

Abstract
Mustard vesicants, including sulfur mustard (2,2′-dichlorodiethyl sulfide, SM) and nitrogen mustard (bis(2-chloroethyl)methylamine, HN2) are cytotoxic blistering agents synthesized for chemical warfare. Because they contain highly reactive electrophilic chloroethyl side chains, they readily react with cellular macromolecules like DNA forming monofunctional and bifunctional adducts. By targeting DNA, mustards can compromise genomic integrity, disrupt the cell cycle, and cause mutations and cytotoxicity. To protect against genotoxicity following exposure to mustards, cells initiate a DNA damage response (DDR). This involves activation of signaling cascades including ATM (ataxia telangiectasia mutated), ATR (ataxia telangiectasia and Rad3-related) and DNA-PKcs (DNA-dependent protein kinase, catalytic subunit). Signaling induced by the DDR leads to the recruitment and activation of repair related proteins such as phospho H2AX and phospho p53 to sites of DNA lesions. Excessive DNA modifications by mustards can overwhelm DNA repair leading to single and double strand DNA breaks, cytotoxicity and tissue damage, sometimes leading to cancer. Herein we summarize DDR signaling pathways induced by SM, HN2 and the half mustard, 2-chloroethyl ethyl sulfide (CEES). At the present time, little is known about how mustard-induced DNA damage leads to the activation of DDR signaling. A better understanding of mechanisms by which mustard vesicants induce the DDR may lead to the development of countermeasures effective in mitigating tissue injury.
Keywords: sulfur mustard, vesicants, DNA damage response, p53, H2AX, cell cycle, nitrogen mustard
1. Introduction
Vesicants, including the bifunctional alkylating agents sulfur mustard (2,2′-dichlorodiethyl sulfide, SM) and nitrogen mustard (bis(2-chloroethyl)methylamine, HN2) and the monofunctional alkylating agent, 2-chloroethyl ethyl sulfide (CEES) or half mustard, target DNA and are mutagenic and cytotoxic (Dacre and Goldman, 1996; Singh et al., 2018; Wang et al., 2012). These agents contain electrophilic chloroethyl side chains that readily react with nucleophilic nitrogen or oxygen atoms in DNA bases (Wang et al., 2012). CEES mainly generates monoadducts on the N7 position of guanine and the N3 position of adenine (Zubel et al., 2019). In addition to monoadducts, bifunctional vesicants also produce intrastrand and interstrand cross-links, as well as DNA-protein cross-links; typical cross-linked products include bis-N7-guanine adducts, N7-guanine-N3-adenine adducts and bis-N3-adenine adducts (Balcome et al., 2004; Fidder et al., 1994; Yue et al., 2014; Zubel et al., 2019). These covalent adducts, especially cross-links, can alter the global structure of DNA and interfere with replication and transcription. Importantly, mustard adducts do not cause DNA strand breaks directly; errors are introduced in damaged DNA by endonucleases that are responsible for replication and repair and this can cause single-strand breaks (SSBs) and double-strand breaks (DSBs). Both SSBs and DSBs have been detected in cells and tissues following exposure to mustards (Jost et al., 2015; Mol et al., 1993). Inaccurate repair of DNA breaks, particularly DSBs, is associated with cell cycle arrest, genomic instability and chromosomal damage (Her and Bunting, 2018).
Mechanisms by which cells repair DNA have been reviewed for many agents that modify DNA including alkylating agents as well as radiation and ultraviolet light (Chatterjee and Walker, 2017; Panahi et al., 2018; Sirbu and Cortez, 2013). In the present review we have overviewed cellular response to mustard vesicants with an emphasis on DNA damage signaling cascades important in repair processes. Precise mechanisms by which many of these signaling pathways are induced in target cells following exposure to vesicants are not known. However, it is clear that DNA damage signaling is required to maintain the integrity of DNA and its proper functioning, an important component of the wound healing process (Panahi et al., 2018).
