

Abstract
Imidazolium salts have shown great promise as anticancer materials. A new imidazolium salt (TPP1), with a triphenylphosphonium substituent, has been synthesized and evaluated for in vitro and in vivo cytotoxicity against bladder cancer. TPP1 was determined to have a GI50 ranging from 200 to 250 μM over a period of 1 h and the ability to effectively inhibit bladder cancer. TPP1 induces apoptosis, and it appears to act as a direct mitochondrial toxin. TPP1 was applied intravesically to a bladder cancer mouse model based on the carcinogen N-butyl-N-(4-hydroxybutyl)nitrosamine (BBN). Cancer selectivity of TPP1 was demonstrated, as BBN-induced tumors exhibited apoptosis but normal adjacent urothelium did not. These results suggest that TPP1 may be a promising intravesical agent for the treatment of bladder cancer.
Keywords: Imidazolium salt, Triphenylphosphonium salt, Bladder cancer, Apoptosis
1. Introduction
Bladder cancer will be diagnosed in the United States in 80,000 patients in 2019, with about 1/3 of these patients harboring high-grade superficial cancer [1,2]. Management of high-grade superficial bladder cancer is significantly more challenging for these patients now that M. bovis Bacillus Calmette Guerin (BCG), the long-time standard-of-care, is in short supply due to the manufacturing discontinuation by Sanofi and difficulty in manufacturing and QC by Astellas, the main producers. With this shortage, 40 years of successful treatment of bladder cancer has been reversed [3]. There have been multiple year + long shortages in the last 5 years and the situation is not expected to improve anytime soon [4–6]. Second-line therapies, such as intravesical gemcitabine, valrubicin, and docetaxel, predictably have little efficacy because they only act at specific points in the cell cycle and cannot practically be retained in the bladder long enough to have an effect. Clinical experience with these approaches in BCG-failure patients results in 70–80% failure rate and often radical cystectomy (removal of the bladder) [7–10]. Radical cystectomy is among the most complicated [11–14] and expensive [15–17] elective surgeries performed. In addition, radical cystectomy requires permanent urinary diversion, typically with a urostomy, which is life-altering. Avoidance of radical cystectomy is therefore likely a high priority for patients and this is reflected in the medical literature [18–22] and actively-enrolling clinical trials (NCT02710734, NCT03609216). Even with BCG treatment, high grade superficial bladder cancer progresses to metastasis or death in about 1/3 of cases [23]. BCG really only reduces the risk of cancer progression (i.e. reduces the need for radical cystectomy) by 27% [24]. The BCG shortage, its relative ineffectiveness, and the ineffectiveness of second line therapies underscore the urgent and genuine need for new intravesical therapies for bladder cancer.
One redeeming quality of the currently available treatments for high-grade superficial bladder cancer is intravesical administration of therapeutics. Systemic absorption, and thus symptoms associated with parenteral and oral administration of chemotherapeutic agents, are greatly reduced in intravesical administration. However, BCG can still cause systemic symptoms such as fever, malaise, and rarely sepsis, and also local symptoms such as urinary urgency/frequency/dysuria by virtue of its nature as a live bacterial immunogenic vaccine [23,25].
A group of compounds known as mitocans target and kill cancer cells through accumulation in and disruption of mitochondria [26]. Targeting mitochondria with toxic compounds swiftly and irreversibly leads to apoptosis and disruption of the production of ATP. Delocalized lipophilic cations (DLCs) are known to accumulate in the mitochondrial inner matrix due to the negative mitochondrial membrane potential (MMP) [27]. The enhanced MMP in cancer cells relative to normal cells contributes to the effectiveness that positively-charged mitocans have in selectively targeting cancerous cells over normal cells [28,29]. For example, previous reports have shown that cationic mitochondrial-targeted dyes such as Rh123 [30] have an increased uptake and retention time in mouse bladder epithelial cells that have been transformed into cancerous cells with mutagens (e.g. dimethylbenzanthracene, benzo[a]pyrene, or butyl-(4-hydroxybutyl)nitrosamine (BBN)) in comparison to normal mouse bladder epithelial cells [31].
Triphenylphosphonium (TPP) salts are a group of DLCs that are known to target mitochondria [32,33]. A classic example of a mitochondrial targeted TPP containing compound is MitoQ10 [34]. When TPP is bound to doxorubicin, it causes doxorubicin to be redirected to the mitochondria and thereby enhancing cytotoxicity in doxorubicin resistant cell lines [35]. In addition, imidazolium salts have been of great interest for their biological activity [36–43]. The focus of the Youngs’ group has been the anticancer potential of imidazolium salts and their extensive structure activity relationships [44–48]. Furthermore, some imidazolium salts have been shown to induce apoptosis and disrupt the MMP of lung cancer cells [48]. Combining the anti-proliferative effect of imidazolium salts with the selectivity of the TPP moiety could afford a compound that is able to target cancer cells and disrupt the mitochondria, leading to cell death. In addition, the cationic nature of the TPP moiety can impart some aqueous solubility to these lipophilic compounds. This would allow for ease of administration as well as the ability for excess compound to exit the bladder. Therefore, TPP imidazolium salts could be developed as a viable alternative to BCG and intravesical chemotherapy based on their combined anti-tumor properties and targeting ability. In this study, we report the synthesis of a compound, TPP1, containing an imidazolium salt core and a TPP moiety, as well as the biological activity of the compound against bladder cancer cell lines and murine models.
2. Results and discussion
2.1. Synthesis and characterization
The synthesis outlined in Scheme 1 shows the synthetic strategy used to produce the triphenylphosphonium (TPP)-substituted benzimidazolium salt (TPP1). 2-(3-hydroxypropyl)-1,3-bis(naphthalen-2-ylmethyl)benzimidazolium bromide (1) was synthesized, using a previously published procedure [46]. To transform the alcohol to a TPP substituent, compound 1 was reacted with thionyl bromide to form the alkyl halide. The resulting 2-(3-bromopropyl) imidazolium salt, 2, was quickly separated from the remaining thionyl bromide, without further characterization, and immediately combined with excess triphenyl phosphine. The substitution of triphenyl phosphine for the alcohol in 1 to form TPP1 was suggested by the 1H NMR spectrum with the observed resonance of the methylene between the naphthalenes and nitrogens having a slight downfield shift from 6.10 ppm (1) to 6.15 ppm (TPP1). For TPP1 the propyl resonances were observed at 4.02 (m), 3.97 (m), and 1.90 ppm (m). The 13C NMR spectrum showed that the resonances in the alkyl region at 20.1 (d, J = 52.1 Hz) and 24.2 (d, J = 21.8 Hz) were split by the 31P nucleus. The 31P NMR spectrum showed a single resonance at 23.3 ppm, which was consistant with a single phosphorus atom present in the imidazolium salt. The Compound was characterized by 1H COSY, 1H{13C} HSQC, and 1H {31P} HSQC (see supplementary information). Single crystal X-ray diffraction analysis (see supplementary information) comfirmed the structure of TPP1 as shown in the thermal ellipsoid plot in Fig. 1.
