

Summary
The enteroinvasive bacterium Shigella flexneri forces its uptake into non-phagocytic host cells through the translocation of T3SS effectors that subvert the actin cytoskeleton. Here, we report de novo actin polymerization after cellular entry around the bacterium-containing vacuole (BCV) leading to the formation of a dynamic actin cocoon. This cocoon is thicker than any described cellular actin structure and functions as a gatekeeper for the cytosolic access of the pathogen. Host CDC42, TOCA-1, N-WASP, WIP, the Arp2/3 complex, cortactin, coronin, and cofilin are recruited to the actin cocoon. They are subverted by T3SS effectors, such as IpgD, IpgB1, and IcsB. IcsB immobilizes components of the actin polymerization machinery at the BCV dependent on its fatty acyltransferase activity. This represents a unique microbial subversion strategy through localized entrapment of host actin regulators causing massive actin assembly. We propose that the cocoon promotes subsequent invasion steps for successful Shigella infection.
Keywords: host-pathogen interactions, intracellular pathogens, Shigella flexneri, vacuolar rupture, actin cytoskeleton, effector proteins, Arp2/3 complex, CDC42, N-WASP, Nε fatty acylation
Graphical Abstract
Highlights
-
•
A thick actin cocoon forms de novo around the Shigella-containing vacuole upon entry
-
•
The effector IcsB entraps host actin regulators at the vacuole by lipidation
-
•
Cdc42, N-WASP, and the Arp2/3 complex are major actin cocoon regulators
-
•
Cocoon formation promotes subsequent Shigella niche formation and dissemination
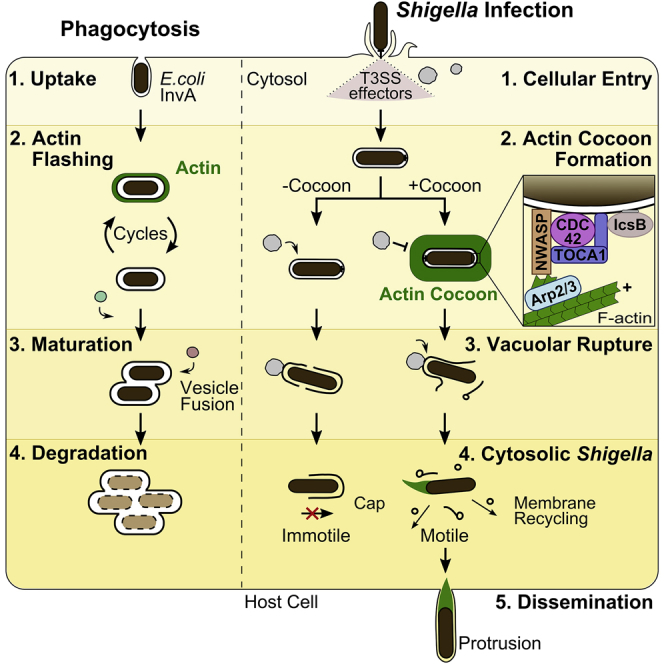
Kühn et al. present the formation of a massive actin cocoon structure around the Shigella-containing vacuole upon bacterial entry in enterocytes. This cocoon is formed by the pathogen-induced localization of host actin regulators at the phagosome. It favors efficient cytosolic access and spread for successful infection.
Introduction
Bacterial pathogens have evolved sophisticated ways to drive infection and to establish their intracellular niches for survival and replication. Especially the cellular actin cytoskeleton is extensively hijacked for bacterial purposes. This cytoskeletal meshwork is controlled by a complex network of actin-binding proteins (ABPs), which nucleate new actin filaments (F-actin) from actin monomers (G-actin) or elongate, maintain, and disassemble existing ones. ABPs are spatiotemporally localized and regulated by Rho GTPases, phospholipids, post-translational modifications, or membrane-bound scaffold (Le Clainche and Carlier, 2008, Rottner et al., 2017, Pollard, 2016). The main F-actin nucleating factor is the Arp2/3 complex (Machesky et al., 1994, Mullins et al., 1998), which generates branched actin meshworks in membrane ruffles and lamellipodia, at phagosomes and intracellular vesicles. Yet efficient F-actin nucleation requires additional nucleation-promoting factors (NPFs) such as N-WASP. N-WASP is itself activated by several factors, such as PI(4,5)P2, the F-BAR scaffold TOCA-1, and the Rho GTPase CDC42 (Ho et al., 2004, Rohatgi et al., 1999, Rottner et al., 2017). Rho GTPases are central regulators of the actin cytoskeleton and switch between an inactive, GDP-bound and an active, GTP-bound state (Vetter and Wittinghofer, 2001). This is controlled by guanine nucleotide-exchange factors (GEFs) promoting GDP dissociation, GTPase-activating proteins (GAPs), and guanine nucleotide dissociation inhibitors (GDIs) (Etienne-Manneville and Hall, 2002, Cherfils and Zeghouf, 2013). In the active state, Rho GTPases interact with effector proteins for cell signaling and to regulate the actin cytoskeleton. Remarkably, bacterial pathogens such as the Gram-negative, enteroinvasive bacterium Shigella flexneri (hereafter Shigella) do not directly modify actin (Kühn and Mannherz, 2017). Instead, Shigella modulates the recruitment and the activation of actin regulators by subverting upstream Rho GTPases, kinases, and phospholipid signaling (Schnupf and Sansonetti, 2019, Schroeder and Hilbi, 2008, Valencia-Gallardo et al., 2015).
Shigella is the causative agent of bacterial dysentery and an important model for intracellular pathogenesis (Schnupf and Sansonetti, 2019). It forces its uptake into non-phagocytic epithelial cells through the translocation of type 3 secretion system (T3SS) effectors. These proteins target the host actin cytoskeleton and endomembrane trafficking to induce cellular entry and to establish an intracellular replicative niche. For cellular entry, thin membrane protrusions make the first contact with bacteria, followed by the initiation of massive actin rearrangements enclosing the entering Shigella (Schroeder and Hilbi, 2008, Valencia-Gallardo et al., 2015, Cossart and Sansonetti, 2004, Romero et al., 2012). After cellular uptake in a tight bacterium-containing vacuole (BCV) (Weiner et al., 2016), Shigella induces its rapid escape for replication into the host cytosol. There, it recruits the host actin nucleation machinery to one of its poles by its virulence factor IcsA to spread from cell to cell (Suzuki et al., 1998, Egile et al., 1999, Gouin et al., 1999). Parallel to its uptake, Shigella induces the formation of infection-associated macropinosomes (IAMs). These IAMs accumulate at the entry site and surround the BCV. They form membrane-membrane contacts with the ruptured BCV, and their presence correlates with efficient rupture (Mellouk et al., 2014, Weiner et al., 2016).
We have recently discovered the formation of a hitherto undescribed actin cytoskeleton structure that assembles around vacuolar Shigella (Ehsani et al., 2012, Mellouk et al., 2014, Weiner et al., 2016). Here, we performed its in-depth characterization, coining it as an “actin cocoon.” We found that this cocoon is thicker than any other cellular actin structure and assembles only after bacterial uptake. We identified the process underlying its formation, namely, the involved bacterial T3SS effectors and a subverted host pathway for actin rearrangements. Finally, we demonstrate that interfering with cocoon formation and disassembly affects Shigella’s capacity to invade the host cytosol.
Results
The Actin Cocoon Assembles In Situ after Cellular Entry around Shigella-Containing Phagosomes, and Its Disassembly Precedes Cytosolic Escape
Actin-GFP transfected HeLa cells were imaged during early infection steps of wild-type (WT), dsRed-expressing Shigella at high spatiotemporal resolution (Figures 1A and 1B). After 2 h, almost all cells were infected, with no further primary infection, and membrane ruffling was shut down. Live imaging revealed the in situ assembly of a thick actin coat-like structure after pathogen entry, as indicated by a massive increase in fluorescence intensity around the BCV (Figures 1A and 1B; Videos S1 and S2). This structure, termed the “actin cocoon,” was distinct from cortical actin and polymerized de novo at the surface of the entire vacuolar membrane. After a fast nucleation phase of 1–3 min, the actin cocoon was maintained until its final disassembly, which was immediately followed by BCV membrane rupture (Figures 1A–1C). All observed actin rearrangements took place in the time span after entry site formation and before the cytosolic spread of Shigella. Phalloidin staining of endogenous actin in fixed experiments revealed the presence of F-actin in the cocoon of Shigella invading HeLa or Caco-2 cells (Figures S1A and S1B).
Figure 1.

The Dynamic Actin Cocoon Polymerizes De Novo after Cellular Entry around Shigella’s BCV and Disassembles before Vacuolar Escape
(A and B) Real time-monitoring of the thick actin cocoon. HeLa cells expressing actin-GFP (green) were infected with Shigella WT DsRed (red) (A; asterisk denotes bacterium with cocoon) or Shigella WT (B; blue dashed line denotes bacterium). t = 0 min: onset of entry site formation.
(C) The actin cocoon needs to at least partially disassemble before vacuolar rupture. Time lapse of Shigella WT infecting HeLa cells expressing actin-GFP (green) and galectin-3-mOrange (red, Gal3). Arrow, newly formed cocoon; arrowhead, moment of rupture.
(D) Time lapses of FRAP experiments of the actin cocoon in comparison with cellular actin structures (see Videos S3, S4, and S5).
(E) Quantification of (D). Plotted are mean values ± 95% confidence interval [CI] and curve fit of at least 3 independent experiments (actin cocoon: n = 40 FRAP curves ; lamellipodial tip: n = 31; stress fibers: n = 75).
(F and G) Actin cocoon formation depends on the time point of bacterial infection and pre-infection. Depicted are percentages of bacteria that successfully escaped into the host cytosol and previously assembled an actin cocoon (+) or not (−) (F). On average, three entry sites formed per cell (Figures S1 and S2), and 71.2% ± 1.75% of invading bacteria (n = 631, 4 individual experiments) had a cocoon. All late invaders assembled a cocoon before vacuolar escape (G). Mean values ± SD of individual experiments (F) or rupture time points of pooled single invaders (G) are plotted. Mann-Whitney test with p < 0.05 as significant: ∗∗∗∗p < 0.0001.
Scale bars: 3 μm. See Figures S1 and S2.
Time lapse of actin rearrangements (green) at the entry site during Shigella WT DsRed (red) infection. Shigella induces massive membrane ruffling around the adhered bacterium and cocoon assembly after uptake. The cocoon is much denser than cellular actin structures and finally disassembled. Transient actin flashing around IAMs is also visible (scale bar: 3 μm).
Time-lapse of actin rearrangements during cellular entry of Shigella WT (scale bar: 3 μm).
To monitor the precise time point of vacuolar rupture in correlation to actin cocoon formation, we used fluorescently labeled galectin-3 as a marker. At the moment of vacuolar membrane damage, galectin-3 molecules are recruited to β-galactosides at the inner leaflet of the phagosomal membrane. We never observed vacuolar rupture by bacteria residing in an intact cocoon. At least a partial disassembly of the actin cocoon always preceded the directly following vacuolar rupture (Figure 1C). We conclude that cocoon disassembly is tightly linked with bacterial release into the host cytosol.
The Actin Cocoon Is Dynamically Re-assembled during Its Lifetime, and Its Formation Depends on the Time Point of Infection
Next, we performed fluorescence recovery after photobleaching (FRAP) measurements to compare the spatiotemporal dynamics of the actin cocoon with cellular stress fibers and the lamellipodium tip (Figures 1D and 1E). We anticipated fluorescence recovery either from F-actin treadmilling and de novo polymerization or to a lesser extent from free diffusion of cytoplasmic G-actin (a very fast saturated process). Strikingly, actin filaments of the cocoon had a high turnover rate, with a half-time of fluorescence recovery (t1/2) of 13.8 ± 0.79 s and a mobile fraction (Fm) of 84.6% ± 1.51% (Figure 1D; Video S3). This revealed constant incorporation of new, unbleached G-actin and thus ongoing F-actin polymerization throughout the cocoon until its final disassembly. The same was observed by imaging with constant partial disassembly and re-assembly of the cocoon (Figure S1C). This points to constant de novo F-actin nucleation or elongation during its entire lifetime. We compared the cocoon with either very dynamic cellular actin structures, like the de novo assembling actin meshwork at lamellipodium tips, or very thick, like cellular stress fibers of F-actin bundles. We found the turnover of the cocoon to be similar to the lamellipodial tip (t1/2 = 10.8 ± 1.36 s, Fm = 70.8% ± 2.03%) and clearly different compared with cellular stress fibers (t1/2 = 7.55 ± 0.74 s, Fm = 36.8% ± 4.20%) (Figure 1E; Video S5).
Fluorescence recovery after photobleaching revealed constant actin turnover of the cocoon during its maintained phase. Fluorescence intensity was monitored 10 s before and 60 s after bleaching (t = 0 s) (scale: 5 μm).
The lamellipodium was photobleached and the recovery at its tip was measured showing de novo actin polymerization. The actin-GFP fluorescence signal was followed 10 s before and 60 s after bleaching (t = 0 s) (scale: 5 μm).
Stress fibers have different actin turnover dynamics compared to the actin cocoon and recover much slower from photobleaching. A stress fiber with a very small mobile fraction is depicted. The actin-GFP fluorescence signal was followed 10 s before and 254 s after bleaching (t = 0 s) (scale: 5 μm).
To quantify Shigella infection with regard to actin cocoon formation and vacuolar rupture, we monitored the successive infection steps of individual bacteria (Figures S1D–S1G). Strikingly, in total 71.2% ± 1.75% of all Shigella assembled a dynamic actin cocoon before cytosolic escape (Figure 1F). Shigella’s probability for cocoon formation was pronounced in cells that had already been infected. This occurred with the same tendency, whether several bacteria entered via the same or via different entry sites (Figures 1F and S1G). We also quantified the time span that individual Shigella required after the start of entry site formation (initial cortical actin rearrangements) to escape into the host cytosol (initial galectin-3 recruitment). All bacteria invading at later time points polymerized an actin cocoon. This is shown by a significant delay in the rupture time point (−cocoon, 9.48 ± 5.63 min; +cocoon, 20.37 ± 13.13 min; p < 0.0001; Figure 1G). Increasing the multiplicity of infection (MOI > 15) increased the infection efficiency, with more bacteria invading via the same entry site, but not the probability of cocoon formation and rupture time (see Figure S2). Taken together, we conclude (1) that cocoon assembly depends on the phagocytic load as well as the order of infection and (2) that all late invading bacteria assemble an actin cocoon.
