

Abstract
Significant advances in biotechnology have led to the development of a number of different mutation-directed therapies. Some of these techniques have matured to a level that has allowed testing in clinical trials, but few have made it to approval by drug-regulatory bodies for the treatment of specific diseases. While there are still various hurdles to be overcome, recent success stories have proven the potential power of mutation-directed therapies and have fueled the hope of finding therapeutics for other genetic disorders. In this review, we summarize the state-of-the-art of various therapeutic approaches and assess their applicability to the genetic disorder neurofibromatosis type I (NF1). NF1 is caused by the loss of function of neurofibromin, a tumor suppressor and downregulator of the Ras signaling pathway. The condition is characterized by a variety of phenotypes and includes symptoms such as skin spots, nervous system tumors, skeletal dysplasia, and others. Hence, depending on the patient, therapeutics may need to target different tissues and cell types. While we also discuss the delivery of therapeutics, in particular via viral vectors and nanoparticles, our main focus is on therapeutic techniques that reconstitute functional neurofibromin, most notably cDNA replacement, CRISPR-based DNA repair, RNA repair, antisense oligonucleotide therapeutics including exon skipping, and nonsense suppression.
Graphical Abstract
We present a comprehensive review that addresses therapeutic approaches to treating specific genetic defects that cause neurofibromatosis type I, including nanoparticle delivery, exon skipping, gene editing, mRNA trans-splicing ribozymes, and non-sense suppression therapeutics. Our multi-faceted approach can be utilized for virtually any rare disease to affect personalized medicine.
Main Text
Neurofibromatosis type I (NF1, OMIM #162200) is one of the most common genetic disorders, occurring in approximately 1:2,000–3,000 births.1,2 It is caused by pathogenic variants in the NF1 gene (NCBI: NG_009018.1), which is located on chromosome 17q11.2. With a length of about 300 kb, it is one of the largest human genes. Precursor mRNA (pre-mRNA) splicing of its 61 exons is complex, with several alternatively spliced exons, alternative splice sites, and potentially regulatory pseudo-exon inclusion.3, 4, 5 The NF1 mRNA transcript variant 2 (NCBI: NM_000267.3) has a length of approximately 12.4 kb and is composed of 57 constitutively expressed exons. Neurofibromin (P21359-2), the protein encoded by NF1, is a tumor suppressor and downregulator of the Ras signaling pathway. It is expressed in many cell types, notably in neurons, Schwann cells, oligodendrocytes, astrocytes, and leukocytes. Loss of function, or lack of neurofibromin, leads to a condition that is characterized by a pleiotropic phenotype affecting the skin (café-au-lait macules, skin fold freckling, hyperpigmentation, cutaneous neurofibromas), the eye (Lisch nodules and optic glioma), skeleton (dysplasias and scoliosis), and peripheral and central nervous system (CNS) (cognitive disabilities, motor delays, gliomas, neurofibromas). Malignant peripheral nerve sheath tumors (MPNSTs) can be observed, with poor prognosis. Almost 2,900 pathogenic variants have been reported in the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php).
To date, NF1 cannot be cured. Treatments attempt to manage the various symptoms, especially NF1-associated tumors and other cancers, using standard cancer therapies. At least a dozen different therapeutics targeting almost as many proteins have been used in clinical trials for NF-associated tumors.6,7 Also, oncolytic viral therapies for treating MPNSTs have shown promising results.8 However, these treatments are largely adaptations of existing therapies against sporadic tumors and do not target the specific growth control pathways dysregulated in NF1. More recently, drug development has focused on targeting key molecules downstream of NF1, most notably the RAF-MEK-ERK signaling cascade downstream of RAS.9, 10, 11, 12, 13, 14 MEK inhibitors such as selumetinib have demonstrated effectiveness for patients who respond and can tolerate treatment; however, not all patients benefit, plexiform neurofibromas do not completely disappear, and there can be significant side effects.11 Hence, therapeutics that address the underlying cause of the disease by restoring neurofibromin function to a level that leads to a non-pathogenic phenotype do not yet exist. Various gene and mRNA targeting strategies have been proposed in the context of other diseases. For example, Zhou et al.15 specifically reviewed many of the RNA-based therapeutics in clinical trials. These and other strategies are worth being evaluated for their therapeutic potential in NF1.
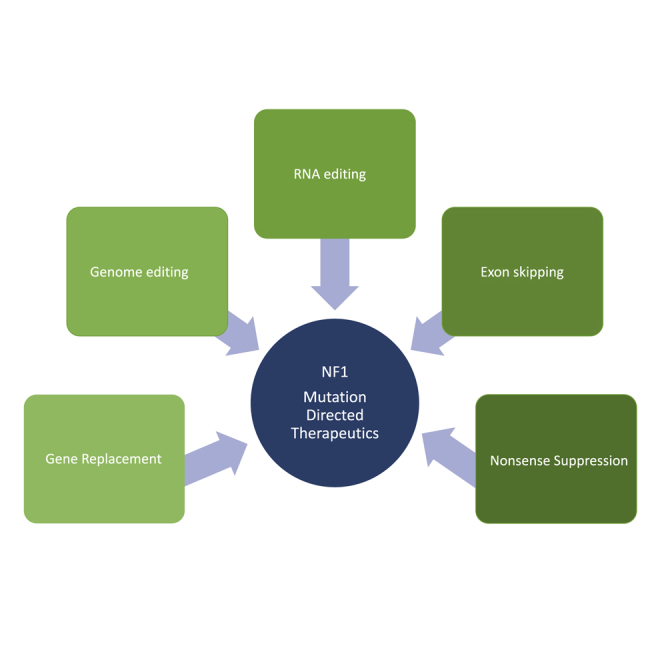
In the following sections we introduce therapeutic strategies to replace or repair the NF1 gene or its transcripts or to inhibit the effect of certain genetic mutations. Table 1 provides a summary of these methods, the types of mutations that can be targeted, advantages and disadvantages, as well as examples of success for each method. We discuss gene replacement by cDNA/mRNA delivery, CRISPR (clustered regularly interspaced short palindromic repeats)-based DNA repair, RNA repair, exon skipping, and nonsense suppression therapies (in combination with inhibition of nonsense-mediated mRNA decay [NMD]) in the context of NF1 and address the pros and cons of these individual approaches. Subsequently, we evaluate data regarding required levels of neurofibromin function. Lastly, we address important aspects of the delivery of any such therapeutic and nanoparticles as a promising delivery vehicle. We anticipate that this review of mutation-directed therapeutics for NF1 may also stimulate similar discussions and active steps toward the development of treatments of other genetic diseases.
Table 1.
Approaches for NF1 Gene Therapy
| Approach | Targeted Mutations | Advantages | Disadvantages/Challenges | Successes | NF1 Status |
|---|---|---|---|---|---|
| Gene replacement | all loss of function mutations | might target the largest mutation spectrum | mNF1 cDNA; efficient delivery using nanoparticles | Luxterna for retinal dystrophy associated with loss of RPE65, and Zolgensma for SMA and loss of SMN1 | development of full-length mNf1 cDNA and development of model systems for testing |
| Genome editing | most small mutations | permanent cell editing | efficiency of editing; non-specific gene editing; delivery using nanoparticles | ex vivo CCR5 deletion to block HIV infection | testing CRISPR-Cas9 and CRISPR Prime in nanoparticles and development of model systems for testing |
| RNA editing | most small mutations in 5′ and 3′ regions | does not change DNA | efficiency of editing, non-permanence; delivery using virus or nanoparticles | β-globin and DMPK repair in vitro | defining high-efficiency splice sites within NF1 and evolving ribozymes and development of model systems for testing |
| Exon skipping | select exons | low toxicity | each exon must be considered/designed separately; delivery using cell-penetrating peptides | Exondys 51 (eteplirsen) and Vyondys 53 for DMD; Spinraza for SMA | definition of selected exons and testing of AOs and development of model systems for testing |
| NST | nonsense ~20% | low toxicity; may be able to repurpose other drugs | efficiency of readthrough; NMD | Ataluran for cystic fibrosis | small molecule drug screens for NSTs and development of model systems for testing |
Approaches for Nucleic Acid Therapies and Their Applicability to NF1
Gene Replacement
Perhaps one of the more straightforward concepts of gene therapy is to supplement a working copy of a gene into a cell with a defective gene. This allows for the treatment of gene deletions or other loss-of-function type mutations. In 2017, Luxturna (voretigene neparvovec) became the first US Food and Drug Administration (FDA)-approved gene replacement therapy for the treatment of patients with confirmed biallelic RPE65 mutation-associated retinal dystrophy that leads to vision loss and may cause complete blindness in certain patients. Luxturna works by delivering a normal copy of the RPE65 gene directly to retinal cells, using a recombinant adeno-associated virus serotype 2 (rAAV2) vector. This therapy is regarded as the first true FDA-approved gene therapy, because the viral treatment is delivered directly to the patient’s body. That said, Luxturna, even though heralded as a therapy that restores vision, is far from being a cure: it has shown to improve vision to some degree in some but not all treated patients. Furthermore, improvements may not be permanent.16 Previous FDA-approved gene therapies (Yescarta [axicabtagene ciloleucel] and Kymriah [tisagenlecleucel]) are called ex vivo treatments, as the cells receiving gene therapy (immune cells) are removed from the body prior to treatment. Zolgensma (onasemnogene abeparvovec) is another therapeutic that uses an AAV to deliver a functional copy of the SMN1 gene to motor neurons in pediatric patients with spinal muscular atrophy (SMA).
Gene replacement for NF1 has been challenging for two primary reasons: the lack of a full-length NF1 cDNA and a delivery system. Since discovery of the NF1 gene in 1990, research efforts have been hindered by the lack of a full-length coding cDNA. This is due to the size of the cDNA and toxicity of the human construct. Despite this, as the GAP-related domain (GRD) was assumed to be the most important segment of NF1, replacement with the GRD has been attempted, but was unsuccessful in several ways. Expression of isolated GRD is unable to rescue overgrowth of neural crest-derived tissues, leading to perinatal lethality in Nf1−/− embryos.17 The GRD rescues endothelial but not neural crest development in Nf1 mice. These results suggest that neurofibromin may possess activities outside of the GRD that modulate neural crest homeostasis and that therapeutic approaches solely aimed at targeting Ras activity may not be sufficient. Most recently, the feasibility of restoring Ras guanosine triphosphatase (GTPase) via exogenous expression of various NF1-GRD constructs, via gene delivery using a panel of AAV vectors in MPNST and human Schwann cells (HSCs), has been explored.18 Several AAV serotypes achieved favorable transduction efficacies, in particular AAV-DJ. A membrane-targeting GRD fused with an H-Ras C-terminal motif containing the palmitoylation sites and CAAX motif (C10) inhibited the Ras pathway and MPNST cells in a NF1-specific manner; however; a transfection efficiency of only 9.8% of cells at a MOI of 5,000 is reported. Since the non-transduced populations exhibit a clear growth advantage, efficacy would require almost all of the MPNST cells or the majority of the NF1-haploid Schwann cells to receive the GRD-C10 transgene. With the current available tools for in vivo gene delivery, this would only be feasible with a drastically improved AAV vector for MPNST or Schwann cells, which could be achieved through protein engineering of the AAV capsids.
We have developed and validated a mouse Nf1 cDNA expression system that allows us to examine the biochemical effects of any Nf1 genetic variant.19 The full-length cDNA sequences of endogenous hNF1 and mNf1 have 92% sequence identity; amino acid sequences share 98% identity.20 A heavily codon-optimized human NF1 cDNA21 (R777-E139 Hs.NF1 [Addgene plasmid #70423]) that is stable in E. coli and non-toxic to human cells has also become available. This human construct is so heavily codon optimized that when the sequence is used for a BLASTn (nucleotide) search, the NF1 gene sequence does not come back as a hit; yet a BLASTp (protein) search comes back with 100% identity. Furthermore, GC content is increased from 43% to 60% in the codon-optimized transcript. Hence, the mNf1 cDNA is more homologous to the endogenous human sequence. Furthermore, codon optimization may have direct implications, as codon usage is thought to affect transcription and translation efficiency as well as mRNA stability and protein folding. Another hNF1 cDNA eliminates the cDNA cloning toxicity by introducing a mini-intron.22
Gene Replacement for NF1. As viral vectors cannot accommodate the size or the NF1 cDNA, we are investigating the ability to package our full-length mouse Nf1 cDNA into nanoparticles for intracellular delivery as described in “Delivery of NF1 Gene Therapeutic” below. As an NF1 therapeutic, this approach would target the largest mutation spectrum.