Scheme 1.

Synthesis of compound TPP1 by direct functionalization of the propanol substituent in 1.
Fig. 1.

Thermal ellipsoid plot of TPP1 with hydrogen atoms, carbon labels, and solvent molecules not shown for clarity. The thermal ellipsoids were drawn to 50% probability.
Previously studied imidazolium salts exhibit very low aqueous solubility [44]. Therefore the aqueous solubility achieved by the substitution of imidazolium salts with a TPP moiety was desirable. The biscationic nature of TPP1 has considerably enhanced the water solubility to 8 mg/mL as compared to previously reported systems such as starting material 1, which has a solubility of <1 mg/mL [46]. While the aqueous solubility is helpful for aiding in the process of TPP1 delivery, it is important to balance that with sufficient lipophilicity to diffuse across the cell membrane.
2.2. Evaluation of TPP1 as an anticancer agent that causes apoptosis
Evaluation of TPP1 in the National Cancer Institute’s (NCI) Developmental Therapeutics Program (DTP) 60 human tumor cell line one-dose assay was utilized to determine preliminary cytotoxicity prior to evaluation against select bladder cancer cell lines. The results of this study showed significant activity against a variety of cancer cells lines over 48 h of drug exposure (see Supplementary Table 1). However, for imidazolium salts to be effective translational intravesical therapies for bladder cancer, they must elicit a cell-killing effect after exposure for 1 h or less. Patients treated intravesically cannot practically retain a drug for long periods, and this likely contributes to the ineffectiveness of intravesical DNA-damaging agents or synthetic toxins, which require prolonged exposure for incorporation into the cell cycle. We therefore assessed the growth inhibition of TPP1 at various concentrations against select bladder cancer cell lines using the Cell-Titer-Glo® assay after 1-h exposures. For most of the cell lines assessed, the GI50 ranged from 200 to 250 μM when exposed to TPP1 for 1 h (Fig. 2A), was higher than the GI50 in the NCI-60 panel, as expected. The cancer cell line growth inhibition by TPP1 was also assessed at a lower exposure times in RT112 cells. The inhibition at a 250 μM (highest calculated GI50) dose was fairly constant when exposed for 5, 15 or 30 min, while the toxicity increased when the exposure time was extended to 1 h (Fig. 2B, blue bars). When RT112 cells were treated with 500 μM TPP1 (200% of highest GI50), the toxicity increased proportionally with exposure time, resulting in complete inhibition after 1 h exposure (Fig. 2B, red bars). Further, increasing the dose of TPP1 to the GI99 concentration (1000 μM) resulted in near-complete inhibition of cell growth for the 15-, 30-and 60-min treatments, with significant growth inhibition at 5 min. Similar results were achieved with RT4 cells (not shown). Thus, for all further in vitro assays, 250 μM and 500 μM concentrations with an exposure time of 1 h were chosen to achieve the highest efficacy.
Fig. 2.
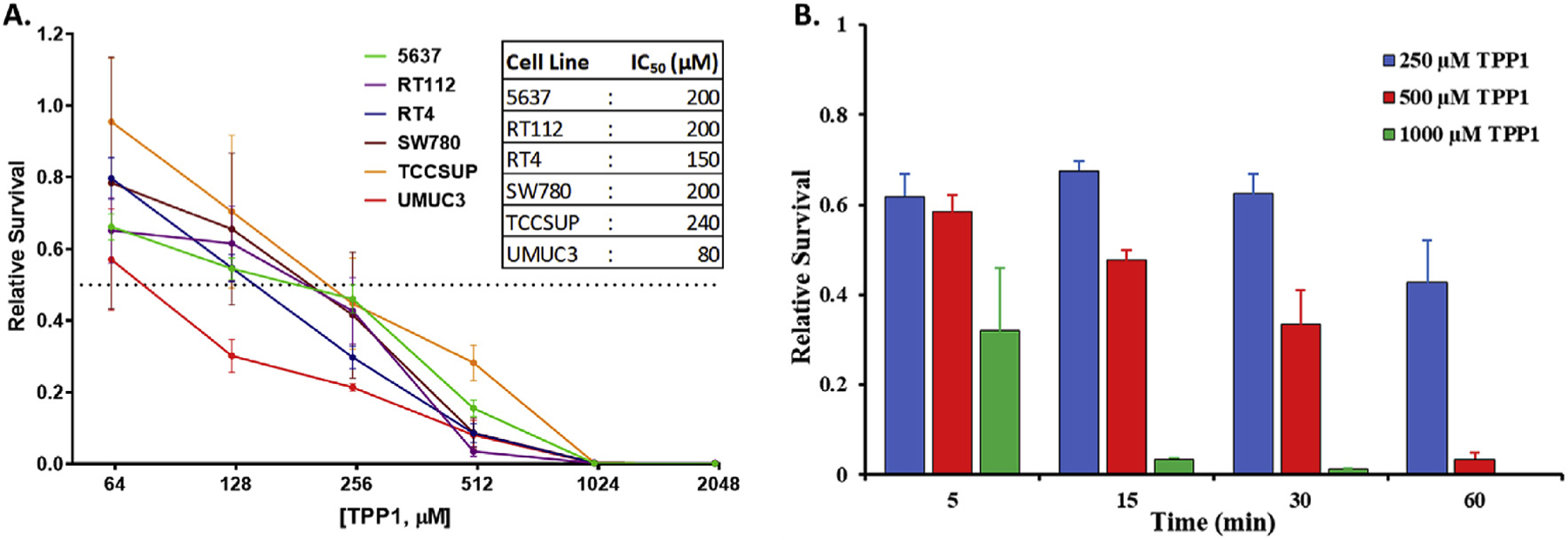
(A) Growth inhibition caused by TPP1 to various bladder cancer cell lines at 1 h exposure time, measured as the relative survival with respect to untreated cells. The dotted line parallel to x-axis represents 50% survival cutoff. Table inset shows the calculated GI50 values of TPP1 for each cell line. (B) Growth inhibition of RT112 cells caused by different concentrations of TPP1 at different exposure times.
The long-term cytotoxicity on select bladder cancer cell lines after exposure to TPP1 was assessed using a colony-forming assay (Fig. 3). No colonies formed after treatment with TPP1 for 1 h at 500 μM or after 250 μM treatment for RT112 and UMUC3 cell lines, while the J82 cell line had few colonies.
Fig. 3.

Colony formation by selected bladder cancer cells 10 days after 1 h TPP1 exposure.
To determine if TPP1 caused growth inhibition or cell death, propidium iodide staining was performed followed by flow cytometry to measure the amount of late phase apoptotic cells (sub-G1 fraction). All cell lines assessed had significant amounts of apoptotic debris with respect to untreated cells (Fig. 4A). RT112 had a lower sub-G1% after 500 μM TPP1 exposure than when treated with 250 μM. This may be due to the extreme and rapid toxicity of the higher dose (see below).
Fig. 4.