The Shigella-Specific Actin Cocoon Represents a Unique Structure
To better understand the nature of actin cocoons, we compared them with well-characterized host actin cytoskeletal structures in parallel to the FRAP experiments (Figure 2A). We measured the fluorescence intensities of the thickest stress fibers per infected cell and normalized each individual measurement against the average stress fiber intensity (see STAR Methods). Our analysis revealed that actin cocoons are much denser than any other actin structure, with on average 7-fold higher fluorescence intensity compared with stress fibers (7.02 ± 1.25, p < 0.0001) and 11.5-fold compared with the lamellipodium tip (0.61 ± 0.10, p < 0.0001) (Figure 2A). Remarkably, the actin turnover dynamics at the Shigella BCV result, although similar to the lamellipodium tip (Figure 1D), in a much thicker actin structure. In line with this, our previously published CLEM datasets of three actin cocoons (Weiner et al., 2016) exhibited a maximum thickness of 350 nm. Additionally, the cocoon differed from short-lasting actin rearrangements around Shigella-induced macropinosomes, which did not differ significantly in fluorescence intensity compared with stress fibers (1.30 ± 0.57; Figure 2A; Video S1), resembling the actin flashing phenomenon (Yam and Theriot, 2004, Liebl and Griffiths, 2009) (see below).
Figure 2.

The Actin Cocoon Is a Shigella-Specific Cytoskeletal Structure
(A) The actin cocoon is thicker than any cellular actin structure and is Shigella specific. Left: scheme of investigated actin structures with stress fibers (Stress F.; red), lamellipodial tip (Lamellip; orange), and actin at BCVs (bacterium-containing vacuole; green) and at IAMs (infection-associated macropinosome; blue). Right: quantification of relative fluorescence intensities of actin-GFP (S. flexneri WT: n = 250; E. coli InvA: n = 236; S. Typhimurium WT: n = 322).
(B) The actin cocoon assembles independently of septin 7 after siRNA-mediated knockdown (KD) (Scr: scramble, control, n = 264 cytosolic bacteria; SEPT7, n = 250; KD 91.7%; see Figure S3).
(C) Time-lapse imaging examples of (A). Top: invading S. Typhimurium did not assemble an actin cocoon. Bottom: cyclic actin flashing around E. coli InvA phagosomes (asterisk) in comparison with cellular stress fibers.
(D) The actin cocoon lifetime of Shigella WT (20.64 ± 13.87 min; n = 95) differs from actin flashing by E. coli InvA (n = 103).
(E and G) Representative time lapses of HeLa cells co-transfected with the PI3P marker 2xFYVE and actin and infected either with Shigella WT (E) or E. coli InvA (G).
(F and H) Quantification of (E) and (G) with cytosolic background correction and normalized to the maximal fluorescence intensity (S. flexneri WT: n = 12; E. coli InvA: n = 12; mean values ± SEM).
Statistical significance: Student’s t test with p < 0.05 as significant: ∗∗∗∗p < 0.0001; ns, not significant. Indicated are mean values ± SD of at least 3 independent experiments. See Figures S3 and S4.
Next, we examined if other cytoskeletal systems, such as septin filaments (Kinoshita et al., 2002, Mavrakis et al., 2014), contribute to the assembly of this unique actin structure. During late infection steps, cytosolic Shigella are trapped in septin-actin cages that restrict bacterial proliferation (Mostowy and Cossart, 2012, Mostowy et al., 2010, Mostowy et al., 2011). We performed knockdown of SEPT7 by RNA interference, a common technique for the efficient inhibition of septin filament formation (Sirianni et al., 2016). This neither prevented cocoon formation nor delayed Shigella’s cytosolic escape (Figure 2B; Figures S3A, S3B, and S3F). Therefore, the actin cocoon around vacuolar Shigella forms independently of septin 7.
Furthermore, we deciphered if the actin cocoon is specific for Shigella or a general mechanism occurring during cell entry. Therefore, we compared it with actin rearrangements during early infections of the closely related bacterium Salmonella enterica serovar Typhimurium (S. Typhimurium). Like Shigella, Salmonellae induce massive membrane ruffling and macropinocytosis during cellular entry by T3SS effector proteins. To the contrary, S. Typhimurium pursue mainly an intravacuolar lifestyle for bacterial replication (Santos and Enninga, 2016). To decipher actin polymerization around entering Salmonella, we followed bacteria-induced actin rearrangements during the first 2 h of infection (Figures 2A and 2C). We barely detected any around Salmonella-containing vacuoles (SCVs) (35-fold fewer, 0.20 ± 0.05) (Figures 2A and 2C). Instead, actin occurred as small, intense dots around the SCV, probably derived from recycling and fusion events for vacuolar maturation. We concluded that Salmonella does not assemble an actin coat-like structure during early invasion steps.
Finally, we examined the host cell contribution for cocoon assembly. Previous work of several groups described short-lasting, repeated cycles of actin polymerization and depolymerization around fully internalized phagosomes. These actin rearrangements, termed “actin flashing,” were identified as downstream consequences of several cellular entry mechanisms independent of cell type and cargo in cells with high phagocytic load (Yam and Theriot, 2004, Liebl and Griffiths, 2009). To compare actin flashes with actin cocoons, we infected HeLa cells with Escherichia coli-expressing Yersinia pseudotuberculosis invasin A (E. coli InvA) as model for canonical phagocytosis. Invasin A binds integrins and is sufficient to transfer Yersinia’s zippering entry to E. coli (Isberg et al., 1987). We observed successive waves of actin flashing around internalized E. coli InvA with 32% lower fluorescence intensity compared with stress fibers (0.68 ± 0.19, p = 0.0056). In line with previous studies (Yam and Theriot, 2004, Liebl and Griffiths, 2009), the duration was 2.0 ± 1.9 min per cycle (Figures 2A, 2C, and 2D). Thus, actin cocoons were distinct from the “actin flashing” phenomenon in several major points. First, the fluorescence intensity of Shigella’s cocoons was on average 10-fold higher (Figure 2A). Second, the cocoon had a 10 times longer lifespan than one actin flash (Figure 2D). Third, we did not identify any consecutive cycles of actin assembly at Shigella’s BCV. Cocoon disassembly was always followed by immediate cytosolic escape, while E. coli InvA phagosomes never ruptured. In conclusion, the actin cocoon around vacuolar Shigella clearly differs from transient actin structures around endocytic compartments.
Actin Cocoon Assembly Precedes Diversion from Canonical Phagosomal Maturation
To obtain more details about Shigella’s vacuolar identity, we investigated the recruitment of markers for canonical phagosome maturation and autophagy to the BCV. We previously realized an altered lipid and protein composition of the two compartments induced during Shigella infection. Although canonical maturation and recycling of IAMs is followed through PI3P, Rab5, Rab7, and Rab11 recruitment (Mellouk et al., 2014, Weiner et al., 2016, Kühn et al., 2017), PI3P was only very transiently detected at the Shigella BCV (Weiner et al., 2016). Remarkably, the actin cocoon forms at the onset of this short lifetime PI3P peak (Figures 2E and 2F). Although PI3P is quickly and irreversible depleted from the BCV, the cocoon remains assembled (Figures 2E and 2F). Cocoon disassembly is followed by immediate vacuolar rupture without the BCV’s becoming PI3P positive again or recruiting Rab7 (Figure 2E; Figure S4A). In contrast, depletion of the thin actin coat around the canonical phagosomes of E. coli InvA, like the last actin flashing peak, was proceeded by a long PI3P peak (Figures 2G and 2H). This is followed by Rab7 enrichment at the E. coli InvA phagosome and PI3P depletion (Figure S4B), indicating phagosomal maturation by vesicle fusion events favoring degradative pathways. In addition, LAMP1 and LC3 are also not recruited to the Shigella BCV or the actin cocoon (Figures S4C and S4D). In conclusion, actin cocoon assembly correlates with the moment when the Shigella vacuole changes its identity, with no Rab7 recruitment and the avoidance of the late endocytic pathway, eventually leading to bacterial degradation (Figure S4E).
The Host Arp2/3 Complex-Dependent Actin Nucleation Machinery Is Recruited to the BCV
In order to identify host factors that initiate actin cocoon formation, we performed an inhibitor screen targeting host actin regulators (Figure 3A). A general complication in examining intracellular, pathogen-induced actin cytoskeleton subversions is that the studied events are downstream of cellular entry. Manipulation will, necessarily, also interfere with invasion efficiency. To overcome this, we used conditions that still enabled pathogen entry and had a measurable effect on cocoon formation (see STAR Methods). Furthermore, we focused only on Shigella that successfully invaded the host cytosol (galectin-3-positive BCV) as an indicator of efficient infection. The readout rupture time can indicate changes in actin cocoon dynamics but is not sufficient, because it includes successive actin-regulated steps from cellular uptake to vacuolar rupture (Figure S1D). For this reason, we analyzed in parallel the percentage of bacteria per cell with actin cocoon (Figure 3B). We observed the strongest impact by the inhibition of the Arp2/3 complex with a 2.5-fold reduction of actin cocoons per cell (+CK666, 29.6% ± 2.76%; p < 0.0001). Although Arp2/3 complex inhibition did not impede bacterial uptake, it was sufficient to prevent F-actin nucleation at the BCV (Figure 3B). Thus, the Arp2/3 complex regulates cocoon assembly. Interestingly, inhibition of PI3 kinases (42.3% ± 0.41%, p < 0.0001) or myosin II (38.1% ± 6.92%, p < 0.0001) significantly reduced the amount of actin cocoons (Figure 3B), and the remaining ones had decreased thickness and a less dense F-actin meshwork, respectively. On the other hand, inhibition of formins or ROCK kinase did not prevent cocoon formation (Figure 3B). Yet Shigella entered delayed into formin-inhibited cells (19.6 ± 13.1 min, p = 0.0007; Figure 3C). Eventually, formins might be rather involved in cocoon maintenance by F-actin elongation than filament nucleation. After inhibition of actin polymerization by cytochalasin D, we barely detected actin rearrangements at the entry site and no cocoon-like structure (Figure 3B), underlining again that cocoon assembly requires de novo F-actin polymerization. We also observed a strongly reduced infection rate, probably caused by impaired actin rearrangements at the entry site and in line with the increased vacuolar rupture time (+DMSO, 16.4 ± 11.4 min; +CytD, 39.6 ± 19.3 min; p < 0.0001). Taken together, these results emphasize first a role of Arp2/3-mediated actin rearrangements around the vacuolar niche of Shigella. Second, preventing or perturbing actin cocoon formation at Shigella’s BCV affects cytosolic access, underlining the importance of the cocoon for intracellular niche formation.
Figure 3.

Host Actin Regulators Are Involved in Actin Cocoon Assembly and Are Recruited to Shigella’s BCV before Vacuolar Escape
(A) Scheme of host proteins inhibited by compounds of the inhibitor screen (B and C; see also Figures 4F, S5F, S6C, and S6D). Blue box, Rho GTPases; black, ABPs and their regulators; gray box, effect on F-actin; red, inhibits cocoon assembly; green, no effect on initial cocoon assembly. CytD, cytochalasin D, an actin polymerization inhibitor; SMIFH2, formin inhibitor; Y27632, ROCK inhibitor; Blebbist, blebbistatin, a myosin II inhibitor; Wortm, wortmannin, a PI3K inhibitor; CK666, Arp2/3 inhibitor; NSC23766, RAC1 GEF inhibitor; ETH1864, RAC1 inhibitor; ML141, CDC42 inhibitor.
(B and C) Quantitative analysis of inhibitor screens identifies host proteins and signaling pathways involved in cocoon assembly (B) and cytosolic access (C) (n = 2,912 total invaders, on average 416 per condition; Ctrl, DMSO control).
(D) Overexpression of seven selected host ABPs that interfere with the timing of vacuolar rupture with galectin-3-mOrange (Ctrl, control; n = 2,641, on average 330 per condition).
(E) Recruitment of host ABPs to Shigella’s BCV before cytosolic escape. Time lapses of HeLa cells expressing fluorescently tagged host proteins and actin or galectin-3 (Gal3; scale bars: 3 μm). Arrow, recruitment to BCV; arrowhead, vacuolar rupture.
Indicated are mean values ± SD of at least 3 independent experiments. Student’s t test with p < 0.05 as significant compared with controls: ∗p < 0.05, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001; ns, not significant.
To identify host factors involved in actin cocoon formation, we overexpressed selected ABPs and their regulators. We hypothesized that their cellular excess interferes with F-actin polymerization and, in case these proteins are involved in cocoon regulation, potentially alters Shigella’s cytosolic access. We first screened ABPs for their recruitment to infection sites, determined afterward the rupture time of positive candidates, and finally monitored their precise subcellular localization (Figures 3D and 3E). Remarkably, cytosolic escape of Shigella invading cells overexpressing TOCA-1 (also known as FNBP1L), N-WASP, WIP, cortactin, villin, coronin-1B, or cofilin-1 was significantly delayed (Figure 3D). N-WASP is an NPF for the Arp2/3 complex stabilized by WIP and activated by CDC42-GTP, TOCA-1, and PI(4,5)P2. TOCA-1 activity itself is regulated by CDC42 and PI(4,5)P2 (Rottner et al., 2017, Ho et al., 2004, Rohatgi et al., 1999). Cortactin binds to F-actin and the Arp2/3 complex and links signaling, cytoskeleton, as well as trafficking proteins to actin filaments (Kirkbride et al., 2011). Overexpression of these ABPs probably caused increased F-actin nucleation at the BCV membrane. Villin bundles F-actin, and its overexpression likely stabilizes actin cocoons. Furthermore, coronin-1B collaborates with cofilin-1 in actin disassembly (Cai et al., 2008, Chan et al., 2011). Overexpression of actin, coronin-1B, or cofilin-1 may boost F-actin turnover and polymerization at Shigella’s BCV. Because cocoons exhibited clear borders with uniformly distributed, exceptionally high fluorescence intensity (Figures 1A–1C), and because of the identified ABPs (Figure 3), we expect a rather stiff, highly bundled, and cross-linked F-actin meshwork.
