

Abstract
The use of allogeneic, pluripotent stem‐cell‐derived immune cells for cancer immunotherapy has been the subject of recent clinical trials. In Japan, investigator‐initiated clinical trials will soon begin for ovarian cancer treatment using human leukocyte antigen (HLA)‐homozygous‐induced pluripotent stem cell (iPSC)‐derived anti–glypican‐3 (GPC3) chimeric antigen receptor (CAR)‐expressing natural killer/innate lymphoid cells (NK/ILC). Using pluripotent stem cells as the source for allogeneic immune cells facilitates stringent quality control of the final product, in terms of efficacy, safety and producibility. In this paper, we describe our methods for the stable, feeder‐free production of CAR‐expressing NK/ILC cells from CAR‐transduced iPSC with clinically relevant scale and materials. The average number of cells that could be differentiated from 1.8‐3.6 × 106 iPSC within 7 weeks was 1.8‐4.0 × 109. These cells showed stable CD45/CD7/CAR expression, effector functions of cytotoxicity and interferon gamma (IFN‐γ) production against GPC3‐expressing tumor cells. When the CAR‐NK/ILC cells were injected into a GPC3‐positive, ovarian‐tumor‐bearing, immunodeficient mouse model, we observed a significant therapeutic effect that prolonged the survival of the animals. When the cells were injected into immunodeficient mice during non–clinical safety tests, no acute systemic toxicity or tumorigenicity of the final product or residual iPSC was observed. In addition, our test results for the CAR‐NK/ILC cells generated with clinical manufacturing standards are encouraging, and these methods should accelerate the development of allogeneic pluripotent stem cell‐based immune cell cancer therapies.
Keywords: chimeric antigen receptor, GPC3, ILC, immunotherapy, iPSC, NK
This translational study aimed to develop anti–GPC3 CAR‐expressing NK/ILC cells derived from HLA‐homozygous iPSC clone as an effective cell therapy against disseminated ovarian tumors and to assess the clinical cell manufacturing and pre–clinical aspects of the therapy, including safety and efficacy. Those aspects of the therapy clarified in the study provide perspective for the planned clinical trial.

1. INTRODUCTION
Natural killer (NK) cells are a subset of innate lymphoid cells (ILC) that have direct cytotoxic effects on pathogenic cells by inducing apoptosis. NK cells sense the “non–self” or “missing‐self” status of target cells using NK activating and NK inhibitory receptors, respectively, and are distinct from cytotoxic T cells. NK cells were first identified in the early 1970s, in both mice 1 and humans, 2 as a novel type of lymphocyte with strong cytotoxicity to tumor cells and are now considered a subset of ILC. 3 For almost half a century since their identification, there have been high expectations for their use in cancer immunotherapy. Despite the huge number of clinical studies involving various types of cancer, NK cell‐based immunotherapies have not demonstrated significant clinical efficacy until very recently. However, the recent development of IL‐15‐containing regimens for ex vivo NK cell culture to increase proliferation and cytotoxic granule production are improving the therapeutic efficacy of NK cell‐based cancer immunotherapies. Moreover, new technologies are being applied to the field, including chimeric antigen receptor (CAR) technology, which has been appropriated to increase antigen specificity in NK cells, 4 , 5 , 6 and pluripotent stem cell technology, which can be used to regenerate ILC, including NK cells, for an unlimited supply of therapeutic cells. 7 , 8 Significantly, these two technologies have been combined to regenerate CAR‐expressing NK cells. 9
We have previously reported the rejuvenation of antigen‐specific T cells from induced pluripotent stem cells (iPSC), 10 and have demonstrated their potential for cancer immunotherapy. In particular, we have used allogeneic HLA homo‐iPS cells, 11 which are not only expected to be a versatile source of products for multiple patients but also a platform for manipulation techniques, such as genome editing, to improve various functions of the final cell product. 12 We have also reported the regeneration of ILC from iPSC by an alternative but direct differentiation pathway from CD7‐positive NK/T cell progenitor cells. 8 , 13 The induced ILC had innate lymphoid‐cell‐like functions, similar to PSC‐derived NK cells reported by other research groups. 14 , 15
Chimeric antigen receptor‐T cells have been shown to have a distinct therapeutic effectiveness against B‐cell malignancies; however, CAR‐T cell therapies are less effective for solid tumors regardless of whether they are primary or metastatic lesions. Glypican‐3 (GPC3) is a cancer‐specific membrane protein that is expressed in hepatoblastoma, hepatocellular carcinoma and clear‐cell carcinoma of the ovary but is hardly expressed in normal tissue. From the viewpoint of on‐target off‐tumor, GPC3 is an excellent target for CAR‐based immunotherapy. Because a phase I clinical trial of anti–GPC3 CAR‐T cell therapy for relapsed refractory liver cancer reported no side effects over grade 3 (clinical trial information: NCT02395250 16 ), we decided to apply GPC3‐CAR to NK/ILC cells to develop a safe and effective treatment for GPC3‐expressing local tumors. We focused on a disseminated clear‐cell carcinoma of the ovary, as the standard chemotherapy is less effective for relapsed or recurrent cases.