(A) Cell cycle analysis of selected bladder cancer cell lines were used to measure the sub-G1 population 24 h after 1 h exposure to various concentrations of TPP1. (B) Western blots revealing that RT112 cells underwent PARP cleavage (c-PARP) and caspase-9 cleavage (c-Casp9) after treatment with TPP1. Ran was used as a loading control.
To confirm that the mechanism of toxicity was apoptosis, RT112 cells and cell debris were collected immediately after or 24 h after the end of treatment and cleaved PARP1 (c-PARP) and cleaved caspase-9 (c-Casp9), both markers of apoptosis, were measured using immunoblot (Fig. 4B). The amount of c-PARP after 500 μM TPP1 treatment was higher immediately after treatment than at 24 h, while the amount of Ran (loading control) was depleted immediately after treatment and further decreased with increasing doses after 24 h. In addition, c-Casp9 was detected after treatment with 250 μM TPP1 immediately after treatment, but was absent in 500 μM TPP1 treated groups. Caspase-9 is cleaved during mitochondrion-triggered apoptosis to initiate the cell death cascade. This suggests that RT112 cells undergo apoptosis with such briskness that the detectable signal was disintegrated after 24 h due to cell fragmentation.
2.3. Establishment of a TPP1 as a mitotoxin
Based on the aforementioned literature showing that delocalized lipophilic cations are mitotoxins, we hypothesized that TPP1 had a similar mechanism of action. However, identification of c-Casp9 is common to all chemotherapeutics which cause apoptosis, even though they may not specifically be mitotoxins. To test our hypothesis, we isolated mitochondria from RT112 cells and treated them with TPP1. Identification of mitotoxicity in the absence of the rest of the cellular components would prove that TPP1 is a direct mitotoxin. We first probed the cytosol/nuclear fraction and the mitochondrial fraction with Lamin A/C (a nucleoprotein) and cytochrome c (a mitochondria-specific protein). This proved that there was no contamination in either fraction (Fig. S11). We measured mitochondrial toxicity by assaying cytochrome c release into the supernatant of the incubation buffer using immunoblot. Cytochrome c expulsion from the mitochondria is an irreversible step in the apoptotic cascade. There was intense release of cytochrome c into the supernatant of mitochondrial suspensions 4 h after TPP1 treatment, with no release into the supernatant after incubation with vehicle, and some release after CCCP exposure (Fig. 5A).
Fig. 5.
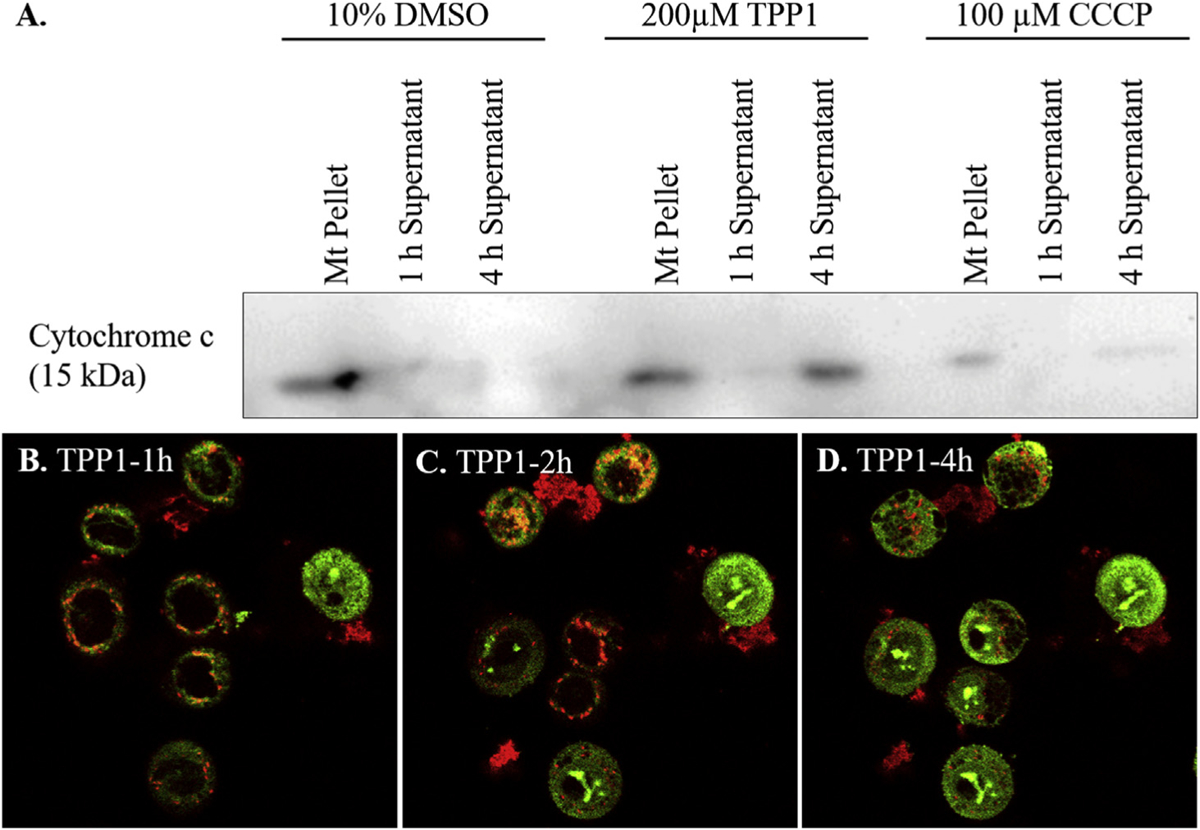
Representative pictures from live cell imaging of JC-1 stained RT112 cells (A) 1 h, (B) 2 h, and (C) 4 h after a treatment with 200 μM TPP1 with cells showing mitochondria-mediated apoptosis of the cells. (D) Immunoblot showing the release of cytochrome c from mitochondrial pellet into supernatant after treatment with vehicle, CCCP (mitochondrial membrane depolarizing agent, positive control) or TPP1.
In order to confirm that TPP1 triggers apoptosis via a mitochondrial pathway, JC-1 staining of RT112 cells after a treatment with 200 μM TPP1 was performed. JC-1 is a mitochondrial dye that reversibly changes color from red to green as the membrane potential decreases. JC-1 stains polarized mitochondria red and depolarized mitochondria green. After treatment of cells with TPP1 for 1 h, cells had red punctate markings which indicated the presence of intact (polarized) mitochondria (Fig. 5B–D). The gradual loss of membrane potential and initiation of apoptosis was observed as the red color slowly shifted from red diffusely to green. As a positive control carbonyl cyanide m-chlorophenyl hydrazone (CCCP) was used, and indeed, CCCP-treated cells also underwent mitochondrial depolarization, but vehicle-treated or untreated cells maintained mitochondrial integrity (Fig. S12).