2. DNA damage responses to vesicants
In response to DNA damage, cells initiate a complex DNA damage response (DDR), a process important in repairing DNA lesions and preventing genotoxicity. The DDR is comprised of DNA damage signaling including activation of cell cycle checkpoints, DNA repair, chromatin remodeling and modulation of gene expression, which are all important in maintaining genome stability (Blackford and Jackson, 2017; Giglia-Mari et al., 2011; Lanz et al., 2019). ATR interacting protein (ATRIP), MRE11– RAD50–NBS1 (MRN) and Ku70/Ku80 complex are among an array of sensor proteins that detect DNA damage (Blackford and Jackson, 2017). These sensors rapidly recruit an array of kinases to the damaged DNA which function to activate DNA damage signaling (Fig 1). Key to initiating DNA damage signaling cascades are phosphatidylinositol 3-kinase-related kinases (PIKKs), in particular, ATM (ataxia telangiectasia mutated), ATR (ataxia telangiectasia and Rad3-related), and DNA-PKcs (DNA-dependent protein kinase, catalytic subunit). These PIKKs bind to DSBs as well as a variety of other DNA lesions, many of which interfere with replication (Blackford and Jackson, 2017; Giglia-Mari et al., 2011). This launching kinase transduces signaling via phosphorylation to a wide variety of adapter/mediator proteins, such as 53BP1 (p53-binding protein 1) and MDC1 (mediator of DNA damage checkpoint 1) for ATM, and TopBP1 (topoisomerase-binding protein 1) and Claspin for ATR. Phosphorylation of effector kinases such as CHK1 and CHK2 transmits signals to regulate many cellular functions including transcription factors, cell cycle regulators, apoptotic mediators, and DNA repair proteins (Giglia-Mari et al., 2011; Lanz et al., 2019).
Fig. 1. Summary of DNA damage signaling following exposure of cells to vesicants.

Mustard vesicants cause DNA damage forming both signal-strand breaks (SSBs) and double-strand breaks (DSBs). In response, cells activate DNA damage response (DDR) signaling pathways. This is initiated by DNA damage sensors such as ATRIP (ATR interacting protein), MRN (MRE11–RAD50–NBS1) complex and the Ku70/Ku80 complex. These proteins are recruited to sites of DNA damage where they trigger DNA damage signaling transducers, including apical phosphatidylinositol 3-kinase-related kinases (PIKKs) such as ATM, ATR, and DNA-PKcs. ATM and DNA-PKcs respond to DNA double strand breaks, while ATR responds to replication protein A (RPA)-coated single strand DNA (ssDNA). These are activated by phosphorylation. ATR and ATM activate downstream cell cycle regulators CHK1 and CHK2, respectively, which in turn signals downstream regulatory proteins such as p53 to regulate cell cycle transit and to activate DNA repair, apoptosis and/or senescence. Activated PIKKs also phosphorylate H2AX, a histone H2A variant, at S139 to promote DNA repair.
Table I summarizes activation of DNA damage response signaling following mustard vesicant exposure in both cells in culture and animal models. SM and its analogs activate ATM, ATR, and DNA-PKcs by stimulating phosphorylation of these proteins on S1981, S428 and S2056, respectively (Inturi et al., 2014; Jan et al., 2019; Jowsey et al., 2012; Sagar et al., 2017; Tewari-Singh et al., 2010). Effectors of apical kinases such as CHK1, CHK2, p53, and H2AX are subsequently activated to retain cells at cell cycle checkpoints and induce DNA damage repair (Black et al., 2010; Goswami et al., 2016; Joseph et al., 2014; Jowsey and Blain, 2015; Kumar et al., 2015; Zhang et al., 2017). Activation of p53, CHK1, and CHK2 checkpoints by mustards slows progression through the cell cycle (Inturi et al., 2014; Jan et al., 2019) and can lead to apoptosis (Sourdeval et al., 2006).
Table 1.
Examples of DNA damage signaling induced by vesicants.