Gene Editing
Direct repair of small DNA or RNA mutations is an alternative to whole-gene replacement. The three prominent systems for DNA repair based on programmable nucleases are zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and CRISPR and CRISPR-associated (Cas) proteins.
The CRISPR-Cas9 system has demonstrated great promise in many biological applications.23 It is an RNA-based programmable nuclease derived from a prokaryotic immune system. The CRISPR-Cas9 nuclease system has emerged as the most promising genome editing technology. The system uses an engineered single guide RNA (sgRNA) to direct the Cas9 nuclease to complementary regions, where Cas9 cleaves the recognized DNA and generates double-stranded breaks (DSBs), leading to insertions or deletions at specific target genomic loci (Figure 1).24,25 Cas9 cleavage induces subsequent DNA repair by either non-homologous end-joining (NHEJ) or homology-directed repair (HDR).23,26 When an externally supplied homologous donor template is provided, HDR may enable precise genome editing and permanent correction of a targeted mutation.27 However, under normal physiological conditions, NHEJ repair, which is an error-prone process and tends to lead to disruption of targeted genes is more than 10-fold more frequent than HDR at DSBs.27 Consequently, without suppressing NHEJ, the HDR-mediated precise genome editing often suffers from low efficiency.
Figure 1.

Nanoparticle Delivery
Delivery of CRISPR-Cas9 gene editing to a target cell through ligand-conjugated nanoparticles is shown. Nanoparticles recognize targeted cells through ligand-receptor interaction and penetrate into cells through endocytosis. After escaping the endosome, nanoparticles release cargo CRISPR-Cas9, which, in turn, translocates into the nucleus and exerts its gene editing function.
Non-specific (“off target”) gene editing is also a major concern when using CRISPR-Cas9 technology. While there are methods for detecting off-target editing mutations and minimizing unintentional cleavage,23,28 improvements in CRISPR-Cas9 targeting specificity will broaden its clinical applications. Recently, a novel tweak to CRISPR technology, prime editing,29 directly writes new genetic information into a specified DNA site using a catalytically impaired Cas9 fused to an engineered reverse transcriptase, programmed with a prime editing guide RNA (pegRNA) that both specifies the target site and encodes the desired edit. Prime editing offers efficiency and product purity advantages over HDR with much lower off-target editing than Cas9 nuclease at known Cas9 off-target sites.
One of the earliest studies utilizing CRISPR-Cas9 for somatic in vivo repair editing was performed in a mouse model for tyrosinemia to correct a mutation in the enzyme fumarylacetoacetate hydrolase in hepatocytes.30 The authors found that delivering vectors encoding Cas9 and the corresponding sgRNA for the mutant fumarylacetoacetate hydrolase allele resulted in stabilization of the protein and reduced hepatocellular toxicity. Additionally, other studies have been able to correct PTEN, dystrophin, and factor IX gene mutations for treatment of hepatocellular carcinoma,31 Duchenne muscular dystrophy (DMD),32 and hemophilia,33 respectively. One of the most promising applications of CRISPR-Cas9 for gene therapy is ex vivo chemokine receptor 5 (CCR5) deletion, which results in almost complete resistance to HIV-1 infection.34 Studies have shown that CCR5 disruption is possible in ex vivo expanded CD4+ T cells and hematopoietic stem/progenitor cells and that long-term therapeutic effects are achievable using CRISPR-Cas9 gene editing.35
Recently, the first human CRISPR-Cas9 clinical trial has been approved in the United States. This trial proposes to isolate, genetically modify, and reinfuse T cells into patients for treatment of multiple myeloma, sarcoma, and melanoma (ClinicalTrials.gov: NCT03399448). Another trial proposes to treat hematopoietic stem cells of sickle cell anemia patients to increase the production of fetal hemoglobin in their red blood cells (ClinicalTrials.gov: NCT03745287). Although it is too early to determine whether these therapies are successful, CRISPR-Cas9-based gene editing has begun to move out of research laboratories into clinics worldwide.
Genome Editing for NF1. Despite the promise, clinical translation of the Cas9-sgRNA technology for disease treatment is contingent on the development of a delivery system that can efficiently and safely target cells (described below).36 While genome editing has not yet been attempted as a therapy for NF1, it would be applicable to most small mutations. NF1 patient-specific cell lines and mouse models have been developed that can be used for studies to investigate the ability and efficiency of the CRISPR-Cas9 system to edit NF1. As discussed below, CRISPR-Cas9 and prime editing could be delivered using synthetic nanoparticles. These nanoparticles can either deliver Cas9 and sgRNA genetic material (as either plasmid DNA or mRNA) or Cas9-sgRNA ribonucleoprotein complexes.37, 38, 39, 40, 41, 42, 43, 44, 45, 46 However, decreasing off-target editing may be especially relevant for NF1 due to the presence of multiple pseudogenes with high homology to some regions of NF1.
Sequence Correction on the RNA Level
Another way to restore function to proteins of mutant genes is correction of the gene transcript or mRNA. Different mechanisms for RNA editing have been proposed, most notably spliceosome-mediated RNA trans-splicing (SMaRT),47 as well as engineered trans-splicing ribozymes.48 These can be used to repair most types of mutations (outside of whole-gene deletions).
SMaRT. The SMaRT approach was first introduced by Puttaraju et al.47 in 1999. The idea is to use the existing splicing machinery in cells in combination with a so-called pre-trans-splicing molecule (PTM) to reprogram or repair endogenous primary transcripts. A PTM is an exogenous RNA molecule that has been genetically engineered for a specific target pre-mRNA. Different PTM designs have been proposed,49 all of which have in common that they contain three key elements: a trans-splicing domain for spliceosome recognition and splicing, one or two binding domains complementary to one or two intronic subsequences of the target’s pre-mRNA, and a coding domain (i.e., an exon) that is used to reprogram the target pre-mRNA. Depending on the PTM design, one of three reprogramming modes can be achieved: (a) a functional 5′ splice site (ss) in the PTM can trans-splice to a specific 3′ splice site in the target pre-mRNA, yielding replacement of the pre-mRNA 5′ end by the coding exon; (2) conversely, a 3′ ss domain can trans-splice to a 5′ ss in the target pre-mRNA, leading to 3′ end replacement; or (3) a PTM with both a 3′ ss and a 5′ ss, with the coding domain in between, can trans-splice to a 5′ ss and 3′ ss in the target pre-mRNA, respectively, thereby producing an mRNA that has an entire coding region replaced. When the PTM is bound to the target RNA, the spliceosome is guided by the splice sites on both molecules and recombines the target pre-mRNA together with the PTM’s coding domain according to the PTM’s mode of action, thereby producing a reprogrammed messenger. As part of a therapy, any PTM would have to be delivered to the nucleus of target cells in order to be able to re-program the pre-mRNA target. Delivery of PTMs in the form of DNA templates, rather than pre-synthesized, has been shown to be suitable.
A limitation with respect to the target’s pre-mRNA is that spliceosome-mediated trans-splicing can only occur at intron-exon junctions, i.e., at all naturally existing splice sites in pre-mRNA. This is a potential disadvantage compared to ribozyme-based trans-splicing, as ribozymes are able to act on both primary transcripts and mature messengers. Alternatively, the splicing machinery is very robust and efficient, in principle allowing for efficient reprogramming.49 More concerning has been the observation that SMaRT-based trans-splicing can lead to non-targeted trans-splicing of endogenous pre-mRNA.50 Such lack of specificity has raised the question of how safe such a form of gene therapy would be, and how to improve the design to reduce off-target effects and unwanted chimeric proteins.
SMaRT-based RNA reprogramming has been implemented in a number of scenarios, including in cultured cells, in xenograft models and in animal models47,49, 50, 51 and addressing a number of diseases, such as cystic fibrosis,51 DMD, Huntington disease, SMA, and hemophilia A, to name a few. We refer the reader to Berger et al.52 for a recent review of SMaRT for gene therapy, including a list of applications.
Engineered Trans-Splicing Ribozymes for (Pre-)mRNA Repair. Ribozymes are RNA molecules with catalytic properties similar to those of protein enzymes.53 Among the most advanced tools for correcting mutant gene transcripts are trans-splicing group I intron ribozymes.48 These are derived from naturally occurring, cis-splicing group I intron ribozymes and re-engineered to specifically recognize a target site on a mutant RNA, splice the RNA at this site, and replace the mutated portion with the corrected sequence.54 This sequence is provided by the trans-splicing ribozymes. Figure 2A depicts the secondary structure of the group I intron ribozyme from Tetrahymena thermophila. Depending on their design, they can replace the RNA’s 3′-portion with their own 3′-tail (Figure 2B),54,55 the RNA’s 5′-portion with their own 5′-tail (Figure 2C),55,56 or create an internal fragment deletion (Figure 2D). The repaired transcript then translates into fully functional protein. Most ribozymes for trans-splicing purposes are based on the group I intron ribozyme from the organism T. thermophila. This is the most extensively studied and most robust group I intron ribozyme for different designs in trans-splicing.48 It has been designed to repair or reprogram aberrant RNAs in various human disease scenarios by replacing the 3′-portion of mRNAs.
Figure 2.

Trans-Splicing Ribozymes
(A) Secondary structure of the group I intron ribozyme from Tetrahymena thermophila, in its re-designed format for 3′-replacement trans-splicing. All splice sites are indicated by black triangles. (A) The ribozyme (black) recognizes the target site on the substrate (red) with its 5′-terminus and a 5′-terminal extension (green) that enhances trans-splicing efficiency. During the trans-splicing reaction (gray arrow) the ribozyme 3′-tail (blue) replaces the substrate 3′-fragment. This reaction can be used to repair genetic mutations on the RNA level if the mutation is located in the substrate 3′-fragment and the ribozyme’s 3′-tail contains the correct sequence. (B–D) Three different modes of trans-splicing ribozymes. All splice sites are indicated by black triangles. (B) 3′-Replacement ribozymes (black) bind to the target site on the substrate (red) with their 5′-terminus. During the trans-splicing reaction their 3′-tail (blue) replaces the 3′-fragment of the substrate. (C) 5′-Replacement ribozymes (black) bind to the target site on the substrate (red) with their 3′-terminus. Their 5′-tail replaces the 5′-fragment of the substrate. (D) Internal fragment deletion ribozymes (black) bind to two target sites on the substrate (red/blue/red) with their 5′-terminus and with their 3′-terminus. Trans-splicing removes the fragment between the splice sites (blue) and joins the flanking portions of the product (red).
Clinically relevant mutations in the mRNAs of β-globin57 and myotonic dystrophy protein kinase (DMPK)58 were repaired to address the genetic diseases sickle cell anemia and myotonic muscular dystrophy. Using engineered ribozymes of this type, mutant β-globin was converted into γ-globin, both in vitro and in erythrocyte precursors derived from sickle cell patients, while DMPK was repaired both in vitro and in human fibroblasts. In addition, mutant canine skeletal muscle chloride channel (cClC-1) mRNA that is causing heritable myotonia was repaired by replacing the mutant mRNA’s 4-kb-long 3′-portion with its corresponding wild-type sequence.59 As a form of anti-cancer therapy, mutations in the mRNAs of p16,60, p53,61,62 and KRas63 were targeted and either repaired (p16, p53) or reprogrammed to induce cell death (KRas). Moreover, trans-splicing group I intron ribozymes have been developed that reprogram hepatitis C virus and dengue virus RNA and induce apoptosis of infected cells, while other types of ribozymes have been proposed for addressing other infectious diseases, most notably HIV. We refer to Lee et al.64 for a recent review of therapeutic applications of trans-splicing group I intron ribozymes to inheritable genetic diseases, infectious diseases, and cancers.