Many intravesical chemotherapeutic drugs are known to exfoliate the bladder urothelium after treatment [49,50]. The exfoliating activity of TPP1 in murine models was assessed by intravesical instillation of 100 μL of either 750 μM or 1500 μM TPP1 in anesthetized female mice for 1 h or vehicle (10% DMSO) and repeating the treatment after 24 h. Mice were sacrificed 24 h after the final treatment. TPP1 treated mice showed a slightly shortened urothelial cell height compared to vehicle treatment, but the layer was still 2 + cell layers thick in most placed and there were no areas where the underlying submucosa was exposed (Fig. 6), despite applying such high doses. Therefore, TPP1 does not appear to exfoliate the urothelium, in contrast to our prior series of compounds [51]. In addition, no inflammation or fibrosis was seen, although longer term studies with repetitive instillations are underway.
Fig. 6.
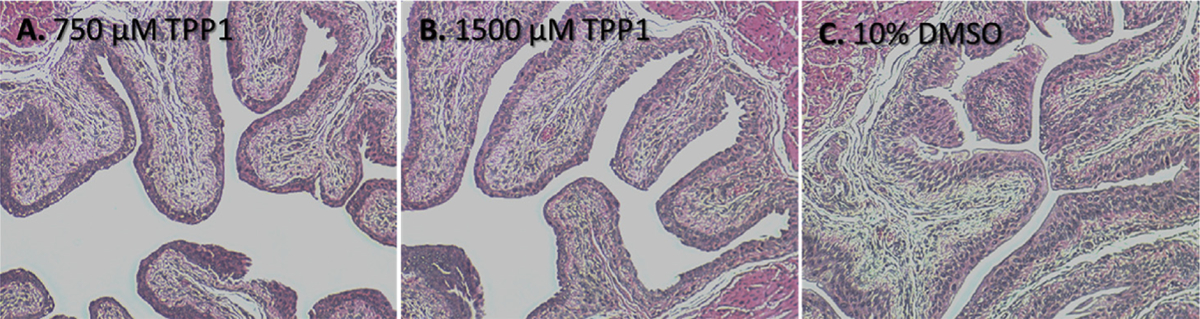
Representative histological images showing the normal bladder epithelium after treatment with 750 μM TPP1 (A) or 1500 μM TPP1 (B) or vehicle (C). Mice were kept under anesthesia for 1 h after instillation of TPP1 or vehicle in the bladder.
Due to the brisk and effective induction of cell death in bladder cancer cell lines, with the absence of histological damage in the normal bladder of murine models after intravesical instillation, we hypothesized that TPP1 would behave as a selective agent for cancerous tissue versus normal tissues in a murine bladder cancer model. To test this hypothesis, tumors were induced in murine bladders by the addition 0.05% BBN in drinking water until a bladder tumor was identified. The presence of tumors in the bladders of the mice were confirmed by excretory μCT urography. Anesthetized mice with bladder tumors were then treated for 1 h with 1500 μM TPP1, and histology and immunohistology was performed to stain the apoptotic marker, cleaved caspase-3. Mice treated with TPP1 showed significantly higher levels of cleaved caspase-3 in the locations that had a significant amount of hyperplasia, dysplasia and necrosis (Fig. 7A) Cleaved caspsase-3 was not appreciably detected in the normal adjacent urothelium in TPP1-treated mice with tumors, (Fig. 7B). Cleaved caspase-3 was also not appreciably detected in the tumor, or in the adjacent normal urothelium of the vehicle control group (Fig. 7C–D). This shows that TPP1 induces apoptosis in cancer cells without harming normal cells in the bladder.
Fig. 7.

Mouse bladder immunohistology with anti-caspase-3 antibody (dark brown) after treatment with TPP1 (A, B) or vehicle (C, D) on BBN-induced mice which harbored bladder tumors. Images A and C represent area of bladder urothelium that harbored hyperplasia/dysplasia (tumor) that were treated with TPP1 and vehicle, respectively, while images B and D represent sections of tumor-adjacent normal urothelium in TPP1-treated and vehicle-treated mice, respectively.
3. Conclusions
In this work, the synthesis of a new compound, TPP1, containing a triphenylphosphonium moiety combined with an imidazolium salt and its cancer-specific toxicity in vitro and in vivo is described. The biscationic nature of TPP1 affords an increase in aqueous solubility, as compared to imidazolium salts alone [44]. Bladder cancer cell lines briefly treated with TPP1 cause irreversible toxicity, demonstrating the ability of TPP1 to exert an apoptotic effect in a rapid manner within an acceptable patient treatment window for use in intravesical administration. Multiple lines of congruent evidence suggests that TPP1 causes apoptosis and that the mechanism of action is mitochondrial toxicity, where the drug depolarizes mitochondria, triggering apoptosis.
Typical intravesical chemotherapies such as mitomycin, thiotepa, and doxorubicin also cause urothelial exfoliation, denudation, and cause nuclear changes in the normal urothelium [50,52,53]. Of great importance is the generation of a compound that can induce cancer-specific cell death in vivo with a limited exposure time for intravesical administration, preferably in the absence of inflammation or bladder pain. The results presented here demonstrate that TPP1 causes apoptotic cell death in bladder cancer cells with limited exposure times in a cancer-specific manner. The apparent rapidity and irreversibility of the drug effect might translate to better patient compliance when translated to clinical settings. Current standard-of-care protocols suggest a 1 h dwell time for BCG or other intravesical chemotherapies. IC99 doses may be adequate for shorter exposure times in patients – a significant potential benefit. Another benefit of TPP1 is its synthetic versatility, as the imidazolium salt can be modified to balance aqueous solubility and anticancer activity. Future work is aimed at the generation of a library of compounds similar to TPP1 as well as further studies to better understand the observed biological activity.
4. Experimental section
4.1. General considerations
All reactions were performed aerobically unless otherwise stated. Solvents and chemical reagents used were unmodified and were purchased from VWR, Fischer Scientific, or Sigma Aldrich. 2-(bromomethyl)naphthalene was purchased from Waterstone Technologies. The starting material 1 was synthesized performed by our previously published procedure [46]. The 1H, 31P and 13C NMR spectra were acquired on Varian 300 MHz, Inova 400 MHz, or Varian 500 MHz instruments. All NMR samples were prepared using DMSO-d6 purchased from Cambridge Isotope Laboratories and were referenced to residual protons of the solvent (DMSO-d6, 1H: 2.50 ppm, 13C: 39.52 ppm). Melting points were obtained on a MelTemp apparatus. Infrared spectroscopy was obtained using a Thermo Scientific ATR-IR Nicolet iS5 FT-IR spectrometer with an iD5 ATR adapter. TPP1 was evaluated for PAINS by using the web-site as follows: http://zinc15.docking.org/patterns/home/ [54].
