

Abstract
MicroRNAs (miRNAs) can act not only as tumor suppressor genes but also as oncogenes. Oncogenic miRNAs (oncomiRs) could therefore provide opportunities for the treatment of human malignancies. Here, we aimed to identify oncomiRs present in oral squamous cell carcinoma (OSCC) and addressed whether targeting these miRNAs might be useful in treatment for cancer. Functional screening for oncomiRs in a human OSCC cell line (GFP‐SAS) was carried out using the miRCURY LNA microRNA Knockdown Library – Human version 12.0. We identified a locked nucleic acid (LNA)/DNA antisense oligonucleotide against miR‐361‐3p (LNA‐miR‐361‐3p) which showed the largest degree of growth inhibition of GFP‐SAS cells. Transfection with a synthetic mimic of mature miR‐361‐3p resulted in an approximately 20% increase in the growth of GFP‐SAS cells. We identified odd‐skipped related 2 (OSR2) as a miR‐361‐3p target gene. Transfection of GFP‐SAS cells with LNA‐miR‐361‐3p caused a significant increase in the expression levels of OSR2. Cotransfection of a OSR2 3′‐UTR luciferase reporter plasmid and LNA‐miR‐361‐3p into GFP‐SAS cells produced higher levels of luciferase activity than in cells cotransfected with the LNA‐nontarget. We assessed the effect of LNA‐miR‐361‐3p on the in vivo growth of GFP‐SAS cells. We found that LNA‐miR‐361‐3p significantly reduced the size of s.c. xenografted GFP‐SAS tumors, compared to the control group treated with LNA‐NT. Finally, we observed that miR‐361‐3p is overexpressed in OSCC tissues. These results suggest that miR‐361‐3p supports the growth of human OSCC cells both in vitro and in vivo and that targeting miR‐361‐3p could be a useful therapeutic approach for patients with OSCC.
Keywords: LNA/DNA antisense oligonucleotide, microRNA, miR‐361‐3p, oncomiR, oral squamous cell carcinoma
This study shows that microRNA (miR)‐361‐3p supports the growth of human oral squamous cell carcinoma (OSCC) cells both in vitro and in vivo and that targeting miR‐361‐3p could be a useful therapeutic approach for patients with OSCC.

1. INTRODUCTION
Oral squamous cell carcinoma (OSCC) is the most frequently occurring cancer among head and neck squamous cell carcinomas (HNSCCs) with a worldwide estimated incidence of more than 300 000 new cases and 145 000 deaths in 2012.1 Oral squamous cell carcinoma has a high potential to invade local tissue and metastasize to lymph nodes and has an approximately 50% mortality within 5 years.2 Despite the increasing knowledge of the pathogenesis of OSCC and advances in chemotherapy, radiotherapy, and surgery, little improvement in the relative survival rate of patients with OSCC has been observed in the past several decades.2 Therefore, novel strategies based on a greater understanding of the pathogenesis of OSCC are needed in order to develop improved therapies.
MicroRNAs (miRNAs) belong to a class of small noncoding RNAs that influence many biological processes through binding to the 3′‐UTR of target mRNAs, mediating either mRNA degradation or translational repression. Thus, miRNAs play crucial roles in the modulation of physiological processes and in pathogenesis.3, 4 Several miRNAs, including miR‐21, miR‐92b, miR‐29b, and miR‐155, have been reported to regulate OSCC proliferation, migration, and invasion.5, 6, 7, 8 Addiction to these oncogenic miRNAs (oncomiRs) could provide therapeutic opportunities for treating human malignancies.
In this study, we aimed to identify novel oncomiRs in human OSCC cells and assessed the possibility of targeting these miRNAs for the treatment of cancer.
2. MATERIALS AND METHODS
2.1. Cells and cell culture
In this study, we used a human OSCC cell line, GFP‐SAS,9 which can form tumors when injected into athymic nude mice. Cells were maintained in DMEM (Wako) supplemented with 10% FBS (BioSource), 100 U/mL penicillin, and 100 µg/mL streptomycin (Wako), referred to here as complete medium, and grown in an incubator with a humidified atmosphere of 95% air and 5% CO2 at 37°C.
