

Abstract
DNA double‐strand break (DSB) is a serious type of DNA damage and is known to trigger multiple responses within cells. In these responses, novel relationships among DSB, DSB repair, and transcription machineries are created. First, transcription is repressed if DSB occurs near or at the transcription site, termed DSB‐induced transcriptional repression, which contributes to DSB repair with the aid of DNA damage‐signaling pathways, ATM‐ or DNA‐PKcs‐signaling pathways. DSB‐induced transcriptional repression is also regulated by transcriptional factors TLP1, NELF, and ENL, as well as chromatin remodeling and organizing factors ZMYND8, CDYL1, PBAF, and cohesin. Second, transcription and RNA promote DSB repair for genome integrity. Transcription factors such as LEDGF, SETD2, and transcriptionally active histone modification, H3K36, facilitate homologous recombination to overcome DSB. At transcriptional active sites, DNA:RNA hybrids, termed R‐loops, which are formed by DSB, are processed by RAD52 and XPG leading to an activation of the homologous recombination pathway. Even in a transcriptionally inactive non‐genic sites, noncoding RNAs that are produced by RNA polymerase II, DICER, and DROSHA, help to recruit DSB repair proteins at the DSB sites. Third, transcriptional activation itself, however, can induce DSB. Transcriptional activation often generates specific DNA structures such as R‐loops and topoisomerase‐induced DSBs, which cause genotoxic stress and may lead to genome instability and consequently to cancer. Thus, transcription and DSB repair machineries interact and cooperate to prevent genome instability and cancer.
Keywords: cancer, DSB repair, genome instability, homologous recombination, transcription
Transcription is repressed under ATM or DNA‐PKcs‐signaling when DSBs are induced near or in transcription sites. DSB induced at transcriptionally active sites can be repaired via HR using transcriptional histone modifications, transcription factors, R‐loop, and transcribed RNA. On the other hand, transcriptional activation is predisposed to generate genotoxic stress, such as R‐loop formed at gene termination and DNA breaks induced by topoisomerase, which are to be repaired by DSB repair factors.
Abbreviations
- AR
androgen receptor
- ATM
ataxia telangiectasia mutated
- BRCA1/2
breast cancer type 1/2
- BRD
Bromodomain
- CDYL1
chromodomain protein, Y chromosome‐like
- CSB
Cockayne syndrome B
- CtIP
C‐terminal binding protein interacting protein
- DNA‐PKcs
DNA‐dependent protein kinase catalytic subunit
- DSB
DNA double‐strand break
- DSIF
DRB sensitivity‐inducing factor
- ENL
eleven nineteen leukemia
- ER
estrogen receptor
- EZH2
enhancer of zeste homolog 2
- HDAC
histone deacetylase
- HR
homologous recombination
- I‐PpoI
an intron‐encoded endonuclease
- IR
Ionizing Radiation
- I‐SceI
an intron‐encoded endonuclease
- LEDGF
lens epithelium‐derived growth factor
- lncRNA
long noncoding RNA
- MLLT1
myeloid/lymphoid or mixed‐lineage leukemia translocated to 1
- NELF‐E
Negative elongation factor E
- NELF‐F
Negative elongation factor F
- NER
nucleotide excision repair
- NHEJ
nonhomologous end‐joining
- NuRD
nucleosome remodeling and deacetylase
- PARP1
poly [ADP‐ribose] polymerase 1
- PBAF
polybromo‐associated BRG‐/BRM‐associated factor
- PHD domain
plant homeodomain
- PRC1
polycomb repressive complex 1
- PRC2
polycomb repressive complex 2
- PWWP domain
Pro‐Trp‐Trp‐Pro domain
- REST
repressor element‐1 silencing transcription factor
- RING1
ring finger protein 2
- RNAPI
RNA polymerase I
- RNAPII
RNA polymerase II
- RPA
replication protein A
- SEC
super elongation complex
- SETD2
SET domain containing 2
- SETX
senataxin
- snRNA
small nuclear ribonucleic acid
- SWI/SNF
SWItch/Sucrose Non‐Fermentable
- TA‐HRR
transcription‐associated homologous recombination repair
- TCR
transcription‐coupled DNA repair
- TDP2
tyrosyl‐DNA phosphodiesterase 2
- TFIIA
transcription factor IIA
- TLP
TBP‐like protein
- TOP2
topoisomerase (DNA) II
- tRNA
transfer ribonucleic acid
- TSS
transcription start site
- USP
ubiquitin specific protease
- UV
ultraviolet
- UVSSA
UV‐sensitive syndrome‐A complementation group
- WWP2
WW Domain Containing E3 Ubiquitin Protein Ligase 2
- XPF
xeroderma pigmentosum, complementation group F
- XPG
xeroderma pigmentosum, complementation group G
- ZMYND8
zinc finger MYND‐type containing 8
1. INFLUENCE OF DSB ON TRANSCRIPTION
DNA damage influences various types of DNA metabolism, most prominently replication and transcription. The relationship between transcription and DNA repair has been most intensively analyzed in TCR, which removes lesions from the template DNA strands of actively transcribed genes repair by NER. 1 , 2 , 3 , 4 UV damage in the transcribed template strand induces stalled RNAPII. The stalled RNAPII is recognized by UVSSA, USP7, and Cockayne syndrome proteins, which results in translocating (including backtracking) stalled RNAPII and activating NER repair. In contrast, DSB represses transcription, termed as DSB‐induced transcriptional repression, under ATM (see Section 1.1 for more detail) or DNA‐PKcs (see Section 1.2 for more detail) signaling pathways (Figures 1 and 2). These signaling pathways control transcription factors (details in 1.3) and chromatin remodeling and factors (details in 1.4) for the repression. Herein, we first discuss the mechanism of DSB‐induced transcriptional repression by referring to the recent findings.
FIGURE 1.
