

Abstract
Serum amyloid A (SAA) is a plasma protein that transports lipids during inflammation. To explore SAA solution conformations and lipid binding mechanism, we used hydrogen-deuterium exchange mass spectrometry, lipoprotein reconstitution, amino acid sequence analysis and molecular dynamics simulations. Solution conformations of lipid-bound and lipid-free mSAA1 at pH~7.4 agreed in details with the crystal structures but also showed important differences. The results revealed that amphipathic α-helices h1 and h3 comprise a lipid-binding site that is partially pre-formed in solution, is stabilized upon binding lipids, and shows lipid-induced folding of h3. This site sequesters apolar ligands via a concave hydrophobic surface in SAA oligomers. The largely disordered/dynamic C-terminal region is conjectured to mediate promiscuous binding of other ligands. The h1-h2 linker region is predicted to form an unexpected β-hairpin that may represent an early amyloidogenic intermediate. The results help establish structural underpinnings for understanding SAA interactions with its key functional ligands, its evolutional conservation, and its transition to amyloid.
Keywords: Hydrogen-deuterium exchange mass spectrometry, molecular dynamics simulations, lipoprotein nanoparticle, intrinsically disordered protein, β-hairpin misfolding intermediate, inflammation and immunity
Graphical Abstract
INTRODUCTION
Serum amyloid A (SAA) is a major acute-phase protein that is a biomarker of inflammation [1, 2] and the protein precursor of amyloid A (AA) that causes AA amyloidosis, a life-threatening complication of chronic inflammation [3, 4]. Elevated SAA is a causative factor for atherosclerosis [5]. SAA comprises a family of ~12 kDa proteins that have been highly evolutionally conserved for at least 5,000,000 years, and is proposed to play an essential role in the inflammatory response [6, 7] (Figure 1). In humans and other mammals, SAA is secreted by the liver into plasma and is also produced by various extrahepatic tissues at the inflammation sites [8, 9]. Hours after the onset of infection or injury, plasma levels of inducible SAA increase nearly a thousand-fold, reaching up to 3 mg/ml, and then rapidly decrease [1, 7, 10, 11]. The beneficial role of these spikes in SAA levels is unclear and probably relates to lipid transport.
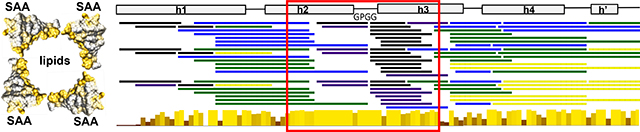
Figure 1.

Predicted and observed secondary structure and amino acid sequence conservation in the SAA protein family. The residue numbers correspond to human SAA1.
(A) Predicted β-sheet structure. Green arrows show residue segments predicted to initiate β-aggregation in SAA, or “amyloid hotspots” [15]; brown arrows show the β-hairpin predicted by MD simulations of the current study.
(B) Linear diagram shows β-sheets observed in the AA amyloid structure, derived from mSAA1.1 (PDB: 6DSO); arrows represent β-strands and lines are turns/loops [34].
(C) Linear diagram depicts secondary structure observed by x-ray crystallography (PDB 4IP9) in lipid-free hSAA1.1 [31]. Rectangles show α-helices h1-h4 and a 3/10 helix h’; first and last residue numbers in each helix are indicated. Lines show turn/coil regions; dashed line indicates the variable h3-h4 linker. Apices 1 and 2 (A1, A2) and vertices 1 and 2 (V1, V2) are indicated, along with their residue numbers (see Figure 2 for detail). Residues numbering is according to human SAA1 that has an additional N-terminal Gly compared to murine SAA1.
(D) Temperature factors of the Cα atoms from the crystal structure of hSAA1.1 (PDB: 4IP9); the B-factors are color-coding as shown, from low to high (blue to red). Similar B-factor distribution was observed in all nine symmetry-unrelated SAA molecules from the four available crystal structures of hSAA1 and mSAA3 (PDB: 4IP8, 4IP9, 4Q5G, 6PY0) in four different space groups; therefore, this B-factor distribution was determined by factors other than lattice contacts. Amino acids are shown for hSAA1. Black arrow shows the major cleavage site that generates AA fragment found in the in vivo deposits.
(E) The conservation table lists amino acid sequences of 19 SAA proteins. The sequence alignment was performed using Clustal Omega server [64] and the results were displayed using Jalview, http://www.jalview.org/ [65]. Conserved residues are color-coded: apolar (blue), polar (green), acidic (purple), basic (red), Pro (yellow), Gly (orange). Yellow box highlights the β-hairpin observed in our MD simulations of mSAA1.
(F) Conservation plot shows more conserved residues in lighter colors; the bar height represents the number of tabulated SAA sequences wherein each residue is conserved.
(G) Consensus plot shows the predominant residues in each position.
Most circulating SAA binds to plasma high-density lipoproteins (HDL) and reroutes HDL transport [12, 13]. Like other HDL proteins, SAA is water-soluble and is transiently released in a structurally labile metabolically active free form [14] that probably is a protein precursor of amyloid [15–17]. In vitro at pH~7, free SAA solubilizes vesicles containing diverse phospholipids, cholesterol, and their mixtures [17–23], the products of lipid oxidation [24], and a lipophilic vitamin retinol [25, 26]. Binding of diverse lipids suggests that SAA can act as a lipid scavenger. Moreover, in vitro SAA can spontaneously solubilize phospholipid bilayers to form HDL-size particles (8–10 nm) that are hydrolyzed by another acute-phase reactant, secretory phospholipase A2. SAA also sequesters toxic products of lipolysis, compelling us to propose that SAA and secretory phospholipase A2 act in synergy to remove cell membrane debris from the sites of injury [27]. In summary, compelling evidence indicates that lipids are the major functional ligands of SAA which regulate its activity and metabolic fate, and that SAA preferentially binds to HDL-size particles in vivo or forms them de novo [27–29]. The current study establishes the structural basis for SAA-lipid interactions in such particles.
In addition to lipids, SAA binds various cell receptors involved in lipid homeostasis and host defense, including CD36, SR-BI, LOX1, RAGE, TLR2 and TLR4, along with other diverse ligands ([9, 11, 29] and references therein). Such promiscuous ligand binding, as well as amyloid formation, probably stem from the dynamic conformation of SAA [21, 23]. In fact, SAA is an intrinsically disordered protein that is substantially unfolded at near-physiological conditions in the absence of bound ligands [30].
Remarkably, x-ray crystal structures of lipid-free human SAA isoform 1.1 (hSAA1.1) and murine SAA isoform 3 (mSAA3) have been determined to ~2 Å resolution in four different crystal forms [25, 26, 31]. All forms present a similar Y-shaped helix bundle with ~75% helical content, comprised of α-helices h1 through h4 followed by a 3/10 helix h’ (Figure 2A). Hooper and colleagues noticed that the protein interior is polar, and proposed that this atypical fold has been evolutionally conserved to bind retinol [25]. The current study determines how the crystal structures relate to the partially disordered protein conformations in solution and in lipoproteins.
Figure 2.

SAA monomer x-ray crystal structure and model.
(A) Cartoon representation of the SAA monomer based on the ~2.2 Å resolution x-ray crystal structures of hSAA1 and mSAA3 [25, 26, 31]. Rectangles show α-helices (h1, h2, h3, h4) and a 3/10 helix (h’), rainbow-colored from the N- to the C-terminus (blue to red). First and last residue numbers in each helix are in italics. Residue numbering is according to hSAA1. Two apices and two vertices are indicated. Dashed line shows variable h3-h4 linker at apex 2.
(B) Space-filling model illustrating surface hydrophobicity of mSAA1 monomer. A homology model of mSAA1 was obtained using the crystal structure of hSAA1 (PDB ID 4IP9) and Swiss model software [66]. Hydrophobic residues are colored yellow. Dotted arc indicates a concave hydrophobic surface whose radius of curvature, r~4.5 nm, ideally fits the HDL surface curvature. Crystal structures of hSAA1 and mSAA3A show a similar hydrophobic surface [21].
Previous studies using SAA truncations and antibody binding have implicated the N-terminal ~10 residues in lipid binding and self-association [18, 31–33]. However, deletion of these residues did not completely eliminate lipid binding, suggesting that other protein regions are involved. We observed that in the crystal structures, the two amphipathic α-helices, h1 and h3, form a large concave hydrophobic surface in SAA monomer (Figure 2B) and proposed that this surface binds HDL and lipids [21]. Recent crystallographic studies supported this idea and showed that a hydrophobic pocket created by h1 and h3 in the well-ordered SAA oligomer binds retinol [26]. The current study combines experimental and computational approaches to elucidate the lipid-binding mechanism of this intrinsically disordered protein. The results provide key insights into the conformation of lipid-free SAA in solution (the structurally labile and metabolically active transient form that is the likely precursor of amyloid) and in model HDL-size lipoproteins (the major circulating form of SAA in vivo).