| Vesicant | System (treatment concentration, exposure time) | Responses | References |
|---|---|---|---|
| CEES (half mustard) | TK6 cells (100 μM, 1–24 h) | ↑ phosphorylation of p53 (S15) and Chk2 (T68) | Jowsey et al., 2009 |
| JB6 and HaCaT cells (0.25–1 mM, 0.5–4 h) | ↑ phosphorylation of ATM (S1981), ATR (S428), CHK1 (S345), CHK2 (T68), Cdc25C (S216), Cdc2 (Y15), and Cdk2 (Y15) | Tewari-Singh et al., 2010 | |
| RAW264.7 cells (0.05–1 mM, 24 h) | ↑ phosphorylation of ATM (S1981), ATR (S428), p53 (S15), and H2AX (S139) | Sagar et al., 2017 | |
| EpiDerm-FT™ (a human skin equivalent) (0.1–1 mM, 15 min-24 h) | ↑ phosphorylation of H2AX (S139) | Black et al., 2010 | |
| Nitrogen mustard (Mechlorethamine) | JB6 cells (0.75 μM, 0.5–24 h) | ↑ phosphorylation of DNA-PK (S2056), H2AX (S139),and p53 (S15) | Inturi et al., 2014 |
| HCE cells (50–100 μM, 12–48 h) | ↑ phosphorylation of H2AX (S139) and p53 (S15) | Goswami et al., 2016 | |
| A549 cells (1–50 μM, 0.5–24 h) | ↑ phosphorylation of ATM (S1981), Chk2 (T68), H2AX (S139), and p53 (S15) | Jan et al., 2019 | |
| SKH1 mouse skin (3.2 mg, 12–120 h) | ↑ phosphorylation of p53 (S15) and H2AX (S139) | Kumar et al, 2015 | |
| Sulfur mustard | TK6 cells (0.1–1 μM, 1–24 h) | ↑ phosphorylation of ATM (S1981), ATR (S428), p53 (S15), CHK1 (S317), CHK2 (T68), ↑ p53 | Jowsey et al., 2012 |
| Hela, HCT 116, HEK 293, SH-SY5Y and A549 cells (25 μM 5 h) | ↑ phosphorylation of CHK1 (S296 and S317) | Jowsey and Blain, 2015 | |
| SKH1 mouse skin (1.4 g/m3 vapor, 6 min) | ↑ phosphorylation of H2AX (S139) | Joseph et al., 2014 | |
| Balb/c mice (bone marrow) (15–40 mg/kg s.c. injection) | ↑ phosphorylation of H2AX (S139) | Zhang et al., 2017 |
It should be noted that DDR mediated signaling is an early and transient event after vesicant exposure. This is supported by earlier studies showing that CEES (1000 μM)-induced H2AX phosphorylation (S139; γH2AX) occurred after 15 min post-exposure and was maximal after 2–6 h in a full-thickness human skin equivalent (EpiDerm-FT™) model (Black et al., 2010). H2AX phosphorylation in this model was sustained for at least 24 h followed by a marked decline after 72 h. Jowsey et al. (2009) also reported that phosphorylation of CHK2 at T68 appeared one h after CEES treatment (100 μM), reached a peak at 4 h, and reduced to control levels after 24 h in TK6 cells. Phosphorylation of other DNA damage signaling molecules including ATM (S1981), ATR (S428), DNA-PKcs (S2056), p53 (S15), and CHK1 (S345) was also increased at early times (30 min - 2 h) in several cell models following treatment with CEES, HN2 and SM (Goswami et al., 2016; Inturi et al., 2014; Jan et al., 2019; Jowsey and Blain, 2015; Jowsey et al., 2009, 2012; Kumar et al., 2015; Sagar et al., 2017; Tewari-Singh et al., 2010).
The predominant pathways for DNA repair after mustard exposure include direct chemical reversal by alkyltransferases (Cheng et al., 2016; Kisby et al., 2009), base excision repair (BER) (Bhat et al., 2006; Jowsey et al., 2009, 2012), nucleotide excision repair (NER) (Jowsey et al., 2012), translesion synthesis (TLS) (Ho et al., 2011; Roy et al., 2016), homologous recombination (HR) (Jowsey et al., 2010, 2012), and non-homologous end joining (NHEJ) (Jowsey et al., 2009, 2012). Mechanisms of DNA repair by these pathways has been reviewed (Panahi et al., 2018). Table II summarizes studies that evaluated these DNA damage repair pathways in response to vesicant exposures. Chemical reversal, BER, and NER are critical pathways to remove vesicant-DNA adducts. TLS is a damage tolerance mechanism responsible for repairing interstrand cross-links (Roy and Scharer, 2016). BER and NER also mediate SSBs repair; DSBs are repaired by HR and NHEJ pathways (Abbotts and Wilson, 2017; Panahi et al., 2018). From these studies it is apparent that DNA damage induced by mustard vesicants activates DDR and a number of repair pathways. This is likely to be important in protecting tissues from SM-induced DNA damage and cytotoxicity. Excessive DNA damage can overwhelm DNA damage repair resulting in toxicity in tissues exposed to SM.
Table 2.
DNA damage and repair pathways activated following mustard vesicant exposure.