This translational study aimed to develop anti–GPC3 CAR‐expressing NK/ILC cells as an effective cell therapy against disseminated ovarian tumors and to assess the clinical cell manufacturing and pre–clinical aspects of the therapy, including safety and efficacy. Those aspects of the therapy clarified in the study provide perspective for the planned clinical trial.
2. MATERIALS AND METHODS
2.1. Experimental model and subject details
2.1.1. Mice
Mice used in this study were 6‐ to 12‐week‐old female NOD‐SCID IL2Rγcnull (NSG) mice purchased from Oriental Bio for non–GLP efficacy studies, or 6‐week‐old female NOD.Cg‐Prkdcscid I12rgtm1Sug/ShiJic (NOG) mice purchased from In Vivo Science (Tokyo, Japan) for GLP safety studies. The mice were housed under controlled conditions, humidity and light/dark cycle in a specific‐pathogen‐free facility. All animal experiments were performed in accordance with the Ethical Review Body at Kyoto University.
2.1.2. Cell lines
JHH7 cells were purchased from JCRB Cell Bank. KOC7c, SK‐Hep‐1 and SK‐Hep‐1 transduced with GPC3 17 were provided by Dr Nakatsura. The mycoplasma status of the cells was routinely checked. JHH7, HepG2, KOC7c and SK‐Hep‐1 were maintained in DMEM supplemented with 10% FBS.
QHJI‐iPSC, an iPSC line generated from blood cells from a healthy individual homozygous for the most common HLA type in Japan, was maintained on iMatrix‐511 (Matrixome)‐coated plates in StemFit AK03 medium (Ajinomoto) under 5% CO2, at 37°C.
2.2. Immunization and construction of immunized antibody phagemid library
To obtain a monoclonal antibody reactive to GPC3, we immunized 6 week‐old SKG/Jcl mice (CLEA Japan) with 100 µL PBS containing 50 µg of soluble recombinant human GPC3 (R & D Systems) for four times and repeated injection with 2.5 × 106 cells of SK‐Hep‐1 overexpressing GPC3 (SK‐Hep‐1‐GPC3) for boost immunizations (SKG mice). Serum titers were evaluated by ELISA and cell‐based ELISA using SK‐Hep‐1‐GPC3. Lymph nodes were harvested from immunized SKG mice with the high antibody titer. Total RNA was extracted from lymph nodes and used as a template for first strand cDNA synthesis with oligo dT. The VH and VL genes were amplified separately and fused with a flexible linker (Gly4Ser)3 by assembly PCR. The resulting single chain Fv (scFv) were inserted into phagemid vector pTZ19R. The scFv‐cp3 phagemid were introduced into Escherichia coli strain DH12S by electroporation. The transformed E. coli were infected with M13KO7 helper phage to generate phage particles displaying scFv‐cp3. Selection of scFv‐cp3 phages was carried out by biopanning using 6 × His‐tagged recombinant GPC3 fixed using a Dynabeads His‐Tag Isolation and Pulldown kit (Veritas). Final biopanning was performed using JHH7 cells. To isolate amino terminus of GPC3 specific antibody, anti–GPC3 antibodies including GC33 and GC199, which have C‐terminus epitope antibodies, were premixed with GPC3‐magnetic beads during biopanning. A sequence of scFv phage clones was analyzed using BigDye ver3.1 (Thermo Fisher) according to the manufacturer’s protocol. Binding affinities of the scFv for human GPC3 were determined by SPR (BIACORE T100) and evaluated by Biacore X100 evaluation software (version.2.0.1), and analyzed using mouse IgG Capture Kit (GE Healthcare) according to the manufacturer’s protocol. In brief, the anti–GPC3 antibody was captured with anti–mouse Fc antibody on a CM5 sensor chip (GE Healthcare) at capture level 100 RU. Thereafter, the interaction with the recombinant GPC3 (R and D systems) was analyzed in a dilution series from 47 to 380 nmol/L using 120‐s association time and 600‐s dissociation time at a flow rate of 60 μL/min at 25°C. Binding curves were evaluated using Biacore X100 evaluation software. A monovalent Langmuir binding model was used to calculate binding kinetic parameters.
