23,49). By flow cytometry, we observed an increase in CD3+CD8−CD4+Ifnγ+ T cells in KC;Foxp3DTR pancreata compared with both KC and KC;CD4−/− mice (Fig. S6D). Analysis of the cytokine profile in sorted T cells from KC;Foxp3DTR pancreata revealed an increase in the Th2 cytokines Il4, Il10 and Il13, and a reduction in Th1 cytokines such as Il2 and Tnfα, and well as Il22 and Il17 in KC;Foxp3DTR mice. Our data is thus consistent with a mixed Th1/Th2 response, but the cytokine profile suggests a prevalence of a Th2 response (50) (Fig. S6E). We then depleted CD4+ T cells from wild type, FoxP3DTR, KC and KC;Foxp3DTR mice. Isotype control IgG was administered to the control cohorts and DT was administered for consistency with previous experiments, as indicated (Fig. 7C). While Treg depletion causes pancreatitis, CD4+ T cell depletion had no effect on wild type mice. Similarly, CD4 depletion did not accelerate carcinogenesis in young KC mice (prior to lesion formation), while Treg depletion promoted widespread metaplasia, PanIN formation and accumulation of fibroinflammatory stroma (Fig. 7D and S6F). Interestingly CD4+ T cell depletion prevented the formation of metaplasia and PanIN in KC;Foxp3DTR mice, as well as ameliorated the pancreatitis in FoxP3DTR mice (Fig. 7D). CD4+ T cell depletion also prevented the extensive immune infiltration and T cell infiltration elicited by Treg depletion (Fig. S6G). Together, our data point to multiple roles for Tregs within the pancreatic cancer microenvironment, that include restraining pathogenic T cell responses caused by other CD4+ T cell subsets.
Discussion
Pancreatic cancer precursor lesions, such as Pancreatic Intraepithelial Neoplasia, are surrounded by a fibroinflammatory stroma that persists in late-stage disease (for review see (4)). While immune cells are prevalent surrounding the lesions, they are mostly suppressive in nature (10). CD4+ T cells, and among those regulatory T cells, are the most abundant T cell population, while CD8+ T cells are rare. Depletion of CD4+ T cells inhibits PanIN formation (17,23); although it is insufficient to activate anti-tumor immunity at later stages (51). Treg depletion is sufficient to induce anti-tumor immunity in melanoma (52). Further, Treg depletion in orthotopically transplanted tumors derived from the KPC model (expressing both mutant Kras and p53 (45)) in syngeneic C57/Bl6 mice causes CD8+ T cell mediated tumor regression (21), a finding that we reproduce here. Here, we generated KC;FoxP3DTR and KPC;FoxP3DTR mice, where Tregs can be depleted at will in the context of spontaneous carcinogenesis. In contrast with previous findings, we show that Treg depletion in mice before or after the development of PanIN caused accelerated neoplastic progression. In invasive tumors in KPC;FoxP3DTR mice, Treg depletion failed to restrain tumor growth. Analysis of the tissue revealed that an increase in CD8+ T cells upon Treg depletion was offset by a compensatory increase in other CD4+ T cells, immunosuppressive myeloid cells, and by reprogramming of the fibroblast population, from predominantly SMA expressing to largely SMA low (Fig. 7E and 7F).
The role of fibroblasts in pancreatic cancer is complex and different studies have provided contradictory results. Depletion of specific fibroblast subsets, such as the FAP+ cell, resulted in a reduction in tumor growth and improved response to immunotherapy (53). Conversely, depletion of SMA+ fibroblasts resulted in tumor promotion (54). A solution to this apparent contradiction came from the observation that heterogeneous fibroblast populations are present in the pancreatic cancer stroma (42,48). The SMA-high myCAF population is predominantly driven by TGFβ and has been described as tumor restraining (42,54). We thus measured TGFβ mRNA levels and found them reduced in the pancreas of Treg depleted mice. Using single cell sequencing, we determined that tumor epithelial cells, myeloid cells and T cells, including Tregs are the sources of TGFβ1 within the tissue. Conversely, epithelial cells and pancreatic fibroblasts express TGFβ receptors. TGFβ signaling in epithelial cells is known to be tumor suppressive at early stages of carcinogenesis (55); thus, a direct effect of TGFβ reduction might partially explain the increase in carcinogenesis upon Treg depletion. We also observed fibroblast reprogramming upon Treg depletion, from tumor-restricting and SMAhigh myCAFs to a tumor-promoting fate, providing in vivo evidence of a phenomenon described in tumor organoids by the Tuveson laboratory (41). Conversely, we did not observe changes in Il1α, a key driver of iCAF differentiation (41). We show that reprogrammed fibroblasts have increased secretion of chemokines that act as chemoattractant for suppressive myeloid cells. In addition to an increase in myeloid cells, Treg depletion also resulted in elevated expression of immune suppressive genes in fibroblasts, such as Arginase 1 and Cd274 (PD-L1). Clinically, fibroblast reprogramming using different approaches – such as vitamin D analogs, inhibition of focal adhesion kinase– as well as remodeling of the extracellular matrix are actively being tested for pancreatic cancer therapy (56–59).
