

ABSTRACT
Cleft lip is one of the most common human birth defects. However, there remain a limited number of mouse models of cleft lip that can be leveraged to characterize the genes and mechanisms that cause this disorder. Crosstalk between epithelial and mesenchymal cells underlies formation of the face and palate, but the basic molecular events mediating this crosstalk remain poorly understood. We previously demonstrated that mice lacking the epithelial-specific splicing factor Esrp1 have fully penetrant bilateral cleft lip and palate. In this study, we further investigated the mechanisms leading to cleft lip as well as cleft palate in both existing and new Esrp1 mutant mouse models. These studies included a detailed transcriptomic analysis of changes in ectoderm and mesenchyme in Esrp1−/− embryos during face formation. We identified altered expression of genes previously implicated in cleft lip and/or palate, including components of multiple signaling pathways. These findings provide the foundation for detailed investigations using Esrp1 mutant disease models to examine gene regulatory networks and pathways that are essential for normal face and palate development – the disruption of which leads to orofacial clefting in human patients.
KEY WORDS: Cleft lip, Cleft palate, Epithelial-mesenchymal crosstalk, Lip morphogenesis
Summary: Alterations in epithelial-mesenchymal crosstalk underlie the lip and palate formation defects caused by ablation of the epithelial-specific splicing factor Esrp1.
INTRODUCTION
Cleft lip with or without cleft palate (CL/P) and cleft palate only (CP or CPO) are among the most common congenital birth defects, affecting approximately 1 in 700 live births, and affected children face a variety of health and psychosocial problems as well as a need for extensive surgical and dental treatments (Dixon et al., 2011). The causes of CL/P and CP are heterogeneous and include both environmental and genetic factors. Although either CL/P or CP can be a component of disease syndromes, most cases are non-syndromic. CL/P is more common in human patients than isolated cleft palate (CP) and these disorders are largely genetically and etiologically distinct (Fraser, 1970; Gritli-Linde, 2008). The proper development of the lip and palate is similar between humans and mice and thus mouse models have served an important role in studies to identify genes and characterize pathways that, when disrupted, lead to CL/P or CP (Gritli-Linde, 2012; Juriloff and Harris, 2008). In mice, formation of the face commences around embryonic day (E) 9.5 and involves the five facial prominences consisting of mostly neural crest-derived mesenchyme and overlying epithelium: the frontonasal prominence (FNP) and the paired maxillary and mandibular prominences (MxP and MdP) (Jiang et al., 2006). The FNP gives rise to the lateral and medial nasal prominences (LNP and MNP) and these prominences grow into close apposition and by E12.5 the nasal and maxillary prominences fuse to form the upper lip and primary palate. Defects in the growth and/or fusion of these prominences result in CL/P. The formation of the secondary palate is a separate developmental process that occurs from E12 to E15.5 when the palatal shelves emerge from the maxillary prominences, elevate, and fuse in the midline (Jiang et al., 2006). Defects in any of these steps can lead to CP. Although CL/P is the more common human clinical presentation, there are many mouse models of CP, but relatively few for CL/P (Gritli-Linde, 2008). Thus, newer mouse models for CL/P are needed to further define genes and pathways involved in CL/P pathogenesis. During craniofacial development, the epithelial cells of the facial prominences and palate provide the signals required for mesenchymal proliferation and patterning. At the same time, the mesenchyme provides feedback to epithelial cells and these reciprocal epithelial-mesenchymal interactions are crucial for normal facial and palatal development (Jiang et al., 2006; Wedden, 1987). These interactions involve Wnt, TGFβ/BMP, Hedgehog and Fgf signaling pathways, and mutations in components of these signaling pathways have been shown to cause CL/P and CP in human patients (Reynolds et al., 2019).
We identified epithelial splicing regulatory proteins 1 and 2 (ESRP1 and ESRP2) as epithelial-specific regulators of multiple target transcripts, including fibroblast growth factor receptor 2 (Fgfr2), dysregulated splicing of which is associated with cleft palate (Bebee et al., 2015; Rice et al., 2004; Warzecha et al., 2009a). Although there is some functional redundancy between these two paralogous proteins, only ESRP1 is essential, as loss of ESRP2 alone does not overtly alter splicing and Esrp2 knockout (KO) mice have no apparent phenotype (Bebee et al., 2015; Warzecha et al., 2009b). In contrast, the loss of ESRP1 alone can lead to substantial alterations in the splicing of numerous target transcripts, whereas the loss of both ESRP1 and ESRP2 is associated with larger changes in splicing. We previously showed that ablation of Esrp1 alone in mice led to fully penetrant bilateral CL/P, adding this splicing factor to the limited number of genes for which ablation leads to this defect in mice (Bebee et al., 2015). We therefore hypothesized that facial development is dependent on ESRP1-regulated splicing events and that studies using these mice have the potential to reveal molecular mechanisms that when dysregulated may lead to CL/P. We carried out a detailed analysis of the defects in facial and palatal development and an extensive analysis of changes in alternative splicing in the epithelial cells of the facial prominences, as well as changes in total gene expression, in both epithelial cells and the underlying mesenchyme. We identified reduced expression of several genes in Esrp1 KO ectoderm, including several canonical Wnts, as well as sonic hedgehog (Shh). These gene expression changes were accompanied by reduced expression of canonical Wnt target genes, transcriptional targets of Shh signaling, and components of the TGFβ/Bmp pathway in adjacent mesenchyme. These observations indicate that ESRP1 plays an important role in epithelial-mesenchymal crosstalk during craniofacial development. We also noted a defect in epithelial cell fusion during lip formation and in palatal explant cultures, indicating that ESRP1 is required for two distinct processes, growth and fusion, that are required for proper lip and palatal formation.
RESULTS
Conditional ablation of Esrp1 in surface ectoderm leads to CL/P
We examined the tissue specificity of ESRP1 in the developing face and palate by generating mice with endogenously FLAG epitope-tagged ESRP1. Analysis of these mice (Esrp1FLAG/FLAG mice, Fig. S1) confirmed that ESRP1 protein is specifically expressed in surface ectoderm at E11.5 as well as in epithelial cells of the secondary palate as previously shown for Esrp1 mRNA (Revil and Jerome-Majewska, 2013; Warzecha et al., 2009a). We generated mice with conditional ablation of Esrp1 in surface ectoderm derived from Esrp1flox/flox mice and transgenic Crect mice, which express Cre specifically in surface ectoderm and derivatives starting at E8.5, a time point prior to lip fusion (Reid et al., 2011). We confirmed relatively efficient conditional ablation in the epidermis of Esrp1flox/flox; Crect+/− E18.5 embryos (Fig. S2). Compared with littermate controls, we noted bilateral CL/P in Esrp1flox/flox; Crect+/− embryos at E18.5 (Fig. 1A). Although the phenotype was less extensive than previously observed in Esrp1−/− mice, there was a clear cleft of the primary palate and failure of the lip processes to come together to form a midline philtrum. We also performed staining for bone and cartilage structures, which confirmed a complete cleft of the secondary palate and a bilateral cleft of the primary palate. The palatine bones were hypoplastic and located to the side and the palatal processes of the maxilla were similarly dysmorphic. The premaxilla was undeveloped with a poor connection to the maxilla and thus extended out in front of the face (Fig. 1B).
Fig. 1.