| Repair pathway | Mediators | System / Alkylating agent | References |
|---|---|---|---|
| Direct chemical reversal | O6-methylguanine-DNA methyltransferase (Mgmt) | Mice Mgmt(−/−) neurons / HN2 | Kisby et al., 2009 |
| 16HBE cells MGMT siRNA knockdown/ HN2 | Cheng et al., 2016 | ||
| Translesion synthesis (TLS) | TLS polymerase | Synthesized major groove DNA interstrand cross-links / NM | Ho et al., 2011 |
| e.g. Pol Z, Pol ζ, Pol η and Pol κ | Synthesized DNA interstrand cross-links / cisplatin, NM | Roy et al., 2016 | |
| Nucleotide excision repair (NER) | Glycosylase, APE1, ligase, DNA polymerase | LB708 cells (NER-deficient cells) / CEES | Jowsey et al., 2009 |
| GM04312 cells (NER-deficient cells) / SM | Jowsey et al., 2012 | ||
| Base excision repair (BER) | Poly-ADP-Ribose-Polymerase (PARP) | PARP (−) HEK cells / SM | Bhat et al., 2006 |
| ligase, Pol β | Hela cells MX inhibition / CEES | Jowsey et al., 2009 | |
| MV1174 cells (BER-deficient cells) / SM | Jowsey et al., 2012 | ||
| Homologous recombination (HR) | ATM signaling kinases | V-C8 CHO cells (HR-deficient cells) / SM | Jowsey et al., 2010 |
| JB6 cells BO2 inhibition / HN2 | Inturi et al., 2014 | ||
| Non-homologous end joining (NHEJ) | DNA-PKcs signaling kinases | TK6 cells NU7026 inhibition / CEES | Jowsey et al., 2009 |
| TK6 cells NU7026 inhibition / SM | Jowsey et al., 2012 | ||
| JB6 cells NU7026 inhibition / HN2 | Inturi et al., 2014 |
Delineating mechanisms of DNA damage and repair following exposure to mustards such as SM, HN2 and CEES is complex as there are multiple pathways causing DNA damage that depend not only on concentration and tissue distribution if studies are performed in vivo, but also because induction of these pathways is often time- and/or cell cycle-dependent. For example, mustards can rapidly damage DNA, however, there is often a mix of early and later onset DNA repair pathways (Inturi et al., 2014; Jan et al., 2019). Cells with high expression of repair proteins may respond immediately to DNA damage, while other cells requiring induction of repair signaling may require up to 24 hours (Inturi et al., 2014). During this time, cells may transit through the cell cycle, a process that may also regulate different DNA repair mechanisms. Alternatively, DNA modifications may hold cells in different phases of the cell cycle and this may alter DNA repair signaling.
3. DNA damage signaling in different phases of the cell cycle
Earlier studies have suggested that DNA damage and repair are modulated by chromatin structure (Mao and Wyrick, 2019). As chromatin undergoes dynamic changes in structure during the cell cycle, this can strongly affect the accessibility of DNA to mustards, as well as the recruitment of repair proteins. This can lead to distinct DNA lesions and repair in different phases of cell cycle. Indeed, the major DNA repair pathways are known to be regulated in a cell cycle phase-dependent manner (Branzei and Foiani, 2008). Both BER and NER are important throughout the cell cycle, it is thought that BER is important in removing damaged nucleotides and repair of SSBs while NER is known to be involved in repairing pyrimidine dimers (Branzei and Foiani, 2008). Earlier studies have also suggested that NER has an essential role in DNA repair during the G1 phase of the cell cycle (Ray et al., 2016). HR repairs DSBs during S and G2 phases of the cell cycle, while NHEJ repairs DNA in all phases of the cell cycle (Her and Bunting, 2018).
Our laboratory and others have shown that alkylating agents can cause selective DNA damage and repair in different phases of the cell cycle (Branzei and Foiani, 2008; Dhuppar and Mazumder, 2018; Escribano-Diaz et al., 2013; Halicka et al., 2005; Jan et al., 2019; Saha et al., 2017). Dhuppar and Mazumder reported that γH2AX levels peaked in the S phase of A549 damaged cells after exposure to 4-nitroquinoline 1-oxide, a known DNA damaging agent (Dhuppar and Mazumder, 2018). Preferentially induction of γH2AX in S phase of the cell cycle was also observed in Hela and HL-60 cells damaged by other DNA damage agents including UV-B (Halicka et al., 2005) and alkylating agent such as HN2 in A549 cells (Jan et al., 2019). In addition, markers for other types of DNA strand breaks, assessed by the accumulation of BRCA1 and RPA foci, were reported to be more pronounced in the S/G2 phase of the cell cycle when compared to the G1 phase after HeLa-Fucci cells or U2OS cells were irradiated with ionizing radiation (Escribano-Diaz et al., 2013; Saha et al., 2017).
Figure 2 shows an example from our laboratories of differential activation of DNA damage signaling in different phases of the cell cycle following exposure of human lung epithelial A549 cells to HN2. Treatment of A549 cells with HN2 leads to cell cycle dependent activation of phospho H2AX (S139) and phospho p53 (S15) (Fig 2) (Jan et al., 2019). Activation of these proteins was significantly greater in cells in S phase when compared to cells in G0/G1 and G2/M. Higher expression of phospho H2AX (S139), a biomarker for DNA DSBs, suggests that cells in S phase contain more DNA lesions and are able to recruit greater amounts of DNA repair proteins. This may be explained by the fact that during S-phase replication, the DNA double helix unwinds and opens to facilitate DNA synthesis. When this occurs, there is increased accessibility of DNA to mustard alkylation and DSBs. Similarly, S phase cells also express greater levels of phospho p53, a DDR that can suppress cell cycle progression allowing time for DNA repair, as well as regulate various repair pathways including BER and NER (Williams and Schumacher, 2016).