To obtain mechanistic insight into the paradoxical Treg depletion driven tumor progression, we sequenced RNA of flow sorted fibroblasts from Treg depleted KC pancreata, and uncovered increased expression in a number of chemokines, most notably Ccl3, Ccl6, Ccl8. CCL3 (or macrophage inflammatory protein 1-alpha, MIP-1-alpha) and CCL8 (or monocyte chemoattractant protein 2, MCP-2), are chemotactic for many different immune cells, including macrophages and other myeloid cells (47). Ccl6 is a potent chemoattractant for macrophages and other immune cells in rodents (60). These three CCLs share a common chemokine receptor, CCR1 (47), expressed by myeloid cells and implicated in myeloid cell recruitment in in rheumatoid arthritis (61) as well as myeloma and colon cancer (62,63). CCR1 inhibition reduced myeloid infiltrates and reduced PanIN progression, supporting the notion that myeloid suppressive cells are a key mediator of immune suppression in pancreatic cancer (11–13,18). Treg depletion also unleashed pathologic T cell responses that we attributed to a Th2-type response based on the cytokine profile of the tissue. Th2 cells, as well as Th17, have been implicated in the promotion of pancreatic carcinogenesis (23,49). Thus, several parallel mechanisms might explain the paradoxical promotion of carcinogenesis upon Treg depletion, and support the notion that Tregs might not be a key mediator of immune suppression of pancreatic cancer (64).
In summary, our work reveals a complex cellular cross-talk within the neoplastic pancreas that includes multiple mechanisms to induce immunosuppression. Currently, CCR2 and CXCR2 inhibition is being tested in clinical trials for pancreatic cancer, based on positive results in preclinical models (65–67). Based on our results, CCR1 blockade should be considered as an additional potential target to inhibit myeloid cell infiltration in the pancreas.
Materials and Methods
Mice
KC;Foxp3DTR mice were generated by crossing KC (Ptf1a-Cre;LSL-KrasG12D) (8) with Foxp3DTR mice (B6.129(Cg)-Foxp3tm3(DTR/GFP)Ayr/J, Jackson Laboratory) (34). KC;CD4−/− mice were generated by crossing KC with CD4-deficient mice (B6.129S6-Cd4tm1Knw/J, Jackson Laboratory).
Animal experiments
For Treg depletion, Foxp3DTR, KC;Foxp3DTR mice were treated with diphtheria toxin (DT) (50 ng/g) (Enzo Life Science) by intraperitoneal injection (i.p.). DT injections were repeated according to the specific experimental design shown in Figures. Control mice lacking Foxp3DTR alleles received the same DT treatments. In KC, KC;Foxp3DTR and KC;CD4−/− mice, mild acute pancreatitis was induced in 4–5-week-old mice by a series of 8 hourly intraperitoneal injections of caerulein (Sigma-Aldrich, 75 μg/kg) over 1-day period. For CCR1 inhibition, mice were subcutaneously dosed with CCR1 antagonist BX471 (Sigma-Aldrich, 50 mg/kg) for 7 days at 12-hour intervals. DMSO was used as vehicle to dissolve BX471 at a concentration of 50 mg/ml.
To establish the orthotopic pancreatic cancer model, 1×105 of 7940B cells (C57BL/6J strain)(46) derived from KPC tumor (Ptf1a-Cre; LSL-KrasG12D; p53flox/+) were injected into Foxp3DTR mice of compatible genetic background. Cells were tested for mycoplasma free by MycoAlertTM PLUS Mycoplasma Detection Kit (Lonza) and passage 15–20 were used for all experiments. For CD8+ T cell depletion, anti-CD8 mAb (BioXcell clone 2.43; 200 μg/mouse) was injected i.p. twice per week. For CD4+ T cell depletion, anti-CD4 mAb (BioXcell clone GK1.5; 200 μg/mouse) was injected i.p. at least every three days.
In vitro macrophage polarization
Mouse bone marrow cells were treated with L929 conditioned media for 5 days for macrophage differentiation; primary mouse pancreatic cancer cell iKras#1 (18) conditioned media was used to further educate macrophages. In addition, LPS or IL4 was used to polarize differentiated macrophages as previously described (68).