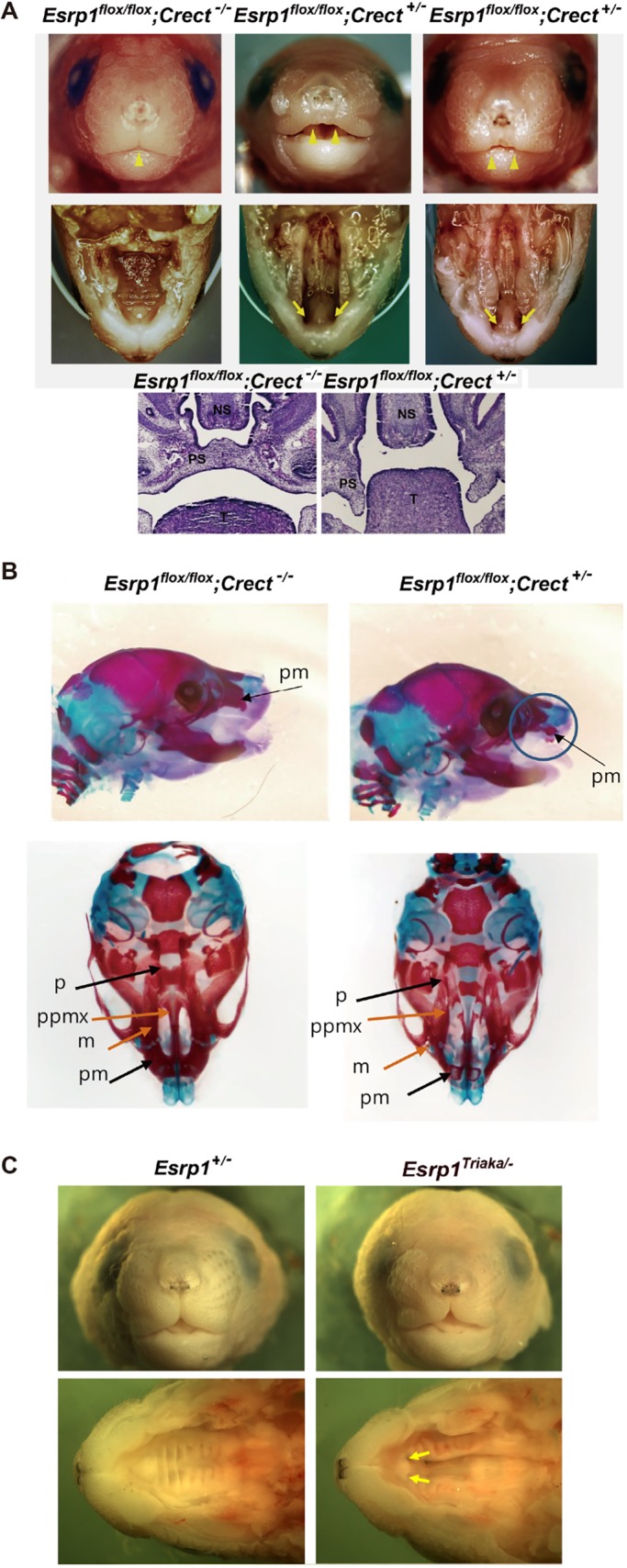
Conditional ablation of Esrp1 in surface ectoderm leads to CL/P. (A) Frontal views of control (Esrp1flox/flox) and Esrp1flox/flox; Crect+/− conditional KO (cKO) E18.5 embryos showing that the midline philtrum observed in control mice (single arrowhead) is absent in cKO mice as the upper lip primordia fail to meet at the midline (double arrowheads). Views of the palate after removal of the mandible show clefting of the secondary palate and primary palate in cKO mutants (arrows). Hematoxylin & Eosin images of coronal sections show hypoplastic palatal processes in Esrp1flox/flox; Crect+/− embryos. NS, nasal septum; PS, palatal shelf; T, tongue. (B) Alizarin Red/Alcian Blue stains of E18.5 embryos viewed from the side (top) and the ventral side with mandible removed (bottom). Defects are apparent in the cKO maxilla (m), palatine (p), premaxilla (pm) and palatal process of the maxilla (ppmx). (C) Frontal and ventral view with lower jaw removed of Esrp1Triaka/− mice without cleft lip or primary palate formation (arrows), but with cleft secondary palate.
In contrast to mice with complete ablation of Esrp1, mice with an N-ethyl-N-nitrosourea-induced point mutation in a highly conserved region of Esrp1 (Esrp1Triaka/Triaka mice) have modestly altered intestinal function, but no craniofacial defects (Mager et al., 2017). Whereas neither Esrp1+/− mice nor Esrp1Triaka/Triaka homozygous animals showed overt craniofacial defects, the faces of compound mutant Esrp1Triaka/− pups (5/5), although showing some hypoplasia compared with littermate controls, had clefting of the secondary soft and hard palate, but no apparent cleft of the lip or primary palate (Fig. 1C). These data show that this null/hypomorph allelic combination is able to generate a model of cleft secondary palate in the absence of cleft primary palate, indicating that these two developmental processes are not linked, but have different sensitivities to the levels of active ESRP1. The observation of a cleft secondary palate in the absence of cleft lip in Esrp1Triaka/− pups strongly indicated that Esrp1 deficiency independently leads to a failure in palate formation.
Esrp1 ablation leads to reduced outgrowth of facial prominences associated with reduced epithelial and mesenchymal cell proliferation
Lip formation results from outgrowth and fusion of the MxP, LNP and MNP and failure in either process can lead to cleft lip. Ki-67 staining of MNP and LNP in sections from E10.5 embryos showed reduced cell proliferation in both epithelial and mesenchymal cells associated with less apparent growth of the two processes towards apposition (Fig. 2A). Evaluation of apoptosis using staining for activated caspase 3 showed no apparent differences between control and KO embryos other than the expected apoptosis in sections in which these MNP and LNP processes were beginning to fuse in control embryos. In Esrp1−/− mice, these processes either did not make contact, or if they did make contact there was no observed fusion and no apparent apoptosis at the sites of contact (Fig. 2B). In one case in which we observed contact between the MNP and LNP in Esrp1−/− mice, we noted that there was persistent expression of E-cadherin (cadherin 1) that was not observed in wild-type (WT) embryos (Fig. 2C). We conclude that a reduction in proliferation of facial prominences contributes to CL/P in Esrp1−/− mice, but that there is also a defect in fusion when these processes make contact. Nonetheless, the reduction in cell proliferation in mesenchyme adjacent to Esrp1-ablated epithelial cells indicated that there was a disruption in a communication pathway from epithelium to mesenchyme during facial development.
Fig. 2.

Esrp1−/− embryos exhibit reduced proliferation of the MNP and LNP and do not fuse. (A) Frontal section showing decreased Ki-67 staining (green) in Esrp1−/− ectoderm and mesenchyme of Esrp1−/− embryos at E10.5 compared with controls. Quantification of Ki-67 is shown from WT (n=3) and Esrp1−/− (n=3) embryos. y-axis shows the percentage of Ki-67-positive cells out of total cells within each section. Error bars indicate s.d. Statistical significance was determined by two-tailed t-test. *P<0.05. (B) Arrows indicate the epithelial seam at the fusion site between MNP and LNP in WT embryos showing the expected apoptosis by activated caspase 3 staining (red). MNP and LNP from two Esrp1−/− embryos did not reveal substantial apoptosis, including one example in which MNP and LNP make contact (bottom right). Nuclei are stained with DAPI (blue). (C) E-cadherin staining (green) showing loss of epithelial cells between MNP and LNP after fusion in WT embryos, but persistence in MNP and LNP making contact in Esrp1−/− embryos. Arrow indicates contact between MNP and LNP, but no fusion. (D) Scanning electron microscopy (SEM) showing normal lip fusion in WT embryos from E10.5 to E12.5. The arrow in the WT image at E10.5 shows the normal formation of the lambdoidal junction. In Esrp1−/− embryos, we observed examples with no apparent contact between MNP and LNP or cases with contact, but none with fusion by E12.5. Panels outlined in red show magnifications of the boxed areas on the left.
We used scanning electron microscopy (SEM) to evaluate lip formation and fusion further in WT and Esrp1−/− embryos at different stages of lip formation. At E10.5, WT embryos showed the onset of fusion between the MNP and LNP, LNP and MxP, and MNP and MxP at the 3-way lambdoid junction. At E11.5 the fusion between these processes was more complete and by E12.5 there was normal development of the upper lip and nasal pit (Fig. 2D). In Esrp1−/− embryos, the LNP and MNP were smaller and a gap remained between them that resulted in a larger nasal pit (Fig. 2D). Compared with WT embryos, the MNP and LNP were further separated from each other, but in some cases there was contact between these processes, but no apparent fusion. There was also no contact observed between the hypoplastic MxP and either MNP or LNP to generate a typical lambdoid junction. By E12.5, there was very little contact noted between any of the prominences other than one example where there was some contact between MNP and LNP deeper into the nasal pit. Taken together, the SEM studies showed that, although there was some reduction in size of the facial prominences, there were some cases where contact occurred between MNP and LNP, but this was not followed by fusion. These observations, together with sections of these prominences in E10.5 embryos, further indicated that reduced proliferation and a defect in fusion underlie the CL/P phenotype.