4.2. Synthesis of TPP1
1,3-bis(naphthalen-2-ylmethyl)-2-(3-hydroxypropyl)-1H-benzimidazol-3-ium bromide, 1,(1.01 g, 1.88 mmol) was suspended in dry dichloromethane (24 mL) and dry DMF (0.5 mL). The suspension was stirred in an ice bath and thionyl bromide (1.50 mL 19.41 mmol) was added resulting in the suspension dissolving. This solution was allowed to warm to room temperature and after 7 h diethyl ether was added (50 mL) producing a thick orange oil. The oil was allowed to settle and the solvent was decanted away. Additional portions (3 × 20 mL) of diethyl ether were added to remove any remaining thionyl bromide. The remaining oil was suspended in dry acetonitrile (50 mL) and triphenylphosphine (4.00 g, 15.27 mmol) was added changing the color from orange to yellow-brown. The reaction was refluxed for 72 h after which the mixture was cooled in an ice bath. The reaction was filtered and the filtrate was collected. Upon removal of solvent under reduced pressure, the product was taken up in dichloromethane and filtered to remove impurities. The product was precipitated from the filtrate with diethyl ether and collected by vacuum filtration. The solid was then washed with water (~15 mL) and recovered by vacuum filtration. The tan solid was stirred in diethyl ether for 24 h, collected by vacuum filtration, and dried under reduced pressure. (0.9896 g, 61.05%) 1H NMR (400 MHz DMSO-d6): δ 7.94–7.92 (m, 2H, Ar), 7.90–7.86 (m, 6H, Ar), 7.82–7.80 (m, 2H, Ar), 7.75–7.72 (m, 3H, Ar), 7.59–7.54 (m, 18H, Ar), 7.47 (d, 2H, Ar), 6.15 (s, 4H, CH2), 4.00 (m, 4H, 2-CH2), 1.88 (m, 2H, CH 2). 13C NMR (125 MHz DMSO-d6): δ 153.5, 134.8 (d, J = 2.8 Hz), 133.2 (d, J = 10.4 Hz), 132.6, 132.4, 131.8, 131.4, 130.0 (d, J = 12.5 Hz), 128.5, 127.6, 126.53, 126.50, 126.4, 125.3, 124.6, 117.6 (d, J = 86.2 Hz), 113.7, 48.8, 24.2 (d, J = 22.3 Hz), 20.5, 20.1 (d, J = 51.2 Hz). 31P NMR (121 MHz DMSO-d6) δ 23.3. ATR-IR: 3054w (C–H, sp2), 2910w (C–H, sp2), 2862w (C–H, sp2), 733w (P–C stretch) cm−1. HRMS: m/z = 351.1528 (theor for [M2+] C50H43N2 P2+ = 351.158); m/z = 701.309 (theor for [M+-H] (Wittig) C50H42N2P+ 701.308). mp = 189–190 °C.
Crystal data for TPP1: C52H51Br2N2O2P, M 926.74, monoclinic, a = 10.8583(2) A, b = 34.0758(6) A, c = 12.9337(13) A, β = 112.0353(10)°, V = 4435.97(13) A3, T = 100(2) K, space group P2(1)/n, Z = 4, 50384 reflections measured, 9015 independent reflections (Rint = 0.1152). The final R1 values were 0.0502 (I > 2σ(I)). The final wR(F2) values were 0.0869 (I > 2σ(I)). The final R1 values were 0.1017 (all data). The final wR(F2) values were 0.1055 (all data).
4.3. Cell titer glo assay
Bladder cancer cell lines were seeded at 10000 cells/well in black-walled 96 well plates and incubated for 24 h. Cells were treated with TPP1 in their respective media (as described by ATCC) at concentrations of 62.5 μM–2000 μM dissolved in two-fold increments or vehicle for designated time, followed by replacement of treatment media with normal growth media. After 24 h, plates with cells were incubated at room temperature for 30 min, followed by addition of 20 μL of CellTiter-Glo® luminescent cell viability assay reagent (Promega) and mixing. After 2 min of incubation at room temperature, luminescence for each plate was measured using IVIS. Relative survival for each treatment group was calculated using vehicle control. All treatments were performed in quadruple.
4.4. Colony forming assay
Cells were seeded at 500 cells/well in 6-well plates and 24 h later treated with 250 μM or 500 μM TPP1 or vehicle for 1 h. Cells were then allowed to grow in normal media for 10 days followed by staining with crystal violet (0.5% crystal violet in 20% MeOH/water) and incubation at 4 °C for 5 min. The wells were then washed several times with DI water, and dried prior to imaging. All the experiments were performed in triplicate.
4.5. Sub-G1 analysis
Cells were seeded in 6-well plates at 300,000 cells/well, in triplicate for each treatment. After 24 h, they were treated with 250 μM or 500 μM TPP1 or vehicle for 1 h. After 24 h growth in normal media, the media and cells were collected and washed with 1% FBS in PBS. Washed cells were suspended in 0.5 mL of PBS and fixed with 5 mL 70% cold ethanol (−20 °C), followed by incubation at 4 °C for 30 min protected from light. Cells were pelleted, washed, incubated at room temperature in phosphate citric acid buffer, repelleted, and resuspended in propidium iodide (PI)/RNase solution and analyzed. The stained cells were analyzed using a BD FACScan™ flow cytometer for sub-G1 populations using the appropriate gating parameters.
4.6. Western blot analysis
RT112 cells were seeded at 300,000 cells/well in a six well plate and 24 h later treated with 250 μM or 500 μM TPP1 or vehicle. Either immediately after treatment or 24 h after treatment, the cells and floating debris were pelleted, washed with PBS, and lysed in RIPA buffer with protease inhibitors. Cell lysate (~30 μg protein) was mixed in the sample buffer containing 50 μM DTT and incubated at 95 °C for 10 min. Proteins were separated using SDS-PAGE and transferred to a nitrocellulose membrane. The membrane was blocked with 2% milk (fat-free), stained with PARP (Cell Signaling Technology, #9542), caspase 9 (Cell Signaling Technology, #9502) and Ran (BD Biosciences, #610340) primary antibodies for 1 h, and washed. HRP-conjugated secondary antibodies (Invitrogen, #31430 and #31460) were added and incubated for 1 h. The bands were detected using an Amersham™ ECL kit (GE Life Sciences) and imaged on FluorChem E Digital Darkroom.
4.7. Live cell imaging
In a 35 mm glass bottom microwell dish (MatTek Corp), 200000 RT112 cells were plated and incubated overnight. The cells were treated with 200 μM TPP1 and incubated for 45 min. JC-1 (EMD Millipore Corp) was added into the media at the final concentration of 10 μg/mL and further incubated for 15 min. The cells were imaged using λEx: 485 nm and λEm: 530 and 590 nm at 15 min intervals for 6 h in Inverted Leica SP8 6 channels confocal microscope.
4.8. Cytochrome c release study
Mitochondria were extracted from 20 million RT112 cells using mitochondria extraction kit for mammalian cells (Thermo Scientific) using manufacturer’s instructions. Extracted mitochondria were treated with 10% DMSO, 100 μM CCCP or 200 μM TPP1 for 1 h at 37 °C. The treated mitochondria were then suspended in mitochondria preservative buffer and incubated at 37 °C. The treated mitochondria were centrifuged to obtain supernatant and pellet after 1 h and 4 h. The pellets were lysed, and each fraction was analyzed for cytochrome c using western blot following the above mentioned protocol.
