

Abstract
Post-transcriptional regulation via small regulatory RNAs (sRNAs) has been implicated in diverse regulatory processes in bacteria, including virulence. One class of sRNAs, termed trans-acting sRNAs, can affect the stability and/or the translational efficiency of regulated transcripts. In this study, we utilized a collaborative approach that employed data from infection with the Borrelia burgdorferi Tn library, coupled with Tn-seq, together with borrelial sRNA and total RNA transcriptomes, to identify an intergenic trans-acting sRNA, which we designate here as ittA for infectivity-associated and tissue-tropic sRNA locus A. The genetic inactivation of ittA resulted in a significant attenuation in infectivity, with decreased spirochetal load in ear, heart, skin and joint tissues. In addition, the ittA mutant did not disseminate to peripheral skin sites or heart tissue, suggesting a role for ittA in regulating a tissue-tropic response. RNA-Seq analysis determined that 19 transcripts were differentially expressed in the ittA mutant relative to its genetic parent, including vraA, bba66, ospD and oms28 (bba74). Subsequent proteomic analyses also showed a significant decrease of OspD and Oms28 (BBA74) proteins. To our knowledge this is the first documented intergenic sRNA that alters the infectivity potential of B. burgdorferi.
Author summary
Lyme disease is a tick-borne infection mediated by the spirochetal bacterium, Borrelia burgdorferi, that is responsible for greater than 300,000 infections in the United States per year. As such, additional knowledge regarding how this pathogen modulates its regulatory armamentarium is needed to understand how B. burgdorferi establishes and maintains infection. The identification and characterization of small, non-coding RNA molecules in living systems, designated as sRNAs, has recalibrated how we view post-transcriptional regulation. Recently, over 1,000 sRNAs were identified in B. burgdorferi. Despite the identification of these sRNAs, we do not understand how they affect infectivity or B. burgdorferi pathogenesis related outcomes. Here, we characterize the ittA B. burgdorferi sRNA and show that it is essential for optimal infection using murine experimental infection as our readout. We also track the effect of this sRNA on the transcriptional and proteomic profile as the first step in providing mechanistic insight into how this important sRNA mediates its regulatory effect.
Introduction
Lyme disease results from the infection by the spirochetal bacterium, Borrelia burgdorferi, and represents the most common vector-borne disease in the United States with an estimated 329,000 cases diagnosed each year [1]. B. burgdorferi is transmitted to mammalian hosts through the bite of infected Ixodes spp. ticks [2–4]. In humans, the infection is characterized by a flu-like illness and, in most instances, is accompanied by a skin lesion denoted as erythema migrans [5,6]. The subsequent infection, if effectively treated with antibiotics early in the development of Lyme disease, can be cleared. If untreated, B. burgdorferi can disseminate throughout the host to distal organs and tissues resulting in multiple pathologies, including carditis, various neuropathies, and arthritis [5,6].
B. burgdorferi oscillates in nature between vastly disparate environments of the tick vector and vertebrate hosts [2–4,7]. The enzootic life cycle of B. burgdorferi initiates by uninfected tick larvae feeding on an infected vertebrate (usually a small mammal or bird), resulting in the acquisition of the spirochete during the resulting blood meal [2–4]. The infected larvae then molt into a nymph and will seek another blood meal. It is at this point that B. burgdorferi infect vertebrate hosts, including dead end hosts such as humans. In order to survive in these changing host environments, B. burgdorferi alters its transcriptional and protein profiles [2,8,9]. Previous studies demonstrated that B. burgdorferi senses and responds to environmental cues such as temperature, pH, and dissolved gases, as well as unidentified host factors, to modulate its gene expression [10–19]. Despite significant insight into these processes, the mechanisms utilized by B. burgdorferi to modulate its gene expression for environmental adaptation continues to be an area of active research.
In B. burgdorferi, several transcriptional regulators have been identified and characterized, as well as a growing list of DNA interacting proteins, which serve to alter borrelial gene expression either directly or indirectly [3,20–33]. Many of these regulators govern, in part, the production of surface proteins involved in borrelial virulence [20,34–37]. In addition to these regulators, recent results indicate that B. burgdorferi produces a battery of small non-coding RNA molecules, designated sRNAs [38–41]. The role and molecular mechanisms of the sRNAs in B. burgdorferi, or their impact on borrelial pathogenesis, is not well understood.
sRNA-mediated post-transcriptional regulation in bacteria commonly involves the alteration of transcript stability or translation efficiency; transcript targets include those that contribute to pathogenesis [42–44]. In addition, sRNAs can bind to proteins and modify protein activity [44–46]. As such, sRNAs encoded in intergenic regions of bacterial genomes are trans-acting regulators that can influence multiple, genetically unlinked transcripts [44,47–50]. These trans-acting sRNAs have partial complementarity to the transcripts they target. The resulting sRNA-mRNA duplex that forms can alter gene expression by multiple mechanisms including affecting mRNA stability, which can lead to the stabilization or the degradation of the mRNA [44,49,51–54]. The sRNA-mRNA duplex can also alter translation initiation, thus affecting translation efficiency [44,49,51–54]. The outcome of trans-acting sRNA regulation is an increase or decrease in production of the encoded protein [44,49,51–54].
Recently, the sRNA transcriptome of B. burgdorferi was reported and 1,005 sRNA species were identified; many of these sRNAs are upregulated at 37°C, a condition that models the mammalian host temperature in vitro [40]. Independently, a Luminex-based procedure for the detection of Signature-tagged mutagenesis (STM) clones of the transposon (Tn) library of B. burgdorferi strain B31, identified several intergenic non-coding regions that, when genetically inactivated, exhibited infectivity deficits [55]. Here we further characterize one of these intergenic sRNAs, that maps to the 17 kilobase linear plasmid (lp17) between bbd18 and bbd21, which was designated as SR0736 [40]. We have renamed SR0736 as ittA for infectivity-associated and tissue-tropic sRNA A. We independently inactivated this locus and showed that mutants lacking ittA are significantly attenuated and do not disseminate to additional skin sites or heart tissue. To our knowledge this is the first intergenic sRNA that has been linked to infectivity and pathogenesis of B. burgdorferi. Taken together, our data suggests that ittA exerts its effect by engaging several unlinked genetic targets and altering their production in a manner that is required for optimal infection and dissemination throughout the host.
Results
Identification of sRNA associated with B. burgdorferi infectivity
Given the importance of sRNAs in other pathogenic bacteria [42,44] and the population of sRNAs in B. burgdorferi that are induced at conditions that mimic the mammalian host temperature in vitro [40], we sought to determine if a subset of intergenic trans-acting sRNAs in B. burgdorferi contributed to borrelial pathogenesis. Initially, we utilized a B. burgdorferi transposon (Tn) mutant library [55], coupled with Tn-seq analysis following mouse infection [56,57], to identify Tn insertions that mapped to intergenic (IG) regions or non-coding regions within the genome. We focused on intergenic Tn mutants that were represented in the initial in vitro grown inoculum used for infection but were substantially reduced following murine infection (Table 1). These data were overlapped with B. burgdorferi strain B31 sRNA annotations [40] to identify the Tn insertions that interrupted sRNAs. We identified eight such Tn mutants and tested their ability to establish infection as individual isolates by infecting C3H/HeN mice at a dose of 104 B. burgdorferi cells. After 21 days, a qualitative assessment of infectivity was determined (Table 2). Five of eight sRNA Tn mutants displayed reduced infectivity relative to the parent strain, 5A18NP1 [58], with a reduction in culture positive sites ranging from 25% to 75% (Table 2). After this initial screen, we focused on the sRNA (SR0736; [40]) that maps between genes bbd18 and bbd21 of linear plasmid 17 (lp17), which demonstrated the most significantly attenuated phenotype of the strains evaluated (Tables 1 and 2). Based on the phenotype observed for B. burgdorferi cells that have a Tn insertion in the SR0736 sRNA, we designated SR0736 as ittA for infection-associated and tissue-tropic sRNA A.
Table 1. Reduced prevalence of candidate sRNA mutants during murine infection, based on Tn-seqa. Genetic mutations that either reduce (guaA, dbpA, pncA) or do not affect infection (bbg22, bbk52, bbb28) are included for comparison.
| Location of Tn insert | sRNA IDb | Input 1c | Input 2c | Input 3c | Output 1d | Output 2d | Output 3d | Avg.Output /Avg.Input |
Comments |
|---|---|---|---|---|---|---|---|---|---|
| cp26; guaA (bbb18) | N/Ae | 1.2032 | 0.7411 | 0.7736 | 0.0009 | 0.0018 | 0.0009 | 0.0013 | Control; attenuated |
| lp54; dbpA (bba24) | N/Ae | 0.0041 | 0.0073 | 0.0084 | NDf | NDf | NDf | 0g | Control; attenuated |
| lp25; pncA (bbe22) | N/Ae | 0.0089 | 0.0045 | 0.0027 | NDf | NDf | 0.0003 | 0.0185 | Control; attenuated |
| lp28-2; bbg22 | N/Ae | 0.0672 | 0.0991 | 0.0927 | 0.1688 | 0.2016 | 0.0267 | 1.53 | Unchanged |
| lp36; bbk52 | N/Ae | 0.00057 | 0.00028 | 0.00038 | 0.00126 | 0.0007 | 0.00051 | 2.01 | Unchanged |
| cp26; bbb28 | N/Ae | 1.5432 | 1.0517 | 1.1083 | 1.3479 | 3.5336 | 4.1401 | 2.44 | Unchanged |
| lp54; IG bba34-bba36 | SR0897 | 0.0201 | 0.0210 | 0.0085 | NDf | NDf | NDf | 0g | 6.5x105 readsh |
| lp54; IG bba66-bba68 | SR0912 | 0.1001 | 0.1327 | 0.2092 | 0.00005 | NDf | 0.00006 | 0.00025 | 6x105 readsh |
| cp26; IG bbb03-bbb04 | SR0948 | 0.2269 | 0.1362 | 0.0982 | 0.0001 | NDf | 0.0006 | 0.0015 | 1.8x104 readsh |
| cp26; IG bbb13-bbb14 | SR0962 | 0.0258 | 0.0107 | 0.0062 | NDf | NDf | NDf | 0g | 3x106 readsh |
| lp17; IG bbd04-bbd05a | SR0725 | 0.0378 | 0.0323 | 0.0145 | NDf | 0.00006 | NDf | 0.00071 | 2x106 readsh |
| lp17; IG bbd18-bbd21 | SR0736/ittA | 0.0244 | 0.0549 | 0.0454 | 0.0001 | NDf | 0.00006 | 0.0013 | 1x105 readsh |
| lp28-3; IG bbh36a-bbh36b | SR0795 | 0.1628 | 0.2209 | 0.1344 | 0.00005 | NDf | 0.00006 | 0.00021 | 1x105 readsh |
| lp38; IG bbj37-bbj41 | SR0869 | 0.0384 | 0.0456 | 0.0337 | NDf | NDf | 0.00006 | 0.00051 | 2.5 x103 readsh |
a Data following a 3 week infection with a library of Tn mutants [55];
b sRNAs from the B. burgdorferi sRNA library [40];
c % of Tn-seq reads from the libraries made from the inoculum used for mouse infection;
d % of Tn-seq reads from the libraries made from bacteria recovered from infected mice; d Output is pooled data from 24 mice;
e N/A = not applicable;
f ND = not detected;
g indicates that no sequences were observed following Tn-seq;
h represents rounded number of reads from sRNA-specific libraries [40].
Table 2. Infectivity of B. burgdorferi intergenic (IG) sRNA transposon mutants relative to their genetic parent clone 5A18NP1a.
| Strainb | sRNA IDc | Genomic location |
Ear | Skind | Lymph Node | Heart | Bladder | Joint | Total sites | % Positive Tissues |
|---|---|---|---|---|---|---|---|---|---|---|
| 5A18NP1 (parent) | N/A | N/A | 4/4 | 4/4 | 4/4 | 4/4 | 4/4 | 4/4 | 24/24 | 100% |
| T05TC355 | SR0897 | lp54; IG bba34-bba36 | 3/4 | 3/4 | 3/4 | 3/4 | 3/4 | 3/4 | 18/24 | 75% |
| T10TC061 | SR0912 | lp54; IG bba66-bba68 | 4/4 | 4/4 | 4/4 | 4/4 | 4/4 | 4/4 | 24/24 | 100% |
| T11TC387 | SR0948 | cp26; IG bbb03-bbb04 | 4/4 | 4/4 | 4/4 | 4/4 | 4/4 | 4/4 | 24/24 | 100% |
| T10TC351 | SR0962 | cp26; IG bbb13-bbb14 | 2/4 | 1/4 | 2/4 | 2/4 | 2/4 | 2/4 | 11/24 | 46% |
| T04TC273 | SR0725 | lp17; IG bbd04-bbd05a | 2/4 | 2/4 | 2/4 | 2/4 | 2/4 | 2/4 | 12/24 | 50% |
| T06TC412 | SR0736 | lp17; IG bbd18-bbd21 | 0/4 | 1/4 | 3/4 | 0/4 | 2/4 | 0/4 | 6/24 | 25% |
| T08TC464 | SR0795 | lp28-3: IG bbh36a-bbh36b | 4/4 | 4/4 | 4/4 | 4/4 | 4/4 | 4/4 | 24/24 | 100% |
| T09TC062 | SR0853 | lp38: IG bbj15a-bbj16 | 3/4 | 2/4 | 3/4 | 3/4 | 3/4 | 3/4 | 17/24 | 71% |
Popitsch et al. identified ittA via deep sequencing and showed that the ittA-encoded sRNA was made at a higher level when B. burgdorferi is grown at 37°C [40]. The sRNA and 5’ end deep sequencing data indicate that ittA is a processed intergenic sRNA (Fig 1) [40,59]. The mapped primary 5’ ends (transcriptional start sites) are upstream from the ittA sRNA with minimal read coverage of the precursor RNA, suggesting the ittA sRNA is initially transcribed as a larger transcript (130 nt) and cleaved or processed into a stable smaller transcript (70 nt) [Fig 1A and 1B]. In addition, the distal 5’ transcriptional start site for ittA overlaps with the transcription start site for bbd18 on the opposite DNA strand (Fig 1A and 1B).
