

Abstract
The cyclin‐dependent kinase (CDK)4/6‐cyclin D1‐Rb‐p16/ink4a pathway is responsible for regulating cell progression past the G1 restriction point during the cell cycle. The development of a majority of human tumors is associated with dysregulation of this pathway, resulting in increased cancer cell proliferation. Both CDK4 and CDK6, well‐validated cancer drug targets, function primarily as catalytic enzymes that mediate the phosphorylation of retinoblastoma protein (Rb). Here, we determined that SPH3643 is a novel potent antiproliferative agent that inhibits CDK4/6 kinase activity. In biochemical assays, SPH3643 showed more potent inhibition of both CDK4 and CDK6 than did 2 published CDK4/6 inhibitors, LY2835219 and palbociclib, and had better selectivity than LY2835219. Further in vitro study revealed that SPH3643 blocked Cdk/Rb signaling by inhibiting the phosphorylation of RbSer780 and arrested the MCF‐7 cancer cells at G0/G1 phase, resulting in marked inhibition of the proliferation of Rb‐positive cancer cell lines. In vivo SPH3643 treatment in mice bearing xenograft tumor models of breast cancer, colon cancer, acute myelocytic leukemia, and glioblastoma resulted in significant decreases in tumor growth. SPH3643 was able to particularly strongly inhibit glioblastoma (U87‐MG) cell growth in the brains of orthotopic carcinoma xenograft mice due to its high degree of intracerebral penetration and significant persistence in this setting. Together these results revealed that SPH3643 is a potent, orally active small‐molecule inhibitor of CDK4/6 with robust anticancer efficacy and a high degree of blood‐brain barrier permeability.
Keywords: breast cancer, CDK4/6 inhibitor, cell cycle, glioblastoma, SPH3643
1. INTRODUCTION
Dysregulation of the cell cycle is a primary hallmark of cancer. 1 The cyclin‐dependent kinases (CDK)4 and CDK6 belong to the retinoblastoma signaling pathway (CDK4/6‐cyclin D1‐Rb‐p16/ink4a), 2 , 3 and are involved in the promotion of the G1‐S phase transition during the cell cycle, 4 , 5 corresponding to the point where cell proliferation becomes independent of mitogens and growth factors. Retinoblastoma‐associated protein (Rb) controls and restricts the cell cycle progression through this G1‐S phase transition. 6 Both CDK4 and CDK6 negatively regulate this Rb protein. As downstream targets of RAS‐dependent kinase, CDK4 and CDK6 recruit Cyclin D to form a CDK4/CDK6‐Cyclin D complex, 7 phosphorylating Rb in the early G1 phase to relieve the transcription repression mediated by certain transcription factors, including those of the E2F family, 8 leading cells to pass through the G1 phase, and to activate downstream target genes that are required for the S phase transition and DNA replication. 5 , 9 Additionally, CDK4/6 can phosphorylate FOXM1 and E2F1 in a cell cycle‐independent manner. 10 , 11 Conversely, natural CDK inhibitors, such as p16/ink4a and p21cip1, can prevent CDK4/6 from binding to D‐type cyclins, 12 , 13 or they can stabilize the formation of the CDK4/6‐cyclin D complex and then prevent the generation of a CDK2‐cyclin E complex. 14 , 15
In many types of cancer, the retinoblastoma pathway has been found to be dysregulated, thereby promoting aberrant cell proliferation. Cyclin‐dependent kinase 4 is amplified or overexpressed in several tumors, including soft tissue sarcomas, glioblastoma, and melanoma, 16 , 17 leading to unrestrained CDK4 activity and increased cell proliferation. 18 , 19 Cyclin‐dependent kinase 6 overexpression has also been documented in lymphomas, leukemia, and melanomas as a consequence of chromosomal translocations. 19 Functional inactivation or genetic loss of function of p‐Rb has been observed in a wide range of malignancies, including breast cancer, non‐small‐cell lung cancer, and malignant glioma. 20 , 21 Amplification (15%‐20%) and overexpression (50%) of the cyclin D1 gene have also been identified in a significant fraction of human breast cancers. 22 , 23 Overexpression of cyclin E, or loss of p27Kip1, occurs at relatively high frequency in breast cancers and both are associated with poor disease outcomes. 24 P16/ink4a, a tumor suppressor gene and a negative regulator of CDK4, is deleted in 38% of melanomas. 18 All this evidence suggests that CDK4 and CDK6 represent ideal therapeutic targets in many cancer types.
To date, 3 CDK4/6 inhibitors had been approved by the FDA. Palbociclib, approved in February 2015, was the first to be announced in treating estrogen receptor (ER)+ metastatic breast cancer. Together with letrozole, palbociclib significantly prolongs patient disease‐free survival to 20.2 months, compared to 10.2 months for letrozole single‐agent treatment. 25 In phase III clinical trials, palbociclib combined with fulvestrant improved progression‐free survival (PFS) from 3.8 months to 9.2 months in patients with hormone receptor (HR)+/human epidermal growth factor receptor (HER)2− metastatic breast cancer. 26 Ribociclib, approved in March of 2017, also showed great therapeutic effect in HR+/HER2− late‐stage or advanced breast cancer. In September 2017, LY2835219 (abemaciclib) was approved by the FDA. LY2835219 plus fulvestrant showed significant improvement to PFS, increasing it to 16.4 months compared to 9.3 months for fulvestrant alone. 27 The combination of LY2835219 plus anastrozole or letrozole significantly improved the objective response rate and PFS in women with ER/progesterone receptor+/HER2− advanced breast cancer. 28
All 3 inhibitors are now primarily used in breast cancer treatment, and are undergoing clinical trials in other tumor types, with the goal of broadening their clinical indications. SPH3643 is a highly selective CDK4/6 inhibitor. By reducing Rb phosphorylation, SPH3643 can impair the proliferation of a broad range of Rb‐positive cancer cell lines in vitro and in vivo with better efficiency and less toxicity. In combination with letrozole, SPH3643 showed better efficiency and less toxicity in mice bearing aromatase‐resistant MCF‐7/ARO tumors. Palbociclib is currently undergoing clinical testing as a potential chemotherapeutic for the treatment of primary or secondary brain tumors. Here SPH3643 was found to efficiently and stably cross the blood‐brain barrier (BBB), yielding significantly elevated levels of this compound in the brain relative to plasma. These results suggest that SPH3643 is a potent CDK4/6 inhibitor well‐suited to treating Rb‐positive cancers, with great potential for the treatment of intracranial tumors.
2. MATERIALS AND METHODS
2.1. Cell lines and culture
MCF‐7, ZR‐75‐1, T‐47D, SK‐OV‐3, A549, NCI‐H292, LN‐18, MV‐4‐11, and U87‐MG cell lines were purchased from ATCC. MDA‐MB‐468, ES‐2, SW620, Colo‐205, and HepG2 cells were ordered from the Shanghai Institutes for Biological Sciences cell bank. MCF‐7/ARO cells which were stably transfected with human aromatase. Cells were maintained at 37°C at 5% CO2 in DMEM or RPMI‐1640 containing 10% FBS (Life Technologies).
2.2. Drugs and Abs
LY2835219 and palbociclib were purchased from Shanghai Kermanda Biotech. SPH3643 was synthesized by Shanghai Pharmaceuticals Holdings. These drugs were dissolved in DMSO at 10 mmol/L and stored at −20°C. Antibodies against Rb, pRbS780, and cyclin D were purchased from Cell Signaling Technology. Antibodies against β‐tubulin and β‐actin were purchased from Sigma.