Next, we performed time-lapse microscopy of Shigella infections in cells co-transfected with the involved host proteins and galectin-3. We followed their recruitment to the BCV and correlated their localization with the time point of cytosolic escape (Figure 3E). N-WASP was recruited to the Shigella-containing vacuole and co-localized there with actin. We did not observe any case of N-WASP recruitment to the BCV without actin assembly or vice versa. In addition, WIP, the Arp2/3 complex component Arp3, and cortactin were recruited to the BCV membrane and all were, like actin (Figure 1C), at least partially depleted before vacuolar rupture (Figure 3E). Coronin-1B was recruited early during infection and cocoon assembly, while cofilin-1 localized around the vacuolar membrane remnant even after rupture (Figure 3E). Taken together, we identified a network of host actin regulators that are recruited to Shigella’s BCV and regulate cytosolic invasion.
Actin Cocoon Formation Depends on CDC42-Activated and Arp2/3 Complex-Mediated F-Actin Nucleation
We next identified the involved upstream Rho GTPase signaling for Arp2/3 complex-mediated actin cocoon assembly combining RNA interference, inhibitors, and loss-of-function mutations. Knockdown of CDC42 and the Arp2/3 complex component ArpC3 resulted in clearly altered entry sites compared with the control (Figures 4A–4C). Although CDC42 knockdown or overexpression of its inactive mutant strongly reduces infection efficiency up to 75% (Mounier et al., 1999, Mellouk et al., 2014), it does not entirely prevent cellular uptake in our experiments (Figures 4B–4D; Figures S5A–S5E). Hence, we considered only bacteria that successfully invaded host cells to focus on the role of the actin cocoon. Strikingly, cocoon formation was completely or strongly abolished by knockdown of CDC42 and ArpC3, respectively (+siRNA [small interfering RNA] CDC42, 2.60% ± 1.84%; +siRNA ArpC3, 16.6% ± 0.47%; Figure 4D). We confirmed the strong delay of Shigella vacuolar rupture in CDC42- and ArpC3-depleted cells (Figures S5D and S5E) (Mellouk et al., 2014). Similar results were obtained in cells treated with the CDC42 inhibitor ML141 (Figure S5F). Interestingly, although knockdown of ARPC3 was very efficient, cocoon formation was not as strongly inhibited as for CDC42 (Figure 4D). Either the remaining amount of Arp2/3 complex was sufficient to assemble some cocoons, or another, CDC42-dependent actin polymerization pathway might be involved, for example, via formins. In addition, we investigated the dependence of CDC42 activity on vacuolar escape by comparing the vacuolar rupture time of invading Shigella in cells overexpressing CDC42 WT, a GDP- or GTP-bound CDC42 mutant. Locking CDC42 in either an active or inactive state significantly delayed the cytosolic escape of Shigella (Figure 4E; Figures S5G–S5J). Thus, cytosolic release of Shigella strongly depends on the ability of CDC42 to function as a molecular switch. Finally, RAC1 is probably not essential for de novo actin cocoon assembly, as we did not observe a significant effect in cocoon assembly and cytosolic escape by its knockdown or inhibition with ETH1864 and NSC23766 (Figure 4F; Figures S6A–S6D). Consequently, cocoon assembly requires CDC42-activated and Arp2/3 complex-mediated F-actin nucleation with CDC42 signaling via N-WASP. Thus, the main NPF of the Arp2/3 complex at the BCV is N-WASP.
Figure 4.

CDC42-Activated F-Actin Nucleation by the Arp2/3 Complex Is Essential for Cocoon Formation
(A–D) Knockdown of CDC42 or the Arp2/3 complex subunit ARPC3 inhibits cocoon assembly. Representative time lapses monitoring actin rearrangements during early Shigella WT infection are presented for the scramble siRNA-treated control (Scr; A), for CDC42 (B; KD 87.1%), and for ARPC3 knockdown (C; KD 95.6%) (scales: 5 μm, 1.05 μm z stack; see Figure S3F). (D) Quantitative analysis of actin cocoon assembly shows strongly reduced cocoon formation (no siRNA [HeLa], n = 374 bacteria; siRNA Scr, n = 481; siRNA CDC42, n = 464; siRNA ARPC3, n = 573).
(E) The ability of CDC42 to cycle between an active and inactive state is required for efficient cytosolic access of Shigella. T17N, inactive, GDP-bound mutant; G12V, active, GTP-bound mutant (n = 1,123 total rupture events; WT, n = 373; T17N, n = 375; G12V, n = 375).
(F) Neither RNAi-mediated RAC1 knockdown nor its inhibition by the RAC1 inhibitor ETH1864 alters actin cocoon assembly (siRNA Scr, n = 386; siRNA RAC1, n = 409; DMSO, n = 385; ETH1864, n = 328).
Depicted are mean values ± SD of 3 independent experiments; p < 0.05 is significant compared with control: ∗∗∗∗p < 0.0001; ns, not significant. See Figures 3A, S3, S5, and S6.
Actin Cocoon Assembly Is Regulated by Shigella T3SS Effector Proteins
To identify which Shigella effectors are involved in actin cocoon regulation, we screened a Shigella mutant library with single T3SS effector deletions (Sidik et al., 2014). Results of nine selected mutants are presented (Figures 5A and 5B). As the amount of entering bacteria per cell varies for different mutants, we focused our analysis on the first invading bacteria of individual strains. Strikingly, single deletions of the bacterial effectors IpgD, IpgB1, and IcsB strongly increased cytoskeletal rearrangements around the BCV. We found up to threefold more ΔicsB residing in an actin-containing structure before cytosolic escape (Figure 5A; Figure S6E). We could also confirm our previous reported finding that IpgD increases cocoon formation of the total invading population (Mellouk et al., 2014). The rupture timing of ΔipgB1, ΔvirA, and ΔicsB was likewise significantly delayed (Figure 5B). IcsA recruits N-WASP to the pole of cytosolic bacteria for actin tail formation (Suzuki et al., 1998, Egile et al., 1999) but is not accessible before vacuolar rupture. Consistent with this, we observed no effect on cocoon assembly in ΔicsA infections (Figure 5A). Thus, IpgD, IpgB1, and IcsB play a role in actin cocoon regulation. Deletion of these Shigella effectors might lead to a misregulation of the cocoon with a disturbed actin turnover.
Figure 5.

The Actin Cocoon Is Regulated by Shigella T3SS Effector Proteins, and Host Actin Regulators Constantly Localize IcsB Dependent at the BCV
(A and B) Infection of HeLa cells expressing actin-GFP and galectin-3-mOrange by Shigella single effector deletion mutants. The first invading bacteria per cell was analyzed with regard to actin cocoon assembly (A) and rupture time (B). (n = 1,085 total counted bacteria; WT, n = 101; ΔipaH7.8, n = 80; ΔvirA, n = 131; ΔospC1, n = 112; ΔicsA, n = 103; ΔipaJ, n = 95; ΔipgD, n = 121; ΔipgB1, n = 116; ΔicsB, n = 120. p < 0.05 is significant compared with WT: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001; ns, not significant).
(C) Representative time lapses of constant CDC42 localization at the BCV during Shigella WT infection.
(D) Quantification of the normalized relative fluorescence intensity of CDC42 recruitment during successive Shigella invasion steps. PM ruffle, fluorescence intensity at plasma membrane ruffles; BCV-start, at the phagocytic cup before scission; BCV-maint, at maintained BCV after cellular uptake; BCV-rupture, at BCV after rupture.
(E) Deletion of IcsB leads to CDC42 removal from the BCV before rupture but does not prevent its initial recruitment to membrane ruffles.
(F) Quantitative analysis of Cdc42 localization at the BCV of successfully invading Shigella with constant (dark blue), transient (light blue), or no (gray) CDC42 localization (n = 311). Shigella WT versus ΔicsB: constant CDC42, p < 0.0001; depleted CDC42, p < 0.001; no CDC42, p < 0.05.
(G) Recruited TOCA-1 is constantly localized at the BCV dependent on IcsB (infection top: Shigella WT; bottom: ΔicsB).
Indicated are mean values ± SD of at least 3 independent experiments, in (A) normalized to Shigella WT. Statistical significance: Indicated are mean values ± SD. one-way ANOVA (D) or two-way ANOVA with Tukey’s multiple-comparisons test (F). Arrow, initial protein recruitment; arrowhead, vacuolar rupture; scale bar: 3 μm. Green bar, protein at BCV; red bar, galectin-3 (Gal-3) at BCV; yellow bar, protein and galectin-3 at BCV remnant; gray bar, no protein or galectin-3. See STAR Methods.
Clustering of Host Actin Regulators at Shigella’s BCV Depends on the Fatty Acyltransferase Activity of the T3SS Effector IcsB
We next questioned if the recruitment of the actin nucleation machinery at the BCV (Figure 3E) depends on specific Shigella effectors. We focused on IcsB, the strongest hit of our T3SS effector screen (Figures 5A and 5B). During canonical phagocytosis, active CDC42 localizes to forming pseudopods but is inactivated and depleted to complete cup closure (Niedergang and Grinstein, 2018, Freeman and Grinstein, 2014, Hoppe and Swanson, 2004). Likewise, CDC42 initiates de novo F-actin rearrangements for Shigella’s cellular uptake (Adam et al., 1996). We confirmed CDC42 recruitment to forming membrane ruffles during Shigella infection. Yet contrary to canonical phagocytosis, CDC42 remained at the BCV after cellular uptake and persisted with galectin-3 at the membrane remnant (91.9% ± 1.56% of cytosolic bacteria; Figures 5C and 5D). Remarkably, we deciphered that this constant localization of CDC42 depends on IcsB (Figure 5E). We followed the successive invasion steps of individual bacteria and quantified the recruitment of CDC42 to their BCV as well as the BCV membrane remnant at the moment of vacuolar rupture. Deletion of IcsB did not interfere with initial CDC42 recruitment to membrane ruffles but led to its depletion from the BCV before vacuolar rupture (Figure 5F). Although the Rho GTPase RAC1 is likewise clustered at the BCV dependent on IcsB (Figures S7A and S7B), it is not essential for cocoon assembly (Figures 4F and S6A–S6D). We detected only a few cases of RhoA recruitment (Figure S7C). As for CDC42, TOCA-1 also constantly localized IcsB dependent at Shigella’s vacuole following initial recruitment (Figure 5G). Noteworthy, although the membrane remnant was quickly recycled in WT Shigella infections, it remained loosely and sticky around cytosolic ΔicsB bacteria (Figures 6E–6G).
Figure 6.

Constant N-WASP Localization at the BCV Depends on Post-translational Lipidation by IcsB, but CDC42 Is Crucial for Its Initial Recruitment
(A and B) The Shigella effector IcsB constantly localizes N-WASP at the BCV during infection after initial IcsB-independent recruitment. Representative time lapses for Shigella WT (A) and ΔicsB (B) are shown.
(C) CDC42 knockdown strongly impairs initial N-WASP recruitment to the BCV of Shigella WT.
(D) Quantification of N-WASP localization at the BCV of invading Shigella with constant (dark green), transient (light green), or no (gray) N-WASP (n = 441). Statistical significance of Shigella WT versus ΔicsB: constant N-WASP localization, p < 0.0001; N-WASP depleted, p < 0.0001; no N-WASP, ns. Shigella WT versus siRNA CDC42: constant N-WASP localization, p < 0.0001; N-WASP depleted, ns; no N-WASP, p < 0.0001.
(E) The complemented Shigella icsB− mutant (icsB−[compl.]) clusters N-WASP like Shigella WT around the BCV.
(F) In contrast, complementation of icsB− with IcsB lacking fatty acyltransferase activity (icsB− [C306A]) does not restore constant N-WASP localization.
(G) Quantification of N-WASP localization at the BCV of invading Shigella, color coded as in (D) (n = 748; WT, n = 138; icsB−, n = 182; icsB−[compl.], n = 136; icsB−[C306A], n = 292). Shigella WT versus icsB−: constant N-WASP localization, p < 0.0001; N-WASP depleted, p < 0.0001; no N-WASP, ns. Shigella WT versus Shigella icsB−(compl.): constant N-WASP localization, ns; N-WASP depleted, ns; no N-WASP, ns. Shigella WT versus Shigella icsB−(C306A): constant N-WASP localization, p < 0.0001; N-WASP depleted, p < 0.0001; no N-WASP, ns.
Statistical significance: two-way ANOVA with Tukey’s multiple-comparisons test. Indicated are mean values ± SD of 3 independent experiments. Arrow, initial N-WASP recruitment; arrowhead, moment of vacuolar rupture; scale bars: 3 μm (A–C) and 5 μm (E). Green bar, N-WASP at BCV; red bar, galectin-3 at BCV; yellow bar, N-WASP and galectin-3 at BCV remnant; gray bar, no N-WASP or galectin-3. See STAR Methods and Figure S7.
Furthermore, we found N-WASP to constantly localize around Shigella’s endocytic compartment in HeLa and Caco-2 cells (Figures 6A and S7D). N-WASP was recruited after cellular entry to 78.6% ± 3.25% of WT bacteria that successfully escaped from the BCV and remained associated with the BCV membrane remnants in 69.2% ± 9.45% of the cases (Figures 6A and 6D). As for CDC42, the permanent localization of N-WASP at the BCV, but not its initial recruitment, was IcsB dependent (Figures 6B and 6D). We further confirmed N-WASP recruitment to be independent of the Shigella virulence factor IcsA (Figure S7E). We hypothesized that CDC42 initiates N-WASP recruitment, as CDC42 localizes during early entry steps at membrane ruffles. Therefore, we knocked down CDC42 and followed the localization of N-WASP during Shigella WT infection (Figure 6C). Interestingly, we found a strong reduction in initial N-WASP recruitment to the BCV (16.67% ± 7.92%), but recruited N-WASP remained localized (14.48% ± 4.84%) (Figure 6D). Residual CDC42 activity was probably sufficient to recruit N-WASP in some rare cases with constant BCV localization because of IcsB.
It has recently been discovered that IcsB post-translationally modifies the actin regulators identified herein by 18-carbon lipidation (Liu et al., 2018). IcsB is an acyltransferase, which catalyzes Nε-fatty acylation of lysine residues in membrane-bound host proteins (Liu et al., 2018). A C306A-IcsB mutant has been described to be crucial for IcsB activity (Liu et al., 2018). Therefore, we analyzed the role of the IcsB enzymatic activity during cocoon formation. Although complementation of the icsB− mutant with IcsB resembled the phenotype of Shigella WT infections, expression of the acyltransferase-inactive IcsB-C306A mutant (icsB− [C306A]) impaired N-WASP clustering at the Shigella vacuole (Figures 6F and 6G; Figures S7F–S7H). Thus, we found that IcsB clusters key factors of the host actin nucleation machinery at the BCV dependent on its fatty acyltransferase enzymatic activity. However, the initial recruitment of the Arp2/3 complex activator N-WASP depends on CDC42 signaling. Because knockdown of CDC42 prevents cocoon formation, the CDC42-mediated activation of N-WASP is crucial for actin cocoon assembly.