Palatal processes show reduced proliferation as well as a defect in fusion during palatogenesis
The SEM and histology studies suggested that CL/P in Esrp1−/− mice was due to defects in both fusion and cell proliferation. We also noted reduced outgrowth of the palatal shelves towards each other, suggesting a defect in cell proliferation. Analysis of secondary palate formation in Esrp1−/− E16.5 embryos using Ki-67 staining and cleaved caspase 3 detection confirmed that the palatal shelves had a proliferation defect without apparent differences in apoptosis compared with WT embryos (Fig. 3A). However, because the palatal shelves did not make contact in vivo in Esrp1−/− mice, we were unable to determine directly whether there was an associated fusion defect. Therefore, to evaluate the defect in palatogenesis in Esrp1−/− mice further we performed palatal organ culture assays. WT and Esrp1−/− palatal shelves were isolated from E13.5 embryos and cultured for up to 72 h. Palatal shelves from WT embryos grew together within 48 h and after adhering underwent fusion with dissolution of the medial epithelial seam (MES) and mesenchymal confluence (n=7/7). Evaluation of palatal cultures from Esrp1−/− mice was complicated by increased fragility and reduced proliferation such that contact between opposing palatal shelves was delayed. Nonetheless, we noted that although the palatal shelves from eight out of 13 Esrp1−/− embryos were able to achieve contact, in seven of the eight palatal cultures in which the palatal processes achieved adherence there was no dissolution of the medial edge epithelial cells and there was a failure to achieve mesenchymal confluence. Only one of the eight Esrp1−/− palatal shelves that made contact showed partial dissolution of the MES and a small region of apparent mesenchymal connection. For one representative control and one Esrp1−/− sample in which the palatal processes made contact, we performed staining for the epithelial marker E-cadherin as well as activated caspase 3 and Ki-67. In the WT cultures, we observed a nearly complete loss of E-cadherin expression in cells at the site of fusion as well as the expected apoptosis at the fusion site. However, in the Esrp1−/− palates that achieved close contact, there was persistence of the MES and an absence of mesenchymal confluence (Fig. 3B; additional examples in Fig. S3). These results demonstrate that, although reduced growth and proliferation of palatal shelves contribute to the cleft palate defect, there is also a defect in fusion and dissolution of the MES. The results from the palatal explant cultures also further verify that the clefting of the secondary palate in Esrp1−/− mice represents an independent defect.
Fig. 3.

Reduced proliferation and a fusion defect contribute to cleft palate in Esrp1−/− mice. (A) Coronal sections stained for E-cadherin (green) in E16.5 control and Esrp1−/− embryos showing the medial edge epithelial seam (MES) becoming discontinuous following fusion in controls whereas palatal shelves failed to elevate in the mutants. Activated caspase 3 staining shows apoptosis at the site of fusion, but no apparent increase in apoptosis in Esrp1−/− embryos, which also show reduced Ki-67 staining for proliferating cells compared with WT in the palatal shelves. Inset shows higher magnification of caspase 3 staining at the site of fusion. Graph shows quantification of the percentage of Ki-67-positive cells out of total cells in WT (n=3) and Esrp1−/− embryos (n=3). Error bars indicate s.d. Statistical significance was determined by two-tailed t-test. *P<0.05. Error bars indicate s.d. (B) Palatal organ culture showing lack of dissolution of the MES and associated apoptosis and reduced proliferation in palatal shelves from Esrp1−/− embryos compared with WT.
Ablation of Esrp1 leads to large-scale changes in splicing in surface ectoderm
The specific expression of ESRP1 in surface ectoderm together with the CL/P defect observed in Esrp1flox/flox; Crect+/− embryos indicated that alterations in the transcriptome of surface ectoderm-derived cells underlie orofacial clefting in Esrp1−/− mice. However, the reduction in cell proliferation observed in the mesenchyme adjacent to epithelial cells also indicated that gene expression alterations in Esrp1−/− epithelial cells induced changes in signaling crosstalk from epithelium to mesenchyme. To investigate the molecular mechanisms that lead to CL/P in Esrp1−/− mice further, we performed RNA sequencing (RNA-Seq) using RNAs collected from both epithelial cells and mesenchymal cells from control and Esrp1−/− embryos. We used a previously described method to separate facial ectoderm and mesenchyme from facial prominences at E12.0, a stage at which lip fusion is underway (Li and Williams, 2013). We prepared RNA from four replicate samples each of ectoderm and mesenchyme in WT and Esrp1−/− embryos and performed RNA-Seq. We used paired end sequencing and obtained an average of 100 million read pairs per replicate. Preliminary analysis of RNA-Seq reads from epithelial and mesenchymal control samples validated that they were derived from relatively pure populations of each cell type using a panel of standard epithelial and mesenchymal cell type-specific markers, including Esrp1 (Fig. 4A). To identify genome-wide alterations in splicing in ectoderm from Esrp1 KO embryos compared with WT controls, we used replicate-based multivariate analysis of transcript splicing (rMATS) (Shen et al., 2014). We also used rMATS to identify global differences in splicing of epithelial cells compared with mesenchymal samples using RNAs from control embryos. In Esrp1 KO epithelial cells, rMATS identified a total of 1467 alternative splicing changes compared with WT epithelial cells, with cassette exons [skipped exons (SE)] representing the largest fraction (Fig. 4B,C, Table S1). Analysis of splicing differences between WT epithelial cells and WT mesenchymal cells identified 2546 splicing events, including many Esrp-regulated events that switch splicing from epithelial to mesenchymal splice variants after Esrp1 ablation (Fig. 4B,D, Table S2). The larger number of splicing differences between epithelial and mesenchymal cells compared with splicing differences between WT and Esrp1−/− epithelial cells is consistent with previous studies by our group and other investigators showing combinatorial regulation of splicing events that are induced during epithelial-mesenchymal transition or that differ between epithelial and mesenchymal cells by several splicing factors, including ESRP1/2, but also other splicing regulators (Braeutigam et al., 2014; Pillman et al., 2018; Shapiro et al., 2011; Venables et al., 2013; Yang et al., 2016). We validated 19 cassette exon (SE) events in WT compared with Esrp1−/− ectoderm that were the most statistically significant and showed the largest absolute change in percent spliced in (PSI) by semi-quantitative RT-PCR (Fig. 4E; Table S1). We also validated six representative examples of splicing differences between control ectoderm and control mesenchyme (Fig. 4F). As expected, a change in alternative splicing of Fgfr2 was identified among alterations in mutually exclusive (MXE) exon splicing in Esrp1−/− ectoderm. This event involves two alternative exons, named IIIb or IIIc, that encode a region in the extracellular ligand-binding domain and with the resulting receptor isoforms, FGFR2-IIIb and FGFR2-IIIc, having different Fgf binding preferences (Zhang et al., 2006). We confirmed this splicing switch by RT-PCR, which showed a switch from nearly complete splicing of the epithelial-specific IIIb exon to predominant splicing of the mesenchymal IIIc exon in Esrp1−/− ectoderm (Fig. 4E). Both global splicing comparisons were subjected to Gene Ontology (GO) analysis, which revealed overlapping enrichment terms for both splicing comparisons, including cytoskeletal organization, cell morphogenesis, and cellular component organization (Fig. 4G,H). Similarly, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis demonstrated an enrichment for terms including adherens junctions, suggesting that alternatively spliced genes contribute to epithelial functions (Fig. 4G,H). KEGG analysis in WT compared with Esrp1−/− ectoderm also showed enrichment for components of the Hippo pathway, which has been implicated in human clefting disorders (Williamson et al., 2014).
Fig. 4.

RNA-Seq identified large-scale alterations in alternative splicing in Esrp1−/− ectoderm compared with WT ectoderm and differences in alternative splicing between WT ectoderm and WT mesenchyme. (A) Expression of epithelial markers (left) and mesenchymal markers (right) validates efficient separation of epithelial cells from mesenchymal cells in WT samples. TPM, transcripts per million. (B) Summary derived from Tables S1 and S2 showing both the total number and percentage of the different types of alternative splicing events in WT versus Esrp1−/− ectoderm (top) and in WT ectoderm versus WT mesenchyme (bottom) identified by rMATS with false discovery rate (FDR)<5%, and |deltaPSI|≥5%. (C) Heatmap representing the skipped exon (SE) splicing changes with increased exon inclusion (red) or decreased exon inclusion (blue). (D) Venn diagram depicting detected SE events identified in WT ectoderm versus Esrp1 KO ectoderm (yellow), WT ectoderm versus WT mesenchyme (green), and those identified in both comparison sets (blue). (E) RT-PCR validations of changes in splicing of cassette exons Esrp1−/− ectoderm (E1KO) compared with WT. The ‘percent spliced in’ (PSI) value indicates the percentage exon inclusion for each event. Also shown is the change in splicing of mutually exclusive exons IIIb and IIIc of Fgfr2. Products containing each exon were distinguished by restriction digests with AvaI and HincII, which cut products containing exon IIIb and IIIc, respectively. (F) Validation of several differences in splicing between control ectoderm and mesenchyme. (G) GO and KEGG pathway enrichment for genes with alternative splicing differences between WT ectoderm and Esrp1−/− ectoderm. (H) GO and KEGG pathway enrichment for genes with splicing differences between WT ectoderm and WT mesenchyme.