2.3. Enzymatic profiling
The BRAF and CRAF enzymatic activities were evaluated using the LanthaScreen technology according to the manufacturer’s protocol (Invitrogen). Checkpoint kinase (CHK)1 and CHK2 activities were determined using the ADP‐Glo kit (Promega) through an Envision2014 Multiple Reader. The activities of 26 additional enzymes including CDK4 and CDK6 were measured with a Caliper EZ Reader II by testing the phosphorylation level of FAM‐labeled peptide substrates. The CDK3 and CDK5 activity was determined using a LANCE Ultra assay (PerkinElmer). The relative inhibition rate of each data point was calculated to obtain the IC50 value for each compound.
2.4. Cell proliferation assay
Cells seeded into 96‐well plates were treated with a range of compound concentrations (1‐104 nM) for 6 days. Inhibition of cell proliferation was then measured using an MTT assay for Colo‐205 and MV4‐11 cells by Synergy H4 at 570 nm. The SRB assay was used for the other 15 cell lines, and optical density (OD) values were measured at 510 nm. Cell growth inhibition rates were calculated as follows: Inhibition rate = (OD value of control well − OD value of drug delivery well)/OD value of control well × 100%. The IC50 values were determined based on dose‐response curves.
For cell proliferation recovery experiments, cells were seeded into 6‐well plates and treated with 100 nM compounds for 6 days. Cells were seeded into 96‐well plates, and further treated with or without drugs for another 6 days. The CellTiter‐Glo assay (Promega) was used to evaluate the cell proliferation rate.
2.5. Western blot analysis
MCF‐7 cells or tissue samples were lysed in a buffer containing phosphatase inhibitors. Proteins were separated and transferred as well as incubated with Abs. Blots were developed with Immobilon Western HRP substrate Luminol reagent (Abcam, Cambridge, UK), and protein bands were detected with a Clinx Science instrument.
2.6. Flow cytometry
Cells were seeded into 6‐well plates and treated with a range of compound concentrations for 24 hours. For cell cycle recovery experiments, cells were washed twice with PBS and then cultured with medium without drugs for another 6, 12, or 24 hours. Cells were harvested and fixed with 70% pre‐iced ethanol. RNase and propidium iodide (BD Biosciences) were added and incubated at 37°C for 30 minutes in the dark. Fluorescence intensity was then measured by flow cytometry (BD FACSCalibur or Beckman CytoFlex). Data were analyzed by Kaluza.
2.7. Senescence assay (β‐galactosidase staining)
According to the manufacturer’s instructions of the Senescence Associated β‐Galactosidase Staining Kit (Cell Signaling Technology), cells were seeded into 6‐well plates and treated with indicated drugs or vehicle for 12 days and then stained by β‐Galactosidase Staining Solution.
2.8. Tumor xenograft studies
The solid tumor models Colo‐205, MCF‐7/ARO, MV4‐11, and U87‐MG were developed from cell lines and maintained in immune‐deficient mice. The mice were 5‐ to 6‐week‐old female BALB/c nude mice obtained from the Shanghai SLAC laboratory animal company (Colo‐205) and the Shanghai Lingchang Bio‐tech Company (MCF‐7/ARO, MV4‐11, and U87‐MG). When the mean tumor volume was approximately 100‐200 mm3, animals were randomized into subgroups according to tumor volume and compounds were administered. Drugs were given by lavage or s.c. injection (letrozole only), once daily, at a volume of 10 mL/kg. SPH3643 was formulated in 0.2% Tween‐80 + 0.5% caboxy methyl cellulose. LY2835219 and palbociclib were formulated in 0.1% Tween‐80. Letrozole was formulated in 20% PEG400 dissolved in normal saline. Tumor volume and body weight were measured twice weekly. At the end of the experiment, or when the tumor volume reached 1500 mm3, tumors were collected and captured after animals were killed using CO2.
All animal studies were carried out in accordance with Association for Assessment and Accreditation of Laboratory Animal Care guidelines. Tumor volumes were estimated based on the formula: Volume = long × width2 × 0.5.
The rejection rate was estimated according to the formula: the tumor growth inhibition value (TGI) (%) = 100 − T/C (%); or, when tumors showed regression: TGI (%) = 100 − (T − T0) / T0 × 100. T and C represented tumor volumes at the end of the experiment, and T0 and C0 were tumor volumes at the beginning of the experiment.
2.9. MCF‐7 CDX model
17β‐Estradiol (0.18 mg) was implanted s.c. into 9‐ to 10‐week‐old BALB/c nude mice 3 days before tumor inoculation. Each mouse was inoculated s.c. with MCF‐7 cells (107) in 0.2 mL PBS supplemented with Matrigel (1:1). Treatments were started on day 7 after inoculation (average tumor size reached approximately 169 mm3). Tumors, plasma, and brains were collected after animals were asphyxiated with CO2.
2.10. U87‐MG‐Luc human glioblastoma orthotopic model
Cell suspensions of 3 × 105 U87‐MG‐Luc cells and 20% Matrigel were slowly injected into a hole approximately 2 mm to the right of the anterior fontanel and 1 mm ahead of the coronal on the skulls of 6‐ to 8‐week‐old female BALB/c nude mice (Shanghai BK Laboratory Animal Co.). On day 8 after tumor cell inoculation, the animals were randomly assigned to groups based on tumor bioluminescence intensity. Tumor bioluminescence was detected weekly and used for the calculation of T/C values (percentages), with T and C representing the relative mean bioluminescence of the treated and control groups, respectively.
2.11. Plasma and brain tissue drug concentration test
SPH3643 and LY2835219 were given continuously for 28 days. Plasma, brain tumor, and brain tissue samples were collected at 2 time points of 1.5 and 24 h after the last treatment, and the drug concentration was determined. However, due to the absence or small brain tumors in most groups of animals, the relevant drug concentration could not be detected in brain tumor samples.
2.12. Bioluminescence measurements
The surgically inoculated mice were weighed and i.p. treated with luciferin (150 mg/kg). Ten to 15 minutes after the luciferin injection, the animals were pre‐anesthetized with a mixture of oxygen and isoflurane. When the animals were completely anesthetized, the mice were moved into the imaging chamber for bioluminescent measurements with an IVIS (Lumina II) imaging system.
2.13. Statistical analyses
Data were expressed as means ± SD or means ± SEM. Student’s t tests were used to analyze between two groups. One‐way ANOVAs were used for comparisons of 3 or more groups. Data were analyzed with SPSS version 17.0 or GraphPad Prism 7.0. The IC50 values were calculated by nonlinear regression analysis of dose‐response curves. P < .05 was considered to be statistically significant.
3. RESULTS
3.1. SPH3643 is a potent and selective CDK4/6 inhibitor
To define the potency and Cdk‐inhibitory activity of SPH3643, this compound was tested with a panel of enzymes in parallel with LY2835219 and palbociclib (Table 1, Figure S1). SPH3643 showed highly selective inhibition of CDK4 (IC50 = 2.20 nmol/L) and CDK6 (IC50 = 8.30 nmol/L), with little or no activity against other Cdk kinases or other assayed protein kinases. SPH3643 showed comparable inhibitory effects on CDK4 as LY2835219, stronger than palbociclib. For CDK6, SPH3643 showed more significant inhibition than the other 2 inhibitors. Consequently, SPH3643 has better potential to serve as a dual CDK4/6 inhibitor.