4.9. Animal experiments
All animal experiments were performed in live animal facility at FCCC strictly following the IACUC protocol (19–03). Bladder tumors were induced in a group of mice by supplementing water with 0.05% N-butyl-N-(4-hydroxybutyl)nitrosamine (BBN). Presence of a bladder mass was confisrmed using excretory μCT urograms. Mice were injected with 75 μL of 1:3 iodixanol (Visipaque™) dissolved in PBS. All mice were anesthetized and maintained at 1–3% isoflurane during treatment. Treatment of female mice was performed by instilling two doses of 100 μL of 1500 μM or 750 μM TPP1 in 10% DMSO/PBS after bladder evacuation using a 24G angiocatheter. Male mice were instilled with 50 μL of 3000 μM or 1500 μM TPP1 in 20% DMSO/PBS. Double the concentration and half the volume was used because the bladder of male mice cannot be evacuated with the catheter. The male mouse bladder was partially evacuated by gentle palpation of the abdomen prior to anesthesia. Confirmation of proper instillation was achieved using a retrograde μCT cystogram, which showed contrast in the bladder only. Male and female control groups received equal volume of vehicle. Two doses were given at an interval of 24 h and 1 h incubation time each. Jelco ProtectIV®Plus 24G x ¾” catheters (Smith Medical) were used for instilling TPP1 or vehicle control and stayed there until the end of the treatment. Bladders were collected 24 h post second treatment, and submitted to FCCC Histology Core Facility for further processing with hematoxylin and eosin, or immunohistochemistry using anti-cleaved caspase-3 antibody and developed with diaminobenzene.
Supplementary Material
Acknowledgments
The authors would like to thank NCI’s DTP for performing the 60 human tumor cell line screening with their one dose assay for TPP1. We are also grateful for the funding provided by the University of Akron’s Department of Chemistry for the purchase of chemical reagents. We thank the National Science Foundation (NSF) for providing funds for the purchase of the NMR instruments (Nos. CHE-0341701 and DMR-0414599) and the X-ray diffractometer (CHE-0840446) used in this work. Furthermore we thank Jason O’Neill and Chrys Wesdemiotis for their mass spectrometry work in this manuscript (NSF: CHE-1808115).
The authors would also like to thank the generosity of Peter Kriendler, Newport Foundation, Bucks County Board of Associates. Additionally we are grateful for the technical assistance from Jim Oesterling of the Fox Chase Histopathology Core, Andrey Efimov from Confocal Facility and Lab Animal Facility.Authors thank Jeremiah Buttram for technical experimental assistance and optimization. PHA is supported by the BCAN Young Investigator Award, and NCI CA218976. U.S. is supported by the Avery Postdoctoral Fellowship. Work was supported by the Fox Chase Center Core Grant (P30 CA006927).
Funding
Funding was recieved for this work.
Abbreviations Used
- GI50
concentration at 50% inhibition of cellular proliferation
- GI99
concentration at 99% inhibition of cellular proliferation
- PARP1
Poly(ADP-Ribose) Polymerase 1
- BBN
N-butyl-N-(4-hydroxybutyl)nitrosamine
- BCG
Bacillus Calmette-Guerin
- QC
quality control
- DLC
delocalized lipophilic cation
- MMP
mitochondrial membrane potential
- TPP
triphenylphosphonium
- NCI
National Cancer Institute
- DTP
Developmental Therapeutics Program
- μCT
micro computed tomography
- DMSO
dimethyl sulfoxide
- PBS
phosphate buffered saline
- PI
propidium iodide
- CCCP
Carbonyl cyanide m-chlorophenyl hydrazine
Footnotes
Intellectual property
We confirm that we have given due consideration to the protection of intellectual property associated with this work and that there are no impediments to publication, including the timing of publication, with respect to intellectual property. In so doing we confirm that we have followed the regulations of our institutions concerning intellectual property.
Declaration of competing interest
No conflict of interest exists.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ejmech.2019.111832.
References
- [1].American Cancer Society, Cancer Facts & Figures, Springer New York, New York, NY, 2019, 10.1007/978-1-4614-8063-1. [DOI] [Google Scholar]
- [2].Apollo A, Ortenzi V, Scatena C, Zavaglia K, Aretini P, Lessi F, Franceschi S, Tomei S, Sepich CA, Viacava P, Mazzanti CM, Naccarato AG, Molecular characterization of low grade and high grade bladder cancer, PLoS One 14 (2019) 1–18, 10.1371/journal.pone.0210635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Morales A, Eidinger D, Bruce AW, Intracavitary Bacillus calmette-guerin in the treatment of superficial bladder tumors, J. Urol 167 (2002) 891–894, 10.1016/j.juro.2016.10.101. [DOI] [PubMed] [Google Scholar]
- [4].Davies BJ, Hwang TJ, Kesselheim AS, Ensuring access to injectable generic drugs-the cae of intravesical BCG for bladder cancer, N. Engl. J. Med 376 (2017) 1401–1403, 10.1056/NEJMp1701294. [DOI] [PubMed] [Google Scholar]
- [5].Messing EM, The BCG shortage, Bladder Cancer 3 (2017) 227–228, 10.3233/blc-179018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bandari J, Maganty A, MacLeod LC, Davies BJ, Manufacturing and the market: rationalizing the shortage of Bacillus calmette-guérin, Eur. Urol. Focus 4 (2018) 481–484, 10.1016/j.euf.2018.06.018. [DOI] [PubMed] [Google Scholar]