Transcription and DNA double‐strand breaks (DSBs) cooperate to prevent genome instability and cancer. 1. Influence of DSBs on transcription. 2. Influence of transcription or its factor on DSB repair. 3. Transcriptional activation‐induced DSBs are associated with cancer development
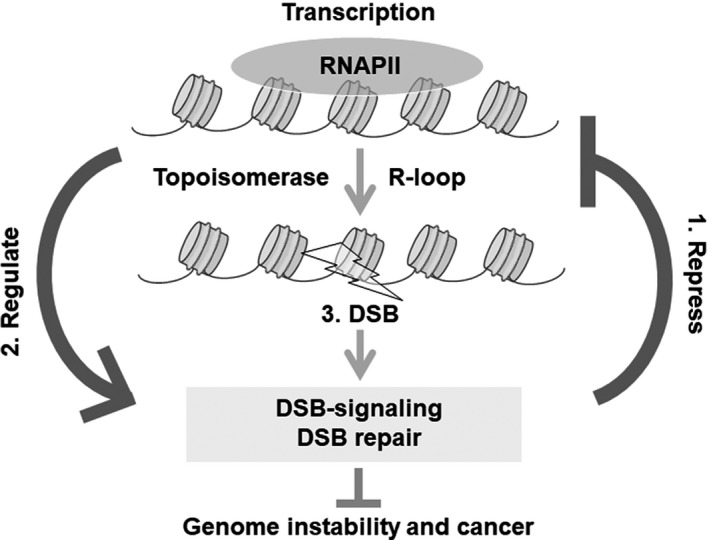
FIGURE 2.
Influence of double‐strand breaks (DSBs) on transcription. DSB‐induced transcriptional repression prevents genome rearrangement and tumorigenesis. (1.1) Ataxia telangiectasia mutated (ATM) and (1.2) DNA‐dependent protein kinase catalytic subunit (DNA‐PKcs) regulate DSB‐induced transcriptional repression. (1.3) Transcription factors also regulate DSB‐induced transcriptional repression. TBP‐like protein (TLP) represses transcription globally upon transcriptional initiation. Negative elongation factor E (NELF‐E) is recruited to RNA polymerase II (RNAPII) in a poly [ADP‐ribose] polymerase 1 (PARP1)‐dependent manner and represses transcription. Eleven nineteen leukemia (ENL) in the super elongation complex recruits polycomb repressive complex 1 (PRC1) of the polycomb complex at transcriptional elongation sites in an ATM‐dependent manner and promotes histone H2A K119/120 ubiquitination and repression. (1.4) Chromatin organizing factors such as histone modification complex, chromatin remodeling complex, and cohesion are also involved in DSB‐induced transcriptional repression. Zinc finger MYND‐type containing 8 (ZMYND8) and the nucleosome remodeling and deacetylase (NuRD) complex are recruited at transcription sites via TIP60‐mediated acetylation of histones to repress transcription. Chromodomain protein, Y chromosome‐like (CDYL1) binds DSBs via H3 K9 methylation and recruits PRC2 at transcription sites to promote H3 K27 methylation for transcriptional repression. Cohesin and polybromo‐associated BRG‐/BRM‐associated factor (PBAF) repress transcription and prevent mis‐rejoining of broken DNA ends to maintain genome stability
1.1. ATM signaling
ATM is reported to be one of the first factors to repress transcription in the proximity of the DSB sites, and this repression is termed DSB‐induced transcriptional repression (Figure 2). ATM represses both RNA polymerase I (RNAPI) and RNAPII‐mediated transcription. Following IR, RNAPI‐mediated ribosomal RNA synthesis in nucleoli was found to be repressed by ATM. 5 Additionally, the I‐PpoI sites in 28S ribosomal RNA in the nucleolus were used to produce DSBs and induce transcriptional repression by ATM (Figure 2). 6 , 7 Furthermore, DSBs outside the nucleolus silenced RNAPI‐mediated rRNA transcription in the nucleolus, suggesting the presence of an RNAPI‐mediated transcriptional silencing in the different (in trans) chromosome in response to DSBs. 7 Conversely, RNAPII‐mediated transcriptional silencing by ATM occurs in the proximity of the same (in cis) chromosome for DSBs. 8 DSBs near the promoter region but not within the gene body trigger ATM‐induced ubiquitination of H2A at transcriptional activation sites, leading to transcriptional silencing in cis. This ATM‐mediated transcriptional silencing reduces phosphorylated RNAPII at Ser2, whereas the total RNAPII levels at transcriptional sites remain unchanged. This suggests that ATM‐dependent transcriptional silencing stalls and maintains RNAPII at transcriptional sites near DSBs. When DSBs are induced within the gene body, DNA‐PKcs, but not ATM, represses the transcription by eliminating RNAPII from the template DNA (see Section 1.2 for more detail). Therefore, DNA‐PKcs is considered to repress transcription within the gene body to avoid collision between RNAPII and DSB repair machinery, whereas ATM only stalls and maintains RNAPII levels by changing the chromatin structure outside the gene body where no collision occurs between them.
Additionally, ATM phosphorylates PBAF, which is a complex belonging to the SWI/SNF chromatin remodeling complexes, ENL/MLLT1 in SEC, and NELF‐E; these phosphorylations promote changes in the repressive chromatin structure and negative transcriptional regulation (see Sections 1.3 and 1.4 for more detail; Figure 2).
1.2. DNA‐PKcs signaling
DNA‐PKcs is also reported to control DSB‐induced transcriptional repression within gene bodies by evicting RNAPII (Figure 2). DSBs induced by I‐PpoI within the gene body of active transcription sites represses RNAPII‐mediated transcription by the enzymatic function of DNA‐PKcs but not ATM at the DSB site. 9 The arrested RNAPII complex was degraded by DNA‐PKcs signaling and the DSB was repaired by NHEJ. Notably, in the absence of DNA‐PKcs, transcription was not hindered by the presence of a DSB. DNA‐PKcs associates with the HECT E3 ubiquitin ligase, WWP2, and recruits it at DSB sites in a transcription‐dependent manner to ubiquitinate RNAPII subunit RPB1 for degradation via the proteasome. Therefore, DNA‐PKcs represses transcription by promoting the eviction of RNAPII from the transcribed template DNA 10 to promote the NHEJ pathway.
1.3. Transcription factors
DSB‐induced transcriptional repression is also regulated by transcription factors working at the transcriptional initiation and elongation stages. During transcription: (1) TLP, which has a function in the initiation stage; and (2) ENL and (3) NELF‐F, which have a function in the elongation stage, have been reported to be involved in DSB‐induced transcriptional repression (Figure 2).