Table 1.
Protein kinase profiling
| Protein kinase |
SPH3643 IC50 (nmol/L) |
LY2835219 IC50 (nmol/L) |
Palbociclib IC50 (nmol/L) |
|---|---|---|---|
| CDK4 | 2.2 | 2.6 | 3.7 |
| CDK6 | 8.3 | 23 | 12 |
| CDK9 | 171 | 52 | 1380 |
| CDK5 | 311.25 | 229 | >3333 |
| CDK2 | 350 | 74 | 8773 |
| CDK1 | 812 | 436 | >10 000 |
| CDK3 | 838.26 | 482.2 | >3333 |
| CDK7 | 862 | 1876 | >10 000 |
| PKCa | 73 | — | — |
| FLT3 | 75 | — | — |
| GSK3β | 88 | — | — |
| JNK3 | 378 | — | — |
| P70S6K | 614 | — | — |
| JNK2 | 832 | — | — |
| TRK‐A | 891 | — | — |
| CHK1 | 1707 | — | — |
| PDGFRb | 1948 | — | — |
| AMPKa1 | 2100 | — | — |
| FLT4 | 2776 | — | — |
| CHK2 | 5666 | — | — |
| AURA | 5754 | — | — |
| AURB | 6974 | — | — |
| FLT1 | 8265 | — | — |
| EGFR | 8315 | — | — |
| KDR | 9518 | — | — |
| PDGFRa | >10 000 | — | — |
| AKT1 | >10 000 | — | — |
| ERK1 | >10 000 | — | — |
| MAPKAPK2 | >10 000 | — | — |
| BRAF | >10 000 | — | — |
| CRAF | >10 000 | — | — |
| FGFR1 | >10 000 | — | — |
IC50 values were calculated by nonlinear regression analysis of dose‐response curves.
—, not present; AUR, Aurora; AMPK, Adenosine 5‘‐monophosphate‐activated protein kinase; CDK, cyclin‐dependent kinase; CHK, checkpoint kinase; EGFR, epidermal growth factor receptor; ERK, extracellular regulated protein kinases; FGFR, fibroblast growth factor receptor; FLT, fms‐related tyrosine kinase; GSK3β, glycogen synthase kinase 3β; JNK, c‐Jun N‐terminal kinase; KDR, kinase insert domain receptor; PDGFR, platelet‐derived growth factor receptor; MAPKAPK, mitogen‐activated protein kinase–activated protein kinase; p70S6K, p70 Ribosomal protein S6 kinase; PKC, protein kinase C; TRK‐A, tyrosine kinase receptor A.
3.2. SHP3643 shows in vitro antitumor activity
Next, the antitumor efficacy of SPH3643 in various cancer cell lines was examined (Table 2, Figure S2). For Rb‐positive breast cancer cells, SPH3643 mediated comparable inhibition of the proliferation of MCF‐7 (IC50 = 203.5 ± 63.8 nmol/L) and T‐47D (IC50 = 628.8 ± 18.5 nmol/L) cells relative to LY2835219, whereas it was more effective than these reference compounds at inhibiting the proliferation of MCF‐7/ARO (IC50 = 178.9 ± 58.9 nmol/L) and ZR‐75‐1 (IC50 = 176.8 ± 26.2 nmol/L) cells. But it showed limited sensitivity on Rb‐negative MDA‐MB‐468 breast cancer cells (IC50 > 5000 nmol/L). We further analyzed a variety of Rb‐positive cell lines, and SPH3643 was more effective than the other 2 compounds in inhibiting the proliferation. Notably, SPH3643 showed greater inhibition of glioblastoma (IC50 = 33.8 ± 6.60 nmol/L) than LY2835219 (IC50 = 467.5 ± 267.6 nmol/L) and palbociclib (IC50 = 253.2 ± 119.0 nmol/L), indicating potential utility for glioblastoma treatment. In summary, SPH3643 showed wide spectrum proliferation inhibition on Rb‐positive cells, which was comparable to LY2835219, but stronger than palbociclib.
Table 2.
Proliferation inhibition by SPH3643 in tumor cells
| Cell line | Cell type | Rb expression | IC50 (nmol/L, mean ± SD) | ||
|---|---|---|---|---|---|
| SPH3643 | LY2835219 | Palbociclib | |||
| MCF‐7 | Breast cancer | Rb++ | 203.5 ± 63.8 | 218.5 ± 8.7 | 370.5 ± 68.9 |
| MCF‐7/ARO | Breast cancer | Rb++ | 178.9 ± 58.9 | 327.2 ± 43.8 | 373.7 ± 48.7 |
| T‐47D | Breast cancer | Rb+ | 628.8 ± 18.5 | 672.1 ± 119.2 | 1115.7 ± 686.4 |
| ZR‐75‐1 | Breast cancer | Rb++ | 176.8 ± 26.2 | 260.5 ± 88.7 | 758.7 ± 79.8 |
| MDA‐MB‐468 | Breast cancer | Rb− | >5000 | >2000 | >5000 |
| ES‐2 | Ovarian cancer | Rb++ | 15.7 ± 0.5 | 27.8 ± 10.3 | 53.4 ± 1.3 |
| SK‐OV‐3 | Ovarian cancer | Rb++ | 576.5 ± 80.1 | 526.3 ± 5.0 | 1979.0 ± 1000.6 |
| SW620 | Colon cancer | Rb+ | 145.0 ± 12.4 | 282.8 ± 42.7 | 267.6 ± 36.8 |
| Colo‐205 a | Colon cancer | Rb++ | 62.5 ± 10.4 | 117.5 ± 18.2 | 110.1 ± 2.5 |
| A549 | Lung cancer | Rb+ | 206.6 ± 52.0 | 263.5 ± 28.4 | 794.7 ± 34.5 |
| NCI‐H292 | Lung cancer | Rb++ | 469.7 ± 39.0 | 902.7 ± 93.8 | 850.1 ± 265.2 |
| Calu‐3 | Lung cancer | Rb++ | 1772.0 ± 185.3 | 1005.0 ± 30.0 | >10 000 |
| U87‐MG | Glioblastoma | Rb++ | 33.8 ± 6.6 | 467.5 ± 267.6 | 253.2 ± 119.0 |
| LN‐18 | Glioblastoma | Rb++ | 136.2 ± 23.7 | 244.4 ± 3.0 | 112.5 ± 25.6 |
| MV‐4‐11 a | Acute myeloid leukemia | Rb++ | 32.6 ± 14.0 | 79.8 ± 35.3 | 43.1 ± 9.5 |
| HepG2 | Hepatocellular carcinoma | Rb++ | 39.4 ± 5.7 | 82.3 ± 20.4 | 60.6 ± 9.0 |
Cells were treated with SPH3643, LY2835219, or palbociclib for 6 d.
Rb, retinoblastoma protein.
Cell viability was determined by MTT assay. Other cells’ cell viability was determined by SRB assay. IC50 values were calculated by nonlinear regression analysis of dose‐response curves (n = 2; mean ± SD).
SPH3643 inhibits Rb expression and phosphorylation without interfering with cyclin D1 expression, leading to a recoverable G1 phase cell cycle arrest and cellular senescence.