2.3. Establishment of lentiviral vector encoding chimeric antigen receptor
The sequence encoding the anti–GPC3 scFv in the VH‐VL orientation was obtained based on the sequence of the Ab (G2 scFV). As shown in Figure 1A, G2 scFv was linked to the human CD8α hinge transmembrane region and the intracellular signaling domains of CD28, CD137 and CD3ζ molecules in tandem to form a CAR construct, then further linked with truncated EGFR by T2A to monitor transgene expression. The extended CAR construct was cloned into a Ubc‐promotor‐modified pLVSIN, to create pLVSIN (G2 CAR) (Clontech).
Figure 1.
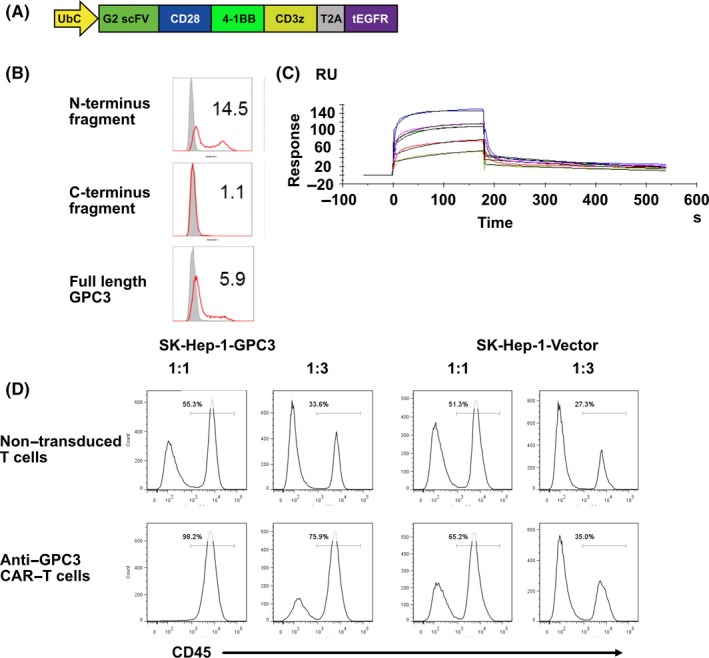
Characterization of third generation chimeric antigen receptor (CAR) with a novel scFv that efficiently binds to the GPC3 N‐terminus. A, Schematic representation of the lentiviral vector expressing G2 CAR. B‐C, Binding specificity of anti–GPC3 antibody B. Anti–GPC3 antibody was bound to GPC3 full‐length and N‐terminus fragment but not C‐terminus domain expressing 293T cells. C, Binding affinity was analysis by multi‐cycle method of SPR measurement. The antibody fixed on CM5 sensor chip was bound to GPC3. D, Cytolytic activity of anti–GPC3 CAR‐T cells specific to SK‐Hep‐1‐GPC3 cells. Anti–GPC3 CAR‐T cells or non–transduced T cells were co–cultured with GPC3‐positive or GPC3‐negative SK‐Hep‐1 cells at 1:1 or 1:3 CAR‐T to target ratios for 48 h. The cultured cells were harvested and analyzed by flow cytometry to detect CD45, a marker expressed on T cells but not target tumor cells
2.4. Generation of G2 CAR‐expressing human T cells by retroviral transduction
We generated G2 CAR‐expressing human T cells by retroviral transduction as previously reported, 18 with some modifications. Briefly, MSGV1 retroviral vector to express G2 CAR was produced by transfection of G2 CAR plasmid into the Ampho packaging cell line, followed by collection of culture supernatants. For transduction of human T cells, peripheral blood mononuclear cells (PBMC) were cultured in the presence of anti–human CD3 mAb and IL‐2, and then further incubated with the supernatants containing G2 CAR retroviral vector in the presence of RetroNectin. We centrifuged and expanded the cells in the presence of IL‐2 to obtain G2 CAR‐T cells.
