

Abstract
Liquid chromatography (LC)-selected reaction monitoring (SRM) is a powerful protein quantification technique in terms of sensitivity, reproducibility, and multiplexing capability. LC-SRM can accurately measure the concentrations of surrogate proteotypic peptides for targeted proteins in complex biological samples by using their stable heavy isotope-labeled counterparts as internal standards. Herein, we describe a step-by-step protocol of the application of LC-SRM to quantify candidate protein biomarkers in human urine.
Keywords: Targeted quantification, LC-SRM, Urine, Biomarker, Stable heavy isotope-labeled peptide, Skyline
1. Introduction
Liquid chromatography coupled with selected reaction monitoring (LC-SRM) has become an increasingly popular technology for accurate quantification of targeted proteins in complex biological samples [1–4]. LC-SRM has recently been recognized as an alternative to immunoassays for protein biomarker verification [5–8]. For developing protein-based clinical tests, voided urine is often a good source, with an advantage over other clinical samples because it is noninvasive and easily accessible in large quantities. However, the verification of protein biomarkers in patient urine is challenging with LC-SRM because the urinary protein concentration is low and varies significantly at the intraindividual and interindividual levels [9, 10]. To address this issue in our LC-SRM workflow of measuring candidate urinary biomarkers, we use a low molecular weight protein cutoff filter to concentrate the proteins from patient urine and either the total urinary protein mass or the urinary creatinine concentration to normalize the urinary protein concentration.
In this chapter, we describe the detailed procedure for the application of LC-SRM to accurately quantify candidate urinary protein biomarkers that were selected based upon our previous discovery data and/or literature reports [10]. The detailed LC-SRM workflow includes urine sample preparation, SRM assay development, LC-SRM quantification, and SRM data analysis.
2. Materials
2.1. Urine Sample Collection and Storage
Urine sample collection is based on recommendations from the Human Kidney and Urine Proteome Project (HKUPP).
Collect 50–100 mL of morning void midstream urine in sterile containers.
Centrifuge at 2000× g for 20 min at room temperature within 1 h of collection.
Separate the supernatant from any particulate matter (including cells and cell debris).
Measure the urinary creatinine level by standard colorimetric assay.
Adjust the pH of the supernatant to be 7.0.
Aliquot the sample into 10 mL aliquots, and store at −80 °C until further analysis.
2.2. Heavy Isotope-Labeled Synthetic Peptides
Crude heavy synthetic peptides labeled with 13C/15N on the C-terminal lysine (K) and arginine (R) (Thermo Fisher Scientific, San Jose, CA).
2.3. Protein Extraction, Digestion, and Cleanup Components
Amicon Ultra centrifugal filtration units (10 kDa molecular weight cutoff, Millipore, Bedford, MS).
Ultracentrifugal filtration chamber exchanging buffer: 50 mM NH4HCO3, pH 8.0.
Bicinchoninic acid (BCA) protein assay kit (Pierce Biotechnology Inc., Rockford, IL).
Solid high-purity urea (Sigma, St. Louis, MO) for denaturing the proteins.
Reducing reagent: 500 mM dithiothreitol (DTT) in water.
Thermomixer (Eppendorf North America, Hauppauge, NY).
Alkylation reagent: 1 M iodoacetamide (IAA) in water.
Digestion buffer: 0.5 M triethylammonium bicarbonate buffer (TEAB).
Trypsin solution: sequencing grade modified porcine trypsin (Promega, Madison, WI) freshly dissolved in digestion buffer to a final concentration of 1 μg/μL.
1 mL solid-phase extraction (SPE) C18 column (Supelco, Bellefonte, PA).
SPE conditioning solution: 100% methanol and 0.1% trifluoroacetic acid (TFA) in water.
SPE washing solution: 0.1% TFA in 5% acetonitrile/95% water (v/v).
SPE eluting solution: 0.1% TFA in 80% acetonitrile/20% water (v/v).
2.4. LC-SRM Components
LC instrumentation: nanoACQUITY UPLC® system equipped with 5 μL injection loop, 100 μm 100 mm BEH 1.7 μm C18 column.