- [7].Skinner EC, Goldman B, Sakr WA, Petrylak DP, Lenz HJ, Lee CT, Wilson SS, Benson M, Lerner SP, Tangen CM, Thompson IM, SWOG S0353: phase II trial of intravesical gemcitabine in patients with nonmuscle invasive bladder cancer and recurrence after 2 prior courses of intravesical bacillus calmette-guérin, J. Urol 190 (2013) 1200–1204, 10.1016/j.juro.2013.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Steinberg RL, Thomas LJ, O’Donnell MA, Nepple KG, Sequential intravesical gemcitabine and docetaxel for the salvage treatment of non-muscle invasive bladder cancer, Bladder Cancer 1 (2015) 65–72, 10.3233/BLC-150008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Milbar N, Kates M, Chappidi MR, Pederzoli F, Yoshida T, Sankin A, Pierorazio PM, Schoenberg MP, Bivalacqua TJ, Oncological outcomes of sequential intravesical gemcitabine and docetaxel in patients with non-muscle invasive bladder cancer, Bladder Cancer 3 (2017) 293–303, 10.3233/BLC-170126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Lightfoot AJ, Breyer BN, Rosevear HM, Erickson BA, Konety BR, O’Donnell MA, Multi-institutional analysis of sequential intravesical gemcitabine and mitomycin C chemotherapy for non-muscle invasive bladder cancer, Urol. Oncol. Semin. Orig. Invest 32 (2014) 35, 10.1016/j.urolonc.2013.01.009, e15–35.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Finks JF, Osborne NH, Birkmeyer JD, Trends in hospital volume and operative mortality for high-risk surgery, N. Engl. J. Med 364 (2011) 2128–2137, 10.1056/NEJMsa1010705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hautmann RE, Hautmann SH, Hautmann O, Complications associated with urinary diversion, Nat. Rev. Urol 8 (2011) 667–677, 10.1038/nrurol.2011.147. [DOI] [PubMed] [Google Scholar]
- [13].Novara G, De Marco V, Aragona M, Boscolo-Berto R, Cavalleri S, Artibani W, Ficarra V, Complications and mortality after radical cystectomy for bladder transitional cell cancer, J. Urol 182 (2009) 914–921, 10.1016/j.juro.2009.05.032. [DOI] [PubMed] [Google Scholar]
- [14].Gregg JR, Cookson MS, Phillips S, Salem S, Chang SS, Clark PE, Davis R, Stimson CJ, Aghazadeh M, Smith JA, Barocas DA, Effect of preoperative nutritional deficiency on mortality after radical cystectomy for bladder cancer, J. Urol 185 (2011) 90–96, 10.1016/j.juro.2010.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Avritscher EBC, Cooksley CD, Grossman HB, Sabichi AL, Hamblin L, Dinney CP, Elting LS, Clinical model of lifetime cost of treating bladder cancer and associated complications, Urology 68 (2006) 549–553, 10.1016/j.urology.2006.03.062. [DOI] [PubMed] [Google Scholar]
- [16].Botteman MF, Pashos CL, Redaelli A, Laskin B, Hauser R, The health economics of bladder cancer, PharmacoEconomics 21 (2003) 1315–1330, 10.1007/BF03262330. [DOI] [PubMed] [Google Scholar]
- [17].Riley GF, Potosky AL, Lubitz JD, Kessler LG, Medicare payments from diagnosis to death for elderly cancer patients by stage at diagnosis, Med. Care 33 (1995) 828–841. [DOI] [PubMed] [Google Scholar]
- [18].Meyer A, Ghandour R, Bergman A, Castaneda C, Wosnitzer M, Hruby G, Benson M, McKiernan J, The natural history of clinically complete responders to neoadjuvant chemotherapy for urothelial carcinoma of the bladder, J. Urol 192 (2014) 696–701, 10.1016/j.juro.2014.03.078. [DOI] [PubMed] [Google Scholar]
- [19].Herr HW, Outcome of patients who refuse cystectomy after receiving neoadjuvant chemotherapy for muscle-invasive bladder cancer, Eur. Urol 54 (2008) 126–132, 10.1016/j.eururo.2007.12.031. [DOI] [PubMed] [Google Scholar]
- [20].Herr HW, Bajorin DF, Scher HI, Neoadjuvant chemotherapy and bladder-sparing surgery for invasive bladder cancer: ten-year outcome, J. Clin. Oncol 16 (1998) 1298–1301, 10.1200/JCO.1998.16.4.1298. [DOI] [PubMed] [Google Scholar]
- [21].Solsona E, Climent MA, Iborra I, Collado A, Rubio J, Ricós JV, Casanova J, Calatrava A, Monrós JL, Bladder preservation in selected patients with muscle-invasive bladder cancer by complete transurethral resection of the bladder plus systemic chemotherapy: long-term follow-up of a phase 2 nonrandomized comparative trial with radical cystectomy, Eur. Urol 55 (2009) 911–921, 10.1016/j.eururo.2008.08.027. [DOI] [PubMed] [Google Scholar]
- [22].Solsona E, Iborra I, Collado A, Rubio-Briones J, Casanova J, Calatrava A, Feasibility of radical transurethral resection as monotherapy for selected patients with muscle invasive bladder cancer, J. Urol 184 (2010) 475–481, 10.1016/j.juro.2010.04.008. [DOI] [PubMed] [Google Scholar]
- [23].Cookson MS, Herr HW, Zhang ZF, Soloway S, Sogani PC, Fair WR, The treated natural history of high risk superficial bladder cancer: 15- year outcome, J. Urol 158 (1997) 62–67, 10.1097/00005392-199707000-00017. [DOI] [PubMed] [Google Scholar]
- [24].Sylvester RJ, van der Meijden APM, Lamm DL, Intravesical bacillus Calmette-Guerin reduces the risk of progression in patients with superficial bladder cancer: a meta-analysis of the published results of randomized clinical trials, J. Urol 168 (2002) 1964–1970, 10.1097/01.ju.0000034450.80198.1c. [DOI] [PubMed] [Google Scholar]
- [25].Liaw F, Tan YY, Hendry D, Systemic BCG-osis following intravesical BCG instillation for bladder carcinoma, Clin. Case Rep 5 (2017) 1569–1572, 10.1002/ccr3.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Neuzil J, Dong LF, Rohlena J, Truksa J, Ralph SJ, Classification of mitocans, anti-cancer drugs acting on mitochondria, Mitochondrion 13 (2013) 199–208, 10.1016/j.mito.2012.07.112. [DOI] [PubMed] [Google Scholar]
- [27].Modica-Napolitano JS, Aprille JR, Delocalized lipophilic cations selectively target the mitochondria of carcinoma cells, Adv. Drug Deliv. Rev 49 (2001) 63–70, 10.1016/S0169-409X(01)00125-9. [DOI] [PubMed] [Google Scholar]
- [28].Davis S, Weiss MJ, Wong JR, Lampidis TJ, Chen LB, Mitochondrial and plasma membrane potentials cause unusual accumulation and retention of rhodamine 123 by human breast adenocarcinoma-derived MCF-7 cells, J. Biol. Chem 260 (1985) 13844–13850. [PubMed] [Google Scholar]
- [29].Modica-Napolitano JS, Aprille JR, Basis for the selective cytotoxicity of rhodamine 123, Cancer Res. 47 (1987) 4361–4365. [PubMed] [Google Scholar]
- [30].Johnson LV, Walsh ML, Chen LB, Localization of mitochondria in living cells with rhodamine 123, Proc. Natl. Acad. Sci 77 (1980) 990–994, 10.1073/pnas.77.2.990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Summerhayes IC, Lampidis TJ, Bernal SD, Nadakavukaren JJ, Nadakavukaren KK, Shepherd EL, Chen LB, Unusual retention of rhodamine 123 by mitochondria in muscle and carcinoma cells, Proc. Natl. Acad. Sci 79 (1982) 5292–5296, 10.1073/pnas.79.17.5292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zielonka J, Joseph J, Sikora A, Hardy M, Ouari O, Vasquez-Vivar J, Cheng G, Lopez M, Kalyanaraman B, Mitochondria-targeted triphenylphosphonium-based compounds: syntheses, mechanisms of action, and therapeutic and diagnostic applications, Chem. Rev 117 (2017) 10043–10120, 10.1021/acs.chemrev.7b00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Smith RAJ, Porteous CM, Gane AM, Murphy MP, Delivery of bioactive molecules to mitochondria in vivo, Proc. Natl. Acad. Sci 100 (2003) 5407–5412, 10.1073/pnas.0931245100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ross MF, Prime TA, Abakumova I, James AM, Porteous CM, Smith RAJ, Murphy MP, Rapid and extensive uptake and activation of hydrophobic triphenylphosphonium cations within cells, Biochem. J 411 (2008) 633–645, 10.1042/BJ20080063. [DOI] [PubMed] [Google Scholar]