2.5. Lymphocyte differentiation from G2‐CAR‐transduced QHJI01s04
G2‐CAR‐transduced QHJI01s04 was differentiated into a hematopoietic precursor through the feeder‐free embryoid body formation method. Undifferentiated T‐iPSC colonies were treated with TrypLE select (Gibco) for 8 minutes and transferred to low‐attachment plates and incubated overnight in Stemfit AK03N containing 10 μmol/L Y‐27632 to allow for the formation of embryoid bodies (EB). The EB were collected and transferred to EB medium (StemPro‐34 [Invitrogen], 2 mmol/L l‐glutamine, 400 μmol/L monothioglycerol, 50 μg/mL ascorbic acid‐2‐ phosphate and insulin‐transferrin‐selenium supplements) and cultured in the presence of 40 ng/mL hBMP‐4, 10 ng/mL hbFGF and 50 ng/mL VEGF. On day 4, the EB were cultured with a cocktail of hematopoietic cytokines (50 ng/mL hSCF, 20 ng/mL hFlt3L, 20 ng/mL hIL‐3 and 30 ng/mL TPO). On day 14 of culture, the differentiated cells were transferred onto FcDLL4‐coated plates and cultured in the presence of a cocktail of T lineage cytokines (10 ng/mL hFlt3L, 5 ng/mL IL‐7). After 21 days of culture, the hematopoietic cells were differentiated into CD7, CD45‐positive lymphocyte progenitor cells (iCAR‐LPC). The iCAR‐LPC were harvested and stimulated to expand with PHA to generate iCAR‐NK/ILC.
2.6. Flow cytometry
Stained cell samples were analyzed using an LSR or FACSAria II Flow Cytometer (BD Biosciences), and the data were processed using FlowJo (Tree Star). For T cell phenotyping, the following antibodies were used: PE‐EGFR (clone AY13; BioLegend), FITC‐CD14 (clone HCD14; BioLegend), PacificBlue‐CD235a (clone HI264; BioLegend), PE/Cy7‐CD34 (clone 4H11; Abcam), APC‐CD43 (clone 1G10; BD Bioscience), BV510‐CD45 (clone HI30; BioLegend), APC‐cy7‐CD3 (clone UCHT1; BioLegend), BV421‐CD4 (clone OKT4; BioLegend), FITC‐CD5 (clone UCHT2; eBioscience), CD7‐APC (clone CD7‐6B7; BioLegend), APC‐CD8a (clone SK1; BioLegend), PerCPcy5.5‐CD8 (clone SK1; BioLegend) and PE‐Cy7‐CD8β (clone SIDI8BEE; eBioscience).
2.7. 51Cr‐release assays
We performed 51Cr‐release assays to evaluate the effector cell cytolytic ability. Target tumor cells were loaded with 1.85 MBq 51Cr for 1 hour, and then 2500 tumor cells were co–incubated with effector cells for 5 hours at effector‐to‐target (E:T) ratios of 40:1 to 5:1. Supernatants were harvested, and 51Cr release was quantified using a beta counter. Percent lysis was calculated as % lysis = (experimental lysis − spontaneous lysis)/(maximal lysis − spontaneous lysis) × 100%, where maximal lysis was induced by incubation in a 2% Triton X‐100 solution.
2.8. Intracellular staining of interferon gamma
The iCAR‐NK/ILC were co–cultured with irradiated SK‐Hep‐1 or SK‐Hep‐1 o/e GPC3 for 5 hours in monensin‐containing medium. After staining of surface antigens, cells were fixed, permeabilized and labelled with an APC‐conjugated anti–IFN gamma antibody.
2.9. Bioluminescence imaging
We detected bioluminescence with a Xenogen IVIS Imaging System (Xenogen), as previously described. We performed imaging 10‐15 minutes after intraperitoneal injection with VivoGlo Luciferin (3 mg/mice; Promega).
2.10. In vivo model
2.10.1. Peritoneum dissemination animal model
A total of 2 × 105 luciferase‐transduced KOC7C cells were injected i.p. into NSG mice followed by i.p. injections of 5 × 106 iCAR‐NK/ILC cells on days 3, 7, 10, 14, 17 and 21. Tumor burden was monitored by in vivo bioluminescence imaging (IVIS 100 Imaging System, Caliper).