Fig 1. Location of SR0736/ittA sRNA on linear plasmid 17 (lp17) and the identification of its primary 5’ transcriptional start sites (TSS).

A. The deep sequencing results of the ittA sRNA are displayed in a coverage map. The negative strand coverage is shown in blue and the positive strand coverage in green. The genomic context is illustrated below the coverage maps: black arrows indicate the annotated ORFs, the yellow box indicates the Northern probe used, and the wavy line is the proposed mature sRNA species as determined by RNA-seq and Northern blot analysis. The red box represents the antibiotic resistance marker that is inserted into the ittA sRNA locus to obtain strain DM103. The sRNA is encoded in the positive strand and has two primary 5’ ends indicating transcriptional start sites (TSS) denoted by red arrows in the 5’ end + track. The sequencing results show the sRNA is processed into its mature form visualized as the “green box” in the sRNA + strand track. B. The primary 5’ TSS of ittA and gene bbd18. The bbd18 gene is encoded on the negative strand and is located upstream of ittA in lp17. The ittA sRNA has two primary 5’ TSS; one of the TSS (nucleotide 11,689) overlaps with the bbd18 TSS (nucleotide 11,691) on the opposite strand, as observed in the 5’end—track. The sequencing results show the sRNA processed into its mature form.
Genetic inactivation of the ittA sRNA in B. burgdorferi.
To independently test the role of the ittA sRNA in borrelial pathogenesis, we genetically interrupted ittA as depicted in Fig 2A. The parent strain is the B31 derivative ML23 that lacks the 25 kb linear plasmid [60]. Due to the absence of lp25, this strain is non-infectious in the murine model of experimental infection. However, when a region of lp25, containing the bbe22 gene, is provided in trans infectivity is restored and provides selective pressure for the maintenance of plasmids encoding it during experimental infection [61,62]. The shuttle vector pBBE22luc used here contains bbe22 and the firefly luciferase reporter that facilitates bioluminescent imaging as reported previously [61,63,64].
Fig 2. Strategy and confirmation of the insertional inactivation of the ittA sRNA.

A. Schematic representation of ittA insertional inactivation strategy. The region of lp17 from the parent strain (ML23) is shown at the top. A PflgB-StrR cassette was inserted into the 3’ end of the ittA sRNA and the mutant (DM103) was obtained following homologous recombination. Following isolation of the mutant strain, the ittA sRNA locus was reintroduced in the complemented strain (DM113). B. Primer pairs P1/P4, P2/P4 and P1/P3 (S7 Table) were used to confirm the presence of the PflgB-StrR in the mutant (ittA:StrR), relative to its parent strain by PCR. C. For the cis complementation of the ittA sRNA mutant strain, the PflgB-StrR cassette and the flanking region were replaced by the ittA sRNA sequence linked to a PflgB-GentR cassette by a double crossover homologous recombination event. The resulting complement strain (Comp) was evaluated by PCR using primer pairs P5/P6, P1/P6 and P5/P4 (S7 Table). Note that the distances between genetic loci are not shown to scale.
The ittA-encoding sRNA was insertionally inactivated in the B. burgdorferi strain ML23 (Fig 2A). Transformants were selected with streptomycin and PCR was employed to distinguish the parent from potential ittA::StrR mutant candidates (Fig 2B). The resulting ittA mutant strain was designated DM103. We then genetically restored the ittA sRNA in cis in strain DM103 by selecting for resistance to gentamicin and then screening for sensitivity to streptomycin (Fig 2A). Candidates predicted to encode ittA were vetted further using PCR (Fig 2C). The ittA complement strain was designated DM113. Following the aforementioned screen of the ittA::StrR mutant strain DM103 and the complement strain DM113, both were transformed with pBBE22luc so they could be tested in the murine experimental model of infection using a firefly luciferase reporter [61,63,64]. Note that all strains maintained plasmid content identical to the parent strain.
To confirm that the ittA mutant and complement strains lacked and restored the ittA sRNA, respectively, both Northern blot and Reverse Transcriptase PCR (RT-PCR) were performed (Fig 3). Notably, the ittA sRNA was detected in the parent strain ML23 and the complement strain DM113 (Comp), but not in the mutant strain DM103 (ittA::StrR), when either Northern blot (Fig 3A) or RT-PCR analysis was employed (Fig 3B).
Fig 3. Confirmation that the ittA is not made in the mutant strain and is restored in the genetic complement.

A. Northern blot of total RNA isolated from the parent strain ML23 (denoted Parent), the sRNA mutant strain (ittA:StrR), and the ittA cis complemented strain (denoted Comp). Detection of 5S from each strain serves as a loading control. B. RT-PCR using purified total RNA from the B. burgdorferi ML23 (Parent), sRNA mutant (ittA:StrR), and genetic complement (Comp) strains. The first three lanes had no reverse transcriptase (RT) added to the reactions (indicated with a “-“) whereas the next three lanes included reverse transcriptase (designated with a “+”). The DNA ladder is shown to the left and base pair values are indicated. The arrow on the right indicates the presence of the sRNA species observed in the parent and complement strains.
Considering the nature of intergenic sRNAs (i.e., between two annotated genes), one concern in deleting the ittA sRNA is the potential polar effect this alteration might have on expression of flanking genes. Here, we focused on the only intact encoding genes in the region, bbd18 and bbd21. When RT-PCR was employed, no qualitative difference in bbd18 or bbd21 transcripts were observed between the parent, mutant, or complement strains (S1 Fig). Note that both bbd19 or bbd20 are no longer annotated as intact ORFs and, consistent with this, no transcript was detected for bbd20 by RT-PCR for any of the strains. Taken together, these data indicate that the inactivation of the ittA sRNA exhibits no polar effects. In addition, the ML23 parent, the ittA mutant, and the cis complement strains all grew similarly, indicating that the loss of the ittA sRNA did not impair replication of these borrelial strains under in vitro growth conditions (S2 Fig).
The loss of ittA attenuates B. burgdorferi infectivity
We then evaluated the loss of the ittA-encoded sRNA on murine infectivity. To spatially and temporally track infection, C3H/HeN mice were inoculated at a 103 dose with the parent B. burgdorferi strain, the ittA mutant, and the complement. Light emission was quantified at the time points indicated in Fig 4. As a background control for luminescence, a single infected mouse was not given the luciferase substrate D-luciferin (leftmost mouse in each panel, Fig 4A). At the dose tested (103 B. burgdorferi cells), no signal is detected for any of the strains prior to day 4. At day 4, a clear signal is observed in mice infected with all three strains except for one mouse infected with the sRNA mutant strain (Fig 4A). Subsequently, the signal increased with a peak at day 7 and then decreased concomitant with the development of the adaptive immune response (Fig 4A; [65]). All strains displayed similar light emission until day 10 when the ittA::StrR strain exhibited a reduction in signal and retained lower light emission through 21 days, e.g., the duration of the infectivity analysis (Fig 4A). Consistent with the images obtained, quantification of in vivo luminescence from the mice revealed significantly lower light emission by the ittA::StrR strain compared to the parent on days 10 and on day 14 of infection (Fig 4B). The complement strain DM113 emitted light comparable to the parent strain on days 7, 14 and 21 (Fig 4B). These results, as assessed by infectivity-based criterion used here, suggest that the ittA complement strain DM113 displays comparable in vivo complementation during experimental infection (Fig 4).
Fig 4. Spatial and temporal infectivity analysis of the ittA mutant.
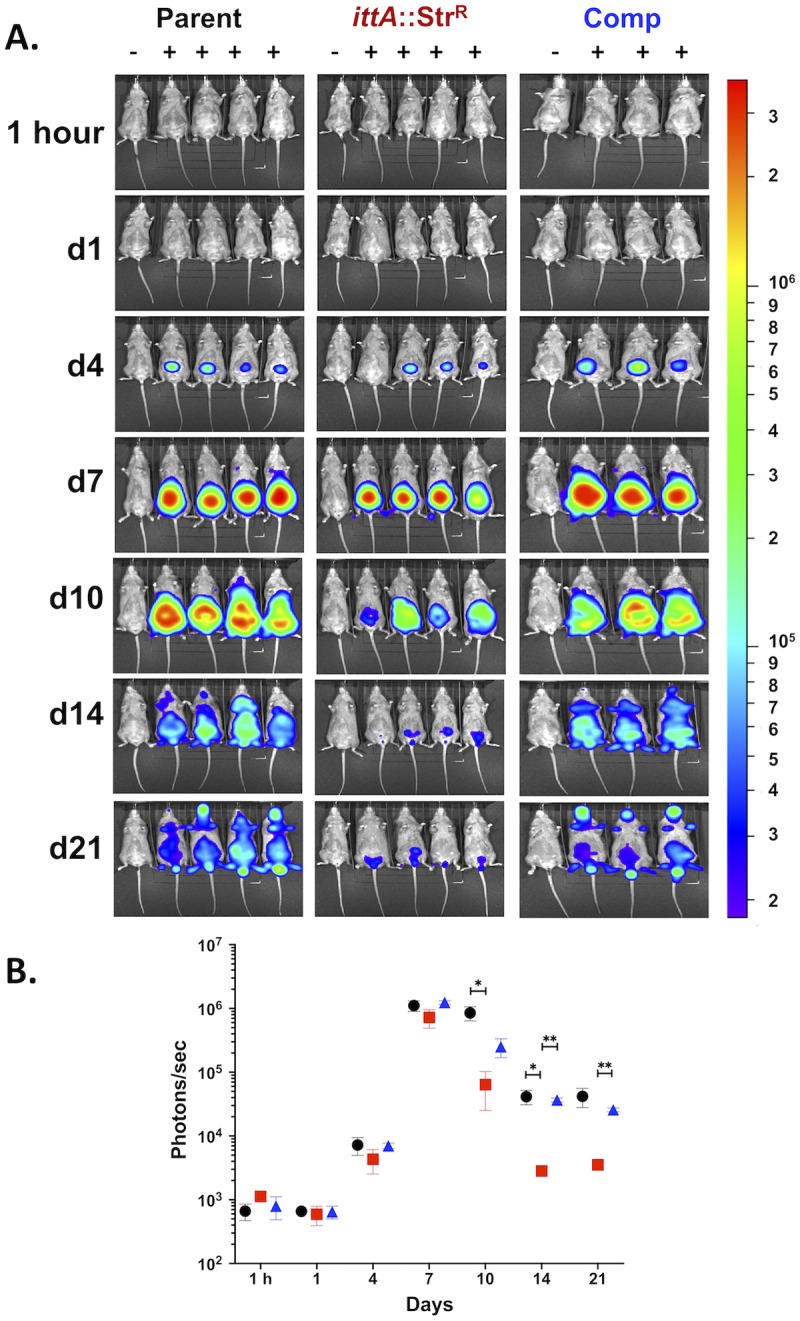
A. The course of infection of bioluminescent B. burgdorferi strains was tracked following the infection of C3H/HeN mice with 103 of each B. burgdorferi isolate. Mice were infected for 21 days total with the parent (n = 5), ittA sRNA mutant (n = 5), and the ittA genetic complement (n = 4) and imaged on the time or day (d) listed on the left. For each image shown, the mouse on the far left (denoted with a ‘−’) was infected with B. burgdorferi but did not receive D-luciferin to serve as a background control. Mice denoted with a ‘+’ were infected with the strain indicated and treated with D-luciferin to promote light emission. All images were normalized to the same scale (in photons/sec; shown on the right). B. Quantification of in vivo luminescence images of mice infected at a dose of 103 of B. burgdorferi. Parent strain ML23/pBBE22luc is depicted as black circles, the ittA sRNA mutant DM103/pBBE22luc as red squares, and the genetic complement strain DM113/pBBE22luc as blue triangles. Each time point represents the average value and the standard error from the four mice given D-luciferin substrate for the parent and sRNA mutant strains and three mice for the complement strain. *p < 0.05; **p < 0.01.
To further assess the phenotype of the ittA mutant and complement, the infected mice were sacrificed after 21 days and tissues cultured to qualitatively score for infection. As shown in Table 3, the ittA mutant that was needle inoculated on the ventral side (abdomen) is impaired in its ability to disseminate to peripheral ear skin and it is attenuated in its ability to disseminate and colonize heart tissue with overall infectivity reduced by 40%. In addition to this qualitative assessment of infection, we also scored for spirochete load using quantitative PCR (qPCR) analysis to enumerate borrelial genome copies relative to murine β-actin copies. As observed in Fig 5, the ittA mutant had significantly lower bacterial burden in all tissues analyzed relative to its parent and complemented strains with the notable exception of the inguinal lymph node. The complemented strain exhibited bacterial burden comparable to the parent strain, thus demonstrating complete in vivo complementation during infection. When taken together with the in vivo imaging data shown in Fig 4, these data suggest that the ittA sRNA is required for optimal tissue tropism and/or dissemination during B. burgdorferi experimental infection.