TLP was shown to repress transcription during the transcriptional initiation stage by negatively regulating the transcription factor TFIIA at its processing. 11 TLP is also required for stabilization of protein p53, which leads to p53‐induced apoptosis and senescence following genotoxic stress. 12 Several TLP mutations have been mapped in the p53‐binding region of TLP in human cancer. TLP knockdown was shown to reduce apoptosis and sensitivity following etoposide treatment compared with control cells. 13 Interestingly, knockdown of TLP did not lead to global transcriptional shutdown and showed increased activation of HR repair compared with control cells. 13 These results suggested that after DSBs TLP represses transcription by inhibiting initiation, and this repression reduces HR repair, preferential repair at transcriptional active sites.
ENL is a component of the SEC that functions in phosphorylation of C‐terminal RNAPII and promotes transcriptional elongation. We previously reported that ENL is involved in DSB‐induced transcriptional repression with BMI1 and RING1B, the E3‐ubiquitin ligase complex in PRC1, when DSB was induced at the promoter region. SEC binds to the RNAPII complex and promotes transcription during the elongation stage in gene expression. 14 , 15 , 16 , 17 , 18 , 19 PRC1 ubiquitinates K119/120 at histone H2A (H2A K119/120 ubiquitination), leading to transcriptional repression during development. 20 , 21 , 22 Thus, ENL and PRC1 have opposite transcriptional functions of activation and repression, respectively. 23 , 24 During transcriptional activation, ENL and PRC1 do not co‐localize and have different functions. However, after DSB induction, ENL is phosphorylated by ATM, leading to increased interaction between PRC1 and ENL. 25 , 26 ENL in the SEC binds to RNAPII to promote elongation, therefore phosphorylated ENL recruits PRC1 at transcriptional elongation sites to facilitate DSB‐induced transcriptional repression by H2A K119/120 ubiquitination.
Negative elongation factor (NELF) cooperates with DSIF to repress transcriptional elongation by promoting RNAPII to pause at the TSS. NELF‐E, a subunit of NELF, was shown to enhance MYC‐signaling and MYC‐induced hepatocellular carcinoma, indicating its role as an oncogenic protein. 27 NELF‐E was shown to be phosphorylated by ATM 28 , 29 , 30 and was rapidly recruited to DSB sites following laser microirradiation. Combined inhibition of ATM and NELF‐E knockdown did not cause synergistic or additive effects on DSB‐induced transcriptional repression, indicating that ATM and NELF‐E may function in the same silencing pathway. However, PARP1, but not ATM, is required for the recruitment of NELF‐E at DSB‐induced transcriptional repression sites and for the interaction between NELF‐E and RNAPII. NELF‐E knockdown led to a decrease in both HR and NHEJ activity. These results suggest that NELF‐E is possibly recruited to ADP‐ribosylated RNAPII to repress transcription cancelling in response to DSBs for its repair. 31
1.4. Chromatin organizing factors
A previous study has shown that chromatin decondensation is prevented at transcription sites following DSB, suggesting that chromatin condensation plays an important role in DSB‐induced transcriptional repression. 9 Recent findings have suggested that when DSB is produced near the promoter region, DSB‐induced transcriptional repression requires: (1) repressive factors, such as CDYL1, PRC1, and PRC2; (2) chromatin remodeling complexes, such as PBAF, one of the SWI/SNF nucleosome remodeling complexes, and ZMYND8 with NuRD complex, histone deacetylation complex; 25 , 32 , 33 and (3) cohesin, which functions to maintain higher order chromatin structures, including condensation (Figure 2). 34
Loss of function of CDYL1 increases oncogene expression, suggesting that it has tumor suppressor activity. 35 CDYL1 protein belongs to the CDY family that contains a chromodomain and enoyl‐CoA hydratase‐like domain. CDYL1 interacts with REST, histone methyltransferase G9a, H3 K9 methylation, PRC2, HDAC1, and HDAC2 35 , 36 , 37 , 38 and functions in transcriptional repression. Following DSB, CDYL1 is recruited to the DNA damage site in a PARP1‐dependent but not in an ATM‐dependent manner. 32 CDYL1 is required for DSB‐induced transcriptional repression by promoting accumulation of EZH2 in the PRC2 complex and H3 K27 methylation at the DSB site. These results suggested that PRC2 as well as PRC1 (see Sections 1.3 for more detail) has a role in DSB‐induced transcriptional repression via CDYL1.
Chromatin remodeling complexes also function in DSB‐induced transcriptional repression. Chromatin remodeling activity and ATM‐mediated phosphorylation of PBAF chromatin remodeling complex were reported to be required for transcriptional silencing and H2A K119/120 ubiquitination in response to DSBs. 39 Furthermore, ZMYND8 interacts with and recruits CHD4, a core component of the NuRD chromatin remodeling and deacetylase complex, to damaged chromatin to promote DSB‐induced transcriptional repression and HR but not NHEJ. ZMYND8 contains PHD, BRD, and PWWP chromatin‐binding domains as well as an MYND domain for protein‐protein interaction; and it represses metastasis‐linked genes, which suppresses invasiveness in prostate cancer cells. 40 ZMYND8 was recruited at DSB sites via the interaction between its BRD domain and histone H4 acetylated by TIP60. Therefore, the interaction between ZMYND8 and histone H4 following DSBs recruits the NuRD chromatin remodeling complex to transcription sites to promote DSB‐induced transcriptional repression.
Cohesin, and cohesin loading factors are required for DSB‐induced transcriptional repression in both G1 and G2 phases, and cancer‐associated mutations of SA2 play an important role in this repression. Furthermore, DSB‐induced transcriptional repression prevents mis‐rejoining of broken DNA ends and genome rearrangements. These results suggest that DSB‐induced transcriptional repression plays a role in preventing tumorigenesis. 34
It should be noted that the transcriptional repressive histone modifications such as H2A K119/120 ubiquitination, methylation of H3 K27 methylation, and negative transcriptional factors, are applied in DSB‐induced transcriptional repression. However, it is interesting that transcriptionally active histone modifications such as the acetylation of H4K16 and transcriptional activation factor ENL, are required for DSB‐induced repression to switch off the transcription in response to DSBs. The link between these transcriptional active and repressive regulators remains unclear. Furthermore, among the factors required for DSB‐induced transcriptional repression, ZMYND8 and CDYL1 promote HR, 32 , 33 TLP reduces HR, 13 and NELF promotes both NHEJ and HR. 31 However, the pathways involved in DSB repair at the sites of DSB‐induced transcriptional repression still remain unclear.