RESULTS
HDX MS of lipid-free SAA at 5 °C reveals local tren ds that parallel the crystal structures
We used recombinant full-length murine SAA isoform 1 (mSAA1, or SAA for brevity); this major isoform binds HDL and forms amyloid in vivo. The residue numbering throughout this paper corresponds to human SAA1 that has an additional N-terminal Gly. The protein was obtained as described in Methods; the amino acid sequence marked “SAA1 mouse” is shown in Figure 1E. The protein was characterized for self-association, secondary structure, and stability using gel electrophoresis, size-exclusion chromatography, mass spectrometry (MS) and circular dichroism (CD) spectroscopy (Figures S1, S2). Lipid-free mSAA1 was 25 ± 5% α-helical at 5 °C and unfolded upon heating to 25 °C with a midpoint Tm=18 °C (Figure S2) [19]. Hydrogen-deuterium exchange (HDX) MS data were recorded as described in Methods using 0.5 mg/ml protein in standard buffer (10 mM sodium phosphate, pH 7.5). The HDX data summary and experimental conditions are reported in Table S1. Linear sequence coverage was 95–100 % with ~5-fold redundancy (Figure S3), facilitating a rigorous analysis by HDX MS.
The HDX MS results at 5 °C revealed large variation s in deuteration across the molecule, as shown in the deuterium uptake plots for representative regions (Figure 3), and in the data summary for all regions (Figure 4A, Supplemental dataset 1). Most regions from the N-terminal half of SAA, which contains h1, h2 and the N-terminal part of h3, showed relatively slow exchange, indicating high protection consistent with the presence of ordered structure. Except for the N-terminal portion of h4 discussed below, the rest of the molecule showed much lower protection from exchange, especially in the C-terminal part of h3 and the C-terminal tail. Therefore, at 5 °C the helical structure in free pr otein must be located mainly in its N-terminal half.
Figure 3.

Deuterium uptake plots for selected regions of lipid-free SAA (blue) and SAA-POPC (red). Error bars for individual time points represent the range for multiple charge states (varies per peptide) from the combined results of multiple replicates (described in Table S1 and in Methods). Residue numbers for representative peptides are indicated. Linear representation of the SAA secondary structure observed by crystallography is shown at the top. Apices 1 and 2 (A1, A2), vertices 1 and 2 1 (V1, V2), and the C-terminal tail (CT) are indicated; Figure 2A shows their locations in the crystal structure. Peptide coverage map for lipid-free SAA and SAA-POPC at 5 °C is shown above the uptake plots. Each bar r epresents a peptic peptide fragment of SAA detected by MS. Selected fragments whose uptake plots are displayed are color-coded.
Figure 4.

Relative deuterium incorporation at 5 °C of all pep tides in lipid-free SAA (A), in SAA-POPC (B), and their difference (C) where subtraction was performed as DSAA-POPC – DSAA. The relative percent deuteration scale for panels A and B is used to color each peptide from black/violet (low uptake, high structural protection) to red (high uptake, low structural protection). The deuteration difference scale for panel C indicates less deuteration (negative numbers and colors) in SAA-POPC. Similar data at 15 °C and 25 ° C are shown in Figures S4, S5.
Despite large difference in the α-helical content of free SAA in solution (~25% at 5 °C) and in the crystals (70–75%) [25, 26, 31], x-ray crystallography and HDX showed very similar trends in the local ordering of the helical and interhelical regions. In the crystals, helices h1 and the h2-h3 segment were substantially more ordered/less dynamic (had lower B-factors) than the C-terminal ~35 residues (Figure 1D). This parallels the relative protection observed by HDX in these regions (Figure 4A).
The four interhelical linkers observed in the crystal structures (Figure 2A), including h1-h2 (apex 1), h2-h3 (vertex 1), h3-h4 (apex 2), and h4-h’ (vertex 2), showed distinct HDX protection in solution. Notably, the h1-h2 linker region of free SAA, which is best represented by the peptide 25–35 (marked A1 in Figure 4A), showed more deuteration (less protection) than the adjacent segments from h1 and h2 at early deuterium-labeling times. The crystal structures also show the h1-h2 linker to be less ordered than its adjacent helices (Figure 1C, D).
The h2-h3 linker (represented by peptides 44–53 and 45–53, marked V1 in Figure 4A) showed comparable or greater protection than the adjacent well-protected segments from h2 and h3. Similarly, the crystal structures showed uniformly low B-factors in h2, h3 and the h2-h3 linker (Figure 1D), including the GPGG motif that formed a tight interhelical turn (Figure 2A) [21]. Hence, despite its potential flexibility, the GPGG motif was well-ordered both in the crystals and in solution.
Unexpectedly, the h3-h4 linker region, which is also potentially flexible due to the presence of conserved G70 and G72, showed greater HDX protection in solution than the adjacent segments from h3 and h4. In our HDX MS analysis, this region (marked A2 in Figures 3 and 4) was best represented by peptides 69–78 and 70–76, which also contains the beginning of h4. The crystal structures showed an opposite trend: the h3-h4 linker, which forms apex 2, together with the beginning of h4 formed the least ordered region in SAA (Figure 1D). Notably, the h3-h4 linker has variable length and composition in SAA proteins (Figure 1E), and the beginning of h4 contains the major cleavage site after residue 76 that generates the N-terminal fragments found in AA amyloid deposits in vivo [3, 34]. Therefore, high structural protection observed in this region by HDX MS was a surprise; MD simulations described below help explain this unexpected observation.
Finally, the h4-h’ linker in residues 89–90 (contained within the peptide 79–94 and marked V2 in Figure 4A) showed slightly greater protection than its adjacent segments. This is consistent with the relatively well-ordered structure in this linker observed in the crystals (Figure 1D), which forms vertex 2 packed against vertex 1 in the middle of the molecule (Figure 2A). However, such packing requires ordered helices h4 and h’, which is apparently not the case in solution given the relatively high deuteration of residues in the C-terminal half of SAA. In free protein in solution the C-terminal half lacks a stable helical structure. Nevertheless, substantial protection in vertex 2 observed by HDX, combined with lower but still meaningful protection in the rest of the C-terminal region of SAA (Figure 4A), indicates that this region is not entirely disordered.
In summary, HDX results at 5 °C indicate that for f ree SAA in solution, most helical structure is located in the N-terminal half, while the C-terminal half is relatively disordered/flexible, except for the beginning of h4. Importantly, despite the large difference in the helical content of free SAA in solution (25–30% α-helix at 5 °C) and in the crystal (70–75%), HDX of free SAA at 5 °C and x-ray crystallography s how similar trends in the local ordering of α-helical and linker regions. Consequently, the x-ray crystal structures depict key aspects of the solution conformation of free SAA. The only discrepancy between the local structural ordering in solution and in the crystals was observed in the h3-h4 linker region at apex 2 (Figure 4A), suggesting that the local solution conformation near this apex deviates from the crystal structure.
Insights from HDX MS analysis of free SAA at 15 °C and 25 °C
To analyze the solution conformation of SAA at temperatures closer to physiological conditions and to monitor protein unfolding, HDX MS was performed at 15 °C where free SAA is in the middle of thermal unfolding, and at 25 °C wh ere thermal unfolding is nearly complete (Figure S2). For any protein, temperature can influence HDX both because the intrinsic rates of exchange go up with temperature [35] and because the protein becomes more dynamic/flexible – and therefore more easily deuterated – as temperature increases. While one cannot use HDX to compare the structural effects of exchange at two temperatures without compensating for changes in the intrinsic HDX rates, one can compare differences in exchange between two states at one temperature (e.g. 5 °C lipid-free vs 5 °C lipid-bound) to difference s in exchange at another temperature (e.g. 15 °C).
The results in Figures 4, S4 and S5 showed that HDX was progressively greater going from 5 °C to 25 °C. When the intrinsic rates of exc hange were considered, by adjusting mathematically the measured exchange at 5 °C to wha t it would be at 15 °C and 25 °C, or by adjusting measured exchange at 15 °C and 25 °C to w hat it would be at 5 °C (see Supplemental Dataset 1), calculated exchange was found to be slower at 15 °C and 25 °C than could be explained by temperature correction alone. That is, SAA was more protected at 15 °C and 25 °C than one would predict mathematically based on t he 5 °C HDX data. This makes sense in light of the CD melting curves (Figure S2) which show that SAA is partially (at 15 °C) or totally (at 25 °C) unfolded, which may influence its oligom erization (see below). HDX was progressively greater, particularly in the h2-h3 region, suggesting its unfolding. In free SAA at 25 °C, most regions showed little protection (Figure S 5 A), consistent with the CD results indicating decreased helical structure (Figure S2). The only regions retaining substantial protection from deuteration at 25 °C were: i) residues 7–35, which encompass parts of h1, h2 and the h1-h2 linker at apex 1, and ii) residues 69–80, which encompass the h3-h4 linker and the beginning of h4 at apex 2. Consequently, high structural protection in these two regions in solution was consistently observed at 5–25 °C, i. e. both below and above thermal unfolding of free SAA. The structural basis for this local protection emerges from our MD simulations below.
A caveat in our experimental analysis of free SAA is its propensity to form oligomers of various sizes in a broad range of solvent conditions and temperatures ([20, 30, 38] and references therein), including those used in our HDX experiments (Figure S1). Since the N-terminal end of h1 is implicated in self-association of free SAA in solution [30, 33], this may contribute to high protection observed in h1 by HDX (Figure 4A). However, since SAA self-association competes with lipid binding at pH~7 [16, 21, 33], it is not expected to significantly influence our HDX MS analysis of SAA-POPC complexes. In fact, binding to lipids stabilizes the structure of SAA, which likely makes it less prone to aggregation. This hypothesis is directly supported by HDX results for SAA-POPC as described next.