Ablation of Esrp1 in ectoderm is associated with altered expression of genes associated with CL/P and disruptions in signaling crosstalk in underlying mesenchyme
We also investigated changes in total transcript levels in Esrp1-ablated epithelial cells, as well as in adjacent mesenchyme. In epithelial cells we identified 167 downregulated genes and 546 upregulated genes (Table S3). Among the downregulated genes we noted six transcripts encoding canonical Wnt ligands, Wnt3, Wnt3a, Wnt4, Wnt7b, Wnt9b and Wnt10b, each of which were downregulated approximately 2-fold, of which several were validated by RT-qPCR (Fig. 5A). We also noted downregulation of Wls, which is required for Wnt secretion. Consistent with these observations, for genes downregulated in Esrp1 KO ectoderm, Wnt signaling pathway was an enriched GO term for biological processes and was also enriched in the KEGG pathway analysis (Fig. 5B). We also noted an increase in both Aldh1a2 and Aldh1a3, which generate retinoic acid and have been implicated in feedback regulation of Wnt signaling during craniofacial development (Osei-Sarfo and Gudas, 2014; Song et al., 2009). We examined whether the downregulation of Wnt ligands in the ectoderm was also associated with a corresponding reduction in targets of canonical Wnt/β-catenin signaling in mesenchyme as previously shown in Wnt9b and Lrp6 KO mice with CL/P (Jin et al., 2012). Interestingly, we identified larger changes in gene expression in mesenchyme of Esrp1−/− mice than in ectoderm, with 3048 upregulated genes and 2396 downregulated genes (Table S3). In addition, we identified genes that were differentially expressed in WT ectoderm compared with WT mesenchyme, which included numerous known epithelial and mesenchymal markers (Table S5). Although Wnt signaling was not among the most enriched GO terms or pathways for genes downregulated in Esrp1−/− mesenchyme compared with WT mesenchyme, we noted numerous canonical Wnt targets among the downregulated genes under the ‘Wnt signaling pathway’ category, several of which were also validated by RT-qPCR along with several other genes encoding components of the Wnt pathway (Fig. 5A). We also noted reduced expression of sonic hedgehog (Shh) in Esrp1−/− ectoderm and a corresponding reduction in expression of the Gli1, Gli2 and Gli3 transcription factors in mesenchyme, with the greatest reduction in Gli2 confirmed by RT-qPCR (Fig. 5A). We also identified several other genes with reduced expression in Esrp1−/− mesenchyme that have previously been implicated in cleft palate, including Bmp7, Tgfb2 and Tgfb3 (Fig. 5A, Table S4) (Kaartinen et al., 1995; Kouskoura et al., 2013; Proetzel et al., 1995; Sanford et al., 1997). Thus, we identified transcriptomic changes in multiple signaling pathways, including several that have previously been shown to play an essential role in lip/palate morphogenesis (Table S6). We validated reduced expression of Wnt9b and Shh in Esrp1−/− mice using in situ hybridization (ISH) in mouse embryos at E10.5-11.5. The reductions in these transcripts were limited to facial ectoderm in Esrp1−/− embryos, consistent with the RNA-Seq data showing that Wnt9b and Shh were specifically expressed in ectoderm (Fig. 5C, Table S3). We also confirmed reduced expression of the canonical Wnt targets Lef1 and Axin2 using both whole-mount and section ISH (Fig. 5D).
Fig. 5.

Esrp1 ablation in ectoderm results in altered expression of components of the Wnt signaling pathway and other signaling pathways implicated in cleft lip and/or cleft palate. (A) qRT-PCR validation of selected changes in total transcript levels in Esrp1−/− ectoderm as well as in adjacent mesenchyme. Error bars indicate s.d. Statistical significance for each comparison was determined by two-tailed t-test. *P<0.05. (B) GO and KEGG pathway enrichment for genes that are downregulated in Esrp1−/− ectoderm. (C) Whole-mount in situ hybridization of E11.5 embryos showing reduced expression of Shh and Wnt9b in epithelial cells of the developing face. (D) Whole-mount and tissue section in situ hybridization showing reduced expression of canonical Wnt target genes Lef1 and Axin2 in Esrp1−/− embryos.
To examine canonical Wnt signaling during facial development in Esrp1−/− mice further, we crossed both WT and Esrp1−/− mice with TCF/Lef:H2B-GFP transgenic reporter mice that express an H2B-EGFP fusion protein under the control of six copies of the TCF/LEF response element (Ferrer-Vaquer et al., 2010). In Esrp1−/− E11.5 embryos, we noted reduced reporter activity in both the ectoderm and mesenchyme of the nasal prominences and MxP, although the reduction in mesenchyme was more pronounced in the MxP than in the MNP or LNP (Fig. 6A). Because of some differences in various Wnt reporter models and to verify changes in canonical Wnt signaling further, we also used Axin2lacZ mice in which lacZ is knocked in at the endogenous Axin2 locus as a second readout for Wnt signaling in WT compared with Esrp1−/− embryos. At E10.5, we noted that although there was no apparent difference in lacZ expression (as determined by β-galactosidase detection) in the LNP, there was pronounced reduction in lacZ in both the ectoderm and mesenchyme of the MNP (Fig. 6B). We also noted reduced lacZ in the MxP at E11.5. Taken together, these observations are consistent with the results from RNA-Seq showing reduced Wnt expression in ectoderm of Esrp1−/− embryos and an associated reduction in canonical Wnt targets. These findings are consistent with a model in which Esrp1 ablation leads to alterations in the epithelial-mesenchymal interactions that underlie normal facial development.
Fig. 6.

Reduced Wnt signaling in Esrp1−/− confirmed in-crosses with WNT/β-catenin signaling reporter mice. (A) Reduced activation of the TCF/Lef1-GFP reporter is observed in ectoderm and mesenchyme of Esrp1−/− MNP, LNP and MXP. (B) Reduced expression of the Axin2-lacZ reporter in LNP and MXP in Esrp1−/− embryos. For A and B, frontal sections are shown. Graphs show quantification of mean fluorescence intensity corrected for area in WT (n=3) and Esrp1−/− embryos (n=3). Error bars indicate s.d. Statistical significance was determined by two-tailed t-test. *P<0.05. Scale bars: 20 µm.
DISCUSSION
Studies using mouse models have played a major role in our understanding of the morphogenetic programs in the face and palate that, when disrupted, lead to CL/P. Mouse models have been particularly informative in defining genes and pathways disruption of which leads to CPO (Li et al., 2017). However, there remain relatively few mouse models that lead to CL/P, such as those involving mutations or deletions of Wnt9b, Lrp6, Bmp4, Bmpr1a, Tfap2a and Pbx1/2 (Ferretti et al., 2011; Green et al., 2015; Gritli-Linde, 2012; Jin et al., 2012; Juriloff and Harris, 2008; Juriloff et al., 2006; Liu et al., 2005; Song et al., 2009). As a result, our understanding of the molecular mechanisms involved in the formation of the lip and primary palate have lagged behind those described for formation of the secondary palate. Our identification of fully penetrant CL/P in Esrp1−/− mice provides a new genetic model that can be exploited to investigate the mechanisms of lip development further. It is notable that although much has been learned about the signaling pathways and transcription factors that are involved in craniofacial development, the role of alternative splicing in lip and/or palate development has been largely unexplored. Prior to our identification of CL/P in Esrp1 KO mice, no studies have identified roles of splicing factors in CL/P. Furthermore, other than the role of a specific splice variant for Fgfr2 (Fgfr2-IIIb), the possibility that the functions of some genes required for face and palate development are splice isoform specific has not generally been considered. There is now firm evidence that, like transcription factors, tissue-specific splicing regulators coordinate programs of alternative splicing involving transcripts that encode proteins that function in biologically coherent pathways (Kalsotra and Cooper, 2011; Lee et al., 2018; Ule et al., 2005; Zhang et al., 2008). Thus, our studies demonstrating that Esrp1 is required for formation of both lip and palate indicates that gene targets of ESRP1 also play essential developmental roles and that mutations in these genes may cause or predispose patients to CL/P.