2.2. Functional screening of miRNA knockdown library
To identify novel oncomiRs in human OSCC cells, the miRCURY LNA microRNA Knockdown Library – Human version 12.0 (Exiqon) was used. We then transfected 918 locked nucleic acid (LNA)/DNA antisense oligonucleotides (ASOs) for specific human mature miRNAs into GFP‐SAS cells. Cells (2 × 103/well) were then seeded into a 96‐well plate in complete medium with 25 nmol/L of each ASO and Lipofectamine RNAiMAX (Thermo Fisher Scientific) in a final volume of 100 µL. After 80 hours, the cell number was evaluated using a WST‐8 assay (CCK‐8; Dojindo).
2.3. Cell growth assay
Cells (2 × 103/well) were seeded into a 96‐well plate in complete medium with 20 nmol/L of a synthetic mimic of human mature miR‐361‐3p and Lipofectamine RNAiMAX (Thermo Fisher Scientific) in a final volume of 100 µL. After 72 hours, the cell number was evaluated using a WST‐8 assay (Dojindo).
2.4. Quantitative RT‐PCR
Total RNA was extracted by lysing cells and tissues after homogenization with a TissueLyser (Qiagen) and ISOGEN (Nippon Gene) according to the manufacturer’s protocol. To analyze miRNA expression, cDNA was synthesized using a miScript II RT Kit (Qiagen), and quantitative RT‐PCR (qRT‐PCR) was then undertaken using a miScript SYBR Green PCR Kit (Qiagen). The PCR amplification was carried out in a 10 µL final reaction volume containing 5 µL of 2× QuantiTect SYBR Green Master Mix, 1 µL of 10× miScript Universal Primer, 1 µL of 10× miScript Primer Assay, 2 µL RNase free water, and 1 µL cDNA. The thermal‐cycling conditions were a PCR initial activation step at 95°C for 15 minutes, followed by 40 cycles of 94°C for 15 seconds and 55°C for 30 seconds. To analyze mRNA expression, we used a One Step SYBR PrimeScript RT‐PCR Kit II (Takara). Polymerase chain reaction amplification was carried out in a 10 μL final reaction mixture containing 5 μL of 2× One Step SYBR RT‐PCR Buffer 4, 0.4 μL PrimeScript One Step Enzyme Mix 2, 0.4 μL forward primer (10 μmol/L), 0.4 μL reverse primer (10 μmol/L), and 100 ng total RNA. Reverse transcription was undertaken at 42°C for 5 minutes, followed by 95°C for 10 seconds. The PCR amplification step consisted of 40 cycles at 95°C for 5 minutes and 60°C for 10 seconds. The relative expression levels of miRNAs and mRNAs were calculated using the ΔCt (cycle threshold) and normalized to the control using the equation 2−ΔΔCt. RNU6B and hydroxymethylbilane synthase (HMBS) were used as internal controls. SYBR Green 1 was detected using ViiA 7 (Thermo Fisher Scientific). The sequences of primers used were as follows: odd‐skipped related 2 (OSR2) forward, 5′‐TTT GCG GCA GAC ACT TTA CCA ‐3′ and reverse, 5′‐TTC CCA CAC TCC TGA CAT TTG A‐3′; and HMBS forward, 5′‐CAT GCA GGC TAC CAT CCA TGT C‐3′ and reverse, 5′‐GTT ACG AGC AGT GAT GCC TAC CAA‐3′.
2.5. Microarray analysis
Total RNA was extracted from cells at 24 hours after the treatment with LNA/DNA ASO against miR‐361‐3p (LNA‐miR‐361‐3p) or miR‐nontarget (LNA‐miR‐NT). We used an Affymetrix Human Genome U219 Array Strip (Affymetrix) according to the manufacturer’s instructions. After washing and staining the array strips, the signal was developed and scanning was undertaken using a GeneAtlas system (Affymetrix). Data analysis was carried out using GeneSpring GX 13 (Agilent Technologies) and a miRNA target filter in the Ingenuity Pathway Analysis (Qiagen). The microarray data have been deposited in Gene Expression Omnibus (experiment no. http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE138587) according to minimum information about microarray experiment guidelines.