LC solvents: mobile phase A, 0.1% formic acid (FA) in water; mobile phase B and weak needle wash, 0.1% FA in 90% acetonitrile/10% water (v/v); strong needle wash, 100% acetonitrile; and seal wash, 20% methanol/80% water (v/v).
MS instrumentation: Thermo Scientific TSQ Vantage triple-stage quadrupole mass spectrometer.
3. Methods
Typically there are five steps in the LC-SRM workflow (see Fig. 1): (1) SRM assay development, (2) urine sample processing, (3) addition of synthetic heavy isotope-labeled peptides into protein digests, (4) LC-SRM analysis, and (5) data analysis using Skyline software [11].
Fig. 1.
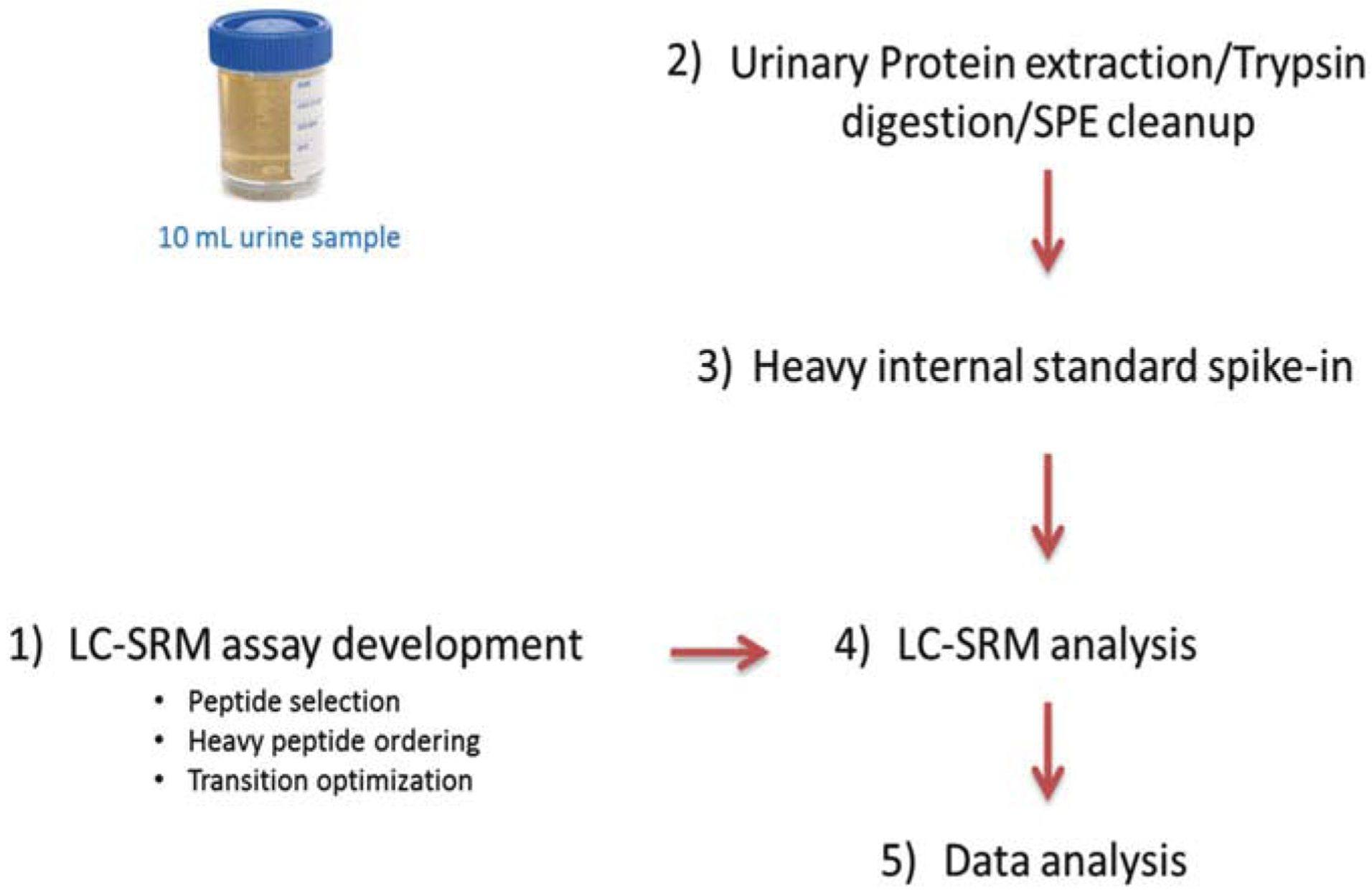
Workflow of LC-SRM quantification of candidate urinary protein biomarkers
3.1. SRM Assay Development
Before performing urine sample analysis, SRM assays for the biomarker proteins of interest need to be developed, which include surrogate peptide selection, purchasing synthetic heavy isotope-labeled peptides, peptide transition selection, and achieving optimal collision energy for each transition.
