

Abstract
CRISPR-Cas9 RNA-guided nucleases are a transformative technology for biology, genetics, and medicine due to the simplicity with which they can be programmed to cleave specific DNA target sites in living cells and organisms. However, to translate these powerful molecular tools into safe, effective clinical applications, it will be of critical importance to carefully define and improve their genome-wide specificities. Here we outline our state-of-the-art understanding of target DNA recognition and cleavage by CRISPR-Cas9 nucleases, methods to determine and improve their specificities, and key considerations for how to evaluate and reduce off-target x effects for research and therapeutic applications.
Introduction
The transformative capability to modify the mammalian genome by homologous recombination1,2 in mouse embryonic stem cells3 was first developed in the laboratories of Mario Capecchi, Martin Evans, and Oliver Smithies for which they were awarded the Nobel Prize in Physiology or Medicine for 20074. Although gene targeting in mouse embryonic stem cells can be successfully achieved with the use of drug-selectable genetic markers, the absolute rates of these homology-directed repair (HDR) events remains low. This limitation contributed to the more restricted use of gene targeting for other cell types or organisms and for therapeutic applications.
The discovery that nuclease-induced targeted DNA double-stranded breaks (DSBs) could stimulate gene targeting by HDR or the formation of variable-length insertions or deletions (indels) by non-homologous end-joining (NHEJ) repair in mammalian cells5 marked a second inflection point in the advancement of genome-modifying capabilities. NHEJ-induced indels can efficiently disrupt genes or genetic elements; by contrast, with a user-supplied homologous ‘donor’ template, HDR can be used to create precise alterations such as point mutations or insertions (Box 1). However, the challenge for many years remained how to create the site-specific DSB required to initiate DNA repair events needed to effect targeted genome editing by NHEJ or HDR.
Box 1. Cas9 target recognition, binding, and cleavage.
Streptococcus pyogenes Cas9 nuclease has been engineered to require only two components: Cas9 protein and a short ~100 nucleotide guide RNA (gRNA) that together form a complex that can recognize and cleave a 20 bp dsDNA target site (protospacer) that is complementary to the 5 end of the gRNA and is adjacent to a protospacer adjacent motif (PAM) of the form NGG (where N can be any nucleotide) (see the figure, part a). The single gRNA transcript is an engineered fusion of naturally occurring CRISPR RNA (crRNA) and transactivating crRNA (tracrRNA)17. The tracrRNA was originally discovered by differential RNA sequencing and found to be an essential component for CRISPR interference in S. pyogenes bacteria92. The target specificity of Cas9 is mediated by nucleic acid interactions between the 20 nucleotides at the 5 end of the gRNA and the protospacer DNA, as well as by protein–DNA interactions between Cas9 protein and the PAM. Upon recognition of a PAM sequence, Cas9 initiates sequential unwinding of the protospacer target site duplex, stabilized by the formation of a triplex R-loop structure between the protospacer DNA and the gRNA82. Sufficient RNA:DNA complementarity between the gRNA and the target DNA strand triggers a conformational change in Cas9 that activates concerted cleavage of the target DNA strand by Cas9’s HNH domain, and of the non-target strand by its RUVC domain87. In vitro, Cas9 nucleases can produce either blunt or 1 bp 5 staggered ends17,71. In mammalian cells, Cas9 nuclease-induced DSBs can be repaired by one of two competing DNA repair pathways: error-prone non-homologous end-joining (NHEJ) resulting in insertions or deletions (indels) that are often exploited to create frame-shift or ‘knock-out’ mutations; or precise homology-directed repair (HDR) which in the presence of a user-supplied donor template is often used for gene correction or ‘knock-in’ (see the figure, part b)
The results of multiple studies strongly suggest that the cleavage specificity of Cas9 nuclease differs from the binding site specificity of catalytically inactive dCas9. For example, chromatin immunoprecipitation followed by sequencing (ChIP-seq) has been used to identify DNA sites bound genome-wide by catalytically inactive Cas9 (dCas9) in human and mouse cells. Analysis of off-target binding sites detected by ChIP-seq have shown that very few of these are cleaved or mutagenized by catalytically active Cas993–95, consistent with the proposed mechanism that more extensive pairing of the gRNA mediates a conformational change that enables Cas9 cleavage93,94. This conformational gating mechanism may in part explain why the extent of protospacer complementarity required is different for efficient cleavage by wild-type Cas9 nuclease (≥17 bp) versus transcriptional activation by dCas9–VP64 fusion proteins (≥12 bp).
Over the past two decades, four major classes of engineered nucleases have been used for genome editing: meganucleases6, zinc-finger nucleases (ZFNs)7, transcription activator-like effector nucleases (TALENs)8, and clustered regularly interspaced short palindromic repeats (CRISPR)-CRISPR associated (Cas) nucleases8–10. Meganucleases are endonucleases that can recognize extended DNA sequences of 14–40 bp through extensive non-modular protein–DNA contacts, but whose target specificities can be difficult to re-engineer6. ZFNs11 and TALENs12,13 are fusions between arrays of ZF or TALE DNA-binding domains and the non-specific, dimerization-dependent FokI nuclease domain14,15. Fusions of meganucleases to transcription activator-like effector arrays (Mega-TALs)16 have also been used to induce DSBs and genome editing. All of these various classes of genome-editing nucleases rely on exclusively on protein–DNA interactions to mediate site-specific recognition of genomic DNA sequence.
By contrast, CRISPR-Cas9 nucleases are RNA-guided endonucleases originally discovered in bacteria that have been broadly and enthusiastically adopted by the scientific community for genome editing in diverse cells and organisms, largely because of the ease with which their target specificity can be reprogrammed. These nucleases consist of two components: a Cas9 protein complexed with a ~100 nucleotide guide RNA (gRNA; often termed single guide RNA (sgRNA) to denote that it is an engineered fusion of naturally occurring bacterial CRISPR RNA (crRNA) and transactivating crRNA (tracrRNA)17). CRISPR–Cas9 nucleases can be engineered to recognize a target DNA site consisting of a protospacer and a protospacer adjacent motif (PAM) sequence17 (Box 1). The sequence-specific recognition of the protospacer DNA is mediated by base-pairing interactions with the gRNA, while recognition of the PAM is achieved by Cas9-mediated protein–DNA interactions (Box 1). Thus, target site specificity can be readily reprogrammed by simply changing the sequence composition of the 5 end of the gRNA. Cas9 nucleases from a number of different bacterial species including Streptococcus pyogenes (SpCas9), Neisseria meningitidis (NmCas9), and Staphylococcus aureus (SaCas9) have all been shown to be robust for inducing efficient genome editing18–23, with SpCas9 being most widely used orthologue to date.
An important challenge for the use of engineered CRISPR–Cas9 for both research and therapeutic applications is the need to identify and minimize off-target mutations induced by these nucleases. A number of novel strategies to both define and improve the genome-wide specificities of CRISPR-Cas9 nucleases have been published over the past few years. In this Review, we first describe the various recently developed approaches for defining nuclease specificity and their associated advantages and limitations. Next, we summarize insights gained from recent studies of Cas9 specificity as well as strategies for improving the genome-wide specificity of Cas9, placing them both in the context of structural and mechanistic studies of Cas9 target site recognition. Finally, we discuss the implications of our current understanding of specificity for both research and therapeutic applications of Cas9. Because broad recent interest has catalyzed a number of studies defining the genome-wide specificity of CRISPR-Cas9 nucleases, the Review focuses on this class of engineered nucleases in particular and not on other genome editing tools (a number of related recent reviews may also be of interest24–30).
Defining CRISPR–Cas9 off-target effects
Although an ideal engineered nuclease would have singular genome-wide specificity, in practice CRISPR–Cas9 has been shown to exhibit off-target cleavage events31–39. Here we discuss early efforts to characterize the specificity of CRISPR-Cas9 nucleases and the subsequent development of more comprehensive and unbiased methods to characterize genome-wide off-target cleavage effects.
Initial identification of off-target effects.
Following the demonstrations that SpCas9 could be simply and robustly programmed to cleave target sites not only in vitro17,40, but also used for efficient genome editing in bacteria17, human cells18–20,41 and one-cell embryos of a whole organism (the zebrafish)21, enthusiasm quickly grew for both research and translational applications of this platform. However, we and others wondered early on whether the RNA-guided nature of this nuclease might have greater potential for off-target effects, especially relative to ZFNs and TALENs, which recognize their target sites using protein–DNA interactions.
To determine whether SpCas9 off-target mutagenesis might occur with any given gRNA, one method that we and others used in early studies was in silico prediction of potential off-target cleavage sites based on similarity to the intended target site followed by targeted experimental asessment of characteristic NHEJ-induced indel mutations at those genomic locations35,36. In one study from our group, potential off-target cleavage sites with 5 or fewer mismatches relative to the intended target sequence were identified, with at least one of those mismatches in the PAM-distal portion of the potential off-target cleavage site35(Fig. 1a). Another study computationally searched for and tested sites with RNA or DNA ‘bulges’ at the RNA:DNA interface38. The main conclusions from these and other initial studies of SpCas9 specificity were that: high-frequency mutagenesis is possible at mismatched sites35,36; in some cases, off-target sites were mutagenized at frequencies comparable to or higher than what was observed at the intended on-target site35; sites with a non-canonical PAM sequence could also be cleaved and mutagenized (e.g., NAG PAMs compared with the canonical NGG PAMs)36; off-target cleavage could be detected at sites with up to 5 mismatches relative to the intended target sequence34–36; and target sites with small 1 bp bulge insertions or deletions in the DNA target strand relative to the gRNA sequence could also be mutated38. Based on the results of studies such as these, some groups developed online tools designed to predict potential off-target sites based on their degree of similarity to the on-target site and other parameters such as the location of mismatches within the protospacer sequences. Examples of such publicly available, web-based tools include the CRISPR Design Tool36 and E-CRISP42.