HDX MS analysis of SAA-POPC complexes at 5 – 25 °C
SAA-POPC particles differing in size from ~10 nm to ~20 nm were reconstituted using 1:10, 1:30 or 1:80 protein:lipid molar ratios as described previously [29]; all particles showed similar secondary structure, thermal stability, and intrinsic Trp fluorescence that reports on SAA-lipid interactions. HDX MS analysis of SAA-POPC in 1:10, 1:30 or 1:80 particles showed that deuterium uptake plots were identical for the particles of different sizes (Figure S6), suggesting a similar protein conformation.
For detailed HDX studies, we selected 1:10 particles, henceforth termed SAA-POPC, which showed the highest temporal stability and were 8–10 nm in size like HDL [29]. The structure and stability of these SAA-POPC particles were characterized by gel filtration and CD spectroscopy (Figures S1, S2). HDX SAA-POPC was measured at 5 °C (Figure 3, Figure 4B), 15 °C, and 25 °C (Figure S4 and Supplemental dataset 1). As described above for free SAA, HDX at higher temperatures would be expected to be faster due to the intrinsic rates of exchange. In fact, the results showed that, while the overall deuteration rate progressively increased from 5 to 25 °C, the site-specific trends remained invariant (panel B in Figures 4, S4, and S5). This was in line with the CD data showing that the protein conformation in SAA-POPC remained invariant upon heating from 5 to 25 °C (Fi gure S2). At all temperatures, the greatest protection in SAA-POPC was detected in two segments, one containing the central part of h1 (residues 7–24) and the other extending from the C-terminal end of h2 to the end of h3 (panel B in Figures 4, S4 and S5). Consequently, these two segments contained the most helical structure in SAA-POPC.
Comparison of HDX MS data of free SAA and SAA-POPC reveals the lipid binding site
Comparison of HDX data for SAA-POPC and free SAA showed that the structural protection increased non-uniformly across the protein molecule upon lipid binding. At 5 °C, the greatest increase was observed in the central and C-terminal part of h3 that showed low protection in free SAA but very high protection in SAA-POPC (Figure 3E, F; Figure 4). At 15 °C and 25 °C, when progressive helical unfolding was o bserved by CD spectroscopy in free SAA but not in SAA-POPC (Figure S2), this difference between SAA-POPC and free SAA was further amplified (Figures S4, S5; Supplemental file 1). These results suggest that the large lipid-induced increase in protection of h3 observed by HDX at 5–25 °C reflects coupled helical folding and lipid binding. Lipid binding stabilizes the helices greatly: h3 unfolds upon heating free SAA from 5 to 25 °C but remains helical on POP C.
The interhelical linkers in SAA-POPC, like in free SAA, showed distinct trends in local protection from HDX at all temperatures explored (Figure 3; panel B in Figures 4, S4 and S5). Interestingly, the h1-h2 linker (A1), which in free SAA showed more exchange (less protection) than the adjacent segments from h1 and h2, showed even more exchange (lower protection) than the adjacent segments in SAA-POPC. Another unexpected finding was that the h1-h2 linker consistently showed a meaningful lipid-induced deprotection (rather than protection) at all temperatures explored (pink regions in panels C of Figures 4, S4 and S5). These results suggest that a more ordered solution conformation in the h1-h2 linker forms in free protein compared to SAA-POPC. The nature of this ordered conformation emerges below from our MD simulations.
Furthermore, lipid binding increased the protection in the h2-h3 linker (V1) at 5 °C (Figure 3D, Figure 4C), and this increase was greatly amplified at higher temperatures. Other protein regions showed small lipid-induced changes in protection at 5–25 °C (Figures 4, S4, S5).
Taken together, these results revealed that both helical and interhelical regions observed by x-ray crystallography show distinct structural protection in free protein in solution as compared to SAA-POPC. Therefore, key aspects of the monomer fold seen in the SAA crystal structure must be retained in solution and on the lipid. Interestingly, h1-h2 linker showed more protection in free SAA as compared to SAA-POPC at 5 – 25 °C (Figures 4, S4, S5), even though free SAA but not SAA-POPC underwent helical unfolding at the higher temperatures (Figure S2). A likely origin of this paradoxical observation emerges below in our MD simulations. Notably, of all linkers, the h2-h3 turn at vertex 1 showed the largest increase in protection upon lipid binding at 5 – 25 °C (Figure 3E, F; panel C i n Figures 4, S4 and S5). The structural basis for this observation is described below.
HDX suggests lipid-induced conformational selection at vertex 1
The mass spectra for each peptic fragment were analyzed for the presence of EX1 and EX2 kinetics, the two kinetic extremes in proteins that are easily observed by HDX MS [37, 38]. In EX2, which is predominant in well-folded globular proteins, local refolding is faster than the HDX reaction, and progressive deuteration occurs upon multiple unfolding and refolding events. EX2 manifests itself as a unimodal spectrum with an isotope distribution that gradually shifts toward higher mass as a function of deuteration time. In EX1, the deuteration occurs after a cooperative or coordinated unfolding event where local refolding is slower than HDX. EX1 manifests itself as a bimodal spectral distribution (when the alternative conformations are separated on the m/z scale) or as spectral broadening (when the two populations are close in m/z), with the lower-mass peak corresponding to the protected conformation and higher-mass peak to the unprotected one. EX1 generally indicates coexistence of alternative conformations.
At 5 °C, most short peptides in free SAA and in SAA -POPC showed EX2 exchange behavior. However, in free SAA, all peptides encompassing the middle of h1 (e. g. peptide 7–18) and a region from the middle of h2 to the middle of h3 (e. g. peptides 45–53 and 54–64) showed EX1 (Figure 5). In SAA-POPC, the mass spectra showed a dominant lower-mass peak representing a large population of well-protected species, while in free SAA the spectra also showed a higher-mass peak representing the unprotected species (Figure 5). At 15 °C, which is close to Tm~18 °C of free SAA (Figure S2), EX1 in these region s of free protein was even more obvious, with two well-resolved peaks suggesting high populations of at least two distinct conformations, one poorly protected (fast exchange) and another more protected (slow exchange) (Figure S7). Upon temperature increase from 5 to 15 °C in free SAA, there was a large shift from a highly protected to the poorly protected conformation (Figures 5, S7), but in SAA-POPC at 15 °C only the highly protected conform ation was observed (Figure S7). These trends continued at 25 °C: in free SAA, HDX MS showed further population shift towards the poorly protected conformation (Figure S7), while CD data showed a largely unfolded helical structure (Figure S2). At all temperatures explored, the highly protected conformation was selectively stabilized in SAA-POPC (Figures 5, S7).
Figure 5.

Mass spectra of selected peptides that display EX1 kinetics at 5 °C. Other peptides that overlap those shown here (see Figs 3 and 4) also displayed EX1 kinetics. Spectra are shown for free SAA (left of each pair) and for SAA-POPC (right of each pair) as a function of the exchange time for peptides 6–17 (blue boxes), 44–52 (green boxes), 53–64 (yellow boxes). The regions showing EX1 in free SAA, color-coded in blue (residues 6–17), green (44–52) and yellow (53–64). Each of these peptides is indicated in the linear protein model (top) and is mapped on the crystal structure (far right). Stars indicate spectra that clearly show two peaks characteristic of EX1 regime. Spectra for a representative peptide showing only EX2 (residues 79–94, orange boxes) are shown for comparison. Figure S7 shows similar data of selected peptides that display EX1 kinetics at 15 °C and at 25 °C.
Importantly, segments from h1 and h2-h3 that clearly showed EX1 for free SAA in solution pack against each other at vertex 1 in the crystal structure (Figure 5, right), which helps explain the coupled conformations of these segments in solution. We conclude that in free SAA at 5–25 °C, the region around vertex 1 displays at least two distinct conformations. During thermal unfolding of free SAA, the poorly protected conformation becomes predominant at the expense of the highly protected conformation (Figures 5, S7). The latter is selectively stabilized upon lipid binding (Figures 5, S7), leading to a lipid-induced increase in the overall structural protection in h1 and h2-h3 region observed at all temperatures explored (Figures 4, S4, S5). We propose that these distinct conformations reflect alternative states of the GPGG motif, one unfolded as expected of this Gly-rich motif, and the other one well-folded and perhaps resembling vertex 1 packing in the crystal structure.
MD simulations of SAA monomer in solution and on the POPC micelle
To gain additional insights into the SAA conformation in solution and on the lipid surface, MD simulations were performed as described in Methods and illustrated in Figure S8. The starting configuration of full-length mSAA1 monomer, termed SAA68, was 68% α-helical and was modeled based on the x-ray crystal structure of hSAA1 (Figure S9 C). To promote helical unfolding and thereby approximate the solution conformation of lipid-free protein, SAA68 was simulated at 370 K for 3.8 μs. We observed a progressive unraveling of h4, h2, N- and C-terminal ends of h1, and the C-terminal part of h3 (Figure 6). This result was supported by experiment as similar regions in free SAA showed low protection from HDX (panel A in Figures 4, S4 and S5), except for the N-terminal end of h1 the protection of which was probably influenced by the protein self-association.
Figure 6.

Low- and high-temperature simulations of lipid free SAA monomer in solution. The starting model, denoted SAA68, was obtained from the x-ray crystal structure of hSAA1 (PDB ID: 4IP8) as shown in Figure S9. The structure was simulated at 310 K for 1 μs and at 370 K for 3.8 μs as described in Methods.
(A) Time evolution of the secondary structure showing ⍺-helices (purple/blue) and β-sheets (orange) over the simulation trajectory at 310 K (left) and 370 K (right panel). The remaining structure (off-white) was turn/coil. Linear secondary structure model based on the hSAA1 crystal structure is color-coded.