The trans-splicing ribozymes described above are all 3′-replacement ribozymes, which means that they replace the 3′-portion of a (mutated) mRNA with their own 3′-tail that contains the “healthy” sequence. However, different re-designs of group I intron ribozymes have allowed replacement of the 5′-terminus of an mRNA,56 or deletion of an internal sequence of a target RNA.65,66 The deletion of internal sequences combines the principles of 3′-replacement and 5′-replacement into one ribozyme. Since there are two ways for substrate recognition in 5′-replacement splicing, there are also two different ways for the removal of internal sequences (reviewed in Müller55). None of the principles that use 5′-replacement is currently advanced far enough for clinical applications. One recognition principle leads to the efficient removal of a single nucleotide but is inefficient for longer sequences.66 The other recognition principle allows the removal of 100-nt-long internal sequences from RNA in bacterial cells but currently has requirements on the substrate sequence that may only be met in very specific substrates.65 However, it may be possible to overcome all of these challenges using combinatorial approaches, which optimize the sequences of these ribozymes and/or their flanking sequences in experiments of directed evolution.55 Therefore, the coming years may bring more efficient ribozymes for the repair of mRNA 3′-termini and mRNA 5′-termini, and of the removal of internal mRNA sequences by either generating new designs of trans-splicing ribozymes or by the evolutionary optimization of existing ribozyme designs.
RNA Editing for NF1. When considering editing NF1 with trans-splicing ribozymes, the size of NF1 is an important factor in determining a suitable delivery method. For several viral vectors such as AAV, the delivered construct size can only be up to 4 kb. Together with the ribozyme size of ~400 nt, this constrains the repair template to ~3.5 kb in length. This means that mutations can be repaired only in the last 3.5 kb of NF1 with a single construct; those in the middle of the gene can currently not be repaired by a single trans-splicing event. Alternatively, nanoparticles may make a suitable delivery vehicle. In addition, it is unclear whether the repair efficiencies of current trans-splicing ribozymes are medically realistic for the treatment of NF1. Current trans-splicing ribozymes require relatively strong transcription promoters, and therefore for expression levels to be effective. For example, the strong bacteriophage T7 RNA polymerase promoter, with the co-expression of T7 RNA polymerase, led to the repair of up to 50% of mRNAs, while a medium-strength RNA polymerase II promoter led to the repair of up to 9% of mRNAs.67 Ribozymes from species other than Tetrahymena, or design-optimized variants of the existing Tetrahymena ribozyme constructs, may be able to overcome these hurdles to clinical applications, not only for 3′-replacement repair, but also for 5′-repair and for the repair of internal mRNA segments.
Antisense Oligonucleotides and Therapeutic Targeting of Disease-Associated Transcripts
Antisense oligonucleotides (AOs) are valuable tools for the manipulation of gene expression. AOs are synthetic polymers, usually of 15–30 deoxynucleotides, with sequence complementary to pre-mRNA transcripts. They can be used to inhibit gene expression, drive exon inclusion, mask cryptic splice sites (CSSs), and skip exons.68 Different chemistries of AOs are associated with various advantages and disadvantages, and several chemistries have been through clinical trials or are under development.69 For example, phosphorodiamidate morpholino oligomer (PMO) chemistry has clear safety profiles.70 2ʹ-O-methoxyethyl phosphorothioate-modified backbones have also been successful in clinical trials.71 Cellular uptake of AOs can be improved through their conjugation to cell-penetrating peptides (CPPs); this has worked particularly well for PMOs.72
There are at least two ways to use AOs to inhibit gene expression. One is to target critical regulatory regions such as the translational start codon of the mature mRNA73, 74, 75, 76 or the polyadenylation signal in the 3′ UTR (untranslated region).77,78 An alternative method for inhibition of gene expression would be to design AOs to skip exons that result in an out-of-frame transcript.79,80 Such AOs mask exonic splice enhancer (ESE) motifs in out-of-frame exons within the pre-mRNA. As the cellular splicing machinery no longer recognizes the exon, it is spliced out with their neighboring introns, resulting in a transcript with a disrupted reading frame. This brings a premature termination codon (PTC) into frame, resulting in NMD of the transcript81 and effective inhibition of protein expression.82
AOs can additionally be used to drive exon inclusion.83, 84, 85 For example, SMA, a severe muscle wasting disease, is caused by mutations in the SMN1 gene, which normally encodes SMN protein that maintains motor neuron function. A second copy of the SMN gene (SMN2) also exists, but due to alternative splicing of the exon 7, the majority of SMN protein produced by SMN2 is truncated and unable to compensate for the loss of SMN1. Increasing full-length SMN protein production by promoting exon 7 inclusion in SMN2 mRNA is therapeutic. This has been mediated through the use of an AO targeting the exonic splice silencer motifs of exon 7.71 This AO, with the tradename Spinraza (nusinersen), received accelerated clinical approval in December 2016 on the basis of its performance in clinical trials.86
The ability of AOs to alter pre-mRNA splicing has also been exploited to correct both gain-of-function and loss-of-function genetic mutations that cause disease. Treatable gain-of-function mutations are those that result in creation of a CSS and maintenance of the transcript reading frame. These CSS mutations can either be within an exon or an intron so that a truncated or longer protein is, respectively, expressed with altered function. AOs can be designed to mask these CSSs so that the correct splicing of the pre-mRNA is restored.87,88 Treatable loss-of-function variants may include pathogenic missense variants and those that alter the transcript reading frame, bringing a PTC into frame so that the transcript undergoes NMD and no functional protein is expressed.
By designing AOs to mask the ESE motifs of an out-of-frame exon that neighbors a loss-of-function deletion,89 it is possible to induce the splicing out of the target (mutated) exon so that the reading frame is restored and truncated but partially functional protein is expressed (Figure 3). This is termed exon skipping and has been applied to a number of diseases, as well as in the manipulation of alternative splicing for proteins that can be expressed as different isoforms.90
Figure 3.

Exon Skipping
Exon skipping is a form of RNA splicing where faulty, misaligned, or targeted coding sections of genetic sequence (exons) are “skipped,” leading to truncated but functional proteins. Skipping is mediated by antisense oligonucleotides (AOs) (denoted in red), which are short, synthetic pieces of modified RNA or DNA that hybridize to RNA via base pairing, causing evasion of the splicing machinery, i.e., skipping. Here, the top line denotes the exon-intron structure of a gene with a mutation in exon 4 (denoted with a red star). When an AO to exon 4 hybridizes, it masks the exon 4 splice site so that the splicing machinery does not recognize it and a transcript is created without it (middle line). This results in a protein that is missing exon 4 (bottom line). If this protein maintains the reading frame and exon 4 does not contain a domain critical for protein function, the product may retain function.
The most advanced exon skipping therapy is the one developed for the severe muscle wasting disease, DMD.91 DMD is caused by an array of loss-of-function mutations of the DMD gene. The lack of dystrophin in skeletal and cardiac muscle results in instability of the muscle fiber membrane, such that with contraction the fiber becomes damaged. Repeat degeneration and regeneration results in exhaustion of the stem (satellite) cell population so that the muscle becomes atrophied and fibers are replaced by inflammatory and fibrotic tissue.92
The DMD gene is the largest in the human body, and the disease is associated with mutation hotspots. Targeted skipping of exon 51 has the highest patient applicability at 15%, and skipping of exon 53 could apply to 8% of patients.93 It has been calculated that overall, 60% of DMD patients could be treated by skipping of a single exon, and a further 23% with skipping of two exons.93 This would require the optimization and clinical work-up of numerous AOs, and for double exon skipping an improvement in efficacy. As of September 2016, an AO targeting exon 51 has been conditionally approved by the FDA. Similar accelerated approval has recently been granted to Vyondys 53 (golodirsen) injection to treat DMD patients who have a confirmed mutation in exon 53. Approval is based on the ability of the AOs to stabilize the disease. The ability to halt the disease in its tracks has been a major step forward for a condition that only had symptomatic medicines available previously.
The therapeutic potential of AOs is promising. AOs could also be used in NF1 to target CSS mutations and for exon skipping. The literature indicates that CSS created by deep intronic mutations within NF1 can be skipped in vitro.94,95 One manuscript describes using antisense morpholino oligomers (AMOs) to successfully target the newly created 5′ splice sites to restore normal splicing in fibroblasts and lymphocyte cell lines with three different deep intronic mutations (c.288+2025T>G, c.5749+332A>G, and c.7908−321C>G). This study showed an AMO-dependent decrease in Ras-GTP levels, which is consistent with the restoration of neurofibromin function. The second study assessed c.3198−314G>A and noted leakiness of the splicing mechanism that generated a proportion of correctly spliced transcripts and demonstrated correction of the splicing defect by using specific AMOs.
Exon Skipping for NF1. While repression of CSSs is limited to a few very specific mutations, constitutive exon skipping offers the possible benefit of skipping over any mutation within the region of interest and potentially helping many patients with different mutations; however, this technology has not yet been applied to NF1. It is possible that exon skipping may lead to the expression of a truncated but functional neurofibromin protein, as has been seen in DMD. The DMD gene has distinct advantages for the application of exon skipping as a therapy. DMD has mutation hotspots that are lacking for NF1. The DMD gene is also associated with a less severe muscle wasting disease called Becker muscular dystrophy (BMD), in which a truncated protein with partial function is expressed. BMD mutations are generally associated with in-frame mutations within regions of the DMD gene that encode for the central rod domain repeats of the dystrophin protein. It would appear that some of these repeats are dispensable.96 For NF1, there is no less severe condition associated with the disease gene, and it is not yet known whether any of the protein motifs are dispensable. Indeed, the function of many of the NF1 protein motifs is not known.
Nonsense Suppression Therapies
Nonsense mutations are single DNA base-pair changes that generate an in-frame PTC in the transcribed mRNA (Figure 4). A PTC reduces protein expression by two mechanisms. First, a PTC prematurely terminates translation of an mRNA, preventing a full-length protein from being generated. The resulting truncated protein is often unstable and/or non-functional. Second, a PTC can trigger NMD, which targets mRNAs containing a PTC for degradation, preventing their translation and further reducing protein levels. These PTC-mediated mechanisms severely reduce the level of full-length, functional protein produced in cells. Nonsense mutations comprise 11% of all disease-causing gene lesions,97 and a subset of patients with many different types of genetic diseases harbor this class of mutation.
Figure 4.

Nonsense Suppression Therapeutics
(A) When normal translation occurs, a full-length functional polypeptide is generated. Translation is terminated when the ribosome encounters a normal stop codon (UAA is denoted in red). (B) When a ribosome encounters a premature termination codon (PTC) that was generated by a nonsense mutation, the ribosome terminates the transcript and a truncated protein is produced. Generally, the truncated transcript undergoes NMD, which lowers the cellular levels of this protein product. (C) Aminoglycosides and other small molecules can induce the ribosome to readthrough a PTC. If this occurs, one of three possible products may be made; these are not mutually exclusive. The correct amino acid may be inserted in place of the PTC; this leads to a full-length functional protein. Alternatively, a different amino acid may be inserted in place of the PTC; this leads to a missense mutation. This missense will either lead to a functional protein or a non-functional protein.
The suppression of translation termination at PTCs is being explored as a potential therapeutic approach to treat protein deficiencies caused by nonsense mutations.98 Nonsense suppression therapy works at the level of protein synthesis. During translation of an mRNA into a protein, codons in the ribosomal acceptor (A) site are normally sampled by aminoacyl-tRNAs until one that carries a complementary anticodon is encountered. The ribosome proofreads codon-anticodon interactions to ensure that the correct amino acid is inserted into the nascent polypeptide. Termination codons serve as signals for the ribosome to stop translating an mRNA and to release the nascent polypeptide. Rather than being recognized by tRNAs, termination codons are recognized by eRF1, which is part of a group of interacting proteins called the termination complex. The termination complex binds to a stop codon and mediates peptide release from the ribosome. Normally, the process of translation termination is highly efficient, where termination is suppressed at a termination codon less than once per 100 translation events.98 However, termination can be suppressed at low levels due to the occasional aberrant binding of an aminoacyl-tRNA to an acceptor site-bound PTC, resulting in the incorporation of an amino acid at the site of the PTC. The suppression of termination at PTCs via amino acid incorporation (also called readthrough) leads to continued translation elongation in the same ribosomal reading frame until the natural termination codon is encountered, resulting in the generation of a full-length protein. The goal of this therapeutic strategy is to restore enough protein function to alleviate genetic diseases that are caused by nonsense mutations. Current strategies to suppress termination at PTCs include the use of suppressor tRNAs or pharmaceuticals that target the translational machinery to induce readthrough.
