

Summary
The inflammasomes control the bioactivity of pro-inflammatory cytokines of the interleukin (IL)-1 family. The inflammasome assembled by NLRP3 has been predominantly studied in homogeneous cell populations in vitro, neglecting the influence of cellular interactions that occur in vivo. Here, we show that platelets boost the inflammasome capacity of human macrophages and neutrophils and are critical for IL-1 production by monocytes. Platelets license NLRP3 transcription, thereby enhancing ASC oligomerization, caspase-1 activity, and IL-1β secretion. Platelets influence IL-1β production in vivo, and blood platelet counts correlate with plasmatic IL-1β levels in malaria. Furthermore, we reveal an enriched platelet gene signature among the highest-expressed transcripts in IL-1β-driven autoinflammatory diseases. The platelet effect is independent of cell-to-cell contact, platelet-derived lipid mediators, purines, nucleic acids, and a host of platelet cytokines, and it involves the triggering of calcium-sensing receptors on macrophages. Hence, platelets provide an additional layer of regulation of inflammasomes and IL-1-driven inflammation.
Keywords: inflammasomes, interleukin-1, platelets, auto-inflammatory diseases, malaria, NLRP3, ASC, Caspase-1, Cell Death, Pyroptosis
Graphical Abstract
Highlights
-
•
Platelets license NLRP3 for inflammasome activattion in innate immune cells
-
•
Platelets are required for optimal monocyte inflammasome activation
-
•
Platelets shape IL-1β in vivo, and platelet counts correlate with IL-1β in plasma
-
•
A constitutive, heat-sensitive soluble platelet-factor boost IL-1β in macrophages
Rolfes et al. show that platelets license NLRP3 expression in immune cells, boost inflammasome responses in vitro, and are critical for full IL-1 responses in vivo. A platelet-gene signature is present in NLRP3-related autoinflammatory diseases. Soluble platelet factors enhance NLRP3 transcription and boost IL-1 in macrophages.
Introduction
An unbalanced production of interleukin-1 (IL-1) cytokines drives the immunopathology of several auto-inflammatory diseases (Sims and Smith, 2010). As nearly all cells express the IL-1 receptor (IL-1R), IL-1 cytokines have the ability to influence innate and adaptive immunity and exert broad effects in the body. IL-1β is unique in the medical literature: while many human inflammatory diseases are caused by a host of cooperative inflammatory factors, mutations in genes controlling the expression of IL-1β cause a spectrum of life-threatening auto-inflammatory syndromes (Broderick et al., 2015). Monotherapies blocking IL-1β activity in patients with auto-inflammatory syndromes result in a rapid and sustained reversal of symptoms and severity (Dinarello et al., 2012) and are currently the first line of intervention against these conditions. A growing bulk of evidence has now characterized other common inflammatory and metabolic conditions as responsive to IL-1β neutralization (Dinarello, 2011, Dinarello and van der Meer, 2013). This was further validated by the results of the CANTOS Study, which showed that canakinumab, a humanized anti-IL-1β monoclonal antibody, significantly reduced the risk for recurrent cardiovascular events (Ridker et al., 2017a) and indicated that IL-1β is associated with increased incidence of fatal lung cancer (Ridker et al., 2017b).
The expression of IL-1 cytokines is tightly regulated. For instance, the production of some members of this family (e.g., IL-1β and IL-18) is restricted to immune cells. Furthermore, upon induction, these proteins are synthesized as inactive precursors in the cytosol, and a series of intracellular events is required for their maturation and release into the extracellular space. These events include the assembly of inflammasomes—intracellular protein complexes formed by a sensor—such as NLRP3, the adaptor protein apoptosis-associated speck-like protein containing a CARD (ASC), and the cysteine protease caspase-1 (Latz et al., 2013). Upon activation, inflammasome sensors recruit ASC, which oligomerizes to form a micron-sized structure termed ASC speck, which recruits and activates caspase-1. Active caspase-1 processes pro-IL-1β and pro-IL-18 into their bioactive forms and drives an inflammatory cell death, termed pyroptosis, through gasdermin-D-induced membrane pore formation and leakage of cytosolic content (Kayagaki et al., 2015, Liu et al., 2016).
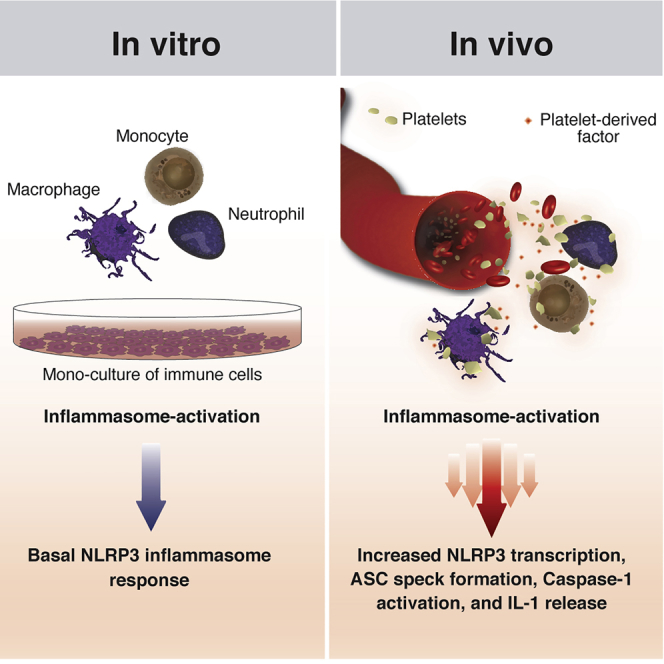
The majority of the studies on inflammasomes were performed in vitro, predominantly in monocultures of macrophages. Although this approach allowed the discovery of molecular mechanisms controlling inflammasomes, it underestimated the influence of other cell populations on the regulation of IL-1 cytokines in vivo. For instance, it has been recently discovered that T cells drive inflammasome-independent IL-1β production by dendritic cells (DCs) (Jain et al., 2020).
Around one trillion platelets (150–450 × 109/L) circulate in the blood of a healthy individual, outnumbering all other leukocytes in the vasculature by several folds. Platelets have been increasingly recognized for their roles in immunity (Carestia et al., 2019, Dann et al., 2018, Kral et al., 2016, Passacquale et al., 2011) and were reported to produce IL-1 cytokines (Allam et al., 2017, Denis et al., 2005, Hawrylowicz et al., 1989, Thornton et al., 2010). More recently, they were reported to assemble inflammasomes (Cornelius et al., 2019, Hottz et al., 2013). Platelets could therefore be relevant cellular sources of IL-1 or extracellular ASC specks in vivo or could alter the inflammatory responses of other immune cells.
Here, we report that platelets are essential for the full inflammasome activation of innate immune cells. Co-culture with platelets not only boosted the NLRP3 activation and production of IL-1α, IL-1β, and IL-18 from human macrophages and neutrophils, but it was also crucial for the optimal production of IL-1 cytokines by human monocytes. Platelets specifically influenced the NLRP3 inflammasome activation of macrophages in trans by enhancing the transcription of NLRP3 and IL-1β. Using a series of complementary techniques that included assays with human platelets and megakaryocytes (MKs), as well as platelets and cells from knockout and transgenic mice, we demonstrated that platelets and MKs do not express the components of the canonical inflammasome (NLRP3, ASC, caspase-1, and IL-1β). Indeed, the effect of platelets on human monocyte-derived macrophages (hMDMs) was independent of IL-1 signaling and did not require direct cell contact, platelet-derived nucleic acids, lipid mediators, or purines, but it could be neutralized by heat inactivation. We further show that platelet depletion attenuated systemic production of IL-1β in vivo. Furthermore, platelet counts and plasma levels of IL-1β were positively correlated in naturally infected malaria patients. Moreover, we observed an enriched platelet signature among the highest-expressed genes in a cohort of pediatric patients with gain-of-function mutations in NLRP3 that cause Muckle-Wells syndrome (MWS) and neonatal-onset multisystem inflammatory disease (NOMID).
In summary, platelets fuel NLRP3 activity of immune cells and shape IL-1 inflammation. Thus, platelet-modifying therapies could have widespread implications for autoinflammatory and autoimmune diseases.
Results
Platelets Boost IL-1 Production in Inflammasome-Activated Immune Cells
The NLRP3 inflammasome is primarily assembled in myeloid cells. Although activated platelets are known to form aggregates with and modulate the functions of immune cells (Allen et al., 2019, Carestia et al., 2019, Dann et al., 2018, Kral et al., 2016, Passacquale et al., 2011), their effects on the inflammasome activation of leukocytes remains unexplored. To address this question, we investigated the influence of platelets on the production of IL-1 cytokines elicited by the bona fide NLRP3 inflammasome activators Nigericin and ATP in mouse and human immune cells. Mouse bone marrow-derived macrophages (BMDMs) (Figure 1A), hMDMs (Figure 1B), human neutrophils (Figure 1C), and human CD14+ monocytes (Figure 1D) were cultured or not with platelets. Co-cultures were left untreated or primed with lipopolysaccharide (LPS), followed by NLRP3 activation. The levels of IL-1β and tumor necrosis factor alpha (TNF-α) were measured in cell-free supernatants. A multiplex cytokine assay was used to additionally measure the production of IL-1α and IL-18, two other members of the IL-1 family, along with several other cytokines and growth factors in hMDMs and human neutrophils (Figures S1A and S1B). Co-culture with platelets boosted the production of IL-1β from mouse and human inflammasome-activated macrophages and human neutrophils (Figures 1A–1C) without significantly altering the release of lactate dehydrogenase (LDH) from activated cells (Figure S2A). Fitting a sigmoidal curve between the IL-1β levels produced in inflammasome-activated macrophages and the platelet-macrophage ratio, we found a strong (R = 0.8886, p = 0.0163) concentration-dependent effect of platelets in these cells (Figure S1C). The addition of platelets also boosted the release of IL-18 and IL-1α from activated hMDMs (Figure S1A). Extending these results, the addition of platelets to neutrophils also enhanced their production of IL-8 (Figure S1B). Platelets alone produced CCL5 (RANTES) and low levels of IL-18 that were close to the detection limit of the assays. Platelets lacked expression of most of the other investigated cytokines (Figures 1, S1A, and S1B), indicating that they do not directly contribute to the cytokines measured in the co-cultures. Notably, the co-culture of platelets with mouse macrophages, human monocytes, and neutrophils, but not hMDMs, resulted in diminished TNF-α production in response to LPS stimulation. These results suggest that despite NFκB being a common transcription factor regulating the production of TNF-α and IL-1 cytokines, platelets differentially regulate the production of these cytokines.
Figure 1.

Platelets Boost Inflammasome Activation of Innate Immune Cells
(A–D) IL-1β and TNF-α levels released by unstimulated (Unstim), LPS-primed, and Nigericin, or ATP-activated (A) WT BMDMs, (B) human macrophages (hMDMs), (C) neutrophils, or (D) monocytes. Cells were cultivated alone (−) or with platelets (+Plts) in the indicated ratios. Data is presented as floating bars (with mean and minimum to maximum values) and pooled from at least three independent experiments with cells and platelets from different donors. Each symbol represents the average from technical triplicates per blood donor or mouse.
See also STAR Methods and Figure S1.
Next, we asked whether platelets also influence the activity of other inflammasomes. For this, we activated AIM2 or NLRC4, respectively, by intracellular delivery of double-stranded DNA (dsDNA) (Poly[dA:dT]) (Figure S1D) or the rod protein of the T3SS apparatus from Salmonella typhimurium (PrgI) (STAR Methods; Figure S1E) in co-cultures of human platelets and macrophages. Stimulation of the AIM2 or NLRC4 inflammasomes in hMDMs resulted in IL-1β secretion, which was not significantly altered by the addition of platelets (Figures S1D and S1E), indicating that the platelet effect was NLRP3-specific. Co-culture with platelets also boosted IL-1β production from hMDMs primed with Toll-like receptor (TLR)2 or TLR7/8 agonists (Pam3Csk4 and R848, respectively) (Figure S1F), indicating that the platelet effect is not exclusively mediated through TLR4. However, blockade of TLR4 signaling on hMDMs with resatorvid (TAK-242) (Matsunaga et al., 2011) partially prevented the effect of platelets (Figure S1G), indicating that the platelet effect is in part orchestrated by TLR4. Together, these data show that platelets boost the production of IL-1 cytokines in NLRP3-activated innate immune cells.
Platelets Are Critical for the Inflammasome Activation of Human Monocytes
Notably, co-culture with platelets did not influence the production of IL-1β by inflammasome-activated monocytes (Figure 1D), likely due to the steady-state presence of contaminating platelets on the preparations of freshly isolated monocytes. To address that, we isolated CD14+ monocytes from peripheral blood using magnetic separation kits, added or not with a platelet removal cocktail (STAR Methods). Cell purity was assessed by fluorescence-activated cell sorting (FACS) (Figure 2A). Platelet depletion efficiently reduced the numbers of free platelets (CD41+CD14–) or platelets associated with monocytes (CD41+CD14+), while enriching the frequency of platelet-free monocytes (CD14+CD41–) (Figures 2A and 2B). Importantly, platelet depletion was not detrimental to monocytes, assessed by LDH release (Figure S2B). However, platelet removal impaired the cytokine response of NLRP3-activated monocytes. Notably, monocyte responses could be restored by the re-addition of autologous platelets (Figures 2C and S2C). These data indicate that platelets are crucial for monocytes to trigger an optimal inflammatory response.
Figure 2.

Platelets Are Critical for the Production of IL-1 Cytokines by Human Primary Monocytes
(A) Representative FACS dot plots of CD41 and CD14 expression in human PBMCs, standard (Std-Mo), or platelet-depleted (PD-Mo) CD14+ monocytes (see STAR Methods).
(B) Quantification of platelets (CD41+CD14−), platelet-monocyte aggregates (CD41+CD14+), and platelet-free monocytes (CD41−CD14+) in PBMCs and isolated monocytes.
(C) IL-1β and TNF-α levels in cell-free supernatants of Std-Mo versus PD-Mo, or in platelet-depleted monocytes replenished with autologous platelets (PD-Mo + Plts, 50:1 Ptl:Cell), and stimulated as indicated.
(D and E) IL-1β levels released by non-canonically activated Std-Mo, PD-Mo, or PD-Mo + Plts, stimulated with LPS (1 μg mL−1) for 16 h (D) or by inflammasome-activated THP-1 s ± platelets (E). Data is presented as floating bars (with mean and minimum to maximum values) and pooled from independent experiments. Each symbol represents the average from technical triplicates per blood donor. Data in (D) shows bar graph with Mean + SD, from a representative of two independent experiments.
See also Figure S2.
Human monocytes can also engage an alternative inflammasome activation, in which LPS alone is sufficient to trigger IL-1β maturation and secretion (Gaidt et al., 2016). To test the effect of platelet removal in alternatively activated monocytes, we stimulated standard or platelet-depleted CD14+ monocytes with LPS (1 μg mL−1) for 16 h. Similar to the effect on the canonical inflammasome (Figure 2C), platelet depletion impaired the IL-1β response from alternatively activated cells (Figure 2D), and the re-addition of platelets rescued their IL-1β production (Figure 2D).
Next, we asked whether co-culture with platelets could also enhance NLRP3 activation in the monocytic cell line THP-1, which has been used extensively to characterize the biology of the inflammasomes. As expected (Gaidt et al., 2016, Gaidt et al., 2017), NLRP3-activated THP-1s were able to produce IL-1β on their own. Nevertheless, co-culture with platelets boosted the production of IL-1β by THP-1s to levels comparable to those produced by primary monocytes (Figure 2E). No IL-1β was detected on platelets cultivated alone. Together, these findings reveal that platelets are critical for optimal inflammasome activation by human monocytes.
Platelets Influence IL-1 Responses In Vivo
As monocytes and macrophages are relevant sources of IL-1, we reasoned that platelets might influence IL-1 responses in vivo. Therefore, we investigated the effects of platelet depletion using a mouse model of systemic IL-1-dependent inflammation triggered by imidazoquinolinone compounds (Kanneganti et al., 2006). We induced thrombocytopenia in mice by intravenous (i.v.) injection of a GPIba monoclonal antibody (αCD42b). Treatment with αCD42b has been widely used in literature (Barrett et al., 2019, Boilard et al., 2010, Sreeramkumar et al., 2014, Xiang et al., 2013) and results in a drop of the blood platelet counts (Figures 3A, S3A, and S3B), with no reported effects on the blood counts of other leukocytes (Barrett et al., 2019, Packham et al., 2014). As previously reported (Kanneganti et al., 2006), intraperitoneal (i.p.) injection of R848 in combination with LPS induced robust IL-1β systemically, measured in sera (Figure 3B), and locally, in peritoneal lavage (PEL) (Figure 3C). Notably, this response was attenuated by platelet depletion. Although levels of IL-1β were significantly lower in the sera of platelet-depleted mice (Figure 3B), we could not detect IL-1β in the PEL of all mice (Figure 3C). Importantly, platelet depletion did not affect the migration of neutrophils and monocytes to the peritoneum (Figures 3D and S3B). These data suggest that the decrease in IL-1β levels was not due to reduced peritoneal leukocyte counts.