(B) Representative molecular structures at indicated simulation times from the 370 K trajectory, from 0 μs (SAA68) to 3.5 μs. The orientation of h1 is similar in all figures. Color-coding: h1 (blue), h2 (teal), h3 (green), residue segment 70–104 (red), and β-sheet (orange).
During the last microsecond of the high-temperature trajectory, the average α-helical content was ~30%, approaching that estimated from the CD spectra of free SAA at 5 °C. This last microsecond was used to extract representative structures to assess the protein conformational ensemble at ambient temperatures. Three structures, collectively termed SAA30, were extracted based on a clustering analysis; each structure was simulated at 310 K, either lipid-free (denoted SAAR1 – SAAR3) or on a POPC micelle (denoted SAA-POPCR1 – SAA-POPCR3) as described in Methods. Figure S10 shows the time evolution for the secondary structure in each simulation.
Figure 7A illustrates external H-bonding between the main chain nitrogens of SAA and water molecules in SAA68 and SAA30, averaged over R1 – R3 runs, and Figure S11A shows such external H-bonding for individual runs of SAA30. The results consistently show that, unlike the rest of the molecule, the central part of h1 (residues 6–24), as well as the h2-h3 linker and most of h3 (residues 50–64) except for its C-terminal part, are protected from the main chain H-bonding with water. This result agrees with HDX MS showing high protection from HDX in h1 and in the N-terminal half of h3 in free SAA (panel A in Figures 4, S4 and S5).
Figure 7.

Main chain hydrogen bonding and secondary structure in SAA monomer in solution. Three representative structures collectively termed SAA30, which were extracted from the last 1 μs of the 370 K trajectory (shown in Figure 6A), were equilibrated at 310 K in the absence (SAA) or presence of a POPC micelle (SAA-POPC) for 5 or 10 μs, respectively. The resulting models, denoted R1-R3, are reported.
(A) Hydrogen bonding of the protein main chain nitrogens to water molecules. Fraction of frames showing H-bonds with water is plotted versus residue number; darker colors indicate greater H-bonding probability. The plots represent an average of R1-R3 models; the results for individual models and their 3D structures are shown in Figure S11. Similar data for the starting model, SAA68, are shown for comparison (top panel).
(B) Location of the α-helices and β-strands in the primary sequence of SAA and SAA-POPC is shown for R1-R3 models. Individual replicates are color-coded.
Figure 7B depicts the secondary structural propensity in SAAR1 – SAAR3 and SAA-POPCR1 – SAA-POPCR3. Lipid-free SAA shows two major highly α-helical regions: the central part of h1 and the residue segment encompassing the h2-h3 linker and most of h3 except for its C-terminal part. Our MD simulations suggest that experimentally observed high protection from exchange in these regions of free SAA results from their stable helical structure. Moreover, MD simulations predict substantial helicity in the h3-h4 linker and at the beginning of h4 spanning residues 71–77 (Figure 7B). This helps explain why this region shows relatively high protection from exchange (e.g. peptide 69–78 in Figures 3G, 4A, S4A, and S5A), even though in all available x-ray crystal structures this variable region is substantially disordered/dynamic (Figure 1D).
Unlike free protein whose average secondary structural content changed little in simulations of SAA30 at 310 K, the helical content in similar simulations of SAA-POPC increased, suggesting lipid-induced helical stabilization (Figure 7). A clear increase was seen in the h2-h3 linker and in the C-terminal part of h3, which regained complete helicity in SAA-POPCR3. This trend agreed with the HDX data showing a large lipid-induced increase in the structural protection of the C-terminal part of h3 upon POPC binding at all temperatures explored (Figures 4C, S4C, S5C). As the helical content in our simulations of SAA-POPCR1 – SAA-POPCR3 has not yet reached the ~50% level observed in SAA-POPC complexes by CD spectroscopy, a direct quantitative comparison of computational and experimental results was not possible. However, the trends predicted in our MD simulations were validated by the CD and HDX MS data. Taken together, these data demonstrate that interactions with the lipid surface stabilized the α-helical structure throughout the SAA molecule and induced helical folding in the C-terminal part of h3.
To identify protein regions that interact with the POPC surface, SAA68 was randomly oriented 5 nm away from the micelle surface and allowed to diffuse and interact with a preformed POPC micelle at 310 K for 1 μs. In the initial orientation, helices h1 and h3 were not facing the micelle. During the first ~50 ns of the simulation, the protein explored various orientations with respect to the micelle before converging to a stable orientation after ~90 ns (Figure 8A). In this orientation, the protein contacts with the phospholipid head groups and acyl chains were formed largely by helices h1 and h3, with minimal contributions from h2 and no contributions from residues 70–104 (Figure 8B). Figure 8C illustrates the protein orientation on the micelle; the protein-lipid contacts involve mainly the hydrophobic faces of h1 and h3. This result, combined with our comparative HDX studies of SAA and SAA-POPC (Figures 4, S4, S5, 5, and S7), supports the conjecture that the hydrophobic faces of h1 and h3 form the lipid binding site in SAA.
Figure 8.

Protein-lipid contacts in SAA-POPC68 model. SAA68 was oriented randomly in respect to the POPC micelle containing 72 lipid molecules, the system was simulated at 310 K for 1 μs (for detail see flowchart in Figure S8), and the last 0.5 μs of the trajectory were used for analysis.
(A) Average protein-lipid distances are plotted as a function of simulation time for individual SAA helices (color coded). Solid lines show distances involving phospholipid head groups, and dotted lines those involving acyl chains.
(B) Final configuration of the SAA-POPC68 system. Protein ribbon diagram shows h1 (blue), h2 (teal), h3 (green), h4 (yellow) and residue segment 70–104 (red). POPC head groups are in pink and acyl chains in grey.
(C) Fraction of frames showing SAA contacts with the POPC head groups (pink) and acyl chains (grey) per residue. A contact is defined as lipid atoms within 5 Å of protein atoms. Average results from R1-R3 simulations are shown.
The h1-h2 linker region can form a β-hairpin
Although the SAA crystal structures contain no β-sheet, our MD simulations predicted an unexpected β-hairpin with β-strands in residues 28–30 and 33–35 encompassing the h1-h2 linker and the beginning of h2 (Figures 7B, S10–S12). The β-hairpin first formed in the 370 K simulations (Figure 6) and was partially retained at 310 K in SAAR1, SAAR2 and SAA-POPCR2 (Figure S10). Structural analysis of SAAR1 and SAAR2 suggests a network of fluctuating interactions including aromatic ring stacking W29-W35 and W29-F36, a cation-π interaction between K30-Y35, formation of ion pairs D33-K34 and K30-D33, and charge-dipole interactions, e. g. K30-G32 carbonyl oxygen (Figure S12). The combined effect of these and other weak interactions confers marginal stability to the observed β-hairpin. The nascent formation of the β-hairpin helps explain why peptide 25–35 in free SAA is better protected from HDX than its adjacent segments (Figure 4A), even though in the crystal structure this region has higher B-factors than the adjacent segments (Figure 1D). Moreover, peptide 25–35 is the only region showing greater protection from exchange in free SAA as compared to SAA-POPC (orange in Figures 4C, S4C, S5C). This experimental observation suggests that the β-hairpin is more likely to form in free SAA than in SAA-POPC.
Additional support for β-sheet formation in this region comes from the fibril structure of mSAA1 determined by cryo-electron microscopy [34]. Strands β4 and β5 in this structure overlap the β-hairpin strands predicted in our simulations (Figure 1A, B). Moreover, a sharp bend between β4 and β5 at residues G32, D33 observed in the fiber structure overlaps the tight turn in the β–hairpin seen in SAAR1 and SAAR2 (Figures S10, S12).
In conjunction, our MD simulations and HDX MS data suggest that residues 27–35 can form a β-hairpin in mSAA1 monomer, while cryo-electron microscopy shows that similar β-strands are retained in amyloid fibrils of this protein. Therefore, this β-hairpin may represent a previously unknown misfolding intermediate on pathway to amyloid formation.
DISCUSSION
Crystal structures depict key aspects of the solution conformations of SAA and SAA-POPC
This study reports solution conformations of SAA ligand-free and in model HDL-size lipoproteins. It establishes the structural basis for understanding how this intrinsically disordered protein binds lipids, and proposes an early step in the amyloidogenic α-helix to β-sheet transition in mSAA1 monomer. Our studies of mSAA1 free in solution or in SAA-POPC complexes agree in remarkable detail with the crystal structures of lipid-free hSAA1 and mSAA3, but also reveal important differences. Structural protection determined by HDX MS in free SAA and in SAA-POPC (Figure 4) shows local trends similar to those in the crystallographic B-factors (Figure 1D). The sole exception is the variable h3-h4 linker discussed below. This similarity was unexpected given large differences in the α-helical content of free SAA (~25% at 5 °C, <10% at 25 °C), SAA-POPC (~50% α-helix at 5–25 °C) (Figure S2), and the crystals (7 0–75% α-helix). We conclude that the crystal structures depict essential aspects of the local conformation of SAA free in solution and bound to lipoproteins. Therefore, the crystal structure serves as a useful starting model for MD simulations, which provide additional insights into the solution conformation of free SAA, its interactions with the lipid surface, and its misfolding and formation of β-structure.