- [35].Han M, Vakili MR, Soleymani Abyaneh H, Molavi O, Lai R, Lavasanifar A, Mitochondrial delivery of doxorubicin via triphenylphosphine modification for overcoming drug resistance in MDA-MB-435/DOX cells, Mol. Pharm 11 (2014) 2640–2649, 10.1021/mp500038g. [DOI] [PubMed] [Google Scholar]
- [36].Cui B, Zheng BL, He K, Zheng QY, Imidazole alkaloids from Lepidium meyenii, J. Nat. Prod 66 (2003) 1101–1103, 10.1021/np030031i. [DOI] [PubMed] [Google Scholar]
- [37].Yang X, Zeng X, Zhang Y, Qing C, Song W, Li L, Zhang H, Synthesis and cytotoxic activities of novel phenacylimidazolium bromides, Bioorg. Med. Chem. Lett 19 (2009) 1892–1895, 10.1016/j.bmcl.2009.02.065. [DOI] [PubMed] [Google Scholar]
- [38].Chen W, Yang X-D, Li Y, Yang L-J, Wang X-Q, Zhang G-L, Zhang H-B, Design, synthesis and cytotoxic activities of novel hybrid compounds between dihydrobenzofuran and imidazole, Org. Biomol. Chem 9 (2011) 4250–4255, 10.1039/c1ob05116d. [DOI] [PubMed] [Google Scholar]
- [39].Wang D, Richter C, Rühling A, Hüwel S, Glorius F, Galla HJ, Anti-tumor activity and cytotoxicity in vitro of novel 4,5-dialkylimidazolium surfactants, Biochem. Biophys. Res. Commun 467 (2015) 1033–1038, 10.1016/j.bbrc.2015.10.015. [DOI] [PubMed] [Google Scholar]
- [40].Wang D, De Jong DH, Rühling A, Lesch V, Shimizu K, Wulff S, Heuer A, Glorius F, Galla HJ, Imidazolium-based lipid analogues and their interaction with phosphatidylcholine membranes, Langmuir 32 (2016) 12579–12592, 10.1021/acs.langmuir.6b02496. [DOI] [PubMed] [Google Scholar]
- [41].Riduan SN, Zhang Y, Imidazolium salts and their polymeric materials for biological applications, Chem. Soc. Rev 42 (2013) 9055–9070, 10.1039/c3cs60169b. [DOI] [PubMed] [Google Scholar]
- [42].Malhotra SV, Kumar V, A profile of the in vitro anti-tumor activity of imidazolium-based ionic liquids, Bioorg. Med. Chem. Lett 20 (2010) 581–585, 10.1016/j.bmcl.2009.11.085. [DOI] [PubMed] [Google Scholar]
- [43].Rühling A, Wang D, Ernst JB, Wulff S, Honeker R, Richter C, Ferry A, Galla HJ, Glorius F, Influence of the headgroup of azolium-based lipids on their biophysical properties and cytotoxicity, Chem. Eur J 23 (2017) 5920–5924, 10.1002/chem.201604182. [DOI] [PubMed] [Google Scholar]
- [44].Wright BD, Deblock MC, Wagers PO, Duah E, Robishaw NK, Shelton KL, Southerland MR, DeBord MA, Kersten KM, McDonald LJ, Stiel JA, Panzner MJ, Tessier CA, Paruchuri S, Youngs WJ, Anti-tumor activity of lipophilic imidazolium salts on select NSCLC cell lines, Med. Chem. Res 24 (2015) 2838–2861, 10.1007/s00044-015-1330-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Shelton KL, DeBord MA, Wagers PO, Southerland MR, Taraboletti A, Robishaw NK, Jackson DP, Tosanovic R, Kofron WG, Tessier CA, Paruchuri S, Shriver LP, Panzner MJ, Youngs WJ, Synthesis, anti-proliferative activity, and toxicity of C4(C5) substituted N,N0-bis(arylmethyl) imidazolium salts, Tetrahedron 72 (2016) 5729–5743, 10.1016/j.tet.2016.07.068. [DOI] [Google Scholar]
- [46].Shelton KL, DeBord MA, Wagers PO, Southerland MR, Williams TM, Robishaw NK, Shriver LP, Tessier CA, Panzner MJ, Youngs WJ, Synthesis, anti-proliferative activity, SAR study, and preliminary in vivo toxicity study of substituted N,N′-bis(arylmethyl)benzimidazolium salts against a panel of non-small cell lung cancer cell lines, Bioorg. Med. Chem 25 (2017) 421–439, 10.1016/j.bmc.2016.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].DeBord MA, Wagers PO, Crabtree SR, Tessier CA, Panzner MJ, Youngs WJ, Synthesis, characterization, and in vitro SAR evaluation of N,N′-bis(arylmethyl)-C2-alkyl substituted imidazolium salts, Bioorg. Med. Chem. Lett 27 (2017) 196–202, 10.1016/j.bmcl.2016.11.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].DeBord MA, Southerland MR, Wagers PO, Tiemann KM, Robishaw NK, Whiddon KT, Konopka MC, Tessier CA, Shriver LP, Paruchuri S, Hunstad DA, Panzner MJ, Youngs WJ, Synthesis, characterization, in vitro SAR and in vivo evaluation of N,N0bisnaphthylmethyl 2-alkyl substituted imidazolium salts against NSCLC, Bioorgan. Med. Chem. Lett 27 (2017) 764–775, 10.1016/j.bmcl.2017.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Pandey R, Jackson JK, Liggins R, Mugabe C, Burt HM, Enhanced taxane uptake into bladder tissues following co-administration with either mitomycin C, doxorubicin or gemcitabine: association to exfoliation processes, BJU Int. 122 (2018) 898–908, 10.1111/bju.14423. [DOI] [PubMed] [Google Scholar]
- [50].Murphy WM, Soloway MS, Crabtree WN, The morphologic effects of mitomycin C in mammalian urinary bladder, Cancer 47 (1981) 2567–2574. [DOI] [PubMed] [Google Scholar]
- [51].Wagers PO, Tiemann KM, Shelton KL, Kofron WG, Panzner MJ, Wooley KL, Youngs WJ, D.a. Hunstad, Imidazolium salts as small-molecule urinary bladder exfoliants in a murine model, Antimicrob. Agents Chemother 59 (2015) 5494–5502, 10.1128/AAC.00881-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Lopez-Beltran A, Changes produced in the urothelium by traditional and newer therapeutic procedures for bladder cancer, J. Clin. Pathol 55 (2002) 641–647, 10.1136/jcp.55.9.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Murphy WM, Soloway MS, Finebaum PJ, Pathological changes associated with topical chemotherapy for superficial bladder cancer, J. Urol 126 (1981) 461–464. [DOI] [PubMed] [Google Scholar]
- [54].Kenny PW, Comment on the ecstasy and agony of assay interference compounds, J. Chem. Inf. Model 57 (2017) 2640–2645, 10.1021/acs.jcim.7b00313. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.