2.6. Luciferase reporter assay
The plasmid, miTarget miRNA 3′ Target Clone (HmiT001838‐MT01; GeneCopoeia) was used in the luciferase reporter assay. GFP‐SAS cells were seeded in 96‐well plates and after 24 hours they were cotransfected with LNA‐miR‐361‐3p (20 nmol/L) and the OSR2 3′‐UTR plasmid (50 ng/well) complexed with Lipofectamine LTX Reagent (Thermo Fisher Scientific), and then incubated for 10 hours. Firefly and Renilla luciferase activities were measured sequentially using the Dual‐Glo Luciferase Assay System (Promega). Results were expressed as relative luciferase activity units measured using a Wallac 1420 ARVO MX/Light (PerkinElmer).
2.7. Western blot analysis
Cells were lysed in 0.5 M EDTA (Dojindo) and 1% NP‐40 (Nacalai Tesque) in PBS (Wako) containing a protease inhibitor cocktail and a phosphatase inhibitor (Roche Diagnostics). The lysates were centrifuged at 15 000 g for 15 minutes at 4°C and the supernatants were electrophoresed on SDS‐polyacrylamide gels and proteins transferred to PVDF membranes (Millipore). The membranes were blocked with 5% nonfat dried milk (Wako) in 1× TBS‐T (25 mmol/L Tris‐HCl, 125 mmol/L NaCl, and 0.1% Tween‐20) (Sigma‐Aldrich) for 1 hour at room temperature. They were then probed with a polyclonal rabbit anti‐OSR2 Ab (Abcam; diluted at 1:1000) or a monoclonal mouse anti‐β‐tubulin Ab (BD; diluted at 1:1000) in 5% nonfat dried milk in 1× TBS‐T for 1 hour at room temperature, followed by treatment with HRP‐conjugated secondary Abs against rabbit or mouse IgG (GE Healthcare) for 1 hour at room temperature. The immune complexes were visualized using an ECL Prime Western Blotting Detection Reagent (GE Healthcare). The density of visualized immune complexes was digitized using a RAS3000 imaging system (Fujifilm).
2.8. Xenograft model and tumor therapy
GFP‐SAS cells complexed with Matrigel (BD) at a density of 2 × 106 cells per 100 µL aliquot were injected s.c. at 2 sites in the flanks of male athymic nude mice (CLEA Japan). One week later, tumor‐bearing nude mice were randomly divided into 2 treatment groups, LNA‐miR‐361‐3p or LNA‐miR‐NT. These ASOs (50 µg) were injected into the xenograft tumors every 3 days. Tumor diameters were measured at regular intervals using digital calipers, and tumor volume (mm3) was calculated using the following formula: length × width × height × 0.523. Sixteen days after the first treatment, the xenografts were dissected and the miR‐361‐3p and OSR2 expression levels were determined by qRT‐PCR. These animal studies were approved by the Ehime University animal care committee.
2.9. Statistical analysis
Student’s t tests were used to determine the significance of differences between groups. Differences with P values of less than .05 were considered statistically significant. Statistical analyses were undertaken using GraphPad Prism software, version 5.04 (GraphPad Software).
3. RESULTS
3.1. Identification of oncomiR candidates in human OSCC cells
We carried out functional screening for novel oncomiRs in the human OSCC cell line GFP‐SAS using miRCURY LNAM microRNA Knockdown Library – Human version 12.0 (Exiqon). A total of 918 LNA/DNA ASOs specific for human mature miRNAs were transfected into the human GFP‐SAS cells. After 80 hours, cell growth was evaluated using a WST‐8 assay (Figure 1A, Table S1). Thirteen of these LNA/DNA ASOs that targeted miRNAs significantly inhibited the growth of GFP‐SAS cells by more than 60% (Figure 1B). Among these, we found the LNA/DNA ASO against miR‐361‐3p (LNA‐miR‐361‐3p) showed the greatest growth inhibition (Figure 1B, C). Next, we confirmed the target specificity of LNA‐miR‐361‐3p by qRT‐PCR and found that LNA‐miR‐361‐3p significantly reduced the expression of miR‐361‐3p (Figure 1D). Cotransfection of a synthetic mimic of mature miR‐361‐3p abrogated the growth inhibitory effect of LNA‐miR‐361‐3p in GFP‐SAS cells (data not shown). Furthermore, transfection of this synthetic mimic of mature miR‐361‐3p resulted in an approximately 20% increase in the growth of GFP‐SAS cells. (Figure 1D). These results indicate that miR‐361‐3p supports the growth of these human OSCC cells.