Helical structure in the N- and C-terminal regions
Previous amino-acid sequence analyses predicted that residues 1–70 contain the most stable helical structure in SAA [21, 39]. Our experiments and simulations verify this prediction and show that in free SAA in solution, most helices are located in two segments, one including a large part of h1 and the other spanning from the end of h2 to the middle of h3. These segments show the greatest protection from HDX in free protein at 5 °C (Figure 4A) and form particularly stable helices in MD simulations (Figure 6, Figure 7). In the crystal structure, these segments are packed together at vertex 1 (Figure 2, Figure 5), perhaps nucleating the native fold.
Unlike the N-terminal region, most of the C-terminal third of SAA lacks a stable secondary structure free in solution and on POPC; the only exception is the h3-h4 linker and the beginning of h4 at apex 2 (Figures 4, S4, S5, Figure 7B). Unexpectedly, both free SAA and SAA-POPC show relatively high HDX protection in peptides 69–78 and 70–76 encompassing apex 2 region (A2 in Figure 3G, Figures 4A, S1A, S2A). In contrast, the crystal structures show high B-factors in this variable region (Figure 1D). MD simulations reveal a likely basis for this discrepancy and predict high helicity in the linker residues 71–77 (RGHEDTM in mSAA1) in free protein and, particularly, in SAA-POPC, while the adjacent segments are unfolded (Figure 7B). This helix is not expected to block the cleavage at the 76–77 junction to release the major AA fragment found in amyloid fibrils in vivo. Direct observation of this short helix and its functional role remain to be determined; this and other conclusions from our MD simulations remain to be verified in future studies. We speculate that such an α-helix may provide a recognition motif for cell receptors and other ligands that are implicated to bind SAA via its C-terminal ~30 residues [21]. It is possible that two strictly conserved acidic residues from this short helix, E74 and D75, attract the electropositive ligand-binding site in cell receptors such as CD36, LOX-1 or SR-BI [40, 41], facilitating SAA-receptor interactions.
Amphipathic α-helices h1 and h3 form an evolutionally conserved lipid binding site
Our results revealed that POPC binding leads to increased protection from HDX across the SAA molecule, most strikingly in the C-terminal half of h3 that shows poor protection in the free protein but high protection in SAA-POPC (Figure 3E, F; Figures 4, S4, S5). This parallels increased helical content in SAA-POPC observed by CD spectroscopy (Figure S2). MD simulations support the observation that, while the helical structure across the SAA molecule increases upon POPC binding, the most substantial increase occurs in the C-terminal part of h3, which is unfolded in free SAA in solution but acquires α-helical structure in SAA-POPC (Figure 7). Consequently, lipid binding induces helical folding in the C-terminal part of h3.
SAA-POPC shows the greatest protection from HDX in h1, h2-h3 linker and h3 (Figures 3E, F; 4B; S4B; S5B). In free SAA, parts of h1 and h2-h3 segments show on average relatively high protection from HDX at 5 °C (Figure 4A) but di splay alternative conformations differing in protection, as indicated by the EX1 exchange kinetics (Figures 5, S7). By contrast, in SAA-POPC this region shifts towards a highly protected conformation, suggesting its involvement in lipid binding (Figures 5, S7). Moreover, MD simulations of SAA68 in the presence of a POPC micelle suggest that the amphipathic helices h1 and h3 directly interact with the lipid via their hydrophobic faces (Figure 8). Additional evidence for lipid binding via these helices comes from the intrinsic fluorescence of the three Trp in mSAA1, which are located in the hydrophobic faces of h1 (W18) and h3 (W52) or in the h1-h2 linker (W29). A blue shift in the wavelength of maximal emission indicates that these tryptophans shift from a largely aqueous to a largely apolar environment upon POPC binding [29]. Taken together, these findings clearly show that the lipid-binding site in SAA contains both h1 and h3, and not just the N-terminal end of h1 as previously thought. Moreover, the hydrophobic cavity in SAA oligomer formed by h1 and h3 was recently shown to bind retinol [26]. These findings substantiate the idea that this site binds diverse apolar ligands of SAA [21].
Importantly, in the crystal structures, the N-terminal and central parts of h1 as well as the central and C-terminal parts of h3 in SAA monomer form a concave hydrophobic face that optimally fits the HDL surface curvature (Figure 2B). This suggests a structural basis for the preferential binding of SAA to HDL, which is its major plasma carrier [21]. The current study supports this conjecture and also establishes the structural basis for de novo formation of HDL-size particles by SAA. This is particularly important as sequestering unprotected lipids and their degradation products into nanoparticles is proposed to be a major evolutionally conserved function of SAA [27].
Interestingly, the protection from HDX observed in SAA-POPC parallels the evolutional conservation in the SAA protein family. Particularly highly conserved is the h2-h3 residue segment (Figure 1F, G), the packing of which against h1 is necessary to form the concave hydrophobic lipid-binding face (Figure 2). This conservation parallels high protection from exchange of the h2-h3 segment observed in SAA-POPC at 5–25 °C (Figures 4B, S4B, S5B). This parallel compels us to postulate that the amino acid sequence encoding for the unique molecular fold of SAA has been evolutionally conserved to encapsulate lipids into self-assembled nanoparticles.
Structural and functional roles of the N- and C-terminal vertices in SAA
The curvature of the lipid-binding face in SAA monomer is defined by the angle between h1 and h3. In the maximally helical state, this angle is critically hinged upon the GPGG motif in the h2-h3 linker (Figure 2, Figure 5). This motif, together with several conserved alanines and glycines located nearby, facilitates a tight packing of h1 against h3 at an angle of ~43°, with unusually short main chain spacing of 3.6 Å between A10 and G50 seen in the crystal structures [21]. The EX1 data at 5 °C reveal that in free prot ein in solution, the region around vertex 1 alternates between highly protected and minimally protected conformations (Figure 5). The population of the latter progressively increases at 15 and at 25 °C upon helical unfolding in free SAA (Figure S7). This suggests that in SAA in solution, the GPGG linker and its adjacent helices are either unordered as expected of this Gly-rich motif, or are very well-ordered as seen in the crystal structures (Figure 1D, Figure 2).
In SAA-POPC, vertex 1 region shifts towards a highly protected conformation (Figures 5, S7). This suggests that lipid binding selectively stabilizes the pre-existing well-ordered structure, reducing the entropic penalty for lipid binding by this intrinsically disordered protein. Taken together, our findings are consistent with a mixed model for lipid binding by SAA. The central and C-terminal part of h3 undergoes coupled folding and binding, in line with a mechanism of “induced fit”. Lipid-induced structural changes in the central part of h1 and the h2-h3 region at vertex 1 are consistent with a “conformational selection” model wherein the well-ordered conformation has low population in the ligand-free state but is selectively stabilized in the ligand-bound state.
Helices h4 and h’ mimic the orientation of helices h2 and h3 in the crystal structure (Figure 2). Like the GPGG motif that forms the h2-h3 turn in vertex 1, the Gly-containing h4-h’ turn in vertex 2 positions h4 against h’ at an angle of ~45° (Figure 2A, Figure 5). Like the h2-h3 linker at V1, the h4-h’ linker at V2 is relatively well ordered in the crystal structures (Figure 1D) and shows relatively high structural protection in free SAA in solution and in SAA-POPC (Figure 4). Moreover, like V1, V2 shows unusually close interhelical main chain spacing in the crystal structure, including a 3.3 Å distance between Cα of Y42 from h2 and carbonyl O of G86 from h4. Notably, Y42 and G86 are 100% evolutionally conserved, while other residues from the h4-h’ linker at V2 are also highly conserved (Figure 1). Similarly, the GPGG linker and other residues facilitating unusually tight helical packing in V1 are 100% conserved [21]. Together, these findings suggest that V1 and V2 play similar structural roles in the N- and C-terminal regions of SAA, conferring its unique molecular fold. We speculate that, similar to the V1 region whose ordered structure is stabilized upon lipid binding via h1 and h3, the ordered structure at V2 may be stabilized upon ligand binding via the C-terminal part of SAA. Future studies of other SAA ligands will test these hypotheses.
A β-hairpin in SAA monomer may initiate amyloid formation
Our MD simulations have predicted an unexpected β-hairpin in mSAA1 monomer in solution (Figures 7B, S10–S12). The HDX MS results at 5–25 °C suggest strongly that the population of this β-hairpin decreases when SAA is in contact with lipid (orange in Figures 4C, S4C, S5C). In other amyloidogenic proteins such as Aβ peptide or α-synuclein, early misfolding intermediates are thought to involve activated protein monomers [42, 43] that can contain transient β-sheet structure such as solvent-exposed β-hairpins [44, 45]. These aggregation-prone conformations can be stabilized upon protein oligomerization and are ultimately converted into intermolecular cross-β-sheet in amyloid. Therefore, sequestering β-hairpin intermediates has been proposed as a strategy to block amyloid formation by various proteins [44, 45].
Our MD simulations suggest the transient formation of a β-hairpin in residues 28–36, with two short β-strands separated by a tight turn at G32, D33 (Figures 7B, S10–S12). In the crystal structure, which lacks β-sheets, residues 28–36 encompass an interhelical linker h1-h2 and the beginning of h2 at apex 1. Several lines of experimental evidence provide support for β-hairpin formation. Firstly, the lipid-induced decrease (rather than increase) in protection from HDX, which was consistently observed only in this region of SAA (pink in Figures 4C, S4C, S5C), suggests that, unlike the helical structure that is stabilized upon lipid binding, the ordered structure at apex 1 is destabilized. Secondly, h1-h2 region is part of the fibril-forming N-terminal segment found in AA deposits in vivo. Thirdly, the two β-strands connected by a tight turn identified in our MD simulations overlap β-strands β4 and β5 (residues 27–29 and 33–38) observed in the amyloid fibril structure formed by a fragment of mSAA1.1 [34] (Figure 1B).