Table 3. Infectivity of the sRNA mutant strain relative to its parent and genetic complementa.
| Strain | Ear | Inoculation site (skin)b | Lymph node | Heart | Bladder | Joint | Total sites | % Positive Tissues |
|---|---|---|---|---|---|---|---|---|
| ML23 pBBE22luc | 10/12 | 11/12 | 12/12 | 11/12 | 12/12 | 12/12 | 68/72 | 94% |
| DM103 pBBE22luc |
1/13 | 12/13 | 13/13 | 1/13 | 9/13 | 11/13 | 47/78 | 60% |
| DM113 pBBE22luc |
12/12 | 10/12 | 12/12 | 12/12 | 12/12 | 12/12 | 70/72 | 97% |
a Dose of 103 per strain of B. burgdorferi tested.
b Skin from abdomen (inoculation site).
Fig 5. Quantitative assessment of B. burgdorferi load from infected mouse tissues.

Quantitative PCR (qPCR) of tissues from mice infected with the parental strain (black circles; n = 5), the ittA mutant (red squares; n = 5), and the genetic complement (blue triangles; n = 4) was used to enumerate borrelial genomic equivalents relative to the murine samples. Mice were infected with 103 dose of B. burgdorferi strains for 21 days. Tissues tested are shown at the bottom: PS for peripheral ear skin; SK for abdomen skin at the site of infection; LN for Lymph node; HT for heart; and JT for the tibiotarsal joint. The results are represented as the number of borrelial recA genomic copies per 106 mouse β-actin copies. The horizontal line in each data set depicts the mean value. Each data point shown represents an independent sample from a single mouse tissue assayed in triplicate and averaged. * p < 0.05; *** p < 0.001.
Transcriptional profile of the ittA mutant
Trans-acting sRNAs often regulate gene expression by base-pairing with target mRNAs, affecting their stability and/or translation. Given the infectivity defect observed, we hypothesized that ittA regulates the expression of gene(s) that are important for infectivity and, specifically, skin and heart tissue colonization. We performed RNA-seq to compare the global transcriptional profile of the parent and the ittA mutant in an unbiased manner under in vitro conditions that mimic mammalian-like conditions. It is important to note that the cultures used for this analysis were also utilized for the subsequent proteomic analysis described below.
In our transcriptional comparison we detected 1,343 transcripts total (S1 Table). From this group, we found 92 transcripts that exhibited more than +/- 1.4-fold change and were statistically significant (Padj <0.05; S2 Table). Further evaluation showed that the ittA mutant exhibited statistically significant, 2-fold change expression of 19 transcripts when compared to the parent (Fig 6; shown as red spots; see S3 Table). Of the 19, 13 transcripts were upregulated in the sRNA mutant strain, including transcripts predicted to be involved in pathogenesis (vraA [bbi16] [66]), as well as bba66, a locus required for effective transmission from the tick vector to mice and expressed during mammalian infection [67,68]. In addition, there were 6 transcripts downregulated in the sRNA mutant strain, including ospD (bbj09), ospA (bba15), and oms28 (bba74) (Fig 6). It is important to note that 73 additional genes showed statistically significance (Padj <0.05) with a fold change range between +/-1.4 and +/-1.9 (Fig 6; shown as gold spots; see S4 Table). It is interesting to note that 64 of the significantly affected transcripts are from genes that are known to be BosR/RpoS regulated and 79.7% of them are upregulated in the ittA mutant. Overall, these results suggest that the ittA sRNA exerts a regulatory effect by either destabilizing or stabilizing several target transcripts or by indirectly altering the expression of these transcripts via an unknown regulatory mechanism. The net effect in the mutant cells lacking ittA is a dysregulation of several genes that may contribute to the decrease in fitness and in a reduction in infectivity potential observed.
Fig 6. Differential gene expression in the parent relative to the ittA mutant.

The volcano plot depicts log2 fold change on the x-axis and False Discovery Rate adjusted p value (q -value) on the y-axis. The parent and ittA mutant strain were grown in biological triplicates in vitro using conditions that induce proteins important for mammalian infection. RNA was purified, and the samples subjected to RNA-seq. Single genes are depicted as dots. Of the transcripts, 19 achieved significance with p-value <0.05 and with greater than 2-fold change in the mutant relative to the parent strain. Of the differentially expressed genes, 13 were downregulated and 6 upregulated in the parent relative to the mutant. Yellow spots (73 total) are transcripts that achieved significance with a p-value <0.05 and a fold change range between +/- 1.4 to 1.9.
qRT-PCR confirms differential expression of transcripts in the ittA mutant
We next utilized quantitative RT-PCR (qRT-PCR) to assess the expression of the candidate genes identified to validate our RNA-seq analysis (Fig 6) using the parent, ittA mutant, and complement strains grown in vitro under mammalian-like conditions. A constitutively expressed gene, flaB, which was not affected by any of strains tested in this study, was used for normalization [17,18]. As a control, we tested ospC and found that this transcript was not affected by the loss of ittA, as predicted. Five genes were tested, bba66, vraA, oms28 (bba74), ospD, and ospA (Fig 7). The qRT-PCR confirmed the upregulation or downregulation observed in the RNA-seq experiment for each gene (Figs 6 and 7) for the parent and ittA mutant. Unexpectedly, only bba66 was restored to wild type levels in the complemented strain (Fig 7). Notably, bbd18 was one of the 19 genes that were differentially regulated. Our qualitative assay demonstrated bbd18 was expressed in the mutant strain, but our RNA-seq data suggest it may be downregulated in the ittA mutant strain. We also attempted to quantify bbd18 transcripts by both qRT-PCR and Northern blot analysis but were not successful using in vitro grown B. burgdorferi. Therefore, it is unclear whether bbd18 expression is altered due to the ittA strain construction or ittA-dependent gene regulation.
Fig 7. Quantification of transcripts by qRT-PCR.
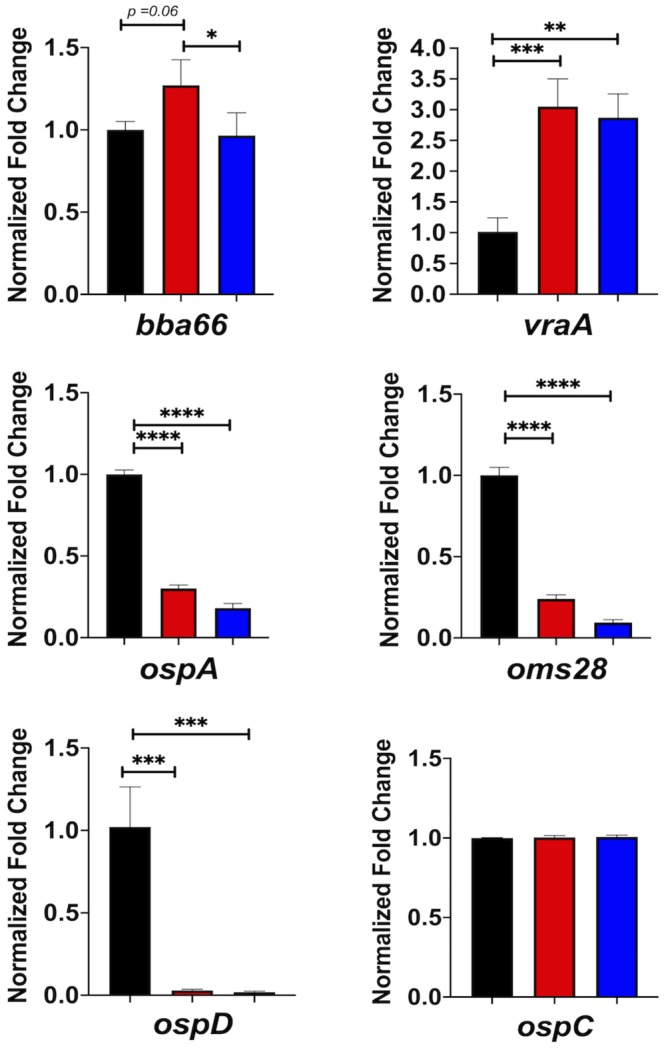
Quantitative RT-PCR (qRT-PCR) of the parent (black bars), ittA mutant (red bars), and complemented strain (blue bars) were performed for a subset of transcripts indicated at the bottom of each panel. The data for all samples were normalized to the endogenous control, flaB, whose transcription was not affected by the conditions used in the experiment. The error bars indicate standard error. Significance is denotated as * p < 0.05, ** p < 0.01, *** p < 0.001. **** p < 0.0001.
A limitation in the RNA expression data is that expression of genes differentially expressed in the ittA mutant was not restored in the complemented mutant. It is possible that ittA is not processed at native levels in the complemented strain. The Northern blot analysis of ittA demonstrated that the major sRNA species is the same size and comparable to steady-state levels relative to the parent strain. However, when the Northern blot was overexposed, several bands unique to the complement sample were detected in addition to the major product (S3 Fig). Notably, several of these larger RNA transcripts differ between the parent and the complement strain, suggesting that these differences may affect the in vitro readouts tested. Furthermore, the downregulation of bbd18 in the ittA mutant strain may be affecting gene regulation independently of ittA.
ittA alters the borrelial proteome
sRNAs often regulate translation initiation by occluding or releasing the ribosome binding site of transcripts [44,48,69]. In addition, sRNAs can bind within coding regions of transcripts resulting in regulatory effects. We hypothesized that ittA might modulate translation efficiency of some transcripts at the post-transcriptional level [44,48,69]. To address this, we used a global proteomic screen to identify and quantify the entire borrelial proteome of both the parent and the ittA mutant under conditions that mimic mammalian-like conditions (Fig 8). A total of 718 proteins were detected and are listed in S5 Table. Of the 718 proteins, 637 were classified with False Discovery Rate (FDR) of 1% and 81 with FDR of 5%.
Fig 8. Proteomic evaluation of the parent relative to the ittA mutant.

Tandem Mass Tags (TMT) was used to determine the relative abundance of the total proteome of three biological replicates of parent and ittA mutant strains grown under conditions that induce proteins important during mammalian infection. Volcano plot depicts log2 fold change (x-axis) and log10 adjusted p-value (y-axis) of proteins identified from parent versus the ittA sRNA mutant strain. Single proteins are plotted as dots. Proteins outside of the red dashed boxes are significant. Red spots have a +/- 2 fold change difference in the parent strain relative to the ittA mutant strain and a p-value < 0.05. Yellow spots are proteins that achieved significance of p-value < 0.05 and a fold change range between +/- 1.4 to 1.9. OspD and Oms28 were found to be significantly higher in abundance in the parent relative to the ittA mutant.
The samples used for proteomic analyses were isolated from the same cultures as used for the RNA-seq analysis shown in Fig 6. From this assessment we found two proteins, OspD and Oms28, that were affected the most by the loss of ittA; specifically, they were decreased 2.3- and 2.1-fold, respectively, in the ittA mutant (Fig 8, red spots). In total, 69 proteins were significantly altered by the loss of ittA, but 67 of these proteins were in the range of fold change between 1.4- to 1.9-fold (Fig 8, gold and red spots, respectively, and S6 Table). Only one additional protein was made at a statistically higher level in the parent strain relative to the ittA mutant (BBG01); all other proteins that fit these criteria were made at higher levels in the ittA mutant (Fig 8 and S6 Table). Interestingly, one of the proteins upregulated with fold change of 1.9 in the ittA mutant is BBA66, as well as several additional RpoS-regulated surface proteins. Here a subset of RpoS-regulated proteins is synthesized at higher levels in the ittA mutant and thus may place the spirochetes at a selective disadvantage since their ectopic production may make them targets for antibody-mediated killing. Presumably this effect is amplified in vivo thereby mediating the phenotype observed. However, the complement corrects for this in vivo by rescuing the mutant in a manner that is not observed in vitro.
Since both the ospD and oms28 transcripts were downregulated in the ittA mutant strain in RNA-seq data and proteomic data (Figs 6 and 8), we hypothesize that ittA stabilizes these transcripts and thus allows for the increased translation of OspD and Oms28. We hypothesize that, in the absence of ittA, the ospD and oms28 transcripts are destabilized, resulting in reduced levels of OspD and Oms28 proteins. To determine levels of OspD and Oms28 proteins in the ittA mutant strain, we performed Western blot analysis of proteins lysates from parent, the ittA mutant, and the complement grown in vitro under mammalian-like conditions probed with polyclonal antibodies directed against OspD and Oms28. We observed lower levels of OspD and Oms28 in the sRNA mutant than the parent, consistent with the proteomic analysis (Fig 9). Of note, the complement did not produce OspD to levels similar to that observed in the parent strain, consistent with the expression of ospD not being restored in the qRT-PCR analysis (Fig 7). For Oms28, the complement produced higher proteins levels than the mutant but not to the same level as the parent. These results demonstrate partial complementation of the ittA mutant relative to the parent strain using both qRT-PCR and Western immunoblot metrics of assessment.
Fig 9. Evaluation of OspD and Oms28 in B. burgdorferi lacking ittA.

Protein lysates were derived from parent, ittA sRNA mutant (ittA:StrR), and complement (Comp) strains grown under conditions that induce proteins important during mammalian infection in vitro and probed against anti-Oms28 (panel A) and anti-OspD (panel B). The production of FlaB was used as a loading control for both immunoblots shown.
Discussion
Small non-coding RNAs (sRNAs) have emerged as an additional mechanism for the regulation of transcript levels or protein function. Specifically, base-pairing sRNAs, such as trans-acting sRNAs, bind to transcripts and modulate their expression by either altering their stability or their ability to be translated [48,50,70]. Alternatively, protein-binding sRNAs can bind to cellular proteins and modify their activity [45,46,49]. Regulation via sRNAs is fast acting and functions as an additional layer in response to environmental signals [44,71]. Despite the significant effect on translation efficiency, it is important to note that the loss of a single sRNA species often has a limited effect on measurable phenotypes. This is likely due to the subtle effect of the sRNA-mRNA interaction and the lack of an absolute effect seen by this event; that is, transcripts are not entirely inhibited or activated depending on the relative abundance of these RNA molecules and the fine-tuning that is associated with the sRNA-mRNA transient interactions [72,73]. Modest phenotypes may also be due to redundancy, as a given mRNA can be regulated by multiple sRNAs; therefore, the elimination of a single sRNA may not drastically alter mRNA turnover or translation [44,72,73]. Nevertheless, sRNAs encoded by pathogenic bacteria are recognized as important players in adaptive responses with some identified as important effectors in regulatory pathways [44,69,74].