Figure 1.
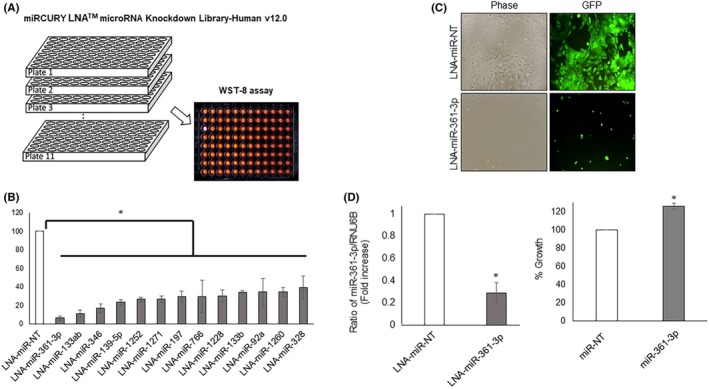
Identification of oncogenic microRNA (oncomiR) candidates in human oral squamous cell carcinoma (OSCC) cells. A, Functional screening for oncomiRs in OSCC cells (GFP‐SAS) was carried out using miRCURY LNA microRNA Knockdown Library – Human version 12.0 (Exiqon). GFP‐SAS cells were transfected with a total of 918 locked nucleic acid (LNA)/DNA antisense oligonucleotides (25 nmol/L) complexed with Lipofectamine RNAiMAX that targeted specific human mature miRNAs. After 80 hours, cell growth was evaluated using a WST‐8 assay. B, C, LNA‐miR‐361‐3p showed the largest growth inhibition. D, LNA‐miR‐361‐3p specifically downregulates the levels of endogenous miR‐361‐3p. Transfection of synthetic mimic of mature miR‐361‐3p (20 nmol/L) into GFP‐SAS cells resulted in an approximately 20% increase in cell growth. *P < .01 compared to control culture. NT, nontarget
3.2. MicroRNA‐361‐3p target genes
We attempted to identify the miR‐361‐3p target genes using a microarray analysis to evaluate the function and transfection efficiency in vivo of LNA‐miR‐361‐3p. The expression levels of 22 genes were markedly upregulated by miR‐361‐3p inhibition in GFP‐SAS cells (Table S2). Among these genes, only the OSR2 mRNA had the target sequence for miR‐361‐3p in its 3′‐UTR (Figure 2A). Cotransfection of an OSR2 3′‐UTR luciferase reporter plasmid and LNA‐miR‐361‐3p into GFP‐SAS cells produced higher luciferase activity than cells cotransfected with LNA‐miR‐NT (Figure 2B). Also, the expression levels of OSR2 mRNA after transfection with LNA‐miR‐361‐3p were significantly increased (Figure 2B). Subsequently, we examined the expression levels of the OSR2 protein by western blot analysis. Overexpression of miR‐361‐3p reduced the expression level of the OSR2 protein, whereas knockdown of miR‐361‐3p enhanced OSR2 protein expression (Figure 2C). These findings suggest that OSR2 is a direct target gene of miR‐361‐3p in GFP‐SAS cells.
Figure 2.