Histopathological analysis
Tissues were fixed overnight in 10% neutral-buffered formalin, embedded in paraffin and sectioned. Hematoxylin and eosin (H&E), Periodic Acid Schiff (PAS), Gomori’s Trichrome, immunohistochemical and immunofluorescent staining were performed on formalin-fixed, paraffin embedded mouse pancreatic tissues as described before (17). Antibodies used are listed in Supplementary Materials Table S1. For immunofluorescence, Alexa Fluor (Invitrogen) secondary antibodies were used. Cell nuclei were counterstained with Prolong Diamond Antifade Mountant with DAPI (Invitrogen). In addition, TSA Plus Fluorescein system (PerkinElmer) was used when double immunofluorescence staining with primary antibodies raised in the same species. Images were taken using Olympus BX53F microscope, Olympus DP80 digital camera, and CellSens Standard software. The confocal images were acquired using Olympus IX-71 confocal microscope with FluoView FV500/IX software. Quantitative analysis for immunofluorescence staining was performed in at least 3 random non-overlapping fields (200× magnification) in each sample using ImageJ software to measure the percentage of positive area. To quantify the positive cell number, we used confocal images at 600× magnification. At least three samples per group were analyzed. Histopathological quantification for H&E staining was performed by a pathologist (WY) on de-identified images as previously described (69).
Opal Multiplexed IHC staining and multispectral imaging
A formalin fixed paraffin embedded tissue microarray containing human PDA, PanIN and chronic pancreatitis was acquired from the University of Michigan. Blocks were sectioned onto slides creating a 5 micron thickness per core. The slides were then processed using the Opal 7 manual kit (PerkinElmer). Images were captured on the Mantra™ Quantitative Pathology Work Station (Perkin Elmer) and analyzed using inForm® Cell Analysis™ software (Perkin Elmer), as previously described (30). For detailed methods, see Supplementary Methods (complex phenotype design for multispectral imaging analysis in Table S2).
Flow cytometry and Fluorescence-activated cell sorting (FACS)
Single-cell suspensions of pancreata were prepared and stained with fluorescently conjugated antibodies as previously described (17). Flow cytometric analysis was performed on a Cyan™ ADP analyzer (Beckman Coulter) and data were analyzed with FlowJo v10 software. FACS was performed using MoFlo Astrio (Beckman Coulter). Antibodies were listed in Supplementary Materials Table S1.
Cytometry by Time-of-Flight (CyTOF)
Single-cell suspensions of mouse pancreata were prepared as previously described (17). Human patient tissues from FNB or surgery were immediately placed into DMEM media supplemented with Y27632 (Rho-Kinase inhibitor) for transport to the laboratory. Whole blood was collected pre-operatively into 2 ten mL EDTA tubes and transported to the laboratory. Tissues were mechanically minced and enzymatically digested with collagenase P (1mg/mL DMEM) and subsequently filtered through a 40 uM mesh to obtain single cells. Up to 1×107 cells was stained with Cell-ID Cisplatin (1.67μM) for 5 minutes at room temperature, and then a Combining Fix and Perm Sensitive Surface Epitopes and Nuclear Antigen Staining protocol was followed according to manufacturer’s instructions (Fluidigm) for mouse samples, and as previously described (70). Analysis was performed using the Premium CytoBank Software (cytobank.org). Additional details are provided in the Supplementary Methods section.
RNA sequencing and Data analysis
Cell lysates of FACS sorted cells were homogenized using QIAshredder, then total RNA samples were isolated using RNeasy Plus Micro Kit (Qiagen), including an on-column DNase treatment using RNase-Free DNase Set (Qiagen) according to manufacturer’s instruction. RNA concentration and quality were determined, and Strand mRNA libraries were prepared by the University of Michigan Sequencing Core. Libraries were sequenced using paired end 50 cycle reads on a HiSeq 4000 (Illumina). Raw data are available at the NCBI’s Gene Expression Omnibus database (GSE120395 and GSE128707).
The raw data were processed and analyzed by the University of Michigan Bioinformatics Core. The quality control was done using FastQC (version v0.11.3, http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc/) for both pre- and post-alignment and raw reads were aligned to the mouse reference genome of UCSC mm10 (http://genome.ucsc.edu/) using Bowtie2 (version 2.2.1) and TopHat (version 2.0.13) of the Tuxedo suite. Gene expression quantitation and differential expression analysis were performed using HTSeq (version 0.6.1) and DESeq2 (version 1.14.1). Differentially expressed genes were defined by false discovery rate (FDR) of 0.05 and fold changes of 1.5 or more. For the secretome data, we used the intersection of MetazSecKB (curated secreted) and UniProt (secreted, reviewed, https://www.uniprot.org/) as of 11/2017. Plots and heatmaps were generated in R/Bioconductor.