3.1.1. Surrogate Peptide Selection for Biomarker Proteins of Interest
The selection of surrogate peptides is a key step for sensitive and accurate quantification of target proteins. There are numerous tryptic peptides for a single protein; the selection step aims to select peptides with the best MS response to represent each protein. The number of observations from the MS/MS shotgun proteomics data repository or a theoretical prediction by protein sequences is typically used to facilitate the selection process. The most frequently used MS/MS data repositories are PeptideAtlas [12] and GPM [13] (see Note 1):
Present the information about the identified peptide sequences and their number of observations in a table similar to Table 1.
Calculate the length of peptide sequence in terms of number of amino acids.
Blast the sequences in the MS-Homology website (http://prospector.ucsf.edu/prospector/cgi-bin/msform.cgi?form=mshomology) for any shared sequences with other proteins in homo sapiens database.
Examine the peptide usefulness based on the length of amino acids within a 6–25 range and without any other complicating factors, such as containing posttranslational modifications (PTMs) or methionine and covering the signal peptide region (see Table 1).
- Rank the peptides by their number of observations in PeptideAtlas [12] and GPM [13] (see Note 2):
- PeptideAtlas: http://www.peptideatlas.org
Select the three best peptides based on the number of observations; the larger the number of observations, the better the candidate for SRM study.
- When there is no MS/MS data available, base the peptide ranking on prediction scores from CONSeQuence [14] and ESP-Predictor [15]; the larger the score, the better the candidate for SRM study:
- CONSeQuence: http://king.smith.man.ac.uk/CONSeQuence
Table 1.
Peptide selections of urine protein biomarker candidate of glucose-6-phosphate isomerase (G6PI)
| Clean sequence | # Obs in PeptideAtlas | # Obs in GPM | Peptide length | Unique peptide | Comments |
|---|---|---|---|---|---|
| TLAQLNPESSLFIIASK | 26 | 80166 | 17 | Yes | Potential surrogate peptide |
| HFVALSTNTTK | 5 | 46316 | 11 | Yes | Potential surrogate peptide |
| VWYVSNIDGTHIAK | 3 | 44475 | 14 | Yes | Potential surrogate peptide |
| ILLANFLAQTEALMR | 29 | 101445 | 15 | Yes | Containing M |
| TFTTQETITNAETAKEWFLQAAK | 4 | 13384 | 23 | Yes | Missed cleavage |
| EWFLQAAK | 3 | 9735 | 8 | Yes | Relatively lower # obs |
| INYTEGR | 3 | 7307 | 7 | Yes | Relatively lower # obs |
| TFTTQETITNAETAK | 2 | 58239 | 15 | Yes | Relatively lower # obs |
| KIEPELDGSAQVTSHDASTNGLINFIK | 2 | 45152 | 27 | Yes | # of amino acids >25 |
| VDHQTGPIVWGEPGTNGQHAFYQLIHQGTK | 2 | 37597 | 30 | Yes | # of amino acids >25 |
| LTPFMLGALVAMYEHK | 2 | 11890 | 16 | Yes | Containing M |
| VKEFGIDPQNMFEFWDWVGGR | 2 | 4067 | 21 | Yes | Containing M |
| SNTPILVDGKDVMPEVNK | 1 | 26371 | 18 | Yes | Containing M |
The three rows in boldface indicate the selected three best peptides
Note: “#Obs” stands for number of observations
3.1.2. Synthetic Heavy Isotope-Labeled Peptides
- The crude synthetic heavy isotope-labeled peptides are purchased based on the selection above with the following criteria:
- Isotopically label the C-terminus with heavy arginine ([12C6, 15N4]-arginine) or lysine ([12C6, 15N2]-lysine).