The CDK4/6 inhibitors have been developed to inhibit Rb phosphorylation, thus preventing cells from proceeding through the G1 restriction point and thereby disrupting cell proliferation. 29 In this study, the MCF‐7 cells were treated with compounds for 24 hours, and then complementary cellular assays were undertaken to monitor Rb phosphorylation and cell cycle status. SPH3643 treatment resulted in a significant decrease in RbSer780 phosphorylation and total Rb protein levels in a dose‐dependent manner, as did LY2835219 and palbociclib, without significantly altering the expression of the CDK4/6 binding partner cyclin D1 (Figure 1A).
Figure 1.
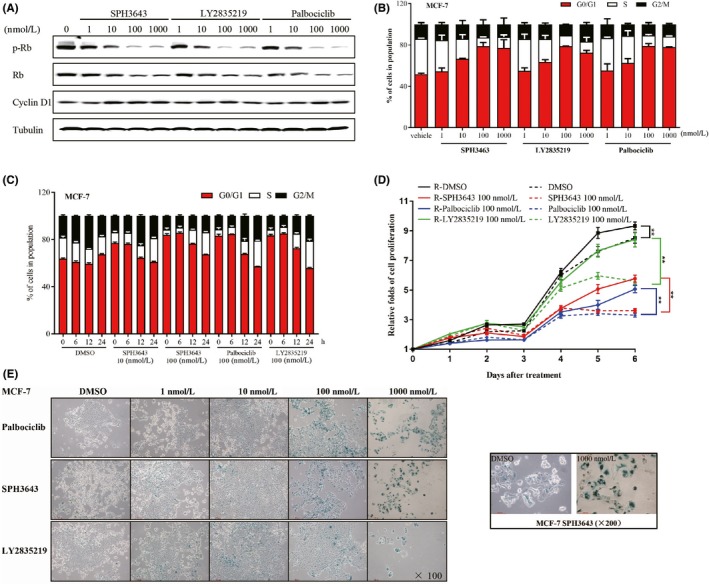
SPH3643 inhibits retinoblastoma protein (Rb) phosphorylation (p‐Rb) without interfering with cyclin D1 expression, mediating a reversible G1 phase cell cycle arrest and cellular senescence. A, After 24 h of treatment, western blotting was used to test indicated Abs. B, DNA content was accessed after 24 h of treatment. C, Reversibility assay. After 24 h of exposure to drugs, cells were further treated with replaced drug‐free medium for 6, 12, and 24 h, after which DNA content was assessed. D, Cell proliferation. MCF‐7 cells were first treated with 100 nM each drug for 6 days, then the cells were treated with or without drugs for another 6 days. Proliferation of MCF‐7 cells was measured each day. E, MCF‐7 cells were treated with drugs at the indicated concentrations for 12 d, and the activity of SA‐β‐galactosidase was determined by SA‐β‐gal staining. Blue indicates positive staining. *P < .05; **P < .01
Treating MCF‐7 cells (Figure 1B) with the 3 drugs resulted in cell cycle arrest at the G1 phase in a dose‐dependent manner as well as in U87‐MG (Figure S3A). This phenotype began to manifest at 10 nM, consistent with the changes in phosphorylation levels of RbSer780. The frequency of cells in the G0/G1 phase increased from 51.85 ± 0.88% to 76.99 ± 8.15% (1000 nmol/L) in SPH3643 treated cells. However, SPH3643 treated cells exhibited minimal cell apoptosis (Figure S4).
Cell cycle arrest can be reversible or irreversible. 30 To test the reversibility of the cell cycle arrest induced by CDK4/6 inhibitors, MCF‐7 cells were first treated with 3 compounds for 24 hours, then the drugs were removed from the medium for another 6, 12, and 24 hours. DNA content was then assessed by flow cytometry. The results showed that the cell cycles could return to a level comparable to that of the control group (Figure 1C). Similar results were also obtained in U87‐MG cells (Figure S3B). Cell proliferation is a direct indicator of the antitumor ability of drugs. To test whether the cell proliferation is reversible, MCF‐7 cells were first treated for 6 days, then the cells were treated with or without drugs for another 6 days. As shown in Figure 1D, when the drug was removed, MCF‐7 cell proliferation was reversible (P < .01).
Cellular senescence was used to test the irreversible cell cycle arrest. After a 12‐day exposure to SPH3643, senescence β‐galactosidase staining was used to investigate whether it could induce senescence. As shown in Figure 1E, SPH3643 could induce cell senescence in MCF‐7 cells (Huh‐7 as a positive control, 31 Figure S5) in a dose‐dependent manner as well as LY2835219 and palbociclib.
Together, these data indicated that SPH3643 significantly inhibited the Rb expression and phosphorylation, which contributed to the reversible G1 phase arrest cell cycle and cellular senescence, and leading to cell proliferation inhibition.
3.3. In vivo antitumor activity in Rb‐positive s.c. tumor xenografts
In vivo antitumor activity of SPH3643 was further assessed. The MCF‐7, MCF‐7/ARO, Colo‐205, MV4‐11, and U87‐MG cell lines, which corresponded to potential clinical indications of SPH3643, were used to generate s.c. xenograft models in nude mice (Table 3, Figure 2A‐F). As shown in Table 3, SPH3463 could inhibit tumor growth in a dose‐dependent manner that was comparable to that of LY2835219 and palbociclib in Colo‐205, MV4‐11, and U87‐MG (Figure 2D‐F) s.c. xenograft models. In addition, SPH3643 was well tolerated by tumor‐bearing mice, and no significant weight loss was observed, indicating no significant toxicity at all doses.
Table 3.
Antitumor activity of SPH3463 in s.c. human tumor xenografts
| Subcutaneous xenograft | Treatment | Method of treatment | Concentration (mg/kg) | TGI (%) | Body weight change (%) |
|---|---|---|---|---|---|
| MCF‐7 | Vehicle | i.g., QD´21 d | — | — | ↑1.6 |
| SPH3643 | 12.5 | 62 | ↓0.9 | ||
| 25 | 82** | ↓2.9 | |||
| 50 | 96**, a | ↓3.3 | |||
| LY2835219 | 50 | 114** | ↓6.0 | ||
| Palbociclib | 50 | 84** | ↓1.4 | ||
| MCF‐7/ARO | Vehicle | i.g., QD´21 d | — | — | ↑15 |
| SPH3643 | 37.5 | 51* | ↓3 | ||
| 75 | 60** | ↑1 | |||
| 150 | 71** | ↓4 | |||
| LY2835219 | 150 | 67** | ↓17 | ||
| Palbociclib | 150 | 67** | ↓11 | ||
| MCF‐7/ARO | Vehicle | i.g., QD´21 d | — | — | ↓1 |
| SPH3643 | 75 | 44** | ↓10 | ||
| LY2835219 | 75 | 49** | ↓12 | ||
| Letrozole | 20 | 45** | ↓1 | ||
| SPH3643 + letrozole | 75 + 20 | 74**,b,c | ↓7 | ||
| LY2835219 + letrozole | 75 + 20 | 62** | ↓13 | ||
| Colo‐205 | Vehicle | i.g., QD´14 d | — | — | ↓11 |
| SPH3643 | 25 | 73** | ↑2 | ||
| 50 | 86** | ↑1 | |||
| 100 | 99** | ↑9 | |||
| LY2835219 | 50 | 82** | ↑5 | ||
| Palbociclib | 50 | 93** | ↑8 | ||
| MV4‐11 | Vehicle | i.g., QD´20 d | — | — | ↑21 |
| SPH3643 | 12.5 | 11 | ↑9 | ||
| 25 | 31* | ↑6 | |||
| 50 | 56**, # | ↑3 | |||
| LY2835219 | 50 | 54**, # | ↑5 | ||
| Palbociclib | 50 | 68** | ↑3 | ||
| U87‐MG | Vehicle | i.g., QD´18 d | — | — | ↑10 |
| SPH3643 | 12.5 | 70** | ↑6 | ||
| 25 | 88** | ↑7 | |||
| 50 | 139**,## | ↑5 | |||
| LY2835219 | 50 | 131**,## | ↓2 | ||
| Palbociclib | 50 | 96** | ↑6 |
*P < .05, treatment vs vehicle; **P < .01, treatment vs vehicle; #P < .05, treatment vs palbociclib in U87‐MG or MV4‐11 cells; ##P < .01, treatment vs palbociclib in U87‐MG or MV4‐11 cells.