Due to the enzootic nature of B. burgdorferi and its ability to quickly adapt to environmental factors encountered during their lifecycle, we hypothesized that B. burgdorferi use sRNAs to affect post-transcriptional regulatory processes that calibrate these responses. Recently, 1,005 sRNAs were identified in B. burgdorferi, suggesting that these spirochetes exploit this type of genetic regulation [40]. However, how B. burgdorferi utilizes these sRNA candidates remains largely unknown. In this study, we inactivated a trans-acting, intergenic (IG) sRNA, designated ittA, and demonstrated that this sRNA is required for optimal infection, as well as dissemination to and/or survival in distal tissues. It was further shown that transcript and protein production are affected when the ittA-encoded sRNA is not expressed in B. burgdorferi. To our knowledge, no other intergenic sRNA from B. burgdorferi has been characterized to this extent.
As a first step to link a B. burgdorferi trans-acting intergenic sRNA to an infectivity phenotype, mice were infected with the transposon library of B. burgdorferi strain B31 [55] and decreased infectivity was scored using Tn-seq [56]. These data were compared against the recently described B. burgdorferi sRNA-specific library to identify intergenic sRNA species that mapped to existing Tn mutants. We tested each identified intergenic sRNA Tn mutant individually using the experimental murine infection model. One sRNA mutant, initially described as SR0736 [40] and renamed ittA herein, exhibited the most severe infectivity defect. Subsequent in vivo imaging of an independently derived mutant in ittA confirmed the attenuated phenotype observed for the qualitative infectivity assessment (Table 3) relative to the parent and complement (Fig 4). The inactivation of ittA affected tissue tropism of B. burgdorferi where spirochetes were cultured out of peripheral ear skin and heart in less than 8% of the samples tested (Table 3). Even though the qualitative assessment of infection showed some degree of colonization in the remaining tissues (except the lymph node), the total bacterial load of B. burgdorferi was significantly lower for the ittA mutant relative to the parent and complement strains (Fig 5). These data indicate that ittA is needed for optimal borrelial colonization and/or dissemination. Furthermore, it is important to note that the ittA complement strain DM113 effectively restored infectivity in a manner indistinguishable from the parent using infection as our readout, indicating that the phenotype of the ittA mutant was due to its absence and not a second site mutation.
To globally assess the role of the ittA sRNA we used both transcriptomic and proteomic approaches to identify transcripts and proteins that are altered in its absence, respectively. Two proteins, OspD and Oms28 (BBA74), were produced at lower levels in the ittA mutant consistent with their respective transcripts being reduced in the mutant background. We hypothesize that the ittA sRNA might bind to the ospD and oms28 transcripts, prevents their degradation, and consequently enhances OspD and Oms28 translation. Whether this sRNA-mRNA interaction occurs and affects the ospD and oms28 transcripts directly, or if this effect is due to other ittA-regulated targets that indirectly affect this process, remains to be determined.
We aimed to validate five of the nineteen transcripts identified as being altered due to the loss of the ittA sRNA: ospA, ospD, oms28, vraA, and bba66. Of these five, bba66 and vraA have been associated with some aspect of mammalian-based virulence; four of these genes, including ospA, ospD, and oms28, as well as bba66, are expressed in the arthropod vector and some have significant phenotypes in this stage of the B. burgdorferi life cycle, particularly ospA and bba66 [67,68,75–81]. OspA is a well characterized surface lipoprotein that functions as an adhesin in the midgut of Ixodes ticks [8,77,78,82]. BBA66 is a lp54 encoded surfaced expressed lipoprotein that is upregulated during nymph blood meal and is highly expressed in the mammal for an extended period of time, suggesting a role in persistence [68]. Needle inoculation of mice with B. burgdorferi bba66 mutants results in lower bacteria burden in joint tissue and significantly lower joint swelling relative to the parent strain, suggesting that BBA66 contributes to borrelial-mediated inflammation [67]. Mutants in bba66 are acquired by larvae and persist through molting, but were significantly impaired in their ability to infect mice when introduced by tick bite compared to that of mice fed upon by ticks seeded with wild type B. burgdorferi, suggesting a role for BBA66 in transmission [67]. Recently, an additional function of BBA66 was found, whereby BBA66 binds to the neuroglial and macrophage protein Daam1 [83]. Daam1 is in the formin family of proteins involved in regulating cytoskeletal reorganization in mammalian cells [84]. A prior study showed that Daam1 co-localized to pseudopods on macrophages that processed B. burgdorferi by coiling phagocytosis [85]. A more recent report showed that BBA66 mediated the attachment of B. burgdorferi to these cells via the Daam1 protein [83]. Interestingly, the bba66 mutant exhibited reduced levels of internalized B. burgdorferi while borrelial cells that produced greater amounts of BBA66 were phagocytosed more efficiently [83]. In the RNA-seq data shown (Fig 6), higher expression of bba66 was observed in the sRNA mutant compared to the parent. In addition, in Fig 8 and S6 Table, the mutant has a 1.9 fold change increase in protein abundance of BBA66 relative to the parent. It is tempting to speculate that the increase of bba66 expression in the sRNA mutant may lead to increased phagocytosis and local clearance of B. burgdorferi by macrophages as a result of BBA66-mediated binding of Daam1. This could help explain the lower bacteria burden in the majority of the mice tissues examined in the sRNA mutant, where BBA66 levels are predicted to be greater (Fig 4). In Fig 10, the predicted secondary structure of ittA and interaction with bba66 is displayed. Whether this sRNA-mRNA interaction occurs and affects bba66 transcript directly remains to be determined. Taken together, the dysregulation of bba66 expression may be an important factor in the phenotype observed for the ittA mutant (Figs 4 and 5).
Fig 10. IntaRNA (Freiburg RNA Tools) predictions of interactions of the ittA sRNA with the bba66 transcript.

A. A predicted secondary structure of the ittA sRNA (mfold). B. Potential interaction of the ittA sRNA (lower) within the coding sequence of bba66 (upper). The red depicts the bba66 stop codon.
In addition to BBA66, several BosR/RpoS-regulated genes and the proteins they encode are present in the ittA mutant in vitro at higher levels than the parent (Figs 6 and 8), suggesting that the loss of this sRNA leads to mis-regulation of surface exposed proteins during mammalian infection. Additional RpoS-regulated targets are subject to mis-regulation in the ittA mutant include OspC, DbpA, DbpB, and BBK32, which all contribute to borrelial pathogenesis [61,86–93]. That these virulence-associated proteins are produced more in the mutant relative to the parent suggests that the defect here may be due to ectopic mis-regulation that place the spirochete at a selective disadvantage. Along these lines, previous studies have shown that the mis-regulation of ospC, in the form of constitutive expression, results in the clearance of B. burgdorferi in experimental infection [86,87]. It is thus likely that a coordinated mis-production of several of these proteins could have a detrimental synergistic effect that clears B. burgdorferi in experimentally infected mice.
One significant and confounding issue stemmed from the incomplete in vitro complementation of the ittA mutant. Notably, while the cis-acting complement restored infectivity to wild type levels as assessed by all in vivo metrics tested, e.g., in vivo imaging as well as both qualitative and quantitative measure of infected tissues, in vitro indicators were not fully reinstated. Surprisingly, during in vitro growth, several of the transcripts and proteins affected by the absence of the ittA sRNA were not restored to wild type levels in the complement. It is possible that factors impacting the genetic context for ittA (e.g. the presence of the downstream gentamicin cassette) or secondary genetic changes in the complemented strain may affect ittA sRNA expression, processing, or stability during in vitro culture conditions. The in vitro complement strain results indicate some caveat to the mutant or complement strain construction (discussed below). However, the in vivo data demonstrate that ittA is an important factor for infectivity. As mentioned previously, additional RNA species containing ittA sequences were detected by Northern blot analysis in the complemented strain. Additional experimentation is planned to resolve this issue.
Of the five targets tested for transcript levels, only bba66 was restored to wild type levels in the complemented strain. A predictive algorithm, IntaRNA (Freiburg RNA tools, [94]) indicated potential ittA binding site within the bba66 transcript with the 5’ loop of the predicted ittA secondary structure. We hypothesize that the ittA-bba66 RNA interaction may lead to RNase-dependent degradation of the bba66 transcript (Fig 10), since bba66 is expressed in higher amounts in the ittA mutant than the parent and complement strains. Further study is needed to determine whether this mechanism is active in the case of bba66 regulation.
At the protein level, we were limited by available antibody reagents and, of the two candidates tested, OspD, and Oms28, only Oms28 appeared partially restored to the levels observed in the parent strain in Western immunoblotting. Although the cis complementation strategy was designed to create as little a difference relative to the parent strain as possible, the addition of an additional antibiotic resistance marker may alter the expression of the ittA sRNA in a manner that yields an atypical regulatory response. In this regard, the Northern blot shown in Fig 3, when exposed longer, showed a unique unprocessed form of the ittA sRNA (S3 Fig). Whether this contributes to the complementation defect remains to be determined. Another possibility is that the overlap of the distal 5’ transcription start site of ittA with the transcriptional start site of bbd18 on the opposite DNA strand (Fig 1) results in some interference that affects the ability of ittA to carry out its regulatory effect(s) or affects the levels of BBD18. The bbd18 transcript was down-regulated in the ittA mutant in our transcriptomics analysis (Fig 6), but no significant difference in protein abundance was observed for BBD18 between the strains (S5 Table). Unfortunately, the transcriptomics data could not be validated for bbd18 because we could not detect the bbd18 transcript by Northern blot or qRT-PCR. BBD18 downregulates RpoS post-transcriptionally when overexpressed [33] and several RpoS-dependent genes are up-regulated in the ittA mutant strain. These RpoS-dependent genes may be affected by decreased levels of BBD18 and/or loss of ittA in the ittA mutant strain. This is consistent with the induction of several RpoS-regulated targets that are made at higher levels in the ittA mutant, including OspC, DbpB, and BBA66 (Fig 8). Why this complementation defect is limited to our in vitro studies but not observed in vivo is not clear. Further studies will be needed to address this experimental conundrum.
In conclusion, sRNA-mediated regulation has been proven to be complex but important in fine-tuning gene expression in many bacteria species [95,96]. Our findings indicate that the ittA sRNA is required for optimal infectivity and that its absence alters the expression and production of a number of genes and proteins, respectively. One possibility posits that ittA may help in a quick adaptive response that alters the translation efficiency in a number of transcripts. The effect of each gene regulated by ittA individually is subtle but collectively the net effect may result in the dysregulation of these targets, yielding a synergistic response that results in an attenuated phenotype. Here, there is an additional layer of complexity, seen in the form of tissue tropism, such that colonization at remote skin and cardiac tissue sites is impaired. This work thus suggests that sRNA-based regulation via ittA is important in maintaining appropriate levels of gene expression that promote B. burgdorferi colonization and dissemination during experimental infection.
Materials and methods
Bacteria strains and culture conditions
Bacterial strains and plasmids used in this study are described in Table 4. Escherichia coli strains were grown aerobically at 37°C in Luria Broth (LB). Concentration of antibiotics used in E. coli are as follows: kanamycin, 50 μg/ml; spectinomycin, 50 μg/ml; and gentamicin, 5 μg/ml. B. burgdorferi strains were grown in BSK-II media supplemented with 6% normal rabbit serum (Pel-Freez Biologicals, Rogers, AR) under conventional microaerophilic conditions at 32°C, pH 7.6, under 1% CO2 atmosphere, or conditions that induce genes and gene products important in the mammalian environment, namely, 37°C, pH 6.8, under 5% CO2 atmosphere. Borrelia burgdorferi B31 ML23 [60] and derivative strains were grown under antibiotic selective pressure, dependent on genetic composition, with either kanamycin at 300 μg/ml, streptomycin at 50 μg/ml, or gentamicin at 50 μg/ml. B. burgdorferi 5A18NP1 [58] was grown in the presence of kanamycin at 300 μg/ml and the B. burgdorferi transposon mutants [55] were grown in the presence of both gentamicin at 50 μg/ml and kanamycin at 300 μg/ml.