PTC Suppression Using Suppressor tRNAs. This strategy utilizes an aminoacyl-tRNA whose anticodon has been altered to become complementary to a stop codon. When the suppressor tRNA is expressed in mammalian cells, an amino acid can be incorporated into a PTC to generate full-length, functional protein. This strategy was shown to suppress termination at nonsense mutations associated with β-thalassemia,99 xeroderma pigmentosum,100 DMD,101, and Ullrich disease102 in mammalian cells. Furthermore, injection of suppressor tRNAs into mouse tissues locally restored protein activity.101
One of the limitations of using suppressor tRNAs to suppress PTCs is that the suppressor tRNAs must be properly charged by specific aminoacyl synthetases and delivered to the ribosome by eukaryotic elongation factor 1α (eEF1α). It is difficult to predict whether the modification of a tRNA will alter its charging or delivery. However, a recent study identified large sets of tRNAs that can efficiently serve as suppressor tRNAs when expressed in mammalian cells or when injected into mouse tissues.103 These studies indicate the potential promise of utilizing suppressor tRNAs to suppress termination at PTCs. However, the safe, effective, and precise delivery of suppressor tRNAs remains a challenge for developing suppressor tRNAs as a therapy for nonsense mutations.
Readthrough Using Pharmaceuticals. The frequency of readthrough can also be increased to restore partial levels of full-length, functional protein through the use of pharmaceuticals that target the translational machinery. Several low-molecular-weight compounds have been identified that induce readthrough of PTCs.98,104 The best characterized readthrough agents are the aminoglycosides and ataluren (also called PTC124 or Translarna). Aminoglycosides, a class of antibiotic, have been shown to bind specifically to the decoding region of the ribosome, which increases amino acid incorporation at PTCs.105 Ataluren is a non-antibiotic compound that was identified from a screen of low-molecular-weight compounds as a readthrough agent that suppresses PTCs in mammalian cells.106 While these compounds have been found to restore significant protein function via readthrough in several disease models,98,107 obstacles associated with each of these compounds may prevent their use for long-term nonsense suppression therapy.
Aminoglycosides bind off-target sites in the kidney and the inner ear, which can lead to toxicity in these organs.108,109 Designer aminoglycosides have recently been generated that are more efficient at suppressing PTCs while also having reduced toxicity.110 Phase 1 safety studies are underway to determine whether one of these new designer aminoglycosides, ELX-02, is safe for clinical use (ClinicalTrials.gov: NCT03309605 and NCT03776539).
Ataluren has been shown to have a favorable safety profile.107 However, the results of a recent clinical trial in which cystic fibrosis patients who harbor nonsense mutations were administered ataluren did not find significant improvements in patient lung function,111 suggesting that ataluren was unable to restore enough CFTR protein function to alleviate this disease. Based on promising preliminary results, ataluren has been granted conditional approval to treat patients with DMD in the European Union (via PTC Therapeutics website: https://www.ptcbio.com/our-pipeline/approved-medicines), and additional studies are in progress to determine whether ataluren is effective in treating DMD in patients who carry nonsense mutations (ClinicalTrial.gov: NCT03179631). Given the potential disadvantages of the current compounds available for nonsense suppression therapy, additional safe, efficacious readthrough compounds are needed for clinical development.
A recent approach that has been explored to enhance the effectiveness of nonsense suppression therapy is to combine readthrough with NMD inhibition. Because a PTC often elicits NMD, which leads to degradation of an mRNA and prevents its translation, inhibiting NMD would increase the level of PTC-containing mRNA for translation and subsequent nonsense suppression.98 Proof-of-principle studies have verified that inhibiting NMD does enhance nonsense suppression, leading to the restoration of greater protein function than by nonsense suppression alone.112,113 Although several NMD inhibitors have been identified, none has yet been assessed in long-term in vivo studies for safety and efficacy.
Context Effects on PTC Suppression. Recent studies have revealed that only a subset of near-cognate aminoacyl-tRNAs, those with an anticodon complementary to two of the three nucleotides of a PTC, are accommodated during PTC suppression.114,115. The amino acids generally incorporated at UAA and UAG include glutamine, tyrosine, and lysine, whereas arginine, tryptophan, and cysteine are generally inserted at UGA.114,115 However, the identity and proportions of amino acids inserted at a PTC during readthrough varies depending on the mRNA sequence context surrounding the PTC and the method of stimulating readthrough. Furthermore, the amino acid that becomes inserted at a PTC can alter the overall stability and/or activity of the protein restored via readthrough.114,115 The local mRNA context surrounding a PTC has also been shown to affect basal levels of readthrough, as well as the susceptibility of a PTC to readthrough by different compounds.116, 117, 118, 119, 120 Context also likely plays a role in the effectiveness of suppressor tRNAs.103 Taken together, these data suggest that the local mRNA context plays a critical role on PTC suppression and is a very important factor to consider when attempting to identify compounds that efficiently readthrough a PTC and restore physiologically relevant protein function within a disease setting.
Readthrough of Normal Stop Codons Is Not Significant. Evidence suggests that global readthrough of natural stop codons does not occur at significant levels with either suppressor tRNAs or with readthrough compounds. Ribosomal profiling was used to examine the ribosomal footprint densities in the 3′ UTRs of mammalian transcripts when cells expressed a suppressor tRNA or when cells were treated with the strong readthrough compound, G418.103 Less than a 2-fold change in 3′ UTR ribosomal density in G418-treated compared to controls was found, suggesting that readthrough of natural stop codons occurred infrequently as a result of PTC readthrough. The 3′ UTR ribosomal density in cells expressing different suppressor tRNAs increased from 2- to 4-fold compared to controls, suggesting that some suppressor tRNAs may suppress termination at natural PTCs, and this is likely to be a consideration when examining different suppressor tRNAs as a potential therapy. Consistent with these results, a study that used two-dimensional electrophoretic analysis to assess global shifts in protein mobility found that the readthrough compound ataluren showed no evidence of natural stop codon readthrough.106 These results support the hypothesis that termination at PTCs is less efficient than at normal stop codons due to differences in the ribonucleoprotein structure that is influenced by the interaction between the termination complex and poly(A) binding protein (PABP), which is bound to the poly(A) tail.121,122
PTC Suppression for NF1. Nonsense mutations are among the most prevalent class of recurring mutations identified in NF1 patients,123 with 20% of NF1 cases caused by germline nonsense mutations.124 This suggests that therapies aimed at overcoming the effects of PTCs could benefit a significant subset of NF1 patients. Both suppressor tRNAs and pharmaceuticals are promising therapies for NF1. Because the level of neurofibromin protein function needed to rescue phenotypes in NF1 patients has not yet been determined (discussed below), it remains unclear whether nonsense suppression therapy will be able to restore enough NF1 function to completely alleviate the disease. Typically, 25%–35% of normal protein function needs to be restored for rescuing phenotypes associated with cystic fibrosis125 or DMD.126 Separate Nf1 mouse models have been developed that carry the recurring R681X and R816X mutations that can be used for future studies to investigate the ability of current and developing nonsense suppression therapies to alleviate NF1.127 With the continued discovery of new readthrough compounds, as well as the identification of novel therapeutic targets for translation and NMD, it is likely that nonsense suppression will eventually be explored as a precision medicine approach for NF1.
Level of Neurofibromin Required to Restore Normal Phenotype
While for some diseases, restoration of even minimal levels of protein function may be therapeutic, it is not known how much neurofibromin might be required to restore a normal or near-normal phenotype. There are multiple mitigating factors and data that influence our thinking about the protein level required for clinical improvement. First, affected individuals with the same mutation, including individuals in the same family, may differ in phenotypic severity, suggesting that the same level of neurofibromin restoration may have different therapeutic effects in one patient versus another, even in the case of two individuals with similar genetic background and an identical NF1 mutation. Second, different heterozygous patient mutations lead to different levels of expression of mutant and normal NF1 alleles and, consequently, different (combined mutant and wild-type) neurofibromin protein levels within patient fibroblasts, ranging from 12% to 89% of normal levels.128 Unfortunately, protein expression levels do not equate to function, as significantly elevated Ras activity (based on 2- to 3-fold increased levels of RAS-GTP) was observed across the entire range of neurofibromin levels in these fibroblasts.128 Third, as described in Li et al.,127 Nf1+/G848R mouse embryonic fibroblasts (MEFs) appear to have normal levels of neurofibromin (100%), while homozygous null G848R MEFs have only about half of the normal levels, resulting in an ~2.5-fold increase in phosphorylated (p-)ERK compared to wild-type cells. Interestingly, this is similar to the effect of another heterozygous knockout allele, Nf1+/Δ4, which also has 50% neurofibromin levels and 2.5-fold increased p-ERK. Despite the reduction in neurofibromin and increase in p-ERK, homozygous G848R mice are phenotypically normal. This suggests that restoration of at least 50% neurofibromin function may rescue some in vivo phenotypes but not p-ERK phenotypes, indicating that outcomes of molecular (or other) endpoint assays may differ depending on the phenotype in question. Fourth, NF1 phenotypes are cell type-specific, and so are NF1 expression levels. Consequently, the amount of functional neurofibromin required may differ based on cell type. A recent study of homozygous G848R mice under glial fibrillary acidic protein (GFAP)-Cre129 found that in brainstem astrocytes 60% of (mutant) neurofibromin is present, enough to maintain many in vivo phenotypes that other Nf1 loss-of-function mutations cannot, including optic nerve volume, lack of hyper-cellularity and nuclear atypia, cell proliferation rates, prevention of optic nerve axonal injury and retinal apoptosis, retinal ganglia cell number, retinal nerve fiber layer thickness, Ras activation in terms of p-Akt, p-ERK, and p-S6 (mTor), and microglial number and function. Fifth, neurofibromin has been shown to form dimers,21,130 but depending on the mutation, a mutated neurofibromin may not be able to form dimers, not even with wild-type neurofibromin. Alternatively, a mutated neurofibromin can affect the functionality of the dimer. In the worst case, mutated neurofibromin sequesters functional neurofibromin, obtained from a normal allele (or a delivered gene), into non-functional dimers. The level of functional dimers needed for a normal or near-normal phenotype is not known, but may again depend on the cell type. Lastly, neurofibromin may well have other functions currently unknown that are not captured by existing assays. Hence, it is also not yet possible to determine how much neurofibromin is required for these unknown functions.
Delivery of NF1 Gene Therapeutic
A major hurdle that limits the clinical translation of gene therapy has been the lack of safe and efficient delivery vector. The safety and efficacy of gene therapies are largely dependent on the vehicle used to deliver the genetic material to the target cells. During the last five decades, various types of vehicles for the delivery of therapeutic genes (or messengers) to target cells have been proposed and investigated.131 So far, however, no delivery technology has emerged that allows simple replacement of any mutated, disease-causing gene with its corresponding reference gene anywhere (or everywhere) in the human body, and without side effects for the patient. It appears that there is no “one-fits-all” technology and, instead, each disease requires its respective fine-tuned vehicle design and administration.
Indeed, as existing approaches show, there are a number of important aspects that any delivery strategy has to consider in the context of the targeted disease and with respect to the therapeutic. These are as follows: (1) type of delivery vehicle: plasmid, viral vector, human artificial chromosomes, engineered capsids, patient-derived exosomes, nanoparticles; (2) site of transduction: ex vivo, i.e., patient-derived cells (e.g., HSCs and T cells) are modified outside the body and then transplanted back, versus in vivo, i.e., the vector is administered to the patient; successful transduction depends on vector and cell membrane properties; and (3) in case of in vivo transduction: vector delivery (targeted injection, systemic delivery). Below we focus on viral vectors and engineered nanoparticles.