Figure 3.

Platelets Influence IL-1 Levels In Vivo
(A) Representative FACS plots and quantification of platelets in the blood of mice treated with a platelet-depleting Ab (αCD42b) or IgG control.
(B and C) IL-1β levels in sera (B) or peritoneal lavage (C) from platelet-depleted mice after i.p. challenge with LPS + R848 (see STAR Methods).
(D) FACS quantification of neutrophils and monocytes in peritoneal lavage of mice treated as in (C).
(E) Correlation between plasma cytokines and blood cell counts or hemoglobin levels in naturally infected malaria patients (n = 78). Red lines, positive correlations; shaded areas, 95% confidence; squared symbols, outliers (see STAR Methods).
(F) Volcano plot of log2 fold change versus significance (p value) highlighting the platelet-signature genes within transcripts of healthy (n = 14) versus pediatric patients (n = 22) with NLRP3 mutations (NLRP3mut) causing MWS/NOMID (Balow et al., 2013).
See also Figure S3.
Platelets were reported to amplify IL-1 inflammation in rheumatoid arthritis (RA) (Boilard et al., 2010) and more recently in atherosclerosis (Barrett et al., 2019), two inflammatory diseases characterized by strong IL-1 inflammation (Di Giovine et al., 1987, Joosten et al., 2009, Mantovani et al., 2019, Ridker et al., 2017a), supporting their role in modulating IL-1 levels in vivo. In line with this hypothesis, a positive correlation between blood platelet counts and plasma IL-1β concentrations was recently reported in a cohort of 500 healthy, Caucasian volunteers (Netea et al., 2016, Tunjungputri et al., 2018). However, blood leukocyte counts were equally correlated with plasma levels of IL-1β in that study, hindering the precise contribution of platelets for the IL-1β concentrations (Tunjungputri et al., 2018). To investigate the relation between platelet counts and IL-1β concentrations in the context of disease, we studied a cohort of human subjects naturally infected with Plasmodium vivax, the predominant cause of malaria in the Brazilian Amazon (Howes et al., 2016). Infections with P. vivax commonly cause pro-inflammatory cytokine imbalance (Andrade et al., 2010), with thrombocytopenia and anemia being the two most commonly associated complications. We found that platelet counts positively correlated with plasma IL-1β concentrations (Spearman’s R = 0.504, p = 0.0028). Despite a positive trend, no correlations were found between platelet counts and IL-1α (R = 0.15, p = 0.37) or TNF-α (R = 0.014, p = 0.917) (Figure 3E). Unexpectedly, IL-18 levels were negatively correlated with platelet counts (R = −0.45, p < 0.0001), which suggests that different mechanisms regulate the production of this cytokine in vivo. Supporting this hypothesis, heme (Li et al., 2014) and oxidized hemoglobin were shown to activate the NLRP3 inflammasome (Nyakundi et al., 2019). Indeed, IL-18 (R = −0.32, p = 0.005) and TNF-α (R = −0.29, p = 0.003) correlated with hemoglobin levels (Figure 3E), suggesting that anemia may be a contributing factor for the regulation of these cytokines, but not for the associations between platelet counts and IL-1β. Importantly, none of the cytokines investigated correlated with leukocyte counts (Figure 3E) in the malaria cohort. As leukocytes are a relevant source in IL-1 cytokines in the blood, these data support that platelets are major contributors in the regulation of IL-1 levels in this context. IL-1β levels also had no correlation with parasitemia (data not shown), suggesting that IL-1β production may not be caused by Plasmodium-associated danger molecules (Dostert et al., 2009).
Next, we asked whether there is a relationship between platelets and inflammation in exclusively IL-1β-driven human autoinflammatory syndromes (Broderick et al., 2015). We generated a platelet gene signature comprising 45 transcripts known to be either platelet-specific or strongly associated with platelet activity from direct comparisons of publicly available gene expression analyses of purified human platelets (Eicher et al., 2016, Rowley et al., 2011). Remarkably, 42 out of 45 platelet-signature genes were upregulated in the whole blood of pediatric patients harboring NLRP3 mutations (NLPR3mut) associated with the IL-1-driven autoinflammatory disorders MWS and NOMID (Balow et al., 2013, Goldbach-Mansky et al., 2006), compared to healthy children (Figure 3F). Importantly, as reported in the original cohort (Goldbach-Mansky et al., 2006), the baseline clinical manifestations and blood cell counts of MWS/NOMID patients were within the pediatric reference ranges, supporting that differences in blood cell counts are likely not responsible for the changes we observed. Collectively, these findings support that platelets influence IL-1β in vivo.
Platelet Effect on Immune Cells Is Independent of Platelet-Derived IL-1 or Platelet Inflammasomes
Previous reports have indicated that platelets express IL-1 cytokines, including IL-1α (Thornton et al., 2010), IL-1β (Denis et al., 2005), and IL-18 (Allam et al., 2017). However, as shown in Figures 1, S1A, and S1B, IL-1α and IL-1β were not detected in monocultures of platelets in our experimental settings. To exclude the possibility that platelets directly contribute to the IL-1 cytokines measured in our assays, we performed co-culture experiments using platelets or BMDMs from mice with targeted genetic deficiency in IL-1 proteins or IL-1 receptors. First, we activated the NLRP3 inflammasome in wild-type (WT) BMDMs co-cultured with platelets from either WT or IL-1β-deficient (Il1b–/–) mice. We found that Il1b–/– platelets were equally able to boost IL-1β production in inflammasome-activated BMDMs as WT platelets (Figure 4A). Similarly, the addition of WT platelets to BMDMs from IL-1R-deficient mice (Il1r–/–) also boosted their IL-1β production upon inflammasome activation (Figure 4B), thus excluding a role for platelet-derived IL-1α in mediating the response. Finally, the addition of platelets to Il-18r–/– BMDMs equally boosted their IL-1β production toward LPS + Nigericin (Figure 4C), indicating that neither the platelet-derived IL-1α/β nor the IL-18 cytokines could be responsible for the platelet effect.
Figure 4.

The Effect Is Independent of Platelet-Derived IL-1 Cytokines or Inflammasomes
(A) IL-1β released by WT BMDMs ± platelets (5:1 ratio) from WT or Il1b–/– mice. Bar graph with Mean and SD from one experiment with three mice.
(B and C) IL-1β levels in cell-free supernatants of WT or Il1r–/– (B) or Il18r–/– (C) BMDMs ± platelets (5:1 ratio) from WT mice.
(D) IL-1β levels in cell-free supernatants of WT, Nlrp3–/–, or Pycard–/– BMDMs ± WT platelets (5:1 ratio). Confocal imaging of hMDMs co-cultured with platelets (50:1 ratio). Cells were pretreated or not with Cytochalasin D before the addition of platelets. Blue (Draq5, nuclei); red (wheat germ agglutinin [WGA], plasma membrane); green (anti-CD41, platelets).
(E) IL-1β released by hMDMs treated as in (D).
(F) Schematics of trans-well and IL-1β released by hMDMs cultured alone, in direct co-cultures with platelets (Well), or in trans-well (insert).
(G and H) IL-1β released by hMDMs ± platelets or platelet (G) and MK (H) supernatants. Graphs show floating bars (with mean and minimum to maximum values) from pooled data from two to four independent experiments. Each symbol represents the average of technical triplicates from different donors.
Recent studies reported the assembly of the NLRP3 inflammasome in platelets and their involvement in the pathogenesis of dengue (Hottz et al., 2013) and sickle cell disease (Vogel et al., 2018). To determine whether the co-activation of platelet NLRP3 could contribute to the IL-1 production in co-cultures, we tested if the addition of WT platelets could compensate for the impaired IL-1 response of Nlrp3–/– or Pycard–/– (Asc–/–) BMDMs. We stimulated LPS-primed Nlrp3–/– or Pycard–/– BMDMs with Nigericin or ATP, alone or in co-culture with WT platelets. As expected, macrophages from Nlrp3–/– and Pycard–/– mice failed to produce IL-1β in response to inflammasome activation (Figure S2D). The addition of WT platelets to Nlrp3–/– or Pycard–/– macrophages had no effect on the IL-1β production by these BMDMs (Figure S2D), indicating that the activation of inflammasomes in platelets is not involved in the amplification of macrophage IL-1β responses. To determine whether the NLRP3/ASC inflammasome is expressed and assembled in platelets, we imaged inflammasome assembly in bone marrow (BM) cells from transgenic (Tg) ASC-mCitrine mice (Tzeng et al., 2016). ASC-mCitrine+ BM cells were stained for leukocytes (CD45), neutrophils (Ly6G), and platelets (CD41) and assessed by flow cytometry and confocal microscopy. While ASC was clearly visualized and assembled into fluorescent specks in inflammasome-activated leukocytes (CD45+), macrophages (CD45+F4/80+), and neutrophils (CD45+Ly6G+), it was not observed in platelets and MKs (CD41+cells) (Figures S3C and S3D). These results were confirmed using flow cytometry and confocal imaging of BM cells from ASC-mCherry knock-in reporter mice (Figure S3E).
Most studies reporting the presence (Brown et al., 2013, Denis et al., 2005) or activity (Hawrylowicz et al., 1989, Kaplanski et al., 1993) of IL-1 cytokines or inflammasome molecules used preparations of washed platelets without assessment of purity, which could not rule out the presence of contaminating leukocytes in their platelet preparations (Pillitteri et al., 2007). We therefore re-evaluated the expression of the canonical inflammasome components in platelets and peripheral blood mononuclear cells (PBMCs) from healthy volunteers. The purity of platelet preparations was assessed by flow cytometry, microscopy, and qPCR using platelet (PF4) and leukocyte markers (CD45, CD14) (Figures S4A–S4E and Table S1). Importantly, both freshly isolated human and mouse platelets remained viable after purification and upregulated P-selectin (CD62P) upon thrombin stimulation (Figures S3F and S4E). Nevertheless, human platelets did not express the inflammasome components NLRP3, ASC, and caspase-1, nor IL-1β at the mRNA (Figure S4D) and protein level (Figures S4F–S4H), although these molecules were promptly detected in PBMCs from the same donors and increased upon LPS stimulation. Furthermore, unlike the human monocytic line THP-1, no expression of IL-1β and inflammasome molecules was detected in the human megakaryocytic cell MEG-01 (Figure S3G), which also failed to secrete mature IL-1β upon inflammasome activation (Figure S3H).
Finally, we re-analyzed publicly available transcription profiles of purified platelets from five independent studies (Devignot et al., 2010, Gnatenko et al., 2005, Londin et al., 2014, Raghavachari et al., 2007, Spivak et al., 2014), including the gene expression profiles of platelets from patients with dengue (Devignot et al., 2010) and sickle cell disease (Raghavachari et al., 2007; Figures S5A–S5E). In all these studies, we re-assessed the expression of IL-1β and the inflammasome components NLRP3, ASC, and caspase-1 using the expression of platelet markers PF4 (CXCL4) or platelet-derived growth factor A (PDGFA) as comparisons (Figures S5A–S5E). None of the NLRP3, ASC, caspase-1, or IL-1β transcripts were detected in human platelets in these studies (Figures S5A–S5E). Together with our previous findings, these observations support the conclusion that human and mouse platelets do not express the components of the canonical NLRP3 inflammasome and are therefore unable to assemble inflammasomes.
The Platelet Effect Is Mediated by a Soluble and Constitutive Factor
Phagocytosis of activated platelets has been shown to regulate platelet and neutrophil function, survival, and differentiation (Badrnya et al., 2014, Chatterjee et al., 2015, Lang et al., 2002, Senzel and Chang, 2013). To determine whether phagocytosis is responsible for the effect of platelets on inflammasome activation, we performed confocal imaging of hMDMs (Figure 4D) and purified blood neutrophils (Figure S2E) incubated with freshly isolated platelets. Co-cultures were stimulated with LPS, followed by inflammasome activation. Both hMDMs (Figure 4D) and neutrophils (Figure S2E) phagocytosed platelets; however, pretreatment of hMDMs with the phagocytosis inhibitor Cytochalasin D did not prevent the platelet-mediated boost of IL-1β (Figure 4E). Furthermore, platelets were able to boost the IL-1β responses of hMDMs in both direct and in trans-well co-cultures (Figure 4F), and cell-free supernatant from resting, LPS-stimulated, or thrombin-activated platelets also enhanced IL-1β production from hMDMs (Figure 4G). Importantly, thrombin had no effect on macrophages stimulated in the absence of platelets (Figure S2F). Thus, the platelet influence on IL-1β production by inflammasome-activated macrophages is independent of cell contact. In contrast, the transfer of platelet supernatants to cultures of human neutrophils or monocytes did not promote a significant increase in the production of IL-1β (Figure S2G; data not shown), suggesting that cell-to-cell contact may be required for the platelet-boosting effects in these cells.
We observed that supernatants from quiescent and activated platelets had similar effects on the inflammasome activation of hMDMs (Figure 4G). To determine whether this capacity of quiescent platelets was due to undetectable activation during isolation, we transferred supernatants from resting MEG-01 to cultures of hMDMs. Similar to platelets, supernatants from resting MEG-01 boosted the inflammasome activity in hMDMs (Figure 4H). This finding points toward the presence of a soluble and constitutively expressed factor secreted by both platelets and MKs, which modulates the inflammasome activity of macrophages.
Platelets License NLRP3 Transcription to Boost Inflammasome Activation on Human Macrophages
In most immune cells, the activation of NLRP3 requires a priming stimulus, such as the engagement of pattern recognition receptors, that initiates the transcription of NLRP3. The amount of NLRP3 available in the cytosol is a key limiting factor regulating its activation. Since we observed that the platelet effect was specific to the activation of the NLRP3 inflammasome (Figures S1D and S1E), we investigated if the platelet effect on macrophages occurs at the transcriptional level during priming or afterward during activation of the NLRP3 inflammasome. To this end, we co-cultured NLRP3-overexpressing immortalized mouse macrophages (NLRP3FiMøs) with platelets and directly stimulated these cells with Nigericin. As the LPS priming signal is also required for the upregulation of pro-IL-1β, we assessed caspase-1 activity using a specific caspase-1 luminescence assay as a readout. The addition of platelets to NLRP3FiMøs did not enhance their caspase-1 activity (Figure 5A), supporting that platelets influence the inflammasome activation of hMDMs by enhancing the transcription of NLRP3. To determine whether platelets modulate the expression of other inflammasome molecules, we evaluated hMDMs cultivated or not with platelet supernatants, which were used to minimize the detection of transcripts arising from platelets. The addition of supernatants from resting platelets to macrophages boosted their expression of IL-1β (Figure 5B and Table S1). Surprisingly, the platelet supernatant was as effective as LPS to induce the expression of NLRP3 and pro-IL-1β protein (Figure S2H, lanes 1 and 3). However, the platelet supernatant did not alter the expression of pro-caspase-1 or ASC (Figure 5B). In conformity with increased NLRP3 availability in the cytosol, exposure to platelets boosted the formation of ASC specks in hMDMs (Figure 5C). Importantly, cells stained with isotype-matched immunoglobulin G (IgG), as well as platelets cultured alone, did not show ASC specks (Figures 5C and S6A). Co-culture with platelets or exposure to their supernatants increased caspase-1 activation and maturation of IL-1β in supernatants of hMDMs (Figures 5D–5F). Again, these proteins were not present in platelets (Figures 5D and 5E; Plts alone). We also detected pro-IL-1β in cell-free supernatants of LPS-primed hMDMs, likely due to cell death during LPS priming. Furthermore, despite lacking pro-IL-1β mRNA (Figure S4D) and protein (Figures 5D, lane 11, and S4G), platelets and their supernatants increased the concentrations of pro-IL-1β even on unstimulated hMDMs (Figure 5D, lanes 3 and 4). Contrary to NLRP3, which requires priming for its transcription, NLRC4 and AIM2 are constitutively expressed in the cytosol of hMDMs, which might explain why platelets had minor effects on the AIM2- or NLRC4-dependent IL-1β secretion (Figures S1D and S1E). Altogether, these findings indicate that platelets boost inflammasome activation by enhancing the transcription of pro-IL-1β and NLRP3 in hMDMs.
Figure 5.

Platelets Enhance NLRP3 and Pro-IL-1 Transcription in Human Macrophages
(A) Caspase-1 activity in cell-free supernatants of NLRP3-overexpressing mouse macrophages ± platelets (50:1 ratio). Cells were left resting (Unstim) or activated with Nigericin (10 μM, 90 min) without LPS priming.
(B) qPCR analysis of the indicated genes in hMDMs ± platelet supernatants.
(C) Confocal imaging and quantification of ASC specks in LPS-primed and Nigericin-activated hMDMs ± platelets (50:1 ratio). Scale bar, 11 μm.
(D and E) Immunoblotting for IL-1β (D) or caspase-1 (E) in cell-free supernatants, as well as β-actin in cell lysates of resting, LPS-primed hMDMs ± platelets (50:1 ratio), or platelet supernatants. Different donors are indicated.