Table 4. Plasmids and strains used in this study.
| E. coli strains | Genotype | Comments/Ref. |
|---|---|---|
| Mach-1-T1 | F- ϕ80(lacZ)ΔM15 ΔlacX74 hsdR(rK-mK+) ΔrecA1398 endA1 tonA | Invitrogen |
| B. burgdorferi strains | ||
| ML23 | B. burgdorferi B31 clonal isolate missing lp25; parent strain. | [60] |
| ML23 pBBE22luc | Clonal isolate of strain B31 lacking lp25; shuttle vector encodes bbe22 and B. burgdorferi codon optimized luc gene under the control of a strong borrelial promoter (PflaB-luc). | [61] |
| DM103 | ML23 IG sRNA ittA::StrR. | This study |
| DM103 pBBE22luc | DM103 background carrying shuttle vector pBBE22luc; StrR and KanR. | This study |
| DM113 | ML23 containing intact sRNA ittA with genetically linked GenR complemented in cis in same genetic location of lp17 (between genes bbd18 and bbd21). | This study |
| DM113 pBBE22luc | DM113 background carrying shuttle vector pBBE22luc; GenR and KanR. | |
| 5A18NP1 | Borrelia burgdorferi B31 clone missing lp28-4 and lp56 with disruption of bbe02::KanR. | [58] |
| Plasmids | ||
| pCR-Blunt | pCR-Blunt vector, KanR, ZeocinR. | Invitrogen |
| pKFSS1 | B. burgdorferi shuttle vector containing PflgB-StrR cassette; SpecR in E. coli, StrR in B. burgdorferi. | [97] |
| pBSV2G | B. burgdorferi shuttle vector containing PflgB-GenR cassette. | [98] |
| pBBE22luc | Borrelial shuttle vector containing bbe22 and B. burgdorferi codon-optimized luc gene under the control of a strong borrelial promoter (PflaB-luc). | [61] |
| pCR2.1Bactin | Murine β-actin gene cloned into pCR2.1 vector; KanR. | [61] |
| pCR2.1recA | B. burgdorferi recA gene cloned into pCR2.1 vector; KanR. | [61] |
| pDM103 | B. burgdorferi IG sRNA ittA mutant construct. Contains sequences 1280 bp upstream of the sRNA, the PflgB-StrR cassette from pKFSS1 inserted into the sRNA sequence, and sequences 1233 bp downstream of the sRNA; pCR-Blunt vector backbone; StrR and KanR. | This study |
| pDM113 | Complement construct of the sRNA ittA located in between genes bbd18 and bbd21. Contains sequences 1359 bp upstream of sRNA, a gentamicin cassette from pBSV2G, and 1171 sequences downstream of the sRNA; pCR-Blunt vector backbone; GenR and KanR. | This study |
Transposon mutagenesis screen
Transposon mutants were obtained from the arrayed B. burgdorferi library [55]. In order to conduct Tn-seq experiments, a single pool containing all of the Tn mutants was generated by combining sub-pools containing 70–80 individual Tn mutants each [55,99,100]. The Tn library was grown for 48 hours in the presence of kanamycin and gentamicin. Cell density was determined by dark-field microscopy. Groups of six 9–14 week old C3H/HeJ mice (Jackson laboratories) were injected in the right flank with 5x105 B. burgdorferi by needle inoculation. As a control to prevent in vitro growth defects from affecting the results of the in vivo screen, 5x105 organisms from the inoculum used to inject the mice were cultured in 12 ml BSK-II broth supplemented with kanamycin and gentamicin. The in vitro control cultures were grown for 3 days to parallel time in culture for the tissue cultures. The bacteria in the culture were collected using centrifugation for 20 minutes at 3,000 x g and the pellet frozen at -80°C.
The mice infected with the transposon library were sacrificed two weeks post-infection. The tibiotarsal joint closer to the inoculation site was removed under aseptic conditions. The tibiotarsal joints from each group of injected mice were cultured together in 12 ml of BSK-II broth supplemented with kanamycin and gentamicin. The cultures were checked daily for growth. When the density of the cultures reached late exponential phase, the bacteria were centrifuged and the pellet frozen as described above.
Genomic DNA was obtained from the frozen bacteria pellets using a DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA) as per the manufacturer’s instructions. For the in vivo samples, genomic DNA obtained from the organ cultures of different groups of infected mice were pooled in relative proportions such that each library for sequencing represented the bacteria recovered from 24 mice. For the in vitro control samples, DNA recovered from the cultures of the inoculums were pooled to match the groups of mice pooled to make each of the in vivo sample libraries. Libraries for sequencing were constructed as described previously [100]. Briefly, an aliquot of the genomic DNA was placed in a 2 ml microfuge tube and sheared by sonication. Cytosine tails (C-tails) were added to 1 μg sheared DNA using terminal deoxynucleotidyl transferase (TdT, Promega, Madison, WI). Transposon containing fragments were amplified in a PCR containing DNA from the TdT reaction as template and primers specific to the ColE1 site on the 5’ end of the transposon, pMargent1 and the C-tail, olj376 (S7 Table). To prepare the DNA for sequencing and further amplify the transposon-genomic DNA junction, a nested PCR reaction was performed using DNA from the first PCR as a template, a primer specific to the transposon end, pMargent2 (S7 Table), and an indexing primer containing the specific sequences required for sequencing on an Illumina platform and where NNNNNN represents a six-base-pair barcode sequence allowing samples to be multiplexed in a single sequencing lane. Within an experiment, a unique indexing primer was used for each individual B. burgdorferi sample. A majority of the PCR products were between 200 and 600 bp. The sequencing libraries made from the in vitro and in vivo samples were pooled at equal concentrations prior to sequencing. The pooled libraries were sequenced on an Illumina HiSeq 2500 at the Tufts University Core Facility as 50 bp single-end reads using the custom sequencing primer pMargent3 and the standard Illumina index primer.
Sequence data analysis was performed as described previously [100]. Briefly, sequenced reads were aligned to the B. burgdorferi B31 genome using the short read aligner Bowtie. A custom script was then used to compile the resulting SAM files into a Microsoft Excel spreadsheet with the number of reads aligned to each site or annotated gene listed. The frequency of transposon mutants with insertions in a particular site or gene in the population was assessed by determining the number of sequence reads aligned to that mutant or gene as a percentage of all reads in a given sample.
Genetic inactivation of the B. burgdorferi ittA intergenic sRNA
The intergenic (IG) small regulatory RNA (sRNA) located between genes bbd18 and bbd21 in lp17 was insertionally inactivated via homologous recombination by replacing the 3’ end of the sRNA (nucleotides 11,820–11,850) with the PflgB-aadA (streptomycin resistant; StrR) antibiotic cassette [97]. This sRNA was designated SR0736 by Popitsch et al. [40]. Based on the data obtained herein, we renamed the SR0736 sRNA ittA. DNA sequences that flanked the ittA sRNA locus were amplified using PCR with PrimeSTAR GXL polymerase (Takara, Mountain View, CA). For the upstream fragment, 1280 bp were amplified using primers US-F and US-SpecR (see S7 Table). The 1266 bp fragment containing PflgB-StrR was PCR amplified from pKFSS1 [97] using the oligonucleotide primers pair US-SpecF and SpecDS-R (S7 Table). An additional 1,233 bp PCR product, which amplified sequences downstream from the ittA sRNA, was engineered with primers SpecDS-F and DS-R (S7 Table). All three fragments had 20 base pair overlap sequences and were assembled by overlap PCR [63,64,101].
For the creation of the sRNA cis complement strain, the PflgB-StrR cassette of the sRNA mutant was replaced on lp17 using the native ittA-containing sequence with a linked PflgB-GentR marker downstream of the sRNA [98]. The US-F and compUS-gentR primers were used to PCR amplify the 1359 bp portion containing ittA (S7 Table). The 983 bp gentamicin cassette from pBSV2G [98] was produced using primers compUS-gentF and compgentDS-R (S7 Table). A 1171 bp region downstream from ittA was amplified using the primers compgent-DSF and compDS-R (S7 Table). As before, all three fragments had 20 base pair overlap sequences and were assembled by NEBuilder HiFi DNA Assembly Master Mix (New England Biolabs, Ipswich, MA). All constructs were verified by Sanger sequencing prior to transformation into the B. burgdorferi strain B31 derivative ML23 [60] or DM103.
Transformation of B. burgdorferi
B. burgdorferi were made competent for DNA transformation as previously described [63,64,91,102]. Prior to transformation via electroporation, all plasmid constructs were linearized with XhoI. Following antibiotic selection for the desired strain, putative transformants were tested for the presence of the genetic constructs using a PCR-based screen using primers from S7 Table. Subsequent mutant or complemented strains were tested for B. burgdorferi strain B31 plasmid content by PCR [18].
Infectivity studies and bioluminescent imaging
Infectivity studies were performed as previously described [61,63,64,91]. Briefly, 8-week-old C3H/HeN female mice were inoculated with 103 organisms of the B. burgdorferi parent strain ML23/pBBE22luc, the sRNA ittA::StrR strain DM103/pBBE22luc, or the genetic complement strain DM113/pBBE22luc, by intradermal injection in the abdomen skin. For the parent and complement strains, twelve mice were infected, for the sRNA inactivation strain, thirteen mice were infected.
The bioluminescent imaging was performed as done previously [61,63,64]. Briefly, five mice were imaged for the parent and mutant strains and four mice were imaged for the complement strain per experiment. The mice were injected intraperitoneally with 5 mg of D-luciferin dissolved in 100 μL of PBS 10 minutes prior to imaging with an IVIS Spectrum live animal imaging system (Caliper Life Sciences, Hopkinton, MA), with the exception of one mouse that was infected with each B. burgdorferi strain tested but did not receive D-luciferin substrate. This mouse served as a negative control for background luminescence [61]. Imaging of the mice was performed 1 hour and at 1, 4, 7, 10 14 and 21 days post-infection [61]. After 21 days, the mice were sacrificed and the ear, abdominal skin, inguinal lymph node, heart, bladder, and tibiotarsal joint tissues were collected from each mouse aseptically for in vitro cultivation. From each of these mice the remaining ear, inguinal lymph node, and tibiotarsal joint tissues were collected for qPCR analysis. For the abdominal skin, an adjacent piece of skin was collected for qPCR relative to that used for the cultivated sample. The heart of each mouse was aseptically divided vertically with a sterile scalpel and one half was used for culture and the other for qPCR. These samples were then prepared for qPCR analysis of B. burgdorferi burden as described previously [61,91].
RNA Isolation for conventional RT-PCR and qRT-PCR
Three independent cultures of B. burgdorferi strains ML23 [60] (parent), the sRNA mutant strain DM103, and the genetic complement strain DM113, were grown to mid-log phase of 5 x107 cells per ml at either mammalian-like conditions, defined as: 37°C, pH 6.8, under 5% CO2 or at conventional microaerophilic conditions of 32°C, pH 7.6, under 1% CO2. The cultures were centrifuged at 4500 x g for 20 minutes at 4°C, washed with 1 ml PBS, and centrifuged at 14,000 rpm for 15 minutes. The pellet was resuspended in 100 μl of sterile water and 300 μl of TRIzol (Invitrogen, Carlsbad, CA) was added prior to employing Direct-zol RNA Miniprep (Zymo Research, Irvine, Ca, USA) for total RNA isolation. The resulting RNA was treated with DNAse I (Roche, Indianapolis, IN) and RNAsin (Promega, San Luis Obispo, CA) to eliminate contaminating DNA and inhibit RNAse activity, respectively. To ensure that there was no contaminating genomic DNA in the cDNA reaction mixture, samples without reverse transcriptase were also included as controls. For conventional RT-PCR, 200 ng of total RNA from B. burgdorferi strains grown under conventional microaerophilic conditions were used to reverse transcribe into cDNA using primer sRNA R (S7 Table) and SuperScript III (Thermo Fisher Scientific, Waltham, MA). Primers sRNA F and sRNA R (S7 Table) were used to amplify ittA. For conventional RT-PCR of genes bbd18 and bbd21, 1 μg of total RNA from B. burgdorferi strains grown under mammalian-like conditions were used for reverse transcription into cDNA using random primers (Thermo Fisher Scientific, Waltham, MA) and SuperScript III (Thermo Fisher Scientific, Waltham, MA). Oligonucleotide primers for bbd18 and bbd21 (S7 Table) were used to amplify the genes.
For qRT-PCR analysis, 1 μg of total RNA from B. burgdorferi strains grown under mammalian-like conditions were used for reverse transcription into cDNA using random primers (Thermo Fisher Scientific, Waltham, MA) and SuperScript III (Thermo Fisher Scientific, Waltham, MA). Oligonucleotide primers from S7 Table were used to amplify specific B. burgdorferi strain B31 targets. Template cDNAs generated by the strains under the same conditions were normalized by using the constitutively expressed flaB gene as previously described [10,18] with the ΔΔCt method, in which the quantity of a given transcript is determined by the equation 2 –ΔΔCt, where Ct is the cycle number of the detection threshold.
Northern blot analysis
Northern blotting of SR0736/ittA was conducted using the same probe described previously [40,103] from RNA extracted from B. burgdorferi strains grown under conditions that mimic mammalian-like conditions.
DNA extraction of B. burgdorferi from infected tissues and qPCR analysis
Total DNA was isolated from ear, abdominal skin, inguinal lymph node, heart, and tibiotarsal joint using Roche High Pure PCR template preparation kit (Roche, Indianapolis, IN) as previously described [63,64,91]. Total DNA of 100 ng was used for each qPCR reaction. Quantitative real-time PCR analysis was conducted using the Applied Biosystems StepOnePlus Real-Time PCR system. B. burgdorferi genome copies and mammalian cell equivalents were determined using oligonucleotide primers in S7 Table.
RNA Sequencing (RNA-seq)
Three independent cultures of B. burgdorferi cells were grown to mid-log phase of 5 x 107 cells per ml at 37°C, pH 6.8 and 5% CO2, e.g., in vitro conditions that mimic mammalian-like infection. Cells were centrifuged at 4,500 x g for 30 minutes at 4°C. Approximately 1 x 109 cells were lysed in 1ml of TRIzol (Invitrogen, Carlsbad, CA) and RNA extracted following manufacturer’s instructions. RNA was checked for quality using the Agilent TapeStation 2200 standard RNA screen tape and quantified using the Qubit 2.0 Broad Range RNA assay. Total RNA was normalized between all samples for sequencing library preparation using the TruSeq Stranded Total RNA library preparation kit with ribosomal depletion. Each sample was uniquely barcoded, then the libraries were pooled at equal concentrations. Library pools were sequenced on a 2 x 75 bp paired-end sequencing run generating ~15 million reads/sample by the Texas A&M Institute for Genome Sciences and Society (TIGSS).