aSPH3643 50 mg/kg vs LY2835219 50 mg/kg.
bSPH3643 + letrozole vs SPH3643 75 mg/kg.
cSPH3643 + letrozole vs LY2835219 + letrozole.
—, not applicable; i.g., intragastric administration; QD, once daily; TGI, the tumor growth inhibition value.
Figure 2.
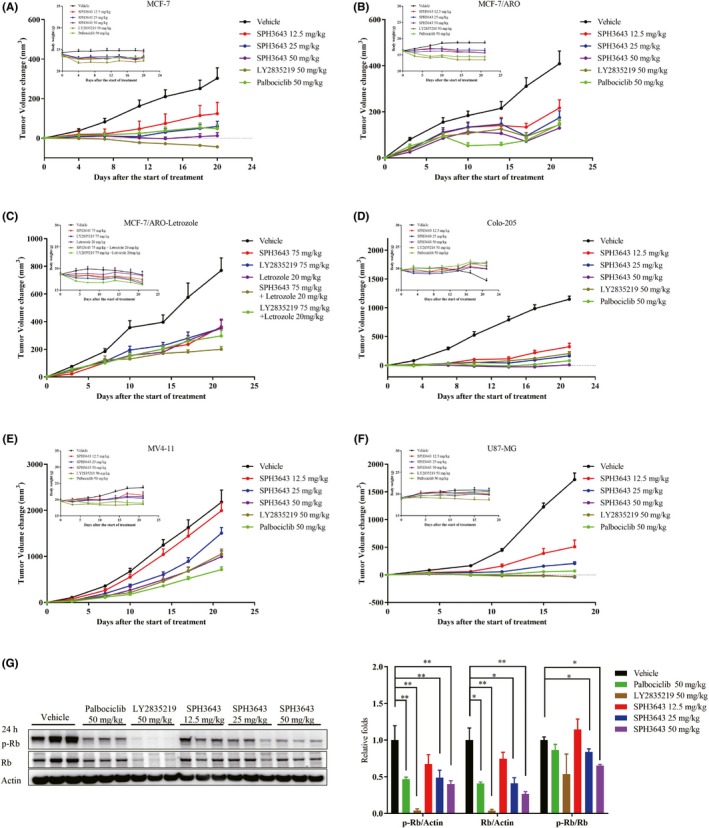
Antitumor effects of SPH3643 in s.c. murine tumor models. Tumor volume changes and body weights of xenograft mice were measured and recorded. A range of doses of SPH3643, LY2835219, and palbociclib were used to treat human xenograft mice bearing the (A) MCF‐7, (B) MCF‐7/ARO, (D) Colo205, (E) MV4‐11, and (F) U87‐MG cell lines. C, SPH3643 and LY2835219, alone or combined with letrozole, were tested in MCF‐7/ARO xenograft mice. G, Therapeutically active doses of SPH3463 in vivo caused the downregulation of retinoblastoma protein (Rb) Ser780 in MCF‐7 xenograft tumor tissues (mean ± SD). Tumor volumes are presented as means ± SEM. p‐Rb, phosphorylated Rb
The MCF‐7 model system (Figure 2A) was explored as a representative system. The mean tumor size of the vehicle‐treated mice reached 703 mm3 (relative tumor volume [RTV] = 403%) at day 20. Compared with the vehicle group, SPH3643 at doses of 25 and 50 mg/kg showed significant antitumor activity, with mean tumor sizes of 266 mm3 (RTV = 159%, TGI = 82%, P < .01) and 188 mm3 (RTV = 112%, TGI = 96%, P < .01), respectively. Palbociclib (50 mg/kg) and LY2835219 (50 mg/kg) also achieved significant efficacy, with mean final tumor sizes of 256 mm3 (RTV = 148%, TGI = 84%, P < .01) and 94 mm3 (RTV = 56%, TGI = 114%, P < .01), respectively. SPH3643 showed similar antitumor efficacy at identical doses as palbociclib (P > .05), less effective than LY2835219 (P < .05). At the end of the experiment, levels of Rb and p‐Rb in tumor tissues were assessed. Western blotting revealed that the expression ratios of p‐Rb and Rb were significantly inhibited by SPH3643 (25 mg/kg and 50 mg/kg) after 24 hours (Figure 2G). In summary, SPH3643 showed dose‐dependent antitumor efficacy against the MCF‐7 xenograft model, and was well tolerated by mice at all tested doses.
Breast cancer patients receiving long‐term endocrine drug treatment develop therapeutic resistance. The CDK4/6 inhibitors have shown promise for treating breast cancer in combination with aromatase inhibitors to overcome endocrine therapy resistance in clinical trials. The MCF‐7/ARO cell line, an aromatase inhibitor‐resistant breast cancer cell line, was selected to assess whether CDK4/6 inhibitors alone or in combination with letrozole can overcome aromatase inhibitor resistance. Subcutaneously transplanted xenograft nude mice were orally treated with 37.5, 75, or 150 mg/kg SPH3643 for 21 days, and tumor growth was significantly inhibited by 51%, 60%, and 71%, respectively (Figure 2B). Subgroups treated with LY2835219 (150 mg/kg) and palbociclib (150 mg/kg) showed similar antitumor efficacy (67%) to the 150 mg/kg SPH3643 dose. After a 21‐day oral treatment, both letrozole (20 mg/kg) and SPH3643 (75 mg/kg) showed significant antitumor activity compared to control (Figure 2C). The combination of SPH3643 and letrozole showed a more effective tumor inhibition rate than either single agent, and the combination with SPH3643 was more effective than the combination with LY2835219 (75 mg/kg) in vivo. These data therefore suggested that SPH3643 combined with letrozole could represent great potential as treatment for breast cancer in postmenopausal patients.