Together, these findings suggest that local helical unfolding around the h1-h2 linker, which is more likely to occur in free SAA than in SAA-POPC, may lead to a transient β-hairpin formation in this region. This is consistent with previous finding that at pH~7, SAA binding to HDL [16] or formation of SAA-POPC complexes protects the protein from forming amyloid [18, 23]. Although the aromatic cluster involving W29, Y35 and F36 probably contributes to the transient stability of the β-hairpin in mSAA1 (Figure S12), the hairpin residues, GWKDGDKYF, have a high charge content. Therefore, this residue segment is not assigned high amyloidogenic propensity by sequence-based bioinformatics tools, such as AmylPred2 or PASTA2, which are used to predict amyloidogenic segments in soluble proteins (Figure 1, top) [15]. However, if formation of this hairpin in monomeric SAA is confirmed, it may represent an aggregation-prone N* state and amyloidogenic intermediate [42, 43] and initiate amyloid formation [44, 45]. If so, our study reveals a limitation in the existing sequence-based amyloid prediction tools. Since h1-h2 linker regions in hSAA1 and mSAA1 differ in their primary sequence (Figure 1) and fibril structure [34], the role of this region in β-structure formation in human vis-à-vis murine protein remains to be determined.
MATERIALS and METHODS
Protein, lipid and lipoproteins
Recombinant full-length murine SAA isoform 1.1 (11.6 kDa, 103 amino acids) was used in all experiments. This isoform is termed mSAA2 in the nomenclature based on the gene sequence [46]. SAA was expressed in E. coli and purified under denaturing conditions to 95% purity as described previously [47]. Protein purity and integrity were assessed by SDS PAGE and by mass spectrometry. Lipid 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC; C16:0, C18:1) was 97%+ pure from Avanti Polar Lipids. All other chemicals were of highest purity analytical grade.
Lyophilized mSAA1, which was stored at 4 °C, was di ssolved in double-distilled H2O; the stock solution was diluted into 10 mM sodium phosphate at pH 7.5 (standard buffer) prior to use. Protein concentrations were determined by UV absorbance at 280 nm using Varian Cary-300 UV-vis spectrophotometer.
Lipoprotein reconstitution and characterization
SAA-containing lipoproteins of controlled size were reconstituted by a cholate dialysis method as previously described [29]. Briefly, POPC film was dispersed in standard buffer and solubilized by adding 1 M sodium deoxycholate. The cholate-solubilized POPC was incubated with SAA at 37 °C for 1 h; SAA to POPC molar ratios in the incubation mixture ranged from 1:10 (to obtain lipoproteins ~10 nm in size) to 1:80 (to obtain lipoproteins ~20 nm in size). The lipoprotein solution was dialyzed extensively against the standard buffer. Lipoprotein size and homogeneity were characterized by non-denaturing PAGE and size-exclusion chromatography, and protein and lipoprotein structure and stability were characterized by CD spectrometry as previously described [29]. The results for SAA and SAA-POPC used in the current study are shown in Figures S1 and S2.
Hydrogen-deuterium exchange mass spectrometry
HDX MS data for free SAA and SAA-POPC were obtained under conditions similar to those previously described [48]. To initiate deuterium labeling of free SAA, 1 μl of 0.5 mg/ml protein in standard buffer (10 mM sodium phosphate, pH 7.5) was diluted 18-fold with labeling buffer (10 mM sodium phosphate, pD 7.5, 99.9% D2O) at 5, 15 or 25 °C. The labeling was quenched by a 2-fold dilution with ice-cold quench buffer (150 mM sodium phosphate, pH 2.3, H2O), at time points ranging from 5 seconds to 4 hours. A similar labeling protocol was followed for SAA-POPC. Other HDX experimental parameters, as agreed upon in [49], are provided in Supplemental Table S1.
Deuterated and control samples were digested with pepsin (10 mg/mL, Sigma P6887; Lot#SLBL1721V) for 5 minutes on ice, and then injected into an M-class Acquity UPLC with HDX technology (Waters). The cooling chamber of the UPLC system, which housed all the chromatographic elements, was held at 0.0 ± 0.1 °C for the entire time of the measurements. Peptides were trapped and desalted on a VanGuard Pre-Column trap [2.1 mm × 5 mm, ACQUITY UPLC BEH C18, 1.7 μm (Waters, 186002346)] for 3 minutes at 100 μL/min, eluted from the trap using a 5%–35% gradient of acetonitrile over 6 minutes at a flow rate of 100 μL/min, and separated using an ACQUITY UPLC HSS T3, 1.8 μm, 1.0 mm × 50 mm column. The back pressure averaged ~12,950 psi at 0 °C and 5% acetonitrile : 95% water. The error of determining the deuterium levels was ± 0.30 Da in this experimental setup. The trap column and analytical column were washed with a blank run before each sample. Mass spectra were acquired using a Waters Synapt G2-Si HDMSE mass spectrometer. The mass spectrometer was calibrated with direct infusion of a solution of glu-fibrinopeptide (Sigma, F3261) at 200 femtomole/μL at a flow rate of 5 μL/min prior to data collection. A conventional electrospray source was used, and the instrument was scanned 0.4 scans/second over the range 50 to 2000 m/z with ion mobility engaged. The instrument configuration was the following: capillary was 3.2 kV, trap collision energy at 4 V, sampling cone at 40 V, source temperature of 80 °C and desolvation temperature of 175 °C. All comparison experiments were done under identical experimental conditions such that deuterium levels were not corrected for back-exchange and are therefore reported as relative. Analyses of SAA alone versus SAA-POPC 1:10 (particles prepared using 1:10 SAA:POPC molar ratio) at 5 °C were performed 4 times, 2 technical replicates each of two different protein batches. Analyses comparing SAA with SAA-POPC 1:10 at 15 °C and 25 °C were performed once at 15 °C and once at 25 °C. Analyses of SAA-POPC lipoproteins obtained using 1:30 or 1:80 SAA:POPC molar ratios were each recorded once at 5 °C for comparison with those obtained using 1:10 mo lar ratio (Figure S6).
Peptides were identified using PLGS 3.0.1 (Waters, RRID: SCR_016664, 720001408EN) using multiple replicates of undeuterated control samples. Raw MS data were imported into DynamX 3.0 (Waters, 720005145EN) and filtered as follows: minimum consecutive products of 2; minimum number of products per amino acid of 0.2, 10 ppm error. Those peptides meeting the filtering criteria were further processed automatically by DynamX followed by manual inspection of all processing. The relative amount of deuterium in each peptide was determined by subtracting the centroid mass of the undeuterated form of each peptide from the deuteratered form, at each time point, for each condition. The error bars in all the deuterium uptake plots represent variation supplied by DynamX software, which is determined for each peptide using different charge states for all technical and biological replicates. Deuterium uptake values were used to generate uptake graphs and difference maps. All HDX MS data have been deposited to the ProteomeXchange Consortium via the PRIDE [50] partner repository with the dataset identifier PXD016391.
Calculations to adjust for changes to the intrinsic rates of exchange at different temperatures were performed as described in [35]. The measured relative percent deuteration at each timepoint for each peptide at 5 °C was adju sted mathematically to what it would be at 15 °C or 25 °C by multiplying by the correction factor s of 2.4087 or 5.4696 (respectively). These calculations and the results are found in Supplemental Dataset 1. Alternatively, the measured exchange at 15 °C and 25 °C was adjusted mathematic ally to what it would be at 5 °C by dividing by the correction factors of 2.4087 or 5.4696 (respectively), also shown in Supplemental Dataset 1. The correction factors for the temperature differences in this study were determined using the Arrhenius equation found in [35].
Molecular dynamics simulations
SAA monomer alone or with a POPC micelle was simulated as outlined in Figure S8. All simulations were performed using the GROMACS 2018.3 package [51], employing the CHARMM36m force field for proteins [52] and CHARMM36 force field for lipids [53]. Structures were rendered using VMD [54]. The analyses were conducted with in-house Python scripts utilizing NumPy, SciPy, and MDAnalysis libraries [55]. Protein secondary structure was assigned using the STRIDE algorithm in VMD [56]. All-atom protein simulations were conducted in an aqueous environment, defined by the TIP3P water model [57], with 150 mM NaCl. Temperature was maintained with the Nosé-Hoover thermostat [58, 59] and pressure was held at 1 atm and Parrinello-Rahman barostat [60]. The initial system configurations were constructed and equilibrated according to the CHARMM-GUI protocols [61]. The 2.2 Å resolution x-ray crystal structure of full-length human SAA1 (PDB ID: 4IP8) was equilibrated at 310 K for 2 μs, and point substitutions at positions differing between hSAA1 and mSAA1 (shown in Figure S9) were introduced to obtain a model of mSAA1.
To analyze free SAA in solution, this model was simulated at 370 K for 3.8 μs to encourage helical unfolding, from 68% α-helix in the starting configuration (SAA68) to under 30% detected by CD spectroscopy in solution at 5 °C. Th e protein structures from the final microsecond of the trajectory, where the average helicity was 30%, were clustered employing GROMOS clustering algorithms [62] to extract three solution structures corresponding to the most populated clusters, collectively termed SAA30. Each of these three representative structures was simulated for 500 ns, and the results (termed SAAR1 - SAAR3) were used for analysis.