- Protect all cysteines by carbamidomethylation (CAM).
Upon receiving the crude heavy peptides, store at −20 °C for further use.
3.1.3. Transition Optimization of Selected Peptides
In a large-scale study, there are often hundreds of surrogate peptides used for targeted protein quantification. To get the best transitions and their optimal collision energies (CEs), the optimization of each individual surrogate peptide by direct infusion is time-consuming. The methods we use here to select the best transitions are based on Orbitrap HCD MS/MS data of the synthetic crude heavy peptides [16].
Prepare a stock solution of crude heavy peptides in a 1.5 mL Eppendorf safe-lock tube at the concentration of 1000 fmol/μL in 0.1% TFA in water as solvent.
Aliquot the stock solution into 100 μL aliquots.
Take one aliquot for assay development, and store the rest at −80 °C for future use.
Prepare a solution of crude heavy peptides at 500 fmol/μL in 30 μL 0.1% FA in water.
Load 5 μL of the above 500 fmol/μL heavy peptide solution onto a LC-MS/MS system, and obtain HCD MS/MS data (see Note 3).
Analyze the data by MSGF+, and import the analysis results into Skyline to build a peptide spectral library (https://skyline.gs.washington.edu/labkey/wiki/home/software/Skyline/page.view?name=building_spectral_libraries for instructions).
Select the top 5 ranked y-ion transitions for each peptide.
Save the Skyline file for further analysis (see Note 4).
3.2. Urine Sample Processing (See Note 5)
Wash the Amicon 15 mL ultracentrifugal filtration device with 10 mL of 50 mM NH4HCO3 (pH 8.0), and spin at 4000 × g for 10 min to remove the trace of glycerine.
Thaw the frozen urine samples on ice.
Load a 10 mL aliquot of the urine samples to the filter chamber, and centrifuge the sample at 4000 × g at 10 °C for 20 min to separate small MW peptides and other pigments (< 10 kDa) from the larger proteins.
Buffer exchange the sample twice by adding 10 mL of 50 mM NH4HCO3 (pH 8.0) to the filter chamber and centrifuging as described in step 1. Pipet the final retentate in the filter chamber to a fresh and labeled 1.5 mL Eppendorf tube.
Adjust the final volume of the retentate to 400 μL with 50 mM NH4HCO3 (pH 8.0).
Determine the protein concentration by BCA protein assay.
Add powdered urea into the tube to a final concentration of 8 M for protein denaturation.
Add 500 mM DTT solution to a final concentration of 10 mM for reduction. Sonicate the sample briefly, and incubate at 37 °C for 1 h with constant shaking in a ThermoMixer (see Note 6).
Add 1 M IAA to a final concentration of 40 mM, and incubate at 37 °C for 1 h in the dark with constant shaking for alkylation (see Note 6).
Dilute sample by ten times with digestion buffer, add trypsin solution at protein/trypsin ratio of 50:1 (w/w), incubate at 37 °C for 3 h, and then add TFA solution to a final concentration of 0.1% to stop the reaction.
Precondition 1 mL SPE C18 columns by slowly passing 3 mL methanol and then 4 mL SPE conditioning buffer through the column.
Load each tryptic digest onto separate SPE C18 columns; pass each sample through, and wash each column with 4 mL of SPE washing buffer.
Elute the peptides from each SPE C18 column with 1 mL of SPE eluting buffer, and dry each sample under a reduced vacuum using a SpeedVac.