2. INFLUENCE OF TRANSCRIPTION AND RNA ON DSB REPAIR
While histone modifications and the chromatin structure influence DSB repair, 41 transcriptional machinery and transcriptional active sites also influence the mechanisms underlying DSB repair (Figure 3). Recent findings have shown that DSB repair utilizes transcriptional histone modifications and transcription factors (see Section 2.1 for more detail). Furthermore, RNA including noncoding RNA functions as a scaffold or DSB repair proteins or as a template during DSB repair (see Section 2.2 for more detail).
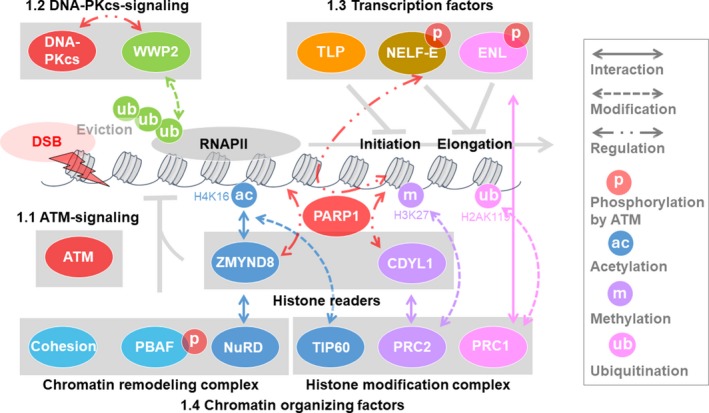
FIGURE 3.
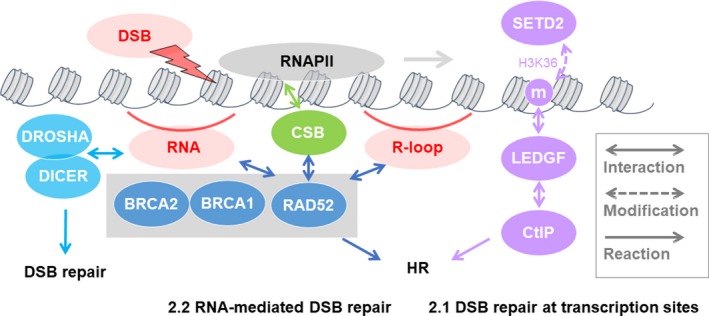
Influence of transcription or transcription factors on double‐strand break (DSB) repair. (2.1) Transcriptionally active regions promote homologous recombination (HR) repair via an interaction between H3 K36 methylation and the lens epithelium‐derived growth factor‐C‐terminal binding protein interacting protein (LEDGF‐CtIP) complex. The histone methyltransferase SET domain containing 2 (SETD2) induces the H3 K36 methylation that is required for the recruitment of RAD51 and replication protein A (RPA) at DSB sites to promote HR. In G2/S‐phase cells, DSB‐induced R‐loops at transcription sites are stabilized and processed by RAD52 and XPG to promote HR. (2.2) RNA also supports DSB repair. RNAs are produced and processed at DSB sites by RNAPII, and DICER and DROSHA recruit DSB repair proteins to promote DSB repair. lncRNAs are also recognized by breast cancer type 1 (BRCA1) to promote HR by recruiting BRCA2 and RNaseH2. RNA could be used as a template of HR in yeast. In G1‐phase cells, CSB recruits HR factors such as RPA, RAD51, and RAD52 and promotes HR at transcription sites
2.1. DSB repair at transcription sites
Several reports have suggested that transcriptionally active regions are preferentially repaired by HR (Figure 3). After DSBs, RAD51 is an HR factor that binds transcriptionally active genes that are associated with active transcriptional histone modifications, such as H3 K36 methylation and H3 K9 acetylation. 42 During transcription, the H3 K36 methylation recruits LEDGF, which is a transcriptional coactivator, and binds methylated histone at the PWWP domain in its N‐terminal region. After DSBs, LEDGF recruits CtIP to damaged chromatin via the H3 K36 methylation to promote end resection. 43 While H3 K36 is reported to be methylated by eight enzymes in human, including SETD2, 44 SETD2 was also shown to be necessary for HR for promoting the recruitment of RPA and RAD51. 45 These findings suggested that LEDGF binds to H3 K36 methylation via SETD2 at transcriptionally active regions, recruits CtIP, and promotes end resection to facilitate HR after DSBs.
Furthermore, at DSBs within transcriptional active sites in G2/S cells, RNA‐DNA hybrid‐containing R‐loops, which are generated by the pausing of RNAPII at the active transcription sites, were shown to accumulate at DSB sites. R‐loops recruited human RAD52 at the DSB sites and were processed by XPG, resulting in promotion of the HR pathway. Therefore, this pathway is described as transcription‐associated homologous recombination repair (TA‐HRR). 46 The defect in the TA‐HHR process increased aberrant NHEJ, and the low expression of RAD52 increased the number of insertions and/or deletions in cancer cells, which suggests that TR‐HRR prevents genome instability.
2.2. RNA‐mediated DSB repair
In the DSB repair process, RNA has been reported to promote DSB repair (Figure 3). DICER and DROSHA (a complex of double‐stranded RNA‐specific endoribonuclease), which produce small double‐stranded RNAs involved in sncRNA (small noncoding RNA), recruit DSB repair proteins at the DSB sites, and these recruitments are inhibited by RNase. 47 , 48 At the DSB sites, DICER and DROSHA are involved in the process of RNA production for DSB repair; and RNAPII is also required for this process.