Figure 1 |. Targeted methods for defining off-target cleavage effects.
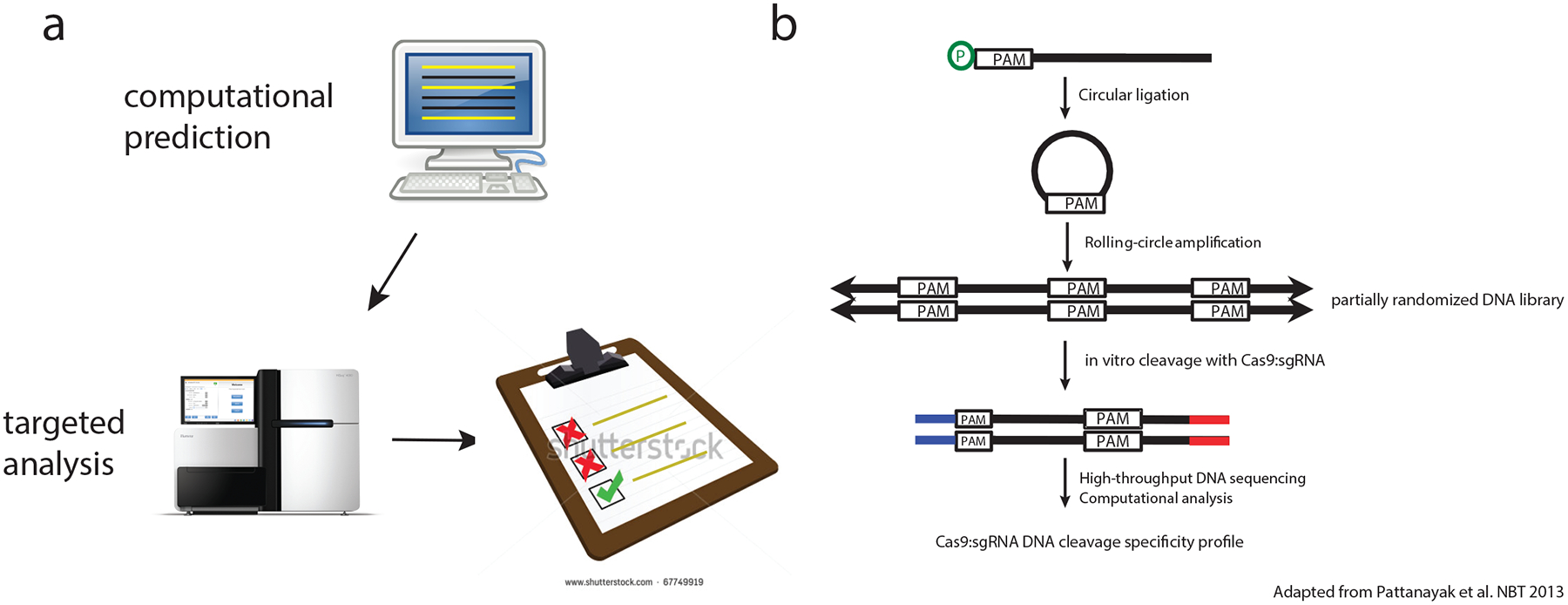
a An early method for identifying off-target effects was computational prediction off off-target sites followed by targeted analysis by mismatch cleavage assay like T7E1 or high-throughput sequencing. The limitation of these approaches are that they are biased by the assumptions made by computational predictions about the sequence features at off-target sites: to narrow the scope of the sites examined, only in silico-predicted sites are interrogated, thus leaving additional genuine off-target sites undetected. b In vitro site selection of partially randomized libraries. Concatameric libraries are generated by rolling circle amplification of circularized oligonucleotide templates. Cleavage of libraries results in members with newly available ends compatible for ligation with adapters (blue and red) for high-throughput sequencing. Part b is adapted from reference34.
At the same time these in silico-directed approaches were being tested, another group performed in vitro interrogation of partially randomized target site libraries34, a method which was first developed to characterize the specificity of ZFNs43 (and, later, TALENs as well44). This approach is based on the circularization of partially degenerate oligonucleotide libraries that are biased to resemble the intended gRNA target sequence, followed by rolling circle amplification, in vitro cleavage by SpCas9, and ligation of adapters to cleaved sites with newly available ends followed by high-throughput sequencing (Fig 1b). The main advantage of using in vitro selection is that large and diverse libraries of sites similar in sequence to the on-target site can be interrogated to define broad cleavage characteristics of a particular SpCas9–gRNA complex. One disadvantage is that the majority of the randomized target sites cleaved do not actually occur in any given genome of interest, leaving open the question of which specific sites are actually cleaved in cells to be edited. Previous studies showed that this limitation can be at least partially addressed by using machine learning algorithms to extract general characteristics of nuclease specificity and to predict and rank the likelihood of cleaving mismatched genomic sites in human cells, a strategy successfully employed with in vitro site selection data characterizing the specificity of ZFNs45.
Although useful for demonstrating that SpCas9 had the potential for inducing off-target mutations, these early studies were not genome-wide in their scope and were not designed to identify sites in an unbiased manner free of assumptions about the sequences of such sites. Bona fide sites that did not fit the computational criteria might never be examined nor discovered. Thus, while useful for establishing that Cas9 off-target mutagenesis was possible, the combined in silico prediction and targeted sequencing approach has fundamental limitations for achieving comprehensive off-target discovery. Even as these initial studies were being published, we and others in the field believed that a more ideal method would be one that identified sites of off-target mutagenesis in a genome-wide unbiased fashion and with high sensitivity (i.e., the ability to detect even low-frequency mutations).
Genome-wide assays for defining off-target cleavage.
A number of methods that enable genome-wide assessment of Cas9 nuclease off-target cleavage effects have recently been described and these can be divided into two broad classes: cell-based and cell-free (in vitro), with each having their respective advantages and limitations.
Cell-based genome-wide assays.
Whole-genome sequencing (WGS) has been proposed as an unbiased method for defining engineered nuclease specificity. Although this method is potentially useful for the analysis of single-cell clones46,47 or non-mosaic F1 animals48 that have been modified by genome editing, it lacks sensitivity as a method for comprehensively defining off-target sites, particularly those that occur with low frequencies in a population of cells49. With existing high-throughput sequencing technologies, it remains impractical to perform WGS on millions (let alone billions) of cellular genomes. At standard WGS depths of 30–50× read coverage, WGS performed on large heterogenous genome-edited cell populations would be expected to be insensitive for detection of all but the highest frequency off-target effects and inadequate for analysis of most currently envisioned genome-editing-based therapeutic strategies49.
Integrase-defective lentiviral vector (IDLV) capture was the first genome-wide approach used to evaluate the specificities of genome-editing nucleases. It was initially applied to engineered ZFNs50 and then later adapted to analyze the specificity of TALENs, CRISPR–Cas9, and mega-TALs51,52. This method is based on capture of IDLVs, which have linear dsDNA genomes, into sites of nuclease-induced DSBs by NHEJ (Fig. 2a). Clustered sites of integrations are recovered by ligation-adapter-mediated-PCR (LAM-PCR) and then mapped using high-throughput sequencing. The advantage of the IDLV capture method is that it can directly identify DSBs that occur in living cells. However, the method has some limitations: it is relatively insensitive due to low absolute integration efficiencies that require positive selection to overcome50; and it has a high background, because IDLVs still retain some capability to randomly integrate into cellular genomes even in the absence of nuclease-induced DSBs50. In addition, because IDLV integration events occur near but not precisely at the site of the nuclease-induced break, it can be challenging to confidently map the exact recognition sequence, particularly for lower-frequency off-target events.
Figure 2 |. Genome-wide methods for defining off-target cleavage effects.