In SAA-POPC simulations, the micelle containing 72 POPC molecules was constructed using Packmol [63] and equilibrated in an aqueous environment for 200 ns, whereupon the protein was placed alongside. In one series of studies, each of the three representative SAA30 structures was simulated with the micelle at 310 K for 1 μs. The resulting structures, termed SAA-POPCR1 to SAA-POPCR3, were used to access the lipid effects on the protein secondary structure (Figure S10; Figure 7B). In other studies, SAA68 was simulated with the POPC micelle at 310 K for 1 μs, and the resulting model, termed SAA-POPC68, was used to assess protein-lipid contacts (Figure 8). The last 500 ns of each simulation were used for analysis.
Supplementary Material
Highlights.
SAA lipid binding mechanism was identified by HDX MS and MD simulations
Helices h1 and h3 form a novel hydrophobic lipid-binding surface
Lipid encapsulation emerges as an evolutionally conserved function of SAA
A β-hairpin encompassing h1-h2 linker of mSAA1 may contribute to amyloid formation
ACKNOWLEDGEMENTS
The authors are indebted to Dr. Shobini Jayaraman for generating Figure S1, for invaluable help with the lipoprotein characterization, and for many useful discussions.
FUNDING This study was supported by the National Institutes of Health grants GM067260, GM135158 (OG), and T32 HL007969 (NF), a research collaboration with the Waters Corporation (JRE), GM107703 (JS), the National Science Foundation CHE-1900416 (JS), the Stewart Amyloidosis Endowment Fund and Weston Visiting Professorship program at the Weizmann Institute of Science, Israel (OG), and the Deutsche Forschungsgemeinschaft grant FA456/15-1 (MF).
Conflict of interest
The authors declare no conflicts of interest. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or any other funding agencies.
ABBREVIATIONS
- SAA
serum amyloid A
- AA
amyloid A
- mSAA1
murine SAA isoform 1
- hSAA1
human SAA isoform 1
- HDL
high-density lipoprotein
- POPC
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- CD
circular dichroism
- HDX
hydrogen-deuterium exchange
- MS
mass spectrometry
- MD
molecular dynamics
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Urieli-Shoval S, Linke RP, Matzner Y, Expression and function of serum amyloid A, a major acute-phase protein, in normal and disease states, Curr. Opin. Hematol 7 (2000) 64–69. [DOI] [PubMed] [Google Scholar]
- 2.Eklund KK, Niemi K, Kovanen PT, Immune functions of serum amyloid A, Crit. Rev. Immunol 32(4) (2012) 335–348. [DOI] [PubMed] [Google Scholar]
- 3.Westermark GT, Fändrich M, Westermark P, AA amyloidosis: pathogenesis and targeted therapy, Annu. Rev. Pathol 10 (2015) 321–344. [DOI] [PubMed] [Google Scholar]
- 4.Papa R, Lachmann HJ, Secondary AA, Amyloidosis, Rheum. Dis. Clin. North. Am 44(4) (2018) 585–603. [DOI] [PubMed] [Google Scholar]
- 5.Shridas P, Tannock LR, Role of serum amyloid A in atherosclerosis, Curr. Opin. Lipidol 30(4) (2019) 320–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Uhlar CM, Burgess CJ, Sharp PM, Whitehead AS, Evolution of the serum amyloid A (SAA) protein superfamily, Genomics 19(2) (1994) 228–235. [DOI] [PubMed] [Google Scholar]
- 7.Sun L, Ye RD, Serum amyloid A1: Structure, function and gene polymorphism, Gene 583(1) (2016) 48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Buck M, Gouwy M, Wang JM, Van Snick J, Opdenakker G, Struyf S, Van Damme J, Structure and expression of different serum amyloid A (SAA) variants and their concentration-dependent functions during host insults, Curr. Med. Chem 23 (2016) 1725–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ye RD, Sun L, Emerging functions of serum amyloid A in inflammation, J. Leukoc. Biol 98(6) (2015) 923–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Uhlar CM, Whitehead AS, Serum amyloid A, the major vertebrate acute-phase reactant, Eur. J. Biochem 265 (1999) 501–523. [DOI] [PubMed] [Google Scholar]
- 11.Jr GH. Sack, Serum amyloid A - a review, Mol. Med 24(1) (2018) 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Benditt EP, Eriksen N, Amyloid protein SAA is associated with high density lipoprotein from human serum, Proc. Natl. Acad. Sci. USA 74 (1977) 4025–4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kisilevsky R, Manley PN, Acute-phase serum amyloid A: perspectives on its physiological and pathological roles, Amyloid 19 (2012) 5–14. [DOI] [PubMed] [Google Scholar]
- 14.Wilson PG, Thompson JC, Shridas P, McNamara PJ, de Beer MC, de Beer FC, Webb NR, Tannock LR, Serum amyloid A is an exchangeable apolipoprotein, Arterioscler. Thromb. Vasc. Biol 38(8) (2018) 1890–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Das M, Gursky O, Amyloid-forming properties of human apolipoproteins: Sequence analyses and structural insights, Adv. Exp. Med. Biol 855 (2015) 175–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Claus S, Meinhardt K, Aumüller T, Puscalau-Girtu I, Linder J, Haupt C, Walther P, Syrovets T, Simmet T, Fändrich M, Cellular mechanism of fibril formation from serum amyloid A1 protein, EMBO Rep. 18(8) (2017) 1352–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jayaraman S, Gantz DL, Haupt C, Fändrich M, Gursky O, Serum amyloid A sequesters diverse phospholipids and their hydrolytic products, hampering fibril formation and proteolysis in a lipid-dependent manner, Chem. Commun 54(28) (2018) 3532–3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liang JS, Schreiber BM, Salmona M, Phillip G, Gonnerman WA, de Beer FC, Sipe JD, Amino terminal region of acute phase, but not constitutive, serum amyloid A (apoSAA) specifically binds and transports cholesterol into aortic smooth muscle and HepG2 cells, J. Lipid Res 37(10) (1996) 2109–2116. [PubMed] [Google Scholar]
- 19.Takase H, Furuchi H, Tanaka M, Yamada T, Matoba K, Iwasaki K, Kawakami T, Mukai T, Characterization of reconstituted high-density lipoprotein particles formed by lipid interactions with human serum amyloid A, Biochim. Biophys. Acta 1842(10) (2014) 1467–1474. [DOI] [PubMed] [Google Scholar]
- 20.Jayaraman S, Haupt C, Gursky O, Thermal transitions in serum amyloid A in solution and on the lipid: implications for structure and stability of acute-phase HDL, J. Lipid Res 56(8) (2015) 1531–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frame NM, Gursky O, Structure of serum amyloid A suggests a mechanism for selective lipoprotein binding and functions: SAA as a hub in macromolecular interaction networks, FEBS Lett. 590(6) (2016) 866–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tanaka M, Nishimura A, Takeshita H, Takase H, Yamada T, Mukai T, Effect of lipid environment on amyloid fibril formation of human serum amyloid A, Chem. Phys. Lipids 202 (2017) 6–12. [DOI] [PubMed] [Google Scholar]
- 23.Jayaraman S, Gantz DL, Haupt C, Gursky O, Serum amyloid A forms stable oligomers that disrupt vesicles at lysosomal pH and contribute to the pathogenesis of reactive amyloidosis, Proc. Natl. Acad. Sci. USA 114(32) (2017) E6507–E6515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jayaraman S, Haupt C, Gursky O, Paradoxical effects of SAA on lipoprotein oxidation suggest a new antioxidant function for SAA, J. Lipid Res 57(12) (2016) 2138–2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Derebe MG, Zlatkov CM, Gattu S, Ruhn KA, Vaishnava S, Diehl GE, MacMillan JB, Williams NS, Hooper LV, Serum amyloid A is a retinol binding protein that transports retinol during bacterial infection, eLife 3 (2014) e03206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu Z, Bang YJ, Ruhn KA, Hooper LV, Molecular basis for retinol binding by serum amyloid A during infection, Proc. Natl. Acad. Sci. USA 116(38) (2019) 19077–19082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jayaraman S, Fändrich M, Gursky O, Synergy between serum amyloid A and secretory phospholipase A2 suggests a vital role for an ancient protein in lipid clearance, eLife 46630.1863(1) (2019) 200–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stonik JA, Remaley AT, Demosky SJ, Neufeld EB, Bocharov A, Brewer HB, Serum amyloid A promotes ABCA1-dependent and ABCA1-independent lipid efflux from cells, Biochem. Biophys. Res. Commun 321(4) (2004) 936–941. [DOI] [PubMed] [Google Scholar]
- 29.Frame NM, Jayaraman S, Gantz DL, Gursky O, Serum amyloid A self-assembles with phospholipids to form stable protein-rich nanoparticles with a distinct structure: A hypothetical function of SAA as a “molecular mop” in immune response, J. Struct. Biol 200(3) (2017) 293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Colón W, Aguilera JJ, Srinivasan S, Intrinsic stability, oligomerization, and amyloidogenicity of HDL-free serum amyloid A, Adv. Exp. Med. Biol 855 (2015) 117–134. [DOI] [PubMed] [Google Scholar]