Fig. 2. Cell cycle-dependent induction of DNA damage response proteins by HN2 in A549 cells.
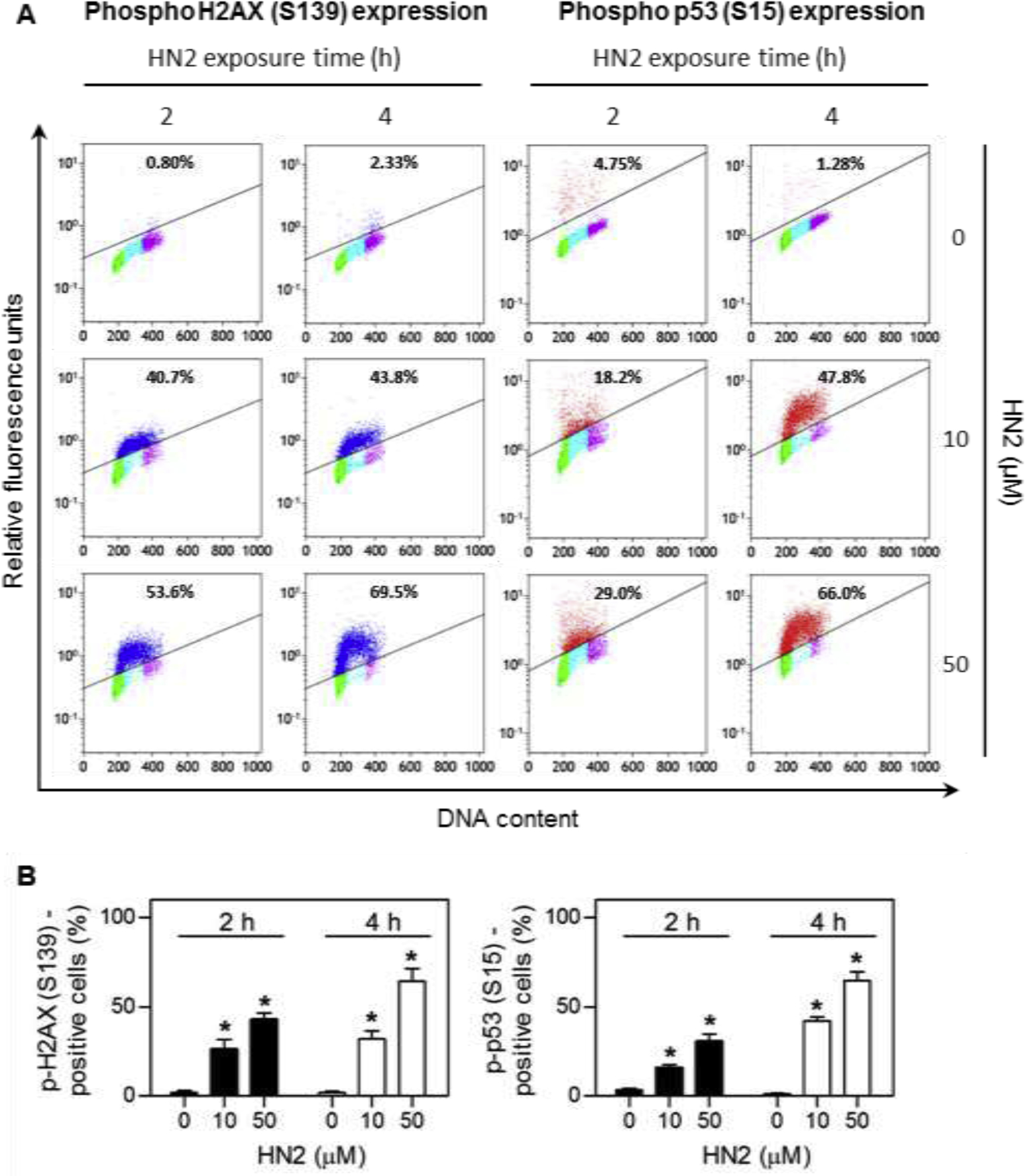
A549 cells were treated with increasing concentrations of HN2 or vehicle control in serum-free medium. After 2 h and 4 h, cells were harvested and stained with Alexa Fluor 488-conjugated phospho H2AX (S139) antibody or Alexa Fluor 647-conjugated phospho p53 (S15) antibody. DNA was then stained with propidium iodide and the cells analyzed by flow cytometry. (A) Representative bivariate distributions of DNA and expression of phospho proteins. Cells shown in blue express phospho H2AX (S139), while cells in red express phospho p53 (S15). Cells that do not express or express very low levels of phospho H2AX or phospho p53 are shown in green for cells in G0/G1 phase, cyan for cells in S phase, and magenta for cells in G2/M phase. The percentages of immunopositive cells are shown at the top of each panel. (B) Relative phospho H2AX and phospho p53 protein expression. Data are presented as means ± SE, n = 4. Data adapted from Jan et al., 2019.