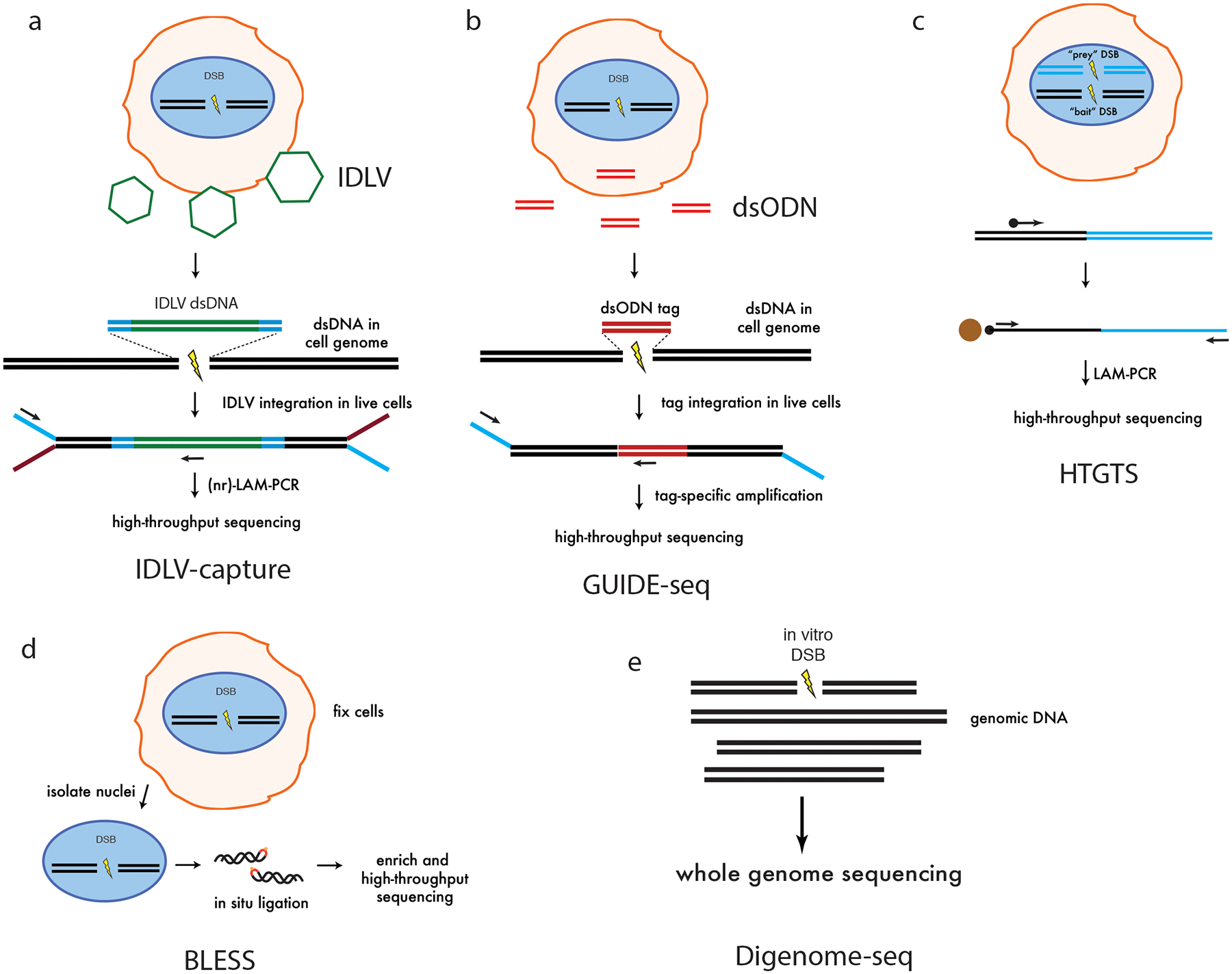
a | IDLV capture. Integration defective lentiviruses (IDLVs; green) are integrated with a selectable marker into sites of nuclease-induced double-stranded breaks (DSBs) in living cells. Integration sites are recovered by ligation-adapter-mediated LAM-PCR, followed by high-throughput sequencing50,52. b | Genome-wide unbiased identification of DSBs enabled by sequencing (GUIDE-seq53). An end-protected short double-stranded oligodeoxynucleotide (dsODN) is efficiently integrated into sites of nuclease-induced DSBs in living cells. This short sequence is used for tag-specific amplification followed by high-throughput sequencing to identify off-target cleavage sites. c | High-throughput genome-wide translocation sequencing (HTGTS56). Two nucleases are expressed in a cell, to generate a ‘prey’ and ‘bait’ DSB. Using a biotinylated primer designed against the bait DSB junction, translocations between ‘prey’ and ‘bait’ are recovered by LAM-PCR for high-throughput equencing. Off-target cleavage sites are identified by analysis of these translocation junctions. d | Breaks labeling, enrichment on streptavidin and next-generation sequencing (BLESS22,57). Nuclease-treated cells are fixed, intact nuclei are isolated, permeabilized, and sequencing adapters are ligated in situ to transient nuclease-induced DSBs. Adapter-ligated fragments are enriched and amplified for high-throughput sequencing. Part d is adapted from reference22. e | Digenome-seq. Genomic DNA is isolated from cells and treated with Cas9 nuclease in vitro. Sequencing adapters are ligated and high-throughput sequencing is performed at standard whole-genome sequencing coverage58.
Genome-wide unbiased identification of DSBs enabled by sequencing (GUIDE-seq) is an assay developed by our group for the sensitive detection of Cas9 off-target cleavage in living cells53. GUIDE-seq is based on the efficient integration of a blunt, end-protected, double-stranded oligodeoxynucleotide (dsODN) tag, followed by tag-specific amplification and high-throughput sequencing. Tag-amplified reads are mapped to a reference genome, and off-target DSB sites located in genomic windows with characteristic bi-directional mapping read signatures are identified (Fig. 2b). GUIDE-seq is highly sensitive and can detect off-target sites that are mutagenized by Cas9–gRNAs with frequencies of 0.1% or lower in a population of cells, even with as few as several million sequencing reads. Importantly, the majority of empirically detected, bona fide Cas9 off-target cleavage sites identified by GUIDE-seq were not predicted by existing web-based computational prediction tools36,42, largely because these algorithms do not consider off-target sites with more than 3 or 4 mismatches. Advantages of GUIDE-seq include its experimental simplicity, the high efficiency and precision with which dsODNs can be captured into DSBs, the quantitative correlation between numbers of GUIDE-seq read counts at a given site with the frequencies of NHEJ-induced mutations in living cells, the detection of repair outcomes of nuclease-induced DSBs in a physiologically relevant cellular context, cumulative detection of tag integration events over time, and the availability of open-source analytical software for downstream bioinformatic analysis54. However, one limitation of GUIDE-seq is the requirement for efficient cellular transfection of the short dsODN tag, which makes it more challenging to use for cell types that cannot be efficiently transfected or for in vivo settings.
High-throughput genome-wide translocation sequencing (HTGTS)55 is another genome-wide method that has been used to identify Cas9 off-target cleavage in live cells56. HTGTS is based on the detection of translocations between a nuclease-induced ‘bait’ DSB and off-target ‘prey’ DSBs (Fig. 2c). The ‘universal donor bait’ that has been used in published studies to date is the on-target site of a previously validated RAG1B gRNA. HTGTS has the advantage that it can, in principle, be applied to analyze the effects of nucleases delivered in vivo because it does not require the introduction of any additional components beyond active Cas9–gRNA nuclease complex. Some limitations of HTGTS are that nuclease-induced translocations represent very rare events that require large numbers of input genomes for detection, that translocations occur more frequently with sites on the same chromosome or chromosomes that are in close nuclear proximity (thereby biasing detection against more distant DSBs), and that estimates of off-target effects may be influenced by positive or negative biological effects of specific translocation products on cells.
Breaks labeling, enrichment on streptavidin and next-generation sequencing (BLESS) is a method for detecting genome-wide nuclease-induced DSBs in fixed cells57. BLESS captures a snapshot of transient DSBs that may exist at a moment in time in a population of cells by direct in situ ligation of a biotinylated hairpin adapter in fixed and permeabalized cell nuclei (Fig. 2d). Advantages of BLESS are that it has been applied to detect DSBs from tissues to which Cas9 nuclease has been delivered in vivo22 and that it does not depend on the endogenous cellular DNA damage repair machinery for detection. Some limitations of BLESS are that it can only capture DSBs present at a specific moment in time; it cannot detect DSBs already cleaved and mutagenized before cells are permeabilized; it also detects background DSBs that can be introduced during fixation or handling; it requires ~10 million cells and the method can be challenging to perform because of the number of technical and specialized protocol steps.
In vitro genome-wide assays.
Digested genome sequencing (Digenome-seq) is an in vitro method for detection of nuclease-induced DSBs in genomic DNA using WGS of Cas9-cleaved genomic DNA58. This method is performed by digesting genomic DNA purified from cells of interest with purified Cas9–gRNA ribonucleoprotein (RNP) complexes in vitro under conditions designed to maximize off-target cleavage. Genomic DNA fragments are then sequenced to high coverage with approximately 500 million reads. Cas9-induced cleavage sites are then identified as sites with a relative enrichment of reads possessing the same start or end mapping positions, in contrast to random DNA breaks created by shearing that occurs during genomic DNA isolation and purification (Fig. 2e). Because this assay is performed in vitro on purified DNA, it is presumably not limited by cell-based factors such as chromatin context, epigenetic factors, subnuclear localization, or fitness effects; in addition, the ability to increase RNP complex concentration to very high levels may better enable detection of even very weakly cleaved sites, thereby enabling detection of additional off-target cleavage at sequences that may otherwise not be found by cell-based methods. One major limitation of Digenome-seq is that because high-throughput sequencing is performed without any enrichment for cleaved sequences, nearly all of the sequencing bandwidth is expended on background reads that are not associated with nuclease-induced DSBs. In all published experiments to date, the HiSeq X10 platform has been used to perform Digenome-seq although a recent report demonstrated that it was possible to multiplex several samples in a single run59. In addition to being sequencing-inefficient, the high background of uninformative reads reduces detectable signal because in WGS by chance many sites will have some reads with uniform ends.
General characteristics of off-target cleavage sites.