Function of the Actin Cocoon in Late Intracellular Infection Steps
We finally deciphered the consequences of actin cocoon assembly on subsequent infection steps following cytosolic release of Shigella from the vacuole. Here, we monitored the successive invasion events for each individual bacterium from cell entry to protrusion for cell-to-cell spread and quantified the fate of the bacterium and its BCV remnant 2 h post-infection (Figures 7A–7D). Here, 78.7% ± 9.40% of the bacteria that assembled an actin cocoon prior to vacuolar rupture were motile (Figure 7B). In contrast, Shigella without cocoon had a high probability of being trapped inside the cytosol either by not starting or by stopped actin tail-mediated movement (62.3% ± 0.24%; Figure 7B). In addition, the majority of Shigella with a cocoon were BCV membrane remnant free (Figure 7C). Bacteria without a cocoon showed difficulties in unwrapping the BCV membrane remnants and were predominantly surrounded by a part of it (68.6% ± 3.91%; Figure 7C). Such remnants or “membrane caps” impaired Shigella spreading, as 69.7% ± 11.8% of them were unable to move at the end of the experiments. In contrast, actin cocoon-supported loss of the BCV membrane fostered successful spreading (73.8% ± 4.03% motile after sticky removal, 100% motile after quick recycling; Figure 7D). Taken together, the actin cocoon supports late cell invasion steps and cell-to-cell spread by aiding to the removal of the BCV membrane remnant, which impairs Shigella’s motility.
Figure 7.

Functional Consequences of Actin Cocoon Formation on Subsequent Infection Steps
(A) Illustration of invading, cytosolic Shigella after vacuolar escape and their fates regarding bacterial movement and membrane recycling.
(B–D) Quantification of the motility of individual cytosolic Shigella (B) and the fate of their BCV membrane remnants (C). Successive infection steps were monitored 2 h post-infection (p.i.) (see STAR Methods; n = 144, mean values ± SD of 6 independent experiments). In most cases, a membrane cap impairs Shigella spreading (trapped), while actin cocoon-supported loss of the BCV remnant fosters dissemination (protrusion, motile) (D).
(E) Model for Shigella-mediated host actin cytoskeleton subversion by injected T3SS effectors to regulate niche integrity. (1) CDC42 is recruited at the forming BCV. (2) TOCA-1 and N-WASP cluster around the BCV after scission. (3a) Arp2/3 complex recruitment and de novo cocoon assembly with constant G-actin turnover. (4) The Arp2/3 complex gets depleted from the BCV during cocoon disassembly, but not CDC42, N-WASP, and TOCA-1. (5) Vacuolar rupture and Shigella cytosolic escape after cocoon disassembly. CDC42, N-WASP, and TOCA-1 remain at the recycled BCV remnant. (3b) In the absence of IcsB, these proteins cluster not at the BCV, leading to an altered, actin-containing structure. (3c) Depletion of CDC42 impairs initial N-WASP recruitment and cocoon formation. (6 and 7) Disturbing actin cocoon regulation (3b) or preventing its formation (3c) interferes with vacuolar rupture.
(F) Host factors and Shigella effectors involved in cocoon formation. Green star, constant IcsB-dependent localization at the BCV (Figures 5 and 6) and reported (Liu et al., 2018) post-translational lipidation.
Discussion
We identified in this study highly dynamic de novo F-actin polymerization of exceptional thickness around the endocytic vacuole of intracellular Shigella. We showed that this actin cocoon assembles after scission at the surface of the entire vacuolar membrane and does not constitute remaining F-actin from cell entry. Compared with previous reported actin rearrangements around internalized phagosomes, we found that the cocoon shares features with the “actin flashing” phenomenon that occurs target- and cell-unspecific around endocytic compartments to protect cells from overloading degradative pathways (Yam and Theriot, 2004, Liebl and Griffiths, 2009). Both phagosomal actin rearrangements have in common (1) the involvement of certain host proteins such as Arp2/3 (Bierne et al., 2001, Yam and Theriot, 2004), (2) assembly at the cell periphery, and (3) preferably at later formed phagosomes. Besides, Shigella’s actin cocoon is clearly distinct and resembles a much longer lasting structure with non-cyclical actin turnover that is much denser than any other cellular actin structure. Its disassembly leads to immediate cytosolic escape of Shigella, while actin flashing is followed by vesicle fusion for phagosome maturation (Figure S4E). Therefore, assembly of these distinct actin structures precedes completely different phagosomal fates: actin cocoon assembly correlates with the onset of altered phagosomal maturation and cumulates in a BCV rupture, while actin flashing leads to fusion and bacterial degradation. We propose that Shigella hijacks the host actin flashing mechanism for its own needs and thus counteracts a cellular response to invasion.
Despite this, the endocytic vacuole of Shigella is intrinsically different from early endocytic compartments with regard to its molecular composition. We identified a network of host actin regulators that are recruited and prolonged localized at the BCV (Figures 7E and 7F). Interestingly, Shigella plays itself an important role in actin cocoon regulation by inducing localized signaling with its effectors IpgD, IpgB1, and IcsB. One strategy to locally induce actin rearrangements is to immobilize key proteins by either (1) increasing their membrane binding affinity or (2) preventing their removal. Both strategies imply subverted host protein localization by Shigella. Here, we present a biological function for the recently discovered fatty acyltransferase activity of IcsB in Shigella infection. We found IcsB to be necessary to cluster CDC42, TOCA-1, and N-WASP at Shigella’s vacuole, dependent on its lipidation activity. Our findings place IcsB as central Shigella effector for the re-localization and entrapment of signaling proteins to regulate cytosolic access. IcsB most likely lipidates its substrates directly at the BCV membrane, as it localizes around intracellular bacteria (Baxt and Goldberg, 2014, Campbell-Valois et al., 2015, Liu et al., 2018). Thus, Shigella seems to “glue” host proteins for localized signaling to its endocytic vacuole by adding an additional membrane binding motif. It remains to be further investigated, if all clustered host proteins are directly lipidated by IcsB, or if their constant localization is an indirect effect of either immobilized binding partners or impaired endomembrane trafficking.
Interestingly, IcsB is not required for initial N-WASP localization at the BCV but the Rho GTPase CDC42. This reveals an unexpected second function of active CDC42 during infection besides cellular uptake: it is essential for efficient actin cocoon assembly, which in turn regulates cytosolic access. However, although the vacuolar immobilization of CDC42 depends on IcsB, its initial recruitment, and eventually activation, may involve other effectors targeting upstream Rho signaling during cellular entry. For instance, IpaC may indirectly increase active CDC42 levels via Src signaling (Tran Van Nhieu et al., 1999, Mounier et al., 2009, Adam et al., 1996). IpgB1 is a membrane-bound Rho GEF at intracellular Shigella with nucleotide exchange activity for RAC1 and, albeit reduced, CDC42 (Ohya et al., 2005, Huang et al., 2009, Weigele et al., 2017). Consistent with this, we also identified IpgB1, but also IpgD, as an actin cocoon regulator (Figures 5A and 5B). How the inositol-phosphatase IpgD, which produces PI5P from PI(4,5)P2 (Niebuhr et al., 2002, Pendaries et al., 2006), is involved in cocoon assembly remains to be addressed. PI5P leads to PI3K pathway activation (Pendaries et al., 2006) and regulates Rac GEF activity (Viaud et al., 2014). Although our results did not indicate an essential contribution of RAC1 in cocoon assembly, it may have a yet unknown role in infection given its strong localization at the BCV.
Furthermore, we deciphered that actin cocoon dynamics need to be tightly regulated. Cocoon disassembly precedes cytosolic escape, and perturbing its proper regulation by interfering with either cocoon assembly or disassembly consequently diminishes cytosolic invasion of Shigella. Remarkably, ΔipgB1 and ΔicsB mutants had more pronounced cocoons, highlighting the interference of these effectors with actin turnover. Impairing the activation and immobilization of Rho GTPases by deletion of these effectors probably causes a misregulation of the actin cocoon ultrastructure with disturbed actin dynamics. In line with this, we found that the ΔicsB BCV differs in its host protein composition from Shigella WT. We propose that this induces the formation of a changed cytoskeletal structure less prone to cytosolic escape (Figure 7E). As Shigella subverts the host actin cytoskeleton via distinct pathways, deletion of major regulators puts an imbalance in this complex, fine-tuned system.
It emerges that several human pathogens assemble varying actin structures around their intracellular niches. It is tempting to speculate that, as in cells, different structures result in different functions. These actin rearrangements have been associated with maintaining vacuolar stability and integrity, trafficking to acquire nutrients, or preventing phagosome maturation (Kühn and Enninga, 2020). Often, F-actin and additional cytoskeletal systems assemble during late infection steps around the replicative niche of pathogens with long vacuolar lifestyles (Kumar and Valdivia, 2008, Méresse et al., 2001, Sukumaran et al., 2011), like the loose actin meshwork around vacuolar Salmonella (Méresse et al., 2001, Vazquez-Torres et al., 2000, Poh et al., 2008, Unsworth et al., 2004).
In contrast, the Shigella actin cocoon is clearly distinct, indicating a different function. We found that first cocoon-surrounded BCVs were stationary, excluding a role in actin-driven motility of endocytic compartments (Merrifield et al., 1999, Taunton et al., 2000). Second, a function in sorting and recycling as indicated by dot-like actin structures (Puthenveedu et al., 2010, Seaman et al., 2013) seems unlikely, as actin assembled homogeneously around entire BCVs. Third, we never detected vesicle fusion events with the BCV, nor recruitment of markers for phagosome maturation (Mellouk et al., 2014, Weiner et al., 2016, Kühn et al., 2017) (Figure S4E), eliminating a function in vesicle fusion. We therefore propose two functions of the actin cocoon during Shigella’s intracellular lifestyle. We previously demonstrated that the cocoon forms between the BCV and surrounding vesicles (Weiner et al., 2016). Thus, we first suggest that the cocoon acts as “gatekeeper” to regulate pathogen entry into the host cytosol. It shields the BCV as physical barrier from the host endomembrane system and prevents fusion with endosomes and lysosomes, while the cocoon alters its phagosomal identity with regard to lipid and protein composition. Interestingly, CDC42 and Arp2/3 have been reported to prevent phagosome maturation by an F-actin coat around the replicative niche of Leishmania donovani (Lodge and Descoteaux, 2005). As cocoon inhibition also delays vacuolar escape, cocoon formation itself may allow proper vacuolar maturation in the presence of macropinosomes. Second, the actin cocoon could prevent host recognition during late infection steps to ensure successful invasion. After vacuolar rupture, it facilitates recycling and removal of the BCV membrane remnants from Shigella. The cocoon and its involved molecular machinery could either support vacuolar rupture or eventually govern short distance endomembrane trafficking. It has been shown that LC3, p62, and ubiquitin are recruited to the cytosolic, galectin-3-positive BCV membrane remnants (Dupont et al., 2009) and that cytosolic Shigella are trapped by GBPs (Wandel et al., 2017). Additionally, a role of IcsB in autophagy escape by preventing LC3 recruitment to bacteria has been reported (Campbell-Valois et al., 2015, Ogawa et al., 2005, Baxt and Goldberg, 2014). We showed that Shigella without cocoon are often surrounded by BCV remnants, causing a high probability of getting trapped inside the cell. We hypothesize that the actin cocoon enables Shigella to lose its “danger” signal, which would mark it for autophagy response and degradation.
In conclusion, we discovered a unique microbial subversion strategy: the entrapment of the host actin nucleation machinery by post-translational modification causes localized signaling at the Shigella vacuole during early infection. This leads to the formation of an exceptionally thick actin structure to ensure niche integrity and cytosolic escape. The dynamic maturation of this structure must be tightly regulated, as either preventing or disturbing it impairs cytosolic escape.