Redissolve the sample with 100 μL water.
Determine the peptide concentration by BCA protein assay.
Store samples at −80 °C freezer until further use.
3.3. Addition of Heavy Peptide Internal Standards
Mix all the heavy isotope-labeled peptides in one 1 mL stock solution at a concentration of 1000 fmol/μL of each peptide using 0.1% TFA in water as solvent.
-
Calculate the volumes needed for LC-SRM analysis:
where VLC-SRM is the volume of solution needed for final LC-SRM analysis (μL), which is typically 20 μL for several injections; Cdigest,LC-SRM is the peptide concentration of final sample (μg/μL) in the final LC-SRM solution; Cdigest original is the peptide concentration (μg/μL) for the original urine protein digest for a given patient sample; Cheavy, LC-SRM is the molar concentration of heavy internal standard peptides (fmol/μL) in the final LC-SRM solutions; and Cheavy peptide stock is the molar concentration of heavy internal standard peptide stock solutions (fmol/μL). Typically, the peptide concentration of the final LC-SRM solutions is 1 μg/μL for 1 μL injection, and the heavy peptide concentration is 100 fmol/μL for crude heavy peptides.
Add the deionized water and urine protein digest sequentially into a Waters glass vial, and shake at 800 × g for 6 min.
Add heavy peptide stock into the same glass vial, and shake at 800 × g for 6 min.
Centrifuge the glass vial at 4000 × g for 2 min to eliminate any air bubbles. The sample is ready for LC-SRM analysis.
3.4. LC-SRM Analysis
The LC-SRM analysis is performed in two steps. The first step is to finalize the LC-SRM assay using the transition list obtained from Orbitrap HCD MS/MS, and the second step is to analyze the urine samples from individual subjects with the finalized method.
3.4.1. LC-SRM Setup
LC-SRM is performed with a nanoACQUITY UPLC system coupled online to a TSQ Vantage triple quadrupole mass spectrometer.
Pack the reversed-phase capillary column, ACQUITY UPLC BEH C18 column, with 1.7 μm particles, 100 mm length × 100 μm i.d.
Degas mobile phases online using a vacuum degasser.
Maintain the LC column temperature at 42 °C.
- Use the following LC gradient:
Time (min) Flow (μL/min) %B 0 0.5 0.5 11 0.5 0.5 11.5 0.4 0.5 *At 13.3 min, end the injection 13.5 0.4 10 17 0.4 15 38 0.4 25 49 0.4 38.5 50 0.4 95 55 0.5 95 60 0.5 95 61 0.5 0.5 *At 70 min, end the method Operate the TSQ Vantage at 1.5 mTorr, and maintain the ESI voltage at 2400 V in positive polarity with a 20 μm i.d. emitter tip. Etch the emitter tips following the previously described methods [17].
Scan the transitions with 0.002 m/z scan width and 0.7 m/z peak widths (FWHM) for both Q1 and Q3.
In nonscheduled LC-SRM method, set the scan/dwell time for each transition to 10 ms, while in scheduled LC-SRM method, set the total cycle time to 1.1 s.
Inject 1 μL (e.g., 1 μg) of protein digest on the LC column.
3.4.2. Finalizing the LC-SRM Method
Analyze one typical urine sample iteratively using nonscheduled LC-SRM with the transitions obtained from Sect. 3.1.3. Each nonscheduled method should contain a maximum of 100 transitions (including both light and heavy peptide transitions). For example, if there are 35 proteins, 105 peptides, and 1050 transitions, it will take 11 nonscheduled LC-SRM methods.
Import the results into the Skyline file.
Select the top three transitions with the highest intensity and lowest interferences.
Draw the peak boundaries of each peptide to achieve retention time (RT) for each peptide.
Export the scheduled LC-SRM method with the average RT of all three transitions.
3.4.3. Analyze the Individual Urine Samples
Randomize all the urine samples and perform the scheduled LC-SRM method.
Run one blank sample (buffer A) between adjacent urine samples to minimize cross-sample contamination.