Published studies using the genome-wide methods described above have yielded a number of important insights into the nature of Cas9-induced off-target cleavage events. Interestingly, our GUIDE-seq study confirmed an earlier observation that some off-target sites were mutagenized at frequencies either comparable to or higher than what is observed even at the intended on-target site when measured by targeted amplification and/or T7E1 mismatch cleavage assay35,53. Genome-wide profiling studies of Cas9 off-target cleavage by GUIDE-seq, HTGTS, IDLV capture, and Digenome-seq all identified sites with up to six mismatches in the protospacer and/or non-canonical PAM sequences52,53,56,58, confirming the assumption that many in the field had made that off-target mutations occur at sites related in sequence to the on-target site and not simply at random genomic sites. Analysis of our GUIDE-seq data also found that off-target mismatches resulting in rU:dG or rG:dT ‘wobble’ base pairing are generally better tolerated than other types of mismatches53. Evidence for off-target cleavage at sites with bulges at the RNA:DNA interface23 has also been confirmed by genome-wide methods such as GUIDE-seq38,53 and Digenome-seq58. Notably, multiple studies have now confirmed that nuclease-induced off-target DSBs can participate in translocations with the on-target site, other off-target sites, or even background genomic DSB hotspots53,56; however, it is worth emphasizing that these events occur with very low frequencies.
Limitations of current off-target prediction algorithms.
As noted above, it it not generally helpful to use computational tools to predict SpCas9 off-target cleavage, in part because of the number of mismatches that can potentially be tolerated in off-target cleavage sites. For example, any given SpCas9 target site will typically have 10,000 or more sites in the human genome that differ by 6 or fewer mismatches just by chance. Existing tools do not accurately predict which of these sites are cleaved and which are not53. In addition, some off-target prediction tools were trained on the basis of mutagenesis frequencies of systematically mismatched gRNAs against a constant target sequence, and these training datasets may be noisy because of potentially confounding effects of differing gRNA expression levels and the loading efficiency of gRNA onto Cas9. Furthermore, of the various published off-target prediction algorithms, none have been prospectively assessed for predictive accuracy with large-scale tests.
Reducing CRISPR–Cas9 off-target effects
Methods for reducing off-target effects.
To date, two general strategies have been proposed to reduce engineered nuclease off-target effects: increasing the specificity of nuclease-mediated target site cleavage (Fig 3; Table 1), or limiting the duration of nuclease expression to minimize the opportunity to accumulate off-target mutations. Additionally, molecular studies are providing mechanistic inights into target recognition and cleavage by Cas9–gRNA complexes, and such understanding could provide further opportunities for rational improvements to specificity.
Figure 3 |. Methods for improving specificity.
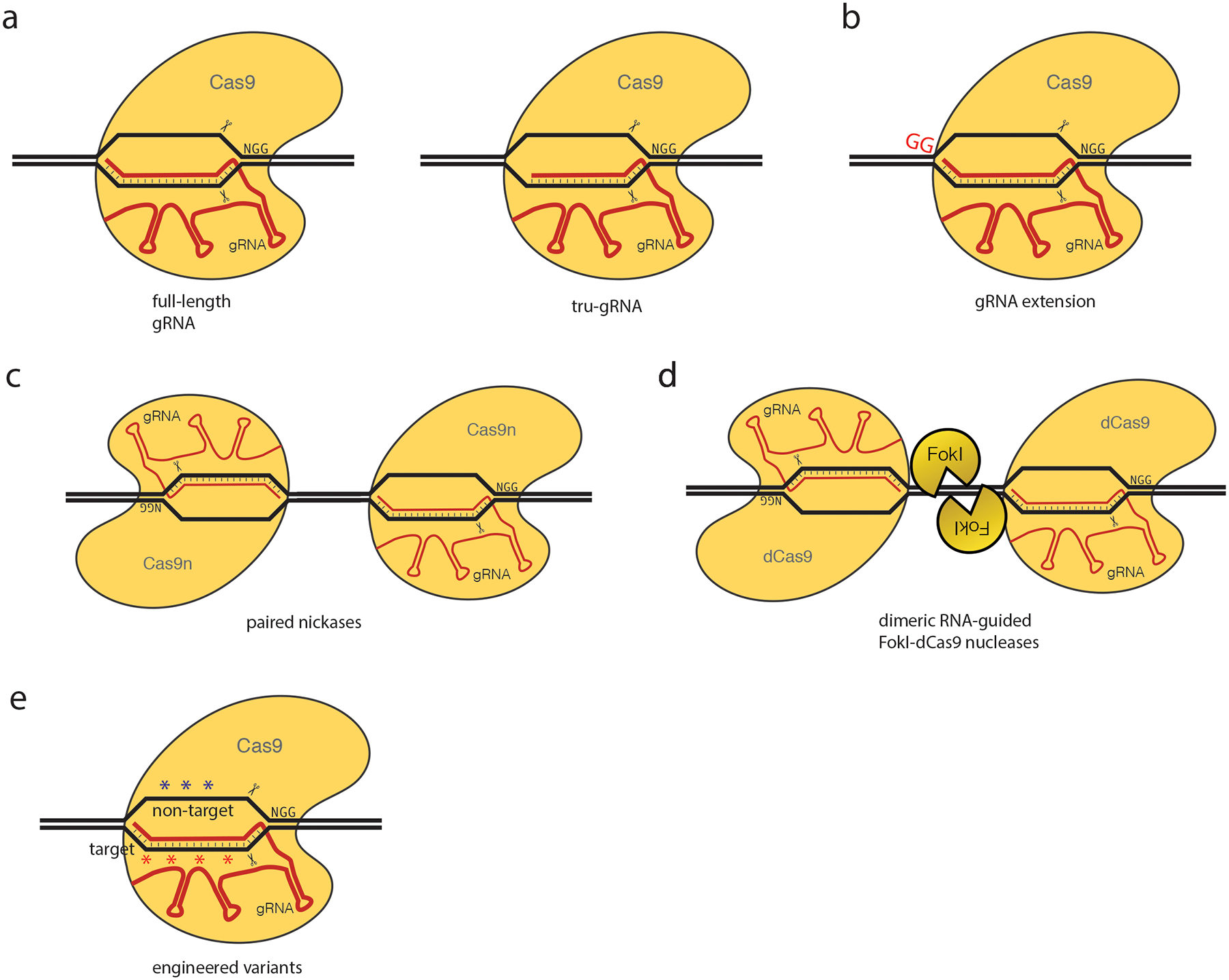
a | Truncated guide RNAs (tru-gRNAs). Cas9 is directed by gRNAs that are truncated by 2–3 nucleotides on the 5 end60. b | gRNA extensions. Cas9 is directed by a gRNA with 2 additional G nucleotides appended to the 5 end32. c | Paired nickases. One of the 2 nuclease domains of Cas9 is catalytically inactivated to make an enzymatically active nickase. Co-localization of a pair of nickases oriented in a ‘PAM-out’ orientation, with each nickase independently nicking one strand, results in efficient DSBs61,62. Part c is adapted from reference61. d | Dimeric RNA-guided FokI-dCas9 nucleases (RFNs). Catalytically inactivated Cas9 (dCas9) is fused to dimerization-dependent FokI non-specific nuclease domain. A pair of FokI-dCas9 monomers oriented in a ‘PAM-out’ orientation mediates efficient DSBs63,64. Part d is adapted from reference63. e | Engineered Cas9 variants. Cas9 variants are engineered with reduced non-specific DNA interations with the target (SpCas9-HF168) or non-target (eSpCas9 1.169) strands. For further details see the main text and Table 1.
Table 1 |.
Comparison of strategies for improving CRISPR–Cas9 specificity
| Strategy | Description | Specificity Characterization |
|---|---|---|
| Truncated guide RNAs (tru-gRNAs)60 (Fig 3a) | Truncate gRNA at the 5 end by 2–3 nucleotides; may reduce excess interaction energy at RNA:DNA interface | Targeted high-throughput sequencing and GUIDE-seq; number of genome-wide off-target sites is reduced; most off-target sites detected have 2 or fewer mismatches compared to intended target site53 |
| Extended gRNAs32 (Fig 3b) | Add 2 G nucleotides to the 5 end of the gRNA. The mechanism of increased specificity is unclear, but may involve stabilization of protein interactions with the 5’ end of the gRNA | Targeted high-throughput sequencing; reduction in off-target effects observed with certain gRNAs; in some cases, on-target activity is also reduced32,58 |
| Paired nickases61,62 (Fig 3c) | Co-localization of paired Cas9 nickases. The requirement for two proximal single-strand breaks (nicks) on opposite DNA strands of the target site (guided by distinct gRNAs) is thought to limit the propensity for off-target nicks of either gRNA-nickase to result in DSBs | Targeted sequencing and HTGTS; number of off-target sites detected is generally reduced; monomeric activity is low but certain sites retain high mutagenesis frequencies32,60,63,61 |
| Dimeric RNA-guided FokI-dCas9 nucleases (RFNs)63,64 (Fig 3d) | Fusion of catalytically inactive Cas9 (dCas9) to dimerization-dependent non-specific FokI nuclease. Similar to paired nickases, generation of DSBs typically requires binding of a pair of separately targeted monomers on opposite DNA strands at the target site, but unlike paired nickases, unpaired monomers are expected to be catalytically inactive because FokI dimerization is required for activity | Targeted high-throughput sequencing of known monomeric and predicted dimeric sites; background or near-background off-target activity detected by high-throughput sequencing; has not yet been fully characterized by genome-wide methods63,64,65 |
| Engineered Cas9 variants (SpCas9-HF1 or eSpCas9 1.1)68,69 (Fig 3e) | Reduce Cas9 non-specific DNA interactions with target (SpCas9-HF1) or non-target strand (eSpCas9 1.1) | Targeted high-throughput sequencing, GUIDE-seq or BLESS; number of detectable genome-wide off-target sites is reduced or eliminated; at certain off-target sites high-frequency mutagenesis remains possible68,69 |
Table Abbreviations
BLESS, breaks labeling, enrichment on streptavidin and next-generation sequencing;
Cas9, CRISPR-associated 9;
GUIDE-seq, genome-wide unbiased identification of DSBs enabled by sequencing;
HTGTS, high-throughput genome-wide translocation sequencing.