STAR★Methods
Key Resources Table
| REAGENT OR RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| polyclonal rabbit anti-Septin 7 | IBL | Cat#18991; RRID: AB_10705434 |
| monoclonal mouse anti-β-actin | Sigma-Aldrich | Cat#A5316; RRID: AB_476743 |
| monoclonal mouse anti-CDC42 | BD Transduction Laboratories | Cat#610929; RRID: AB_398244 |
| monoclonal mouse anti-ArpC3 p21-Arc Clone26 | BD Transduction Laboratories | Cat#612234; RRID: AB_399557 |
| monoclonal mouse anti-RAC1 clone 23A8 | Abcam | Cat# ab33186; RRID: AB_777598 |
| anti-mouse horseradish peroxidase-conjugated | Bio-Rad | Cat#170-6516; RRID: AB_11125547 |
| anti-rabbit horseradish peroxidase-conjugated | Bio-Rad | Cat#170-6515; RRID: AB_11125142 |
| Bacterial strains | ||
| Shigella flexneri (WT) 5a M90T-Sm virulence plasmid pWR100 (GenBank#CM001474.1, GenBank#AL391753.1) | Philippe Sansonetti John R. Rohde | (Onodera et al., 2012) |
| S. flexneri icsB- (non-polar icsB mutant) | Claude Parsot | N/A |
| S. flexneri icsB-(compl.) (icsB- mutant carrying pUC8::icsB-ipgA expressing IcsB and its chaperone IpgA) | Claude Parsot | N/A |
| S. flexneri icsB-(C306A) (icsB- mutant carrying pUC8::icsB(C306A)-ipgA expressing enzymatic inactive IcsB-C306A and its chaperone IpgA) | This work | N/A |
| S. flexneri (WT) mCherry (expressing mCherry and Afa-I (E. coli pIL22)) | Guy Tran Van Nhieu | N/A |
| S. flexneri (WT) (expressing Afa-I (afaE gene) from E. coli) | John R. Rohde | (Labigne-Roussel et al., 1984) |
| S. flexneri (WT) DsRed (expressing dsRed and Afa-I) | Yuen-Yan Chang | N/A |
| S. flexneri ΔicsA (ΔicsA::tetRA expressing Afa-I) | John R. Rohde | (Sidik et al., 2014) |
| S. flexneri ΔipgB1 (ΔipgB1::tetRA expressing Afa-I) | John R. Rohde | (Sidik et al., 2014) |
| S. flexneri ΔipgD (ΔipgD::tetRA expressing Afa-I) | John R. Rohde | (Sidik et al., 2014) |
| S. flexneri ΔipaJ (ΔipaJ::tetRA expressing Afa-I) | John R. Rohde | (Sidik et al., 2014) |
| S. flexneri ΔospG (ΔospG::tetRA expressing Afa-I) | John R. Rohde | (Sidik et al., 2014) |
| S. flexneri ΔospC1 (ΔospC1::tetRA expressing Afa-I) | John R. Rohde | (Sidik et al., 2014) |
| S. flexneri ΔvirA (ΔvirA::tetRA expressing Afa-I) | John R. Rohde | (Sidik et al., 2014) |
| S. flexneri ΔipaH7.8 (ΔipaH7.8::tetRA expressing Afa-I) | John R. Rohde | (Sidik et al., 2014) |
| Escherichia coli DH5α | Thermo Fisher Scientific | Cat#18265017 |
| Escherichia coli InvA (expressing Yersinia invasin A) | Guy Tran Van Nhieu | N/A |
| Salmonella enterica serovar Typhimurium (S. Typhimurium) SL1344 / pGG2-dsRed (WT) | ATCC | ATCC SL1344 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| X-tremeGENE 9 DNA transfection reagent | Roche | Cat#6365779001 |
| Lipofectamine RNAiMAX | Invitrogen | Cat#13778030 |
| DAPI | Thermo Fisher Scientific | Cat#D1306 |
| Alexa Fluor 647 Phalloidin | Thermo Fisher | Cat#A22287 |
| Paraformaldehyde 16% | Electron Microscopy Sciences | Cat#15710 |
| Ampicillin | Sigma-Aldrich | Cat#A9393 |
| Kanamycin | Sigma-Aldrich | Cat#K1377 |
| Isopropyl-β-D-thiogalactoside (IPTG) | Sigma-Aldrich | Cat#10724815001 |
| Poly-L-lysine | Sigma-Aldrich | Cat#P4707 |
| Agarose | Invitrogen | Cat#16500-500 |
| Dulbecco’s Modified Eagle’s Medium, High Glucose, GlutaMax | Thermo Fisher Scientific | Cat#10566016 |
| Fetal Bovine Serum | Sigma-Aldrich | Cat#F7524 |
| DPBS | Thermo Fisher Scientific | Cat#14190144 |
| MEM Non-Essential Amino Acids Solution | Thermo Fisher Scientific | Cat#11140050 |
| HEPES | Thermo Fisher Scientific | Cat#15630080 |
| 0.05% Trypsin-EDTA | Thermo Fisher Scientific | Cat#25300054 |
| ProLong Gold Antifade Mountant | ThermoFisher Scientific | Cat#P10144 |
| FluoroBrite DMEM | ThermoFisher Scientific | Cat#A1896701 |
| Complete Protease Inhibitor | Roche | Cat#11836170001 |
| RIPA buffer | ThermoFisher Scientific | Cat#89901 |
| NuPAGE 4-12% Bis-Tris Protein Gels | Thermo Fisher Scientific | Cat#NP0321BOX |
| Micro BCA™ Protein Assay Kit | Thermo Fisher Scientific | Cat#23235 |
| Trans-Blot® Turbo RTA Mini Nitrocellulose Transfer Kit | Bio-Rad | Cat#1704270 |
| Trans-Blot© Turbo Transfer System | Bio-Rad | Cat#1704150 |
| SuperSignal West Pico PLUS Chemiluminescent Substrate | Thermo Scientific | Cat#34577 |
| QIAprep Spin Miniprep Kit | QIAGEN | Cat# 27106 |
| Q5® Site Directed mutagenesis kit | New Englands Biolabs | Cat#E0554S |
| Inhibitors | ||
| Cytochalasin D | Enzo Life Sciences Inc | Cat#BML-T109-0001 |
| SMIFH2 | Sigma | Cat#S4826 |
| Y-27632 | BD biosciences | Cat#562822 |
| NSC23766 | Calbiochem | Cat#553502 |
| (±)-Blebbistatin | BioVision | Cat#BV-2405-5 |
| Wortmannin | Enzo Life Sciences Inc | Cat#BML-ST415-0001 |
| CK666 | Sigma | Cat#SML0006 |
| ETH1864 | Tocris Bioscience | Cat#3872 |
| ML141 | Tocris Bioscience | Cat#71203-35-5 |
| Experimental Models: Cell Lines | ||
| HeLa Human cervix carcinoma epithelial cell line clone CCL-2 | ATCC | Cat#11033106 |
| Caco-2 TC7 cells | Philippe Sansonetti | N/A |
| HeLa cells stably expressing galectin-3-mOrange | Patricia Latour-Lambert | N/A |
| Oligonucleotides | ||
| ON-TARGETplus Non-targeting Control Pool | Dharmacon | Cat#D-001810-10-05 |
| SEPT7 siRNA | Ambion | Cat#s2743 |
| ON-TARGETplus SMARTpool CDC42 | Dharmacon | Cat#L-005057-00-0005 |
| ON-TARGETplus SMARTpool ARPC3 | Dharmacon | Cat#L-005284-00-0005 |
| ON-TARGETplus SMARTpool RAC1 | Dharmacon | Cat#L-003560-00-0005 |
| Recombinant DNA | ||
| pmCherryC1-Arp3 | Christian Merrifield | (Taylor et al., 2011) Addgene Cat#27682 |
| pmCherryC1- Cortactin | Christian Merrifield | (Taylor et al., 2011) Addgene Cat#27676 |
| pmCherryC1-Cofilin | Christian Merrifield | (Taylor et al., 2011) Addgene Cat#27687 |
| pEYFP-C1-Villin | Sylvie Robine | (Revenu et al., 2007) |
| pEGFP-C1-TOCA-1 | Jennifer Gallop | N/A |
| pGFP-Cortactin | Kenneth Yamada | Addgene Cat#50728 |
| mEmerald-N-WASP-C-18 | Michael Davidson | Addgene Cat#54199 |
| mEmerald-Coronin1B | Michael Davidson | Addgene Cat#54050 |
| pmCherry-C1-WIP | Anna Huttenlocher | Addgene Cat#29573 |
| pEGFP-Actin | (Mounier et al., 2009) | N/A |
| mCherry-Actin | Dominique Lallemand | N/A |
| pOrange-C3-Actin | (Ehsani et al., 2012) | N/A |
| pEGFP-Galectin-3 | (Paz et al., 2010) | N/A |
| pOrange-Galectin-3 | (Ray et al., 2010) | N/A |
| mTurquoise-Galectin-3 | Noelia Lopez-Montero | N/A |
| pmCherry-CDC42 | Sandrine Etienne-Manneville | N/A |
| pEYFP-CDC42 | Joel Swanson | (Hoppe and Swanson, 2004) |
| GFP-CDC42-V12 | Sandrine Etienne-Manneville | N/A |
| GFP-CDC42-N17 | Sandrine Etienne-Manneville | N/A |
| pcDNA3-EGFP-RAC1 | Klaus Hahn (Kraynov et al., 2000) | Addgene Cat#13719 |
| pEYFP-RHOA | Joel Swanson | (Hoppe and Swanson, 2004) |
| pGFP-2XFYVE | Philippe Benaroch | N/A |
| pmCherry-2xFYVE | Harald Stenmark and Kay Oliver Schink | N/A |
| pEYFP-LAMP1 | Walther Mothes | Addgene Cat#1816 |
| pEGFP-LC3B | Thomas Wollert | N/A |
| pmCherry-RAB7 | Michael Davidson | Addgene Cat#55127 |
| Software and Algorithms | ||
| FIJI | NIH | https://fiji.schttps://imagej.net/ImageJ |
| Volocity 6.3 PerkinElmer | PerkinElmer | N/A |
| NIS-Elements Microscope Imaging Software | Nikon | N/A |
| GraphPad Prism | GraphPad Software v6.0, La Jolla, USA | https://www.graphpad.com/ |
| Excel | Microsoft | N/A |
| ICY | Institut Pasteur | http://icy.bioimageanalysis.org/ |
| Inkscape | Version 0.92.3 | https://inkscape.org/ |
| Other | ||
| Zeiss LSM780 Elyra PS1 microscope equipped with the followings: | Zeiss | N/A |
| 63x/1.4 oil Plan Apo objective | Zeiss | N/A |
| EMCCD Andor Ixon 887 1K camera | Andor | N/A |
| Perkin Elmer UltraView spinning disk confocal microscope equipped with the followings: | Perkin Elmer | N/A |
| 60 × /1.3 oil objective | Nikon | N/A |
| PSU C910-50 camera | Hamamatsu | N/A |
| Inverted widefield microscope equipped with the followings: | Nikon | N/A |
| 20x/0.5NA air objective | Nikon | N/A |
| CoolSnap2 camera | Roeper Scientific | N/A |
| DeltaVision Elite epifluorescence microscope | GE Healthcare | N/A |
| 96-well cell culture microplates with clear flat bottom | Greiner Bio One International | Cat#655090 |
| Precision cover glasses thickness No. 1.5H | Marienfeld Superior | Cat#0117580 |
| 35 mm glass bottom μ-Dishes | Ibidi | Cat#81158 |
| Tissue culture flask 75 | TPP | Cat#90076 |
| Costar® 6-well Clear TC-treated Multiple Well Plates | Corning | Cat#3516 |
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jost Enninga (jost.enninga@pasteur.fr).
Materials Availability
We are glad to share all reagents generated in this study without restriction. Please contact the lead contact (jost.enninga@pasteur.fr) without restriction.
Code Availability
The published article includes all datasets analyzed during this study. Please contact the lead author in case you require the original source data, for example time-lapse series that cannot be added to a public database.
Experimental Model and Subject Details
HeLa and Caco-2 Cells
Human epithelial HeLa cells (clone CCL-2, ATCC #11033106), HeLa cells stably expressing galectin-3-mOrange (Patricia Latour-Lambert), and intestinal epithelial Caco-2 TC7 cells (kindly provided by P. Sansonetti) were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Thermo Fisher Scientific #10566016) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS, Sigma-Aldrich #F7524) at 37°C, 5% (HeLa) or 10% (Caco-2) CO2. Cell lines were checked negative for mycoplasma.
Bacterial strains and culture
The following streptomycin-resistant Shigella flexneri 5a M90T-Sm (GenBank #CM001474.1) derived strains harboring the pWR100 virulence plasmid (GenBank #AL391753.1) were used: wild-type M90T (WT), M90T (WT) containing the afimbrile adhesin E gene (afaE) from E. coli (Labigne-Roussel et al., 1984), M90T (WT) expressing dsRed and AfaI (Yuen-Yan Chang). The screened library of S. flexneri single deletion T3SS effector mutants was kindly provided by John R. Rohde (Dalhousie University) as part of a pWR100 collection originating from M90T (WT) AfaI (Sidik et al., 2014). We used Salmonella enterica serovar Typhimurium (S. Typhimurium) strain SL1344 pGG2-dsRed (WT) expressing dsRed. The E. coli InvA strain containing the Yersinia invasin A (Isberg et al., 1987) and S. flexneri M90T (WT) expressing mCherry and AfaI (E. coli plasmid pIL22) were provided by Guy Tran Van Nhieu. S. flexneri icsB- and S. flexneri icsB-(compl.) (pUC8::icsB-ipgA, expressing IcsB and its chaperone IpgA) were provided by Claude Parsot. S. flexneri strains were grown in trypticase soy broth (TCSB) with 50 μg/ml ampicillin, and cultured at 37°C on TCSB agar including 0.01% congo red to select for functional T3SS system. S. Typhimurium was grown at 37°C in lysogeny broth (LB) medium supplemented with 0.3 M NaCl and 50 μg/ml ampicillin, while E. coli InvA was cultured in LB medium with 100 μg/ml ampicillin. E. coli DH5α and derivatives were grown at 37°C in LB medium, when needed the medium was supplemented with 100 μg/ml ampicillin.
Method Details
Bacterial infection and immunocytochemistry
HeLa cells were used for all experiments except Figures S1B and S7B. All infection assays were performed in EM buffer (120 mM NaCl, 7 mM KCl, 1.8 mM CaCl2, 0.8 mM MgCl2, 5 mM glucose, 25 mM HEPES, pH 7.3). Actin cocoon formation and vacuolar escape was not affected by EM buffer compared to FluoroBrite DMEM (Thermo Fisher Scientific #A1896701) supplemented with 10% heat-inactivated FBS and 4 mM GlutaMAX (Thermo Fisher Scientific #35050061). For Shigella, infection cultures in TCBS plus antibiotics (and ITPG 2 mM for complementation assays) were inoculated with a 1:100 dilution of an overnight culture and incubated at 37°C to an optical density (OD 600 nm) of 0.6-0.7. Then, bacteria were washed in PBS and resuspended in EM buffer. Strains without Afa-I adhesin were incubated for 20 min with poly-L-lysine (Sigma, #P4707) as described previously (Weiner et al., 2016). All Shigella-containing experiments were performed with Afa-I strains, besides experiments shown in Figures 6E–6G and S7F–S7H, which were performed with poly-L-lysine-coated Shigella. For complementation, the infection cultures of Shigella icsB-(compl.) and icsB-(C306A) strains were grown in 2 mM ITPG before invasion assays. In general, bacterial dilutions were prepared for the following MOIs: live imaging 96-well format MOI 5-150 (normally MOI in range 15-40 to exclude effects of phagocytic load), live imaging 35 mm glass bottom μ-Dishes (Ibidi, #81158) MOI 15 and poly-L-lysine treated bacteria MOI 50. Other bacterial pathogens, like Salmonella or E. coli InvA, were grown similarly in their corresponding medium. Salmonella infections at MOI 50 were started maximal 10 min after spinning down the bacteria in pre-heated Eppendorf tubes and washing in pre-heated EM buffer. E. coli InvA were added to imaged cells at an MOI of 50. For fixed experiments, Shigella (WT) DsRed adhered for 10 minutes at 20°C (MOI 50), before incubation at 37°C for 45 min or 60 min. Samples were washed three times with PBS, fixed in cold 2% PFA, and stained with Alexa Fluor 647 Phalloidin (Thermo Fisher Scientific, #A22287) for 45 min.