(F) Caspase-1 activity in cell-free supernatants of hMDMs treated as in (C). Graphs show floating bars (with mean and minimum to maximum values) from pooled data from three or four independent experiments. Each symbol represents the average of technical triplicates from different donors.
The Platelet-Mediated Amplification of Inflammasome Activity Requires Calcium and Can Be Prevented by Heat Inactivation
Platelets are sources of lipid mediators (Hinz et al., 2016) such as prostaglandins (COX-1/2-derived mediators), which regulate LPS-induced pro-IL-1β transcription (Zasłona et al., 2017) and inhibit TNF-α in macrophages (Chandra et al., 1995, Xiang et al., 2013, Zasłona et al., 2017). We therefore inhibited COX-1/2 by pre-treating platelets with aspirin (Xiang et al., 2013). Aspirin had no effect on platelet ability to boost IL-1β production in hMDMs (Figure 6A). Similarly, pre-treatment of platelets with Zileuton (Zt), an inhibitor of leukotriene synthesis, did not block the platelet effect. Of note, aspirin and Zt had no effect on IL-1β production when used directly on hMDMs (Figure 6A).
Figure 6.

A Platelet-Derived Ca2+-Dependent Protein Boosts NLRP3 Activity in Human Macrophages
(A) IL-1β released by resting (Unstim), LPS-primed, and Nigericin-activated hMDMs ± aspirin or Zileuton (Zt) pre-treated platelets (50:1 ratio)
(B and C) IL-1β levels released by unstimulated or LPS-primed hMDMs ± platelets. Cells were incubated with Benzonase (B) or Apyrase (C) before LPS priming and inflammasome activation.
(D) IL-1β levels in co-cultures of platelets and hMDMs added with the indicated concentrations of ADP or equivalent dilutions in water.
(E and F) IL-1β released by hMDMs stimulated as in (A) and co-cultured with heat-inactivated (HI), fixed platelets, or fresch or HI platelet supernatants.
(G) IL-1β released by hMDMs activated as in (A) and added with the indicated recombinant proteins.
(H and I) IL-1β levels in co-cultures of platelets and hMDMs added with BAPTA in Ca2+-free medium (H) or BAPTA AM (I) in normal RPMI medium before activation.
(J) IL-1β levels released by inflammasome-activated hMDMs incubated with the indicated concentrations of calcium chloride (CaCl2) in Ca2+-free medium. Graphs show floating bars (with mean and minimum to maximum values) from pooled data from two to four independent experiments. Each symbol represents the average of technical triplicates from different donors.
See also Figure S6.
Activated platelets can also release nucleic acids and nucleosides that display inflammatory properties (Qin et al., 2016). However, degradation of free nucleic acids by Benzonase (Bz) did not prevent the effect of platelet co-culture on IL-1β production (Figure 6B). As platelets store other purines including ADP and ATP, we added apyrase (Apy) to the co-culture of hMDMs with platelets. Apyrase did not alter the effect of platelets on the inflammasome activity of hMDMs (Figure 6C). Likewise, the direct addition of extracellular ADP to inflammasome-activated hMDMs also failed to recapitulate the platelet effect (Figure 6D). This is in line with our findings that platelets boost macrophage IL-1β production through transcriptional regulation during the priming phase of inflammasome activation (Figures 5A and 5B). Altogether, our findings suggest that the platelet-derived factor responsible for the inflammasome boosting does not belong to the classes of nucleic acids, purines, or derivatives of arachidonic acid.
Therefore, we speculated whether the platelet-derived factor could be a protein. Ratifying this hypothesis, both heat inactivation and fixation of platelets (Figure 6E) or platelet supernatants (Figure 6F) extinguished their effects on inflammasome activation in hMDMs. Multiple cytokines were detectable in supernatants of resting and activated platelets in our assays (Figure S1A), including CCL5 (RANTES), CXCL12 (SDF1α), IL-18, and PDGF-BB. Additional literature reported that platelets are cellular sources of CD40L, PF4, brain-derived neurotrophic factor (BDNF), P-selectin, and CXCL7 (Kral et al., 2016, Semple et al., 2011). After the direct addition of human recombinant proteins to hMDMs (Figures 6G, S6B, and S6C), inhibitors or blocking antibodies against platelet-derived cytokines (Figures S6D and S6E), or co-cultures involving cells from animals deficient for chemokine receptors (Figure S6F), we excluded a role for CXCL1, CCL5, CXCL12, CXCL7, PDGF-BB, epidermal growth factor (EGF), vascular endothelial growth factor (VEGFA), CD40L, PF4, BDNF, and P-selectin as contributors to the platelet effect.
The failure of the above-mentioned α-granule-derived proteins to boost IL-1β production in hMDMs suggested that molecules contained in the dense granules of platelets could mediate the IL-1β boosting in activated hMDMs. Platelet-dense granules are rich stores of serotonin (5-HT) and Ca2+. However, we found that neither the addition of 5-HT (Figure S6G) nor the blockade of its uptake by hMDMs with fluoxetine (Du et al., 2016, Marcinkiewcz et al., 2016) altered IL-1β production by inflammasome-activated hMDMs (Figure S6H).
We next examined the effect of chelating extracellular or intracellular Ca2+ with 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA) and BAPTA-AM, respectively. Notably, treatment of co-cultures with BAPTA, but not with the membrane-permeable BAPTA-AM, prevented the platelet effect without interfering with the basal response of hMDMs (Figures 6H and 6I). However, the addition of calcium chloride (CaCl2) to inflammasome-activated hMDMs did not evoke additional IL-1β secretion (Figure S6I), suggesting that Ca2+ is required but not sufficient for the platelet effect.
Cells sense extracellular Ca2+ primarily through calcium sensing receptors (CaSR), which are G-protein-coupled receptors (GPCRs). To test the role of CaSR, we pre-treated hMDMs with the CaSR-selective inhibitor NPS2143. We found that NPS2143 blocked the platelet effect on hMDMs (Figure 6J). Of note, recapitulating the platelet effect, allosteric activation of CaSR with the calcimimetic compound R568 boosted the IL-1β response of inflammasome-activated hMDMs (Figure 6K). These findings indicate that a heat-sensitive protein that induces calcium signaling via CaSR is involved in the platelet-mediated increase of IL-1β from inflammasome-activated hMDMs.
A Combination of Platelet-Derived Proteins Might Boost Inflammasome Activation of Macrophages
Next, we assessed the protein secretome of platelets and MKs and identified additional candidates similarly secreted by these cells that could mediate the IL-1 boosting effect (Figures 7A, 7B, S7A, and S7B). Supernatants from quiescent or LPS-treated human platelets (Figure 7A; n = 4), as well as from MEG-01 cells (Figure 7B, n = 3) were evaluated by liquid chromatography-tandem mass spectrometry (LC-MS). As our findings in Figures 4G and 4H indicated the presence of a factor ubiquitously present in the supernatants of resting and activated platelets, we directed further analysis to proteins that remained unchanged (fold change > −1.5 and < 1.5) following LPS stimulation in both cell types. Among the proteins similarly secreted by platelet and MKs, we identified members of the TGF-β and S100 family and thrombospondin-1 (THBS1), which figured as the most abundant protein in platelet supernatants (Figures 7A and 7B). Notably, TGF-β, S100, and THBS1 require Ca2+ for their activity (Bertheloot and Latz, 2017, Cailotto et al., 2011, Misenheimer and Mosher, 1995). Additionally, in line with our findings that TLR4 signaling is partially involved in the platelet effect (Figure S1G), THBS1 can activate TLR4 (Li et al., 2013), can regulate IL-1β in macrophages (Stein et al., 2016), and is necessary for the full activity of TGF-β (Crawford et al., 1998). We therefore tested the effect of these proteins on the platelet-mediated boosting of inflammasomes. We tested the addition of recombinant human TGF-β (rhTGF-b1) to inflammasome-activated hMDMs. Consistent with previous reports of an antagonistic effect of TGF-β on TNF-α signaling (Vaday et al., 2001, Verrecchia and Mauviel, 2004, Yamane et al., 2003), the addition of rhTGF-b1 to hMDMs inhibited their production of TNF-α triggered by LPS (Figure S7C). However, rhTGF-b1 did not influence the IL-1β response (Figure 7C). Likewise, selective inhibition of the TGF-β receptor by pre-treating hMDMs with SB431542 (Matsuyama et al., 2003) did not prevent the platelet effect (Figure 7D), despite preventing the downregulation of TNF-α (Figure S7C). These data indicate that TGF-β is likely not the platelet-derived factor enhancing the NLRP3 activity of hMDMs, but it might underlie the regulation of TNF-α.
Figure 7.

Platelets Have Broad Effects on Macrophages That May Contribute to Boosting of Inflammasome Activation
(A and B) Proteomics of cell-free supernatants from resting (Unstim) or LPS-treated (A) human platelets (n = 4) or MK (B) (n = 3). Darker points show proteins with log2 fold ≥ 2 between LPS and Unstim.
(C–G) IL-1β released by resting or inflammasome-activated hMDMs added with (C) rhTGF-β1 or (D) pre-treated with SB-431542, (E) added with rhS100A8/9, (F) pre-treated with TAK242 (0.5 μg mL−1) and an αRAGE mAb (10 μg mL−1), or (G) added with rhTHBS1.
(H) IL-1β released by inflammasome-activated BMDMs cultured with WT or Thbs1−/− platelet supernatants.
(I) IL-1β and TNF-α levels released by R848-primed and Nigericin-activated hMDMs ± platelets. hMDMs were pre-treated with the CD36 inhibitor SSO. Graphs show floating bars (with mean and minimum to maximum values) from pooled data from two to four independent experiments
See also Figure S7.
S100 proteins are important regulators of Ca2+ homeostasis, and platelet-derived S100A8/9 has been shown to induce inflammation (Lood et al., 2016, Wang et al., 2014). S100A8/9 is enriched in platelets and binds to membrane receptors such as TLR4, receptor for advanced glycation endproducts (RAGE), and CD36 (Bertheloot and Latz, 2017). TLR4 and CD36 are also known receptors for THBS1 (Li et al., 2013, Stein et al., 2016). However, we found that addition of neither rhTHBS1 (Figures 7G and S7E) nor rhS100A8/9 (Figure 7E) to hMDMs influenced their basal response to LPS + Nigericin. Of note, rhS100A8/9 induced TNF-α secretion from hMDMs (Figure S7D). Furthermore, supernatants from Thbs1−/− platelets were as efficient as WT platelets in boosting IL-1β release and caspase-1 activity from inflammasome-activated BMDMs (Figure 7H), and RAGE inhibition antibodies had no effect on the platelet boosting of IL-1β (Figures 7F and S7G). These findings led us to conclude that platelet-derived THBS1 and S100A8/A9 are not, individually, responsible for the platelet effect on macrophages.
As THBS1 and S100 proteins bind to CD36, we tested the effect of CD36 inhibition with Sulfosuccinimidyl oleate (SSO) (Kuda et al., 2013). SSO interfered with IL-1β and TNF-α release (Figure 7I) and caspase-1 activity (Figure S7F) in both the presence and absence of platelets. Nevertheless, even in the presence of SSO, platelets increased both the release of IL-1β and the activity of caspase-1 from macrophages compared to SSO-pre-treated macrophages in monoculture.
As single inhibition of TLR4, RAGE, and CD36 did not completely abrogate the platelet effect, we speculated that a combined activity of platelet-derived S100 and THBS1 could mediate the boosting of IL-1β on hMDMs. An experimental strategy to block TLR4/RAGE and CD36 resulted in complete inhibition of IL-1β, TNF-α, and caspase-1 activity of hMDMs (Figure S7G), thus precluding us to conclude whether these proteins are synergistically involved in the platelet-mediated enhancement of IL-1β response of hMDMs. Hence, additional work is required to delineate the mechanisms by which platelets affect the inflammasome activation of hMDMs.
Discussion
Here, we report that the interaction with platelets licenses NLRP3, potentiates inflammasome activation and production of IL-1 cytokines by innate immune cells, and is critical for NLRP3-activation in human monocytes. The platelet effect is mediated by a yet unidentified soluble factor (or factors), which enhances NLRP3 transcription and boosts inflammasome activation. Hence, our work identifies platelets as additional players in governing the production of IL-1 cytokines and emphasizes that full inflammasome activity in vivo is too complex to be modeled by in vitro monocultures of single-cell populations.
Platelets are key players in inflammation (Carestia et al., 2019, Dann et al., 2018, Kral et al., 2016, Passacquale et al., 2011) and express TLRs that sense infection and initiate inflammatory responses (Andonegui et al., 2005). While platelets are known to react to LPS (Cognasse et al., 2008, Andonegui et al., 2005), they do not constitutively express membrane CD14 (Damien et al., 2015) and require soluble CD14 from plasma for TLR4 signaling (Brown and McIntyre, 2011, Damien et al., 2015).
Our work adds to a growing bulk of evidence that platelet parameters, such as their blood counts, can be additional causes of variation in the concentrations of circulating cytokines (Hu et al., 2018, Schirmer et al., 2018, Tunjungputri et al., 2018). Platelets secrete a large array of proteins (Coppinger et al., 2004, Maynard et al., 2007), which affect leukocytes in different ways (Kral et al., 2016, Sreeramkumar et al., 2014). They have also been proposed as sources of IL-1 cytokines and, more recently, to assemble inflammasomes (Cornelius et al., 2019, Hottz et al., 2013, Vogel et al., 2018).
Our work challenges these findings with a series of complementary techniques that demonstrate that platelets lack expression of NLRP3, ASC, and caspase-1 and are therefore incapable of assembling inflammasomes. Those included experiments using platelets and BMDMs from mice lacking IL-1 proteins or their receptors (Figures 4A–4C); mice lacking inflammasome molecules (Figure S2D), megakaryocytes, and platelets from humans (Figures S3F, S3G, and S4); and transgenic or knockin inflammasome reporter mice (Figures S3C–S3E), as well as a MS-based proteomics analysis of supernatants from human platelets and MK cell line (Figures 7 and S7). Our conclusions were further supported by a meta-analysis of transcriptomics from five independent studies with human platelets (Figure S5). We believe that the discrepancies are likely because the studies reporting inflammasomes on platelets relied on fluorescence assessment of inflammasome components, using antibodies of weak specificity (Beilharz et al., 2016), and without quantitative measurement of proteins in platelet lysates by immunoblotting. Likewise, IL-1 expression on platelets is reported with conflicting results in literature. Some studies describe expression of IL-1 in activated platelets (Denis et al., 2005, Lindemann et al., 2001, Thornton et al., 2010), while others find IL-1 activity in bioassays (Hawrylowicz et al., 1989, Kaplanski et al., 1993), with no evidence of the cytokine itself. Notably, throughout all our experiments, we assessed the purity (leukocyte contamination CD14 and/or CD45; Figures S3C–S3E, S4D, and S4F) as well as activation capacity (ability to upregulate CD62p upon thrombin stimulation; Figures S3F and S4E) of our platelet preparations. These important controls were not performed in the aforementioned studies. Hence, it cannot be ruled out that their platelet preparations are free of contaminating leukocytes (Pillitteri et al., 2007). For example, a preparation of 5 × 108 platelets, commonly used in previous studies for RNA isolation, with a 98% purity (assessed by FACS) could easily contain ∼2 × 106 contaminating leukocytes, which could account for a significant portion of RNA within a sample. Supporting this, IL-1β expression on platelet preparation is closely correlated to the presence of contaminating leukocytes (Pillitteri et al., 2007).
In line with our findings, platelets have been reported to amplify IL-1β-mediated inflammation in RA (Boilard et al., 2010), an IL-1-driven disease (Dinarello, 2011), and a recent study demonstrated pro-atherogenic effects of platelets by and skewing monocytes into a pro-inflammatory macrophage phenotype (Barrett et al., 2019). Platelet depletion in mice with RA (Boilard et al., 2010) or in Ldlr–/– mice fed a western diet (Barrett et al., 2019) attenuated inflammation and improved clinical outcomes. However, it was not possible in these studies to distinguish whether these effects were caused by decreased platelet-derived cytokines or by the influence of platelets on other immune cells, as we report here.
Our data confirm their relevance to human physiology and disease, as high blood platelet counts are found in patients (Ciccarelli et al., 2014) and mouse models of IL-1-driven auto-inflammatory disorders (Bonar et al., 2012). In Kawasaki disease (KD), an acute systemic vasculitis in children for which thrombocytosis is a common feature, human and experimental mouse models have determined a critical role of IL-1β in the cardiovascular pathogenesis. Furthermore, thrombocytosis and platelet activation are predictive of the development of coronary aneurysm in KD (Burns et al., 2017, Lee et al., 2012b).