SpCas9-HF1, Streptococcus pyogenes Cas9 high-fidelity variant 1
eSpCas9 1.1, enhanced Streptococcus pyogenes Cas9 version 1.1
Increasing CRISPR–Cas9 specificity.
One somewhat counterintuitive method for increasing the cleavage specificity of Cas9 is to use truncated gRNAs (tru-gRNAs), which are shortened by 2–3 nucleotides at their 5 ends (i.e., in the region of RNA:DNA complementarity furthest away from the PAM) (Fig. 3a). Targeted sequencing comparing Cas9 directed by full-length gRNAs versus tru-gRNAs revealed that this strategy reduced nuclease-induced mutagenesis frequencies at known off-target sites of the full-length gRNA in human cells by 5,000-fold or more60. Analysis of the specificity profiles of Cas9 and various tru-gRNAs by GUIDE-seq found that the numbers of off-target cleavage sites detected genome-wide were generally reduced by ~2–5 fold compared with Cas9 directed by matched full-length gRNAs for the same on-target sites53. However, it is important to note that not all off-target effects were reduced to undetectable levels (as judged by GUIDE-seq or targeted deep sequencing) and that a small number of new off-target sites were created as well. Nonetheless, most tru-gRNA off-target sites detected by GUIDE-seq had mismatches at only one or two positions, supporting the hypothesis that tolerance for mismatches at off-target sites is reduced with Cas9 directed by tru-gRNAs. One possible mechanism to explain how tru-gRNAs work is that they reduce excess potential interaction energy at the RNA:DNA interface so that a Cas9–gRNA complex can still efficiently cleave its on-target site but now has reduced tolerance for mismatched off-target sites.
A related method that has been reported to reduce Cas9-induced off-target effects is to use gRNAs with two additional G nucleotides at the 5 end32 (Fig. 3b). However, in some cases, these longer gRNAs can reduce the on-target activity of Cas9 relative to matched standard length gRNAs32,58. The mechanism behind the reduction in off-target effects observed with certain gRNAs is unclear, but one possibility is that disruption of stabilizing protein interactions with the 5 end of the gRNA may be involved. Genome-wide analysis has not yet been performed with these longer gRNAs to define their global impacts on specificity in cells.
Another strategy proposed to reduce off-target effects is to use paired Cas9 nickases (Cas9n), a mutated version of Cas9 in which one of the two nuclease domains (RuvC or HNH) has been catalytically inactivated (e.g., by introduction of a D10A or H840A mutation) (Fig. 3c). Paired nickases can be directed by two gRNAs targeted to neighboring sites to create offset nicks, which can induce indel mutations61,62. Targeted sequencing has shown that paired nickases can reduce off-target mutations induced by one of the two gRNAs with monomeric Cas9 nuclease, with observed reductions in off-target mutations of 50–1,500 fold in human cells61. However, an important caveat to consider is that it remains unknown whether the second gRNA might also induce off-target effects with Cas9n elsewhere in the genome. Although a nickase is generally less efficient at inducing indel mutations than a nuclease (most likely due to efficient cellular nick repair), Cas9n has been shown to induce high-frequency indel and point mutagnesis at certain target sites32,60,63, presumably because some nicks at certain genomic sites can also be converted into DSBs. However, at present, there is no clear understanding of why this happens at some sites and not others. Nonetheless, the clear implication of this is that the off-target effects of each of the gRNAs in the pair needs to be considered. The specificity profile of paired Cas9 nickases directed against a site in the RAG1 gene has been characterized by HTGTS and this revealed only a few nuclease-related translocation junctions detected genome-wide; however, it is unclear how efficiently translocations would occur with the lesions induced by paired or single nickases. Interestingly, end-protected dsODNs do not integrate efficiently into paired-nickase on-target sites (J.K.J. and colleagues unpublished observations), preventing the direct application of GUIDE-seq to characterize the specificity of this system and suggesting that the mechanism of paired-nickase induced break repair by NHEJ may be distinct from that observed with Cas9 nuclease-induced DSBs. Another side effect of Cas9 nickases is that at certain target sites an increased frequency of point mutations has been observed63, an effect that will be even more challenging than indels to detect on a genome-wide scale.
We and another group recently created dimerization-dependent Cas9-based nucleases that require two co-localized proteins for enzymatic activity63,64. This strategy for reducing off-target effects avoids an important limitation of the paired nickase approach: the use of Cas9n enzymes that are active as monomers. This framework was created by fusing catalytically inactive or “dead” Cas9 (dSpCas9) with the dimerization-dependent FokI nuclease domain14 to create a dimeric RNA-guided FokI–dCas9 nuclease (RFN) architecture requiring recognition of extended double-length target sites for efficient cleavage63,64 (Fig. 3d). These fusions are analogous to earlier dimeric ZFN and TALEN architectures. Amino-terminal fusions of FokI to dSpCas9 can recognize two 20-nucleotide ‘half-sites’ in a ‘PAM-out’ orientation separated by a 13–18 bp spacer and efficiently cleave in this intervening region. It is also possible to combine the orthogonal strategies of truncating the 5 end of gRNAs together with dimerization-dependent RFNs to create tru-RFNs, which exhibit further reduced residual undesirable monomeric cleavage behavior65. Interestingly, tru-RFNs induce mutations efficiently with pairs of gRNAs each truncated at the 5 end by only one nucleotide, in contrast to monomeric Cas9 which is generally tolerant of 2–3 nucleotides of truncation. Although dimeric RFNs directed by two gRNAs have demonstrated mutation frequencies comparable to background at known off-target sites of monomeric Cas9 directed by each individual gRNA of the pair, the specificity of this system remains to be fully evaluated by unbiased, genome-wide methods. Some further optimization will be required to apply GUIDE-seq to analyze the genome-wide specificity of RFNs, because the efficiency of dsODN integration into RFN-cleaved sites is proportionally lower than observed with wild-type Cas965. In addition to the original studies conducted in human cells, dimeric RFNs have been used successfully to generate knockout mutant mice66,67. The major advantages of RFNs are: recognition of an extended double-length target site and dimerization-dependent cleavage mediated by the well-characterized FokI nuclease domain, with background levels of mutations observed at the known monomeric off-target sites examined. Some disadvantages of RFNs are a more restricted targeting range in comparison to monomeric Cas9, the theoretical possibility of monomeric off-target activity via reruitment of another monomer from solution, the size of DNA necessary to encode these fusions proteins (~4.8 kb), which is too large to fit into certain therapeutically relevant vectors such as adeno-associated virus (AAV), which have a packaging limit of ~4.2 kb excluding the inverted terminal repeats (ITRs).
Most recently, our group and another have engineered two different variants of monomeric S. pyogenes Cas9 that exhibit improved genome-wide specificities68,69. Our group introduced alanine substitutions at four residues in SpCas9 known from previously published crystal structures70–72 to mediate non-specific contacts with the phosphate backbone of the target DNA strand (which interacts with the gRNA) to create SpCas9-HF1 (high-fidelity variant 1)68. Relative to wild-type SpCas9, the SpCas9-HF1 variant exhibited comparable on-target activities at on-target sites with ~85% of gRNAs tested. More strikingly, when tested with seven different gRNAs targeted to non-repetitive sequences that induced off-target mutations with wild-type SpCas9, the SpCas9-HF1 variant induced no or, in one case, only a single off-target site detectable by GUIDE-seq. Another group has described a different SpCas9 variant bearing alanine substitutions at three positions predicted to interact with the non-target DNA strand69. This variant, named eSpCas9 1.1 (for enhanced SpCas9 version 1.1) also showed robust on-target activities comparable to that observed with wild-type SpCas9. Testing of eSpCas9 1.1 with two gRNAs revealed that it reduced all or most off-target effects detectable with wild-type SpCas9 by BLESS, suggesting that this variant is capable of improving genome-wide specificity. It will be of interest in the future to perform direct comparisons of SpCas9-HF1 and eSpCas9 1.1 using the same gRNAs in the same cell types. Furthermore, as these Cas9 variants were engineered by reducing non-specific protein interactions to different strands of DNA, it is possible that functional mutations could be combined to further improve specificity. These high-fidelity engineered SpCas9 variants provide large gains in genome-wide specificity that can be realized without changing the targeting range or size of DNA needed to encode the required Cas9 nuclease variant and the single gRNA. One limitation of these variants is that high-frequency mutagenesis can still be observed at certain off-target sites, suggesting that there may remain some room to further improve specificity.
Limiting the duration of Cas9 activity to improve specificity.
There are multiple ways to deliver Cas9 and a gRNA for genome editing. One commonly used method, particularly in early studies, was to transfect plasmid DNA vectors that express Cas9 and gRNA. However, in this format, Cas9 protein and gRNA transcripts likely persist for an extended time and therefore have a greater window in which they can presumably cause unwanted off-target mutagenesis.