Delivery with Viral Vectors. The idea of using viral vectors as vehicles to deliver a drug or a gene to patient cells has been discussed since the 1970s.132 Currently, engineered viral vectors, such as AAVs,133 adenoviruses,134 and lentiviruses,135 are widely used as the main delivery method for gene therapy due to their high efficacy to hijack the host cell and replicate the desired genetic material. However, viral-based delivery methods are limited in the size of the package they can carry and have been shown to result in high immunogenicity and cytotoxicity in the target tissue.136,137 Primarily due to size limitations, they are not suitable for NF1 gene replacement.
Novel Vectors: Nanoparticles for Delivery. Given the safety, efficiency, and size limitations of viral vectors, non-viral synthetic nanoparticle-based vectors are becoming increasingly attractive. While traditionally they have suffered from low efficiency or high toxicity,137, 138, 139 the recent progress in developing novel nanoparticles that can deliver gene therapy with high efficiency and limited toxicity has made this approach appealing.140, 141, 142 New generation lipid nanoparticles such as BAMEA-O16B boast high efficiency of in vitro transfection (90% in HEK293 cells) and high in vivo efficacy after intravenous injection, with the ability to knock down protein activity levels to 20% of nontreatment levels.143
Mechanistically, once nanoparticles enter the bloodstream, they are coated with plasma proteins from physiological solutions and internalized by cells via endocytosis (Figure 1). Endocytosis efficiency of nanoparticles is dependent on a wide variety of factors, including the size, shape, and surface chemistry of the nanoparticle, as well as the type of cell with which the nanoparticle is interacting. Before they are degraded in the lysosome, the nanoparticles need to escape from the endosome into the cytoplasm, where the cargo is released. In this example, CRISPR-Cas9 then penetrates into the nucleus via nuclear-localization-signal peptides on Cas9. Once in the nucleus, the Cas9-sgRNA complex can search for the target DNA locus and induce specified deletions or modifications for gene editing (Figure 1).
Although nanoparticles are promising non-viral delivery methods, there are still major hurdles that must be addressed before they can reach their full potential. A key challenge is designing nanoparticles that are able to effectively deliver their cargo to the target location past extracellular and intracellular obstacles. The biodistribution, circulation, and internalization of nanoparticles are all features that must be optimized to ensure the greatest delivery efficiency. Additionally, immune responses and cytotoxicity elicited from their payload may limit their therapeutic potential. While coating with albumin, dextran, or polyethylene glycol (PEG) may reduce these adverse effects, nanoparticle uptake and biological activity may also be diminished. Another challenge is designing materials that can effectively encapsulate and deliver both proteins and nucleic acids. While many nanoparticles use multiple components to achieve this, simpler formulations can increase their translational potential.
Gene Replacement Using Nanoparticles. Synthetic nanoparticles, particularly polymeric nanoparticles, encapsulate cargo genetic materials through electrostatic interaction and entanglement, and, therefore, may deliver DNA without size limitation.140,144 As such, synthetic nanoparticles have been extensively explored for delivery of gene replacement therapy for various diseases, such as the CFTR gene for treatment of cystic fibrosis, and IL-12 gene for various cancers.145 Among them, a CFTR gene replacement clinical trial has entered the phase IIB stage.146
Genome Editing Using Nanoparticles. In current experimental studies and clinical tests, CRISPR-Cas9 is often delivered using viral vectors.147 In addition to safety concerns,148 the viral approach does not allow co-delivery of donor DNA for repair and is unsuitable for delivery of the precise genome editing system.149 To overcome these limitations, non-viral approaches using synthetic nanoparticles are promising.38, 39, 40, 41, 42, 43, 44, 45,140 To date, various synthetic nanoparticles, such as gold nanoparticles, lipid nanoparticles, and lipid-templated hydrogel nanoparticles, have been developed for CRISPR-Cas9 delivery, with some of them having demonstrated excellent efficiency in vivo.38, 39, 40, 41, 42, 43, 44, 45, 46,143 Despite this success, many of those reported nanoparticles suffer from various limitations, such as consisting of multiple components and thus bearing a high degree of complexity, and containing non-biodegradable materials and thus posing potential side effects.
Conclusions and Outlook
There are a variety of mutation-directed therapeutics that may potentially be used to treat NF1. Each is at a different stage of clinical development and each has unique advantages and challenges. Moreover, each is applicable to different types of mutations, and no one therapeutic is likely to treat all patients with NF1. It is more likely that a suite of therapeutics will be required for the treatment of NF1. Unresolved issues involve delivery to the correct cells at the correct time and efficiency. Furthermore, it is unknown how much neurofibromin is required to prevent disease phenotypes, and this level may vary based on cell type. We are attempting to overcome some of these challenges and unknowns to implement mutation-directed therapeutics for NF1.
Conflicts of Interest
B.R.K. is the Chair of the Children’s Tumor Foundation Medical Advisory Committee, the Chair of the External Advisory Committee for NTAP and also for the NF Research Initiative, and a member of advisory committees for AstraZeneca and Springworks. G.D. has consultancies with Sarepta Biopharma, AskBio, and RegenX. R.A.K. is a lead advisor for Infixion. D.M.B. is a consultant for PTC Therapeutics, Inc. D.W., R.A.K., B.R.K., A.L., L.P., and G.D. are inventors on US Provisional Patent Application No. 62/903,521 (Exon skipping to treat neurofibromatosis type 1).
Acknowledgments
This work was partially supported through the Gilbert Family Foundation’s Gene Therapy Initiative grants 563635 (to A.L.), 564070 (to R.A.K.), 563676 (to D.M.B.), and 563624 (to D.W.).
References
- 1.Friedman J.M., Gutmann D.H., MacCollin M., Riccardi V.M., editors. Neurofibromatosis: Phenotype, Natural History, and Pathogenesis. Third Edition. Johns Hopkins University Press; 1999. [Google Scholar]
- 2.Kallionpää R.A., Uusitalo E., Leppävirta J., Pöyhönen M., Peltonen S., Peltonen J. Prevalence of neurofibromatosis type 1 in the Finnish population. Genet. Med. 2018;20:1082–1086. doi: 10.1038/gim.2017.215. [DOI] [PubMed] [Google Scholar]
- 3.Buratti E., Baralle D. Exon skipping mutations in neurofibromatosis. Methods Mol. Biol. 2012;867:65–76. doi: 10.1007/978-1-61779-767-5_5. [DOI] [PubMed] [Google Scholar]
- 4.Vandenbroucke I., Callens T., De Paepe A., Messiaen L. Complex splicing pattern generates great diversity in human NF1 transcripts. BMC Genomics. 2002;3:13. doi: 10.1186/1471-2164-3-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thomson S.A., Wallace M.R. RT-PCR splicing analysis of the NF1 open reading frame. Hum. Genet. 2002;110:495–502. doi: 10.1007/s00439-002-0714-6. [DOI] [PubMed] [Google Scholar]
- 6.Lin A.L., Gutmann D.H. Advances in the treatment of neurofibromatosis-associated tumours. Nat. Rev. Clin. Oncol. 2013;10:616–624. doi: 10.1038/nrclinonc.2013.144. [DOI] [PubMed] [Google Scholar]
- 7.Gutmann D.H., Blakeley J.O., Korf B.R., Packer R.J. Optimizing biologically targeted clinical trials for neurofibromatosis. Expert Opin. Investig. Drugs. 2013;22:443–462. doi: 10.1517/13543784.2013.772979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Antoszczyk S., Rabkin S.D. Prospect and progress of oncolytic viruses for treating peripheral nerve sheath tumors. Expert Opin. Orphan Drugs. 2016;4:129–138. doi: 10.1517/21678707.2016.1128322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baker N.M., Der C.J. Cancer: drug for an “undruggable” protein. Nature. 2013;497:577–578. doi: 10.1038/nature12248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim A., Dombi E., Tepas K., Fox E., Martin S., Wolters P., Balis F.M., Jayaprakash N., Turkbey B., Muradyan N. Phase I trial and pharmacokinetic study of sorafenib in children with neurofibromatosis type I and plexiform neurofibromas. Pediatr. Blood Cancer. 2013;60:396–401. doi: 10.1002/pbc.24281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dombi E., Baldwin A., Marcus L.J., Fisher M.J., Weiss B., Kim A., Whitcomb P., Martin S., Aschbacher-Smith L.E., Rizvi T.A. Activity of selumetinib in neurofibromatosis type 1-related plexiform neurofibromas. N. Engl. J. Med. 2016;375:2550–2560. doi: 10.1056/NEJMoa1605943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jessen W.J., Miller S.J., Jousma E., Wu J., Rizvi T.A., Brundage M.E., Eaves D., Widemann B., Kim M.O., Dombi E. MEK inhibition exhibits efficacy in human and mouse neurofibromatosis tumors. J. Clin. Invest. 2013;123:340–347. doi: 10.1172/JCI60578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Watson A.L., Anderson L.K., Greeley A.D., Keng V.W., Rahrmann E.P., Halfond A.L., Powell N.M., Collins M.H., Rizvi T., Moertel C.L. Co-targeting the MAPK and PI3K/AKT/mTOR pathways in two genetically engineered mouse models of Schwann cell tumors reduces tumor grade and multiplicity. Oncotarget. 2014;5:1502–1514. doi: 10.18632/oncotarget.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jousma E., Rizvi T.A., Wu J., Janhofer D., Dombi E., Dunn R.S., Kim M.O., Masters A.R., Jones D.R., Cripe T.P., Ratner N. Preclinical assessments of the MEK inhibitor PD-0325901 in a mouse model of Neurofibromatosis type 1. Pediatr. Blood Cancer. 2015;62:1709–1716. doi: 10.1002/pbc.25546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou L.Y., Qin Z., Zhu Y.H., He Z.Y., Xu T. Current RNA-based therapeutics in clinical trials. Curr. Gene Ther. 2019;19:172–196. doi: 10.2174/1566523219666190719100526. [DOI] [PubMed] [Google Scholar]
- 16.Darrow J.J. Luxturna: FDA documents reveal the value of a costly gene therapy. Drug Discov. Today. 2019;24:949–954. doi: 10.1016/j.drudis.2019.01.019. [DOI] [PubMed] [Google Scholar]
- 17.Ismat F.A., Xu J., Lu M.M., Epstein J.A. The neurofibromin GAP-related domain rescues endothelial but not neural crest development in Nf1 mice. J. Clin. Invest. 2006;116:2378–2384. doi: 10.1172/JCI28341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bai R.Y., Esposito D., Tam A.J., McCormick F., Riggins G.J., Wade Clapp D., Staedtke V. Feasibility of using NF1-GRD and AAV for gene replacement therapy in NF1-associated tumors. Gene Ther. 2019;26:277–286. doi: 10.1038/s41434-019-0080-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wallis D., Li K., Lui H., Hu K., Chen M.J., Li J., Kang J., Das S., Korf B.R., Kesterson R.A. Neurofibromin (NF1) genetic variant structure-function analyses using a full-length mouse cDNA. Hum. Mutat. 2018;39:816–821. doi: 10.1002/humu.23421. [DOI] [PubMed] [Google Scholar]