4. Summary and conclusions
Alkylation of DNA is an important mechanism by which mustards exert toxicity in target tissues. These modifications interfere with DNA replication and repair which leads to cytotoxicity and tissue injury. DDR signaling is a key process by which cells repair DNA damage and is critical for maintaining tissue homeostasis. This pathway is mediated by intracellular sensors and transducers of DNA damage which in turn, recruit DNA repair proteins to sites of DNA alkylation. Excessive DNA damage caused by mustard vesicants can result in SSBs and DSBs during the DNA repair process contributing to toxicity. Modifications to DNA and subsequent repair are cell cycle dependent; however, this depends on the ability of mustards to modify DNA during DNA synthesis. DDR signaling and repair may depend on the site of injury and time following exposure to the vesicant. Further studies are required to better characterize underlying mechanistic links between DNA damage signaling kinases and DNA repair pathways in cells following exposure to mustards. This will be important for the identification of pathways in the DDR response that may be targeted to prevent SSBs and DSBs potentially leading to the development of drugs that will mitigate toxicity.
Highlights.
Mustard vesicants such as sulfur mustard and nitrogen mustard are highly reactive with DNA
Mustards can form monofunctional and bifunctional adducts
Cells initiate a DNA damage response to protect against genotoxicity from mustards
The DNA damage response activates signaling cascades important in DNA repair
Inaccurate DNA repair can lead to single and double strand DNA breaks that contribute to cytotoxicity
Acknowledgements
This work was supported by National Institute of Health GrantsU54AR055073, R01ES004738, and P30ES005022.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Abbotts R, Wilson DM 3rd, 2017. Coordination of DNA single strand break repair. Free Radic. Biol. Med 107, 228–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balcome S, Park S, Quirk Dorr DR, Hafner L, Phillips L, Tretyakova N, 2004. Adenine-containing DNA-DNA cross-links of antitumor nitrogen mustards. Chem. Res. Toxicol 17, 950–962. [DOI] [PubMed] [Google Scholar]
- Bhat KR, Benton BJ, Ray R, 2006. Poly (ADP-ribose) polymerase (PARP) is essential for sulfur mustard-induced DNA damage repair, but has no role in DNA ligase activation. J. Appl. Toxicol 26, 452–457. [DOI] [PubMed] [Google Scholar]
- Black AT, Hayden PJ, Casillas RP, Heck DE, Gerecke DR, Sinko PJ, Laskin DL, Laskin JD, 2010. Expression of proliferative and inflammatory markers in a full-thickness human skin equivalent following exposure to the model sulfur mustard vesicant, 2-chloroethyl ethyl sulfide. Toxicol. Appl. Pharmacol 249, 178–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackford AN, Jackson SP, 2017. ATM, ATR, and DNA-PK: the trinity at the heart of the DNA damage response. Mol. Cell 66, 801–817. [DOI] [PubMed] [Google Scholar]
- Branzei D, Foiani M, 2008. Regulation of DNA repair throughout the cell cycle. Nat. Rev. Mol. Cell Biol 9, 297–308. [DOI] [PubMed] [Google Scholar]
- Chatterjee N, Walker GC, 2017. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen 58, 235–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J, Ye F, Dan G, Zhao Y, Wang B, Zhao J, Sai Y, Zou Z, 2016. Bifunctional alkylating agent-mediated MGMT-DNA cross-linking and its proteolytic cleavage in 16HBE cells. Toxicol. Appl. Pharmacol 305, 267–273. [DOI] [PubMed] [Google Scholar]
- Dacre JC, Goldman M, 1996. Toxicology and pharmacology of the chemical warfare agent sulfur mustard. Pharmacol. Rev 48, 289–326. [PubMed] [Google Scholar]
- Dhuppar S, Mazumder A, 2018. Measuring cell cycle-dependent DNA damage responses and p53 regulation on a cell-by-cell basis from image analysis. Cell Cycle 17, 1358–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escribano-Diaz C, Orthwein A, Fradet-Turcotte A, Xing M, Young JT, Tkac J, Cook MA, Rosebrock AP, Munro M, Canny MD, Xu D, Durocher D, 2013. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol. Cell 49, 872–883. [DOI] [PubMed] [Google Scholar]