After sequencing, a total of approximately 90.7 million 75 bp paired-end raw sequencing reads were checked to trim any adapter sequences and low quality bases using Trimmomatic [104]. Reads were scanned with a sliding window of 5 bp, cutting when the average quality per base drops below 20, then trimming reads at the beginning and end if base quality drops below 20, and finally dropping reads if the read length is less than 50. Thereafter, approximately 90 million filtered reads (99.2%) were mapped to the Borrelia burgdorferi strain B31 genome assembly (accession: GCF_000008685.2) using HISAT version 2.1.0 [105]. Average mapping rate was about 85.8% (S8 Table). Transcript wise counts were generated using featureCounts tool from the SUBREAD package [106]. In total, the transcripts of 1,343 B. burgdorferi genes were tracked in this analysis. Differentially expressed genes were identified using a 5% False Discovery Rate threshold and a 2-fold change cut off with DESeq2 [107].
The data accumulated from the RNA-seq analysis are available at BioProject via accession number PRJNA565255.
Tandem Mass Tags (TMT)
Three independent cultures of B. burgdorferi cells were grown to mid-log phase of 5 x 107 cells per ml at 37°C, pH 6.8 and 5% CO2 (note: the cells used here were the same as those used for RNA-seq). Cells were centrifuged at 4,500 x g for 30 minutes at 4°C and washed twice with 10ml of cold PBS. Cells were resuspended in 50 mM triethylammonium bicarbonate (TEAB) and 5% SDS. Samples were quantified using Pierce BCA protein assay kit (Thermo Fisher Scientific, Waltham, MA) and 1.2 mg of protein was used for Tandem Mass tags (TMT) experiment. For this approach, protein extracts were isolated from the cells, reduced, alkylated, and proteolytically digested overnight. Samples were labeled with the TMT reagents in a 6-plex experiment and combined before sample fractionation and clean up. Labeled samples were analyzed by high-resolution Orbitrap LC-MS/MS. Identification and quantification of proteins was performed using Proteome Discoverer 2.2 software. The University of Texas Southwestern proteomics core performed the TMT analysis. A total of 718 B. burgdorferi proteins were detected in this analysis based on False Discovery Rates of less than 5%.
The TMT raw data was deposited to MassIVE using Proteomic Xchange Consortium with the data set number of PXD015685.
SDS PAGE and immunoblotting
Borrelia burgdorferi protein lysates were resolved on a 12.5% polyacrylamide gel, transferred to a PVDF membrane, and blocked using non-fat powdered milk as done previously [91,108]. Primary antibodies were used at the following dilutions: anti-Oms28 at 1:1000; anti-OspD at 1:5,000 and anti-FlaB at 1:5,000. Secondary antibodies with horseradish peroxidase (HRP) conjugates were used to detect immunocomplexes, specifically, anti-mouse Ig-HRP (Invitrogen, Carlsbad, CA, USA) or anti-rabbit Ig-HRP (GE Healthcare, Chicago, IL, USA) both diluted to 1:5,000. The membranes were washed extensively in PBS, 0.2% Tween-20, and developed using the Western Lighting Chemiluminescent Reagent plus system (Perkin Elmer, Waltham, MA, USA).
Statistical analysis
For real-time qPCR analysis, multiple unpaired t-test, one per tissue, were performed to analyze the strains and corrected for multiple comparisons using the Holm-Sidak method. For quantitative reverse transcription PCR (qRT-PCR), one-way ANOVA was performed to analyze the strains. For the analysis of in vivo luminescence of mice, two-way ANOVA was performed. For the proteome volcano plot, multiple unpaired t-test, one for each protein identified in the 1% False Discovery Rate (637 proteins), were performed between the strains and corrected for multiple comparisons by using the Holm-Sidak method. Significance was accepted when the p-values were less than 0.05 for all statistical analyses employed.
RNA target and structure prediction
The coding sequence of gene bba66 was subjected to RNA binding predictions using IntaRNA (Freiburg RNA Tools; [94] against the processed ittA sequence. Mfold was used to predict the secondary structure of ittA [109].
Ethics statement
Animal experiments were performed in accordance to National Institute of Health (NIH) Guide for Care and Use of Laboratory Animals. Animal experiments also followed the guidelines of the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC). Approval for animal procedures was given by Tufts University and Texas A&M University Institutional Animal Care and Use Committees (IACUC; protocols B2018-98 [Tufts] and 2015–0367 [Texas A&M]). Mice were euthanized in manner that conforms to the guidelines put forth by the AVMA and was approved by Tufts University and Texas A&M University IACUC.
Supporting information
Data for each figure is shown in individual tabs.
(XLSX)
Deseq2 analysis was employed to assess the RNA-seq of the parent and the ittA mutant strains. A total of 1,343 transcripts were expressed in the parent and ittA mutant when grown in vitro under mammalian-like conditions (see Methods for details).
(XLSM)
Deseq2 analysis was employed to analyze the RNA-seq of the parent and ittA mutant strains grown in vitro under mammalian-like conditions. A total of 92 transcripts were differentially expressed between the groups at the +/-1.4-fold change cutoff.
(XLSX)
Deseq2 analysis was employed to analyze the RNA-seq of the parent and ittA mutant strains grown in vitro under mammalian-like conditions. A total of 19 transcripts were differentially expressed between the groups at the +/- 2-fold change cutoff.
(XLSX)
Deseq2 analysis was employed to analyze the RNA-seq of parent and ittA mutant strains grown in vitro under mammalian-like conditions. A total of 73 transcripts were differentially expressed between the groups at the +/- 1.4 to 1.9-fold change cutoff.
(XLSX)
A total of 718 proteins were identified by employing Tandem Mass Tags (TMT) proteomics technologies to the parent and ittA mutant strains, grown under mammalian-like conditions.
(XLSX)
A total of 69 proteins were identified with a fold change cutoff of +/- 1.4 and with an adjusted p-value of <0.05 between the groups.
(XLSX)
(XLSX)
Three biological replicates of the parent and the ittA mutant strains were grown in vitro under mammalian-like conditions and were subjected to RNA-seq analysis.
(XLSX)
The parent, sRNA mutant (ittA::StrR) and complement (Comp) strains were grown in vitro and total RNA was purified from each. Oligonucleotide primers specific for bbd18 and bbd21 were used without (-) and with (+) added reverse transcriptase (RT). The DNA ladder is shown at the left and the corresponding base pair values are indicated.
(TIF)
The B. burgdorferi parent strain, the ittA sRNA mutant (ittA::StrR) and the sRNA complement strain (Comp) were grown in conventional microaerophilic conditions of 32°C, 1% CO2, pH 7.6 in triplicate in BSK-II media and enumerated by dark field microscopy daily out to day 9. No significant differences in growth were observed. Similar growth kinetics were observed between these three strains when the cells were grown at conditions of 37°C, 5% CO2 and pH 6.8. Data points shown reflect average value with standard error.
(TIF)
Three biological replicates of the B. burgdorferi parent strain, the sRNA mutant (ittA::StrR) and the sRNA complement strain (Comp), were grown in mammalian-like conditions, RNA was purified and the ittA probe was used for Northern blot analysis at longer exposure. The ittA mutant does not expressed ittA, as expected. The stable processed form of ittA is observed as the dark band underneath the 100 nucleotide marker. The parent and complement strains expression of ittA is comparable, but between 800 and 300 nucleotides, the complement strain exhibits additional bands that are missing in the parent strain and could possibly contribute to the partial complementation of the strain in vitro. The marker is shown in nucleotides at the left of the blot.
(TIF)
Acknowledgments
We thank Lauren Weise, Parmida Tehranchi, Kristen Sanchez, and Alexandra Powell for excellent technical assistance for the work done at Texas A&M University. We would like to thank Andrew Hillhouse and Kranti Konganti from the Texas A&M Institute for Genome Sciences & Society (TIGSS) for their help with RNA-seq and the subsequent data analysis. We also want to extend our gratitude to the University of Texas Southwestern Proteomics core, specifically Andrew Lemoff and Mohammad Goodarzi, for their guidance through the TMT analysis.
Data Availability
All RNA-seq data files will be made available from the BioProject and database accession number(s) PRJNA565255 following publication. For the TMT/Proteomic data, this was deposited in MassIVE using the Proteomic Xchange Consortium with the data set number of PXD015685.
Funding Statement
This work was supported by a Graduate Diversity Excellence Fellowship from Texas A&M University (to DM-P) as well as a Genetics Fellow Award from the Program in Genetics at Texas A&M University (to DM-P). This work was funded by Public Health Service Grants AI042345 (to JTS), AI123672 (to JH, ML, TL, and JTS), AI103905 and AI111317 (to LTH and TL), AI131656 (to LTH, JTS, JH, and TL), AI037277 and AI059048 (to SJN and TL), AI119572 (to TL), and AI098388 (to ET). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Nelson CA, Saha S, Kugeler KJ, Delorey MJ, Shankar MB, Hinckley AF, et al. Incidence of Clinician-Diagnosed Lyme Disease, United States, 2005–2010. Emerging Infectious Diseases. 2015. September;21(9):1625–31. 10.3201/eid2109.150417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Radolf JD, Caimano MJ, Stevenson B, Hu LT. Of ticks, mice and men: understanding the dual-host lifestyle of Lyme disease spirochaetes. Nat Rev Microbiol. 2012. January 9;10(2):87–99. 10.1038/nrmicro2714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Samuels DS. Gene regulation in Borrelia burgdorferi. Annu Rev Microbiol. 2011;65:479–99. 10.1146/annurev.micro.112408.134040 [DOI] [PubMed] [Google Scholar]
- 4.Tilly K, Rosa PA, Stewart PE. Biology of Infection with Borrelia burgdorferi. Infect Dis Clin North Am. 2008. June;22(2):217–34. 10.1016/j.idc.2007.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shapiro ED. Borrelia burgdorferi (Lyme disease). Pediatr Rev. 2014. December;35(12):500–9. 10.1542/pir.35-12-500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Steere AC, Coburn J, Glickstein L. The emergence of Lyme disease. J Clin Invest. 2004. April 15;113(8):1093–101. 10.1172/JCI21681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kurtenbach K, Hanincová K, Tsao JI, Margos G, Fish D, Ogden NH. Fundamental processes in the evolutionary ecology of Lyme borreliosis. Nat Rev Micro. 2006. September;4(9):660–9. [DOI] [PubMed] [Google Scholar]
- 8.Samuels DS. Gene regulation in Borrelia burgdorferi. Annu Rev Microbiol. 2011;65:479–99. 10.1146/annurev.micro.112408.134040 [DOI] [PubMed] [Google Scholar]
- 9.Brisson D, Drecktrah D, Eggers CH, Samuels DS. Genetics of Borrelia burgdorferi. Annual Review of Genetics. 2012;46(1):515–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brooks CS, Hefty PS, Jolliff SE, Akins DR. Global Analysis of Borrelia burgdorferi Genes Regulated by Mammalian Host-Specific Signals. Infect Immun. 2003. June;71(6):3371–83. 10.1128/IAI.71.6.3371-3383.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Akins DR, Bourell KW, Caimano MJ, Norgard MV, Radolf JD. A new animal model for studying Lyme disease spirochetes in a mammalian host-adapted state. J Clin Invest. 1998. May 15;101(10):2240–50. 10.1172/JCI2325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carroll JA, Garon CF, Schwan TG. Effects of environmental pH on membrane proteins in Borrelia burgdorferi. Infect Immun. 1999. July;67(7):3181–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ojaimi C, Brooks C, Casjens S, Rosa P, Elias A, Barbour A, et al. Profiling of Temperature-Induced Changes in Borrelia burgdorferi Gene Expression by Using Whole Genome Arrays. Infect Immun. 2003. April;71(4):1689–705. 10.1128/IAI.71.4.1689-1705.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Revel AT, Talaat AM, Norgard MV. DNA microarray analysis of differential gene expression in Borrelia burgdorferi, the Lyme disease spirochete. PNAS. 2002. February 5;99(3):1562–7. 10.1073/pnas.032667699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stevenson B, Schwan TG, Rosa PA. Temperature-related differential expression of antigens in the Lyme disease spirochete, Borrelia burgdorferi. Infect Immun. 1995. November;63(11):4535–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carroll JA, Cordova RM, Garon CF. Identification of 11 pH-regulated genes in Borrelia burgdorferi localizing to linear plasmids. Infect Immun. 2000. December;68(12):6677–84. 10.1128/iai.68.12.6677-6684.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hyde JA, Trzeciakowski JP, Skare JT. Borrelia burgdorferi Alters Its Gene Expression and Antigenic Profile in Response to CO2 Levels. J Bacteriol. 2007. January;189(2):437–45. 10.1128/JB.01109-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seshu J, Boylan JA, Gherardini FC, Skare JT. Dissolved Oxygen Levels Alter Gene Expression and Antigen Profiles in Borrelia burgdorferi. Infection and Immunity. 2004. March;72(3):1580 10.1128/IAI.72.3.1580-1586.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tokarz R, Anderton JM, Katona LI, Benach JL. Combined Effects of Blood and Temperature Shift on Borrelia burgdorferi Gene Expression as Determined by Whole Genome DNA Array. Infect Immun. 