As both IL-1α and IL-1β induce thrombocytosis in mice (Kimura et al., 1990, Nishimura et al., 2015, Trinh et al., 2015), platelets could help feed an inflammatory loop by potentiating IL-1 signaling, which in turn triggers platelet biogenesis. However, whether thrombocytosis is a marker of disease or a contributor to pathogenesis remains to be investigated. Platelet counts have been correlated with the concentrations of other cytokines that affect platelet production in immune thrombocytopenia (ITP) and other diseases. Those include CD40L, CXCL5, CCL5, EGF, VEGF, granulocyte-macrophage colony-stimulating factor (GM-CSF), interferon (IFN)γ, monocyte chemoattractant protein 1 (MCP-1), IL-8, and PDGF-BB (Feng et al., 2012, Pourcelot et al., 2014). Thus, the contribution of platelets to cytokine production in human biology warrants further investigation.
Notably, co-culture with platelets impaired the LPS-induced TNF-α production in several immune cells. This observation is in line with previous studies (Aslam et al., 2006, Gudbrandsdottir et al., 2013, Xiang et al., 2013) and may be explained by the reported ability of platelets to sequester TNF-α and IL-6 released from LPS-stimulated monocytes (Carestia et al., 2019). Platelets have a protective role in sepsis through the regulation of TNF-α and IL-6 via the secretion of a COX1/PGE2/EP4-dependent lipid mediator (Xiang et al., 2013). In line with these findings, we observed that TNF-α is regulated via a platelet-derived COX1-dependent lipid mediator and is related to TGFβ signaling.
Despite our efforts, we were unable to identify a unique platelet factor that boosts the inflammasome activation of innate immune cells. However, we show that the regulation of IL-1β production is likely mediated through a constitutively expressed protein factor that engages a CaSR on primary human macrophages. This is consistent with previous reports of roles for CaSR on inflammasome activation (Lee et al., 2012a, Rossol et al., 2012). These findings will need further validation using genetic deletion of these receptors. It is possible that a combination of factors acting in synergy and affecting different pathways underlie the platelet effect.
Our findings also strengthen recent discoveries of a direct link between the mammalian immune and coagulation systems (Burzynski et al., 2019, Wu et al., 2019, Yang et al., 2019) by showing a direct participation of platelets in the inflammasome output of innate immune cells.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-human Caspase-1 (p20), mAb (Bally-1) | Adipogen International | AG-20B-0048-C100; RRID:AB_2490257 |
| Anti-human CD14-APC (61D3) | Thermo Fisher Scientific | 17-0149-42; RRID:AB_10669167 |
| Anti-human CD41 FITC (HIP8) | Thermo Fisher Scientific | 11-0419-42; RRID:AB_10718234 |
| Anti-human CD45 PE (2D1) | Thermo Fisher Scientific | 12-9459-42; RRID:AB_10718238 |
| Anti-human CD66b | Thermo Fisher Scientific | 11-0666-42; RRID:AB_2572461 |
| Anti-human NAP2 (CXCL7) | PeproTech | 300-14-10; RRID:AB_147531 |
| Anti-human RAGE | R&D Systems | MAB11451; RRID:AB_2224344 |
| Anti-mouse CD41 eFluor450 (eBioMWReg30) | Thermo Fisher Scientific | 48-0411-82; RRID:AB_1582238 |
| Anti-mouse CD45 PE (30-F11) | Thermo Fisher Scientific | 12-0451-83; RRID:AB_465669 |
| Anti-human CXCL12 / SDF-1a | R&D Systems | AF-310-SP; RRID:AB_354461 |
| Anti-human IL-1 beta / IL-1F2 Antibody, goat | R&D Systems | AF-201-NA; RRID:AB_354387 |
| Anti-mouse IRDye 680 | Li-Cor Biosciences | 925-68080; RRID:AB_2814917 |
| Anti-mouse Ly6G-AF488 (RB6-8C5) | Thermo Fisher Scientific | 53-5931-82; RRID:AB_469918 |
| Anti-mouse Ly6G-APC (1A8-ly6g) | Thermo Fisher Scientific | 17-9668-82; RRID:AB_2573307 |
| Anti-mouse NLRP3/NALP3, mAb (Cryo-2) | Adipogen International | AG-20B-0014; RRID:AB_2490202 |
| Anti-human/mouse CD62p (P-selectin) APC (Psel.KO.2.3) | Thermo Fisher Scientific | 12-0628-42; RRID:AB_10804401 |
| Anti-human/mouse Beta-Actin (Host: mouse) | Li-Cor Biosciences | 926-42212; RRID:AB_2756372 |
| Anti-human/mouse Beta-Actin (Host: rabbit) | Li-Cor Biosciences | 926-42210; RRID:AB_1850027 |
| Anti-human/mouse Beta-Tubulin (Host: rabbit) | Li-Cor Biosciences | 926-42211; RRID:AB_1850029 |
| Anti-rabbit IRDye 800 | Li-Cor Biosciences | 926-32213; RRID:AB_621848 |
| FcR Blocking Reagent, human | Miltenyi Biotech | 130-059-901 |
| FcR Blocking Reagent, mouse | Miltenyi Biotech | 130-092-575 |
| Mouse IgG1 K Iso Control APC (P3.6.2.8.1) | Thermo Fisher Scientific | 17-4714-42; RRID:AB_1603315 |
| Mouse IgG1 K Iso Control FITC (P3.6.2.8.1) | Thermo Fisher Scientific | 11-4714-42; RRID:AB_10596964 |
| Mouse IgG1 K Iso Control PE (P3.6.2.8.1) | Thermo Fisher Scientific | 12-4714-42 RRID:AB_1944423 |
| Normal Goat IgG control | R&D systems | AB-108-c; RRID:AB_354267 |
| OneComp eBeads | Thermo Fisher Scientific | 01-1111-42 |
| Polyclonal anti-GPIb alpha | Emfret Analytics | R300; RRID:AB_2721041 |
| Polyclonal non-immune rat immunoglobulins | Emfret Analytics | C301; RRID:AB_2734715 |
| Purified anti-ASC (TMS-1) Antibody | BioLegend | 653902; RRID:AB_2561778 |
| Purified anti-human CD42b Antibody (HIP1) | BioLegend | 303902; RRID:AB_314382 |
| Purified Mouse IgG, k Isotype | BioLegend | 401402; RRID:AB_2801451 |
| Rat Anti-mouse CD14-FITC (Sa14-2) | BioLegend | 123308; RRID:AB_940580 |
| Rat IgG1 K Iso Control eFluor450 (eBRG1) | Thermo Fisher Scientific | 48-4301-82; RRID:AB_1271984 |
| Rat IgG2a K Iso Control APC (eBR2a) | Thermo Fisher Scientific | 17-4321-81; RRID:AB_470181 |
| Rat IgG2b, K Iso Control PE (eB149/10H5) | Thermo Fisher Scientific | 12-4031-82; RRID:AB_470042 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 2-mercaptoethanol | Sigma-Aldrich | M6250-250ML |
| Acetylsalicylic acid (Aspirin) | Sigma-Aldrich | A5376-100G |
| Acid-Citrate-dextrose solution (ACD) | Sigma-Aldrich | C3821-50ML |
| Adenosine 5-diphosphate sodium salt (ADP) | Sigma-Aldrich | A2754-500MG |
| Adenosine triphosphate (ATP) | Sigma-Aldrich | A2382 |
| Apyrase from potatoes, High Activity, ATPase > 600 units/mg protein | Sigma-Aldrich | A2230-100UN |
| BAPTA AM | Enzo Life Sciences Inc | BML-CA411-0025 |
| BAPTA, Tetrasodium salt | Santa Cruz Biotechnology | sc-278716 |
| Benzonase® Nuclease | Sigma-Aldrich | E1014-5KU |
| Bovine Serum Albumin (BSA) | Sigma-Aldrich | A9647 |
| Calcium chloride | Sigma-Aldrich | 21115-100ML |
| Chloroform | Merck | 1.02445.1000 |
| cOmplete EDTA-free protease inhibitor Cocktail Tablets | Sigma-Aldrich | 4693132001 |
| CRID3 (CP-456773-02, MCCP90) | Sigma-Aldrich | PZ0280 |
| Cutasept® F Haut-Desinfiziens | Hartmann | 9803640 |
| Cytochalasin D | Sigma-Aldrich | C8273-1MG |
| D-(+)-Glucose solution | Sigma-Aldrich | G7021-100 g |
| Di-Sodium hydrogen phosphate (Na2HPO4) | Sigma-Aldrich | 255793 |
| Dimethyl sulfoxide (DMSO), cell culture grade | PAN-Biotech | P60-36720100 |
| Dithiothreitol (DTT) | Thermo Fisher Scientific | Y00147 |
| dNTP Mix (10 nM each) | Thermo Fisher Scientific | R0192 |
| DRAQ5 | Thermo Fisher Scientific | 65-0880-96 |
| Ethanol (EtOH) absolute | AppliChem | 147194.1214 |
| Fetal Bovine Serum (FBS) | Thermo Fisher Scientific | 26140079 |
| Ficoll-Paque PLUS | GE Healthcare | 17-1440-03 |
| GIBCO Dulbecco’s Dulbecco’s Modified Eagle Medium (DMEM) | Thermo Fisher Scientific | 41965039 |
| GIBCO Dulbecco’s Phosphate Buffered Saline (PBS) | Thermo Fisher Scientific | 14190144 |
| GIBCO GlutaMAX 100X | Thermo Fisher Scientific | 35050061 |
| GIBCO Penicillin-Streptomycin (10,000 U/mL) | Thermo Fisher Scientific | 15140122 |
| GIBCO RPMI 1640 Medium | Thermo Fisher Scientific | 21875091 |
| GIBCO Sodium Pyruvate 100mM | Thermo Fisher Scientific | 11360070 |
| GIBCO UltraPure 0.5M EDTA, pH 8.0 | Thermo Fisher Scientific | 15575020 |
| Hoechst 34580 | Thermo Fisher Scientific | H21486 |
| L929 cell line supernatant | Gift from Prof. Latz and Ms. Engels (University of Bonn) | N/A |
| Lipopolysaccharide (LPS), ultrapure, E. coli 0111:B4 | Invivogen | tlrl-3pelps |
| Magnesium chloride (MgCl2) solution | Sigma- Aldrich | M1028-100ML |
| Methanol | Merck | 1.06007.2500 |
| Met-RANTES trifluoroacetate salt, human | Bachem | H-6544.0050 |
| Nigericin, free acid | Invitrogen | N1495 |
| Oligo(dT)18 primer | Thermo Fisher Scientific | SO132 |
| PageRuler™ Plus Prestained Protein Ladder, 10 to 250kDa | Thermo Fisher Scientific | 26619 |
| Pam3Cysk4 | InvivoGen | P2200.0005 |
| PhosSTOP Easypack Phasphatase Inhibitor Cocktail Tablets | Sigma-Aldrich | 04906837001 |
| Piperazine-1,4-bis(2ethanesulfonic acid) (PIPES) | Sigma-Aldrich | P6757-25G |
| Poly-L-Lysine solution | Sigma-Aldrich | P4707 |
| Potassium chloride (KCl) | Sigma-Aldrich | P9327-100ML |
| (LFn-)PrgI fusion protein | Gift from Prof. Geyer and Dr. Fußhöller (University of Bonn) | N/A |
| Prostaglandin E1 (PGE-1) | Sigma-Aldrich | P5515-1MG |
| Protective antigen (PA), recombinant from Bacillus anthracis | List Biological Laboratories | 171C |
| R848 | Invivogen | tlrl-r848-5 |
| Recombinant human BDNF | Tocris Bioscience | 2837 |
| Recombinant human CD40L / CD154, soluble | Enzo Life Sciences | ALX-522-015-C010 |
| Recombinant human EGF | Sigma-Aldrich | E9644 |
| Recombinant human GM-CSF | Immunotools | 11343125 |
| Recombinant human GRO-alpha / MGSA (CXCL1) | PeproTech | 300-11-5UG |
| Recombinant human NAP-1 (CXCL7) | PeproTech | 300-14-10 |
| Recombinant human P-selectin | R&D systems | ADP3 |
| Recombinant human PF4 (CXCL4) | PeproTech | 300-16-5UG |
| Recombinant human PGDF-BB | BioLegend | 577304 |
| Recombinant human PLGF-1 (PIGF-1) | Thermo Fisher Scientific | PHG0296 |
| Recombinant human RANTES (CCL5) | Thermo Fisher Scientific | PHC1054 |
| Recombinant human S100A8/S100A9 heterodimer | R&D Systems | 8226-S8-050 |
| Recombinant human SDF-alpha (CXCL12) | PeproTech | 300-28a-10UG |
| Recombinant human TGF-b1 | PeproTech | 100-21C-2UG |
| Recombinant human THBS-1 | Sigma-Aldrich | ECM002-50UG |
| Recombinant human VEGF | Thermo Fisher Scientific | PHC9394 |
| SB431542 | Invivogen | inh-sb43 |
| Sodium chloride (NaCl) | Sigma- Aldrich | S5150-1L |
| Sodium deoxycholate | Sigma- Aldrich | D-6750-10G |
| Sodium dodecyl sulfate (SDS) | Sigma- Aldrich | 75746-1KG |
| Sodium hydroxide (NaOH) | Sigma- Aldrich | S2770-100ML |
| Sulfo-N-succinimidyl Oleate sodium (SSO) | BioMol | Cay11211-5 |
| TAK242 (Resatorvid) | Invivogen | tlrl-cli95 |
| Thrombin from human plasma | Sigma-Aldrich | T6884-100UN |
| Triton X-100 | Carl Roth | 3051.3 |
| Tween® 20 | Sigma- Aldrich | P2287 |
| UK5099 | BioMol | Cay16980-5 |
| Wheat Germ Agglutinin (WGA) Alexa Fluor 555 | Thermo Fisher Scientific | W32464 |
| Zileuton | Tocris | 3308 |
| GIBCO Opti-MEM | Thermo Fisher Scientific | 31985070 |
| Lipofectamine 2000 Transfection Reagent | Thermo Fisher Scientific | 11668019 |
| Poly [dA:dT] | Sigma | P0883 |
| Critical Commercial Assays | ||
| Alexa Fluor 647 Antibody labeling kit | Thermo Fisher Scientific | A20186 |
| Caspase-Glo® 1 Inflammsome Assay | Promega | G9952 |
| CD14 MicroBeads, human | Miltenyi Biotech | 130-050-201 |
| EasySep Direct Human Neutrophil Isolation Kit | STEMCELL Technologies | 19666 |
| EasySep Human Monocyte Isolation Kit | Stemcell | 19359 |
| Human IL1 beta HTRF kit | Cisbio | 62IL1PEB |
| Human TNF alpha HTRF kit | Cisbio | 62TNFPEB |
| Maxima SYBR Green/ROX qPCR Master Mix | Thermo Fisher Scientific | K0221 |
| Mouse IL1 beta HTRF kit | Cisbio | 63ADK010PEB |
| Mouse IL6 HTRF kit | Cisbio | 63ADK043PEB |
| Mouse TNF alpha HTRF kit | Cisbio | 6FMTNPEB |
| Pierce BCA Protein Assay Kit | Thermo Fisher Scientific | 23225 |
| Pierce LDH Cytotoxicity Assay Kit | Thermo Fisher Scientific | B3026-88954 |
| ProcartaPlex Human Cytokine/Chemokine/Growth Factor Panel 1 45plex | Thermo Fisher Scientific | EPX450-12171-901 |
| RNase-Free DNase Set | QIAGEN | 79254 |
| RNeasy Mini Kit | QIAGEN | 74106 |
| S-Monovette 9NC tubes | Sarstedt | 01.1606.001 |
| S-Monovette K3EDTA tubes | Sarstedt | 02.1066.001 |
| SuperScript III First-Strand Synthesis SuperMix | Thermo Fisher Scientific | 18080-400 |
| Deposited Data | ||
| Transcription profiling of whole blood from pediatric patients Muckle-Wells disease and healthy childreen | Balow et al., 2013 | Accession Nr.: GSE43553 |
| Transcription profiling of human platelets from patients with essential thrombocythemia | Gnatenko et al., 2005 | Accession Nr.: E-GEOD-2006 |
| Transcription profiling of human platelets from patients with dengue shock syndrome | Devignot et al., 2010 | Accession Nr.: GEOD17924 |
| Transcription profiling of human platelets from patients with sickle cell disease | Raghavachari et al., 2007 | Accession Nr.: E-GEOD-11524 |
| Transcription profiling of human platelets from patients with sepsis-induced acute lung injury (ALI) | Freishtat et al., 2009 | Accession Nr.: E-GEOD-10361 |
| Transcription profiling of human platelets from patients with Polycythemia Vera (PV) | Spivak et al., 2014 | Accession Nr.: E-GEOD-47018 |
| Raw Mass Spectrometry Data Files | This paper | Data are available via ProteomeXchange: PXD017976 |
| Experimental Models: Cell Lines | ||
| NLRP3-FLAG overexpressing immortalized NLRP3 KO | (Franklin et al., 2014, Stutz et al., 2013) | |
| THP-1 | ATCC | ATCC TIB-202 |
| MEG01 | ATCC | CRL2021 |
| Experimental Models: Organisms/Strains | ||
| Mus musculus, wild-type (C57BL/6j) | The Jackson Laboratory | Stock 000664 |
| Mus musculus, ASC-mCitrine Tg (B6.Cg-Gt(ROSA)26Sortm1.1(CAG-Pycard/mCitrine∗,-CD2∗)Dtg/J) | The Jackson Laboratory | Stock 030744 |
| Mus musculus, Nlrp3–/– mice (B6.B6N-Nlrp3tm1Vmd) | Mariathasan et al., 2006 | N/A |
| Mus musculus, Asc–/– mice (B6.129-Pycardtm1Vmd) | Mariathasan et al., 2004 | N/A |
| Mus musculus, Il1b–/– mice (B6.129S2-Il1btm1Dch) | Shornick et al., 1996 | N/A |
| Mus musculus, Il1r–/– mice (B6.129S7-Il1r1tm1Imx/J) | The Jackson Laboratory | Stock 003245 |
| Mus musculus, Il18r–/– mice (B6.129P2-Il18r1tm1Aki/J) | The Jackson Laboratory | Stock 004131 |
| Mus musculus, Cd36–/– mice (B6.129S1-Cd36tm1Mfe/J) | The Jackson Laboratory | Stock 019006 |
| Mus musculus, Tsp1–/– mice (B6.129S2-Thbs1(tm1Hyn)/J) | The Jackson Laboratory | Stock 006141 |
| Mus musculus, Cxcr3–/– mice (B6.129P2-Cxcr3tm1Dgen/J) | The Jackson Laboratory | Stock: 005796 |
| Mus musculus, Ccr5–/– mice (B6.129P2-Ccr5tm1Kuz/J) | The Jackson Laboratory | Stock: 005427 |
| The knock in ASC-mCherry reporter mouse is available upon request | Eicke Latz, Institute of Innate Immunity | N/A |
| Oligonucleotides | ||
| hACTB-Fwd: 5′-ccaccatgtaccctggcatt-3′ | Metabion | N/A |
| hACTB-Rev: 5′-cggagtacttgcgctcagga-3′ | Metabion | N/A |
| hNLRP3-Fwd: 5′-tcggagacaaggggatcaaa-3′ | Metabion | N/A |
| hNLRP3-Rev: 5′-agcagcagtgtgacgtgagg-3′ | Metabion | N/A |
| hCD14-Fwd: 5′-gagctcagaggttcggaaga-3′ | Metabion | N/A |
| hCD14-Rev: 5′-cttcatcgtccagctcacaa-3′ | Metabion | N/A |
| hPYCARD-Fwd: 5′-gagctcaccgctaacgtgct-3′ | Metabion | N/A |
| hPYCARD-Rev: 5′-actgaggaggggcctggat-3′ | Metabion | N/A |
| hPF4- Fwd: 5′-ctgaagaagatggggacctg-3′ | Metabion | N/A |
| hPF4- Rev: 5′-gtggctatcagttgggcagt-3′ | Metabion | N/A |
| hCASP1-Fwd: 5′-acaacccagctatgcccaca-3′ | Metabion | N/A |
| hCASP1-Rev: 5′-gtgcggcttgacttgtccat-3′ | Metabion | N/A |
| hBP1BA-Fwd: 5′-ctgctctttgcctctgtggt-3′ | Metabion | N/A |
| hBP1BA-Rev: 5′-ctccaggtgtgtggtttgtg-3′ | Metabion | N/A |
| hIL1B-Fwd: 5′-tgggcagactcaaattccagct-3′ | Metabion | N/A |
| Software and Algorithms | ||
| GraphPad Prism 8.1.2 (227) | https://www.graphpad.com | RRID:SCR_002798 |
| FlowJo 10.4 | https://www.flowjo.com | RRID:SCR_008520 |
| Volocity 3D Image Analysis Software v6.0.1 | https://www.perkinelmer.com | RRID:SCR_002668 |
Resources Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Bernardo S Franklin (franklin@uni-bonn.de).