Cas9 and gRNA delivered as RNPs and electroporation have been shown to have a shorter half-life and to induce mutations with lower frequencies at off-target sites with 1–2 mismatches relative to the on-target site. By Western blotting, one study73 showed that Cas9 protein is rapidly degraded within 24 hours when delivered by RNP, whereas Cas9 protein continued to be expressed from a plasmid for several days. At known off-target sites of three gRNAs, RNP delivery improved the ratio of on-target to off-target cleavage in comparison to plasmid transfection by up to 13-fold in a transformed human cancer cell line73. Cationic lipids have also been shown to be capable of delivering Cas9–gRNA complexes, presumably because of the highly anionic properties of the gRNA74. Using this approach, ratios of on-target to off-target activity at known off-target cleavage sites of three gRNAs could be improved by up to 20-fold in human cells. These results are consistent with previous studies demonstrating that delivery of ZFNs to cells as protein reduced mutation frequencies at off-target sites in human cells75.
Inducible Cas9 architectures allow Cas9 to become active only after a supplied stimulus, and thus provide another means for limiting the time of activity and reducing off-target effects. Three groups independently designed split Cas9 mutants based on known structural information76–78. One example is a split Cas9 whose two domains are induced to dimerize only in the presence of rapamycin by fusing them with the FRB (FKBP rapamycin binding) and FK506 binding protein 12 (FKBP) protein domains. This split Cas9 has been shown to reduce off-target effects at known off-target cleavage sites when expressed for long periods of time from a stably integrated low-copy lentiviral vector77. Another engineered split Cas9 protein reduced auto-assembly of the protein complex by pairing with gRNAs missing 3 hairpins76. A different inducible Cas9 architecture, where Cas9 protein is initially catalytically repressed by the insertion of a drug-inducible intein, has been shown to improve specificity by up to 25-fold in human cells79. Finally, a photoactivatable Cas9 has also been recently described, based on split Cas9 fused to photoinducible dimerization domains called Magnets78. One advantage of this optogenetic system to control Cas9 is that it is reversible, in contrast to other approaches such as rapamycin- or intein-based systems.
Insights from structural, biochemical and functional studies.
Several crystal structures of SpCas9 (apo71, pre-complexed with gRNA80, and complexed with gRNA and the target strand without72 or with70 the PAM) provide insight into the conformational changes that accompany Cas9 target site recognition. Cas9 adopts a bilobed architecture with a PAM-interacting and nuclease (NUC) lobe, and an alpha-helical recognition (REC) lobe that interacts with the gRNA71,72. While in its apo form Cas9 adopts an auto-inhibited conformation71 with initial electron microscopy (EM) reconstruction experiments of apo-Cas9 and Cas9–RNA suggesting that binding of the crRNA:tracrRNA by Cas9 induces a large conformational shift in the relative positioning of the REC and NUC lobes71.
Biochemical studies using a single-molecule DNA curtains assay suggest that SpCas9 interrogates target sites by three-dimensional diffusion, interacting primarily with sites containing PAM motifs. Heterochromatic regions can be accessed, but with lower probabilities of being sampled, as assessed by single-molecule studies of dSpCas981. Competitive cleavage assays with different types of mismatched competitors are consistent with a sequential unwinding model beginning from the PAM-proximal end of the target site82. These studies are also in line with observations from genome-wide off-target cleavage analysis in human cells which find that off-target cleavage and mutagenesis occur preferentially, but not exclusively, at sites with PAM-distal mismatches and canonical PAM sequences53.
PAM interactions are important for high-affinity Cas9 binding to target DNA17. Cas9 bound to the gRNA is pre-organized to make PAM-interacting contacts80. Cas9 protein interactions with the PAM on the non-target strand mediate a conformational change and initiate sequential unwinding of the immediately adjacent base pairs in the target DNA duplex70. The mechanism of unwinding is not restricted to a particular PAM sequence because Cas9 variants with altered PAM recognition preferences have been engineered using a combination of protein evolution, bacterial selection, and rational design83,84 or by rational design alone85. This is consistent with observations that the attenuated activity of a Cas9 variant with weakened PAM-binding affinity can be rescued by fusion of an engineered zinc finger DNA-binding array86.
Recent studies using fluorescence resonance energy transfer (FRET) to study Cas9 conformational dynamics have clarified some of the differing requirements observed for Cas9 binding and cleavage (Box 1). Interactions between Cas9 and both the 5 and 3 ends of the guide trigger the first conformational rearrangement that occurs upon Cas9 loading of the gRNA82. Recognition of DNA with sufficient target site complementarity drives a second conformational change in the HNH domain that triggers cleavage in concert with the RuvC domain. Interestingly, the conformational change in the HNH domain seems to be driven by the extent of RNA:DNA heteroduplex formation at the PAM-distal end of the target site, providing the mechanistic basis of a proofreading mechanism for conformational gating of target site cleavage specificity87. A recent structure of Cas9 crystallized in an active form in complex with a 30-bp dsDNA target, confirmed the proposed HNH conformational rearrangement88.
Two recent studies found that the extent of gRNA protospacer complementarity required for efficient cleavage by Cas9 nuclease and transcriptional activation by dCas9 fusions to transcription activation domains are different. These studies confirmed previous observations that gRNAs with 17 or more nucleotides of protospacer complementarity could mediate efficient cleavage by Cas9 nuclease whereas those with shorter lengths did not60. However, these studies also found that only 14 or more nucleotides of complementarity were necessary for DNA binding by dCas989,90 Interestingly, engineered SpCas9-HF1 directed by tru-gRNAs with 17–18 nucleotides complementarity had significantly reduced activity in human cells compared to wild-type Cas968, suggesting that these two strategies of engineering variants with reduced non-specific DNA contacts and gRNA truncation may improve specificity via a similar mechanism of decreasing interaction energy of the protein–gRNA complex or R-loop stability. (See Box 1 for additional discussion on the differences between binding and cleavage for CRISPR–Cas9 nucleases).
In contrast to ZFNs and TALENs, Cas9 mediates the unwinding of its target DNA prior to cleavage of the separated strands by RUVC and HNH domains. One interesting implication of Cas9’s mode of target site binding is that positions of the unwound DNA strands that remain exposed may be susceptible to cellular deamination, resulting in the efficient introduction of point mutations that have been observed most prominently with Cas9 nickases63.
Implications for research and therapeutic applications
With the wealth of methods available for detecting and reducing off-target effects, it can be difficult to know which specific methods to use in different experimental and therapeutic contexts. Here we offer some broad suggestions to help guide users in their choice of an appropriate approach to contend with off-target effects for their particular project or application.
Due to limitations of current high-throughput sequencing technologies in terms of both bandwidth and read-length, it is not possible to comprehensively sequence large numbers of cellular genomes. Although methods like GUIDE-seq, BLESS, HTGTS, and Digenome-seq do a more complete job of finding off-target effects than early approaches in the field, it is also not currently possible to prove that these methods are comprehensive in their coverage. In order to more thoroughly assess genome-wide specificity methods, it will be necessary to develop more highly sensitive methods that can detect mutations below the floor of ~0.1% frequency imposed by current high-throughput sequencing technologies.
The manner and extent to which the genome-wide specificity needs to be characterized is dependent on the intended application: research discovery or therapeutics. For most research applications, the primary concern is whether off-target mutations might confound the interpretation of biological phenotypes observed with introduction of a genome editing event at the on-target site. However, because no method for finding off-target effects has been proven to be fully comprehensive, the application of any or all of the existing genome-wide detection methods to a given experimental system would be still insufficient to completely rule out the confounding effect of an off-target mutation. Thus, our recommendation is that researchers instead perform control experiments that argue against a confounding off-target effect. Examples of such controls would include use of multiple gRNAs to introduce the same mutation; this control should be effective because data from genome-wide methods have now clearly established that off-target sites are not random but clearly related in sequence to the gRNA on-target site. Alternatively, checking that the original pre-editing phenotype is restored following genetic reversion experiments (perhaps performed using genome editing) and/or rescue by genetic complementation can reduce the risk of coming to mistaken conclusions based on confounding off-target effects. These types of redundant functional assays are simple safeguards that can be used in situations in which a biological phenotype appears to be induced by an on-target genome editing event.
For clinical applications of engineered nucleases on large cell populations, safety is obviously the paramount concern. Ideally, the most sensitive, unbiased, genome-wide method(s) should be used to identify all potential off-target cleavage sites of a candidate therapeutic nuclease. Genome-wide coverage is important to ensure that no mutations are missed. Using unbiased approaches is also critical because all experiments to date show that existing in silico approaches alone miss off-target sites and because off-target mutations can occur at sites with single-nucleotide polymorphisms that differ from the genomic reference sequence91. This latter point highlights the need to directly assess each patient’s individual genome for each therapeutic nuclease. High sensitivity is important because even low-frequency events can potentially lead to deleterious outcomes and because most therapeutic strategies being considered for genome-editing nucleases would modify millions to billions of cells. Existing methods can only reliably detect muations with frequencies as low as 0.1% so even greater sensitivity is certainly needed. Finally, it is also important to note that state-ofthe-art assessment of genomic off-target effects will likely only be part of a larger safety evaluation required to quantify the risk associated with any given nuclease.