2004. September;72(9):5419–32. 10.1128/IAI.72.9.5419-5432.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hübner A, Yang X, Nolen DM, Popova TG, Cabello FC, Norgard MV. Expression of Borrelia burgdorferi OspC and DbpA is controlled by a RpoN–RpoS regulatory pathway. Proc Natl Acad Sci U S A. 2001. October 23;98(22):12724–9. 10.1073/pnas.231442498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang XF, Alani SM, Norgard MV. The response regulator Rrp2 is essential for the expression of major membrane lipoproteins in Borrelia burgdorferi. Proc Natl Acad Sci U S A. 2003. September 16;100(19):11001–6. 10.1073/pnas.1834315100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Samuels DS, Radolf JD. Who is the BosR around here anyway? Mol Microbiol. 2009. December;74(6):1295–9. 10.1111/j.1365-2958.2009.06971.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hyde JA, Shaw DK, Smith R, Trzeciakowski JP, Skare JT. The BosR regulatory protein of Borrelia burgdorferi interfaces with the RpoS regulatory pathway and modulates both the oxidative stress response and pathogenic properties of the Lyme disease spirochete. Mol Microbiol. 2009. December;74(6):1344–55. 10.1111/j.1365-2958.2009.06951.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sanjuan E, Esteve-Gassent MD, Maruskova M, Seshu J. Overexpression of CsrA (BB0184) alters the morphology and antigen profiles of Borrelia burgdorferi. Infect Immun. 2009. November;77(11):5149–62. 10.1128/IAI.00673-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karna SLR, Sanjuan E, Esteve-Gassent MD, Miller CL, Maruskova M, Seshu J. CsrA Modulates Levels of Lipoproteins and Key Regulators of Gene Expression Critical for Pathogenic Mechanisms of Borrelia burgdorferi. Infection and Immunity. 2011. February 1;79(2):732–44. 10.1128/IAI.00882-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sze CW, Li C. Inactivation of bb0184, which encodes carbon storage regulator A, represses the infectivity of Borrelia burgdorferi. Infect Immun. 2011. March;79(3):1270–9. 10.1128/IAI.00871-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miller CL, Karna SLR, Seshu J. Borrelia host adaptation Regulator (BadR) regulates rpoS to modulate host adaptation and virulence factors in Borrelia burgdorferi. Molecular Microbiology. 2013. April;88(1):105–24. 10.1111/mmi.12171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Drecktrah D, Lybecker M, Popitsch N, Rescheneder P, Hall LS, Samuels DS. The Borrelia burgdorferi RelA/SpoT Homolog and Stringent Response Regulate Survival in the Tick Vector and Global Gene Expression during Starvation. PLOS Pathogens. 2015. September 15;11(9):e1005160 10.1371/journal.ppat.1005160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fisher MA, Grimm D, Henion AK, Elias AF, Stewart PE, Rosa PA, et al. Borrelia burgdorferi sigma54 is required for mammalian infection and vector transmission but not for tick colonization. Proc Natl Acad Sci USA. 2005. April 5;102(14):5162–7. 10.1073/pnas.0408536102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bugrysheva J, Dobrikova EY, Godfrey HP, Sartakova ML, Cabello FC. Modulation of Borrelia burgdorferi Stringent Response and Gene Expression during Extracellular Growth with Tick Cells. Infect Immun. 2002. June;70(6):3061–7. 10.1128/IAI.70.6.3061-3067.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Savage CR, Jutras BL, Bestor A, Tilly K, Rosa PA, Tourand Y, et al. Borrelia burgdorferi SpoVG DNA- and RNA-Binding Protein Modulates the Physiology of the Lyme Disease Spirochete. Journal of Bacteriology. 2018. June 15;200(12):e00033–18. 10.1128/JB.00033-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ouyang Z, Deka RK, Norgard MV. BosR (BB0647) controls the RpoN-RpoS regulatory pathway and virulence expression in Borrelia burgdorferi by a novel DNA-binding mechanism. Plos Pathogens. 2011. February 10;7(2):e1001272–e1001272. 10.1371/journal.ppat.1001272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dulebohn DP, Hayes BM, Rosa PA. Global Repression of Host-Associated Genes of the Lyme Disease Spirochete through Post-Transcriptional Modulation of the Alternative Sigma Factor RpoS. PLOS ONE. 2014. March 26;9(3):e93141 10.1371/journal.pone.0093141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Caimano MJ, Eggers CH, Hazlett KRO, Radolf JD. RpoS Is Not Central to the General Stress Response in Borrelia burgdorferi but Does Control Expression of One or More Essential Virulence Determinants. Infect Immun. 2004. November 1;72(11):6433–45. 10.1128/IAI.72.11.6433-6445.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burtnick MN, Downey JS, Brett PJ, Boylan JA, Frye JG, Hoover TR, et al. Insights into the complex regulation of rpoS in Borrelia burgdorferi. Mol Microbiol. 2007. July 1;65(2):277–93. 10.1111/j.1365-2958.2007.05813.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Caimano MJ, Iyer R, Eggers CH, Gonzalez C, Morton EA, Gilbert MA, et al. Analysis of the RpoS regulon in Borrelia burgdorferi in response to mammalian host signals provides insight into RpoS function during the enzootic cycle. Mol Microbiol. 2007. September;65(5):1193–217. 10.1111/j.1365-2958.2007.05860.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stevenson B, Seshu J. Regulation of Gene and Protein Expression in the Lyme Disease Spirochete In: Adler B, editor. Spirochete Biology: The Post Genomic Era [Internet]. Cham: Springer International Publishing; 2018. p. 83–112. 10.1007/82_2017_49 [DOI] [PubMed] [Google Scholar]
- 38.Östberg Y, Bunikis I, Bergström S, Johansson J. The Etiological Agent of Lyme Disease, Borrelia burgdorferi, Appears To Contain Only a Few Small RNA Molecules. J Bacteriol. 2004. December 15;186(24):8472–7. 10.1128/JB.186.24.8472-8477.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arnold WK, Savage CR, Brissette CA, Seshu J, Livny J, Stevenson B. RNA-Seq of Borrelia burgdorferi in Multiple Phases of Growth Reveals Insights into the Dynamics of Gene Expression, Transcriptome Architecture, and Noncoding RNAs. PLOS ONE. 2016. October 5;11(10):e0164165 10.1371/journal.pone.0164165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Popitsch N, Bilusic I, Rescheneder P, Schroeder R, Lybecker M. Temperature-dependent sRNA transcriptome of the Lyme disease spirochete. BMC Genomics. 2017;18:28 10.1186/s12864-016-3398-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Casselli T, Crowley MA, Highland MA, Tourand Y, Bankhead T. A Small Intergenic Region of lp17 is Required for Evasion of Adaptive Immunity and Induction of Pathology by the Lyme Disease Spirochete. Cellular Microbiology [Internet]. 2019. April 3 [cited 2019 Apr 9]; https://onlinelibrary.wiley.com/doi/abs/10.1111/cmi.13029 [DOI] [PubMed] [Google Scholar]
- 42.Papenfort K, Vogel J. Regulatory RNA in bacterial pathogens. Cell Host Microbe. 2010. July 22;8(1):116–27. 10.1016/j.chom.2010.06.008 [DOI] [PubMed] [Google Scholar]
- 43.Pitman S, Cho KH. The Mechanisms of Virulence Regulation by Small Noncoding RNAs in Low GC Gram-Positive Pathogens. Int J Mol Sci. 2015. December 14;16(12):29797–814. 10.3390/ijms161226194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Storz G, Vogel J, Wassarman KM. Regulation by small RNAs in bacteria: expanding frontiers. Mol Cell. 2011. September 16;43(6):880–91. 10.1016/j.molcel.2011.08.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Babitzke P, Lai Y-J, Renda AJ, Romeo T. Posttranscription Initiation Control of Gene Expression Mediated by Bacterial RNA-Binding Proteins. Annu Rev Microbiol. 2019. September 8;73(1):43–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Elke eVan Assche, Sandra eVan Puyvelde, Jozef eVanderleyden, Hans P. Steenackers. RNA-binding proteins involved in post-transcriptional regulation in bacteria. Frontiers in Microbiology. 2015; [DOI] [PMC free article] [PubMed]
- 47.Ahmed W, Hafeez MA, Mahmood S. Identification and functional characterization of bacterial small non-coding RNAs and their target: A review. Gene Reports. 2018. March 1;10:167–76. [Google Scholar]
- 48.Nitzan M, Rehani R, Margalit H. Integration of Bacterial Small RNAs in Regulatory Networks. Annu Rev Biophys. 2017. May 22;46(1):131–48. [DOI] [PubMed] [Google Scholar]
- 49.Gottesman S, Storz G. Bacterial Small RNA Regulators: Versatile Roles and Rapidly Evolving Variations. Cold Spring Harb Perspect Biol. 2011. December 1;3(12):a003798 10.1101/cshperspect.a003798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dutta T, Srivastava S. Small RNA-mediated regulation in bacteria: A growing palette of diverse mechanisms. Gene. 2018. May 20;656:60–72. 10.1016/j.gene.2018.02.068 [DOI] [PubMed] [Google Scholar]
- 51.Barik A, Das S. A comparative study of sequence- and structure-based features of small RNAs and other RNAs of bacteria. RNA Biol. 2017. November 3;1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Melamed S, Peer A, Faigenbaum-Romm R, Gatt YE, Reiss N, Bar A, et al. Global Mapping of Small RNA-Target Interactions in Bacteria. Molecular Cell. 2016. September 1;63(5):884–97. 10.1016/j.molcel.2016.07.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Updegrove TB, Shabalina SA, Storz G. How do base-pairing small RNAs evolve? FEMS Microbiol Rev. 2015. May;39(3):379–91. 10.1093/femsre/fuv014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pichon C, Felden B. Proteins that interact with bacterial small RNA regulators. FEMS Microbiol Rev. 2007. September 1;31(5):614–25. 10.1111/j.1574-6976.2007.00079.x [DOI] [PubMed] [Google Scholar]
- 55.Lin T, Gao L, Zhang C, Odeh E, Jacobs MB, Coutte L, et al. Analysis of an Ordered, Comprehensive STM Mutant Library in Infectious Borrelia burgdorferi: Insights into the Genes Required for Mouse Infectivity. PLOS ONE. 2012. October 25;7(10):e47532 10.1371/journal.pone.0047532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tao eLin, Erin B Troy, Linden T Hu, Lihui eGao, Steven J Norris. Transposon mutagenesis as an approach to improved understanding of Borrelia pathogenesis and biology. Frontiers in Cellular and Infection Microbiology. 2014; [DOI] [PMC free article] [PubMed]
- 57.Troy EB, Lin T, Gao L, Lazinski DW, Camilli A, Norris SJ, et al. Understanding Barriers to Borrelia burgdorferi Dissemination during Infection Using Massively Parallel Sequencing. Infect Immun. 2013. July 1;81(7):2347–57. 10.1128/IAI.00266-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kawabata H, Norris SJ, Watanabe H. BBE02 Disruption Mutants of Borrelia burgdorferi B31 Have a Highly Transformable, Infectious Phenotype. Infect Immun. 2004. December;72(12):7147–54. 10.1128/IAI.72.12.7147-7154.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Adams PP, Flores Avile C, Popitsch N, Bilusic I, Schroeder R, Lybecker M, et al. In vivo expression technology and 5΄ end mapping of the Borrelia burgdorferi transcriptome identify novel RNAs expressed during mammalian infection. Nucleic Acids Research. 2016. November 29;45(2):775–92. 10.1093/nar/gkw1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Labandeira-Rey M, Skare JT. Decreased Infectivity in Borrelia burgdorferi Strain B31 Is Associated with Loss of Linear Plasmid 25 or 28–1. Infect Immun. 2001. January;69(1):446–55. 10.1128/IAI.69.1.446-455.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hyde JA, Weening EH, Chang M, Trzeciakowski JP, Höök M, Cirillo JD, et al. Bioluminescent imaging of Borrelia burgdorferi in vivo demonstrates that the fibronectin binding protein BBK32 is required for optimal infectivity. Mol Microbiol. 2011. October;82(1):99–113. 10.1111/j.1365-2958.2011.07801.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Purser JE, Lawrenz MB, Caimano MJ, Howell JK, Radolf JD, Norris SJ. A plasmid-encoded nicotinamidase (PncA) is essential for infectivity of Borrelia burgdorferi in a mammalian host. Molecular Microbiology. 2003. May 1;48(3):753–64. 10.1046/j.1365-2958.2003.03452.x [DOI] [PubMed] [Google Scholar]
- 63.Wager B, Shaw DK, Groshong AM, Blevins JS, Skare JT. BB0744 Affects Tissue Tropism and Spatial Distribution of Borrelia burgdorferi. Infect Immun. 2015. September;83(9):3693–703. 10.1128/IAI.00828-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhi H, Weening EH, Barbu EM, Hyde JA, Höök M, Skare JT. The BBA33 lipoprotein binds collagen and impacts Borrelia burgdorferi pathogenesis. Mol Microbiol. 2015. April;96(1):68–83. 10.1111/mmi.12921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Labandeira-Rey M, Seshu J, Skare JT. The Absence of Linear Plasmid 25 or 28–1 of Borrelia burgdorferi Dramatically Alters the Kinetics of Experimental Infection via Distinct Mechanisms. Infection and Immunity. 2003. August 1;71(8):4608–13. 10.1128/IAI.71.8.4608-4613.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Labandeira-Rey M, Baker EA, Skare JT. VraA (BBI16) Protein of Borrelia burgdorferi Is a Surface-Exposed Antigen with a Repetitive Motif That Confers Partial Protection against Experimental Lyme Borreliosis. Infect Immun. 2001. March 1;69(3):1409–19. 10.1128/IAI.69.3.1409-1419.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Patton TG, Brandt KS, Nolder C, Clifton DR, Carroll JA, Gilmore RD. Borrelia burgdorferi bba66 Gene Inactivation Results in Attenuated Mouse Infection by Tick Transmission. Infection and Immunity. 2013. July 1;81(7):2488–98. 10.1128/IAI.00140-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gilmore RD, Howison RR, Schmit VL, Nowalk AJ, Clifton DR, Nolder C, et al. Temporal Expression Analysis of the Borrelia burgdorferi Paralogous Gene Family 54 Genes BBA64, BBA65, and BBA66 during Persistent Infection in Mice. Infection and Immunity. 2007. June 1;75(6):2753–64. 10.1128/IAI.00037-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Beisel CL, Storz G. Base pairing small RNAs and their roles in global regulatory networks. FEMS Microbiol Rev. 2010. September;34(5):866–82. 10.1111/j.1574-6976.2010.00241.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Michaux C, Verneuil N, Hartke A, Giard J-C. Physiological roles of small RNA molecules. Microbiology. 2014;160(6):1007–19. [DOI] [PubMed] [Google Scholar]