3.4. SPH3643 can cross the BBB to target intracranial tumors
The BBB limits the entry of external substances, making it difficult for therapeutic agents to enter the brain. To further test the ability of SPH3643 to treat brain tumors, a U87‐MG‐Luc human glioblastoma orthotopic model was used. The results showed that SPH3643 was well tolerated by the animals (37.5, 75, and 150 mg/kg) (Figure 3A). As shown in Figure 3B and Table 4, the median survival time for LY2835219 (75 mg/kg) and palbociclib (75 mg/kg) treated animals was 42 ± 2 and 35 ± 10 days, increasing the life span by 40.00% and 16.67% compared with vehicle (P both <.01). SPH3643 (75 and 150 mg/kg) significantly extended the survival time in a dose‐dependent manner. The median survival time of mice treated with SPH3643 (75 mg/kg) was 39 days, a 30.00% increased life span compared to vehicle (P < .01). SPH3643 (150 mg/kg) treatment led to a median survival time of 55 days, an 83.33% increase of life span (P < .01). The analysis of bioluminescence (Figure 3C) suggested that SPH3643 (75 and 150 mg/kg) showed potent tumor growth inhibitory effects compared with the vehicle (P < .01 for both). At an identical dosage, SPH3643 showed a comparative effect with LY2835219 and palbociclib.
Figure 3.
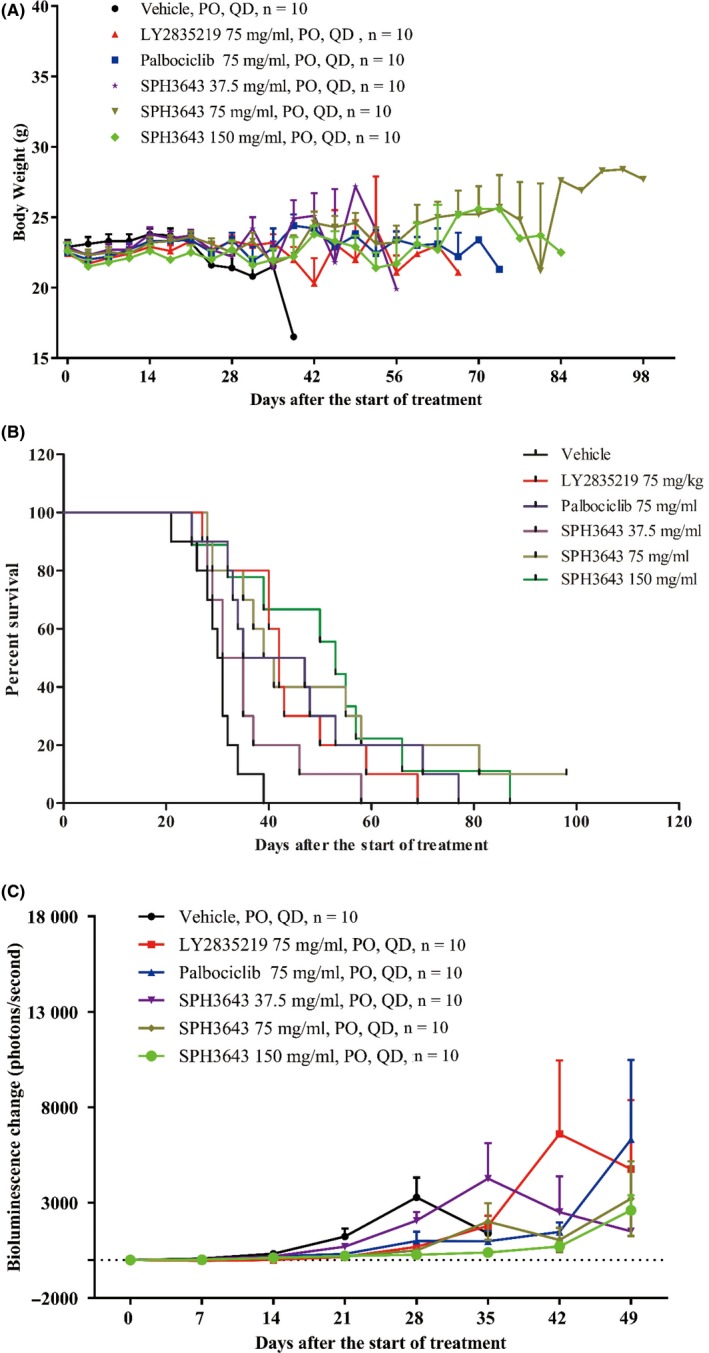
Antitumor effects of SPH3643 in U87‐MG intracranial murine tumor models. Body weight (A) and Kaplan‐Meier curves for the overall survival (B) of the intracranial U87‐MG xenografts treated with a range of doses of SPH3643 and with single doses of the cyclin‐dependent kinase inhibitors LY2835219 and palbociclib. C, Tumor bioluminescence changes of the mice in different treatment groups. Tumor growth in these mice was assessed based on total bioluminescent signal and on the treatment‐to‐control ratio in these intracranial U87‐MG model mice. Data are presented as means ± SEM. PO, per os; QD, once daily
Table 4.
Effects of LY2835219, palbociclib, and SPH3643 on the survival of U87‐MG‐Luc human glioblastoma orthotopic model in female BALB/c nude mice
| Treatment group | Median survival time (d) a | 95% confidence interval | Increase life span (%) | Log‐rank P value b | |
|---|---|---|---|---|---|
| Lower bound | Upper bound | ||||
| Vehicle | 30 ± 1 | 27.934 | 32.066 | — | — |
| LY2835219 (75 mg/kg) | 42 ± 2 | 38.964 | 45.036 | 40.00 | .0003** |
| Palbociclib (75 mg/kg) | 35 ± 10 | 14.856 | 55.144 | 16.67 | .0034** |
| SPH3643 (37.5 mg/kg) | 31 ± 2 | 26.351 | 35.649 | 3.33 | .1187 |
| SPH3643 (75 mg/kg) | 39 ± 3 | 32.802 | 45.198 | 30.00 | .0028** |
| SPH3643 (150 mg/kg) | 55 ± 3 | 49.156 | 60.844 | 83.33 | .0012** |
—, not applicable.
Mean ± SEM.
P value obtained by comparison of treatment groups with vehicle groups.
P < .01.
To explore the BBB permeability, blood and brain tissues were collected at 1.5 or 24 hours after the final dose to measure drug concentrations (Figure 4A‐C). SPH3643 was already at high levels in the brain at 1.5 hours (1126.67 ± 60.48 ng/g, 5671.67 ± 1705.04 ng/g, and 9900.00 ± 1719.74 ng/g), and the brain‐plasma ratio was 1.55 ± 0.26, 5.16 ± 1.64, and 5.92 ± 1.45, respectively. High brain concentration of SPH3643 was still detectable after 24 hours of exposure (1551.67 ± 514.45 ng/g, 8050.00 ± 661.44 ng/g, and 13 583.33 ± 2010.18 ng/g) and the brain‐plasma ratio remained at 4.02 ± 1.14, 9.84 ± 2.16, and 7.28 ± 0.36, respectively. In comparison, ratios for LY2835219 were only 0.26 ± 0.13 at 1.5 h and 0.27 ± 0.08 at 24 hours, and the levels of LY2835219 in the plasma and brain were markedly lower than SPH3643 at 24 hours, indicating a relatively quick elimination rate of LY2835219. In conclusion, SPH3643 could across the BBB more efficiently with longer persistence, making it an excellent candidate for brain tumor indications.
Figure 4.