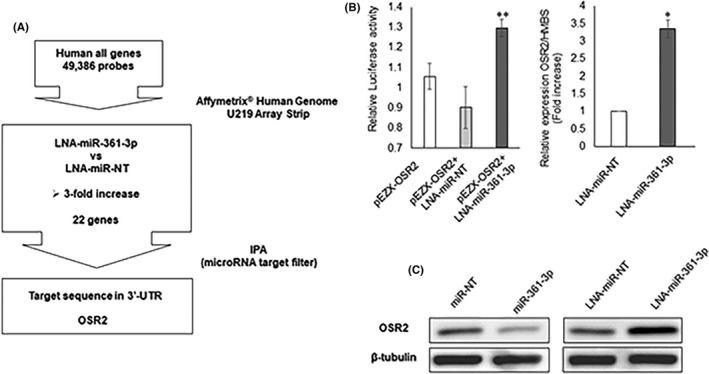
Identification of microRNA (miR)‐361‐3p target genes. A, We identified odd‐skipped related 2 (OSR2) as a target gene for miR‐361‐3p through the use of a microarray, GeneSpring GX, and the microRNA target filter in Ingenuity Pathway Analysis (IPA). B, Cotransfection of the OSR2 3′‐UTR luciferase reporter plasmid (pEZX‐OSR2) and locked nucleic acid (LNA)‐miR‐361‐3p into GFP‐SAS cells produced higher luciferase activity levels than cells cotransfected with LNA‐miR‐nontarget (NT). Expression of OSR2 mRNA after transfection with LNA‐miR‐361‐3p was higher than after transfection of LNA‐miR‐NT. C, Overexpression of miR‐361‐3p reduces the expression levels of the OSR2 protein whereas knockdown of miR‐361‐3p enhances OSR2 protein expression. *P < .01, **P < .05 compared to control
3.3. Effect of LNA‐miR‐361‐3p on in vivo growth of human OSCC cells
We assessed the effect of LNA‐miR‐361‐3p on the in vivo growth of GFP‐SAS cells. Using a xenograft model of GFP‐SAS cells in nude mice, we injected LNA‐miR‐361‐3p into the xenograft tumors every 3 days. We found that treatment with LNA‐miR‐361‐3p significantly reduced the size of the s.c. xenografted GFP‐SAS tumors, compared to the control tumors treated with LNA‐miR‐NT (Figure 3A). The expression levels of miR‐361‐3p and OSR2 in the excised tumors were then examined by qRT‐PCR. LNA‐miR‐361‐3p suppressed the levels of miR‐361‐3p and induced the expression of OSR2 compared with control tumors (Figure 3B, C).
Figure 3.
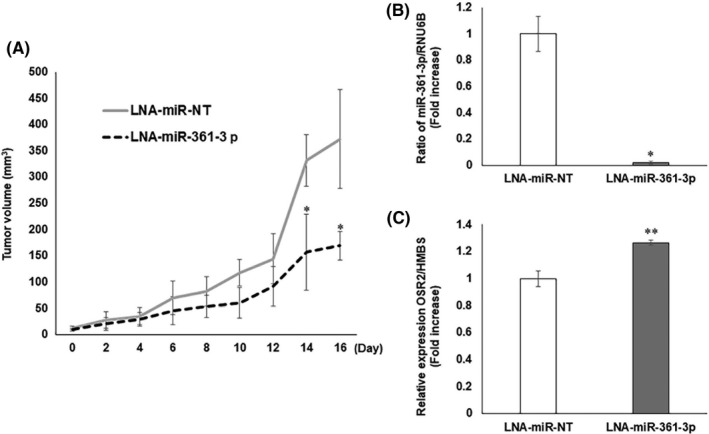
In vivo growth inhibitory effect of locked nucleic acid (LNA)‐miR‐361‐3p. A, LNA‐miR‐361‐3p was injected every 3 d to nude mice bearing s.c. xenografted GFP‐SAS tumors. LNA‐miR‐361‐3p significantly reduced the size of the s.c. xenografted GFP‐SAS tumors, compared with the LNA‐miR‐NT control group. B, C, Expression of miR‐361‐3p and OSR2 in excised tumors was examined by quantitative RT‐PCR. LNA‐miR‐361‐3p markedly suppressed the expression of endogenous miR‐361‐3p and induced the expression of OSR2 mRNA. *P < .01, **P < .05 compared to control. NT, nontarget
3.4. Overexpression of miR‐361‐3p in OSCC tissues
Finally, we assessed the expression of miR‐361‐3p in 18 primary OSCC and 2 normal oral mucosa tissues by qRT‐PCR. The expression of miR‐361‐3p was normalized by RNU6B expression as an internal control. We found that the expression levels of miR‐361‐3p were higher in OSCC tissues than in normal tissues (Figure 4).
Figure 4.