Materials Availability
This study did not generate new unique reagents. The knockin ASC-mCherry reporter mouse is available upon request.
Data and Code Availability
The mass spectrometry proteomics data are available via ProteomeXchange: PXD017976.
Experimental Model and Subject Details
Study Subjects
Peripheral blood was obtained by venipuncture of healthy volunteers after signature of informed consent, and approval of the study by the Ethics Committee of the University of Bonn (Protocol #282/17), and in accordance with the Declaration of Helsinki.
Malaria patients naturally infected with Plasmodium vivax in the Amazon area of Cuiaba (Mato Grosso, Brazil) were invited to participate in the study. Seventy-eight individuals (aged 18 - 78 years old) who sought care at the Julio Muller Hospital, whose thick blood smear was positive for P. vivax were included. Another 9 healthy volunteers from the same endemic area who tested negative for Plasmodium infection were recruited and served as healthy donor controls. Exclusion criteria included: (i) refuse or inability to sign the informed consent; (ii) age < 18 years; (ii) pregnant women; (ii) mixed infection with P. falciparum or P. malariae, tested by both microscopic examination and a nested-PCR; (iv) any other comorbiditythat could be traced. Clinical and demographical data were acquired through a standardized questionnaire, and the hematological profiles were assessed by automated complete blood count carried out at the site hematology facility. Plasma samples were isolated immediately after blood sampling and stored at −80°C until use. The study was approved by the Ethical Review Board of the René Rachou Research Center, FIOCRUZ, Brazilian Ministry of Health (Reporter CEPSH/CPqRR N. 05/2008 and N. 01/2018).
All participants were instructed about the objectives of the study and signed an informed consent in accordance with guidelines for human research, as specified by the Brazilian National Council of Health (Resolution 196/96). Patients diagnosed with P. vivax malaria were treated according to the standard protocols recommended by the National Malaria Control Program (chloroquine + primaquine).
Mice
Mice were housed under standard conditions at 22°C and a 12 h light-dark cycle with free access to food and water. Animal care, handling and experimentation was approved by the ethical committee in Bonn: LANUV-NRW #84-02.04.2016.A487), and in Ribeirão Preto, São Paulo (149/2019). Male 8 – 12 week old C57BL/6j mice were used for experimentation. Male and female 8 week old C57BL/6j, Nlrp3–/–, Asc–/–, Casp-1–/–, Il1b–/–, IL1r–/–, IL18r–/–, Tsp1–/–, Cd36–/–, Cxcr3–/–, Ccr5–/–, and ASC-mCitrine Tg mice were used as source of primary cells.
Cell lines
The monocytic cell line THP-1 (ATCC TIB-202), and the megakaryocytic cell line MEG-01 (ATCC CRL-2021) were cultured in RPMI-1640 medium supplemented with 10% FBS, 1% Penicillin/Streptomycin and GlutaMAX (1x) at 37°C with 5% CO2.
Methods Details
Data Presentation
Unless indicated otherwise (in Figure legends), all graphs are represented as Floating Bars (with mean, and minimum to maximum values) and are built from pooled data from a minimum of two independent experiments (biological replicates), performed in triplicates (technical replicates) with immune cells or platelets from different donors. For experiments performed less than 3 times, bar graphs with mean and standard deviation (SD) are shown. Each symbol represents the average from 3 technical replicates per each donor, experiment, or mouse. Symbols are coded (●, □, ▴, ▾, etc.) to indicate donors so readers can track the internal variability between different donors, mice, or experiments. Dots are semi-transparent, with darker symbols indicating overlapping points.
Generation of human primary macrophages
Buffy coats from healthy donors were obtained according to protocols accepted by the institutional review board at the University of Bonn (local ethics votes Lfd. Nr. 075/14). Primary human macrophages were obtained through differentiation of CD14+ monocytes in a medium complemented with 500 U ml-1 rhGM-CSF (Immunotools) for 3 days. In brief, human peripheral blood mononuclear cells (PBMCs) were obtained from buffy coats of healthy donors by density gradient centrifugation in Ficoll-Paque PLUS (Healthcare). PBMCs were incubated at 4°C with magnetic microbeads conjugated to monoclonal anti-human CD14 antibodies according to the manufacturer’s instructions (Miltenyi Biotech). CD14+ monocytes were thereby magnetically labeled and isolated using a MACS column placed in a magnetic field. CD14+monocytes were cultivated in complete medium (RPMI1640 medium with 10% FBS, 1% Penicillin-Streptomycin, 1% GlutaMAX and 1% Sodium Pyruvate) complemented with 500 U ml-1 rhGM-CSF at a concentration of 2x106 ml-1 in 6-well plates to generate monocyte-derived macrophages. Cells were harvested at day 3, counted using a hemocytometer and seeded at a concentration of 1x105/well in complete medium complemented with 125 U ml-1 rhGM-CSF in 96-well flat-bottom plates and incubated overnight for experiments on the next day.
Generation of mouse bone marrow macrophages
Mice were anaesthetized and sacrificed by cervical dislocation. Femur and tibia from hind limbs were removed and the bones were briefly disinfected with 70% ethanol. The bone marrow cavity was flushed with PBS and the cell suspension was filtered through a 70 μm cell strainer before centrifugation at 400 x g for 5 minutes. Cells were resuspended in DMEM supplemented with 20% L929 supernatant and cultured for 6 days to differentiate into macrophages (BMDMs). On day six, cells were harvested using cold PBS containing 5 mM EDTA and 2% FBS and scraping. After centrifugation at 350 x g for 5 minutes, the BMDM were seeded at 1x105/well in DMEM with 20% L929 supernatants in flat-bottom 96-well plates and incubated overnight for experiments on the next day.
Isolation of CD14+ human Monocytes
Venous blood was collected in S-Monovette® K3EDTA tubes and PBMCs were obtained by density gradient centrifugation in Ficoll-Paque PLUS. Monocytes were isolated from PBMCs using the EasySepTM Human Monocyte Isolation Kit according to the manufacturer instructions (STEMCELL Technologies™). PBMCs were washed twice with PBS complemented with 2% FBS and 1mM EDTA before incubated with the supplied monocyte isolation cocktail and the platelet removal cocktail for 5 minutes. Magnetic beads were added to this suspension for another 5 minutes before magnetic separation in an EasySepTM Magnet. After 2.5 minutes of incubation, the enriched suspension was poured into a new tube. The isolated monocytes were counted using a hemocytometer and resuspended in RPMI medium at a concentration of 1x106 ml-1. The purity of the purified monocytes was assessed by flow cytometry using CD14 (monocyte) and CD41 (platelet) markers.
Isolation of Human Neutrophils
Venous blood was collected in S-Monovette® K3EDTA tubes and neutrophils were isolated using the EasySep™ Direct Human Neutrophil Isolation Kit according to the manufacturer instructions (STEMCELL Technologies™). Whole blood was incubated with the Neutrophil Isolation Cocktail and RapidSpheres™ for 5 minutes and diluted with neutrophil isolation buffer (1mM EDTA in PBS). After 5 minutes of incubation in the EasySep™ Magnet, the enriched cell suspension was poured into a new tube and incubated again with RapidSpheresTM for another 5 minutes, followed by a second, and third round of magnetic separation. The obtained neutrophils were counted using a hemocytometer and pelleted by centrifugation at 350 x g for 5 minutes. Cells were resuspended in RPMI-1640 medium supplemented with 10% FBS, 1% GlutaMAX and 1% Penicillin-Streptomycin. Neutrophil suspension was adjusted to 1x106 ml-1 and 100 μL (1x105cells/well) were seeded in a 96-well round-bottom plate. The purity of the purified neutrophils was assessed by flow cytometry using CD66b (neutrophil marker) and CD41 (platelet marker).
Isolation of Human Platelets
Human platelets were isolated as previously described (Alard et al., 2015) with slight modifications. In brief, venous blood was drawn into S-Monovette® 9NC collection tubes. The blood was centrifuged for 5 minutes at 330 x g without brake to obtain platelet-rich plasma (PRP). All following centrifugation steps were performed without brake and in the presence of 200 nM PGE1 to inhibit platelet activation. PRP was transferred to a new tube and diluted 1:1 with phosphate-buffered saline (PBS) to reduce leukocyte contamination and centrifuged for 10 minutes at 240 x g. Platelets were pelleted by centrifugation at 430 x g for 15 minutes and washed once with PBS. Total platelets were counted using a hemocytometer and resuspended in RPMI medium to a concentration of 1x108 ml-1 unless otherwise indicated. The purity of the purified platelets was assessed by flow cytometry using CD45 (leukocyte) and CD41 (platelet) markers.
Isolation of Mouse Platelets
Blood was drawn by puncturing the vena facialis of anaesthetized mice. Blood from mice of the same genotype were pooled in a sterile 5 mL polystyrene tube containing one-sixth blood volume of pre-warmed citrate-dextrose solution (ACD). PRP was prepared by centrifugation at 330 x g for 5 minutes without break. All following centrifugation steps were performed without brake. PRP was transferred to a new tube and diluted in twice as much volume of PIPES/saline/glucose (PSG) buffer with the final concentration of 1.5 μM PGE1. The suspension was centrifuged at 240 x g for 10 minutes to reduce leukocyte and erythrocyte contamination. The supernatant was transferred into a tube with PGE1 in a final concentration of 0.7 μM in PSG buffer. The platelets were pelleted by centrifugation at 1000 x g for 5 min, washed with 1.5 μM PGE1 in PSG buffer. The washed platelets were resuspended in DMEM, counted in a hemocytometer, and the platelet suspension was adjusted to 5x106 ml-1 unless otherwise indicated. Purity and viability of the prepared platelets were assessed by flow cytometry.
Purity Assessment of Cells Populations
Samples of isolated neutrophils and platelets were analyzed for purity, platelet pre-activation and platelet viability after each experiment. Isolated murine and human platelets were activated with 0.5 or 1 U ml-1 thrombin respectively for 30 min. Cells were blocked with 1:10 mouse or human Fc blocking reagent for 10 minutes at room temperature (RT). The samples were stained with fluorochrome-conjugated monoclonal anti-mouse or anti-human Ig antibodies against CD41/CD41, CD62p, CD14, CD45 or CD66b as indicated for 30 minutes in the dark. Cells were washed and resuspended in flow cytometry buffer (1% FBS in PBS) for analysis. Compensation beads (OneComp Beads) and isotype controls were prepared in the same way. Flow cytometry was performed with a Macs Quant® Analyzer10 (Miltenyi Biotech) and analyzed using the Flowtop software (Tree Star). The applied gating strategy was based on doublet discrimination and isotype-matched control antibodies.
Platelet depletion and LPS + R848 model
Platelet depletion was performed in male, 6-10 week-old C57BL/6j mice by intravenous injection of an anti-CD42b (GPIb-alpha) monoclonal antibody or its isotype IgG control (Emfret Analytics, 2 μg/g animal, 100 μL). 6 hours after injection, a drop of blood was taken from the tail vein to confirm the efficiency of platelet depletion by flow cytometry. Next, the animals were intraperitoneally injected with 200 μg of R848 (Resiquimod, Invivogen) or a combination of 200 μg of R848 and 200 μg of LPS (E.coli O111:B4, Invivogen) in equal volumes (100 μL) as described (Kanneganti et al., 2006). The collection of samples was conducted 6 hours after the inflammatory stimuli. Blood was centrifuged for 10 minutes at 5000 rpm, 4°C for collection of serum. Peritoneal lavage was centrifuged for 5 minutes at 500 g, 4°C and the cells from the peritoneal lavage were fixed and stained for phenotyping. Blood and cell-free peritoneal exudate were used for the cytokine evaluation (R&D Systems).
Immuno-staining of Whole Bone Marrow Cells
Wild-type, ASC-mCitine Tg or ASC-mCherry knock in reporter mice were anesthetized with isoflurane and sacrificed by cervical dislocation. Femurs and tibias from hind limbs were removed and the bones were briefly disinfected with 70% ethanol. The bone marrow cavity was flushed with PBS and the cell suspension was filtered through a 70 μm cell strainer before centrifugation (400 x g, 5 minutes). Cells were resuspended in staining buffer and blocked with 1:10 mouse FcR blocking reagent for 10 minutes at RT before staining with fluorochrome-conjugated anti-mouse CD41 (platelet marker), Ly6G (neutrophil marker), CD14 (monocyte marker) or CD45 (leukocyte marker) (1:5 dilution each). Staining was performed for 30 min at room temperature. Cells were washed twice in PBS and resuspended in staining buffer before flow cytometry analysis or confocal microscopy imaging. Compensation beads (OneComp eBeads) and isotype controls were prepared in the same way. Flow cytometry was performed on MacsQuant® VYB and analyzed using FlowJo. The gating strategy was based on doublet discrimination and isotype-matched control antibodies or unstained controls.
Generation of Platelet and MK supernatants
After platelet isolation, the cell suspension was adjusted to 5x107 platelets ml-1 (for human experiments) or 5x106 platelets ml-1 (for mouse BMDM experiments), in order to fit the macrophage:platelets proportion of 1:50 (human) or 1:5 (mouse). For the generation of supernatants from the megakaryocytic cell line MEG-01, 1x106 cells ml-1 were used. Next, platelets or MEG-01 were left untreated (Unstim) or incubated at 37°C with LPS (200 ng ml-1) or thrombin (1.0 U ml-1) for 3 hours. After stimulation, the platelet cell suspension was centrifuged for 10 minutes at 3000 x g for the generation of the cell-free supernatants. MEG-01 cells were first centrifuged for 7 minutes at 170 x g and the obtained supernatants were centrifuged again for 10 minutes at 3000 x g. The absence of cells was confirmed by microscopic visualization in a hemocytometer. The supernatants were immediately used to stimulate hMDMs in RPMI, or immediately frozen at −80°C until further use.