In addition to the above sncRNAs, long noncoding RNA (lncRNA) have been reported to be involved in DSB repair. In S/G2‐phase cells, DNA damage induces transcription of lncRNA at the DNA end of DSBs both within and outside gene bodies, and lncRNAs pair with the resected DNA ends to form DNA:RNA hybrids. 49 These lncRNA‐mediated DNA:RNA hybrids are recognized by BRCA1 and lead to the promotion of HR by recruiting BRCA2 and RNaseH2 at DSB sites. Both lncRNA and TA‐HRR (described in Section 2.1) mediated DNA:RNA hybrids (R‐loop) mediated by recruit HR factors and promote HR, but the difference between them is transcriptional dependency. The recruitment of DSB repair factors by lncRNA‐mediated DNA:RNA hybrids could occur at non‐genic sites, namely transcriptionally inactive sites by the induction of I‐PpoI. However, TA‐HRR can occur at only transcriptionally active sites. Therefore, TA‐HRR and lncRNA‐mediated DNA:RNA hybrids can cover through transcriptionally active sites and inactive sites for genome stability. It was recently reported that DSBs within the gene body at transcriptionally active sites could recover transcription and produce RNAs, whereas DSBs at a promoter proximal region could not. 50 It is interesting to know whether RNAs that are produced by recovery of transcription in gene bodies are involved in DSB repair.
Furthermore, RNA has been shown to serve as a template for DNA synthesis in bacteria and humans (Figure 3). 51 , 52 , 53 In the chromosomes in human cells, I‐SceI‐induced DSBs in GFP can be repaired using RNA‐containing oligos. 53 In yeast, synthetic RNA oligonucleotides could act as templates for DSB repair. 51 Moreover, RNAs that were transcribed in a different chromosome (in trans) or in the proximity of the same chromosome (in cis) were shown to become a template for HR and that RAD52 facilitated HR using the transcribed RNA in cis. 54 In vitro, yeast and human RAD52 also efficiently catalyzes the annealing of RNA to DNA, suggesting that in human cells, RAD52 protein can promote transcribed RNA‐mediated HR.
The abovementioned mechanism could enable cells to repair DSB by HR even in the G1 phase when sister chromatids do not exist. In the G0/G1 phase, NER factor, CSB, and recruited HR factors, such as RPA1, RAD51C, RAD51, and RAD52, promote HR at site‐specific DNA strand breaks produced by oxidative damage (Figure 3). 55 Furthermore, the inhibitor of transcription sensitized WT cells but not CSB‐deficient cells to IR. These results suggest that CSB contributes to cell survival to promote HR at the active transcription sites. Although it remains unclear how homologous pairing during HR occurs in the G1 phase, there is the possibility that transcribed RNA at transcription sites are used as a template for HR.
Many uncertainties about the mechanism of DSB repair at transcription sites still exist. While RNAPII has been reported to be inhibited by DSBs inside and outside of transcriptional regions under ATM and DNA‐PKcs‐signaling, other findings showed that RNAPII with DICER and DROSHA promotes RNA production at DSB sites. Thus, it remains unclear how RNAPII is regulated at DSB sites. Furthermore, if HR occurs preferentially at transcriptional activation sites, it remains unclear how in G1 phase of cells DNA damage at transcriptional activation sites is repaired by HR and what is the template of homologous pairing during HR. Further research is needed to understand the mechanism of DSB repair at transcription sites and the contribution of RNA in the process.
3. INFLUENCE OF TRANSCRIPTION‐INDUCED DNA DAMAGE ON CANCER DEVELOPMENT
3.1. R‐loop at transcription sites
R‐loops lead to the generation of genotoxic stress if they cannot be resolved and repaired by HR factors (Figure 4). During transcriptional activation, R‐loops are usually formed at transcriptional termination regions, and they promote the recruitment of HR factors at transcription sites, 56 suggesting that active transcription could lead to genome instability without HR factors. Indeed, these genotoxic R‐loops accumulate at transcriptionally active regions in BRCA1 and BRCA2‐deficient cells, 56 , 57 and unresolved R‐loops cause nicking in single‐strand DNA, DNA breaks, and/or other forms of DNA damage. 58 , 59 BRCA1 and BRCA2 are HR factors that are involved in DSB end resection and homologous pairing in the HR pathway, respectively. BRCA1 recruits SETX, which is a RNA/DNA helicase that is involved in TCR and in the processing of RNAs such as tRNAs and sncRNAs, 59 , 60 at transcription termination pause sites of highly transcribed genes to suppress R‐loop‐associated DNA damage. 57 In the absence of SETX, R‐loops are processed into DSBs by the NER endonucleases XPF and XPG. 59 Therefore, the R‐loop is a key factor that leads to the HR pathway during active transcription or after DSB induction at transcriptional active region (described in Section 2.1).In addition to the function in the HR pathway, BRCA1 is involved in transcription and TCR. BRCA1 interacts with and ubiquitinates RNAPII, and is involved in TCR through the polyubiquitination of CSB. 61 , 62 , 63 ; Therefore, BRCA1 may always localize near transcriptional machinery and protect transcription from various types of DNA damage.
FIGURE 4.

Transcription‐induced double‐strand break (DSB) and DNA breaks are associated with cancer development. (3.1) During transcriptional activation, R‐loops generated by transcriptional terminal sites or RNA polymerase II (RNAPII) pausing sites are resolved and repaired by senataxin (SETX) and breast cancer type 1 (BRCA1). (3.2) Furthermore, androgen receptor (AR)‐induced DSB at the promoter region by topoisomerase (DNA) II (TOP2) leads to oncogenic TMPRSS2‐ERG translocation. Estrogen receptor (ER) also induces DSBs at promoter regions by TOP2. Tyrosyl‐DNA phosphodiesterase 2 (TDP2) removes TOP2 covalently bound to DNA to repair cleavage via BRCA1‐mediated nonhomologous end‐joining (NHEJ). Loss of BRCA1 increases DSBs in TDP2‐knockout breast cancer cells, suggesting that it may suppress tumorigenesis in ER‐dependent tissues
3.2. Topoisomerase‐induced DSB at transcription sites
Topoisomerases are thought to be required during transcription to relax the supercoiled DNA formed in front of and behind the transcription machinery and during DNA replication (Figure 4). 64 , 65 TOP2 forms DSBs via its strand‐cleaving activity at the promoter of estrogen‐inducible genes with the ER for transcriptional activation. 66 , 67 These DSBs are generated during the S‐phase and may be repaired by HR. 67
TOP2‐β is also recruited at the promoter region of AR target genes and induces DSBs to promote gene expression. 68 The TOP2‐induced DSBs are transient and are re‐ligated immediately but allow an intact DNA duplex to pass through DSB for resolution of topological stress to promote transcriptional activation. During the transiently produced DSB, TOP2 is bound covalently to DNA ends. Thus, if re‐ligation fails, DSBs remains at stable TOP2‐DNA complex. 69 Such DSBs frequently induce chromosome rearrangement including translocation. For example, TOP2‐β and AR were shown to co‐localize at the TMPRSS2‐ERG genomic breakpoint, generating oncogenic TMPRSS2‐ERG translocation. 68 Therefore, TOP2‐induced DSBs at transcription sites could generate genotoxic stress and cause cancer.