Our finding of bilateral CL/P in Esrp1flox/flox; Crect+/− embryos indicated that ablation of Esrp1 in early surface ectoderm and derivatives underlies cleft lip and cleft palate. Therefore, identification of transcriptomic changes in this cell population is key to understanding the mechanisms leading to this defect. We considered the possibility that the cleft palate observed in Esrp1−/− and Esrp1flox/flox; Crect+/− mice embryos might be a consequence of cleft primary palate extending into the secondary palate. However, mice with compound ablation of Esrp1 and a hypomorphic Esrp1 Triaka allele (Esrp1Triaka/− mice) had no cleft lip or primary palate but a cleft secondary palate, indicating that Esrp1 is independently required for correct secondary palate formation (Mager et al., 2017) (Fig. 1D). These studies establish that ESRP1 is required for both lip and palate development and that both Esrp1 mutant models can be used to characterize mechanisms that are essential for both developmental processes.
We examined proliferation using Ki-67 staining and identified reduced staining in epithelial and mesenchymal cells of the MNP and LNP. There was also reduced Ki-67 staining in the palatal shelves of Esrp1−/− embryos at E16.5. We did not observe a notable difference in staining for the apoptotic marker caspase 3, in either facial or palatine processes, indicating that a reduction in proliferation underlies the reduced size of both the nasal processes as well as palatal shelves. Of note, we also found that the palatal shelves did not elevate in either Esrp1−/− or Esrp1flox/flox; Crect+/− embryos (see also Fig. 1A). However, at this stage we cannot be certain whether there is also a defect in palatal elevation independent of the palatal hypoplasia that contributes to cleft secondary palate.
We used a time series analysis of both WT and Esrp1−/− embryos during several stages of lip formation. Despite reduced proliferation, we noted several stages at which the MNP and LNP in Esrp1−/− embryos were able to make contact and adhere, but this did not appear to lead to fusion by E12.5, at which point fusion was complete between the LNP, MNP and MxP in WT embryos. Although we were unable to successfully complete ex vivo facial explant cultures to verify a fusion defect between MNP, LNP and MxP, our studies using ex vivo palatal explants demonstrated that a fusion defect in Esrp1−/− embryos contributes to cleft palate. As it is believed that the mechanisms of lip and palate fusion during development are similar (Jiang et al., 2006; Ray and Niswander, 2012), we suggest that the fusion defect observed in palatal explants, taken together with our histology and SEM time course, also indicate a defect in fusion during lip and primary palate formation.
We carried out in-depth RNA-Seq analysis to identify alternative splicing changes in Esrp1−/− ectoderm and to define differences in splicing between WT ectoderm and mesenchyme. Numerous changes in splicing were identified in KO compared with WT ectoderm, including some events that had previously been identified in Esrp1−/− epidermis, but the greater sequencing depth in this study identified a greater number of splicing changes than our previous studies. We first identified ESRP1 (and its paralog ESRP2) in a screen for regulators of Fgfr2 splicing and, not surprisingly, one of the largest changes in splicing was a nearly complete switch in Fgfr2 isoforms from Fgfr2-IIIb to Fgfr2-IIIc in Esrp1-ablated ectoderm. This nearly complete change in splicing of Fgfr2 was in contrast to our analysis in E18.5 epidermis that examined changes in splicing in both Esrp1−/− and Esrp1−/−;Esrp2−/− (double KO, or DKO) tissue, in which there was no change in Fgfr2 splicing unless both Esrp1 and Esrp2 were ablated (Bebee et al., 2015). This observation likely reflects the lower expression levels of both Esrp1 and Esrp2 in E12.0 surface ectoderm compared with E18.5 epidermis, including a significantly lower Esrp2 expression level compared with Esrp1 in ectoderm (see Table S3). We noted similar examples in which deletion of Esrp1 alone caused greater changes in splicing in ectoderm compared with the epidermis of Esrp1−/− mice and suspect that compensation by higher Esrp2 levels in other tissues prevents most of the other major defects described in Esrp1−/−;Esrp2−/− mice. We did note additional craniofacial abnormalities in Esrp1−/−;Esrp2−/− mice, including mandibular defects that were not seen in Esrp1−/− mice, indicating that ESRP1-regulated splicing by both paralogs plays broader roles in craniofacial development (Bebee et al., 2015). A previous study demonstrated cleft palate, but not cleft lip, in mice in which the epithelial Fgfr2 exon IIIb was deleted (Rice et al., 2004). However, deletion of exon IIIb in these mice did not default to splicing of exon IIIc in epithelial cells, but caused skipping of both exons and a frameshift that effectively resulted in no Fgfr2 expression in epithelial cells (De Moerlooze et al., 2000). In contrast, ablation of Esrp1 induces a switch in isoforms, such that ectopic FGFR2-IIIc in epithelial cells can still respond to Fgf ligands to sustain Fgf signaling, as we demonstrated in a prior study (Rohacek et al., 2017). These observations strongly suggest that altered splicing of Fgfr2 does not account for the cleft lip observed in Esrp1−/− mice and is also unlikely, by itself, to cause cleft palate.
Although we identified large numbers of splicing changes in Esrp1−/− compared with WT ectoderm, we also identified differences in splicing between WT ectoderm and WT mesenchyme. These analyses identified a large number of differences in splicing between these cell populations, which included many ESRP1-regulated events. We note that although numerous investigations have identified distinct epithelial and mesenchymal markers at the whole transcript or protein level, there remain limited examples in which large-scale differences in splicing between these cell populations have been identified (Venables et al., 2013). The analysis presented here thus provides another resource to identify how different splice isoforms influence epithelial-mesenchymal crosstalk, as previously described for Fgfr2 (De Moerlooze et al., 2000; Warzecha et al., 2009a).
Identification of changes in total transcript levels in Esrp1−/− ectoderm compared with controls revealed substantial numbers of genes that were upregulated or downregulated and there was little, if any, overlap between these genes and those that demonstrated changes in splicing. Among these genes, we noted decreases in the expression level of canonical Wnts, including Wnt9b, as well as Shh, components of two pathways that have previously been shown to be essential for lip and/or palate development (Jin et al., 2012; Lan and Jiang, 2009; Lipinski et al., 2010). These alterations are associated with corresponding downregulation of both canonical Wnt as well as Shh-regulated targets in adjacent mesenchyme, suggesting that a reduction in a communication pathway from ectoderm to mesenchyme leads to reduced mesenchymal proliferation. Alterations in canonical Wnt signaling have been implicated in mouse models, including CL/P in mice with ablation of Wnt9b and Lrp6 (Carroll et al., 2005; Ferretti et al., 2011; Jin et al., 2012; Juriloff et al., 2006; Song et al., 2009), as well as human cases of syndromic and non-syndromic CL/P (Reynolds et al., 2019). The reduced mesenchymal proliferation observed in the mesenchyme of Esrp1−/− mice is similar to that demonstrated in both Lrp6−/− mice and Wnt9b−/− mice, suggesting that a decrease in Wnt signaling may, at least in part, contribute to reduced mesenchymal proliferation and failed approximation of the MNP, LNP and MxP in Esrp1−/− mice. However, Wnt9b−/− mice did not have a defect in fusion of the nasal prominences and MxP in explant cultures, whereas Esrp1−/− mice have a fusion defect in addition to reduced mesenchymal proliferation. Hence, we suspect that alterations in other genes and pathways prevent epithelial fusion in Esrp1−/− mice.
Previous studies have identified crosstalk between Wnt and hedgehog signaling, and ablation of Shh in palatal epithelial cells or in utero treatment with Shh inhibitors have been shown to cause cleft palate and CL/P, respectively (Lan and Jiang, 2009; Lipinski et al., 2010; Reynolds et al., 2019; Rice et al., 2004). It is thus possible that reduction in Shh expression in ectoderm might also contribute to CL/P in Esrp1 KO mice. We noted that, in addition to reductions in Gli transcription factors, there was also reduced expression of Foxf1a, Foxf2 and Osr2 in Esrp1−/− mesenchyme, which were previously shown to be downregulated in mesenchyme when Shh signaling from epithelial to mesenchyme cells caused cleft palate through ablation of Smo in mesenchyme (Lan and Jiang, 2009). These findings are also potentially relevant to CL/P as another study showed that inhibition of Shh signaling with the inhibitor cyclopamine caused cleft lip that was also associated with a reduction of Gli1 and Foxf2 in mesenchyme of MNP (Everson et al., 2017). A recent study using single cell RNA-Seq to identify subpopulations of cell types present at the lambdoidal junction where the MNP, LNP and MxP fuse characterized distinct and dynamic expression patterns in subsets of both ectodermal and mesenchymal cells during lip fusion (Li et al., 2019). This analysis revealed that, although canonical Wnts were specifically expressed in the ectoderm of these processes, they were excluded from the fusion zone once these processes made contact, consistent with a lack of requirement for Wnts (at least for Wnt9b) for fusion and dissolution of the epithelial seam. This study also identified Fgf10 among the genes that are highly expressed in mesenchymal cells at the fusion zone. During palate formation, mesenchymal Fgf10 was previously shown to induce Shh expression in adjacent epithelial cells via the Fgfr2-IIIb isoform, and conditional ablation of Shh in palatal epithelial cells leads to cleft palate (Lan and Jiang, 2009). A switch in Fgfr2 splicing in Esrp1−/− palatal epithelium would render it unresponsive to mesenchymal FGF10, suggesting that the Fgfr2 splicing switch may be one factor leading to reduced Shh expression in epithelial cells of the facial processes and likely also in the epithelial cells of the palate. In addition to Fgf10, the aforementioned single cell RNA-Seq analysis also showed a reduction of Tgfb2 in the fusion zone of both ectoderm and mesenchyme (Li et al., 2019). It is therefore tempting to speculate that the reduction in Tgfb2 observed in mesenchyme adjacent to Esrp1−/− ectoderm may contribute to the observed fusion defect. However, there may also be combinatorial effects of ectodermal genes that are upregulated upon Esrp1 ablation, including Dkk1, Aldh1a3 and Sfrs2, which have been described as Wnt inhibitors.