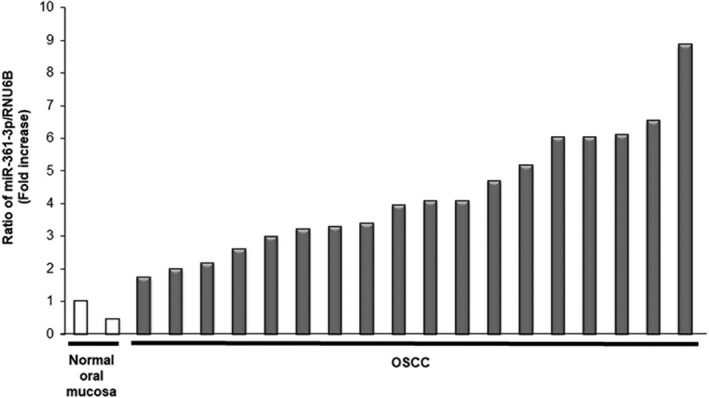
Expression of microRNA (miR)‐361‐3p in oral squamous cell carcinoma (OSCC) tissues. We assessed the expression of miR‐361‐3p in 18 primary OSCC and 2 normal oral mucosal tissues by quantitative RT‐PCR. miR‐361‐3p was overexpressed in all OSCC tissues
Furthermore, we evaluated the association between miR‐361‐3p expression in tumors from 18 OSCC patients and their clinicopathologic parameters. We categorized miR‐361‐3p expression as high or low by the median value, and then examined the association between miR‐361‐3p expression and the clinicopathologic parameters of the OSCC patients. No significant relationship was observed (Table 1).
Table 1.
Association between microRNA (miR)‐361‐3p expression in tumors from patients with oral squamous cell carcinoma and their clinicopathologic parameters
| Parameter | Low miR‐361‐3p (n = 9), N (%) | High miR‐361‐3p (n = 9), N (%) | P value |
|---|---|---|---|
| Sex | |||
| Male | 9 (100) | 7 (77.8) | .177 |
| Female | 0 (0) | 2 (22.2) | |
| Primary tumor site | |||
| Tongue | 3 (33.3) | 2 (22.2) | .375 |
| Maxillary gingiva | 4 (44.4) | 2 (22.2) | |
| Mandibular gingiva | 1 (11.1) | 2 (22.2) | |
| Buccal mucosa | 1 (11.1) | 2 (22.2) | |
| Floor of mouth | 0 (0) | 1 (11.1) | |
| Differentiation | |||
| Well | 5 (55.6) | 5 (55.6) | 1.000 |
| Moderate | 3 (33.3) | 3 (33.3) | |
| Poor | 1 (11.1) | 1 (11.1) | |
| TNM stage | |||
| I‐II | 6 (66.7) | 3 (33.3) | .222 |
| III‐IV | 3 (33.3) | 6 (66.7) | |
| Recurrence/metastasis | |||
| No | 6 (66.7) | 7 (77.8) | 1.000 |
| Yes | 3 (33.3) | 2 (22.2) | |
4. DISCUSSION
MicroRNAs are often found to function as oncogenes or tumor suppressor genes,10 and are thus implicated in the development and progression of human malignancies. Several previous reports have shown that miR‐361‐3p suppresses the growth of human retinoblastoma11 and non‐small‐cell lung cancer (NSCLC)12 cells, suggesting it is a tumor‐suppressive miRNA. However, here we found that miR‐361‐3p is overexpressed in OSCC tissues and that targeting miR‐361‐3p using LNA/DNA ASO inhibits the growth of human OSCC cells both in vitro and in vivo. These results suggest that miR‐361‐3p is an oncomiR that supports the malignant phenotype in OSCC.
Two mature miRNAs, miR‐361‐5p and miR‐361‐3p, are produced from the miR‐361 precursor. Differential expression of miR‐361‐5p has been linked to bleomycin‐induced pulmonary fibrosis13 and fatty acid‐mediated insulin resistance14 in mouse models. On the other hand, miR‐361‐3p regulates the proliferation, migration, and invasion of human lung15 and prostate16 cancer cells. An inhibitor of miR‐361‐3p potently decreased the viability of human NSCLC cells with different genetic backgrounds through S phase arrest and caspase‐3 activation.15 Moreover, high levels of miR‐361‐3p expression have been found to be associated with advanced tumor stage and shorter overall survival in pancreatic ductal adenocarcinomas and are positively correlated with metastasis.17 Consistent with our results, these findings indicate that miR‐361‐3p functions as an oncomiR.