- 20.Anastasaki C., Le L.Q., Kesterson R.A., Gutmann D.H. Updated nomenclature for human and mouse neurofibromatosis type 1 genes. Neurol. Genet. 2017;3:e169. doi: 10.1212/NXG.0000000000000169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sherekar M., Han S.W., Ghirlando R., Messing S., Drew M., Rabara D., Waybright T., Juneja P., O’Neill H., Stanley C.B. Biochemical and structural analyses reveal that the tumor suppressor neurofibromin (NF1) forms a high-affinity dimer. J. Biol. Chem. 2020;295:1105–1119. doi: 10.1074/jbc.RA119.010934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cui Y., Morrison H. Construction of cloning-friendly minigenes for mammalian expression of full-length human NF1 isoforms. Hum. Mutat. 2019;40:187–192. doi: 10.1002/humu.23681. [DOI] [PubMed] [Google Scholar]
- 23.Heidenreich M., Zhang F. Applications of CRISPR-Cas systems in neuroscience. Nat. Rev. Neurosci. 2016;17:36–44. doi: 10.1038/nrn.2015.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guitart J.R., Jr., Johnson J.L., Chien W.W. Research techniques made simple: the application of CRISPR-Cas9 and genome editing in investigative dermatology. J. Invest. Dermatol. 2016;136:e87–e93. doi: 10.1016/j.jid.2016.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morgan M.A., Lawrence T.S. Molecular pathways: overcoming radiation resistance by targeting DNA damage response pathways. Clin. Cancer Res. 2015;21:2898–2904. doi: 10.1158/1078-0432.CCR-13-3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Doench J.G., Fusi N., Sullender M., Hegde M., Vaimberg E.W., Donovan K.F., Smith I., Tothova Z., Wilen C., Orchard R. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016;34:184–191. doi: 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anzalone A.V., Randolph P.B., Davis J.R., Sousa A.A., Koblan L.W., Levy J.M., Chen P.J., Wilson C., Newby G.A., Raguram A., Liu D.R. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576:149–157. doi: 10.1038/s41586-019-1711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yin H., Xue W., Chen S., Bogorad R.L., Benedetti E., Grompe M., Koteliansky V., Sharp P.A., Jacks T., Anderson D.G. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat. Biotechnol. 2014;32:551–553. doi: 10.1038/nbt.2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang D., Mou H., Li S., Li Y., Hough S., Tran K., Li J., Yin H., Anderson D.G., Sontheimer E.J. Adenovirus-mediated somatic genome editing of Pten by CRISPR/Cas9 in mouse liver in spite of Cas9-specific immune responses. Hum. Gene Ther. 2015;26:432–442. doi: 10.1089/hum.2015.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Long C., McAnally J.R., Shelton J.M., Mireault A.A., Bassel-Duby R., Olson E.N. Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA. Science. 2014;345:1184–1188. doi: 10.1126/science.1254445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guan Y., Ma Y., Li Q., Sun Z., Ma L., Wu L., Wang L., Zeng L., Shao Y., Chen Y. CRISPR/Cas9-mediated somatic correction of a novel coagulator factor IX gene mutation ameliorates hemophilia in mouse. EMBO Mol. Med. 2016;8:477–488. doi: 10.15252/emmm.201506039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cannon P., June C. Chemokine receptor 5 knockout strategies. Curr. Opin. HIV AIDS. 2011;6:74–79. doi: 10.1097/COH.0b013e32834122d7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu L., Yang H., Gao Y., Chen Z., Xie L., Liu Y., Liu Y., Wang X., Li H., Lai W. CRISPR/Cas9-mediated CCR5 ablation in human hematopoietic stem/progenitor cells confers HIV-1 resistance in vivo. Mol. Ther. 2017;25:1782–1789. doi: 10.1016/j.ymthe.2017.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nelson C.E., Gersbach C.A. Engineering delivery vehicles for genome editing. Annu. Rev. Chem. Biomol. Eng. 2016;7:637–662. doi: 10.1146/annurev-chembioeng-080615-034711. [DOI] [PubMed] [Google Scholar]
- 37.Chen Z., Liu F., Chen Y., Liu J., Wang X., Chen A.T., Deng G., Zhang H., Liu J., Hong Z., Zhou J. Targeted delivery of CRISPR/Cas9-mediated cancer gene therapy via liposome-templated hydrogel nanoparticles. Adv. Funct. Mater. 2017;27:1703036. doi: 10.1002/adfm.201703036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zuris J.A., Thompson D.B., Shu Y., Guilinger J.P., Bessen J.L., Hu J.H., Maeder M.L., Joung J.K., Chen Z.Y., Liu D.R. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat. Biotechnol. 2015;33:73–80. doi: 10.1038/nbt.3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zuckermann M., Hovestadt V., Knobbe-Thomsen C.B., Zapatka M., Northcott P.A., Schramm K., Belic J., Jones D.T., Tschida B., Moriarity B. Somatic CRISPR/Cas9-mediated tumour suppressor disruption enables versatile brain tumour modelling. Nat. Commun. 2015;6:7391. doi: 10.1038/ncomms8391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jiang C., Mei M., Li B., Zhu X., Zu W., Tian Y., Wang Q., Guo Y., Dong Y., Tan X. A non-viral CRISPR/Cas9 delivery system for therapeutically targeting HBV DNA and pcsk9 in vivo. Cell Res. 2017;27:440–443. doi: 10.1038/cr.2017.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li L., Song L., Liu X., Yang X., Li X., He T., Wang N., Yang S., Yu C., Yin T. Artificial virus delivers CRISPR-Cas9 system for genome editing of cells in mice. ACS Nano. 2017;11:95–111. doi: 10.1021/acsnano.6b04261. [DOI] [PubMed] [Google Scholar]
- 42.Miller J.B., Zhang S., Kos P., Xiong H., Zhou K., Perelman S.S., Zhu H., Siegwart D.J. Non-viral CRISPR/Cas gene editing in vitro and in vivo enabled by synthetic nanoparticle co-delivery of Cas9 mRNA and sgRNA. Angew. Chem. Int. Ed. Engl. 2017;56:1059–1063. doi: 10.1002/anie.201610209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mout R., Ray M., Yesilbag Tonga G., Lee Y.W., Tay T., Sasaki K., Rotello V.M. Direct cytosolic delivery of CRISPR/Cas9-ribonucleoprotein for efficient gene editing. ACS Nano. 2017;11:2452–2458. doi: 10.1021/acsnano.6b07600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sun W., Ji W., Hall J.M., Hu Q., Wang C., Beisel C.L., Gu Z. Self-assembled DNA nanoclews for the efficient delivery of CRISPR-Cas9 for genome editing. Angew. Chem. Int. Ed. Engl. 2015;54:12029–12033. doi: 10.1002/anie.201506030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang M., Zuris J.A., Meng F., Rees H., Sun S., Deng P., Han Y., Gao X., Pouli D., Wu Q. Efficient delivery of genome-editing proteins using bioreducible lipid nanoparticles. Proc. Natl. Acad. Sci. USA. 2016;113:2868–2873. doi: 10.1073/pnas.1520244113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee K., Conboy M., Park H.M., Jiang F., Kim H.J., Dewitt M.A., Mackley V.A., Chang K., Rao A., Skinner C. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nat. Biomed. Eng. 2017;1:889–901. doi: 10.1038/s41551-017-0137-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Puttaraju M., Jamison S.F., Mansfield S.G., Garcia-Blanco M.A., Mitchell L.G. Spliceosome-mediated RNA trans-splicing as a tool for gene therapy. Nat. Biotechnol. 1999;17:246–252. doi: 10.1038/6986. [DOI] [PubMed] [Google Scholar]
- 48.Fiskaa T., Birgisdottir A.B. RNA reprogramming and repair based on trans-splicing group I ribozymes. N. Biotechnol. 2010;27:194–203. doi: 10.1016/j.nbt.2010.02.013. [DOI] [PubMed] [Google Scholar]
- 49.Garcia-Blanco M.A. Messenger RNA reprogramming by spliceosome-mediated RNA trans-splicing. J. Clin. Invest. 2003;112:474–480. doi: 10.1172/JCI19462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kikumori T., Cote G.J., Gagel R.F. Promiscuity of pre-mRNA spliceosome-mediated trans splicing: a problem for gene therapy? Hum. Gene Ther. 2001;12:1429–1441. doi: 10.1089/104303401750298580. [DOI] [PubMed] [Google Scholar]
- 51.Liu X., Jiang Q., Mansfield S.G., Puttaraju M., Zhang Y., Zhou W., Cohn J.A., Garcia-Blanco M.A., Mitchell L.G., Engelhardt J.F. Partial correction of endogenous ΔF508 CFTR in human cystic fibrosis airway epithelia by spliceosome-mediated RNA trans-splicing. Nat. Biotechnol. 2002;20:47–52. doi: 10.1038/nbt0102-47. [DOI] [PubMed] [Google Scholar]
- 52.Berger A., Maire S., Gaillard M.C., Sahel J.A., Hantraye P., Bemelmans A.P. mRNA trans-splicing in gene therapy for genetic diseases. Wiley Interdiscip. Rev. RNA. 2016;7:487–498. doi: 10.1002/wrna.1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cech T.R. Ribozymes, the first 20 years. Biochem. Soc. Trans. 2002;30:1162–1166. doi: 10.1042/bst0301162. [DOI] [PubMed] [Google Scholar]
- 54.Sullenger B.A., Cech T.R. Ribozyme-mediated repair of defective mRNA by targeted, trans-splicing. Nature. 1994;371:619–622. doi: 10.1038/371619a0. [DOI] [PubMed] [Google Scholar]
- 55.Müller U.F. Design and experimental evolution of trans-splicing group I intron ribozymes. Molecules. 2017;22:75. doi: 10.3390/molecules22010075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Alexander R.C., Baum D.A., Testa S.M. 5′ Transcript replacement in vitro catalyzed by a group I intron-derived ribozyme. Biochemistry. 2005;44:7796–7804. doi: 10.1021/bi047284a. [DOI] [PubMed] [Google Scholar]
- 57.Lan N., Howrey R.P., Lee S.W., Smith C.A., Sullenger B.A. Ribozyme-mediated repair of sickle β-globin mRNAs in erythrocyte precursors. Science. 1998;280:1593–1596. doi: 10.1126/science.280.5369.1593. [DOI] [PubMed] [Google Scholar]
- 58.Phylactou L.A., Darrah C., Wood M.J. Ribozyme-mediated trans-splicing of a trinucleotide repeat. Nat. Genet. 1998;18:378–381. doi: 10.1038/ng0498-378. [DOI] [PubMed] [Google Scholar]
- 59.Rogers C.S., Vanoye C.G., Sullenger B.A., George A.L., Jr. Functional repair of a mutant chloride channel using a trans-splicing ribozyme. J. Clin. Invest. 2002;110:1783–1789. doi: 10.1172/JCI200216481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kastanos E., Hjiantoniou E., Phylactou L.A. Restoration of protein synthesis in pancreatic cancer cells by trans-splicing ribozymes. Biochem. Biophys. Res. Commun. 2004;322:930–934. doi: 10.1016/j.bbrc.2004.07.203. [DOI] [PubMed] [Google Scholar]
- 61.Shin K.S., Sullenger B.A., Lee S.W. Ribozyme-mediated induction of apoptosis in human cancer cells by targeted repair of mutant p53 RNA. Mol. Ther. 2004;10:365–372. doi: 10.1016/j.ymthe.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 62.Watanabe T., Sullenger B.A. Induction of wild-type p53 activity in human cancer cells by ribozymes that repair mutant p53 transcripts. Proc. Natl. Acad. Sci. USA. 2000;97:8490–8494. doi: 10.1073/pnas.150104097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim S.J., Kim J.H., Yang B., Jeong J.S., Lee S.W. Specific and efficient regression of cancers harboring KRAS mutation by targeted RNA replacement. Mol. Ther. 2017;25:356–367. doi: 10.1016/j.ymthe.2016.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee C.H., Han S.R., Lee S.W. Therapeutic applications of group I intron-based trans-splicing ribozymes. Wiley Interdiscip. Rev. RNA. 2018;9:e1466. doi: 10.1002/wrna.1466. [DOI] [PubMed] [Google Scholar]