3.5. Data Analysis
Since both the concentrations of heavy internal standards and the loading amounts are the same across all samples, the peak area ratios between endogenous transitions and heavy internal standard transitions will represent the molar ratios between the amounts of endogenous and heavy internal standard peptides. The peak area ratios can be calculated using Skyline software [11], especially for large-scale studies.
Import the LC-SRM datasets into the Skyline file.
Manually examine the peak boundaries of all the peptides for each individual dataset.
Evaluate whether the peptide is detected and which transition to use for the peak area ratio calculation. The detection of endogenous peptides is mainly examined by their signal-to-noise (S/N) ratios and the agreement between the relative intensities of the three transitions of the endogenous peptides and that of the heavy isotope-labeled internal standard. As you can see in Fig. 2, the S/N ratios of all three transitions of endogenous peptide A are more than 10, and the relative abundance of all three transitions of the endogenous peptide A is in good agreement with that of its internal standard, so we can confirm that the endogenous peptide A is clearly detected in the urine sample. We can either use the best transition, the transition with highest SRM response and lowest noise-to-signal ratio, in this case, the Blue transition, or the average peak area ratios of the three transitions to quantify the protein. In comparison, the endogenous peptide B is not detected because the signals of all three transitions of endogenous peptide B are close to their noises. In the case of peptide C, a significant level of interferences is observed. The S/N ratio of the Blue transition of endogenous peptide C is more than 10, but the S/N ratios of the other two transitions, both Purple and Red, are close to 1. Furthermore, the relative intensity between these three transitions of endogenous peptide C is not comparable with that of the internal standard. In that case, the endogenous peptide C is still considered detected, but only the best transition, the Blue transition, can be used for the protein quantification.
Export the results with “Peptide Sequence,” “Precursor Mz,” “Product Mz,” “Dataset Name,” and “Area Ratio.” (The “Area Ratio” is the peak area ratio for each individual transition between endogenous and internal standard peptides.)