Mechanistically it has been shown that overexpression of miR‐361‐3p promotes activation of the ERK pathway, which is widely known to be connected with epithelial‐mesenchymal transition (EMT).17, 18, 19, 20 The ERK pathway is also required for transforming growth factor‐β1/H‐Ras induction of the Snail transcription factor,21 which is recognized for its involvement in the EMT by directly binding to the E‐cadherin/CDH1 gene promoter.22 Extracellular signal‐regulated kinase is a member of the MAPK family that is well known to play pivotal roles in carcinogenesis.23 Mitogen‐activated protein kinases are key downstream signaling molecules that are regulated by the activity of the epidermal growth factor receptor (EGFR) in a number of cancers.24, 25 Up to 90% of HNSCCs, including OSCC, are known to overexpress the EGFR and this leads to excessive activation of the EGFR signaling pathway.26, 27 In clinical practice, the anti‐EGFR mAb cetuximab is used to treat OSCC patients. Compared with platinum‐fluorouracil (PF) chemotherapy alone, cetuximab plus PF chemotherapy significantly improved overall and progression‐free survival in patients with recurrent or metastatic HNSCC, especially in OSCC patients.28 However, despite GFP‐SAS cells used in this study showing constitutive activation of ERK1/2, inhibition of miR‐361‐3p had no effect on their phosphorylation or expression in vitro and in vivo (data not shown). If growth inhibition by targeting miR‐361‐3p is independent of the ERK1/2 pathway in OSCC, combination with cetuximab could be effective.
In summary, miR‐361‐3p is likely to play a significant role in the growth of human OSCC cells and that targeting miR‐361‐3p may be a useful therapeutic approach for patients with OSCC.
CONFLICT OF INTEREST
The authors have no conflicts of interest to declare.
Supporting information
Table S1
Table S2
ACKNOWLEDGMENTS
This work was supported by JSPS KAKENHI Grant Number 24390457.
Ogawa H, Nakashiro K‐I, Tokuzen N, Kuribayashi N, Goda H, Uchida D. MicroRNA‐361‐3p is a potent therapeutic target for oral squamous cell carcinoma. Cancer Sci. 2020;111:1645–1651. 10.1111/cas.14359
REFERENCES
- 1. Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359‐386. [DOI] [PubMed] [Google Scholar]
- 2. Gupta S, Kong W, Peng Y, Miao Q, Mackillop WJ. Temporal trends in the incidence and survival of cancers of the upper aerodigestive tract in Ontario and the United States. Int J Cancer. 2009;125:2159‐2165. [DOI] [PubMed] [Google Scholar]
- 3. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2014;166:281‐297. [DOI] [PubMed] [Google Scholar]
- 4. Selbach M, Schwanhausser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N. Widespread changes in protein synthesis induced by microRNAs. Nature. 2008;455:58‐63. [DOI] [PubMed] [Google Scholar]
- 5. Reis PP, Tomenson M, Cervigne NK, et al. Programmed cell death 4 loss increases tumor cell invasion and is regulated by miR‐21 in oral squamous cell carcinoma. Mol Cancer. 2010;10:238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liu Z, Diep C, Mao T, et al. MicroRNA‐92b promotes tumor growth and activation of NF‐κB signaling via regulation of NLK in oral squamous cell carcinoma. Oncol Rep. 2015;34:2961‐2968. [DOI] [PubMed] [Google Scholar]
- 7. Yang CN, Deng YT, Tang JY, et al. MicroRNA‐29b regulates migration in oral squamous cell carcinoma and its clinical significance. Oral Oncol. 2015;51:170‐177. [DOI] [PubMed] [Google Scholar]
- 8. Rather MI, Nagashri MN, Swamy SS, Gopinath KS, Kumar A. Oncogenic microRNA‐155 down‐regulates tumor suppressor CDC73 and promotes oral squamous cell carcinoma cell proliferation: implications for cancer therapeutics. J Biol Chem. 2013;288:608‐618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shintani S, Mihara M, Nakahara Y, Aida T, Tachikawa T, Hamakawa H. Lymph node metastasis of oral cancer visualized in live tissue by green fluorescent protein expression. Oral Oncol. 2002;38:664‐669. [DOI] [PubMed] [Google Scholar]