Because no single method is currently expected to be definitive or comprehensive in its assessment of a therapeutic CRISPR–Cas9 nuclease, it is our opinion that the best strategy may be to use multiple, partially redundant approaches to assess off-target effects until such an ideal method is developed. The choice of which of the existing methods to use is driven in part by the strengths and weaknesses of these different approaches and any technical constraints imposed by the target cells or tissue of interest. For example, if the cells can be efficiently transfected with the required short end-protected dsODN tag, GUIDE-seq provides an effective and simple method for off-target discovery requiring relatively fewer input genomes for sensitive detection because of the high efficiency of integration. IDLV capture could be used in cases where viral transduction is more practical than dsODN transfection, although the sensitivity of this method may be lower. HTGTS is the only cell-based method that can track accumulated DNA repair outcomes for in vivo nuclease delivery. BLESS is best suited for discovery of transient DSBs at a particular moment in time, and thus might be useful for kinetic studies of nuclease cleavage. Finally, in vitro methods like Digenome-seq for discovering nuclease-induced off-target effects may have some advantages over cell-based methods such as not being subject to biological fitness effects and presumably being unaffected by cell-specific chromatin state or epigenetic modifications. However, current in vitro methods use virtually all sequencing bandwidth on unrelated sequences and thus will require further improvements to maximize detection sensitivity.
Future directions
Great progress has been made with methods to determine CRISPR–Cas9-induced off-target mutations but more work remains to be done. Improving the sensitivity of mutation detection assays is an area that needs particular attention. At present, technical limitations with the error rate of high-throughput sequencing technologies hamper their use to confirm off-target effects below the level of 0.1%. In addition, a more complete understanding of the impacts of chromatin state and gene expression levels on target site editing of genome-editing nucleases is needed. In the long-term, big data sets that delineate the genome-wide off-target effects of large numbers of different gRNAs may provide greater capabilities to predict off-target sites in silico, enabling the development of more effective computational tools. Direct comparisons of the various methods available for defining and improving genome-wide specificity using the same gRNAs in the same cellular contexts would go a long way toward defining and understanding the strengths and limitations of each of these different approaches. Additional data and improvements in technology will in turn help to increase confidence in the safety of therapeutic strategies built on genome-editing nucleases.
Glossary definitions
- BLESS
Breaks labeling, enrichment on streptavidin and next-generation sequencing. Cell-based method for genome-wide discovery of nuclease-induced DSBs based on cell fixing, nuclei isolation, in situ ligation, enrichment, and high-throughput sequencing.
- Bulge
An gap in base pairing between target DNA or gRNA at an RNA-guided nuclease target site.
- Cas9 Nickase
Engineered variant of Cas9 where one of the two nuclease domains has been catalytically inactivated, resulting in the nicking of only one DNA strand and leaving the other strand intact.
- CRISPR
Clustered Regularly Short Interspaced Palindromic Repeats. Components of a system of bacterial adaptive immunity.
- crRNA
(CRISPR RNA). small RNA containing sequence complementarity to protospacer and a short repetitive sequence with sequence complementarity to tracrRNA.
- Digenome-seq
(Digested genome sequencing.) In vitro method for detecting Cas9 cleavage of genomic DNA by whole genome sequencing.
- DNA Curtains Assay
A single-molecule assay for visualization of protein interactions with individual dna strands or ‘curtains’.
- dsODN
Double-stranded oligodeoxynucleotide; used as an integrated genetic tag in GUIDE-seq.
- GUIDE-seq
Genome-wide Unbiased Identification of DSBs Enabled by Sequencing. A cell-based method for genome-wide discovery of nuclease-induced DSBs based on efficient tag integration, tag-specific amplification, and high-throughput sequencing.
- High-throughput sequencing
A method for sequencing populations of DNA molecules, typically with short (<300bp) reads that have error rates an order of magnitude or more higher than standard long-read Sanger sequencing.
- Homology-directed Repair
DNA repair pathways that depend on seequence homology to initiate repair. A user-supplied ‘donor’ template can be used to introduce precise alterations of choice with this repair pathway.
- HTGTS
High-throughput genome-wide translocation sequencing, a method applied to detect nuclease-induced off-target DSBs by observation of translocation junctions.
- Non-homologous end-joining
DNA repair repair pathway where DSB ends are directly ligated together without a requirement for homology. Variable length insertion or deletion mutations can frequently occur as a consequence of NHEJ-mediated DSB repair.
- Point Mutation
Genetic change of a single DNA base pair.
- Protospacer
Target sequence for CRISPR interference, flanked by CRISPR repeats.
- Protospacer Adjacent Motif (PAM)
Sequence required to licence Cas9 for cleavage, adjacent to the target sequence or protospacer.
- Rolling Circle Amplification
A method for generating many concatemerized copies of a circular template using a strand-displacing polymerase.
- tracRNA
trans-activating crRNA, a small trans-encoded RNA that has a portion of sequence complementarity with the crRNA and is required for Cas9 nuclease activity
Footnotes
Competing interests statement:
JKJ is a consultant for Horizon Discovery. JKJ has financial interests in Editas Medicine, Hera Testing Laboratories, Poseida Therapeutics, and Transposagen Biopharmaceuticals. JKJ’s interests were reviewed and are managed by Massachusetts General Hospital and Partners HealthCare in accordance with their conflict of interest policies.
SQT and JKJ are co-founders of Beacon Genomics, a company that is commercializing methods for determining nuclease specificity.
References
- 1.Doetschman T et al. Targetted correction of a mutant HPRT gene in mouse embryonic stem cells. Nature 330, 576–578 (1987). [DOI] [PubMed] [Google Scholar]
- 2.Thomas KR & Capecchi MR Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell 51, 503–512 (1987). [DOI] [PubMed] [Google Scholar]
- 3.Evans MJ & Kaufman MH Establishment in culture of pluripotential cells from mouse embryos. Nature 292, 154–156 (1981). [DOI] [PubMed] [Google Scholar]
- 4.The 2007 Nobel Prize in Physiology or Medicine - Press Release. nobelprize.org at <http://www.nobelprize.org/nobel_prizes/medicine/laureates/2007/press.html>
- 5.Rouet P, Smih F & Jasin M Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease. Mol Cell Biol 14, 8096–8106 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study first demonstrated that nuclease-induced DSBs could stimulate homologous recombination.
- 6.Silva G et al. Meganucleases and other tools for targeted genome engineering: perspectives and challenges for gene therapy. Curr Gene Ther 11, 11–27 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Urnov FD, Rebar EJ, Holmes MC, Zhang HS & Gregory PD Genome editing with engineered zinc finger nucleases. Nature 11, 636–646 (2010). [DOI] [PubMed] [Google Scholar]
- 8.Sander JD & Joung JK CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol 32, 347–355 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hsu PD, Lander ES & Zhang F Development and applications of CRISPR-Cas9 for genome engineering. Cell 157, 1262–1278 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Doudna JA & Charpentier E Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 346, 1258096 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Kim YG, Cha J & Chandrasegaran S Hybrid restriction enzymes: zinc finger fusions to FokI cleavage domain. Proc Natl Acad Sci USA 93, 1156–1160 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cermak T et al. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res 39, e82 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li T et al. Modularly assembled designer TAL effector nucleases for targeted gene knockout and gene replacement in eukaryotes. Nucleic Acids Res (2011). doi: 10.1093/nar/gkr188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bitinaite J, Wah DA, Aggarwal AK & Schildkraut I FokI dimerization is required for DNA cleavage. Proc Natl Acad Sci USA 95, 10570–10575 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wah DA, Bitinaite J, Schildkraut I & Aggarwal AK Structure of FokI has implications for DNA cleavage. Proc Natl Acad Sci USA 95, 10564–10569 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boissel S et al. megaTALs: a rare-cleaving nuclease architecture for therapeutic genome engineering. Nucleic Acids Res 42, 2591–2601 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jinek M et al. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 337, 816–821 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; This was the first published study to demonstrate that the target specificity of Cas9 could be programmed, using an engineered single gRNA (a fusion of naturally occuring crRNA and tracrRNA).
- 18.Mali P et al. RNA-guided human genome engineering via Cas9. Science 339, 823–826 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cong L et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jinek M et al. RNA-programmed genome editing in human cells | eLife. Elife 2, e00471 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hwang WY et al. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat Biotechnol 31, 227–229 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ran FA et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 520, 186–191 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hou Z et al. Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proc Natl Acad Sci USA 110, 15644–15649 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang X-H, Tee LY, Wang X-G, Huang Q-S & Yang S-H Off-target Effects in CRISPR/Cas9-mediated Genome Engineering. Mol Ther Nucleic Acids 4, e264 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pattanayak V, Guilinger JP & Liu DR Determining the specificities of TALENs, Cas9, and other genome-editing enzymes. Methods in Enzymology 546, 47–78 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.O’Geen H, Yu AS & Segal DJ How specific is CRISPR/Cas9 really? Curr Opin Chem Biol 29, 72–78 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.LaFountaine JS, Fathe K & Smyth HDC Delivery and therapeutic applications of gene editing technologies ZFNs, TALENs, and CRISPR/Cas9. Int J Pharm 494, 180–194 (2015). [DOI] [PubMed] [Google Scholar]
- 28.Jamal M et al. Keeping CRISPR/Cas on-Target. Curr Issues Mol Biol 20, 1–20 (2015). [PubMed] [Google Scholar]
- 29.Ishida K, Gee P & Hotta A Minimizing off-Target Mutagenesis Risks Caused by Programmable Nucleases. Int J Mol Sci 16, 24751–24771 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bolukbasi MF, Gupta A & Wolfe SA Creating and evaluating accurate CRISPR-Cas9 scalpels for genomic surgery. Nat Meth 13, 41–50 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Yang H et al. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell 154, 1370–1379 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cho SW et al. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res 24, 132–141 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liang X et al. Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. Journal of Biotechnology 208, 44–53 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Pattanayak V et al. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat Biotechnol 31, 839–843 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fu Y et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells : Nature Biotechnology : Nature Publishing Group. Nat Biotechnol 31, 822–826 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; This was the first study to report high-frequency off-target CRISPR-Cas9 mutagenesis.