-
Calculate the concentration of protein in urine using the following equation. The equation is based on the assumption that there is the same peptide recovery across all of the urinary proteins, including targeted proteins (see Note 7):
where L/H is the peak area ratio of endogenous (light) and heavy internal standard peptides; Cheavy, LC-SRM is the molar concentration of heavy internal standard peptides (fmol/μL) in the final LC-SRM solutions; MW is the molecular weight of targeted protein (Da or g/mol); Cdigest,LC-SRM is the mass con centration of total protein digest (μg/μL) in the final LC-SRM solutions; and Cprotein is mass concentration of total protein (μg/μL) in the 400 μL concentrated retentate from 10 mL of original urine.
-
Normalize the protein concentration by either total urinary protein mass or urinary creatinine concentration:or
Fig. 2.
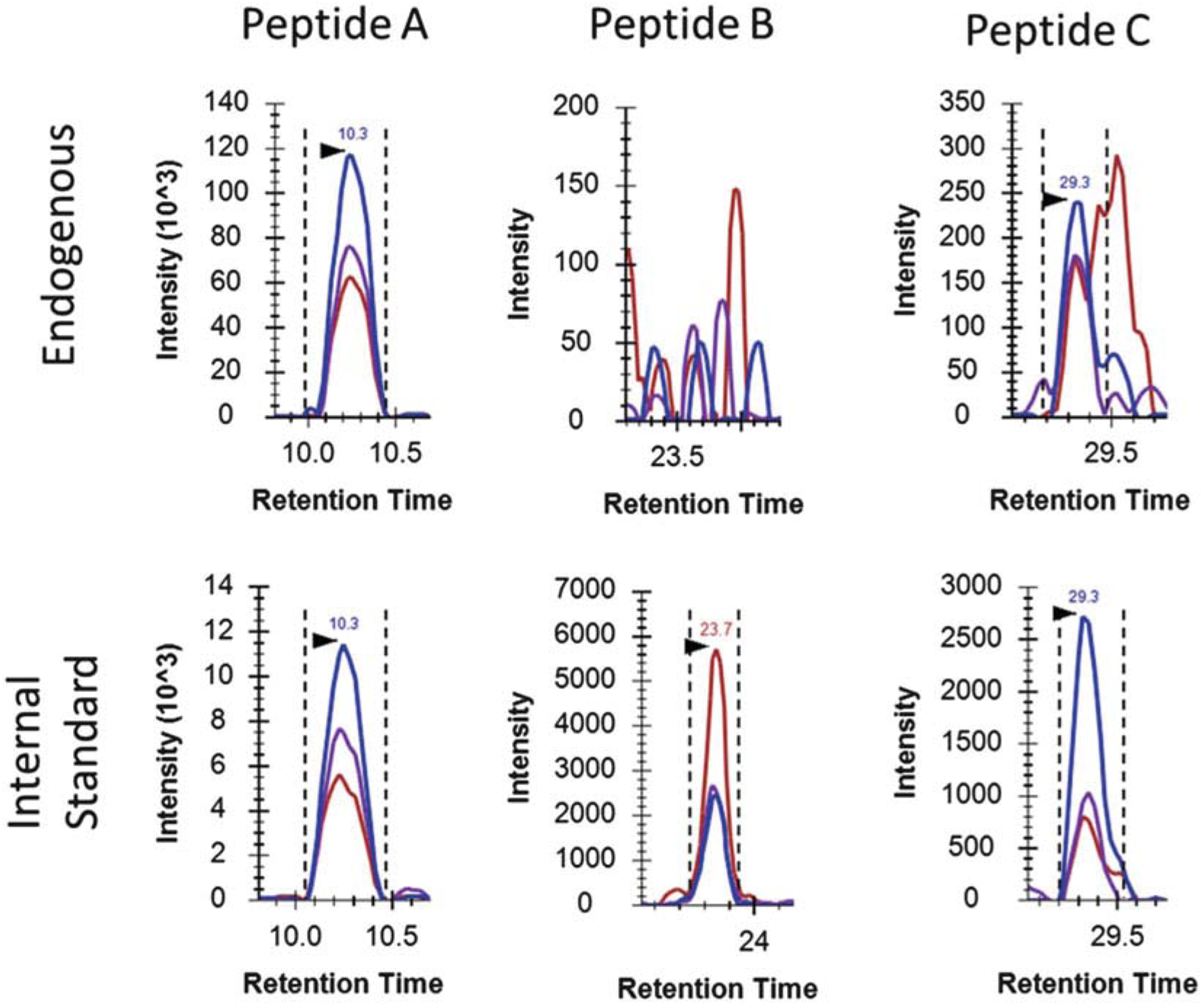
Extracted ion chromatograms (XICs) of three peptides (A, B, C) in a urine sample. The dotted lines demonstrate the peak boundaries, while the arrows indicate the retention times. Peptide A shows confident detection and quantification, while peptide B lacks of clear signals, and peptide C suffers from matrix interferences
4. Notes
In the PeptideAtlas MS/MS data repository, look for “Human Urine” since we are working with urine samples.
In general, the number of observations in PeptideAtlas provides more accurate information than those in GPM.
The detailed LC-MS/MS operation was described in our recent paper [16].
The optimal collision energy (CE) for each y-ion transition was determined using Skyline software [11].
Unless otherwise stated, all solutions should be prepared in deionized water with a resistivity of 18.2 MΩ cm.
DTT and iodoacetamide solution should be made fresh for each operation of digestion.
The internal standard peptides are in crude quality, so the protein concentration obtained is only relative concentration instead of absolute concentration.
Acknowledgments
Portions of the research were supported by NIH grant P41GM103493 and R01DK083447. The experimental work described herein was performed in the Environmental Molecular Sciences Laboratory, Pacific Northwest National Laboratory, a national scientific user facility sponsored by the DOE under contract DE-AC05-76RL0 1830.