- 31.Lu J, Yu Y, Zhu I, Cheng Y, Sun PD, Structural mechanism of serum amyloid A-mediated inflammatory amyloidosis, Proc. Natl. Acad. Sci. USA 111(14) (2014) 5189–5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Patel H, Bramall J, Waters H, De Beer MC, Woo P, Expression of recombinant human serum amyloid A in mammalian cells and demonstration of the region necessary for high-density lipoprotein binding and amyloid fibril formation by site-directed mutagenesis, Biochem. J 318 (3) (1996) 1041–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang L, Colón W, The interaction between apolipoprotein serum amyloid A and high-density lipoprotein, Biochem. Biophys. Res. Commun 317(1) (2004) 157–361. [DOI] [PubMed] [Google Scholar]
- 34.Liberta F, Loerch S, Rennegarbe M, Schierhorn A, Westermark P, Westermark GT, Hazenberg BPC, Grigorieff N, Fändrich M, Schmidt M, Cryo-EM fibril structures from systemic AA amyloidosis reveal the species complementarity of pathological amyloids, Nat. Commun 10(1) (2019) 1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Engen JR, Wales TE, Shi X, Hydrogen exchange mass spectrometry for conformational analysis of proteins, (2011) In “Encyclopedia of Analytical Chemistry”, John Wiley: Meyers Chichester. R. A., Editor. DOI 10.1002/9780470027318.a9201. [DOI] [Google Scholar]
- 36.Patke S, Srinivasan S, Maheshwari R, Srivastava SK, J Aguilera J., Colón W, Kane RS, Characterization of the oligomerization and aggregation of human Serum Amyloid A, PLoS One 8(6) (2013) e64974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ferraro DM, Lazo N, Robertson AD, EX1 hydrogen exchange and protein folding, Biochemistry 43(3) (2004) 587–594. [DOI] [PubMed] [Google Scholar]
- 38.Weis DD, Wales TE, Engen JR, Hotchko M, Ten Eyck LF, Identification and characterization of EX1 kinetics in H/D exchange mass spectrometry by peak width analysis, J. Am. Soc. Mass. Spectrom 17(11) (2006) 1498–1509. [DOI] [PubMed] [Google Scholar]
- 39.Stevens FJ Hypothetical structure of human serum amyloid A protein, Amyloid 11 (2004) 71–80. [DOI] [PubMed] [Google Scholar]
- 40.Neculai D, Schwake M, Ravichandran M, Zunke F, Collins RF, Peters J, Neculai M, Plumb J, Loppnau P, Pizarro JC, Seitova A, Trimble WS, Saftig P, S Grinstein., S. Dhe-Paganon, Structure of LIMP-2 provides functional insights with implications for SR-BI and CD36, Nature 504(7478) (2013) 172–176. [DOI] [PubMed] [Google Scholar]
- 41.Conrad KS, Cheng TW, Ysselstein D, Heybrock S, Hoth LR, Chrunyk BA, Am Ende CW, Krainc D, Schwake M, Saftig P, Liu S, Qiu X, Ehlers MD, Lysosomal integral membrane protein-2 as a phospholipid receptor revealed by biophysical and cellular studies, Nat. Commun 8(1) (2017) 1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Straub JE, Thirumalai D, Principles governing oligomer formation in amyloidogenic peptides, Curr. Opin. Struct. Biol 20(2) (2010) 187–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Straub JE, Thirumalai D, Toward a molecular theory of early and late events in monomer to amyloid fibril formation, Annu. Rev. Phys Chem 62 (2011) 437–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hoyer W, Grönwall C, Jonsson A, Ståhl S, Härd T Stabilization of a β-hairpin in monomeric Alzheimer’s amyloid-β peptide inhibits amyloid formation, Proc. Natl. Acad. Sci. USA 105(13) (2008) 5099–5104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Orr AA, Shaykhalishahi H, Mirecka EA, Jonnalagadda SVR, Hoyer W, Tamamis P, Elucidating the multi-targeted anti-amyloid activity and enhanced islet amyloid polypeptide binding of β-wrapins, Comput. Chem. Eng 116 (2018) 322–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sipe J Revised nomenclature for serum amyloid A (SAA). Nomenclature Committee of the International Society of Amyloidosis, Part 2, Amyloid 6(1) (1999) 67–70. [DOI] [PubMed] [Google Scholar]
- 47.Kollmer M, K Meinhardt, Haupt C, Liberta F, Wulff M, Linder J, Handl L, Heinrich L, Loos C, Schmidt M, Syrovets T, Simmet T, Westermark P, Westermark GT, Horn U, Schmidt V, Walther P, Fändrich M, Electron tomography reveals the fibril structure and lipid interactions in amyloid deposits. Proc. Natl. Acad. Sci. USA 113(20) (2016) 5604–5609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Das M, Wilson CJ, Mei X, Wales TE, Engen JR, Gursky O, Structural stability and local dynamics in disease-causing mutants of human apolipoprotein A-I: What makes the protein amyloidogenic? J. Mol. Biol 428(2 Pt B) (2016) 449–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Masson GR, Burke JE, Ahn NG, Anand GS, Borchers C, Brier S, Bou-Assaf GM, Engen JR, Englander SW, Faber J, Garlish R, Griffin PR, Gross ML, Guttman M, Hamuro Y, Heck AJR, Houde D, Iacob RE, Jørgensen TJD, Kaltashov IA, Klinman JP, Konermann L, Man P, Mayne L, Pascal BD, Reichmann D, Skehel M, Snijder J, Strutzenberg TS, Underbakke ES, Wagner C, Wales TE, Walters BT, Weis DD, Wilson DJ, Wintrode PL, Zhang Z, Zheng J, Schriemer DC, Rand KD Recommendations for performing, interpreting and reporting hydrogen deuterium exchange mass spectrometry (HDX-MS) experiments, Nat. Methods 16(7) (2019) 595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Maye r G., Eisenacher M, Pérez E, Uszkoreit J, Pfeuffer J, Sachsenberg T, Yilma z S., Tiwary S, Cox J, Audain E, Walzer M, Jarnuczak AF, Ternent T, Brazma A, Vizcaíno JA, The PRIDE database and related tools and resources in 2019: improving support for quantification data, Nucleic Acids Res. 47(D1) (2019) D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Linda h E, Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers, SoftwareX 1–2 (2015) 19–25. [Google Scholar]
- 52.Huang J, Rauscher S, Nawrocki G, Ran T, Feig M, de Groot BL, Grubmüller H, MacKerell AD, CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nature Methods 14(1) (2017) 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Klauda JB, Venable RM, Freites JA, O’Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, Jr AD. MacKerell, R.W. Pastor, Update of the CHARMM all-atom additive force Field for lipids: validation on six lipid types, J. Phys. Chem B 114(23) (2010) 7830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Humphrey W, Dalke A, Schulten K, VMD: Visual molecular dynamics, J. Mol. Graphics 14(1) (1996) 33–38. [DOI] [PubMed] [Google Scholar]
- 55.Gowers R, Linke M, Barnoud J, Reddy T, Melo M, Seyle r S., Domanski J, Dotson DL, Buchouxk S, Kenney IM, Beckstein O, MDAnalysis: A Python package for the rapid analysis of molecular dynamics simulations, Proceedings of the 15th Python in Science Conference (Scipy) (2018) 98–105. [Google Scholar]
- 56.Heinig M, Frishman D, STRIDE: a web server for secondary structure assignment from known atomic coordinates of proteins, Nucl. Acids Res 32 (2004) W500–W502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML, Comparison of simple potential functions for simulating liquid water, J. Chem. Phys 79(2) (1983) 926–935. [Google Scholar]
- 58.Hoover WG, Canonical dynamics: Equilibrium phase-space distributions, Phys. Rev. A 31(3) (1985) 1695–1697. [DOI] [PubMed] [Google Scholar]
- 59.Nosé S, A unified formulation of the constant temperature molecular dynamics methods, J. Chem. Phys 81(1) (1984) 511–519. [Google Scholar]
- 60.Parrinello M, Rahman A, Polymorphic transitions in single crystals: A new molecular dynamics method, J. App. Phys 52(12) (1981) 7182–7190. [Google Scholar]
- 61.Jo S, Cheng X, Lee J, Kim S, Park S-J, Patel DS, Beaven AH, Lee KI, Rui H, Park S, Lee HS, Roux B, MacKerell AD Jr., Klauda JB, Qi Y, Im W, CHARMM-GUI 10 years for biomolecular modeling and simulation, J. Comp. Chem 38(15) (2017) 1114–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Daura X, Gademann K, Jaun B, Seebac h D., van Gunsteren WF, Mark AE, Peptide folding: when simulation meets experiment, Angewandte Chemie Int. 38(1–2) (1999) 236–240. [Google Scholar]
- 63.Martínez L, Andrade R, Birgin EG, Martínez JM, PACKMOL: A package for building initial configurations for molecular dynamics simulations, J. Comp. Chem 30(13) (2009) 2157–2164. [DOI] [PubMed] [Google Scholar]
- 64.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, D Thompson J., Higgins DG, Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega, Mol. Syst. Biol 7 (2011) 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barto n G.J., Jalview Version 2 - a multiple sequence alignment editor and analysis workbench, Bioinformatics 25 (9) (2009) 1189–1191. http://www.jalview.org/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Waterhouse A, Bertoni M., Bienert S, Studer G, Tauriello G, umienny GR, Heer FT, de Beer TAP, Rempfer C, Bordoli L, Lepore R, Schwede T, SWISS-MODEL: homology modelling of protein structures and complexes, Nucleic Acids Res. 46(W1) (2018) W296–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.