Although our results suggest that reductions in Wnt signaling may be one factor contributing to CL/P in Esrp1−/− mice, an unresolved question remains regarding how Esrp1 ablation in ectoderm leads to coordinated changes in the expression of several canonical Wnts. Ablation of the transcription factors Pbx1 and Pbx2 was previously shown to lead to CL/P through reduced expression of Wnt9b and Wnt3. However, we did not identify changes in the expression of Pbx1, Pbx2 or Pbx3 in Esrp1−/− ectoderm. Although there is a vast literature describing numerous Wnt target genes in different contexts, there is limited understanding of how the Wnt genes themselves are transcriptionally regulated. We suspect that changes in transcriptional regulation that result from Esrp1 ablation are indirect, possibly caused by alterations in signaling pathways that regulate transcription factor expression and/or activity.
A major challenge for future studies will be to decipher the molecular mechanisms by which loss of Esrp1 leads to alterations in components of numerous pathways that have been linked to clefting disorders (Table S6). These include, but are not limited to, Wnt, Shh, TGFβ, BMP, Fgf and Hippo pathways. Future studies are needed to define the relative contributions of alterations in each of these signaling pathways in CL/P in these mice. The transcriptomic analysis presented here will hopefully provide a resource that can be used by the community to explore further the molecular mechanisms that lead to CL/P as well as Esrp1-regulated targets that merit further investigations as possible disease-related genes.
MATERIALS AND METHODS
Mouse strains
Generation of Esrp1 KO (Esrp1−/−) and conditional Esrp1 (Esrp1flox/flox) were described previously (Bebee et al., 2015) as was the Crect strain (Reid et al., 2011). Axin2lacZ (Manuylov et al., 2008) and TCF/Lef1:H2B-GFP (Ferrer-Vaquer et al., 2010) strains were purchased from the Jackson Laboratory. Esrp1FLAG/FLAG mice were generated in mouse V6.5 embryonic stem cells by the Penn Transgenic and Chimeric mouse core facility using electroporation of an mRNA encoding Cas9, an sgRNA targeting the ATG start codon, and an oligonucleotide repair template encoding two tandem copies of the FLAG epitope tag. Esrp1Triaka mice were described previously (Mager et al., 2017). Relevant strains were interbred from embryo isolation and females were examined in the morning for presence of a vaginal plug; the presence of a plug was designated E0.5. Genomic DNA for genotyping was derived from tail biopsies and genotyping was performed using standard procedures for these strains. Both male and female mice and embryos were used in this study. All animal procedures and experiments were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Pennsylvania.
Scanning electron microscopy
Mouse embryos were harvested at either E10.5, E11.5 or E12.5. The heads were fixed in Karnovsky's solution and a portion of the unfixed body including the tail was saved from each embryo for genotyping. Fixed head samples were dehydrated through a graded ethanol series and placed in Freon (1,1,2-trichloro-1,2,2 trifluoroethane) (Recycle & Reuse Industries, Mansfield, TX, USA) for critical-point drying. The samples were mounted on aluminum stubs with clay and sputter-coated with gold in an Argon atmosphere, using a Denton Vacuum Desk II Cold Sputter Etch Unit (Denton Vacuum). The heads were then viewed under a Quanta 600 FEG Mark II scanning electron microscope (FEI).
Isolation of ectoderm and mesenchyme for RNA harvest
E12.0 mouse embryos were dissected, and the ectodermal and mesenchymal tissue layers of the prominences were separated and collected for WT and Esrp1−/− mice for a total of four pools of six or seven mice in each category. The facial ectoderm and mesenchyme of each embryo was separated using a previously described surgical protocol (Li and Williams, 2013). RNA harvest of collected tissue was described previously (Bebee et al., 2015). For synthesis of cDNA, 300 ng of total RNA was used for ectoderm and mesenchyme samples, oligodT primer and SuperScript3 reverse transcriptase (Invitrogen).
Real-time RT-PCR and RT-PCR
Real-time RT-PCR and RT-PCR were performed as described (Bebee et al., 2015; Lee et al., 2018). Briefly, real-time RT-PCR was quantified using ImageQuant TL, version 7.0. Splicing ratios are represented as PSI for cassette exons and were normalized to RT-PCR product size. Real-time RT-PCR and RT-PCR primer sequences are listed in Table S7.
RNA sequencing and data analysis
Total RNA from ectodermal and mesenchymal samples was used for RNA-Seq at the Penn Next Generation Sequencing Core (NGSC) facility as previously described with the exception that we obtained 150 bp paired end reads (Bebee et al., 2015). The average number of read pairs was approximately 100 million read pairs per replicate. Identification of changes in alternative splicing in ectoderm samples was carried out using rMATS as previously described (Shen et al., 2014). Differential gene expression analysis was carried out by the Penn NGSC using EdgeR. Gene expression values were measured by Kallisto (v0.43.0) with mm10 gencode vM13 as the transcriptome index. Gene Ontology and pathway analysis was conducted using DAVID (Huang et al., 2007).
Skeletal analysis
E18.5 embryo heads were skinned, then fixed in 4% paraformaldehyde overnight. For cartilage and bone staining, E18.5 embryos were stained with Alizarin Red and Alcian Blue for examination of bone and cartilage structure, as previously described (Bebee et al., 2015).
Histology and immunofluorescence
Embryos from E10.5 to E14.5 were harvested and fixed overnight at 4% in paraformaldehyde and fixed in paraffin. Paraffin sections were deparaffinized in xylene and rehydrated using a graded ethanol series. For standard histology, sections were stained with Hematoxylin & Eosin. For immunofluorescence, antigen retrieval was performed using unmasking solution (Vector Laboratories) in a humidified chamber. Samples were blocked with 5% bovine serum albumin and 3% sheep serum in phosphate-buffered saline for 1 h at room temperature. Primary antibodies were incubated overnight at 4°C. After washing with PBST, secondary antibodies were applied for 30 min at room temperature, followed by another wash. Samples were mounted with Prolong Gold antifade reagent with DAPI (Invitrogen) and images were taken using an Olympus BX43. Samples were incubated with primary antibodies against FLAG-M2 (Sigma-Aldrich, F1804, 1:1000), E-cadherin (BD Biosciences, 610181, 1:500), cleaved caspase 3 (Cell Signaling Technology, 9664S, 1:500), Ki-67 (Abcam, Ab16667, 1:500), GFP (Abcam, A-11122, 1:1000), β-galactosidase (Abcam, ab9361, 1:1000) and secondary antibodies rabbit IgG Alexa 488 (Life Technologies, A24922, 1:500) and mouse IgG Alexa 594 (Life Technologies, A24921, 1:500). Quantification of fluorescence on immunostained slides was carried out using ImageJ with comparisons between tissues corrected for area.
Whole-mount and section in situ hybridization
In situ hybridization experiments were carried out as previously described (Moorman et al., 2001; Warzecha et al., 2009a) using digoxigenin-labeled riboprobes for Axin2, Lef1, Wnt9b and Shh. The signal was detected with alkaline phosphatase and then color developed with BM-Purple AP substrate (Roche, 11442074001).
Palatal organ culture
A pair of unfused palatal shelves were dissected out from an E14 mouse embryo under a dissecting microscope and placed on autoclaved nitrocellulose membrane with the oral side facing down. The palatal shelves were correctly oriented as in vivo and gently pushed against each other to ensure contact between the medial edge epithelia. Palatal shelves on a membrane were rested on a wire grid in a 12-well culture plate and cultured for 72 h in DMEM containing 10% fetal bovine serum. Tail DNA isolated from each embryo was used for genotyping. The organ culture was then processed for histology and immunofluorescence labeling as described in the Histology and immunofluorescence section.