References
- 1.Lange V, Picotti P, Domon B, Aebersold R (2008) Selected reaction monitoring for quantitative proteomics: a tutorial. Mol Syst Biol 4:222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Picotti P, Rinner O, Stallmach R, Dautel F, Farrah T, Domon B, Wenschuh H, Aebersold R (2010) High-throughput generation of selected reaction-monitoring assays for proteins and proteomes. Nat Methods 7(1):4. [DOI] [PubMed] [Google Scholar]
- 3.Cox J, Mann M (2011) Quantitative, high-resolution proteomics for data-driven systems biology. Annu Rev Biochem 80:27. [DOI] [PubMed] [Google Scholar]
- 4.Shi T, Su D, Liu T, Tang K, Camp DG II, Qian WJ, Smith RD (2012) Advancing the sensitivity of selected reaction monitoring-based targeted quantitative proteomics. Proteomics 12 (8):19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rifai N, Gillette MA, Carr SA (2006) Protein biomarker discovery and validation: the long and uncertain path to clinical utility. Nat Biotechnol 24(8):13. [DOI] [PubMed] [Google Scholar]
- 6.Whiteaker JR, Zhao L, Anderson L, Paulovich AG (2010) An automated and multiplexed method for high throughput peptide immunoaffinity enrichment and multiple reaction monitoring mass spectrometry-based quantification of protein biomarkers. Mol Cell Proteomics 9(1):13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tang HY, Beer LA, Barnhart KT, Speicher DW (2011) Rapid verification of candidate serological biomarkers using gel-based, label-free multiple reaction monitoring. J Proteomie Res 10 (9):13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Whiteaker JR, Lin C, Kennedy J, Hou L, Trute M, Sokal I, Yan P, Schoenherr RM, Zhao L, Voytovich UJ, Kelly-Spratt KS, Krasnoselsky A, Gafken PR, Hogan JM, Jones LA, Wang P, Amon L, Chodosh LA, Nelson PS, McIntosh MW, Kemp CJ, Paulovich AG (2011) A targeted proteomics-based pipeline for verification of biomarkers in plasma. Nat Biotechnol 29(7):10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Selevsek N, Matondo M, Sanchez Carbayo M, Aebersold R, Domon B (2011) Systematic quantification of peptides/proteins in urine using selected reaction monitoring. Proteomics 11(6):13. [DOI] [PubMed] [Google Scholar]
- 10.Sigdel TK, Salomonis N, Nicora CD, Ryu S, He J, Dinh V, Orton DJ, Moore RJ, Hsieh SC, Dai H, Thien-Vu M, Xiao W, Smith RD, Qian WJ, Camp DG 2nd, Sarwal MM (2014) The identification of novel potential injury mechanisms and candidate biomarkers in renal allograft rejection by quantitative proteomics. Mol Cell Proteomics 13(2):11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, MacCoss MJ (2010) Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26(7):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.EW Deutsch HL, Aebersold R (2008) PeptideAtlas: a resource for target selection for emerging targeted proteomics workflows. EMBO Rep 9 (5):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.R Craig JC, Beavis RC (2004) Open source system for analyzing, validating, and storing protein identification data. J Proteomie Res 3 (6):9. [DOI] [PubMed] [Google Scholar]
- 14.Eyers CE, Lawless C, Wedge DC, Lau KW, Gaskell SJ, Hubbard SJ (2011) CONSeQuence: prediction of reference peptides for absolute quantitative proteomics using consensus machine learning approaches. Mol Cell Proteomics 10(11):M110.003384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fusaro VA, Mani DR, Mesirov JP, Carr SA (2009) Prediction of high-responding peptides for targeted protein assays by mass spectrometry. Nat Biotechnol 27:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu C, Shi T, Brown JN, He J, Gao Y, Fillmore TL, Shukla AK, Moore RJ, Camp DG II, Rod-land KD, Qian WJ, Liu T, Smith RD (2014) Expediting SRM assay development for large-scale targeted proteomics experiments. J Proteome Res 13(10):9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kelly RT, Page JS, Luo Q, Moore RJ, Orton DJ, Tang K, Smith RD (2006) Chemically etched open tubular and monolithic emitters for nanoelectrospray ionization mass spectrometry. Anal Chem 78(22):6. [DOI] [PMC free article] [PubMed] [Google Scholar]