- Fidder A, Moes GW, Scheffer AG, van der Schans GP, Baan RA, de Jong LP, Benschop HP, 1994. Synthesis, characterization, and quantitation of the major adducts formed between sulfur mustard and DNA of calf thymus and human blood. Chem. Res. Toxicol 7, 199–204. [DOI] [PubMed] [Google Scholar]
- Giglia-Mari G, Zotter A, Vermeulen W, 2011. DNA damage response. Cold Spring Harb. Perspect. Biol 3, a000745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami DG, Tewari-Singh N, Dhar D, Kumar D, Agarwal C, Ammar DA, Kant R, Enzenauer RW, Petrash JM, Agarwal R, 2016. Nitrogen mustard-induced corneal injury involves DNA damage and pathways related to inflammation, epithelial-stromal separation, and neovascularization. Cornea 35, 257–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halicka HD, Huang X, Traganos F, King MA, Dai W, Darzynkiewicz Z, 2005. Histone H2AX phosphorylation after cell irradiation with UV-B: relationship to cell cycle phase and induction of apoptosis. Cell Cycle 4, 339–345. [PubMed] [Google Scholar]
- Her J, Bunting SF, 2018. How cells ensure correct repair of DNA double-strand breaks. J. Biol. Chem 293, 10502–10511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho TV, Guainazzi A, Derkunt SB, Enoiu M, Scharer OD, 2011. Structure-dependent bypass of DNA interstrand crosslinks by translesion synthesis polymerases. Nucleic Acids Res. 39, 7455–7464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inturi S, Tewari-Singh N, Agarwal C, White CW, Agarwal R, 2014. Activation of DNA damage repair pathways in response to nitrogen mustard-induced DNA damage and toxicity in skin keratinocytes. Mutat. Res 763–764, 53–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan YH, Heck DE, Laskin DL, Laskin JD, 2019. Sulfur mustard analog mechlorethamine (bis(2-chloroethyl)methylamine) modulates cell cycle progression via the DNA damage response in human lung epithelial A549 cells. Chem. Res. Toxicol 32, 1123–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph LB, Heck DE, Cervelli JA, Composto GM, Babin MC, Casillas RP, Sinko PJ, Gerecke DR, Laskin DL, Laskin JD, 2014. Structural changes in hair follicles and sebaceous glands of hairless mice following exposure to sulfur mustard. Exp. Mol. Pathol 96, 316–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jost P, Svobodova H, Stetina R, 2015. Induction and repair of DNA cross-links induced by sulfur mustard in the A-549 cell line followed by a comet assay. Chem. Biol. Interact 237, 31–37. [DOI] [PubMed] [Google Scholar]
- Jowsey PA, Blain PG, 2015. Checkpoint kinase 1 is activated and promotes cell survival after exposure to sulphur mustard. Toxicol. Lett 232, 413–421. [DOI] [PubMed] [Google Scholar]
- Jowsey PA, Williams FM, Blain PG, 2009. DNA damage, signalling and repair after exposure of cells to the sulphur mustard analogue 2-chloroethyl ethyl sulphide. Toxicology 257, 105–112. [DOI] [PubMed] [Google Scholar]
- Jowsey PA, Williams FM, Blain PG, 2010. The role of homologous recombination in the cellular response to sulphur mustard. Toxicol. Lett 197, 12–18. [DOI] [PubMed] [Google Scholar]
- Jowsey PA, Williams FM, Blain PG, 2012. DNA damage responses in cells exposed to sulphur mustard. Toxicol. Lett 209, 1–10. [DOI] [PubMed] [Google Scholar]
- Kisby GE, Olivas A, Park T, Churchwell M, Doerge D, Samson LD, Gerson SL, Turker MS, 2009. DNA repair modulates the vulnerability of the developing brain to alkylating agents. DNA Repair (Amst) 8, 400–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar D, Tewari-Singh N, Agarwal C, Jain AK, Inturi S, Kant R, White CW, Agarwal R, 2015. Nitrogen mustard exposure of murine skin induces DNA damage, oxidative stress and activation of MAPK/Akt-AP1 pathway leading to induction of inflammatory and proteolytic mediators. Toxicol. Lett 235, 161–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanz MC, Dibitetto D, Smolka MB, 2019. DNA damage kinase signaling: checkpoint and repair at 30 years. EMBO J. 38, e101801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao P, Wyrick JJ, 2019. Organization of DNA damage, excision repair, and mutagenesis in chromatin: a genomic perspective. DNA Repair (Amst) 81, 102645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mol MA, van der Schans GP, Lohman PH, 1993. Quantification of sulfur mustard-induced DNA interstrand cross-links and single-strand breaks in cultured human epidermal keratinocytes. Mutat. Res 294, 235–245. [DOI] [PubMed] [Google Scholar]