- 65.Amini Z.N., Olson K.E., Müller U.F. Spliceozymes: ribozymes that remove introns from pre-mRNAs in trans. PLoS ONE. 2014;9:e101932. doi: 10.1371/journal.pone.0101932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bell M.A., Johnson A.K., Testa S.M. Ribozyme-catalyzed excision of targeted sequences from within RNAs. Biochemistry. 2002;41:15327–15333. doi: 10.1021/bi0267386. [DOI] [PubMed] [Google Scholar]
- 67.Byun J., Lan N., Long M., Sullenger B.A. Efficient and specific repair of sickle β-globin RNA by trans-splicing ribozymes. RNA. 2003;9:1254–1263. doi: 10.1261/rna.5450203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Levin A.A. Treating disease at the RNA level with oligonucleotides. N. Engl. J. Med. 2019;380:57–70. doi: 10.1056/NEJMra1705346. [DOI] [PubMed] [Google Scholar]
- 69.Khvorova A., Watts J.K. The chemical evolution of oligonucleotide therapies of clinical utility. Nat. Biotechnol. 2017;35:238–248. doi: 10.1038/nbt.3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Amantana A., Iversen P.L. Pharmacokinetics and biodistribution of phosphorodiamidate morpholino antisense oligomers. Curr. Opin. Pharmacol. 2005;5:550–555. doi: 10.1016/j.coph.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 71.Neil E.E., Bisaccia E.K. Nusinersen: a novel antisense oligonucleotide for the treatment of spinal muscular atrophy. J. Pediatr. Pharmacol. Ther. 2019;24:194–203. doi: 10.5863/1551-6776-24.3.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.McClorey G., Banerjee S. Cell-penetrating peptides to enhance delivery of oligonucleotide-based therapeutics. Biomedicines. 2018;6:51. doi: 10.3390/biomedicines6020051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Itoh M., Nakaura M., Imanishi T., Obika S. Target gene knockdown by 2′,4′-BNA/LNA antisense oligonucleotides in zebrafish. Nucleic Acid Ther. 2014;24:186–191. doi: 10.1089/nat.2013.0464. [DOI] [PubMed] [Google Scholar]
- 74.Hildner K.M., Schirmacher P., Atreya I., Dittmayer M., Bartsch B., Galle P.R., Wirtz S., Neurath M.F. Targeting of the transcription factor STAT4 by antisense phosphorothioate oligonucleotides suppresses collagen-induced arthritis. J. Immunol. 2007;178:3427–3436. doi: 10.4049/jimmunol.178.6.3427. [DOI] [PubMed] [Google Scholar]
- 75.Dryselius R., Aswasti S.K., Rajarao G.K., Nielsen P.E., Good L. The translation start codon region is sensitive to antisense PNA inhibition in Escherichia coli. Oligonucleotides. 2003;13:427–433. doi: 10.1089/154545703322860753. [DOI] [PubMed] [Google Scholar]
- 76.Braasch D.A., Liu Y., Corey D.R. Antisense inhibition of gene expression in cells by oligonucleotides incorporating locked nucleic acids: effect of mRNA target sequence and chimera design. Nucleic Acids Res. 2002;30:5160–5167. doi: 10.1093/nar/gkf651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Marsollier A.C., Ciszewski L., Mariot V., Popplewell L., Voit T., Dickson G., Dumonceaux J. Antisense targeting of 3′ end elements involved in DUX4 mRNA processing is an efficient therapeutic strategy for facioscapulohumeral dystrophy: a new gene-silencing approach. Hum. Mol. Genet. 2016;25:1468–1478. doi: 10.1093/hmg/ddw015. [DOI] [PubMed] [Google Scholar]
- 78.Golshirazi G., Ciszewski L., Lu-Nguyen N., Popplewell L. Antisense oligonucleotide targeting of 3′-UTR of mRNA for expression knockdown. Methods Mol. Biol. 2018;1828:91–124. doi: 10.1007/978-1-4939-8651-4_6. [DOI] [PubMed] [Google Scholar]
- 79.Lin J., Lee J.H.J., Paramasivam K., Pathak E., Wang Z., Pramono Z.A.D., Lim B., Wee K.B., Surana U. Induced-decay of glycine decarboxylase transcripts as an anticancer therapeutic strategy for non-small-cell lung carcinoma. Mol. Ther. Nucleic Acids. 2017;9:263–273. doi: 10.1016/j.omtn.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kang J.K., Malerba A., Popplewell L., Foster K., Dickson G. Antisense-induced myostatin exon skipping leads to muscle hypertrophy in mice following octa-guanidine morpholino oligomer treatment. Mol. Ther. 2011;19:159–164. doi: 10.1038/mt.2010.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kurosaki T., Maquat L.E. Nonsense-mediated mRNA decay in humans at a glance. J. Cell Sci. 2016;129:461–467. doi: 10.1242/jcs.181008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ward A.J., Norrbom M., Chun S., Bennett C.F., Rigo F. Nonsense-mediated decay as a terminating mechanism for antisense oligonucleotides. Nucleic Acids Res. 2014;42:5871–5879. doi: 10.1093/nar/gku184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.van der Wal E., Bergsma A.J., Pijnenburg J.M., van der Ploeg A.T., Pijnappel W.W.M.P. Antisense oligonucleotides promote exon inclusion and correct the common c.-32-13T>G GAA splicing variant in Pompe disease. Mol. Ther. Nucleic Acids. 2017;7:90–100. doi: 10.1016/j.omtn.2017.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Igreja S., Clarke L.A., Botelho H.M., Marques L., Amaral M.D. Correction of a cystic fibrosis splicing mutation by antisense oligonucleotides. Hum. Mutat. 2016;37:209–215. doi: 10.1002/humu.22931. [DOI] [PubMed] [Google Scholar]
- 85.Scalet D., Balestra D., Rohban S., Bovolenta M., Perrone D., Bernardi F., Campaner S., Pinotti M. Exploring splicing-switching molecules for Seckel syndrome therapy. Biochim. Biophys. Acta Mol. Basis Dis. 2017;1863:15–20. doi: 10.1016/j.bbadis.2016.09.011. [DOI] [PubMed] [Google Scholar]
- 86.Hoy S.M. Nusinersen: first global approval. Drugs. 2017;77:473–479. doi: 10.1007/s40265-017-0711-7. [DOI] [PubMed] [Google Scholar]
- 87.Bergsma A.J., In ‘t Groen S.L., Verheijen F.W., van der Ploeg A.T., Pijnappel W. From cryptic toward canonical pre-mRNA splicing in Pompe disease: a pipeline for the development of antisense oligonucleotides. Mol. Ther. Nucleic Acids. 2016;5:e361. doi: 10.1038/mtna.2016.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bonifert T., Gonzalez Menendez I., Battke F., Theurer Y., Synofzik M., Schöls L., Wissinger B. Antisense oligonucleotide mediated splice correction of a deep intronic mutation in OPA1. Mol. Ther. Nucleic Acids. 2016;5:e390. doi: 10.1038/mtna.2016.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Popplewell L.J., Malerba A., Dickson G. Optimizing antisense oligonucleotides using phosphorodiamidate morpholino oligomers. Methods Mol. Biol. 2012;867:143–167. doi: 10.1007/978-1-61779-767-5_10. [DOI] [PubMed] [Google Scholar]
- 90.Li D., Mastaglia F.L., Fletcher S., Wilton S.D. Precision medicine through antisense oligonucleotide-mediated exon skipping. Trends Pharmacol. Sci. 2018;39:982–994. doi: 10.1016/j.tips.2018.09.001. [DOI] [PubMed] [Google Scholar]
- 91.Syed Y.Y. Eteplirsen: first global approval. Drugs. 2016;76:1699–1704. doi: 10.1007/s40265-016-0657-1. [DOI] [PubMed] [Google Scholar]
- 92.Guiraud S., Aartsma-Rus A., Vieira N.M., Davies K.E., van Ommen G.J., Kunkel L.M. The pathogenesis and therapy of muscular dystrophies. Annu. Rev. Genomics Hum. Genet. 2015;16:281–308. doi: 10.1146/annurev-genom-090314-025003. [DOI] [PubMed] [Google Scholar]
- 93.Aartsma-Rus A., Fokkema I., Verschuuren J., Ginjaar I., van Deutekom J., van Ommen G.J., den Dunnen J.T. Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum. Mutat. 2009;30:293–299. doi: 10.1002/humu.20918. [DOI] [PubMed] [Google Scholar]
- 94.Fernández-Rodríguez J., Castellsagué J., Benito L., Benavente Y., Capellá G., Blanco I., Serra E., Lázaro C. A mild neurofibromatosis type 1 phenotype produced by the combination of the benign nature of a leaky NF1-splice mutation and the presence of a complex mosaicism. Hum. Mutat. 2011;32:705–709. doi: 10.1002/humu.21500. [DOI] [PubMed] [Google Scholar]
- 95.Pros E., Fernández-Rodríguez J., Canet B., Benito L., Sánchez A., Benavides A., Ramos F.J., López-Ariztegui M.A., Capellá G., Blanco I. Antisense therapeutics for neurofibromatosis type 1 caused by deep intronic mutations. Hum. Mutat. 2009;30:454–462. doi: 10.1002/humu.20933. [DOI] [PubMed] [Google Scholar]
- 96.Cross R.A., Stewart M., Kendrick-Jones J. Structural predictions for the central domain of dystrophin. FEBS Lett. 1990;262:87–92. doi: 10.1016/0014-5793(90)80160-k. [DOI] [PubMed] [Google Scholar]
- 97.Mort M., Ivanov D., Cooper D.N., Chuzhanova N.A. A meta-analysis of nonsense mutations causing human genetic disease. Hum. Mutat. 2008;29:1037–1047. doi: 10.1002/humu.20763. [DOI] [PubMed] [Google Scholar]
- 98.Keeling K.M., Xue X., Gunn G., Bedwell D.M. Therapeutics based on stop codon readthrough. Annu. Rev. Genomics Hum. Genet. 2014;15:371–394. doi: 10.1146/annurev-genom-091212-153527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Temple G.F., Dozy A.M., Roy K.L., Kan Y.W. Construction of a functional human suppressor tRNA gene: an approach to gene therapy for β-thalassaemia. Nature. 1982;296:537–540. doi: 10.1038/296537a0. [DOI] [PubMed] [Google Scholar]
- 100.Panchal R.G., Wang S., McDermott J., Link C.J., Jr. Partial functional correction of xeroderma pigmentosum group A cells by suppressor tRNA. Hum. Gene Ther. 1999;10:2209–2219. doi: 10.1089/10430349950017194. [DOI] [PubMed] [Google Scholar]
- 101.Kiselev A.V., Ostapenko O.V., Rogozhkina E.V., Kholod N.S., Seit Nebi A.S., Baranov A.N., Lesina E.A., Ivashchenko T.E., Sabetskiĭ V.A., Shavlovskiĭ M.M. [Suppression of nonsense mutations in the dystrophin gene by a suppressor tRNA gene] Mol. Biol. (Mosk.) 2002;36:43–47. [PubMed] [Google Scholar]
- 102.Sako Y., Usuki F., Suga H. A novel therapeutic approach for genetic diseases by introduction of suppressor tRNA. Nucleic Acids Symp. Ser. (Oxf.) 2006;50:239–240. doi: 10.1093/nass/nrl119. [DOI] [PubMed] [Google Scholar]
- 103.Lueck J.D., Yoon J.S., Perales-Puchalt A., Mackey A.L., Infield D.T., Behlke M.A., Pope M.R., Weiner D.B., Skach W.R., McCray P.B., Jr., Ahern C.A. Engineered transfer RNAs for suppression of premature termination codons. Nat. Commun. 2019;10:822. doi: 10.1038/s41467-019-08329-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lee H.L., Dougherty J.P. Pharmaceutical therapies to recode nonsense mutations in inherited diseases. Pharmacol. Ther. 2012;136:227–266. doi: 10.1016/j.pharmthera.2012.07.007. [DOI] [PubMed] [Google Scholar]
- 105.Lynch S.R., Puglisi J.D. Structure of a eukaryotic decoding region A-site RNA. J. Mol. Biol. 2001;306:1023–1035. doi: 10.1006/jmbi.2000.4419. [DOI] [PubMed] [Google Scholar]
- 106.Welch E.M., Barton E.R., Zhuo J., Tomizawa Y., Friesen W.J., Trifillis P., Paushkin S., Patel M., Trotta C.R., Hwang S. PTC124 targets genetic disorders caused by nonsense mutations. Nature. 2007;447:87–91. doi: 10.1038/nature05756. [DOI] [PubMed] [Google Scholar]