Supplementary Material
Acknowledgements
We thank Natoya Peart, Eric C. Liao, and Shannon Carroll for critical review of the manuscript. We are grateful to the Transgenic and Chimeric Mouse Core of the University of Pennsylvania for generation of Esrp1FLAG/FLAG mice (supported by NIH center grants P30DK050306, P30DK019525, and P30CA016520). We thank Lukas F. Mager and Lester Thoo (University of Bern) for organizing neonatal Triaka mice.
Footnotes
Competing interests
Y.X. is a scientific cofounder of Panorama Medicine. All other authors declare no competing interests.
Author contributions
Conceptualization: S.L., T.W., R.P.C.; Methodology: M.J.S., H.L., I.S., Z.Z., Y.X., H.-D.N., T.W., R.P.C.; Software: Z.Z., Y.X.; Validation: H.-D.N.; Formal analysis: S.L., Z.Z., H.-D.N., T.W., R.P.C.; Investigation: S.L., M.J.S., I.S., P.K., H.-D.N., T.W., R.P.C.; Resources: P.K., T.W., R.P.C.; Data curation: S.L., Z.Z., Y.X., H.-D.N., R.P.C.; Writing - original draft: S.L., R.P.C.; Writing - review & editing: S.L., M.J.S., P.K., T.W., R.P.C.; Supervision: T.W., R.P.C.; Funding acquisition: T.W., R.P.C.
Funding
This work was supported by the National Institutes of Health (R56-DE024749, R01-DE024749, P30-AR050950 to R.P.C.; 1U01DE024429 to T.W.). Deposited in PMC for release after 12 months.
Data availability
The RNA-seq data have been deposited into the NCBI Gene Expression Omnibus under accession number GSE144853.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.187369.supplemental
Peer review history
The peer review history is available online at https://dev.biologists.org/lookup/doi/10.1242/dev.187369.reviewer-comments.pdf
References
- Bebee T. W., Park J. W., Sheridan K. I., Warzecha C. C., Cieply B. W., Rohacek A. M., Xing Y. and Carstens R. P. (2015). The splicing regulators Esrp1 and Esrp2 direct an epithelial splicing program essential for mammalian development. eLife 4, e08954 10.7554/eLife.08954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braeutigam C., Rago L., Rolke A., Waldmeier L., Christofori G. and Winter J. (2014). The RNA-binding protein Rbfox2: an essential regulator of EMT-driven alternative splicing and a mediator of cellular invasion. Oncogene 33, 1082-1092. 10.1038/onc.2013.50 [DOI] [PubMed] [Google Scholar]
- Carroll T. J., Park J.-S., Hayashi S., Majumdar A. and McMahon A. P. (2005). Wnt9b plays a central role in the regulation of mesenchymal to epithelial transitions underlying organogenesis of the mammalian urogenital system. Dev. Cell 9, 283-292. 10.1016/j.devcel.2005.05.016 [DOI] [PubMed] [Google Scholar]
- De Moerlooze L., Spencer-Dene B., Revest J. M., Hajihosseini M., Rosewell I. and Dickson C. (2000). An important role for the IIIb isoform of fibroblast growth factor receptor 2 (FGFR2) in mesenchymal-epithelial signalling during mouse organogenesis. Development 127, 483-492.10631169 [Google Scholar]
- Dixon M. J., Marazita M. L., Beaty T. H. and Murray J. C. (2011). Cleft lip and palate: understanding genetic and environmental influences. Nat. Rev. Genet. 12, 167-178. 10.1038/nrg2933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everson J. L., Fink D. M., Yoon J. W., Leslie E. J., Kietzman H. W., Ansen-Wilson L. J., Chung H. M., Walterhouse D. O., Marazita M. L. and Lipinski R. J. (2017). Sonic hedgehog regulation of Foxf2 promotes cranial neural crest mesenchyme proliferation and is disrupted in cleft lip morphogenesis. Development 144, 2082-2091. 10.1242/dev.149930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer-Vaquer A., Piliszek A., Tian G., Aho R. J., Dufort D. and Hadjantonakis A.-K. (2010). A sensitive and bright single-cell resolution live imaging reporter of Wnt/β-catenin signaling in the mouse. BMC Dev. Biol. 10, 121 10.1186/1471-213X-10-121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferretti E., Li B., Zewdu R., Wells V., Hebert J. M., Karner C., Anderson M. J., Williams T., Dixon J., Dixon M. J. et al. (2011). A conserved Pbx-Wnt-p63-Irf6 regulatory module controls face morphogenesis by promoting epithelial apoptosis. Dev. Cell 21, 627-641. 10.1016/j.devcel.2011.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser F. C. (1970). The genetics of cleft lip and cleft palate. Am. J. Hum. Genet. 22, 336-352. [PMC free article] [PubMed] [Google Scholar]
- Green R. M., Feng W., Phang T., Fish J. L., Li H., Spritz R. A., Marcucio R. S., Hooper J., Jamniczky H., Hallgrimsson B. et al. (2015). Tfap2a-dependent changes in mouse facial morphology result in clefting that can be ameliorated by a reduction in Fgf8 gene dosage. Dis. Model. Mech. 8, 31-43. 10.1242/dmm.017616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gritli-Linde A. (2008). The etiopathogenesis of cleft lip and cleft palate: usefulness and caveats of mouse models. Curr. Top. Dev. Biol. 84, 37-138. 10.1016/S0070-2153(08)00602-9 [DOI] [PubMed] [Google Scholar]
- Gritli-Linde A. (2012). The mouse as a developmental model for cleft lip and palate research. Front. Oral Biol. 16, 32-51. 10.1159/000337523 [DOI] [PubMed] [Google Scholar]
- Huang D. W., Sherman B. T., Tan Q., Kir J., Liu D., Bryant D., Guo Y., Stephens R., Baseler M. W., Lane H. C. et al. (2007). DAVID Bioinformatics Resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 35, W169-W175. 10.1093/nar/gkm415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang R., Bush J. O. and Lidral A. C. (2006). Development of the upper lip: morphogenetic and molecular mechanisms. Dev. Dyn. 235, 1152-1166. 10.1002/dvdy.20646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y.-R., Han X. H., Taketo M. M. and Yoon J. K. (2012). Wnt9b-dependent FGF signaling is crucial for outgrowth of the nasal and maxillary processes during upper jaw and lip development. Development 139, 1821-1830. 10.1242/dev.075796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juriloff D. M. and Harris M. J. (2008). Mouse genetic models of cleft lip with or without cleft palate. Birth Defects Res. A Clin. Mol. Teratol. 82, 63-77. 10.1002/bdra.20430 [DOI] [PubMed] [Google Scholar]
- Juriloff D. M., Harris M. J., McMahon A. P., Carroll T. J. and Lidral A. C. (2006). Wnt9b is the mutated gene involved in multifactorial nonsyndromic cleft lip with or without cleft palate in A/WySn mice, as confirmed by a genetic complementation test. Birth Defects Res. A Clin. Mol. Teratol. 76, 574-579. 10.1002/bdra.20302 [DOI] [PubMed] [Google Scholar]
- Kaartinen V., Voncken J. W., Shuler C., Warburton D., Bu D., Heisterkamp N. and Groffen J. (1995). Abnormal lung development and cleft palate in mice lacking TGF-beta 3 indicates defects of epithelial-mesenchymal interaction. Nat. Genet. 11, 415-421. 10.1038/ng1295-415 [DOI] [PubMed] [Google Scholar]
- Kalsotra A. and Cooper T. A. (2011). Functional consequences of developmentally regulated alternative splicing. Nat. Rev. Genet. 12, 715-729. 10.1038/nrg3052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouskoura T., Kozlova A., Alexiou M., Blumer S., Zouvelou V., Katsaros C., Chiquet M., Mitsiadis T. A. and Graf D. (2013). The etiology of cleft palate formation in BMP7-deficient mice. PLoS ONE 8, e59463 10.1371/journal.pone.0059463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan Y. and Jiang R. (2009). Sonic hedgehog signaling regulates reciprocal epithelial-mesenchymal interactions controlling palatal outgrowth. Development 136, 1387-1396. 10.1242/dev.028167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S., Cieply B., Yang Y., Peart N., Glaser C., Chan P. and Carstens R. P. (2018). Esrp1-regulated splicing of Arhgef11 isoforms is required for epithelial tight junction integrity. Cell Rep. 25, 2417-2430.e2415. 10.1016/j.celrep.2018.10.097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. and Williams T. (2013). Separation of mouse embryonic facial ectoderm and mesenchyme. J. Vis. Exp. 74, e50248 10.3791/50248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C., Lan Y. and Jiang R. (2017). Molecular and cellular mechanisms of palate development. J. Dent. Res. 96, 1184-1191. 10.1177/0022034517703580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Jones K. L., Hooper J. E. and Williams T. (2019). The molecular anatomy of mammalian upper lip and primary palate fusion at single cell resolution. Development 146, dev174888 10.1242/dev.174888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski R. J., Song C., Sulik K. K., Everson J. L., Gipp J. J., Yan D., Bushman W. and Rowland I. J. (2010). Cleft lip and palate results from Hedgehog signaling antagonism in the mouse: phenotypic characterization and clinical implications. Birth Defects Res. A Clin. Mol. Teratol. 88, 232-240. 10.1002/bdra.20656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W., Sun X., Braut A., Mishina Y., Behringer R. R., Mina M. and Martin J. F. (2005). Distinct functions for Bmp signaling in lip and palate fusion in mice. Development 132, 1453-1461. 10.1242/dev.01676 [DOI] [PubMed] [Google Scholar]