- Panahi Y, Fattahi A, Nejabati HR, Abroon S, Latifi Z, Akbarzadeh A, Ghasemnejad T, 2018. DNA repair mechanisms in response to genotoxicity of warfare agent sulfur mustard. Environ. Toxicol. Pharmacol 58, 230–236. [DOI] [PubMed] [Google Scholar]
- Ray A, Blevins C, Wani G, Wani AA, 2016. ATR- and ATM-Mediated DNA Damage Response Is Dependent on Excision Repair Assembly during G1 but Not in S Phase of Cell Cycle. PLoS One 11, e0159344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy U, Mukherjee S, Sharma A, Frank EG, Scharer OD, 2016. The structure and duplex context of DNA interstrand crosslinks affects the activity of DNA polymerase eta. Nucleic Acids Res. 44, 7281–7291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagar S, Parida SR, Sabnam S, Rizwan H, Pal S, Swain MM, Pal A, 2017. Increasing NO level regulates apoptosis and inflammation in macrophages after 2-chloroethyl ethyl sulphide challenge. Int. J. Biochem. Cell Biol 83, 1–14. [DOI] [PubMed] [Google Scholar]
- Saha J, Wang SY, Davis AJ, 2017. Examining DNA double-strand break repair in a cell cycle-dependent manner. Methods Enzymol. 591, 97–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh RK, Kumar S, Prasad DN, Bhardwaj TR, 2018. Therapeutic journery of nitrogen mustard as alkylating anticancer agents: Historic to future perspectives. Eur. J. Med. Chem 151, 401–433. [DOI] [PubMed] [Google Scholar]
- Sirbu BM, Cortez D, 2013. DNA damage response: three levels of DNA repair regulation. Cold Spring Harb. Perspect. Biol 5, a012724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sourdeval M, Lemaire C, Deniaud A, Taysse L, Daulon S, Breton P, Brenner C, Boisvieux-Ulrich E, Marano F, 2006. Inhibition of caspase-dependent mitochondrial permeability transition protects airway epithelial cells against mustard-induced apoptosis. Apoptosis 11, 1545–1559. [DOI] [PubMed] [Google Scholar]
- Tewari-Singh N, Gu M, Agarwal C, White CW, Agarwal R, 2010. Biological and molecular mechanisms of sulfur mustard analogue-induced toxicity in JB6 and HaCaT cells: possible role of ataxia telangiectasia-mutated/ataxia telangiectasia-Rad3-related cell cycle checkpoint pathway. Chem. Res. Toxicol 23, 1034–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang QQ, Begum RA, Day VW, Bowman-James K, 2012. Sulfur, oxygen, and nitrogen mustards: stability and reactivity. Org. Biomol. Chem 10, 8786–8793. [DOI] [PubMed] [Google Scholar]
- Williams AB, Schumacher B, 2016. p53 in the DNA-damage-repair process. Cold Spring Harb. Perspect. Med 6, a026070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue L, Wei Y, Chen J, Shi H, Liu Q, Zhang Y, He J, Guo L, Zhang T, Xie J, Peng S, 2014. Abundance of four sulfur mustard-DNA adducts ex vivo and in vivo revealed by simultaneous quantification in stable isotope dilution-ultrahigh performance liquid chromatography-tandem mass spectrometry. Chem. Res. Toxicol 27, 490–500. [DOI] [PubMed] [Google Scholar]
- Zhang X, Mei Y, Wang T, Liu F, Jiang N, Zhou W, Zhang Y, 2017. Early oxidative stress, DNA damage and inflammation resulting from subcutaneous injection of sulfur mustard into mice. Environ. Toxicol. Pharmacol 55, 68–73. [DOI] [PubMed] [Google Scholar]
- Zubel T, Hochgesand S, John H, Steinritz D, Schmidt A, Burkle A, Mangerich A, 2019. A mass spectrometric platform for the quantitation of sulfur mustard-induced nucleic acid adducts as mechanistically relevant biomarkers of exposure. Arch. Toxicol 93, 61–79. [DOI] [PubMed] [Google Scholar]