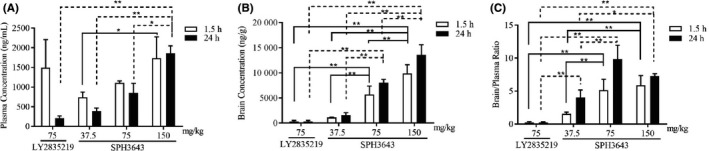
Brain penetration of SPH3643 in intracranial glioblastoma xenografts. Mean plasma (A) and brain (B) pharmacokinetic profiles and the brain : plasma ratios (C) of SPH3643, LY2835219, and palbociclib after oral treatment for 1.5‐24 h. *P < .05, **P < .01
4. DISCUSSION
In this study, we described SPH3643, a novel small‐molecule compound with high selectivity for CDK4/6 and broad antitumor activity in vitro and in vivo, due to its ability to arrest cells at the G1 phase by decreasing p‐Rb levels, and having stronger ability across the BBB and persistence in brain.
Breast cancer is the most common cancer in women. Approximately 80% of patients suffer from HR+ breast cancer. Endocrine therapy is the main treatment method in these patients. Unlike chemotherapy, this approach has less severe and more manageable adverse reactions, however, in the long term, patients develop therapeutic resistance. Cyclin‐dependent kinase 4/6 inhibitors have shown promise for treating breast cancer in combination with aromatase inhibitors, which have been used in the context of endocrine therapy in women with ER+ breast tumors. Resistance to aromatase inhibitors, however, has limited the usage of this therapy in many patients. Surprisingly, in an aromatase inhibitor resistance model, SPH3643 was able to repress tumor cell growth more effectively on its own or in combination with letrozole more readily than 2 other published inhibitors. This suggests that SPH3643 represents an ideal candidate compound for treating postmenopausal women with advanced breast cancer.
Glioblastoma is the most common primary tumor affecting the central nervous system, accounting for approximately 40% of central nervous system tumors and 78% of malignant central nervous system tumors in adults. It also remains the most malignant and fatal form of brain cancer, with adult and pediatric glioblastoma having a 5‐year survival rate of only 10%. Following standard treatment (including surgery, radiotherapy, and chemotherapy), 70% of glioblastoma patients presented with further tumor dissemination accompanied by serious side‐effects including cognitive decline and stroke. There is thus an urgent need for safer and more effective treatments for this deadly form of cancer.
Recently, there has been increasing interest in CDK4/6 inhibitors that can cross the BBB, with studies specifically testing these compounds against glioblastoma cell lines. 32 However, a phase II trial of palbociclib failed to achieve single‐agent efficacy in adult patients with recurrent Rb‐positive glioblastoma. 33 Therefore, there are still clinically relevant differences between clinical trials and animal trials. Only compounds that can effectively cross the BBB and mediate intracerebral efficacy have the potential to improve treatment of glioblastoma. Promisingly, in a mouse xenograft model of malignant glioblastoma, SPH3643 was able to more effectively mediate tumor regression and extend the life span of mice than LY2835219 or palbociclib. SPH3643 was also better able to cross the BBB. Furthermore, mice were able to tolerate SPH3643 without substantial weight loss, suggesting that this compound can be safely dosed in vivo without significant adverse events. For the treatment of brain tumors and metastases, SPH3643 thus represents a promising therapeutic candidate.
In xenograft tumor experiments, tumors showed signs of regression in animals treated with high‐dose CDK4/6 inhibitors, suggesting that these inhibitors had beneficial effects beyond merely arresting cells at the G1 stage. Selective CDK4/6 inhibitors have been previously reported to not only induce tumor cell cycle arrest, but to also promote antitumor immunity. 34 Researchers have proposed that these CDK4/6 inhibitors activate tumor cell expression of endogenous retroviral elements, thus increasing intracellular levels of double‐stranded RNA, which in turn stimulates production of type III interferons and thereby enhances tumor antigen presentation. These compounds can also promote antitumor immunity through markedly suppressing the proliferation of regulatory T cells. These findings suggest the potential for combinations of immunotherapy and CDK4/6 inhibitors.
In addition to breast cancer, studies regarding the efficacy of Cdk inhibitors are ongoing in other tumor types, including KRAS mutant tumors. Notably, CDK4/6 has been shown to trigger the death of KRAS mutant lung cancer cells in animal experiments, such that KRAS mutant tumor cell proliferation is blocked after loss of CDK4/6, whereas this does not occur in KRAS WT tumor cells. 35 Although phase III clinical trials pursuing these results were ultimately terminated, CDK4/6 inhibitors still show the potential for treatment of tumors with KRAS mutations. 36 , 37 Another report has shown CDK4/6 inhibitor efficacy in pancreatic cancers with KRAS mutation, suggesting that the combination of RAF and CDK4/6 inhibitors could represent a novel treatment strategy for KRAS G12R mutant pancreatic cancer. 38 Further unpublished data also revealed the effective activity of a candidate CDK4/6 inhibitor in KRAS mutant tumors, although more work will be needed to validate these findings.
In conclusion, SPH3643 is a potent, orally active small‐molecule inhibitor of CDK4/6 inhibitor with broad‐spectrum antitumor effect against Rb‐positive tumor, potent antitumor efficacy, higher safety, and a high degree of BBB permeability, indicating great potential in the treatment of brain metastases and tumors of the central nervous system.
CONFLICT OF INTEREST
The authors declare no potential conflicts of interest.
Supporting information
Fig S1‐S5
ACKNOWLEDGMENTS
This work was supported by the Program of Shanghai Subject Chief Scientist (B type) (15XD1523600) and Shanghai Science and Technology Innovation Action Plan (15431903400).
Liao X, Hong Y, Mao Y, et al. SPH3643: A novel cyclin‐dependent kinase 4/6 inhibitor with good anticancer efficacy and strong blood‐brain barrier permeability. Cancer Sci. 2020;111:1761–1773. 10.1111/cas.14367
Contributor Information
GuangXin Xia, Email: xiagx@sphchina.com.
YanJun Liu, Email: liuyj@sphchina.com.