Single-cell RNA sequencing
Orthotopic pancreatic cancer model was established by injecting 5×104 of iKras*F1 cells (FVB/N strain) derived from iKras*;p53* tumor (Ptf1aCre;TetO-KrasG12D;R26rtTa-IRES-EGFP;p53R172H/+)(32) into wild type mice of compatible genetic background. PanIN lesions were induced in iKras* (Ptf1aCre;TetO-KrasG12D;R26rtTa-IRES-EGFP) mice by caerulein and doxycycline as previously described (31). Single-cell suspensions of pancreatic tumors were derived as previously described (17). Dead cells were removed using MACS® Dead Cell Removal Kit (Miltenyi Biotec Inc.). Single-cell cDNA library was prepared and sequenced at the University of Michigan Sequencing Core using the 10x Genomics. Samples were run using paired end 50 cycle reads on HiSeq 4000 (Illumina) to the depth of 100,000 reads.
The raw data were processed and analyzed by the University of Michigan DNA Sequencing Core. R package Seurat version 2.3.4 was used for single cell RNA-seq data analysis similarly as previous described (71). Data were initially filtered to only include all cells with at least 200 genes and all genes in greater than 3 cells. Data were initially normalized using the NormalizeData function with a scale factor of 10,000 and the LogNormalize normalization method. Variable genes were identified using the FindVariableFeatures function. Data were assigned a cell cycle score using the CellCycleScoring function and a cell cycle difference was calculated by subtracting the S phase score from the G2M score. Data were scaled, centered and batch corrected using linear regression on the counts, the cell cycle score difference and run ID using the ScaleData function. Principal Component Analysis (PCA) was run with the RunPCA function using the previously defined variable genes. Violin plots were then used to filter data according to user-defined criteria. Cell clusters were identified via the FindClusters function using a resolution of 1.2 for all samples and t-distributed Stochastic Neighbor Embedding (t-SNE) clustering algorithms were performed. FindMarkers table was created and clusters were defined by user-defined criteria. Raw data are available at the NCBI’s Gene Expression Omnibus database (GSE 140628).
TCGA Data Analysis
The human pancreatic adenocarcinoma RNA-Seq data from the cancer genome atlas (TCGA) was downloaded from cBioportal (https://www.cbioportal.org/datasets). The tumor samples were ranked based on FOXP3 expression and assigned into FOXP3 low (n=89) or FOXP3 high (n=88) groups. The differentially expressed genes between the two groups were determined using limma bioconductor package with voom function in R software (https://www.r-project.org/).
Quantitative RT-PCR
Total RNA samples were isolated from frozen pancreatic tissues using PureLink™ RNA Mini Kit (Invitrogen) or from cells using RNeasy Plus Micro Kit (Qiagen). High Capacity cDNA Reverse Transcription kit (Applied Biosystems) was used to reverse-transcripted total RNA into cDNA. Quantitative PCR was prepared with 1X SYBR Green PCR Master Mix (Applied Biosystems) and various primers (primer sequences are listed in Supplementary Materials Table S3). All primers were optimized for amplification under reaction conditions as follows: 95°C 10mins, followed by 40 cycles of 95°C 15 secs and 60°C 1min. Melt curve analysis was performed for all samples after the completion of the amplification protocol. Cyclophilin A was used as the housekeeping gene expression control.
Statistics
Graphpad Prism 7 software was used for all statistical analysis. All data were presented as means ± standard error (SEM). Intergroup comparisons were performed using Two-tailed unpaired t-test, and p<0.05 was considered statistically significant. Pearson correlation coefficients were used to measure R and R2. Comparison between genotypes using the individually defined immune subpopulations was corrected for multiple comparisons with the Benjamini-Hochberg method with an FDR of 0.1.
Study approval
All animal studies were conducted in compliance with the guidelines of the Institutional Animal Care & Use Committee (IACUC) at the University of Michigan. Patient selection/sample procurement: patients over the age of 18 referred for diagnostic endoscopic ultrasound of a pancreas mass lesion suspected of PDAC were consented according to IRB HUM00041280. Up to 2 extra passes were taken for research after biopsy obtained for clinical use. Surgical specimens were obtained from patients referred for Whipple or distal pancreatectomy according to IRB HUM000025339. Written informed consent forms were obtained from the patients, and the studies were conducted in accordance with recognized ethical guidelines. Human patient studies were approved by Institutional Review Boards of the University of Michigan Medical School.
Supplementary Material
Significance.
Here, we describe an unexpected crosstalk between Tregs and fibroblasts in pancreatic cancer. Treg depletion resulted in differentiation of inflammatory fibroblast subsets, in turn driving infiltration of myeloid cells through CCR1, thus uncovering a potentially new therapeutic approach to relieve immunosuppression in pancreatic cancer.