Stimulation of THP-1 and MEG-01 cells
For stimulation assays, THP-1 cells were treated overnight with 100 nM phorbol 12-myristate 13-acetate, then were primed for 2 h with 1 μg ml-1 of LPS and were subsequently activated with 10 μM of Nigericin.
Stimulation assays
Seeded BMDMs, human macrophages or neutrophils were centrifuged at 350 x g for 5 minutes prior to replacing the supernatant by fresh, serum-free DMEM (for BMDMs) or RPMI (for human neutrophils and macrophages) as control or platelet suspension. For NLRP3 stimulation, cells were primed with 200 ng ml-1 LPS for 3 hours and activated with 10 μM Nigericin or 5 mM ATP for 90 minutes unless otherwise indicated. Human monocytes were primed with 2 ng ml-1 of LPS before being activated with Nigericin (10 μM, 90 min).
For the stimulation of the NLRC4 inflammasome, human macrophages were primed with 200 ng ml-1 LPS for 3 hours before 2 μg ml-1 LFn-PrgI and 0.5 μg ml-1 PA (from Bacillus anthracis) were added to the culture medium for 2 hours (Zhao et al., 2011).
For the stimulation of the AIM2 inflammasome, human GM-CSF-derived macrophages were primed with LPS (200 ng ml-1, 3 hours), followed by stimulation with transfected Poly(dA:dT). Briefly, vesicles were formed using a 1:50 dilution of the transfection agent Lipofectamine 2000 in OptiMEM. After 15 minutes of incubation, the empty vesicles were combined in 1:1 proportion of 4 μg ml-1 Poly(dA:dT), and further incubated for 15 minutes. The dsDNA-containing vesicles were added directly on the cell culture for a final volume of 100 μl of medium (final stimulatory concentration of 1 μg ml-1). Cells were stimulated for 3 hours before cell-free supernatants were harvested for cytokine measurements.
R848 and Pam3Cysk4 were also used for priming in some experiments at 10 μM and 1 μg ml-1 respectively. After stimulation, cells were centrifuged and cell-free supernatants were collected to measure cytokine levels by HTRF. In experiments where the activity of COX1/2 and LOX was inhibited, platelets were incubated with Aspirin, or Zileuton (both at 100 μM) for 60 min at 37°C, before their addition to hMDMs. SB-431542 (10 μM) was added to the macrophage culture 60 minutes before stimulation. The drugs were diluted in RPMI 1640, without supplements.
Trans-well assays
HMDMs were seeded at a concentration of 1 × 105/100 μL in HTS 0.4 μm Trans-well 96-well plates on the day prior to the experiment. Platelets were added to the upper wells at a concentration of 6.25 × 107 in 80 μL of DMEM. After addition of platelets, inflammasome activation was carried out as described above and the levels of IL-1β and TNFα were assessed in cell-free supernatants by HTRF.
Calcium chelation experiments
The calcium chelator BAPTA or the cell-permeable calcium chelator BAPTA-AM (see STAR Methods) were used to examine extracellular and intracellular calcium involvement respectively. hMDMs were seeded in RPMI medium (for BAPTA-AM treatment) or in calcium-free medium (for BAPTA treatment) with or without platelets as described before. Cells were primed with 200 ng ml-1 LPS for 3 hours before the addition of either 0.5 mM BAPTA or 5 μM BAPTA -AM. Then, cells were stimulated with 10 μM Nigericin for NLRP3 Inflammasome activation. Cell-free supernatants were analyzed afterward by HTRF for cytokine secretion.
Quantitative PCR
Total RNA containing small RNAs from purified human platelets, or PBMCs was purified using the miRNeasy kit (QIAGEN), DNA was digested with DNase I (QIAGEN), and cDNA was generated by using 500 ng RNA with SuperScript III Reverse Transcriptase kit (Thermo Fisher) following the manufacturer’s instructions. cDNA was diluted (1:10 for PBMCs, and 1:3 for platelets) and qPCR was performed with Maxima SYBR Green on a QuantStudio 6 Flex RT PCR machine (Thermo Fisher) for 40 cycles and followed by a melt curve analysis for off-target products. The primers used were ACTB-fwd-ccaccatgtaccctggcatt, ACTB-rev-cggagtacttgcgctcagga, NLRP3-fwd-tcggagacaaggggatcaaa, NLRP3-rev-agcagcagtgtgacgtgagg, CD14-fwd-gagctcagaggttcggaaga, CD14-rev-cttcatcgtccagctcacaa, PYCARD-fwd-gagctcaccgctaacgtgct, PYCARD-rev-actgaggaggggcctggat, PF4-fwd-ctgaagaagatggggacctg, PF4-rev-gtggctatcagttgggcagt, CASP1-fwd-acaacccagctatgcccaca, CASP1-rev-gtgcggcttgacttgtccat, GP1BA-fwd-ctgctctttgcctctgtggt, GP1BA-rev-ctccaggtgtgtggtttgtg, IL1B-fwd-tgggcagactcaaattccagct, IL1B-rev-ctgtacctgtcctgcgtgttga.
Imaging and quantification of ASC specks
To examine ASC specking, hMDMs were seeded at a concentration of 2 × 105/well in 8-well IBIDI μ-slide one day prior to the experiment. Freshly isolated platelets were added to hMDMs on the day of the experiment at a concentration of 5 × 107 ml-1. Cells were stimulated with 200 ng ml-1 LPS for 3 hours and the NLRP3 inflammasome was activated with 10 μM Nigericin for 45 minutes. Afterward, the plate was centrifuged (500 x g, 5 minutes) and the cells were fixed with 4% paraformaldehyde in PBS for 10 minutes on ice. After two washes with PBS, cells were blocked with 1:10 human FcR blocking reagent for 10 minutes at room temperature and stained with directly labeled anti-ASC-647 or the same amount of directly labeled IgG1 control and incubated overnight at 4°C in the dark (section 3.1.6). On the next day, cells were washed twice with permeabilization buffer (500 x g, 5 minutes) before staining with the DNA dye Hoechst (1:3000 dilution) in PBS for 10 minutes at RT in the dark. After two subsequent washes with PBS, cells were resuspended in PBS and imaged using an Observer.Z1 epifluorescence microscope, 20 × objective (dry, PlanApochromat, NA 0.8; ZEISS, Oberkochen, Germany), Axiocam 506 mono, and ZEN Blue software (ZEISS, Oberkochen, Germany). Representative images were taken using a Leica TCS SP5 SMD confocal system. The quantification of ASC specks per nuclei was carried out using the image analyzer software CellProfiler 3.0 (Open source software: https://cellprofiler.org). A pipeline was constructed to convert the fluorescent images into binary code. Through this conversion, the nuclei and ASC specks were identified and quantified. ASC specks were identified using the EnhanceFeature tool.
Cytokine measurements
Levels of human, or murine IL-1β, IL-6, and TNFα in cell-free supernatants were quantified using commercially available HTRF® (homogeneous time-resolved fluorescence) kits. The HTRF was performed according to manufacturer instructions (Cisbio). Multiplex Cytokine array was used for the detection of several human cytokines and growth factors according to the manufacturer’s instructions (Thermo Fisher).
Caspase-1 activity
Caspase-Glo® 1 Inflammasome Assay (Promega) provides a luminogenic caspase-1 substrate, Z-WEHD-amino luciferin, in a lytic reagent optimized for caspase-1 activity and luciferase activity. The reaction buffer was mixed with the cell-free supernatant and the luminescent signal was evaluated 30 minutes later.
Lactate dehydrogenase (LDH) assay
The extracellular lactate dehydrogenase (LDH) concentrations in cell-free supernatants were assessed using the Pierce LDH Cytotoxicity Assay, according to the manufacturer’s instructions. Samples were measured at an absorbance of 490 nm using a Spectramax i3 plate reader (ThermoFisher). Control hMDMs cultured at the same conditions as the samples were lysed with the provided Lysis buffer (10x) and served as positive control for maximal LDH release.
Confocal laser scanning microscopy
Platelets and immune cells were imaged in a Leica TCS SP5 SMD confocal system (Leica Microsystems, Wetzlar, Germany). Images were acquired using a 63X objective, with a numerical aperture of 1.2, and analyzed using the Volocity 6.01 software (PerkinElmer, Waltham, Massachusetts, U.S.A.).
Meta-analysis of microarray data
Pre-processed microarray data of platelets and whole peripheral blood cells under steady state and certain disease states (GSE2006, GSE11524, GSE10361, GSE50858, GSE47018, GSE17924) were down-loaded from the Gene Expression Omnibus (GEO) data base (https://www.ncbi.nlm.nih.gov/geo/). Expression values for the house-keeping gene GAPDH and platelet- as well as inflammasome-associated genes (CXCL4/PFA4, PDGFA, IL1B, PYCARD, NLRP3, TIMP1 CASP1, ARG2, TP2A, SELENB1) were extracted for each dataset. In case multiple probe sets for GAPDH were present on the microarray chip, a mean expression value for GAPDH was calculated. The expression values for probe sets of platelet and inflammasome-associated genes were normalized for each dataset according to the respective GAPDH mean expression value. Log2 transformed normalized expression data were plotted as bar charts by the Tidyverse package in R (v3.4.2).
Proteomics (LC-MS)
Suspensions of freshly isolated human platelets were adjusted to a concentration of 5x107 ml-1 in RPMI. Platelets were left untreated, or stimulated with 200 ng ml-1 LPS, 1 U ml-1 Thrombin for 3 hours. Platelets were pelleted by centrifugation (3000 x g, 10 minutes) and the cell-free supernatants were harvested. Cell-free supernatants were added with 1x complete protease inhibitor cocktail prior to freezing at −80°C. Proteomics analysis was carried out at the CECAD/CMMC Proteomics Core Facility (University Cologne, Germany) on a Q Exactive Plus Orbitrap (Thermo Scientific) mass spectrometer that was coupled to an EASY-nLC (Thermo Scientific). Briefly, peptides were loaded in 0.1% formic acid in water onto an in-house packed analytical column (50 cm ó 75 μm I.D., filled with 2.7 μm Poroshell EC120 C18, Agilent) and were chromatographically separated at a constant flow rate of 250 nl/minute with the following gradient: 3%–4% solvent B (0.1% formic acid in 80 % acetonitrile) within 1 minute, 4%–27% solvent B within 119 minute, 27%–50% solvent B within 19 minutes, 50%–95% solvent B within 1 minutes, followed by washing and equilibration of the columns. The mass spectrometer was operated in data-dependent acquisition mode. All mass spectrometric raw data were processed by the CECAD/CMMC Proteomics Core Facility using Maxquant (version 1.5.3.8) with default parameters. Briefly, MS2 spectra were analyzed against the Uniprot HUMAN. fasta (downloaded at: 16.6.2017) database, including a list of common contaminants. False discovery rates on protein and PSM level were estimated by the target-decoy approach to 1% FDR for both. The minimal peptide length was determined to be 7 amino acids and carbamidomethylation at cysteine residues was considered as a fixed modification. Oxidation and acetylation were included as variable modifications. For the analysis, the match-between runs option was enabled. Label-free quantification (LFQ) was activated using default settings. Figures were assembled using the R Tidyverse package. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [1] partner repository with the dataset identifier PXD017976.
Quantification and Statistical Analysis
Statistical analyses were performed with GraphPad Prism Version 7.0f. All statistical analysis were preceded by Normality tests, followed by the recommended parametric or nonparametric tests. For most in vitro experiments with several groups (hMDMs alone versus with Platelets, stimulated or not), P values were determined by two-way ANOVA with Tukey’s or Sidak’s or multiple comparison tests. For in vivo experiments, in which two groups of mice are compared (n = 6/group), (Figure 3) we used two-tailed Unpaired t test to calculate differences. Correlations between cytokines and cell counts were computed with Two-tail Spearman. Outliers were detected using the ROUT method, and were excluded from the statistical analysis, but not removed from the figures. Additional statistical details are given in the respective figure legends, when appropriate.
Acknowledgments
We are grateful to Cornelia Rohland, Cleyson O. Barros, Vanessa Borges, and Juliana E. Toller-Kawahisa for experimental assistance and Matthew S. Mangan for helpful discussions. We thank Feng Shao, Matthias Geyer, and David Fußhöller for the reagents and purified proteins to activate NLRC4. We also thank the Microscopy Core Facility (Medical Faculty of University Bonn) for their instruments and services. This study was funded by the European Research Council (PLAT-IL-1, 714175). B.S.F. and E.L. are further supported by grants from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) (SFBTRR57) and the DFG Germany's Excellence Strategy - EXC 2151 - 390873048.
Author Contributions
Conceptualization, B.S.F.; Investigation, B.S.F., L.S.R., V.R., I.H., L.B., N.R., S.M., M.L.S.S., M.R., H.J.S., C.M.d.S.S., and C.W.d.S.W.; Data Analysis, B.S.F. and S.V.S.; Resources, B.S.F., F.d.Q.C., M.A., C.J.F., T.P., and E.L.; Software, B.S.F. and S.V.S.; Writing – Original Draft, B.S.F.; Revisions, M.A., M.N.R., V.R., D.B., and L.S.R.; Visualization, M.A., V.R., L.S.R., and I.H.; Supervision, B.S.F. and L.H.C.; Project Administration, B.S.F.; Funding Acquisition, B.S.F.
Declaration of Interests
E.L. is co-founder and consultant of IFM Therapeutics.
Published: May 12, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107615.
Contributor Information
Lucas Secchim Ribeiro, Email: ribeiro@uni-bonn.de.
Moshe Arditi, Email: moshe.arditi@cshs.org.
Bernardo Simoes Franklin, Email: franklin@uni-bonn.de.