Plasmids, inhibitors, siRNAs, and transfection
The following plasmids were kindly provided by our colleagues: Arp3-pmCherryC1, Cortactin-pmCherryC1, as well as Cofilin-pmCherryC1 from Christian Merrifield (Taylor et al., 2011), CDC42-mCherry, CDC42-V12-GFP, as well as CDC42-N17-GFP from Sandrine Etienne-Manneville, pEYFP-C1-Villin from Sylvie Robine (Revenu et al., 2007), pEGFP-C1-TOCA-1 from Jennifer Gallop, pGFP-Cortactin from Kenneth Yamada, mEmerald-N-Wasp-C-18, mEmerald-Coronin1B and pmCherry-RAB7 from Michael Davidson, pmCherry-C1-WIP from Anna Huttenlocher, pEYFP-CDC42 and pEYFP-RHOA from Joel Swanson (Hoppe and Swanson, 2004), pcDNA3-EGFP-RAC1 from Klaus Hahn (Kraynov et al., 2000), pGFP-2XFYVE from Philippe Benarock, pmCherry-2xFYVE from Harald Stenmark, pEYFP-LAMP1 from Walther Mothes, and pEGFP-LC3 from Thomas Wollert. The plasmids pEGFP-C3-Actin, pOrange-C3-Actin, pEGFP-N1-Galectin-3, and pOrange-Galectin-3 have been described previously (Paz et al., 2010, Mounier et al., 2009) (Ehsani et al., 2012) (Ray et al., 2010). All plasmid constructs were verified by sequencing. For quantitative screening experiments (Figures 1F, 1G, 3B–3D, 4E, 4F, 5A, 5B, S1E–S1G, S2, S5F–S5J, S6C, and S6D), HeLa cells were seeded at a density of 7000 cells/well 24 h prior to transfection into 96-well plates (Greiner, #655090). Cells were then transfected with the respective plasmids using X-tremeGENE 9 DNA transfection reagent (Roche, #6365779001) for 48 h (24 h for TOCA-1 and CDC42).
pUC8::icsB(C306A)-ipgA construction: Mutagenesis to inactivate IcsB enzymatic activity in the pUC derivative was performed using the Q5® Site Directed mutagenesis kit (New Englands Biolabs #E0554S) as suggested by the manufacturer. Mutagenesis primers were icsB_C306_F (ATCTGAAAACgcTGCTGGTATGGCAC) and icsB_C306_R (TTACTGATTAATTTATAATTTGCC). First, plasmid was divergently amplified using mutagenesis primers and Pfu polymerase from the kit. Next, the template plasmid was digested with DpnI and the PCR product phosphorylated and ligated using the KLD enzyme mix according to the manufacturer instructions. Ligation products were transformed in chemocompentent E. coli DH5α and transformants were selected in LB agar supplemented with 100 μg/mL of ampicillin. Plasmids were purified using the QIAprep Spin Miniprep Kit according to the manufacturer instructions and verified by sequencing.
In inhibitor screens, the following compounds and concentrations (DMSO stock solutions dissolved in EM buffer) were applied: 1 μM Cytochalasin D (Enzo Life Sciences Inc, #BML-T109-0001), 10 μM SMIFH2 (Sigma-Aldrich, #S4826), 5 μM ML141 (Tocris Bioscience, #71203-35-5; of note: we observed solubility limitations with this inhibitor, therefore the effective concentration will be lower), 25 μM ETH1864 (Tocris Bioscience, #3872), 15 μM Y-27632 (BD biosciences, #562822), 50 μM NSC23766 (Calbiochem, #553502), 15 μM (±)-Blebbistatin (BioVision, #BV-2405-5), 10 μM Wortmannin (Enzo Life Sciences Inc, #BML-ST415-0001), 200 μM CK666 (Sigma, #SML0006). For each inhibitor, working concentrations were identified that did not affect cell viability in the duration of the experiment, but showed clear effects on either cell shape or actin rearrangements during infection. In general, cells were pre-incubated for 40 min at 37°C in EM buffer containing the corresponding inhibitor. Afterward, bacteria were added in EM buffer with the same inhibitor concentration and imaged immediately. For Cytochalasin D, cells were not pre-incubated and the inhibitor was added with the bacteria.
Following siRNAs were used in siRNA transfections: 50 nM SEPT7 (ID #s2743, Ambion), 10 nM ON-TARGETplus SMARTpool siRNAs from Dharmacon for CDC42 (#L-005057-00-0005), ARPC3 (#L-005284-00-0005), RAC1 (#L-003560-00-0005) as well as Non-targeting pool as negative control (#D-001810-10-05). For quantitative analysis (Figures 2B, 4D, 4F, S5A–S5E, and S6B), HeLa cells were seeded at a density of 5000 cells/well 24 h prior to transfection into 96-well plates. Reverse siRNA transfection was performed using the Lipofectamine RNAiMAX Transfection Reagent (Invitrogen, #13778030) for 72 h. After 48 h, transfection mixes were removed and cells were transfected for 24 h with pEGFP-C3-actin and pOrange-Galectin3 plasmids using X-tremeGENE 9 DNA transfection reagent. In parallel, upscaled samples for western blot quantification were prepared in 6-well plates and for live imaging at the DeltaVision wide-field microscope in 35 mm glass bottom μ-Dishes (Ibidi, #81158) (Figures 4A–4C, 6C, 6D, S3A, and S6A).
Light microscopy
For quantitative screening experiments (Figures 1F, 1G, 2B, 3B–3D, 4D–4F, 5A, 5B, S1E–S1G, S2, S5, and S6B–S6D), image acquisition was performed using an inverted epifluorescence Nikon Ti-E widefield microscope with a 20x (0.5 Numerical Aperture (NA), 2.1 Working Distance (WD)) N-Plan air objective, an automatic programmable xy stage, and the Nikon perfect focus system. Cells were imaged every 1-2 min for 2-3 h inside a 37°C heating chamber. Shigella (WT) infections were monitored as control in each experiment. To detect host protein recruitment to Shigella-induced endocytic compartments (BCV, IAMs), time-lapse movies of high resolution were recorded at 37°C on a DeltaVision Elite (GE Healthcare) widefield microscope using a 60 × /1.42 NA oil objective and a step size of 0.25-0.35 μm (Figures 1A–1C, 2A, 2C–2H, 3E, 4A–4C, 5C–5G, 6, 7B, S1C, S2A, S4, S6A, S6E, and S7; Videos S1 and S2). Imaging was performed for 2-3 h after adding the bacteria and the frequency of image acquisition was adjusted to the dynamics of the investigated event. Images were subsequently deconvolved using an integrated deconvolution analysis software (DeltaVision Elite). Bleach correction of illustrated images was performed using Fiji (http://fiji.sc). Fixed samples were imaged using a Perkin Elmer UltraView spinning disc confocal microscope, with a 60X × /1.3 N.A. oil objective and a Z step size of 0.3 μm (Figures S1A and S1B).
FRAP measurement and data analysis
Live-cell FRAP experiments (Figures 1D and 1E; Videos S3, S4, and S5) were performed at an inverted Perkin Elmer UltraView VOX confocal spinning disk microscope equipped with a FRAP module and Volocity software. Images were acquired using a 60X × /1.3 N.A. oil objective with a single Z plane. Cells expressing actin-GFP in 35 mm glass bottom culture dishes were imaged at 37°C in EM buffer. Cells were infected with Shigella (WT) DsRed and bacterial presence was controlled before bleaching of each single cocoon structure. We monitored the fluorescence of GFP-actin using low intensity laser excitation (488 nm) (pre-bleach scans). A circular region was photobleached with the same laser excitation at high intensity (decrease of the fluorescence into the ROI by 60%–80%), and fluorescence recovery monitored over time (post-bleach scans). 10 pre-bleach images were recorded at 1 s intervals, followed by a single bleaching pulse. One cocoon per cell was bleached with only half-bleached ones considered for analysis. The duration of post-bleach imaging was adjusted to the duration of fluorescence recovery. Actin turnover in the lamellipodium occurs through de novo actin polymerization at the lamellipodial tip with retrograde flow away from the leading edge and in stress fibers through the incorporation of new actin monomers or filaments. Stress fibers were bleached away from focal adhesions. Thus, actin turnover inside the filaments is expected to result from incorporation of unbleached actin from the cytoplasmic pool. In the lamellipodium, a ROI for analysis was selected at the lamellipodium tip inside the photobleached region following the retrograde flow of de novo polymerization below the plasma membrane. Raw data were fitted with simFRAP plugin in ImageJ. For each acquisition reference cell, photobleached cell and bleached area are selected to extract the mobile fraction Fm. Data were plotted and analyzed in Prism version 6 (GraphPad software).
Antibodies and western blotting
Primary antibodies used were polyclonal rabbit anti-Septin 7 (IBL, #18991), and monoclonal mouse anti-β-actin (Sigma-Aldrich, #A5316), anti-CDC42 (BD Transduction Laboratories, #610929), anti-RAC1 (Abcam, Cat# ab33186) as well as anti-ArpC3 p21-Arc (Clone26, #612234, BD Transduction Laboratories). Secondary antibodies were anti-mouse (Bio-Rad, #170-6516) and anti-rabbit (Bio-Rad, #170-6515) horseradish peroxidase-conjugated. To quantify knockdown efficiency by immunoblotting, siRNA treated HeLa cells were lysed with RIPA buffer containing protease inhibitor (Roche, #11836170001) for 30 min at 4°C. Protein quantification of lysates was done with the Micro BCA™ Protein Assay Kit (Thermo Fisher Scientific, #23235) and 10 μg of total protein was loaded into NuPAGE 4%–12% Bis-Tris Protein Gels (Thermo Fisher Scientific, #NP0321BOX) or 12% SDS-PAGE gels. Proteins were transferred to a nitrocellulose membrane using the Trans-Blot® Turbo RTA Mini Nitrocellulose Transfer Kit (Bio-Rad, #1704270) and Trans-Blot© Turbo Transfer System (Bio-Rad, #1704150) or tank blotting system in Towbin Buffer (Tris 25 mM, Glycine 190 mM, 20% Methanol pH 8.3) during 1h at 300 mA at 4°C. Antibody detection was carried out with the SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Fischer Scientific, #34577) with actin as loading control.
Quantification and Statistical Analysis
Time lapse microscopy series were analyzed using Icy (http://icy.bioimageanalysis.org/), Volocity 6.3 (PerkinElmer), or Fiji (http://fiji.sc) to determine starting points of foci formation, actin cocoon assembly, actin cocoon lifetime, vacuolar rupture time, and localization of host proteins. At least three biologically independent experiments per condition were performed. Quantitative data are mean values with error bars indicating ± Standard Deviation (SD) if not mentioned differently. Statistical analysis was performed in GraphPad Prism version 6. If not indicated differently, statistical significance was determined using a two-tailed Student’s t test (Mann-Whitney) or a one-way ANOVA followed by Dunnett’s multiple comparison test. (ns) not significant, p < 0.05 was considered as significant: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Quantitative image analysis
Since almost all invading wild-type Shigella escaped into the host cytoplasm in the duration of the experiment, we focused in our analysis on Shigella that successfully invaded the cytosol (galectin-3 recruitment to BCV membrane remnant) as indicator for efficient infection. Time-lapse of Shigella WT infecting HeLa cells co-expressing actin-GFP and galectin-3-mOrange allowed correlation and precise timing of actin dynamics with vacuolar rupture. Actin cocoon assembly was defined as the de novo formation of an actin-enriched structure around the BCV membrane with clear borders that assembled de novo after cellular entry and before vacuolar rupture for a duration of > 2 min. The comparison of the overall percentage of bacteria that assemble an actin cocoon before vacuolar rupture did represent a solid readout to investigate e.g., the effect of host protein inhibitors or Shigella mutant strains. Cases in which cocoons could not be clearly distinguished from surrounding membrane ruffles or IAMs were excluded from analysis (0%–2% per experiment, within the error range). The presence or absence of actin cocoons was investigated. Changes in dynamics, shape, or relative fluorescence intensities were not taken into account. This study focused on primary infections and early invasion steps. Secondary infections usually lack the massive membrane ruffling of primary infections and were not taken into account.
The vacuolar rupture time was used as supporting readout and was defined as time span between the onset of initial membrane ruffling and the first appearance of galectin-3 recruitment to the BCV membrane remnant. This includes the successive (actin-dependent) invasion steps of cellular uptake, actin cocoon formation, cocoon maintenance as well as its disassembly, and cytosolic escape of Shigella (Figure S1D). Since other endocytic compartments than the BCV (e.g., E. coli InvA phagosomes, IAMs) were never galectin-3-positive, its recruitment was used as marker for Shigella entering the cytosol. All rupture events were considered including statistic outliers. Significance was likewise confirmed for the geometrical mean and datasets excluding statistical outliers. In screens with varying infection efficiencies (e.g., Shigella mutants), only the first invader per cell was taken into account.
To compare the actin cocoon with cellular or pathogen-induced actin structures (Figure 2A), we normalized the effect of extensive membrane ruffling at the bacterial entry site and of varying actin-GFP expression levels in different cells. We measured the fluorescence intensity of on average the 10 most intense cellular actin stress fibers (mainly ventral and dorsal stress fibers, not in close proximity to focal adhesions) of each infected HeLa cell. The average stress fiber intensity was used to normalize the fluorescence intensity of each individual actin structure measurement per cell. In addition, each single measurement was corrected for the cytosolic actin-GFP signal in its immediate vicinity. E. coli InvA is a model for canonical phagocytosis with actin assembly and disassembly cycles around phagosomes (actin flashing). The related enteropathogenic Salmonella (S. Typhimurium) injects some T3SS effector protein homologs to Shigella. Data obtained for E. coli InvA and Salmonella infections were analyzed equally. The maximum intensity of actin assembly was considered. The lifetime of actin coats around Shigella’s BCV or E. coli’s phagosome (Figure 2C) was measured as the time interval between initial actin assembly and its complete disassembly. For E. coli InvA phagosomes, one cycle of complete actin assembly and disassembly was considered.