Acknowledgments:
We thank Dr. Gregory Beatty (University of Pennsylvania) for generously sharing the primary mouse pancreatic cancer cell line 7940B. The Ptf1a-Cre mouse was generous gift from Dr. Chris Wright (Vanderbilt University). The CK19 antibody was obtained from the Iowa Developmental Hybridoma Bank. We thank Carlos Espinoza, Sion Yang and Jeanine M. Ruggeri for technical supports. Special thanks to Daniel Long for prompt histopathology services. Funding: This work was supported by the Cancer Moonshot Initiative (U01CA-224145) to MPM and HCC. This project was also supported by NIH/NCI grants R01CA151588, R01CA198074, the University of Michigan Cancer Center Support Grant (NCI P30CA046592), the American Cancer Society to MPM and NCI-R50CA232985 to YZ. This project was also supported by 3-P30-CA-046592-28-S2 (Administrative Supplement to the Cancer Center Core Grant) to HCC and MPdM. FB was funded by the Association of Academic Surgery Joel Roslyn Award. TLF was funded by K08CA201581. NS is a recipient of the American Cancer Society Postdoctoral Award. ZCN was supported by Michigan Postdoctoral Pioneer Program, University of Michigan Medical School. RM was supported by T-32-GM007315; SBK was supported by T32-GM113900; NS, CJH, VS and KD were supported by T32-CA009676. AVD was supported by Rackham Merit Fellowship Program. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. This work was made possible by the Tissue and Molecular Pathology and the Flow Cytometry Shared resources at the Rogel Cancer Center and the University of Michigan DNA Sequencing Core. CyTOF samples were run at the University of Rochester Medical Center Flow Cytometry Shared Resource and at the Indiana University Simon Cancer Center Flow Cytometry service.
Footnotes
Competing interests: The authors declare no competing interests.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin 2018;68(1):7–30 doi 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 2.Vonderheide RH. The Immune Revolution: A Case for Priming, Not Checkpoint. Cancer Cell 2018;33(4):563–9 doi 10.1016/j.ccell.2018.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012;366(26):2455–65 doi 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hezel AF, Kimmelman AC, Stanger BZ, Bardeesy N, Depinho RA. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev 2006;20(10):1218–49 doi 10.1101/gad.1415606. [DOI] [PubMed] [Google Scholar]
- 5.Ying H, Dey P, Yao W, Kimmelman AC, Draetta GF, Maitra A, et al. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev 2016;30(4):355–85 doi 10.1101/gad.275776.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin WC, Mansour J, et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun 2015;6:6744 doi 10.1038/ncomms7744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kanda M, Matthaei H, Wu J, Hong SM, Yu J, Borges M, et al. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology 2012;142(4):730–3 e9 doi 10.1053/j.gastro.2011.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003;4(6):437–50. [DOI] [PubMed] [Google Scholar]
- 9.Aguirre AJ, Bardeesy N, Sinha M, Lopez L, Tuveson DA, Horner J, et al. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev 2003;17(24):3112–26 doi 10.1101/gad.1158703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clark CE, Hingorani SR, Mick R, Combs C, Tuveson DA, Vonderheide RH. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res 2007;67(19):9518–27 10.1158/0008-5472.CAN-07-0175. [pii] [DOI] [PubMed] [Google Scholar]
- 11.Stromnes IM, Brockenbrough JS, Izeradjene K, Carlson MA, Cuevas C, Simmons RM, et al. Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut 2014;63(11):1769–81 doi 10.1136/gutjnl-2013-306271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res 2013;73(3):1128–41 doi 10.1158/0008-5472.CAN-12-27310008-5472.CAN-12-2731 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res 2014;74(18):5057–69 doi 10.1158/0008-5472.CAN-13-3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Griesmann H, Drexel C, Milosevic N, Sipos B, Rosendahl J, Gress TM, et al. Pharmacological macrophage inhibition decreases metastasis formation in a genetic model of pancreatic cancer. Gut 2017;66(7):1278–85 doi 10.1136/gutjnl-2015-310049. [DOI] [PubMed] [Google Scholar]
- 15.Di Caro G, Cortese N, Castino GF, Grizzi F, Gavazzi F, Ridolfi C, et al. Dual prognostic significance of tumour-associated macrophages in human pancreatic adenocarcinoma treated or untreated with chemotherapy. Gut 2016;65(10):1710–20 doi 10.1136/gutjnl-2015-309193. [DOI] [PubMed] [Google Scholar]