- 71.Dutcher HA, Raghavan R. Origin, Evolution, and Loss of Bacterial Small RNAs. Microbiol Spectr. 2018. April;6(2):UNSP RWR-0004-2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Barquist L, Vogel J. Accelerating Discovery and Functional Analysis of Small RNAs with New Technologies. Annual Review of Genetics. 2015;49(1):367–94. [DOI] [PubMed] [Google Scholar]
- 73.Lybecker MC, Samuels DS. Small RNAs of Borrelia burgdorferi: Characterizing Functional Regulators in a Sea of sRNAs. Yale J Biol Med. 2017. June 23;90(2):317–23. [PMC free article] [PubMed] [Google Scholar]
- 74.Caldelari I, Chao Y, Romby P, Vogel J. RNA-Mediated Regulation in Pathogenic Bacteria. Cold Spring Harbor Perspectives in Medicine. 2013. September 1;3(9):a010298–a010298. 10.1101/cshperspect.a010298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gilmore RD, Howison RR, Schmit VL, Carroll JA. Borrelia burgdorferi expression of the bba64, bba65, bba66, and bba73 genes in tissues during persistent infection in mice. Microb Pathog. 2008. December;45(5–6):355–60. 10.1016/j.micpath.2008.08.006 [DOI] [PubMed] [Google Scholar]
- 76.Liang FT, Nelson FK, Fikrig E. Molecular adaptation of Borrelia burgdorferi in the murine host. J Exp Med. 2002. July 15;196(2):275–80. 10.1084/jem.20020770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Caimano MJ, Eggers CH, Gonzalez CA, Radolf JD. Alternate Sigma Factor RpoS Is Required for the In Vivo-Specific Repression of Borrelia burgdorferi Plasmid lp54-Borne ospA and lp6.6 Genes. J Bacteriol. 2005. November;187(22):7845–52. 10.1128/JB.187.22.7845-7852.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yang XF, Pal U, Alani SM, Fikrig E, Norgard MV. Essential Role for OspA/B in the Life Cycle of the Lyme Disease Spirochete. J Exp Med. 2004. March 1;199(5):641–8. 10.1084/jem.20031960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stewart PE, Bestor A, Cullen JN, Rosa PA. A Tightly Regulated Surface Protein of Borrelia burgdorferi Is Not Essential to the Mouse-Tick Infectious Cycle. Infect Immun. 2008. May;76(5):1970–8. 10.1128/IAI.00714-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mulay VB, Caimano MJ, Iyer R, Dunham-Ems S, Liveris D, Petzke MM, et al. Borrelia burgdorferi bba74 Is Expressed Exclusively during Tick Feeding and Is Regulated by Both Arthropod- and Mammalian Host-Specific Signals. J Bacteriol. 2009. April;191(8):2783–94. 10.1128/JB.01802-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ouyang Z, Narasimhan S, Neelakanta G, Kumar M, Pal U, Fikrig E, et al. Activation of the RpoN-RpoS regulatory pathway during the enzootic life cycle of Borrelia burgdorferi. BMC Microbiology. 2012;12:44 10.1186/1471-2180-12-44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pal U, Li X, Wang T, Montgomery RR, Ramamoorthi N, Desilva AM, et al. TROSPA, an Ixodes scapularis receptor for Borrelia burgdorferi. Cell. 2004. November 12;119(4):457–68. 10.1016/j.cell.2004.10.027 [DOI] [PubMed] [Google Scholar]
- 83.Williams SK, Weiner ZP, Gilmore RD. Human neuroglial cells internalize Borrelia burgdorferi by coiling phagocytosis mediated by Daam1. PLOS ONE. 2018. May 10;13(5):e0197413 10.1371/journal.pone.0197413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liu W, Sato A, Khadka D, Bharti R, Diaz H, Runnels LW, et al. Mechanism of activation of the Formin protein Daam1. PNAS. 2008. January 8;105(1):210–5. 10.1073/pnas.0707277105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hoffmann A-K, Naj X, Linder S. Daam1 is a regulator of filopodia formation and phagocytic uptake of Borrelia burgdorferi by primary human macrophages. The FASEB Journal. 2014. April 2;28(7):3075–89. 10.1096/fj.13-247049 [DOI] [PubMed] [Google Scholar]
- 86.Xu Q, Seemanapalli SV, McShan K, Liang FT. Constitutive expression of outer surface protein C diminishes the ability of Borrelia burgdorferi to evade specific humoral immunity. Infect Immun. 2006. September;74(9):5177–84. 10.1128/IAI.00713-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Xu Q, McShan K, Liang FT. Identification of an ospC operator critical for immune evasion of Borrelia burgdorferi. Molecular Microbiology. 2007. April 1;64(1):220–31. 10.1111/j.1365-2958.2007.05636.x [DOI] [PubMed] [Google Scholar]
- 88.Grimm D, Tilly K, Byram R, Stewart PE, Krum JG, Bueschel DM, et al. Outer-surface protein C of the Lyme disease spirochete: a protein induced in ticks for infection of mammals. Proc Natl Acad Sci USA. 2004. March 2;101(9):3142–7. 10.1073/pnas.0306845101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pal U, Yang X, Chen M, Bockenstedt LK, Anderson JF, Flavell RA, et al. OspC facilitates Borrelia burgdorferi invasion of Ixodes scapularis salivary glands. J Clin Invest. 2004. January 15;113(2):220–30. 10.1172/JCI19894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Xu Q, Seemanaplli SV, McShan K, Liang FT. Increasing the Interaction of Borrelia burgdorferi with Decorin Significantly Reduces the 50 Percent Infectious Dose and Severely Impairs Dissemination. Infection and Immunity. 2007. September 1;75(9):4272–81. 10.1128/IAI.00560-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Weening EH, Parveen N, Trzeciakowski JP, Leong JM, Höök M, Skare JT. Borrelia burgdorferi Lacking DbpBA Exhibits an Early Survival Defect during Experimental Infection. Infect Immun. 2008. December;76(12):5694–705. 10.1128/IAI.00690-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Seshu J, Esteve-Gassent MD, Labandeira-Rey M, Kim JH, Trzeciakowski JP, Höök M, et al. Inactivation of the fibronectin-binding adhesin gene bbk32 significantly attenuates the infectivity potential of Borrelia burgdorferi. Molecular Microbiology. 2006. March 1;59(5):1591–601. 10.1111/j.1365-2958.2005.05042.x [DOI] [PubMed] [Google Scholar]
- 93.Garcia BL, Zhi H, Wager B, Höök M, Skare JT. Borrelia burgdorferi BBK32 Inhibits the Classical Pathway by Blocking Activation of the C1 Complement Complex. Plos Pathogens. 2016. January 25;12(1):e1005404–e1005404. 10.1371/journal.ppat.1005404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Busch A, Richter AS, Backofen R. IntaRNA: efficient prediction of bacterial sRNA targets incorporating target site accessibility and seed regions. Bioinformatics. 2008. December 15;24(24):2849–56. 10.1093/bioinformatics/btn544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Waters LS, Storz G. Regulatory RNAs in Bacteria. Cell. 2009. February 20;136(4):615–28. 10.1016/j.cell.2009.01.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ternan N. Small regulatory RNA molecules in bacteria. OA Microbiology. 2013. December 1; 1(1):1. [Google Scholar]
- 97.Frank KL, Bundle SF, Kresge ME, Eggers CH, Samuels DS. aadA Confers Streptomycin Resistance in Borrelia burgdorferi. Journal of Bacteriology. 2003. November 15;185(22):6723–7. 10.1128/JB.185.22.6723-6727.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Elias AF, Bono JL, Kupko JJ, Stewart PE, Krum JG, Rosa PA. New antibiotic resistance cassettes suitable for genetic studies in Borrelia burgdorferi. J Mol Microbiol Biotechnol. 2003;6(1):29–40. 10.1159/000073406 [DOI] [PubMed] [Google Scholar]
- 99.Ramsey ME, Hyde JA, Medina-Perez DN, Lin T, Gao L, Lundt ME, et al. A high-throughput genetic screen identifies previously uncharacterized Borrelia burgdorferi genes important for resistance against reactive oxygen and nitrogen species. PLOS Pathogens. 2017. February 17;13(2):e1006225 10.1371/journal.ppat.1006225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Troy EB, Lin T, Gao L, Lazinski DW, Lundt M, Camilli A, et al. Global Tn-seq Analysis of Carbohydrate Utilization and Vertebrate Infectivity of Borrelia burgdorferi. Mol Microbiol. 2016. September;101(6):1003–23. 10.1111/mmi.13437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989. April 15;77(1):51–9. 10.1016/0378-1119(89)90358-2 [DOI] [PubMed] [Google Scholar]
- 102.Samuels DS, Drecktrah D, Hall LS. Genetic Transformation and Complementation In: Pal U, Buyuktanir O, editors. Borrelia burgdorferi: Methods and Protocols [Internet]. New York, NY: Springer New York; 2018. p. 183–200. 10.1007/978-1-4939-7383-5_15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lybecker M, Zimmermann B, Bilusic I, Tukhtubaeva N, Schroeder R. The double-stranded transcriptome of Escherichia coli. PNAS. 2014. February 25;111(8):3134–9. 10.1073/pnas.1315974111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014. August 1;30(15):2114–20. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nature Methods. 2015. April;12(4):357–60. 10.1038/nmeth.3317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014. April 1;30(7):923–30. 10.1093/bioinformatics/btt656 [DOI] [PubMed] [Google Scholar]
- 107.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology. 2014;15(12):550–550. 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Seshu J, Boylan JA, Hyde JA, Swingle KL, Gherardini FC, Skare JT. A conservative amino acid change alters the function of BosR, the redox regulator of Borrelia burgdorferi. Molecular Microbiology. 2004;54(5):1352–63. 10.1111/j.1365-2958.2004.04352.x [DOI] [PubMed] [Google Scholar]
- 109.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003. July 1;31(13):3406–15. 10.1093/nar/gkg595 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data for each figure is shown in individual tabs.
(XLSX)
Deseq2 analysis was employed to assess the RNA-seq of the parent and the ittA mutant strains. A total of 1,343 transcripts were expressed in the parent and ittA mutant when grown in vitro under mammalian-like conditions (see Methods for details).
(XLSM)
Deseq2 analysis was employed to analyze the RNA-seq of the parent and ittA mutant strains grown in vitro under mammalian-like conditions. A total of 92 transcripts were differentially expressed between the groups at the +/-1.4-fold change cutoff.
(XLSX)
Deseq2 analysis was employed to analyze the RNA-seq of the parent and ittA mutant strains grown in vitro under mammalian-like conditions. A total of 19 transcripts were differentially expressed between the groups at the +/- 2-fold change cutoff.
(XLSX)
Deseq2 analysis was employed to analyze the RNA-seq of parent and ittA mutant strains grown in vitro under mammalian-like conditions. A total of 73 transcripts were differentially expressed between the groups at the +/- 1.4 to 1.9-fold change cutoff.
(XLSX)
A total of 718 proteins were identified by employing Tandem Mass Tags (TMT) proteomics technologies to the parent and ittA mutant strains, grown under mammalian-like conditions.
(XLSX)
A total of 69 proteins were identified with a fold change cutoff of +/- 1.4 and with an adjusted p-value of <0.05 between the groups.
(XLSX)
(XLSX)
Three biological replicates of the parent and the ittA mutant strains were grown in vitro under mammalian-like conditions and were subjected to RNA-seq analysis.
(XLSX)
The parent, sRNA mutant (ittA::StrR) and complement (Comp) strains were grown in vitro and total RNA was purified from each. Oligonucleotide primers specific for bbd18 and bbd21 were used without (-) and with (+) added reverse transcriptase (RT). The DNA ladder is shown at the left and the corresponding base pair values are indicated.
(TIF)
The B. burgdorferi parent strain, the ittA sRNA mutant (ittA::StrR) and the sRNA complement strain (Comp) were grown in conventional microaerophilic conditions of 32°C, 1% CO2, pH 7.6 in triplicate in BSK-II media and enumerated by dark field microscopy daily out to day 9. No significant differences in growth were observed. Similar growth kinetics were observed between these three strains when the cells were grown at conditions of 37°C, 5% CO2 and pH 6.8. Data points shown reflect average value with standard error.
(TIF)
Three biological replicates of the B. burgdorferi parent strain, the sRNA mutant (ittA::StrR) and the sRNA complement strain (Comp), were grown in mammalian-like conditions, RNA was purified and the ittA probe was used for Northern blot analysis at longer exposure. The ittA mutant does not expressed ittA, as expected. The stable processed form of ittA is observed as the dark band underneath the 100 nucleotide marker. The parent and complement strains expression of ittA is comparable, but between 800 and 300 nucleotides, the complement strain exhibits additional bands that are missing in the parent strain and could possibly contribute to the partial complementation of the strain in vitro. The marker is shown in nucleotides at the left of the blot.
(TIF)
Data Availability Statement
All RNA-seq data files will be made available from the BioProject and database accession number(s) PRJNA565255 following publication. For the TMT/Proteomic data, this was deposited in MassIVE using the Proteomic Xchange Consortium with the data set number of PXD015685.