REFERENCES
- 1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646‐674. [DOI] [PubMed] [Google Scholar]
- 2. Ezhevsky SA, Ho A, Becker‐Hapak M, Davis PK, Dowdy SF. Differential regulation of retinoblastoma tumor suppressor protein by G(1) cyclin‐dependent kinase complexes in vivo. Mol Cell Biol. 2001;21(14):4773‐4784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bartkova J, Lukas J, Guldberg P, et al. The p16‐cyclin D/Cdk4‐pRb pathway as a functional unit frequently altered in melanoma pathogenesis. Cancer Res. 1996;56(23):5475‐5483. [PubMed] [Google Scholar]
- 4. O'Leary B, Finn RS, Turner NC. Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol. 2016;13(7):417‐430. [DOI] [PubMed] [Google Scholar]
- 5. Harbour JW, Luo RX, Dei Santi A, Postigo AA, Dean DC. Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell. 1999;98(6):859‐869. [DOI] [PubMed] [Google Scholar]
- 6. Cobrinik D, Dowdy SF, Hinds PW, Mittnacht S, Weinberg RA. The retinoblastoma protein and the regulation of cell cycling. Trends Biochem Sci. 1992;17(8):312‐315. [DOI] [PubMed] [Google Scholar]
- 7. Kato J, Matsushime H, Hiebert SW, Ewen ME, Sherr CJ. Direct binding of cyclin D to the retinoblastoma gene product (pRb) and pRb phosphorylation by the cyclin D‐dependent kinase CDK4. Genes Dev. 1993;7(3):331‐342. [DOI] [PubMed] [Google Scholar]
- 8. Ezhevsky SA, Nagahara H, Vocero‐Akbani AM, Gius DR, Wei MC, Dowdy SF. Hypo‐phosphorylation of the retinoblastoma protein (pRb) by cyclin D:Cdk4/6 complexes results in active pRb. Proc Natl Acad Sci USA. 1997;94(20):10699‐10704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dyson N. The regulation of E2F by pRB‐family proteins. Genes Dev. 1998;12(15):2245‐2262. [DOI] [PubMed] [Google Scholar]
- 10. Anders L, Ke N, Hydbring P, et al. A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells. Cancer Cell. 2011;20(5):620‐634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fagan R, Flint KJ, Jones N. Phosphorylation of E2F–1 modulates its interaction with the retinoblastoma gene product and the adenoviral E4 19 kDa protein. Cell. 1994;78(5):799‐811. [DOI] [PubMed] [Google Scholar]
- 12. Ortega S, Malumbres M, Barbacid M. Cyclin D‐dependent kinases, INK4 inhibitors and cancer. Biochim Biophys Acta. 2002;1602(1):73‐87. [DOI] [PubMed] [Google Scholar]
- 13. Pavletich NP. Mechanisms of cyclin‐dependent kinase regulation: structures of Cdks, their cyclin activators, and Cip and INK4 inhibitors. J Mol Biol. 1999;287(5):821‐828. [DOI] [PubMed] [Google Scholar]
- 14. Cheng M, Olivier P, Diehl JA, et al. The p21(Cip1) and p27(Kip1) CDK 'inhibitors' are essential activators of cyclin D‐dependent kinases in murine fibroblasts. EMBO J. 1999;18(6):1571‐1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sugimoto M, Martin N, Wilks DP, et al. Activation of cyclin D1‐kinase in murine fibroblasts lacking both p21(Cip1) and p27(Kip1). Oncogene. 2002;21(53):8067‐8074. [DOI] [PubMed] [Google Scholar]
- 16. Barretina J, Taylor BS, Banerji S, et al. Subtype‐specific genomic alterations define new targets for soft‐tissue sarcoma therapy. Nat Genet. 2010;42(8):715‐721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Reifenberger G, Reifenberger J, Ichimura K, Collins VP. Amplification at 12q13‐14 in human malignant gliomas is frequently accompanied by loss of heterozygosity at loci proximal and distal to the amplification site. Cancer Res. 1995;55(4):731‐734. [PubMed] [Google Scholar]
- 18. Yadav V, Burke TF, Huber L, et al. The CDK4/6 inhibitor LY2835219 overcomes vemurafenib resistance resulting from MAPK reactivation and cyclin D1 upregulation. Mol Cancer Ther. 2014;13(10):2253‐2263. [DOI] [PubMed] [Google Scholar]
- 19. Malumbres M, Barbacid M. Cell cycle kinases in cancer. Curr Opin Genet Dev. 2007;17(1):60‐65. [DOI] [PubMed] [Google Scholar]
- 20. Borg A, Zhang QX, Alm P, Olsson H, Sellberg G. The retinoblastoma gene in breast cancer: allele loss is not correlated with loss of gene protein expression. Cancer Res. 1992;52(10):2991‐2994. [PubMed] [Google Scholar]
- 21. Paggi MG, Baldi A, Bonetto F, Giordano A. Retinoblastoma protein family in cell cycle and cancer: a review. J Cell Biochem. 1996;62(3):418‐430. [DOI] [PubMed] [Google Scholar]
- 22. Dickson C, Fantl V, Gillett C, et al. Amplification of chromosome band 11q13 and a role for cyclin D1 in human breast cancer. Cancer Lett. 1995;90(1):43‐50. [DOI] [PubMed] [Google Scholar]
- 23. Gillett C, Fantl V, Smith R, et al. Amplification and overexpression of cyclin D1 in breast cancer detected by immunohistochemical staining. Cancer Res. 1994;54(7):1812‐1817. [PubMed] [Google Scholar]
- 24. Dean JL, Thangavel C, McClendon AK, Reed CA, Knudsen ES. Therapeutic CDK4/6 inhibition in breast cancer: key mechanisms of response and failure. Oncogene. 2010;29(28):4018‐4032. [DOI] [PubMed] [Google Scholar]
- 25. Finn RS, Crown JP, Lang I, et al. The cyclin‐dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first‐line treatment of oestrogen receptor‐positive, HER2‐negative, advanced breast cancer (PALOMA‐1/TRIO‐18): a randomised phase 2 study. Lancet Oncol. 2015;16(1):25‐35. [DOI] [PubMed] [Google Scholar]
- 26. Cristofanilli M, Turner NC, Bondarenko I, et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone‐receptor‐positive, HER2‐negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA‐3): final analysis of the multicentre, double‐blind, phase 3 randomised controlled trial. Lancet Oncol. 2016;17(4):425‐439. [DOI] [PubMed] [Google Scholar]
- 27. Sledge GW Jr, Toi M, Neven P, et al. MONARCH 2: Abemaciclib in combination with fulvestrant in women with HR+/HER2‐ advanced breast cancer who had progressed while receiving endocrine therapy. J Clin Oncol. 2017;35(25):2875‐2884. [DOI] [PubMed] [Google Scholar]
- 28. Goetz MP, Toi M, Campone M, et al. MONARCH 3: Abemaciclib as initial therapy for advanced breast cancer. J Clin Oncol. 2017;35(32):3638‐3646. [DOI] [PubMed] [Google Scholar]
- 29. Lundberg AS, Weinberg RA. Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin‐cdk complexes. Mol Cell Biol. 1998;18:753‐761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Campisi J, d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8(9):729‐740. [DOI] [PubMed] [Google Scholar]
- 31. Bollard J, Miguela V, Ruiz de Galarreta M, et al. Palbociclib (PD‐0332991), a selective CDK4/6 inhibitor, restricts tumour growth in preclinical models of hepatocellular carcinoma. Gut. 2017;66(7):1286‐1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yin L, Li H, Liu W, et al. A highly potent CDK4/6 inhibitor was rationally designed to overcome blood brain barrier in glioblastoma therapy. Eur J Med Chem. 2018;144:1‐28. [DOI] [PubMed] [Google Scholar]
- 33. Taylor JW, Parikh M, Phillips JJ, et al. Phase‐2 trial of palbociclib in adult patients with recurrent RB1‐positive glioblastoma. J Neurooncol. 2018;140:477‐483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Goel S, DeCristo MJ, Watt AC, et al. CDK4/6 inhibition triggers anti‐tumour immunity. Nature. 2017;548(7668):471‐475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang XH, Cheng Y, Shin JY, Kim JO, Oh JE, Kang JH. A CDK4/6 inhibitor enhances cytotoxicity of paclitaxel in lung adenocarcinoma cells harboring mutant KRAS as well as wild‐type KRAS. Cancer Biol Ther. 2013;14(7):597‐605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee MS, Helms TL, Feng N, et al. Efficacy of the combination of MEK and CDK4/6 inhibitors in vitro and in vivo in KRAS mutant colorectal cancer models. Oncotarget. 2016;7:39595‐39608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ziemke EK, Dosch JS, Maust JD, et al. Sensitivity of KRAS‐mutant colorectal cancers to combination therapy that cotargets MEK and CDK4/6. Clin Cancer Res. 2016;22(2):405‐414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lee T, Kim K, Lee J, et al. Antitumor activity of sorafenib plus CDK4/6 inhibitor in pancreatic patient derived cell with KRAS mutation. J Cancer. 2018;9(18):3394‐3399. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S5