- 16.Liou GY, Doppler H, Necela B, Edenfield B, Zhang L, Dawson DW, et al. Mutant KRAS-induced expression of ICAM-1 in pancreatic acinar cells causes attraction of macrophages to expedite the formation of precancerous lesions. Cancer Discov 2015;5(1):52–63 doi 10.1158/2159-8290.CD-14-0474 2159–8290.CD-14–0474 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Y, Yan W, Mathew E, Bednar F, Wan S, Collins MA, et al. CD4+ T lymphocyte ablation prevents pancreatic carcinogenesis in mice. Cancer Immunol Res 2014;2(5):423–35 doi 10.1158/2326-6066.CIR-14-0016-T 2326–6066.CIR-14–0016-T [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Y, Velez-Delgado A, Mathew E, Li D, Mendez FM, Flannagan K, et al. Myeloid cells are required for PD-1/PD-L1 checkpoint activation and the establishment of an immunosuppressive environment in pancreatic cancer. Gut 2017;66(1):124–36 doi 10.1136/gutjnl-2016-312078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Daley D, Zambirinis CP, Seifert L, Akkad N, Mohan N, Werba G, et al. gammadelta T Cells Support Pancreatic Oncogenesis by Restraining alphabeta T Cell Activation. Cell 2016;166(6):1485–99 e15 doi 10.1016/j.cell.2016.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Winograd R, Byrne KT, Evans RA, Odorizzi PM, Meyer AR, Bajor DL, et al. Induction of T-cell Immunity Overcomes Complete Resistance to PD-1 and CTLA-4 Blockade and Improves Survival in Pancreatic Carcinoma. Cancer Immunol Res 2015;3(4):399–411 doi 10.1158/2326-6066.CIR-14-0215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jang JE, Hajdu CH, Liot C, Miller G, Dustin ML, Bar-Sagi D. Crosstalk between Regulatory T Cells and Tumor-Associated Dendritic Cells Negates Anti-tumor Immunity in Pancreatic Cancer. Cell Rep 2017;20(3):558–71 doi 10.1016/j.celrep.2017.06.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gunderson AJ, Kaneda MM, Tsujikawa T, Nguyen AV, Affara NI, Ruffell B, et al. Bruton Tyrosine Kinase-Dependent Immune Cell Cross-talk Drives Pancreas Cancer. Cancer Discov 2016;6(3):270–85 doi 10.1158/2159-8290.CD-15-0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McAllister F, Bailey JM, Alsina J, Nirschl CJ, Sharma R, Fan H, et al. Oncogenic Kras activates a hematopoietic-to-epithelial IL-17 signaling axis in preinvasive pancreatic neoplasia. Cancer Cell 2014;25(5):621–37 doi 10.1016/j.ccr.2014.03.014S1535-6108(14)00122-6 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Balachandran VP, Luksza M, Zhao JN, Makarov V, Moral JA, Remark R, et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 2017;551(7681):512–6 doi 10.1038/nature24462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liyanage UK, Moore TT, Joo HG, Tanaka Y, Herrmann V, Doherty G, et al. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J Immunol 2002;169(5):2756–61 doi 10.4049/jimmunol.169.5.2756. [DOI] [PubMed] [Google Scholar]
- 26.Hiraoka N, Onozato K, Kosuge T, Hirohashi S. Prevalence of FOXP3+ regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clin Cancer Res 2006;12(18):5423–34 10.1158/1078-0432.CCR-06-0369 [pii]. [DOI] [PubMed] [Google Scholar]
- 27.Tang Y, Xu X, Guo S, Zhang C, Tian Y, Ni B, et al. An increased abundance of tumor-infiltrating regulatory T cells is correlated with the progression and prognosis of pancreatic ductal adenocarcinoma. PLoS One 2014;9(3):e91551 doi 10.1371/journal.pone.0091551 PONE-D-13–42001 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Amir el AD, Davis KL, Tadmor MD, Simonds EF, Levine JH, Bendall SC, et al. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat Biotechnol 2013;31(6):545–52 doi 10.1038/nbt.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Van Gassen S, Callebaut B, Van Helden MJ, Lambrecht BN, Demeester P, Dhaene T, et al. FlowSOM: Using self-organizing maps for visualization and interpretation of cytometry data. Cytometry A 2015;87(7):636–45 doi 10.1002/cyto.a.22625. [DOI] [PubMed] [Google Scholar]
- 30.Lazarus J, Maj T, Smith JJ, Perusina Lanfranca M, Rao A, D’Angelica MI, et al. Spatial and phenotypic immune profiling of metastatic colon cancer. JCI Insight 2018;3(22) doi 10.1172/jci.insight.121932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Collins MA, Bednar F, Zhang Y, Brisset JC, Galban S, Galban CJ, et al. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest 2012;122(2):639–53 doi 10.1172/JCI5922759227 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Collins MA, Brisset JC, Zhang Y, Bednar F, Pierre J, Heist KA, et al. Metastatic pancreatic cancer is dependent on oncogenic Kras in mice. PLoS One 2012;7(12):e49707 doi 10.1371/journal.pone.0049707PONE-D-12-09212 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol 2010;10(7):490–500 doi 10.1038/nri2785. [DOI] [PubMed] [Google Scholar]