The mechanism of DSB repair at stable TOP2‐DNA complex has been reported. Tyrosyl‐DNA phosphodiesterase 2 (TDP2) removes TOP2 from DNA to repair the DSBs and is required for AR‐mediated transcription and expression of neuronal genes. 70 Recently, ER‐induced DSBs were reported to be increased in TDP2‐knockout human breast cancer cells during the G1 phase, and BRCA1 was recruited at the DSB sites. Loss of BRCA1 causes prolonged DSBs after exposure to estrogen, suggesting its need for the repair of the ER‐induced DSBs via NHEJ and, therefore, prominent DSBs were formed in NHEJ‐deficient mice. 71 Thus, BRCA1 may suppress tumorigenesis in ER‐dependent tissues by repairing ER‐dependent DSBs that are generated by topoisomerase.
The above study explained how BRCA1 functions against female‐organ‐specific carcinogenesis to repair DSBs produced in ER‐dependent proliferating cells. Further studies are needed to understand whether BRCA2, which is also associated with hereditary breast cancer, has the same function as that of BRCA1.
4. CONCLUSION
Interactive relationships among, DSB, DSB repair, and transcription have recently been proposed for the protection of genetic information from genotoxic stress. When DSBs are induced in the proximity of or within the gene body during active transcription, ATM or DNA‐PKcs control the transcription. DSBs occurring outside the gene body during transcription induce transcriptional repression by ATM, which prevents genome rearrangement and tumorigenesis. DSBs within the gene body during transcription induce transcriptional repression and NHEJ pathway by DNA‐PKcs to avoid the collision of transcription and repair machinery to promote NHEJ.
In contrast to the above finding, DSB induced at transcriptionally active sites can be repaired via HR using transcriptional histone modifications, transcription factors, R‐loop, and transcribed RNA. The finding that transcribed RNA is used as a template for homologous pairing suggests that the HR pathway occurs more frequently, even in the G1 phase, when sister chromatids do not exist. Because HR is a process to preserve genome stability to eliminate DSB, defects of factors involved in the HR may give rise to tumorigenesis.
Conversely, activation of transcription is predisposed to generate genotoxic stress, such as the R‐loop formed at gene termination and DNA breaks induced by topoisomerase, which are to be repaired by DSB repair factors. BRCA1, which is associated with hereditary breast cancer and ovarian cancer, is also involved in preventing the formation of R‐loops, the repair of topoisomerase‐mediated DSB, and HR at transcription active sites, suggesting that BRCA1 is a key factor required to ensure the safety of the transcriptional machinery and genome stability. Thus, it is essential to elucidate the link between transcription and DSB repair as it plays an important role in preventing genome stability and cancer.
CONFLICT OF INTEREST
The authors declare no conflicts of interest for this article.
ACKNOWLEDGMENTS
This work was supported by JSPS KAKENHI Grant Numbers 17K19615; Practical Research for Innovative Cancer Control from Japan Agency for Medical Research and Development (AMED: 19cm0106605h0003), as well as grants from the Takeda Science Foundation, Princess Takamatsu Cancer Research Fund and the Naito Foundation.
Ui A, Chiba N, Yasui A. Relationship among DNA double‐strand break (DSB), DSB repair, and transcription prevents genome instability and cancer. Cancer Sci. 2020;111:1443–1451. 10.1111/cas.14404
REFERENCES
- 1. Bohr VA, Smith CA, Okumoto DS, Hanawalt PC. DNA‐repair in an active gene ‐ removal of pyrimidine dimers from the Dhfr gene of Cho cells is much more efficient than in the genome overall. Cell. 1985;40:359‐369. [DOI] [PubMed] [Google Scholar]
- 2. Marteijn JA, Lans H, Vermeulen W, Hoeijmakers JHJ. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat Rev Mol Cell Biol. 2014;15:465‐481. [DOI] [PubMed] [Google Scholar]
- 3. Mullenders L. DNA damage mediated transcription arrest: Step back to go forward. DNA Repair. 2015;36:28‐35. [DOI] [PubMed] [Google Scholar]
- 4. Spivak G, Hanawalt PC. Photosensitive human syndromes. Mutat Res. 2015;776:24‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kruhlak M, Crouch EE, Orlov M, et al. The ATM repair pathway inhibits RNA polymerase I transcription in response to chromosome breaks. Nature. 2007;447:730‐734. [DOI] [PubMed] [Google Scholar]
- 6. Harding SM, Boiarsky JA, Greenberg RA. ATM dependent silencing links nucleolar chromatin reorganization to DNA damage recognition. Cell Rep. 2015;13:251‐259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Larsen DH, Hari F, Clapperton JA, et al. The NBS1‐Treacle complex controls ribosomal RNA transcription in response to DNA damage. Nat Cell Biol. 2014;16:792‐803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shanbhag NM, Rafalska‐Metcalf IU, Balane‐Bolivar C, Janicki SM, Greenberg RA. ATM‐dependent chromatin changes silence transcription in cis to DNA double‐strand breaks. Cell. 2010;141:970‐981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pankotai T, Bonhomme C, Chen D, Soutoglou E. DNAPKcs‐dependent arrest of RNA polymerase II transcription in the presence of DNA breaks. Nat Struct Mol Biol. 2012;19:276‐282. [DOI] [PubMed] [Google Scholar]