Supplemental Information
References
- Alard J.-E., Ortega-Gomez A., Wichapong K., Bongiovanni D., Horckmans M., Megens R.T.A., Leoni G., Ferraro B., Rossaint J., Paulin N. Recruitment of classical monocytes can be inhibited by disturbing heteromers of neutrophil HNP1 and platelet CCL5. Sci. Transl. Med. 2015;7:317ra196. doi: 10.1126/scitranslmed.aad5330. [DOI] [PubMed] [Google Scholar]
- Allam O., Samarani S., Jenabian M.-A., Routy J.-P., Tremblay C., Amre D., Ahmad A. Differential synthesis and release of IL-18 and IL-18 Binding Protein from human platelets and their implications for HIV infection. Cytokine. 2017;90:144–154. doi: 10.1016/j.cyto.2016.10.016. [DOI] [PubMed] [Google Scholar]
- Allen N., Barrett T.J., Guo Y., Nardi M., Ramkhelawon B., Rockman C.B., Hochman J.S., Berger J.S. Circulating monocyte-platelet aggregates are a robust marker of platelet activity in cardiovascular disease. Atherosclerosis. 2019;282:11–18. doi: 10.1016/j.atherosclerosis.2018.12.029. [DOI] [PubMed] [Google Scholar]
- Andonegui G., Kerfoot S.M., McNagny K., Ebbert K.V., Patel K.D., Kubes P. Platelets express functional Toll-like receptor-4. Blood. 2005;106:2417–2423. doi: 10.1182/blood-2005-03-0916. [DOI] [PubMed] [Google Scholar]
- Andrade B.B., Reis-Filho A., Souza-Neto S.M., Clarêncio J., Camargo L.M.A., Barral A., Barral-Netto M. Severe Plasmodium vivax malaria exhibits marked inflammatory imbalance. Malar. J. 2010;9:13. doi: 10.1186/1475-2875-9-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aslam R., Speck E.R., Kim M., Crow A.R., Bang K.W., Nestel F.P., Ni H., Lazarus A.H., Freedman J., Semple J.W. Platelet Toll-like receptor expression modulates lipopolysaccharide-induced thrombocytopenia and tumor necrosis factor-alpha production in vivo. Blood. 2006;107:637–641. doi: 10.1182/blood-2005-06-2202. [DOI] [PubMed] [Google Scholar]
- Badrnya S., Schrottmaier W.C., Kral J.B., Yaiw K.-C., Volf I., Schabbauer G., Söderberg-Nauclér C., Assinger A. Platelets mediate oxidized low-density lipoprotein-induced monocyte extravasation and foam cell formation. Arterioscler. Thromb. Vasc. Biol. 2014;34:571–580. doi: 10.1161/ATVBAHA.113.302919. [DOI] [PubMed] [Google Scholar]
- Balow J.E.J., Jr., Ryan J.G., Chae J.J., Booty M.G., Bulua A., Stone D., Sun H.-W., Greene J., Barham B., Goldbach-Mansky R. Microarray-based gene expression profiling in patients with cryopyrin-associated periodic syndromes defines a disease-related signature and IL-1-responsive transcripts. Ann. Rheum. Dis. 2013;72:1064–1070. doi: 10.1136/annrheumdis-2012-202082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett T.J., Schlegel M., Zhou F., Gorenchtein M., Bolstorff J., Moore K.J., Fisher E.A., Berger J.S. Platelet regulation of myeloid suppressor of cytokine signaling 3 accelerates atherosclerosis. Sci. Transl. Med. 2019;11:eaax0481. doi: 10.1126/scitranslmed.aax0481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beilharz M., De Nardo D., Latz E., Franklin B.S. Measuring NLR Oligomerization II: Detection of ASC Speck Formation by Confocal Microscopy and Immunofluorescence. Methods Mol. Biol. 2016;1417:145–158. doi: 10.1007/978-1-4939-3566-6_9. [DOI] [PubMed] [Google Scholar]
- Bertheloot D., Latz E. HMGB1, IL-1α, IL-33 and S100 proteins: dual-function alarmins. Cell. Mol. Immunol. 2017;14:43–64. doi: 10.1038/cmi.2016.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boilard E., Nigrovic P.A., Larabee K., Watts G.F.M., Coblyn J.S., Weinblatt M.E., Massarotti E.M., Remold-O’Donnell E., Farndale R.W., Ware J., Lee D.M. Platelets amplify inflammation in arthritis via collagen-dependent microparticle production. Science. 2010;327:580–583. doi: 10.1126/science.1181928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonar S.L., Brydges S.D., Mueller J.L., McGeough M.D., Pena C., Chen D., Grimston S.K., Hickman-Brecks C.L., Ravindran S., McAlinden A. Constitutively activated NLRP3 inflammasome causes inflammation and abnormal skeletal development in mice. PLoS ONE. 2012;7:e35979. doi: 10.1371/journal.pone.0035979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broderick L., De Nardo D., Franklin B.S., Hoffman H.M., Latz E. The inflammasomes and autoinflammatory syndromes. Annu. Rev. Pathol. 2015;10:395–424. doi: 10.1146/annurev-pathol-012414-040431. [DOI] [PubMed] [Google Scholar]
- Brown G.T., McIntyre T.M. Lipopolysaccharide signaling without a nucleus: kinase cascades stimulate platelet shedding of proinflammatory IL-1β-rich microparticles. J. Immunol. 2011;186:5489–5496. doi: 10.4049/jimmunol.1001623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown G.T., Narayanan P., Li W., Silverstein R.L., McIntyre T.M. Lipopolysaccharide stimulates platelets through an IL-1β autocrine loop. J. Immunol. 2013;191:5196–5203. doi: 10.4049/jimmunol.1300354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns J.C., Koné-Paut I., Kuijpers T., Shimizu C., Tremoulet A., Arditi M. Review: Found in Translation: International Initiatives Pursuing Interleukin-1 Blockade for Treatment of Acute Kawasaki Disease. Arthritis Rheumatol. 2017;69:268–276. doi: 10.1002/art.39975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burzynski L.C., Humphry M., Pyrillou K., Wiggins K.A., Chan J.N.E., Figg N., Kitt L.L., Summers C., Tatham K.C., Martin P.B. The Coagulation and Immune Systems Are Directly Linked through the Activation of Interleukin-1α by Thrombin. Immunity. 2019;50:1033–1042. doi: 10.1016/j.immuni.2019.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cailotto F., Reboul P., Sebillaud S., Netter P., Jouzeau J.-Y., Bianchi A. Calcium input potentiates the transforming growth factor (TGF)-beta1-dependent signaling to promote the export of inorganic pyrophosphate by articular chondrocyte. J. Biol. Chem. 2011;286:19215–19228. doi: 10.1074/jbc.M110.175448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carestia A., Mena H.A., Olexen C.M., Ortiz Wilczyñski J.M., Negrotto S., Errasti A.E., Gómez R.M., Jenne C.N., Carrera Silva E.A., Schattner M. Platelets Promote Macrophage Polarization toward Pro-inflammatory Phenotype and Increase Survival of Septic Mice. Cell Rep. 2019;28:896–908.e5. doi: 10.1016/j.celrep.2019.06.062. [DOI] [PubMed] [Google Scholar]
- Chandra G., Cogswell J.P., Miller L.R., Godlevski M.M., Stinnett S.W., Noel S.L., Kadwell S.H., Kost T.A., Gray J.G. Cyclic AMP signaling pathways are important in IL-1 beta transcriptional regulation. J. Immunol. 1995;155:4535–4543. [PubMed] [Google Scholar]
- Chatterjee M., von Ungern-Sternberg S.N., Seizer P., Schlegel F., Büttcher M., Sindhu N.A., Müller S., Mack A., Gawaz M. Platelet-derived CXCL12 regulates monocyte function, survival, differentiation into macrophages and foam cells through differential involvement of CXCR4-CXCR7. Cell Death Dis. 2015;6:e1989. doi: 10.1038/cddis.2015.233. e1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccarelli F., De Martinis M., Ginaldi L. An update on autoinflammatory diseases. Curr. Med. Chem. 2014;21:261–269. doi: 10.2174/09298673113206660303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cognasse F., Hamzeh-Cognasse H., Lafarge S., Delezay O., Pozzetto B., McNicol A., Garraud O. Toll-like receptor 4 ligand can differentially modulate the release of cytokines by human platelets. Br. J. Haematol. 2008;141:84–91. doi: 10.1111/j.1365-2141.2008.06999.x. [DOI] [PubMed] [Google Scholar]
- Coppinger J.A., Cagney G., Toomey S., Kislinger T., Belton O., McRedmond J.P., Cahill D.J., Emili A., Fitzgerald D.J., Maguire P.B. Characterization of the proteins released from activated platelets leads to localization of novel platelet proteins in human atherosclerotic lesions. Blood. 2004;103:2096–2104. doi: 10.1182/blood-2003-08-2804. [DOI] [PubMed] [Google Scholar]
- Cornelius D.C., Baik C.H., Travis O.K., White D.L., Young C.M., Austin Pierce W., Shields C.A., Poudel B., Williams J.M. NLRP3 inflammasome activation in platelets in response to sepsis. Physiol. Rep. 2019;7:e14073. doi: 10.14814/phy2.14073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford S.E., Stellmach V., Murphy-Ullrich J.E., Ribeiro S.M., Lawler J., Hynes R.O., Boivin G.P., Bouck N. Thrombospondin-1 is a major activator of TGF-beta1 in vivo. Cell. 1998;93:1159–1170. doi: 10.1016/s0092-8674(00)81460-9. [DOI] [PubMed] [Google Scholar]
- Damien P., Cognasse F., Eyraud M.-A., Arthaud C.-A., Pozzetto B., Garraud O., Hamzeh-Cognasse H. LPS stimulation of purified human platelets is partly dependent on plasma soluble CD14 to secrete their main secreted product, soluble-CD40-Ligand. BMC Immunol. 2015;16:3. doi: 10.1186/s12865-015-0067-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dann R., Hadi T., Montenont E., Boytard L., Alebrahim D., Feinstein J., Allen N., Simon R., Barone K., Uryu K. Platelet-Derived MRP-14 Induces Monocyte Activation in Patients With Symptomatic Peripheral Artery Disease. J. Am. Coll. Cardiol. 2018;71:53–65. doi: 10.1016/j.jacc.2017.10.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denis M.M., Tolley N.D., Bunting M., Schwertz H., Jiang H., Lindemann S., Yost C.C., Rubner F.J., Albertine K.H., Swoboda K.J. Escaping the nuclear confines: signal-dependent pre-mRNA splicing in anucleate platelets. Cell. 2005;122:379–391. doi: 10.1016/j.cell.2005.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devignot S., Sapet C., Duong V., Bergon A., Rihet P., Ong S., Lorn P.T., Chroeung N., Ngeav S., Tolou H.J. Genome-wide expression profiling deciphers host responses altered during dengue shock syndrome and reveals the role of innate immunity in severe dengue. PLoS ONE. 2010;5:e11671. doi: 10.1371/journal.pone.0011671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giovine F.S., Malawista S.E., Nuki G., Duff G.W. Interleukin 1 (IL 1) as a mediator of crystal arthritis. Stimulation of T cell and synovial fibroblast mitogenesis by urate crystal-induced IL 1. J. Immunol. 1987;138:3213–3218. [PubMed] [Google Scholar]
- Dinarello C.A. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood. 2011;117:3720–3732. doi: 10.1182/blood-2010-07-273417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello C.A., van der Meer J.W.M. Treating inflammation by blocking interleukin-1 in humans. Semin. Immunol. 2013;25:469–484. doi: 10.1016/j.smim.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello C.A., Simon A., van der Meer J.W.M. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat. Rev. Drug Discov. 2012;11:633–652. doi: 10.1038/nrd3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dostert C., Guarda G., Romero J.F., Menu P., Gross O., Tardivel A., Suva M.-L., Stehle J.-C., Kopf M., Stamenkovic I. Malarial hemozoin is a Nalp3 inflammasome activating danger signal. PLoS ONE. 2009;4:e6510. doi: 10.1371/journal.pone.0006510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du R.-H., Tan J., Sun X.-Y., Lu M., Ding J.-H., Hu G. Fluoxetine Inhibits NLRP3 Inflammasome Activation: Implication in Depression. Int. J. Neuropsychopharmacol. 2016;19:pyw037. doi: 10.1093/ijnp/pyw037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eicher J.D., Wakabayashi Y., Vitseva O., Esa N., Yang Y., Zhu J., Freedman J.E., McManus D.D., Johnson A.D. Characterization of the platelet transcriptome by RNA sequencing in patients with acute myocardial infarction. Platelets. 2016;27:230–239. doi: 10.3109/09537104.2015.1083543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng X., Scheinberg P., Samsel L., Rios O., Chen J., McCoy J.P., Jr., Ghanima W., Bussel J.B., Young N.S. Decreased plasma cytokines are associated with low platelet counts in aplastic anemia and immune thrombocytopenic purpura. J. Thromb. Haemost. 2012;10:1616–1623. doi: 10.1111/j.1538-7836.2012.04757.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaidt M.M., Ebert T.S., Chauhan D., Schmidt T., Schmid-Burgk J.L., Rapino F., Robertson A.A.B., Cooper M.A., Graf T., Hornung V. Human Monocytes Engage an Alternative Inflammasome Pathway. Immunity. 2016;44:833–846. doi: 10.1016/j.immuni.2016.01.012. [DOI] [PubMed] [Google Scholar]
- Franklin B.S., Bossaller L., De Nardo D., Ratter J.M., Stutz A., Engels G., Brenker C., Nordhoff M., Mirandola S.R., Al-Amoudi A., Mangan M.S., Zimmer S., Monks B.G., Fricke M., Schmidt R.E., Espevik T., Jones B., Jarnicki A.G., Hansbro P.M., Busto P., Marshak-Rothstein A., Hornemann S., Aguzzi A., Kastenmüller W., Latz E. The adaptor ASC has extracellular and “prionoid” activities that propagate inflammation. Nat. Immunol. 2014;15:727–737. doi: 10.1038/ni.2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freishtat R.J., Natale J., Benton A.S., Cohen J., Sharron M., Wiles A.A., Ngor W.-M., Mojgani B., Bradbury M., Degnan A., Sachdeva R., Debiase L.M., Ghimbovschi S., Chow M., Bunag C., Kristosturyan E., Hoffman E.P. Sepsis alters the megakaryocyte-platelet transcriptional axis resulting in granzyme B-mediated lymphotoxicity. Am. J. Respir. Crit. Care Med. 2009;179:467–473. doi: 10.1164/rccm.200807-1085OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaidt M.M., Ebert T.S., Chauhan D., Ramshorn K., Pinci F., Zuber S., O’Duill F., Schmid-Burgk J.L., Hoss F., Buhmann R. The DNA Inflammasome in Human Myeloid Cells Is Initiated by a STING-Cell Death Program Upstream of NLRP3. Cell. 2017;171:1110–1124.e18. doi: 10.1016/j.cell.2017.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnatenko D.V., Cupit L.D., Huang E.C., Dhundale A., Perrotta P.L., Bahou W.F. Platelets express steroidogenic 17beta-hydroxysteroid dehydrogenases. Distinct profiles predict the essential thrombocythemic phenotype. Thromb. Haemost. 2005;94:412–421. doi: 10.1160/TH05-01-0037. [DOI] [PubMed] [Google Scholar]
- Goldbach-Mansky R., Dailey N.J., Canna S.W., Gelabert A., Jones J., Rubin B.I., Kim H.J., Brewer C., Zalewski C., Wiggs E. Neonatal-onset multisystem inflammatory disease responsive to interleukin-1beta inhibition. N. Engl. J. Med. 2006;355:581–592. doi: 10.1056/NEJMoa055137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudbrandsdottir S., Hasselbalch H.C., Nielsen C.H. Activated platelets enhance IL-10 secretion and reduce TNF-α secretion by monocytes. J. Immunol. 2013;191:4059–4067. doi: 10.4049/jimmunol.1201103. [DOI] [PubMed] [Google Scholar]
- Hawrylowicz C.M., Santoro S.A., Platt F.M., Unanue E.R. Activated platelets express IL-1 activity. J. Immunol. 1989;143:4015–4018. [PubMed] [Google Scholar]
- Hinz C., Aldrovandi M., Uhlson C., Marnett L.J., Longhurst H.J., Warner T.D., Alam S., Slatter D.A., Lauder S.N., Allen-Redpath K. Human Platelets Utilize Cycloxygenase-1 to Generate Dioxolane A3, a Neutrophil-activating Eicosanoid. J. Biol. Chem. 2016;291:13448–13464. doi: 10.1074/jbc.M115.700609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hottz E.D., Lopes J.F., Freitas C., Valls-de-Souza R., Oliveira M.F., Bozza M.T., Da Poian A.T., Weyrich A.S., Zimmerman G.A., Bozza F.A., Bozza P.T. Platelets mediate increased endothelium permeability in dengue through NLRP3-inflammasome activation. Blood. 2013;122:3405–3414. doi: 10.1182/blood-2013-05-504449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howes R.E., Battle K.E., Mendis K.N., Smith D.L., Cibulskis R.E., Baird J.K., Hay S.I. Global Epidemiology of Plasmodium vivax. Am. J. Trop. Med. Hyg. 2016;95(6, Suppl):15–34. doi: 10.4269/ajtmh.16-0141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L.-F., Wu T., Wang B., Wei Y.-Y., Kong Q.-X., Ye Y., Yin H.-F., Li J.-B. The Regulation of Seventeen Inflammatory Mediators are Associated with Patient Outcomes in Severe Fever with Thrombocytopenia Syndrome. Sci. Rep. 2018;8:159. doi: 10.1038/s41598-017-18616-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain A., Irizarry-Caro R.A., Chawla A.S., Philip N.H., Carroll K.R., Katz J.D., Oberst A., Chervonsky A.V., Pasare C. T cells instruct dendritic cells to produce inflammasome-independent IL-1β and cause autoimmunity. Nat. Immunol. 2020;21:65–74. doi: 10.1038/s41590-019-0559-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joosten L.A.B., Netea M.G., Fantuzzi G., Koenders M.I., Helsen M.M.A., Sparrer H., Pham C.T., van der Meer J.W.M., Dinarello C.A., van den Berg W.B. Inflammatory arthritis in caspase 1 gene-deficient mice: contribution of proteinase 3 to caspase 1-independent production of bioactive interleukin-1β. Arthritis Rheum. 2009;60:3651–3662. doi: 10.1002/art.25006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanneganti T.-D., Ozören N., Body-Malapel M., Amer A., Park J.-H., Franchi L., Whitfield J., Barchet W., Colonna M., Vandenabeele P. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature. 2006;440:233–236. doi: 10.1038/nature04517. [DOI] [PubMed] [Google Scholar]