- 34.Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol 2007;8(2):191–7 doi 10.1038/ni1428. [DOI] [PubMed] [Google Scholar]
- 35.Guerra C, Schuhmacher AJ, Canamero M, Grippo PJ, Verdaguer L, Perez-Gallego L, et al. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 2007;11(3):291–302 doi 10.1016/j.ccr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 36.Carriere C, Young AL, Gunn JR, Longnecker DS, Korc M. Acute pancreatitis markedly accelerates pancreatic cancer progression in mice expressing oncogenic Kras. Biochem Biophys Res Commun 2009;382(3):561–5 doi 10.1016/j.bbrc.2009.03.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morris JPt, Wang SC, Hebrok M. KRAS, Hedgehog, Wnt and the twisted developmental biology of pancreatic ductal adenocarcinoma. Nat Rev Cancer 2010;10(10):683–95 doi 10.1038/nrc2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ardito CM, Gruner BM, Takeuchi KK, Lubeseder-Martellato C, Teichmann N, Mazur PK, et al. EGF receptor is required for KRAS-induced pancreatic tumorigenesis. Cancer Cell 2012;22(3):304–17 doi 10.1016/j.ccr.2012.07.024S1535-6108(12)00337-6 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guerra C, Collado M, Navas C, Schuhmacher AJ, Hernandez-Porras I, Canamero M, et al. Pancreatitis-induced inflammation contributes to pancreatic cancer by inhibiting oncogene-induced senescence. Cancer Cell 2011;19(6):728–39 doi 10.1016/j.ccr.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morris JPt, Cano DA, Sekine S, Wang SC, Hebrok M. Beta-catenin blocks Kras-dependent reprogramming of acini into pancreatic cancer precursor lesions in mice. J Clin Invest 2010;120(2):508–20 doi 10.1172/JCI40045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Biffi G, Oni TE, Spielman B, Hao Y, Elyada E, Park Y, et al. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFbeta to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov 2019;9(2):282–301 doi 10.1158/2159-8290.CD-18-0710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ohlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med 2017;214(3):579–96 doi 10.1084/jem.20162024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res 2004;64(16):5839–49 doi 10.1158/0008-5472.CAN-04-0465. [DOI] [PubMed] [Google Scholar]
- 44.Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell 2016;167(3):829–42 e13 doi 10.1016/j.cell.2016.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005;7(5):469–83. [DOI] [PubMed] [Google Scholar]
- 46.Long KB, Gladney WL, Tooker GM, Graham K, Fraietta JA, Beatty GL. IFNgamma and CCL2 Cooperate to Redirect Tumor-Infiltrating Monocytes to Degrade Fibrosis and Enhance Chemotherapy Efficacy in Pancreatic Carcinoma. Cancer Discov 2016;6(4):400–13 doi 10.1158/2159-8290.CD-15-1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol 2014;32:659–702 doi 10.1146/annurev-immunol-032713-120145. [DOI] [PubMed] [Google Scholar]
- 48.Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov 2019;9(8):1102–23 doi 10.1158/2159-8290.CD-19-0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ochi A, Nguyen AH, Bedrosian AS, Mushlin HM, Zarbakhsh S, Barilla R, et al. MyD88 inhibition amplifies dendritic cell capacity to promote pancreatic carcinogenesis via Th2 cells. J Exp Med 2012;209(9):1671–87 doi 10.1084/jem.20111706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.De Monte L, Reni M, Tassi E, Clavenna D, Papa I, Recalde H, et al. Intratumor T helper type 2 cell infiltrate correlates with cancer-associated fibroblast thymic stromal lymphopoietin production and reduced survival in pancreatic cancer. J Exp Med 2011;208(3):469–78 doi 10.1084/jem.20101876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Evans RA, Diamond MS, Rech AJ, Chao T, Richardson MW, Lin JH, et al. Lack of immunoediting in murine pancreatic cancer reversed with neoantigen. JCI Insight 2016;1(14) doi 10.1172/jci.insight.88328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shabaneh TB, Molodtsov AK, Steinberg SM, Zhang P, Torres GM, Mohamed GA, et al. Oncogenic BRAF(V600E) Governs Regulatory T-cell Recruitment during Melanoma Tumorigenesis. Cancer Res 2018;78(17):5038–49 doi 10.1158/0008-5472.CAN-18-0365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A 2013;110(50):20212–7 doi 10.1073/pnas.1320318110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014;25(6):719–34 doi 10.1016/j.ccr.