- 10. Caron P, Pankotai T, Wiegant WW, et al. WWP2 ubiquitylates RNA polymerase II for DNA‐PK‐dependent transcription arrest and repair at DNA breaks. Genes Dev. 2019;33:684‐704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Suzuki H, Isogai M, Maeda R, Ura K, Tamura TA. TBP‐like protein (TLP) interferes with Taspase1‐mediated processing of TFIIA and represses TATA box gene expression. Nucleic Acids Res. 2015;43:6285‐6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Maeda R, Tamashiro H, Takano K, et al. TBP‐like protein (TLP) disrupts the p53‐MDM2 interaction and induces long‐lasting p53 activation. J Biol Chem. 2017;292:3201‐3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Suzuki H, Okamoto‐Katsuyama M, Suwa T, et al. TLP‐mediated global transcriptional repression after double‐strand DNA breaks slows down DNA repair and induces apoptosis. Sci Rep. 2019;9:4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. He N, Chan CK, Sobhian B, et al. Human polymerase‐associated factor complex (PAFc) connects the Super Elongation Complex (SEC) to RNA polymerase II on chromatin. Proc Natl Acad Sci USA. 2011;108:E636‐E645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Smith E, Lin CQ, Shilatifard A. The super elongation complex (SEC) and MLL in development and disease. Genes Dev. 2011;25:661‐672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Luo ZJ, Lin CQ, Shilatifard A. The super elongation complex (SEC) family in transcriptional control. Nat Rev Mol Cell Biol. 2012;13:543‐547. [DOI] [PubMed] [Google Scholar]
- 17. Sobhian B, Laguette N, Yatim A, et al. HIV‐1 Tat assembles a multifunctional transcription elongation complex and stably associates with the 7SK snRNP. Mol Cell. 2010;38:439‐451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yokoyama A, Lin M, Naresh A, Kitabayashi I, Cleary ML. A higher‐order complex containing AF4 and ENL family proteins with P‐TEFb facilitates oncogenic and physiologic MLL‐dependent transcription. Cancer Cell. 2010;17:198‐212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhou Q, Li TD, Price DH. RNA polymerase II elongation control. Annu Rev Biochem. 2012;81(81):119‐143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang H, Wang L, Erdjument‐Bromage H, et al. Role of histone H2A ubiquitination in polycomb silencing. Nature. 2004;431:873‐878. [DOI] [PubMed] [Google Scholar]
- 21. Buchwald G, van der Stoop P, Weichenrieder O, et al. Structure and E3‐ligase activity of the Ring‐Ring complex of polycomb proteins Bmi1 and Ring1b. EMBO J. 2006;25:2465‐2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cao R, Tsukada Y, Zhang Y. Role of Bmi‐1 and Ring1A in H2A ubiquitylation and Hox gene silencing. Mol Cell. 2005;20:845‐854. [DOI] [PubMed] [Google Scholar]
- 23. Mills AA. Throwing the cancer switch: reciprocal roles of polycomb and trithorax proteins. Nat Rev Cancer. 2010;10:669‐682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Steffen PA, Ringrose L. What are memories made of? How Polycomb and Trithorax proteins mediate epigenetic memory. Nat Rev Mol Cell Biol. 2014;15:340‐356. [DOI] [PubMed] [Google Scholar]
- 25. Ui A, Nagaura Y, Yasui A. Transcriptional elongation factor ENL phosphorylated by ATM recruits polycomb and switches off transcription for DSB repair. Mol Cell. 2015;58:468‐482. [DOI] [PubMed] [Google Scholar]
- 26. Ui A, Yasui A. Collaboration of MLLT1/ENL, Polycomb and ATM for transcription and genome integrity. Nucleus. 2016;7:138‐145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dang H, Takai A, Forgues M, et al. Oncogenic activation of the RNA binding protein NELFE and MYC signaling in hepatocellular carcinoma. Cancer Cell. 2017;32:101‐114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Matsuoka S, Ballif BA, Smogorzewska A, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160‐1166. [DOI] [PubMed] [Google Scholar]
- 29. Bennetzen MV, Larsen DH, Bunkenborg J, et al. Site‐specific phosphorylation dynamics of the nuclear proteome during the DNA damage response. Mol Cell Proteomics. 2010;9:1314‐1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Elia A, Boardman A, Wang D, et al. Quantitative proteomic atlas of ubiquitination and acetylation in the DNA damage response. Mol Cell. 2015;59:867‐881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Awwad SW, Abu‐Zhayia ER, Guttmann‐Raviv N, Ayoub N. NELF‐E is recruited to DNA double‐strand break sites to promote transcriptional repression and repair. EMBO Rep. 2017;18:745‐764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Abu‐Zhayia ER, Awwad SW, Ben‐Oz BM, Khoury‐Haddad H, Ayoub N. CDYL1 fosters double‐strand break‐induced transcription silencing and promotes homology‐directed repair. J Mol Cell Biol. 2018;10:341‐357. [DOI] [PubMed] [Google Scholar]
- 33. Chen Y, Zhang BO, Bao L, et al. ZMYND8 acetylation mediates HIF‐dependent breast cancer progression and metastasis. J Clin Invest. 2018;128:1937‐1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Meisenberg C, Pinder SI, Hopkins SR, et al. Repression of transcription at DNA breaks requires cohesin throughout interphase and prevents genome instability. Mol Cell. 2019;73:212‐223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mulligan P, Westbrook TF, Ottinger M, et al. CDYL bridges REST and histone methyltransferases for gene repression and suppression of cellular transformation. Mol Cell. 2008;32:718‐726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Caron C, Pivot‐Pajot C, van Grunsven LA, et al. Cdyl: a new transcriptional co‐repressor. EMBO Rep. 2003;4:877‐882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Franz H, Mosch K, Soeroes S, Urlaub H, Fischle W. Multimerization and H3K9me3 binding are required for CDYL1b heterochromatin association. J Biol Chem. 2009;284:35049‐35059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang YU, Yang X, Gui B, et al. Corepressor protein CDYL functions as a molecular bridge between polycomb repressor complex 2 and repressive chromatin mark trimethylated histone lysine 27. J Biol Chem. 2011;286:42414‐42425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kakarougkas A, Ismail A, Chambers A, et al. Requirement for PBAF in transcriptional repression and repair at DNA breaks in actively transcribed regions of chromatin. Mol Cell. 2014;55:723‐732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li NA, Li Y, Lv J, et al. ZMYND8 reads the dual histone mark H3K4me1‐H3K14ac to antagonize the expression of metastasis‐linked genes. Mol Cell. 2016;63:470‐484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bennett G, Papamichos‐Chronakis M, Peterson CL. DNA repair choice defines a common pathway for recruitment of chromatin regulators. Nat Commun. 2013;4:2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Aymard F, Bugler B, Schmidt CK, et al. Transcriptionally active chromatin recruits homologous recombination at DNA double‐strand breaks. Nat Struct Mol Biol. 2014;21:366‐374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Daugaard M, Baude A, Fugger K, et al. LEDGF (p75) promotes DNA‐end resection and homologous recombination. Nat Struct Mol Biol. 2012;19:803‐810. [DOI] [PubMed] [Google Scholar]