- Mager L. F., Koelzer V. H., Stuber R., Thoo L., Keller I., Koeck I., Langenegger M., Simillion C., Pfister S. P., Faderl M. et al. (2017). The ESRP1-GPR137 axis contributes to intestinal pathogenesis. eLife 6, e28366 10.7554/eLife.28366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuylov N. L., Smagulova F. O., Leach L. and Tevosian S. G. (2008). Ovarian development in mice requires the GATA4-FOG2 transcription complex. Development 135, 3731-3743. 10.1242/dev.024653 [DOI] [PubMed] [Google Scholar]
- Moorman A. F. M., Houweling A. C., de Boer P. A. J. and Christoffels V. M. (2001). Sensitive nonradioactive detection of mRNA in tissue sections: novel application of the whole-mount in situ hybridization protocol. J. Histochem. Cytochem. 49, 1-8. 10.1177/002215540104900101 [DOI] [PubMed] [Google Scholar]
- Osei-Sarfo K. and Gudas L. J. (2014). Retinoic acid suppresses the canonical Wnt signaling pathway in embryonic stem cells and activates the noncanonical Wnt signaling pathway. Stem Cells 32, 2061-2071. 10.1002/stem.1706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillman K. A., Phillips C. A., Roslan S., Toubia J., Dredge B. K., Bert A. G., Lumb R., Neumann D. P., Li X., Conn S. J. et al. (2018). miR-200/375 control epithelial plasticity-associated alternative splicing by repressing the RNA-binding protein Quaking. EMBO J. 37, e99016 10.15252/embj.201899016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proetzel G., Pawlowski S. A., Wiles M. V., Yin M., Boivin G. P., Howles P. N., Ding J., Ferguson M. W. J. and Doetschman T. (1995). Transforming growth factor-β3 is required for secondary palate fusion. Nat. Genet. 11, 409-414. 10.1038/ng1295-409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray H. J. and Niswander L. (2012). Mechanisms of tissue fusion during development. Development 139, 1701-1711. 10.1242/dev.068338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid B. S., Yang H., Melvin V. S., Taketo M. M. and Williams T. (2011). Ectodermal Wnt/β-catenin signaling shapes the mouse face. Dev. Biol. 349, 261-269. 10.1016/j.ydbio.2010.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revil T. and Jerome-Majewska L. A. (2013). During embryogenesis, esrp1 expression is restricted to a subset of epithelial cells and is associated with splicing of a number of developmentally important genes. Dev. Dyn. 242, 281-290. 10.1002/dvdy.23918 [DOI] [PubMed] [Google Scholar]
- Reynolds K., Kumari P., Sepulveda Rincon L., Gu R., Ji Y., Kumar S. and Zhou C. J. (2019). Wnt signaling in orofacial clefts: crosstalk, pathogenesis and models. Dis. Model. Mech. 12, dmm037051 10.1242/dmm.037051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice R., Spencer-Dene B., Connor E. C., Gritli-Linde A., McMahon A. P., Dickson C., Thesleff I. and Rice D. P. C. (2004). Disruption of Fgf10/Fgfr2b-coordinated epithelial-mesenchymal interactions causes cleft palate. J. Clin. Invest. 113, 1692-1700. 10.1172/JCI20384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohacek A. M., Bebee T. W., Tilton R. K., Radens C. M., McDermott-Roe C., Peart N., Kaur M., Zaykaner M., Cieply B., Musunuru K. et al. (2017). ESRP1 mutations cause hearing loss due to defects in alternative splicing that disrupt cochlear development. Dev. Cell 43, 318-331.e315. 10.1016/j.devcel.2017.09.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanford L. P., Ormsby I., Gittenberger-de Groot A. C., Sariola H., Friedman R., Boivin G. P., Cardell E. L. and Doetschman T. (1997). TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development 124, 2659-2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro I. M., Cheng A. W., Flytzanis N. C., Balsamo M., Condeelis J. S., Oktay M. H., Burge C. B. and Gertler F. B. (2011). An EMT-driven alternative splicing program occurs in human breast cancer and modulates cellular phenotype. PLoS Genet. 7, e1002218 10.1371/journal.pgen.1002218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S., Park J. W., Lu Z.-X., Lin L., Henry M. D., Wu Y. N., Zhou Q. and Xing Y. (2014). rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. USA 111, E5593-E5601. 10.1073/pnas.1419161111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L., Li Y., Wang K., Wang Y.-Z., Molotkov A., Gao L., Zhao T., Yamagami T., Wang Y., Gan Q. et al. (2009). Lrp6-mediated canonical Wnt signaling is required for lip formation and fusion. Development 136, 3161-3171. 10.1242/dev.037440 [DOI] [PubMed] [Google Scholar]
- Ule J., Ule A., Spencer J., Williams A., Hu J.-S., Cline M., Wang H., Clark T., Fraser C., Ruggiu M. et al. (2005). Nova regulates brain-specific splicing to shape the synapse. Nat. Genet. 37, 844-852. 10.1038/ng1610 [DOI] [PubMed] [Google Scholar]
- Venables J. P., Brosseau J.-P., Gadea G., Klinck R., Prinos P., Beaulieu J.-F., Lapointe E., Durand M., Thibault P., Tremblay K. et al. (2013). RBFOX2 is an important regulator of mesenchymal tissue-specific splicing in both normal and cancer tissues. Mol. Cell. Biol. 33, 396-405. 10.1128/MCB.01174-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warzecha C. C., Sato T. K., Nabet B., Hogenesch J. B. and Carstens R. P. (2009a). ESRP1 and ESRP2 are epithelial cell-type-specific regulators of FGFR2 splicing. Mol. Cell 33, 591-601. 10.1016/j.molcel.2009.01.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warzecha C. C., Shen S., Xing Y. and Carstens R. P. (2009b). The epithelial splicing factors ESRP1 and ESRP2 positively and negatively regulate diverse types of alternative splicing events. RNA Biol. 6, 546-562. 10.4161/rna.6.5.9606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedden S. E. (1987). Epithelial-mesenchymal interactions in the development of chick facial primordia and the target of retinoid action. Development 99, 341-351. [DOI] [PubMed] [Google Scholar]
- Williamson K. A., Rainger J., Floyd J. A. B., Ansari M., Meynert A., Aldridge K. V., Rainger J. K., Anderson C. A., Moore A. T., Hurles M. E. et al. (2014). Heterozygous loss-of-function mutations in YAP1 cause both isolated and syndromic optic fissure closure defects. Am. J. Hum. Genet. 94, 295-302. 10.1016/j.ajhg.2014.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Park J. W., Bebee T. W., Warzecha C. C., Guo Y., Shang X., Xing Y. and Carstens R. P. (2016). Determination of a comprehensive alternative splicing regulatory network and combinatorial regulation by key factors during the epithelial-to-mesenchymal transition. Mol. Cell. Biol. 36, 1704-1719. 10.1128/MCB.00019-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Ibrahimi O. A., Olsen S. K., Umemori H., Mohammadi M. and Ornitz D. M. (2006). Receptor specificity of the fibroblast growth factor family. The complete mammalian FGF family. J. Biol. Chem. 281, 15694-15700. 10.1074/jbc.M601252200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C., Zhang Z., Castle J., Sun S., Johnson J., Krainer A. R. and Zhang M. Q. (2008). Defining the regulatory network of the tissue-specific splicing factors Fox-1 and Fox-2. Genes Dev. 22, 2550-2563. 10.1101/gad.1703108 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.