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ijichi H, Chytil A, Gorska AE, Aakre ME, Fujitani Y, Fujitani S, et al. Aggressive pancreatic ductal adenocarcinoma in mice caused by pancreas-specific blockade of transforming growth factor-beta signaling in cooperation with active Kras expression. Genes Dev 2006;20(22):3147–60 doi 10.1101/gad.1475506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sherman MH, Yu RT, Engle DD, Ding N, Atkins AR, Tiriac H, et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 2014;159(1):80–93 doi 10.1016/j.cell.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med 2016;22(8):851–60 doi 10.1038/nm.4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012;21(3):418–29 doi 10.1016/j.ccr.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jacobetz MA, Chan DS, Neesse A, Bapiro TE, Cook N, Frese KK, et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 2013;62(1):112–20 doi 10.1136/gutjnl-2012-302529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Alexeev V, Donahue A, Uitto J, Igoucheva O. Chemotaxis-driven disease-site targeting of therapeutic adult stem cells in dystrophic epidermolysis bullosa. Stem Cell Res Ther 2016;7(1):124 doi 10.1186/s13287-016-0388-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lebre MC, Vergunst CE, Choi IY, Aarrass S, Oliveira AS, Wyant T, et al. Why CCR2 and CCR5 blockade failed and why CCR1 blockade might still be effective in the treatment of rheumatoid arthritis. PLoS One 2011;6(7):e21772 doi 10.1371/journal.pone.0021772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dairaghi DJ, Oyajobi BO, Gupta A, McCluskey B, Miao S, Powers JP, et al. CCR1 blockade reduces tumor burden and osteolysis in vivo in a mouse model of myeloma bone disease. Blood 2012;120(7):1449–57 doi 10.1182/blood-2011-10-384784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kitamura T, Fujishita T, Loetscher P, Revesz L, Hashida H, Kizaka-Kondoh S, et al. Inactivation of chemokine (C-C motif) receptor 1 (CCR1) suppresses colon cancer liver metastasis by blocking accumulation of immature myeloid cells in a mouse model. Proc Natl Acad Sci U S A 2010;107(29):13063–8 doi 10.1073/pnas.1002372107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Barilla RM, Diskin B, Caso RC, Lee KB, Mohan N, Buttar C, et al. Specialized dendritic cells induce tumor-promoting IL-10(+)IL-17(+) FoxP3(neg) regulatory CD4(+) T cells in pancreatic carcinoma. Nat Commun 2019;10(1):1424 doi 10.1038/s41467-019-09416-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sanford DE, Belt BA, Panni RZ, Mayer A, Deshpande AD, Carpenter D, et al. Inflammatory monocyte mobilization decreases patient survival in pancreatic cancer: a role for targeting the CCL2/CCR2 axis. Clin Cancer Res 2013;19(13):3404–15 doi 10.1158/1078-0432.CCR-13-0525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nywening TM, Wang-Gillam A, Sanford DE, Belt BA, Panni RZ, Cusworth BM, et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol 2016;17(5):651–62 doi 10.1016/S1470-2045(16)00078-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nywening TM, Belt BA, Cullinan DR, Panni RZ, Han BJ, Sanford DE, et al. Targeting both tumour-associated CXCR2(+) neutrophils and CCR2(+) macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut 2018;67(6):1112–23 doi 10.1136/gutjnl-2017-313738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Halbrook CJ, Pontious C, Kovalenko I, Lapienyte L, Dreyer S, Lee HJ, et al. Macrophage-Released Pyrimidines Inhibit Gemcitabine Therapy in Pancreatic Cancer. Cell Metab 2019;29(6):1390–9 e6 doi 10.1016/j.cmet.2019.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang Y, Morris JPt, Yan W, Schofield HK, Gurney A, Simeone DM, et al. Canonical wnt signaling is required for pancreatic carcinogenesis. Cancer Res 2013;73(15):4909–22 doi 10.1158/0008-5472.CAN-12-43840008-5472.CAN-12-4384 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bertaux-Skeirik N, Wunderlich M, Teal E, Chakrabarti J, Biesiada J, Mahe M, et al. CD44 variant isoform 9 emerges in response to injury and contributes to the regeneration of the gastric epithelium. J Pathol 2017;242(4):463–75 doi 10.1002/path.4918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hosein AN, Huang H, Wang Z, Parmar K, Du W, Huang J, et al. Cellular heterogeneity during mouse pancreatic ductal adenocarcinoma progression at single-cell resolution. JCI Insight 2019. doi 10.1172/jci.insight.129212. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.