- Kaplanski G., Porat R., Aiura K., Erban J.K., Gelfand J.A., Dinarello C.A. Activated platelets induce endothelial secretion of interleukin-8 in vitro via an interleukin-1-mediated event. Blood. 1993;81:2492–2495. [PubMed] [Google Scholar]
- Kayagaki N., Stowe I.B., Lee B.L., O’Rourke K., Anderson K., Warming S., Cuellar T., Haley B., Roose-Girma M., Phung Q.T. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666–671. doi: 10.1038/nature15541. [DOI] [PubMed] [Google Scholar]
- Kimura H., Ishibashi T., Shikama Y., Okano A., Akiyama Y., Uchida T., Maruyama Y. Interleukin-1 beta (IL-1 beta) induces thrombocytosis in mice: possible implication of IL-6. Blood. 1990;76:2493–2500. [PubMed] [Google Scholar]
- Kral J.B., Schrottmaier W.C., Salzmann M., Assinger A. Platelet Interaction with Innate Immune Cells. Transfus. Med. Hemother. 2016;43:78–88. doi: 10.1159/000444807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuda O., Pietka T.A., Demianova Z., Kudova E., Cvacka J., Kopecky J., Abumrad N.A. Sulfo-N-succinimidyl oleate (SSO) inhibits fatty acid uptake and signaling for intracellular calcium via binding CD36 lysine 164: SSO also inhibits oxidized low density lipoprotein uptake by macrophages. J. Biol. Chem. 2013;288:15547–15555. doi: 10.1074/jbc.M113.473298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang D., Dohle F., Terstesse M., Bangen P., August C., Pauels H.-G., Heidenreich S. Down-regulation of monocyte apoptosis by phagocytosis of platelets: involvement of a caspase-9, caspase-3, and heat shock protein 70-dependent pathway. J. Immunol. 2002;168:6152–6158. doi: 10.4049/jimmunol.168.12.6152. [DOI] [PubMed] [Google Scholar]
- Latz E., Xiao T.S., Stutz A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013;13:397–411. doi: 10.1038/nri3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G.-S., Subramanian N., Kim A.I., Aksentijevich I., Goldbach-Mansky R., Sacks D.B., Germain R.N., Kastner D.L., Chae J.J. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature. 2012;492:123–127. doi: 10.1038/nature11588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y., Schulte D.J., Shimada K., Chen S., Crother T.R., Chiba N., Fishbein M.C., Lehman T.J.A., Arditi M. Interleukin-1β is crucial for the induction of coronary artery inflammation in a mouse model of Kawasaki disease. Circulation. 2012;125:1542–1550. doi: 10.1161/CIRCULATIONAHA.111.072769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Qi X., Tong X., Wang S. Thrombospondin 1 activates the macrophage Toll-like receptor 4 pathway. Cell. Mol. Immunol. 2013;10:506–512. doi: 10.1038/cmi.2013.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q., Fu W., Yao J., Ji Z., Wang Y., Zhou Z., Yan J., Li W. Heme induces IL-1β secretion through activating NLRP3 in kidney inflammation. Cell Biochem. Biophys. 2014;69:495–502. doi: 10.1007/s12013-014-9823-9. [DOI] [PubMed] [Google Scholar]
- Lindemann S., Tolley N.D., Dixon D.A., McIntyre T.M., Prescott S.M., Zimmerman G.A., Weyrich A.S. Activated platelets mediate inflammatory signaling by regulated interleukin 1β synthesis. J. Cell Biol. 2001;154:485–490. doi: 10.1083/jcb.200105058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Zhang Z., Ruan J., Pan Y., Magupalli V.G., Wu H., Lieberman J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535:153–158. doi: 10.1038/nature18629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Londin E.R., Hatzimichael E., Loher P., Edelstein L., Shaw C., Delgrosso K., Fortina P., Bray P.F., McKenzie S.E., Rigoutsos I. The human platelet: strong transcriptome correlations among individuals associate weakly with the platelet proteome. Biol. Direct. 2014;9:3. doi: 10.1186/1745-6150-9-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lood C., Tydén H., Gullstrand B., Jönsen A., Källberg E., Mörgelin M., Kahn R., Gunnarsson I., Leanderson T., Ivars F. Platelet-Derived S100A8/A9 and Cardiovascular Disease in Systemic Lupus Erythematosus. Arthritis Rheumatol. 2016;68:1970–1980. doi: 10.1002/art.39656. [DOI] [PubMed] [Google Scholar]
- Mantovani A., Dinarello C.A., Molgora M., Garlanda C. Interleukin-1 and Related Cytokines in the Regulation of Inflammation and Immunity. Immunity. 2019;50:778–795. doi: 10.1016/j.immuni.2019.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcinkiewcz C.A., Mazzone C.M., D’Agostino G., Halladay L.R., Hardaway J.A., DiBerto J.F., Navarro M., Burnham N., Cristiano C., Dorrier C.E. Serotonin engages an anxiety and fear-promoting circuit in the extended amygdala. Nature. 2016;537:97–101. doi: 10.1038/nature19318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariathasan S., Newton K., Monack D.M., Vucic D., French D.M., Lee W.P., Roose-Girma M., Erickson S., Dixit V.M. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature. 2004;430:213–218. doi: 10.1038/nature02664. [DOI] [PubMed] [Google Scholar]
- Mariathasan S., Weiss D.S., Newton K., McBride J., O’Rourke K., Roose-Girma M., Lee W.P., Weinrauch Y., Monack D.M., Dixit V.M. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- Matsunaga N., Tsuchimori N., Matsumoto T., Ii M. TAK-242 (resatorvid), a small-molecule inhibitor of Toll-like receptor (TLR) 4 signaling, binds selectively to TLR4 and interferes with interactions between TLR4 and its adaptor molecules. Mol. Pharmacol. 2011;79:34–41. doi: 10.1124/mol.110.068064. [DOI] [PubMed] [Google Scholar]
- Matsuyama S., Iwadate M., Kondo M., Saitoh M., Hanyu A., Shimizu K., Aburatani H., Mishima H.K., Imamura T., Miyazono K., Miyazawa K. SB-431542 and Gleevec inhibit transforming growth factor-beta-induced proliferation of human osteosarcoma cells. Cancer Res. 2003;63:7791–7798. [PubMed] [Google Scholar]
- Maynard D.M., Heijnen H.F.G., Horne M.K., White J.G., Gahl W.A. Proteomic analysis of platelet alpha-granules using mass spectrometry. J. Thromb. Haemost. 2007;5:1945–1955. doi: 10.1111/j.1538-7836.2007.02690.x. [DOI] [PubMed] [Google Scholar]
- Misenheimer T.M., Mosher D.F. Calcium ion binding to thrombospondin 1. J. Biol. Chem. 1995;270:1729–1733. doi: 10.1074/jbc.270.4.1729. [DOI] [PubMed] [Google Scholar]
- Netea M.G., Joosten L.A.B., Li Y., Kumar V., Oosting M., Smeekens S., Jaeger M., Ter Horst R., Schirmer M., Vlamakis H. Understanding human immune function using the resources from the Human Functional Genomics Project. Nat. Med. 2016;22:831–833. doi: 10.1038/nm.4140. [DOI] [PubMed] [Google Scholar]
- Nishimura S., Nagasaki M., Kunishima S., Sawaguchi A., Sakata A., Sakaguchi H., Ohmori T., Manabe I., Italiano J.E., Jr., Ryu T. IL-1α induces thrombopoiesis through megakaryocyte rupture in response to acute platelet needs. J. Cell Biol. 2015;209:453–466. doi: 10.1083/jcb.201410052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyakundi B.B., Tóth A., Balogh E., Nagy B., Erdei J., Ryffel B., Paragh G., Cordero M.D., Jeney V. Oxidized hemoglobin forms contribute to NLRP3 inflammasome-driven IL-1β production upon intravascular hemolysis. Biochim Biophys Acta Mol Basis Dis. 2019;1865:464–475. doi: 10.1016/j.bbadis.2018.10.030. [DOI] [PubMed] [Google Scholar]
- Packham I.M., Watson S.P., Bicknell R., Egginton S. In vivo evidence for platelet-induced physiological angiogenesis by a COX driven mechanism. PLoS ONE. 2014;9:e107503. doi: 10.1371/journal.pone.0107503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passacquale G., Vamadevan P., Pereira L., Hamid C., Corrigall V., Ferro A. Monocyte-platelet interaction induces a pro-inflammatory phenotype in circulating monocytes. PLoS ONE. 2011;6:e25595. doi: 10.1371/journal.pone.0025595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillitteri D., Bassus S., Boller K., Mahnel R., Scholz T., Westrup D., Wegert W., Kirchmaier C.M., Kirchmaier C.M. Thrombin-induced interleukin 1β synthesis in platelet suspensions: impact of contaminating leukocytes. Platelets. 2007;18:119–127. doi: 10.1080/09537100600800792. [DOI] [PubMed] [Google Scholar]
- Pourcelot E., Trocme C., Mondet J., Bailly S., Toussaint B., Mossuz P. Cytokine profiles in polycythemia vera and essential thrombocythemia patients: clinical implications. Exp. Hematol. 2014;42:360–368. doi: 10.1016/j.exphem.2014.01.006. [DOI] [PubMed] [Google Scholar]
- Qin C., Gu J., Hu J., Qian H., Fei X., Li Y., Liu R., Meng W. Platelets activation is associated with elevated plasma mitochondrial DNA during cardiopulmonary bypass. J. Cardiothorac. Surg. 2016;11:90. doi: 10.1186/s13019-016-0481-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavachari N., Xu X., Harris A., Villagra J., Logun C., Barb J., Solomon M.A., Suffredini A.F., Danner R.L., Kato G. Amplified expression profiling of platelet transcriptome reveals changes in arginine metabolic pathways in patients with sickle cell disease. Circulation. 2007;115:1551–1562. doi: 10.1161/CIRCULATIONAHA.106.658641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridker P.M., Everett B.M., Thuren T., MacFadyen J.G., Chang W.H., Ballantyne C., Fonseca F., Nicolau J., Koenig W., Anker S.D., CANTOS Trial Group Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017;377:1119–1131. doi: 10.1056/NEJMoa1707914. [DOI] [PubMed] [Google Scholar]
- Ridker P.M., MacFadyen J.G., Thuren T., Everett B.M., Libby P., Glynn R.J., Varigos J., Siostrzonek P., Sinnaeve P., Fonseca F., CANTOS Trial Group Effect of interleukin-1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet. 2017;390:1833–1842. doi: 10.1016/S0140-6736(17)32247-X. [DOI] [PubMed] [Google Scholar]
- Rossol M., Pierer M., Raulien N., Quandt D., Meusch U., Rothe K., Schubert K., Schöneberg T., Schaefer M., Krügel U. Extracellular Ca2+ is a danger signal activating the NLRP3 inflammasome through G protein-coupled calcium sensing receptors. Nat. Commun. 2012;3:1329. doi: 10.1038/ncomms2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowley J.W., Oler A.J., Tolley N.D., Hunter B.N., Low E.N., Nix D.A., Yost C.C., Zimmerman G.A., Weyrich A.S. Genome-wide RNA-seq analysis of human and mouse platelet transcriptomes. Blood. 2011;118:e101–e111. doi: 10.1182/blood-2011-03-339705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirmer M., Kumar V., Netea M.G., Xavier R.J. The causes and consequences of variation in human cytokine production in health. Curr. Opin. Immunol. 2018;54:50–58. doi: 10.1016/j.coi.2018.05.012. [DOI] [PubMed] [Google Scholar]
- Semple J.W., Italiano J.E., Jr., Freedman J. Platelets and the immune continuum. Nat. Rev. Immunol. 2011;11:264–274. doi: 10.1038/nri2956. [DOI] [PubMed] [Google Scholar]
- Senzel L., Chang C. Platelet phagocytosis by neutrophils. Blood. 2013;122:1543. doi: 10.1182/blood-2013-03-490854. [DOI] [PubMed] [Google Scholar]
- Shornick L.P., De Togni P., Mariathasan S., Goellner J., Strauss-Schoenberger J., Karr R.W., Ferguson T.A., Chaplin D.D. Mice deficient in IL-1beta manifest impaired contact hypersensitivity to trinitrochlorobenzone. J. Exp. Med. 1996;183:1427–1436. doi: 10.1084/jem.183.4.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims J.E., Smith D.E. The IL-1 family: regulators of immunity. Nat. Rev. Immunol. 2010;10:89–102. doi: 10.1038/nri2691. [DOI] [PubMed] [Google Scholar]
- Spivak J.L., Considine M., Williams D.M., Talbot C.C., Jr., Rogers O., Moliterno A.R., Jie C., Ochs M.F. Two clinical phenotypes in polycythemia vera. N. Engl. J. Med. 2014;371:808–817. doi: 10.1056/NEJMoa1403141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreeramkumar V., Adrover J.M., Ballesteros I., Cuartero M.I., Rossaint J., Bilbao I., Nácher M., Pitaval C., Radovanovic I., Fukui Y. Neutrophils scan for activated platelets to initiate inflammation. Science. 2014;346:1234–1238. doi: 10.1126/science.1256478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein E.V., Miller T.W., Ivins-O’Keefe K., Kaur S., Roberts D.D. Secreted Thrombospondin-1 Regulates Macrophage Interleukin-1β Production and Activation through CD47. Sci. Rep. 2016;6:19684. doi: 10.1038/srep19684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutz A., Horvath G.L., Monks B.G., Latz E. ASC speck formation as a readout for inflammasome activation. Methods Mol. Biol. 2013;1040:91–101. doi: 10.1007/978-1-62703-523-1_8. [DOI] [PubMed] [Google Scholar]
- Thornton P., McColl B.W., Greenhalgh A., Denes A., Allan S.M., Rothwell N.J. Platelet interleukin-1alpha drives cerebrovascular inflammation. Blood. 2010;115:3632–3639. doi: 10.1182/blood-2009-11-252643. [DOI] [PubMed] [Google Scholar]
- Trinh B.Q., Barengo N., Kim S.B., Lee J.-S., Zweidler-McKay P.A., Naora H. The homeobox gene DLX4 regulates erythro-megakaryocytic differentiation by stimulating IL-1β and NF-κB signaling. J. Cell Sci. 2015;128:3055–3067. doi: 10.1242/jcs.168187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunjungputri R.N., Li Y., de Groot P.G., Dinarello C.A., Smeekens S.P., Jaeger M., Doppenberg-Oosting M., Cruijsen M., Lemmers H., Toenhake-Dijkstra H. The Inter-Relationship of Platelets with Interleukin-1β-Mediated Inflammation in Humans. Thromb. Haemost. 2018;118:2112–2125. doi: 10.1055/s-0038-1675603. [DOI] [PubMed] [Google Scholar]
- Tzeng T.-C., Schattgen S., Monks B., Wang D., Cerny A., Latz E., Fitzgerald K., Golenbock D.T. A Fluorescent Reporter Mouse for Inflammasome Assembly Demonstrates an Important Role for Cell-Bound and Free ASC Specks during In Vivo Infection. Cell Rep. 2016;16:571–582. doi: 10.1016/j.celrep.2016.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaday G.G., Schor H., Rahat M.A., Lahat N., Lider O. Transforming growth factor-beta suppresses tumor necrosis factor alpha-induced matrix metalloproteinase-9 expression in monocytes. J. Leukoc. Biol. 2001;69:613–621. [PubMed] [Google Scholar]
- Verrecchia F., Mauviel A. TGF-beta and TNF-alpha: antagonistic cytokines controlling type I collagen gene expression. Cell. Signal. 2004;16:873–880. doi: 10.1016/j.cellsig.2004.02.007. [DOI] [PubMed] [Google Scholar]
- Vogel S., Arora T., Wang X., Mendelsohn L., Nichols J., Allen D., Shet A.S., Combs C.A., Quezado Z.M.N., Thein S.L. The platelet NLRP3 inflammasome is upregulated in sickle cell disease via HMGB1/TLR4 and Bruton tyrosine kinase. Blood Adv. 2018;2:2672–2680. doi: 10.1182/bloodadvances.2018021709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Fang C., Gao H., Bilodeau M.L., Zhang Z., Croce K., Liu S., Morooka T., Sakuma M., Nakajima K. Platelet-derived S100 family member myeloid-related protein-14 regulates thrombosis. J. Clin. Invest. 2014;124:2160–2171. doi: 10.1172/JCI70966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C., Lu W., Zhang Y., Zhang G., Shi X., Hisada Y., Grover S.P., Zhang X., Li L., Xiang B. Inflammasome Activation Triggers Blood Clotting and Host Death through Pyroptosis. Immunity. 2019;50:1401–1411.e4. doi: 10.1016/j.immuni.2019.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang B., Zhang G., Guo L., Li X.-A., Morris A.J., Daugherty A., Whiteheart S.W., Smyth S.S., Li Z. Platelets protect from septic shock by inhibiting macrophage-dependent inflammation via the cyclooxygenase 1 signalling pathway. Nat. Commun. 2013;4:2657. doi: 10.1038/ncomms3657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamane K., Ihn H., Asano Y., Jinnin M., Tamaki K. Antagonistic effects of TNF-alpha on TGF-beta signaling through down-regulation of TGF-beta receptor type II in human dermal fibroblasts. J. Immunol. 2003;171:3855–3862. doi: 10.4049/jimmunol.171.7.3855. [DOI] [PubMed] [Google Scholar]
- Yang X., Cheng X., Tang Y., Qiu X., Wang Y., Kang H., Wu J., Wang Z., Liu Y., Chen F. Bacterial Endotoxin Activates the Coagulation Cascade through Gasdermin D-Dependent Phosphatidylserine Exposure. Immunity. 2019;51:983–996.e6. doi: 10.1016/j.immuni.2019.11.005. [DOI] [PubMed] [Google Scholar]
- Zasłona Z., Pålsson-McDermott E.M., Menon D., Haneklaus M., Flis E., Prendeville H., Corcoran S.E., Peters-Golden M., O’Neill L.A.J. The Induction of Pro-IL-1β by Lipopolysaccharide Requires Endogenous Prostaglandin E2 Production. J. Immunol. 2017;198:3558–3564. doi: 10.4049/jimmunol.1602072. [DOI] [PubMed] [Google Scholar]
- Zhao Y., Yang J., Shi J., Gong Y.-N., Lu Q., Xu H., Liu L., Shao F. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature. 2011;477:596–600. doi: 10.1038/nature10510. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data are available via ProteomeXchange: PXD017976.