- 36.Hsu PD et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 31, 827–832 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cradick TJ, Fine EJ, Antico CJ & Bao G CRISPR/Cas9 systems targeting β-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Res 41, 9584–9592 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin Y et al. CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Res 42, 7473–7485 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang H et al. The CRISPR/Cas9 system produces specific and homozygous targeted gene editing in rice in one generation. Plant Biotechnol J 12, 797–807 (2014). [DOI] [PubMed] [Google Scholar]
- 40.Gasiunas G, Barrangou R, Horvath P & Siksnys V Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci USA 109, E2579–86 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cho SW, Kim S, Kim JM & Kim J-S Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol 31, 230–232 (2013). [DOI] [PubMed] [Google Scholar]
- 42.Heigwer F, Kerr G & Boutros M E-CRISP: fast CRISPR target site identification. Nat Meth 11, 122–123 (2014). [DOI] [PubMed] [Google Scholar]
- 43.Pattanayak V, Ramirez CL, Joung JK & Liu DR Revealing off-target cleavage specificities of zinc-finger nucleases by in vitro selection. Nat Meth (2011). doi: 10.1038/nmeth.1670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guilinger JP et al. Broad specificity profiling of TALENs results in engineered nucleases with improved DNA-cleavage specificity. Nat Meth 11, 429–435 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sander JD et al. In silico abstraction of zinc finger nuclease cleavage profiles reveals an expanded landscape of off-target sites. Nucleic Acids Res 41, e181 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smith C et al. Whole-Genome Sequencing Analysis Reveals High Specificity of CRISPR/Cas9 and TALEN-Based Genome Editing in Human iPSCs. Cell Stem Cell 15, 12–13 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Veres A et al. Low Incidence of Off-Target Mutations in Individual CRISPR-Cas9 and TALEN Targeted Human Stem Cell Clones Detected by Whole-Genome Sequencing. Cell Stem Cell 15, 27–30 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Iyer V et al. Off-target mutations are rare in Cas9-modified mice. Nat Meth 12, 479–479 (2015). [DOI] [PubMed] [Google Scholar]
- 49.Tsai SQ & Joung JK What’s Changed with Genome Editing? Cell Stem Cell 15, 3–4 (2014). [DOI] [PubMed] [Google Scholar]
- 50.Gabriel R et al. An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat Biotechnol 29, 816–823 (2011). [DOI] [PubMed] [Google Scholar]; This is the first report of an unbiased genome-wide analysis of nuclease specificity, by analyzing IDLV capture into ZFN-induced DSBs.
- 51.Osborn MJ et al. Evaluation of TCR Gene Editing achieved by TALENs, CRISPR/Cas9 and megaTAL nucleases. Mol Ther (2015). doi: 10.1038/mt.2015.197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang X et al. Unbiased detection of off-target cleavage by CRISPR-Cas9 and TALENs using integrase-defective lentiviral vectors. Nat Biotechnol (2015). doi: 10.1038/nbt.3127 [DOI] [PubMed] [Google Scholar]
- 53.Tsai SQ et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol 33, 187–197 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study describes the GUIDE-seq method for identifying Cas9 off-target cleavage sites in living cells.
- 54.Tsai SQ, Topkar VV, Joung JK & Aryee MJ guideseq: open-source software for analysis of GUIDE-seq data. Nat Biotechnol [DOI] [PubMed] [Google Scholar]
- 55.Chiarle R et al. Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell 147, 107–119 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Frock RL et al. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat Biotechnol 33, 179–186 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study describes the HTGTS method for identifying Cas9 off-target cleavage by translocation sequencing.
- 57.Crosetto N et al. Nucleotide-resolution DNA double-strand break mapping by next-generation sequencing. Nat Meth 10, 361–365 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; This is the original study describing the BLESS method, which was later adapted to analyze CRISPR-Cas9 off-target cleavage.
- 58.Kim D et al. Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat Meth (2015). doi: 10.1038/nmeth.3284 [DOI] [PubMed] [Google Scholar]; This study describes Digenome-seq, an in vitro genome-wide method for identifying CRISPR-Cas9 off-target cleavage sites.
- 59.Kim D, Kim S, Kim S, Park J & Kim J-S Genome-wide target specificities of CRISPR-Cas9 nucleases revealed by multiplex Digenome-seq. Genome Res gr.199588.115 (2016). doi: 10.1101/gr.199588.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fu Y, Sander JD, Reyon D, Cascio VM & Joung JK Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol (2014). doi: 10.1038/nbt.2808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ran FA et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154, 1380–1389 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mali P et al. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat Biotechnol – (2013). doi: 10.1038/nbt.2675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tsai SQ et al. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat Biotechnol 32, 569–576 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Guilinger JP, Thompson DB & Liu DR Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nat Biotechnol 32, 577–582 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wyvekens N, Topkar VV, Khayter C, Joung JK & Tsai SQ Dimeric CRISPR RNA-Guided FokI-dCas9 Nucleases Directed by Truncated gRNAs for Highly Specific Genome Editing. Hum Gene Ther 26, 425–431 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hara S et al. Generation of mutant mice via the CRISPR/Cas9 system using FokI-dCas9. Sci Rep 5, 11221 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nakagawa Y et al. Production of knockout mice by DNA microinjection of various CRISPR/Cas9 vectors into freeze-thawed fertilized oocytes. BMC Biotechnol 15, 33 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kleinstiver BP et al. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature (2016). doi: 10.1038/nature16526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Slaymaker IM et al. Rationally engineered Cas9 nucleases with improved specificity. Science 351, 84–88 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Anders C, Niewoehner O, Duerst A & Jinek M Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature (2014). doi: 10.1038/nature13579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jinek M et al. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science 343, 1247997–1247997 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nishimasu H et al. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 156, 935–949 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kim S, Kim D, Cho SW, Kim J & Kim JS Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res (2014). doi: 10.1101/gr.171322.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zuris JA et al. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat Biotechnol 33, 73–80 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gaj T, Guo J, Kato Y, Sirk SJ & Barbas CF Targeted gene knockout by direct delivery of zinc-finger nuclease proteins. Nat Meth (2012). doi: 10.1038/nmeth.2030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wright AV et al. Rational design of a split-Cas9 enzyme complex. Proc Natl Acad Sci USA 112, 2984–2989 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zetsche B, Volz SE & Zhang F A split-Cas9 architecture for inducible genome editing and transcription modulation. Nat Biotechnol 1–3 (2015). doi: 10.1038/nbt.3149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nihongaki Y, Kawano F, Nakajima T & Sato M Photoactivatable CRISPR-Cas9 for optogenetic genome editing. Nat Biotechnol 33, 755–760 (2015). [DOI] [PubMed] [Google Scholar]
- 79.Davis KM, Pattanayak V, Thompson DB, Zuris JA & Liu DR Small molecule-triggered Cas9 protein with improved genome-editing specificity. Nature Chemical Biology 11, 316–318 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jiang F, Zhou K, Ma L, Gressel S & Doudna JA A Cas9-guide RNA complex preorganized for target DNA recognition. Science 348, 1477–1481 (2015). [DOI] [PubMed] [Google Scholar]
- 81.Knight SC et al. Dynamics of CRISPR-Cas9 genome interrogation in living cells. Science 350, 823–826 (2015). [DOI] [PubMed] [Google Scholar]
- 82.Sternberg SH, Redding S, Jinek M, Greene EC & Doudna JA DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 507, 62–67 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kleinstiver BP et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature 523, 481–485 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kleinstiver BP et al. Broadening the targeting range of Staphylococcus aureus CRISPR-Cas9 by modifying PAM recognition. Nat Biotechnol (2015). doi: 10.1038/nbt.3404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hirano H et al. Structure and Engineering of Francisella novicida Cas9. Cell 1–13 (2016). doi: 10.1016/j.cell.2016.01.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bolukbasi MF et al. DNA-binding-domain fusions enhance the targeting range and precision of Cas9. Nat Meth (2015). doi: 10.1038/nmeth.3624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sternberg SH, LaFrance B, Kaplan M & Doudna JA Conformational control of DNA target cleavage by CRISPR-Cas9. Nature 527, 110–113 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jiang F et al. Structures of a CRISPR-Cas9 R-loop complex primed for DNA cleavage. Science aad8282 (2016). doi: 10.1126/science.aad8282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kiani S et al. Cas9 gRNA engineering for genome editing, activation and repression. Nat Meth 12, 1051–1054 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dahlman JE et al. Orthogonal gene knockout and activation with a catalytically active Cas9 nuclease. Nat Biotechnol 1–4 (2015). doi: 10.1038/nbt.3390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yang L et al. Targeted and genome-wide sequencing reveal single nucleotide variations impacting specificity of Cas9 in human stem cells. Nature Communications 5, 5507 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Deltcheva E et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471, 602–607 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wu X et al. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat Biotechnol 32, 670–676 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cencic R et al. Protospacer adjacent motif (PAM)-distal sequences engage CRISPR Cas9 DNA target cleavage. PLoS ONE 9, e109213 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.O’Geen H, Henry IM, Bhakta MS, Meckler JF & Segal DJ A genome-wide analysis of Cas9 binding specificity using ChIP-seq and targeted sequence capture. Nucleic Acids Res 43, 3389–3404 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]