To quantify and correlate the PI3P peak with either the actin cocoon at Shigella’s BCV or actin flashing at phagosomes, we monitored over time the recruitment of the fluorescently labeled PI3P probe 2xFYVE and actin to the corresponding endocytic compartments (Figures 2E–2H). Each single measurement was corrected for the cytosolic background signal in its immediate vicinity. Data were normalized by dividing the fluorescence intensities of each 2xFYVE and actin time point by the respective maximum intensity. Individual experiments were aligned to the time point of maximum relative fluorescence intensity of the short-lasting 2xFYVE peak for Shigella, and the short-lasting actin peak for E. coli InvA. The variation of the actin signal at later time points for Shigella (Figure 2F) illustrates differences in actin cocoon dynamics.
All siRNA experiments were performed in parallel with scramble siRNA controls and HeLa cell controls. Septin 7 is an essential septin filament component and its knockdown by RNA interference efficiently inhibits septin filament assembly (Sirianni et al., 2016). Knockdown of ArpC3 was shown previously to be involved in Shigella invasion (Mellouk et al., 2014). For CDC42 siRNA-mediated knockdown or overexpression of CDC42 mutants, in parallel to the total population we also analyzed the behavior of only the first invading bacterium per cell (Figures S5D and S5E). Both analyses gave consistent results. Successful knockdown of CDC42 and ARPC3 by siRNA was also visible by altered actin cytoskeleton rearrangements at entry sites.
The intensity of host protein recruitment (Figures 5C–5G, 6, and S7) was quantified using Fiji. To investigate levels of CDC42 (UniProt# P60953-1) recruitment during Shigella infection, the mean fluorescence intensity of a ROI comprising either only the plasma membrane ruffle that formed the endocytic compartment, or the BCV membrane was measured (Figure 5D). All values were normalized to the initial signal of plasma membrane-recruited CDC42 to measure changes of CDC42 localization at the BCV in successive infection steps. We also analyzed the IcsB-dependent localization of CDC42 and N-WASP around vacuolar Shigella (Figures 6F, 7D, and 7F). Both proteins were considered constantly localized if they, after initial recruitment during early infection steps, remained associated with the galectin-3-stained membrane remnant after vacuolar rupture. This indicated that both proteins localized at BCV membrane remnants after rupture, but we observed different recycling behaviors for example for N-WASP and galectin-3. Proteins were defined as depleted in case they were initially recruited to the intact BCV, but disappeared before vacuolar rupture. CDC42 and N-WASP were specified as not recruited, if no localization at the vacuolar membrane was observed after scission of newly formed phagosomes. This does not exclude e.g., initial CDC42 recruitment to the plasma membrane during membrane ruffling. We considered only bacteria that successfully entered the host cytosol as indicated by galectin-3 (Gal-3) recruitment.
To analyze how actin cocoon assembly affects the fate of individual bacteria after vacuolar rupture, we quantified time lapses from HeLa cells co-expressing actin and galectin-3. The cytosolic fluorescence background from labeled proteins, marker recruitment to the bacterium during successive infection steps, and the characteristic shape of Shigella (individual actin tail, spreading behavior, replication state and size) is sufficient to track single bacteria inside the cell. We followed the infections steps of individual bacteria for 2 hr and monitored the time point of rupture, start and stopped movement, the kind of motility, the kind of vacuolar membrane recycling, and the fate of the bacteria after 2 hr infection. As ‘protrusion’, we defined actin tail-driven membrane protrusion for cell-to-cell spread and as ‘motile’ the intracellular movement by actin tail. Bacteria were defined as ‘trapped’, if after initial spreading they stopped movement, actin tail formation and growth for replication (or few cases that were never motile after rupture). With regard to the fate of the vacuolar membrane remnant, we defined ‘quick recycling’ if the membrane was quickly recycled away from a not yet moving bacterium. ‘Sticky’ defined cases where the bacterium lost the membrane remnant mainly by moving away from it and leaving it behind. As membrane ‘cap’ were all cases classified where bacteria remain surrounded by a part of the BCV during the entire infection. In most cases, bacteria were surrounded by half of the vacuolar membrane, which allowed moving with the cap by actin tail through the cytosol, but in some cases the ruptured BCV surrounded the bacteria completely.
Acknowledgments
For tools and fruitful discussions, we are grateful to John Rohde, Petra Janning, Emmanuel Lemichez, Amel Mettouchi, Claude Parsot, Guy Tran Van Nhieu, Christian Merrifield, Sandrine Etienne-Manneville, Serge Mostowy, Allon Weiner, Yuen-Yan Chang, and Patricia Latour-Lambert. We thank Noelia López-Montero and Laurent Audry for technical support and members of the BCI department for critical discussions. This work was supported by grants from the European Research Council (ERC) (EndoSubvert) to J.E., Agence Nationale de la Recherche (ANR) to J.E. (StopBugEntry) and to C.Z. (ANR-16-CE16-0019-01, NEUROTUNN). J.E. is a member of the LabEx consortium IBEID and MilieuInterieur. S.K. is a recipient of a Pasteur-Roux fellowship.
Author Contributions
Conceptualization and Project Administration, S.K. and J.E.; Methodology and Visualization, S.K.; Investigation, S.K. and J.B.; Experimental Help during Revision, M.G. and C.V.; Formal Analysis, S.K. and J.B.; FRAP Analysis, S.L.; Resources, J.E. and C.Z.; Writing – Original Draft, S.K.; Writing – Review & Editing, S.K. and J.E.; Supervision, S.K., L.B., and J.E.; Funding Acquisition, S.K., C.Z., and J.E. All authors discussed the results and commented on the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: May 12, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107638.
Supplemental Information
References
- Adam T., Giry M., Boquet P., Sansonetti P. Rho-dependent membrane folding causes Shigella entry into epithelial cells. EMBO J. 1996;15:3315–3321. [PMC free article] [PubMed] [Google Scholar]
- Baxt L.A., Goldberg M.B. Host and bacterial proteins that repress recruitment of LC3 to Shigella early during infection. PLoS ONE. 2014;9:e94653. doi: 10.1371/journal.pone.0094653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierne H., Gouin E., Roux P., Caroni P., Yin H.L., Cossart P. A role for cofilin and LIM kinase in Listeria-induced phagocytosis. J. Cell Biol. 2001;155:101–112. doi: 10.1083/jcb.200104037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai L., Makhov A.M., Schafer D.A., Bear J.E. Coronin 1B antagonizes cortactin and remodels Arp2/3-containing actin branches in lamellipodia. Cell. 2008;134:828–842. doi: 10.1016/j.cell.2008.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell-Valois F.X., Sachse M., Sansonetti P.J., Parsot C. Escape of actively secreting Shigella flexneri from ATG8/LC3-positive vacuoles formed during cell-to-cell spread is facilitated by IcsB and VirA. MBio. 2015;6:e02567-14. doi: 10.1128/mBio.02567-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K.T., Creed S.J., Bear J.E. Unraveling the enigma: progress towards understanding the coronin family of actin regulators. Trends Cell Biol. 2011;21:481–488. doi: 10.1016/j.tcb.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherfils J., Zeghouf M. Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol. Rev. 2013;93:269–309. doi: 10.1152/physrev.00003.2012. [DOI] [PubMed] [Google Scholar]
- Cossart P., Sansonetti P.J. Bacterial invasion: the paradigms of enteroinvasive pathogens. Science. 2004;304:242–248. doi: 10.1126/science.1090124. [DOI] [PubMed] [Google Scholar]
- Dupont N., Lacas-Gervais S., Bertout J., Paz I., Freche B., Van Nhieu G.T., van der Goot F.G., Sansonetti P.J., Lafont F. Shigella phagocytic vacuolar membrane remnants participate in the cellular response to pathogen invasion and are regulated by autophagy. Cell Host Microbe. 2009;6:137–149. doi: 10.1016/j.chom.2009.07.005. [DOI] [PubMed] [Google Scholar]
- Egile C., Loisel T.P., Laurent V., Li R., Pantaloni D., Sansonetti P.J., Carlier M.F. Activation of the CDC42 effector N-WASP by the Shigella flexneri IcsA protein promotes actin nucleation by Arp2/3 complex and bacterial actin-based motility. J. Cell Biol. 1999;146:1319–1332. doi: 10.1083/jcb.146.6.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehsani S., Santos J.C., Rodrigues C.D., Henriques R., Audry L., Zimmer C., Sansonetti P., Tran Van Nhieu G., Enninga J. Hierarchies of host factor dynamics at the entry site of Shigella flexneri during host cell invasion. Infect. Immun. 2012;80:2548–2557. doi: 10.1128/IAI.06391-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etienne-Manneville S., Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- Freeman S.A., Grinstein S. Phagocytosis: receptors, signal integration, and the cytoskeleton. Immunol. Rev. 2014;262:193–215. doi: 10.1111/imr.12212. [DOI] [PubMed] [Google Scholar]
- Gouin E., Gantelet H., Egile C., Lasa I., Ohayon H., Villiers V., Gounon P., Sansonetti P.J., Cossart P. A comparative study of the actin-based motilities of the pathogenic bacteria Listeria monocytogenes, Shigella flexneri and Rickettsia conorii. J. Cell Sci. 1999;112:1697–1708. doi: 10.1242/jcs.112.11.1697. [DOI] [PubMed] [Google Scholar]
- Ho H.Y., Rohatgi R., Lebensohn A.M., Le Ma, Li J., Gygi S.P., Kirschner M.W. Toca-1 mediates Cdc42-dependent actin nucleation by activating the N-WASP-WIP complex. Cell. 2004;118:203–216. doi: 10.1016/j.cell.2004.06.027. [DOI] [PubMed] [Google Scholar]
- Hoppe A.D., Swanson J.A. Cdc42, Rac1, and Rac2 display distinct patterns of activation during phagocytosis. Mol. Biol. Cell. 2004;15:3509–3519. doi: 10.1091/mbc.E03-11-0847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z., Sutton S.E., Wallenfang A.J., Orchard R.C., Wu X., Feng Y., Chai J., Alto N.M. Structural insights into host GTPase isoform selection by a family of bacterial GEF mimics. Nat. Struct. Mol. Biol. 2009;16:853–860. doi: 10.1038/nsmb.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isberg R.R., Voorhis D.L., Falkow S. Identification of invasin: a protein that allows enteric bacteria to penetrate cultured mammalian cells. Cell. 1987;50:769–778. doi: 10.1016/0092-8674(87)90335-7. [DOI] [PubMed] [Google Scholar]
- Kinoshita M., Field C.M., Coughlin M.L., Straight A.F., Mitchison T.J. Self- and actin-templated assembly of mammalian septins. Dev. Cell. 2002;3:791–802. doi: 10.1016/s1534-5807(02)00366-0. [DOI] [PubMed] [Google Scholar]
- Kirkbride K.C., Sung B.H., Sinha S., Weaver A.M. Cortactin: a multifunctional regulator of cellular invasiveness. Cell Adhes. Migr. 2011;5:187–198. doi: 10.4161/cam.5.2.14773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraynov V.S., Chamberlain C., Bokoch G.M., Schwartz M.A., Slabaugh S., Hahn K.M. Localized Rac activation dynamics visualized in living cells. Science. 2000;290:333–337. doi: 10.1126/science.290.5490.333. [DOI] [PubMed] [Google Scholar]
- Kühn S., Enninga J. The actin comet guides the way: how Listeria actin subversion has impacted cell biology, infection biology and structural biology. Cell. Microbiol. 2020;22:e13190. doi: 10.1111/cmi.13190. [DOI] [PubMed] [Google Scholar]
- Kühn S., Mannherz H.G. Actin: structure, function, dynamics, and interactions with bacterial toxins. Curr. Top. Microbiol. Immunol. 2017;399:1–34. doi: 10.1007/82_2016_45. [DOI] [PubMed] [Google Scholar]
- Kühn S., Lopez-Montero N., Chang Y.Y., Sartori-Rupp A., Enninga J. Imaging macropinosomes during Shigella infections. Methods. 2017;127:12–22. doi: 10.1016/j.ymeth.2017.05.007. [DOI] [PubMed] [Google Scholar]
- Kumar Y., Valdivia R.H. Actin and intermediate filaments stabilize the Chlamydia trachomatis vacuole by forming dynamic structural scaffolds. Cell Host Microbe. 2008;4:159–169. doi: 10.1016/j.chom.2008.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labigne-Roussel A.F., Lark D., Schoolnik G., Falkow S. Cloning and expression of an afimbrial adhesin (AFA-I) responsible for P blood group-independent, mannose-resistant hemagglutination from a pyelonephritic Escherichia coli strain. Infect. Immun. 1984;46:251–259. doi: 10.1128/iai.46.1.251-259.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Clainche C., Carlier M.F. Regulation of actin assembly associated with protrusion and adhesion in cell migration. Physiol. Rev. 2008;88:489–513. doi: 10.1152/physrev.00021.2007. [DOI] [PubMed] [Google Scholar]
- Liebl D., Griffiths G. Transient assembly of F-actin by phagosomes delays phagosome fusion with lysosomes in cargo-overloaded macrophages. J. Cell Sci. 2009;122:2935–2945. doi: 10.1242/jcs.048355. [DOI] [PubMed] [Google Scholar]
- Liu W., Zhou Y., Peng T., Zhou P., Ding X., Li Z., Zhong H., Xu Y., Chen S., Hang H.C., Shao F. Nε-fatty acylation of multiple membrane-associated proteins by Shigella IcsB effector to modulate host function. Nat. Microbiol. 2018;3:996–1009. doi: 10.1038/s41564-018-0215-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge R., Descoteaux A. Leishmania donovani promastigotes induce periphagosomal F-actin accumulation through retention of the GTPase Cdc42. Cell. Microbiol. 2005;7:1647–1658. doi: 10.1111/j.1462-5822.2005.00582.x. [DOI] [PubMed] [Google Scholar]