- 10. Esquela‐Kerscher A, Slack FJ. Oncomirs ‐ microRNAs with a role in cancer. Nat Rev Cancer. 2006;6:259‐269. [DOI] [PubMed] [Google Scholar]
- 11. Zhao D, Cui Z. MicroRNA‐361‐3p regulates retinoblastoma cell proliferation and stemness by targeting hedgehog signaling. Exp Ther Med. 2019;17:1154‐1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen W, Wang J, Liu S, et al. MicroRNA‐361‐3p suppresses tumor cell proliferation and metastasis by directly targeting SH2B1 in NSCLC. J Exp Clin Cancer Res. 2016;35:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xie T, Liang J, Guo R, Liu N, Noble PW, Jiang D. Comprehensive microRNA analysis in bleomycin‐induced pulmonary fibrosis identifies multiple sites of molecular regulation. Physiol Genomics. 2011;43:479‐487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li ZY, Na HM, Peng G, Pu J, Liu P. Alteration of microRNA expression correlates to fatty acid‐mediated insulin resistance in mouse myoblasts. Mol Biosyst. 2011;7:871‐877. [DOI] [PubMed] [Google Scholar]
- 15. Du L, Borkowski R, Zhao Z, et al. A high‐throughput screen identifies miRNA inhibitors regulating lung cancer cell survival and response to paclitaxel. RNA Biol. 2013;10:1700‐1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fletcher CE, Sulpice E, Combe S, et al. Androgen receptor‐modulatory microRNAs provide insight into therapy resistance and therapeutic targets in advanced prostate cancer. Oncogene. 2019;38:5700‐5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hu J, Li L, Chen H, et al. MiR‐361‐3p regulates ERK1/2‐induced EMT via DUSP2 mRNA degradation in pancreatic ductal adenocarcinoma. Cell Death Dis. 2018;9:807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xie L, Law BK, Chytil AM, Brown KA, Aakre ME, Moses HL. Activation of the Erk pathway is required for TGF‐beta1‐induced EMT in vitro. Neoplasia. 2004;6:603‐610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Buonato JM, Lazzara MJ. ERK1/2 blockade prevents epithelial‐mesenchymal transition in lung cancer cells and promotes their sensitivity to EGFR inhibition. Cancer Res. 2014;74:309‐319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Grände M, Franzen A, Karlsson JO, Ericson LE, Heldin NE, Nilsson M. Transforming growth factor‐beta and epidermal growth factor synergistically stimulate epithelial to mesenchymal transition (EMT) through a MEK‐dependent mechanism in primary cultured pig thyrocytes. J. Cell Sci. 2002;115:4227‐4236. [DOI] [PubMed] [Google Scholar]
- 21. Peinado H, Quintanilla M, Cano A. Transforming growth factor beta‐1 induces snail transcription factor in epithelial cell lines: mechanisms for epithelial mesenchymal transitions. J Biol Chem. 2003;278:21113‐21123. [DOI] [PubMed] [Google Scholar]
- 22. Muqbil I, Wu J, Aboukameel A, Mohammed RM, Azmi AS. Snail nuclear transport: the gateways regulating epithelial‐to‐mesenchymal transition? Semin Cancer Biol. 2014;27:39‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mansour SJ, Matten WT, Hermann AS, et al. Transformation of mammalian cells by constitutively active MAP kinase kinase. Science. 1994;265:966‐970. [DOI] [PubMed] [Google Scholar]
- 24. Roux PP, Blenis J. ERK and p38 MAPK‐activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev. 2004;68:320‐344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37‐40. [DOI] [PubMed] [Google Scholar]
- 26. Temam S, Kawaguchi H, El‐Naggar AK, et al. Epidermal growth factor receptor copy number alterations correlate with poor clinical outcome in patients with head and neck squamous cancer. J Clin Oncol. 2007;25:2164‐2170. [DOI] [PubMed] [Google Scholar]
- 27. Kalyankrishna S, Grandis JR. Epidermal growth factor receptor biology in head and neck cancer. J Clin Oncol. 2006;24:2666‐2672. [DOI] [PubMed] [Google Scholar]
- 28. Vermorken JB, Mesia R, Rivera F, et al. Platinum‐based chemotherapy plus cetuximab in head and neck cancer. N Engl J Med. 2008;359:1116‐1127. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Table S2