- 44. Wagner EJ, Carpenter PB. Understanding the language of Lys36 methylation at histone H3. Nat Rev Mol Cell Biol. 2012;13:115‐126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pfister S, Ahrabi S, Zalmas L‐P, et al. SETD2‐dependent histone H3K36 trimethylation is required for homologous recombination repair and genome stability. Cell Rep. 2014;7:2006‐2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yasuhara T, Kato R, Hagiwara Y, et al. Human Rad52 promotes XPG‐mediated R‐loop processing to initiate transcription‐associated homologous recombination repair. Cell. 2018;175:558‐570. [DOI] [PubMed] [Google Scholar]
- 47. Francia S, Michelini F, Saxena A, et al. Site‐specific DICER and DROSHA RNA products control the DNA‐damage response. Nature. 2012;488:231‐235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Francia S, Cabrini M, Matti V, Oldani A, d'Adda di Fagagna F. DICER, DROSHA and DNA damage response RNAs are necessary for the secondary recruitment of DNA damage response factors. J Cell Sci. 2016;129:1468‐1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. D'Alessandro G, Whelan DR, Howard SM, et al. BRCA2 controls DNA:RNA hybrid level at DSBs by mediating RNase H2 recruitment. Nat Commun. 2018;9:5376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vítor AC, Sridhara SC, Sabino JC, et al. Single‐molecule imaging of transcription at damaged chromatin. Sci Adv. 2019;5:eaau1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Storici F, Bebenek K, Kunkel TA, Gordenin DA, Resnick MA. RNA‐templated DNA repair. Nature. 2007;447:338‐341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nowacki M, Vijayan V, Zhou YI, et al. RNA‐mediated epigenetic programming of a genome‐rearrangement pathway. Nature. 2008;451:153‐158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Shen Y, Nandi P, Taylor MB, et al. RNA‐driven genetic changes in bacteria and in human cells. Mutat Res. 2011;717:91‐98. [DOI] [PubMed] [Google Scholar]
- 54. Keskin H, Shen Y, Huang F, et al. Transcript‐RNA‐templated DNA recombination and repair. Nature. 2014;515:436‐439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wei L, Nakajima S, Böhm S, et al. DNA damage during the G0/G1 phase triggers RNA‐templated, Cockayne syndrome B‐dependent homologous recombination. Proc Natl Acad Sci USA. 2015;112:E3495‐E3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Skourti‐Stathaki K, Kamieniarz‐Gdula K, Proudfoot NJ. R‐loops induce repressive chromatin marks over mammalian gene terminators. Nature. 2014;516:436‐439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hatchi E, Skourti‐Stathaki K, Ventz S, et al. BRCA1 recruitment to transcriptional pause sites is required for R‐loop‐driven DNA damage repair. Mol Cell. 2015;57:636‐647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wimberly H, Shee C, Thornton PC, et al. R‐loops and nicks initiate DNA breakage and genome instability in non‐growing Escherichia coli . Nat Commun. 2013;4:2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sollier J, Stork C, García‐Rubio ML, et al. Transcription‐coupled nucleotide excision repair factors promote R‐loop‐induced genome instability. Mol Cell. 2014;56:777‐785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Cohen S, Puget N, Lin Y‐L, et al. Senataxin resolves RNA:DNA hybrids forming at DNA double‐strand breaks to prevent translocations. Nat Commun. 2018;9:533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Chiba N, Parvin JD. The BRCA1 and BARD1 association with the RNA polymerase II holoenzyme. Cancer Res. 2002;62:4222‐4228. [PubMed] [Google Scholar]
- 62. Starita LM, Horwitz AA, Keogh M‐C, et al. BRCA1/BARD1 ubiquitinate phosphorylated RNA polymerase II. J Biol Chem. 2005;280:24498‐24505. [DOI] [PubMed] [Google Scholar]
- 63. Wei L, Lan LI, Yasui A, et al. BRCA1 contributes to transcription‐coupled repair of DNA damage through polyubiquitination and degradation of Cockayne syndrome B protein. Cancer Sci. 2011;102:1840‐1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Liu LF, Wang JC. Supercoiling of the DNA template during transcription. Proc Natl Acad Sci USA. 1987;84:7024‐7027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. McClendon AK, Rodriguez AC, Osheroff N. Human topoisomerase IIalpha rapidly relaxes positively supercoiled DNA: implications for enzyme action ahead of replication forks. J Biol Chem. 2005;280:39337‐39345. [DOI] [PubMed] [Google Scholar]
- 66. Ju BG, Lunyak VV, Perissi V, et al. A topoisomerase IIbeta‐mediated dsDNA break required for regulated transcription. Science. 2006;312:1798‐1802. [DOI] [PubMed] [Google Scholar]
- 67. Williamson LM, Lees‐Miller SP. Estrogen receptor alpha‐mediated transcription induces cell cycle‐dependent DNA double‐strand breaks. Carcinogenesis. 2011;32:279‐285. [DOI] [PubMed] [Google Scholar]
- 68. Haffner MC, Aryee MJ, Toubaji A, et al. Androgen‐induced TOP2B‐mediated double‐strand breaks and prostate cancer gene rearrangements. Nat Genet. 2010;42:668‐675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Pommier Y, Sun Y, Huang SN, Nitiss JL. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat Rev Mol Cell Biol. 2016;17:703‐721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Cortes Ledesma F, El Khamisy SF, Zuma MC, Osborn K, Caldecott KW. A human 5'‐tyrosyl DNA phosphodiesterase that repairs topoisomerase‐mediated DNA damage. Nature. 2009;461:674‐678. [DOI] [PubMed] [Google Scholar]
- 71. Sasanuma H, Tsuda M, Morimoto S, et al. BRCA1 ensures genome integrity by eliminating estrogen‐induced pathological topoisomerase II‐DNA complexes. Proc Natl Acad Sci USA. 2018;115:E10642‐E10651. [DOI] [PMC free article] [PubMed] [Google Scholar]