- Machesky L.M., Atkinson S.J., Ampe C., Vandekerckhove J., Pollard T.D. Purification of a cortical complex containing two unconventional actins from Acanthamoeba by affinity chromatography on profilin-agarose. J. Cell Biol. 1994;127:107–115. doi: 10.1083/jcb.127.1.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavrakis M., Azou-Gros Y., Tsai F.C., Alvarado J., Bertin A., Iv F., Kress A., Brasselet S., Koenderink G.H., Lecuit T. Septins promote F-actin ring formation by crosslinking actin filaments into curved bundles. Nat. Cell Biol. 2014;16:322–334. doi: 10.1038/ncb2921. [DOI] [PubMed] [Google Scholar]
- Mellouk N., Weiner A., Aulner N., Schmitt C., Elbaum M., Shorte S.L., Danckaert A., Enninga J. Shigella subverts the host recycling compartment to rupture its vacuole. Cell Host Microbe. 2014;16:517–530. doi: 10.1016/j.chom.2014.09.005. [DOI] [PubMed] [Google Scholar]
- Méresse S., Unsworth K.E., Habermann A., Griffiths G., Fang F., Martínez-Lorenzo M.J., Waterman S.R., Gorvel J.P., Holden D.W. Remodelling of the actin cytoskeleton is essential for replication of intravacuolar Salmonella. Cell. Microbiol. 2001;3:567–577. doi: 10.1046/j.1462-5822.2001.00141.x. [DOI] [PubMed] [Google Scholar]
- Merrifield C.J., Moss S.E., Ballestrem C., Imhof B.A., Giese G., Wunderlich I., Almers W. Endocytic vesicles move at the tips of actin tails in cultured mast cells. Nat. Cell Biol. 1999;1:72–74. doi: 10.1038/9048. [DOI] [PubMed] [Google Scholar]
- Mostowy S., Cossart P. Bacterial autophagy: restriction or promotion of bacterial replication? Trends Cell Biol. 2012;22:283–291. doi: 10.1016/j.tcb.2012.03.006. [DOI] [PubMed] [Google Scholar]
- Mostowy S., Bonazzi M., Hamon M.A., Tham T.N., Mallet A., Lelek M., Gouin E., Demangel C., Brosch R., Zimmer C. Entrapment of intracytosolic bacteria by septin cage-like structures. Cell Host Microbe. 2010;8:433–444. doi: 10.1016/j.chom.2010.10.009. [DOI] [PubMed] [Google Scholar]
- Mostowy S., Sancho-Shimizu V., Hamon M.A., Simeone R., Brosch R., Johansen T., Cossart P. p62 and NDP52 proteins target intracytosolic Shigella and Listeria to different autophagy pathways. J. Biol. Chem. 2011;286:26987–26995. doi: 10.1074/jbc.M111.223610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mounier J., Laurent V., Hall A., Fort P., Carlier M.F., Sansonetti P.J., Egile C. Rho family GTPases control entry of Shigella flexneri into epithelial cells but not intracellular motility. J. Cell Sci. 1999;112:2069–2080. doi: 10.1242/jcs.112.13.2069. [DOI] [PubMed] [Google Scholar]
- Mounier J., Popoff M.R., Enninga J., Frame M.C., Sansonetti P.J., Van Nhieu G.T. The IpaC carboxyterminal effector domain mediates Src-dependent actin polymerization during Shigella invasion of epithelial cells. PLoS Pathog. 2009;5:e1000271. doi: 10.1371/journal.ppat.1000271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins R.D., Heuser J.A., Pollard T.D. The interaction of Arp2/3 complex with actin: nucleation, high affinity pointed end capping, and formation of branching networks of filaments. Proc. Natl. Acad. Sci. USA. 1998;95:6181–6186. doi: 10.1073/pnas.95.11.6181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niebuhr K., Giuriato S., Pedron T., Philpott D.J., Gaits F., Sable J., Sheetz M.P., Parsot C., Sansonetti P.J., Payrastre B. Conversion of PtdIns(4,5)P(2) into PtdIns(5)P by the S.flexneri effector IpgD reorganizes host cell morphology. EMBO J. 2002;21:5069–5078. doi: 10.1093/emboj/cdf522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedergang F., Grinstein S. How to build a phagosome: new concepts for an old process. Curr. Opin. Cell Biol. 2018;50:57–63. doi: 10.1016/j.ceb.2018.01.009. [DOI] [PubMed] [Google Scholar]
- Ogawa M., Yoshimori T., Suzuki T., Sagara H., Mizushima N., Sasakawa C. Escape of intracellular Shigella from autophagy. Science. 2005;307:727–731. doi: 10.1126/science.1106036. [DOI] [PubMed] [Google Scholar]
- Ohya K., Handa Y., Ogawa M., Suzuki M., Sasakawa C. IpgB1 is a novel Shigella effector protein involved in bacterial invasion of host cells. Its activity to promote membrane ruffling via Rac1 and Cdc42 activation. J. Biol. Chem. 2005;280:24022–24034. doi: 10.1074/jbc.M502509200. [DOI] [PubMed] [Google Scholar]
- Onodera N.T., Ryu J., Durbic T., Nislow C., Archibald J.M., Rohde J.R. Genome sequence of Shigella flexneri serotype 5a strain M90T Sm. J. Bacteriol. 2012;194:3022. doi: 10.1128/JB.00393-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz I., Sachse M., Dupont N., Mounier J., Cederfur C., Enninga J., Leffler H., Poirier F., Prevost M.C., Lafont F., Sansonetti P. Galectin-3, a marker for vacuole lysis by invasive pathogens. Cell. Microbiol. 2010;12:530–544. doi: 10.1111/j.1462-5822.2009.01415.x. [DOI] [PubMed] [Google Scholar]
- Pendaries C., Tronchère H., Arbibe L., Mounier J., Gozani O., Cantley L., Fry M.J., Gaits-Iacovoni F., Sansonetti P.J., Payrastre B. PtdIns5P activates the host cell PI3-kinase/Akt pathway during Shigella flexneri infection. EMBO J. 2006;25:1024–1034. doi: 10.1038/sj.emboj.7601001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poh J., Odendall C., Spanos A., Boyle C., Liu M., Freemont P., Holden D.W. SteC is a Salmonella kinase required for SPI-2-dependent F-actin remodelling. Cell. Microbiol. 2008;10:20–30. doi: 10.1111/j.1462-5822.2007.01010.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard T.D. Actin and actin-binding proteins. Cold Spring Harb. Perspect. Biol. 2016;8:a018226. doi: 10.1101/cshperspect.a018226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puthenveedu M.A., Lauffer B., Temkin P., Vistein R., Carlton P., Thorn K., Taunton J., Weiner O.D., Parton R.G., von Zastrow M. Sequence-dependent sorting of recycling proteins by actin-stabilized endosomal microdomains. Cell. 2010;143:761–773. doi: 10.1016/j.cell.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray K., Bobard A., Danckaert A., Paz-Haftel I., Clair C., Ehsani S., Tang C., Sansonetti P., Tran G.V., Enninga J. Tracking the dynamic interplay between bacterial and host factors during pathogen-induced vacuole rupture in real time. Cell. Microbiol. 2010;12:545–556. doi: 10.1111/j.1462-5822.2010.01428.x. [DOI] [PubMed] [Google Scholar]
- Revenu C., Courtois M., Michelot A., Sykes C., Louvard D., Robine S. Villin severing activity enhances actin-based motility in vivo. Mol. Biol. Cell. 2007;18:827–838. doi: 10.1091/mbc.E06-05-0423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohatgi R., Ma L., Miki H., Lopez M., Kirchhausen T., Takenawa T., Kirschner M.W. The interaction between N-WASP and the Arp2/3 complex links Cdc42-dependent signals to actin assembly. Cell. 1999;97:221–231. doi: 10.1016/s0092-8674(00)80732-1. [DOI] [PubMed] [Google Scholar]
- Romero S., Quatela A., Bornschlögl T., Guadagnini S., Bassereau P., Tran Van Nhieu G. Filopodium retraction is controlled by adhesion to its tip. J. Cell Sci. 2012;125:4999–5004. doi: 10.1242/jcs.104778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottner K., Faix J., Bogdan S., Linder S., Kerkhoff E. Actin assembly mechanisms at a glance. J. Cell Sci. 2017;130:3427–3435. doi: 10.1242/jcs.206433. [DOI] [PubMed] [Google Scholar]
- Santos J.C., Enninga J. At the crossroads: communication of bacteria-containing vacuoles with host organelles. Cell. Microbiol. 2016;18:330–339. doi: 10.1111/cmi.12567. [DOI] [PubMed] [Google Scholar]
- Schnupf P., Sansonetti P.J. Shigella pathogenesis: new insights through advanced methodologies. Microbiol. Spectr. 2019;7(2) doi: 10.1128/microbiolspec.bai-0023-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder G.N., Hilbi H. Molecular pathogenesis of Shigella spp.: controlling host cell signaling, invasion, and death by type III secretion. Clin. Microbiol. Rev. 2008;21:134–156. doi: 10.1128/CMR.00032-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaman M.N., Gautreau A., Billadeau D.D. Retromer-mediated endosomal protein sorting: all WASHed up! Trends Cell Biol. 2013;23:522–528. doi: 10.1016/j.tcb.2013.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidik S., Kottwitz H., Benjamin J., Ryu J., Jarrar A., Garduno R., Rohde J.R. A Shigella flexneri virulence plasmid encoded factor controls production of outer membrane vesicles. G3 (Bethesda) 2014;4:2493–2503. doi: 10.1534/g3.114.014381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirianni A., Krokowski S., Lobato-Márquez D., Buranyi S., Pfanzelter J., Galea D., Willis A., Culley S., Henriques R., Larrouy-Maumus G. Mitochondria mediate septin cage assembly to promote autophagy of Shigella. EMBO Rep. 2016;17:1029–1043. doi: 10.15252/embr.201541832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukumaran B., Mastronunzio J.E., Narasimhan S., Fankhauser S., Uchil P.D., Levy R., Graham M., Colpitts T.M., Lesser C.F., Fikrig E. Anaplasma phagocytophilum AptA modulates Erk1/2 signalling. Cell. Microbiol. 2011;13:47–61. doi: 10.1111/j.1462-5822.2010.01516.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T., Miki H., Takenawa T., Sasakawa C. Neural Wiskott-Aldrich syndrome protein is implicated in the actin-based motility of Shigella flexneri. EMBO J. 1998;17:2767–2776. doi: 10.1093/emboj/17.10.2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taunton J., Rowning B.A., Coughlin M.L., Wu M., Moon R.T., Mitchison T.J., Larabell C.A. Actin-dependent propulsion of endosomes and lysosomes by recruitment of N-WASP. J. Cell Biol. 2000;148:519–530. doi: 10.1083/jcb.148.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor M.J., Perrais D., Merrifield C.J. A high precision survey of the molecular dynamics of mammalian clathrin-mediated endocytosis. PLoS Biol. 2011;9:e1000604. doi: 10.1371/journal.pbio.1000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran Van Nhieu G., Caron E., Hall A., Sansonetti P.J. IpaC induces actin polymerization and filopodia formation during Shigella entry into epithelial cells. EMBO J. 1999;18:3249–3262. doi: 10.1093/emboj/18.12.3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unsworth K.E., Way M., McNiven M., Machesky L., Holden D.W. Analysis of the mechanisms of Salmonella-induced actin assembly during invasion of host cells and intracellular replication. Cell. Microbiol. 2004;6:1041–1055. doi: 10.1111/j.1462-5822.2004.00417.x. [DOI] [PubMed] [Google Scholar]
- Valencia-Gallardo C.M., Carayol N., Tran Van Nhieu G. Cytoskeletal mechanics during Shigella invasion and dissemination in epithelial cells. Cell. Microbiol. 2015;17:174–182. doi: 10.1111/cmi.12400. [DOI] [PubMed] [Google Scholar]
- Vazquez-Torres A., Xu Y., Jones-Carson J., Holden D.W., Lucia S.M., Dinauer M.C., Mastroeni P., Fang F.C. Salmonella pathogenicity island 2-dependent evasion of the phagocyte NADPH oxidase. Science. 2000;287:1655–1658. doi: 10.1126/science.287.5458.1655. [DOI] [PubMed] [Google Scholar]
- Vetter I.R., Wittinghofer A. The guanine nucleotide-binding switch in three dimensions. Science. 2001;294:1299–1304. doi: 10.1126/science.1062023. [DOI] [PubMed] [Google Scholar]
- Viaud J., Lagarrigue F., Ramel D., Allart S., Chicanne G., Ceccato L., Courilleau D., Xuereb J.M., Pertz O., Payrastre B., Gaits-Iacovoni F. Phosphatidylinositol 5-phosphate regulates invasion through binding and activation of Tiam1. Nat. Commun. 2014;5:4080. doi: 10.1038/ncomms5080. [DOI] [PubMed] [Google Scholar]
- Wandel M.P., Pathe C., Werner E.I., Ellison C.J., Boyle K.B., Von Der Malsburg A., Rohde J., Randow F. GBPs inhibit motility of Shigella flexneri but are targeted for degradation by the bacterial ubiquitin ligase IpaH9.8. Cell Host Microbe. 2017;22:507–518.e5. doi: 10.1016/j.chom.2017.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigele B.A., Orchard R.C., Jimenez A., Cox G.W., Alto N.M. A systematic exploration of the interactions between bacterial effector proteins and host cell membranes. Nat. Commun. 2017;8:532. doi: 10.1038/s41467-017-00700-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner A., Mellouk N., Lopez-Montero N., Chang Y.Y., Souque C., Schmitt C., Enninga J. Macropinosomes are key players in early Shigella invasion and vacuolar escape in epithelial cells. PLoS Pathog. 2016;12:e1005602. doi: 10.1371/journal.ppat.1005602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yam P.T., Theriot J.A. Repeated cycles of rapid actin assembly and disassembly on epithelial cell phagosomes. Mol. Biol. Cell. 2004;15:5647–5658. doi: 10.1091/mbc.E04-06-0509. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Time lapse of actin rearrangements (green) at the entry site during Shigella WT DsRed (red) infection. Shigella induces massive membrane ruffling around the adhered bacterium and cocoon assembly after uptake. The cocoon is much denser than cellular actin structures and finally disassembled. Transient actin flashing around IAMs is also visible (scale bar: 3 μm).
Time-lapse of actin rearrangements during cellular entry of Shigella WT (scale bar: 3 μm).
Fluorescence recovery after photobleaching revealed constant actin turnover of the cocoon during its maintained phase. Fluorescence intensity was monitored 10 s before and 60 s after bleaching (t = 0 s) (scale: 5 μm).
The lamellipodium was photobleached and the recovery at its tip was measured showing de novo actin polymerization. The actin-GFP fluorescence signal was followed 10 s before and 60 s after bleaching (t = 0 s) (scale: 5 μm).
Stress fibers have different actin turnover dynamics compared to the actin cocoon and recover much slower from photobleaching. A stress fiber with a very small mobile fraction is depicted. The actin-GFP fluorescence signal was followed 10 s before and 254 s after bleaching (t = 0 s) (scale: 5 μm).