- 107.Peltz S.W., Morsy M., Welch E.M., Jacobson A. Ataluren as an agent for therapeutic nonsense suppression. Annu. Rev. Med. 2013;64:407–425. doi: 10.1146/annurev-med-120611-144851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lopez-Novoa J.M., Quiros Y., Vicente L., Morales A.I., Lopez-Hernandez F.J. New insights into the mechanism of aminoglycoside nephrotoxicity: an integrative point of view. Kidney Int. 2011;79:33–45. doi: 10.1038/ki.2010.337. [DOI] [PubMed] [Google Scholar]
- 109.Huth M.E., Ricci A.J., Cheng A.G. Mechanisms of aminoglycoside ototoxicity and targets of hair cell protection. Int. J. Otolaryngol. 2011;2011:937861. doi: 10.1155/2011/937861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Shalev M., Baasov T. When proteins start to make sense: fine-tuning aminoglycosides for PTC suppression therapy. MedChemComm. 2014;5:1092–1105. doi: 10.1039/C4MD00081A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kerem E., Konstan M.W., De Boeck K., Accurso F.J., Sermet-Gaudelus I., Wilschanski M., Elborn J.S., Melotti P., Bronsveld I., Fajac I., Cystic Fibrosis Ataluren Study Group Ataluren for the treatment of nonsense-mutation cystic fibrosis: a randomised, double-blind, placebo-controlled phase 3 trial. Lancet Respir. Med. 2014;2:539–547. doi: 10.1016/S2213-2600(14)70100-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Keeling K.M., Wang D., Dai Y., Murugesan S., Chenna B., Clark J., Belakhov V., Kandasamy J., Velu S.E., Baasov T., Bedwell D.M. Attenuation of nonsense-mediated mRNA decay enhances in vivo nonsense suppression. PLoS ONE. 2013;8:e60478. doi: 10.1371/journal.pone.0060478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Linde L., Boelz S., Nissim-Rafinia M., Oren Y.S., Wilschanski M., Yaacov Y., Virgilis D., Neu-Yilik G., Kulozik A.E., Kerem E., Kerem B. Nonsense-mediated mRNA decay affects nonsense transcript levels and governs response of cystic fibrosis patients to gentamicin. J. Clin. Invest. 2007;117:683–692. doi: 10.1172/JCI28523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Roy B., Friesen W.J., Tomizawa Y., Leszyk J.D., Zhuo J., Johnson B., Dakka J., Trotta C.R., Xue X., Mutyam V. Ataluren stimulates ribosomal selection of near-cognate tRNAs to promote nonsense suppression. Proc. Natl. Acad. Sci. USA. 2016;113:12508–12513. doi: 10.1073/pnas.1605336113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Xue X., Mutyam V., Thakerar A., Mobley J., Bridges R.J., Rowe S.M., Keeling K.M., Bedwell D.M. Identification of the amino acids inserted during suppression of CFTR nonsense mutations and determination of their functional consequences. Hum. Mol. Genet. 2017;26:3116–3129. doi: 10.1093/hmg/ddx196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Manuvakhova M., Keeling K., Bedwell D.M. Aminoglycoside antibiotics mediate context-dependent suppression of termination codons in a mammalian translation system. RNA. 2000;6:1044–1055. doi: 10.1017/s1355838200000716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Pavlov M.Y., Freistroffer D.V., Dincbas V., MacDougall J., Buckingham R.H., Ehrenberg M. A direct estimation of the context effect on the efficiency of termination. J. Mol. Biol. 1998;284:579–590. doi: 10.1006/jmbi.1998.2220. [DOI] [PubMed] [Google Scholar]
- 118.Cassan M., Rousset J.P. UAG readthrough in mammalian cells: effect of upstream and downstream stop codon contexts reveal different signals. BMC Mol. Biol. 2001;2:3. doi: 10.1186/1471-2199-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Namy O., Hatin I., Rousset J.P. Impact of the six nucleotides downstream of the stop codon on translation termination. EMBO Rep. 2001;2:787–793. doi: 10.1093/embo-reports/kve176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Keeling K.M., Bedwell D.M. Clinically relevant aminoglycosides can suppress disease-associated premature stop mutations in the IDUA and P53 cDNAs in a mammalian translation system. J. Mol. Med. (Berl.) 2002;80:367–376. doi: 10.1007/s00109-001-0317-z. [DOI] [PubMed] [Google Scholar]
- 121.Sachs M.S., Wang Z., Gaba A., Fang P., Belk J., Ganesan R., Amrani N., Jacobson A. Toeprint analysis of the positioning of translation apparatus components at initiation and termination codons of fungal mRNAs. Methods. 2002;26:105–114. doi: 10.1016/S1046-2023(02)00013-0. [DOI] [PubMed] [Google Scholar]
- 122.Ivanov A., Mikhailova T., Eliseev B., Yeramala L., Sokolova E., Susorov D., Shuvalov A., Schaffitzel C., Alkalaeva E. PABP enhances release factor recruitment and stop codon recognition during translation termination. Nucleic Acids Res. 2016;44:7766–7776. doi: 10.1093/nar/gkw635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ars E., Kruyer H., Morell M., Pros E., Serra E., Ravella A., Estivill X., Lázaro C. Recurrent mutations in the NF1 gene are common among neurofibromatosis type 1 patients. J. Med. Genet. 2003;40:e82. doi: 10.1136/jmg.40.6.e82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Messiaen L., Wimmer K. NF1 mutational spectrum. In: Kaufmann D., editor. Neurofibromatoses. Karger; 2008. pp. 63–77. [Google Scholar]
- 125.Kerem E. Pharmacologic therapy for stop mutations: how much CFTR activity is enough? Curr. Opin. Pulm. Med. 2004;10:547–552. doi: 10.1097/01.mcp.0000141247.22078.46. [DOI] [PubMed] [Google Scholar]
- 126.Chamberlain J. Dystrophin levels required for correction of Duchenne muscular dystrophy. Basic Appl. Myol. 1997;7:251–255. [Google Scholar]
- 127.Li K., Turner A.N., Chen M., Brosius S.N., Schoeb T.R., Messiaen L.M., Bedwell D.M., Zinn K.R., Anastasaki C., Gutmann D.H. Mice with missense and nonsense NF1 mutations display divergent phenotypes compared with human neurofibromatosis type I. Dis. Model. Mech. 2016;9:759–767. doi: 10.1242/dmm.025783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Anastasaki C., Woo A.S., Messiaen L.M., Gutmann D.H. Elucidating the impact of neurofibromatosis-1 germline mutations on neurofibromin function and dopamine-based learning. Hum. Mol. Genet. 2015;24:3518–3528. doi: 10.1093/hmg/ddv103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Toonen J.A., Anastasaki C., Smithson L.J., Gianino S.M., Li K., Kesterson R.A., Gutmann D.H. NF1 germline mutation differentially dictates optic glioma formation and growth in neurofibromatosis-1. Hum. Mol. Genet. 2016;25:1703–1713. doi: 10.1093/hmg/ddw039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Carnes R.M., Kesterson R.A., Korf B.R., Mobley J.A., Wallis D. Affinity purification of NF1 protein-protein interactors identifies keratins and neurofibromin itself as binding partners. Genes (Basel) 2019;10:650. doi: 10.3390/genes10090650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Seow Y., Wood M.J. Biological gene delivery vehicles: beyond viral vectors. Mol. Ther. 2009;17:767–777. doi: 10.1038/mt.2009.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Friedmann T., Roblin R. Gene therapy for human genetic disease? Science. 1972;175:949–955. doi: 10.1126/science.175.4025.949. [DOI] [PubMed] [Google Scholar]
- 133.Daya S., Berns K.I. Gene therapy using adeno-associated virus vectors. Clin. Microbiol. Rev. 2008;21:583–593. doi: 10.1128/CMR.00008-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lion T. Adenovirus infections in immunocompetent and immunocompromised patients. Clin. Microbiol. Rev. 2014;27:441–462. doi: 10.1128/CMR.00116-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Milone M.C., O’Doherty U. Clinical use of lentiviral vectors. Leukemia. 2018;32:1529–1541. doi: 10.1038/s41375-018-0106-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Viral and non-viral gene delivery and its role in pluripotent stem cell engineering. Drug Discov. Today. Technol. 2008;5:e105–e148. doi: 10.1016/j.ddtec.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 137.Zhou J., Atsina K.B., Himes B.T., Strohbehn G.W., Saltzman W.M. Novel delivery strategies for glioblastoma. Cancer J. 2012;18:89–99. doi: 10.1097/PPO.0b013e318244d8ae. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lv H., Zhang S., Wang B., Cui S., Yan J. Toxicity of cationic lipids and cationic polymers in gene delivery. J. Control. Release. 2006;114:100–109. doi: 10.1016/j.jconrel.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 139.Gao X., Yao L., Song Q., Zhu L., Xia Z., Xia H., Jiang X., Chen J., Chen H. The association of autophagy with polyethylenimine-induced cytotoxicity in nephritic and hepatic cell lines. Biomaterials. 2011;32:8613–8625. doi: 10.1016/j.biomaterials.2011.07.047. [DOI] [PubMed] [Google Scholar]
- 140.Zhou J., Liu J., Cheng C.J., Patel T.R., Weller C.E., Piepmeier J.M., Jiang Z., Saltzman W.M. Biodegradable poly(amine-co-ester) terpolymers for targeted gene delivery. Nat. Mater. 2011;11:82–90. doi: 10.1038/nmat3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Han L., Kong D.K., Zheng M.Q., Murikinati S., Ma C., Yuan P., Li L., Tian D., Cai Q., Ye C. Increased nanoparticle delivery to brain tumors by autocatalytic priming for improved treatment and imaging. ACS Nano. 2016;10:4209–4218. doi: 10.1021/acsnano.5b07573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Yin H., Kanasty R.L., Eltoukhy A.A., Vegas A.J., Dorkin J.R., Anderson D.G. Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 2014;15:541–555. doi: 10.1038/nrg3763. [DOI] [PubMed] [Google Scholar]
- 143.Liu J., Chang J., Jiang Y., Meng X., Sun T., Mao L., Xu Q., Wang M. Fast and efficient CRISPR/Cas9 genome editing in vivo enabled by bioreducible lipid and messenger RNA nanoparticles. Adv. Mater. 2019;31:e1902575. doi: 10.1002/adma.201902575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Davis M.E. Non-viral gene delivery systems. Curr. Opin. Biotechnol. 2002;13:128–131. doi: 10.1016/s0958-1669(02)00294-x. [DOI] [PubMed] [Google Scholar]
- 145.Liang X., Liu L., Wei Y.Q., Gao G.P., Wei X.W. Clinical evaluations of toxicity and efficacy of nanoparticle-mediated gene therapy. Hum. Gene Ther. 2018;29:1227–1234. doi: 10.1089/hum.2018.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Alton E.W., Boyd A.C., Cheng S.H., Cunningham S., Davies J.C., Gill D.R., Griesenbach U., Higgins T., Hyde S.C., Innes J.A. A randomised, double-blind, placebo-controlled phase IIB clinical trial of repeated application of gene therapy in patients with cystic fibrosis. Thorax. 2013;68:1075–1077. doi: 10.1136/thoraxjnl-2013-203309. [DOI] [PubMed] [Google Scholar]
- 147.Senís E., Fatouros C., Große S., Wiedtke E., Niopek D., Mueller A.K., Börner K., Grimm D. CRISPR/Cas9-mediated genome engineering: an adeno-associated viral (AAV) vector toolbox. Biotechnol. J. 2014;9:1402–1412. doi: 10.1002/biot.201400046. [DOI] [PubMed] [Google Scholar]
- 148.Li S.D., Huang L. Non-viral is superior to viral gene delivery. J. Control. Release. 2007;123:181–183. doi: 10.1016/j.jconrel.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 149.Remy S., Tesson L., Menoret S., Usal C., De Cian A., Thepenier V., Thinard R., Baron D., Charpentier M., Renaud J.B. Efficient gene targeting by homology-directed repair in rat zygotes using TALE nucleases. Genome Res. 2014;24:1371–1383. doi: 10.1101/gr.171538.113. [DOI] [PMC free article] [PubMed] [Google Scholar]