2.10.2. General toxicity test model
A total of 6 × 107 iCAR‐ILC was administered to the NOG mice on days 0, 3, 7, 10, 14 and 17, and the mice were killed on day 20. The following organs/tissues were collected from all animals, fixed with 10% phosphate buffered formalin solution, embedded in paraffin, sectioned, and hematoxylin/eosin stained (H/E): cerebrum, cerebellum, spinal cord (chest), sciatic nerve, eyeball, optic nerve, harderian gland, pituitary gland, thyroid, parathyroid, adrenal gland, spleen, heart, thoracic aorta, trachea, lung (including bronchi), tongue, esophagus, stomach, duodenum, jejunum, ileum, cecum, colon, rectum, submandibular gland, sublingual gland, liver, gallbladder, pancreas, kidney, bladder, ovary, uterus, sputum, mammary gland (groin), sternum (including bone marrow), femur (including knee joint and bone marrow), femoral skeletal muscle, skin (groin), thymus, submandibular lymph node and mesenteric lymph node.
2.10.3. Tumorgenicity test model
A total of 1 × 107 iCAR‐NK/ILC or 3.6 × 105 G2 CAR QHJI‐iPSC #22 was administered to NOG mice on day 0, and these mice were killed 6 months later. The procedures used were as for the general toxicity test model.
3. RESULTS
3.1. Characterization of third generation CAR with a novel scFv that efficiently binds to GPC3 N‐terminus
Anti–GPC3 monoclonal antibodies were obtained using the GPC3 immunized antibody library and phage display method. N‐terminus specific target binding of the monoclonal antibodies was characterized using GPC3 full length and N‐terminus fragments of GPC3 expressing cell lines and analyzed by FCM. As indicated in Figure 1B,C, an anti–GPC3 antibody (Ab) was newly identified as the N‐terminus of GPC3 specific Ab whose binding affinity was 3.4 × 10−8 mol/L by SPR. The single chain Fc of the Ab (G2 scFv) designed from the amino acid sequence of the Ab was inserted into the third generation CAR, 19 which is composed of the human CD8α hinge transmembrane region and the intracellular signaling domains of CD28, CD137 and CD3ζ molecules. To confirm therapeutic potential of the scFv in CAR, a retroviral vector encoding G2 scFv CAR was transduced to human primary T cells to generate G2 CAR‐T cells, then they were co–cultured with GPC3‐positive or GPC3‐negative SK‐Hep‐1 cell lines at a ratio of 1:1 or 1:3 CAR‐T to tumor cells to evaluate CAR‐mediated target specific cytotoxicity. After 48 hours, CD45‐negative tumor cells were almost completely eliminated at a 1:1 ratio or significantly reduced at a 1:3 ratio when G2 CAR‐T cells were co–cultured with GPC3‐positive target cells (Figure 1D). The elimination of tumor cells was not observed for GPC3‐negative target cells, indicating the GPC3‐specific cytolytic activity of anti–GPC3 CAR‐T cells.
3.2. Anti–GPC3 CAR‐modification and selection of an HLA‐homozygous iPSC clone to establish a master cell bank as a source for iPSC‐derived CAR‐expressing NK/ILC cells
Since effective binding and cytotoxic function of G2 scFv CAR to target GPC3‐expressing cells were confirmed, we applied the CAR to iPSC‐derived NK/ILC cells. For the purpose of avoiding genetic engineering by insertional viral vectors at a later stage of manufacturing, we tried to establish a master cell bank (MCB) of an iPSC clone that efficiently expresses anti–GPC3 CAR. We first transduced G2 scFv CAR and EGFR harboring lentiviral vector (LVSIN [G2CAR], Figure 1A) into iPSC clones, named QHJI01s04, which were generated from blood mononuclear cells of a homozygous healthy donor for Japan’s most frequent HLA. Clone QHJI01s04 was generated under clinically relevant conditions in an institutional clinical cell processing facility. QHJI01s04 was transduced with LVSIN (G2CAR) by spin infection that resulted in a 71.9% transduction efficacy of CAR, then G2 CAR‐transduced QHJI01s04 was cloned by limiting dilution. Because expression of tEGFR varied from iPSC clone to iPSC clone, we selected the six clones expressing the highest levels of tEGFR and pluripotency markers Tra1‐60 and SSEA4 (Figure S1). Then, we differentiated all the clones into CD7‐positive T/NK‐lineage cells to select the clones that could best maintain CAR expression during the entire differentiation process.
The differentiation procedure was composed of three steps, outlined in Figure 2A: (a) the hematopoietic progenitor cell differentiation step, (b) the lymphoid progenitor cell (LPC) differentiation step and (c) the NK/ILC‐like cell generation and expansion step. In step 1, almost all the clones differentiated to CD34‐ and CD43‐double positive hematopoietic progenitor cells (Figure S2). In step 2, almost all cells differentiated to CD45‐ and CD7‐double positive but CD4‐ and CD8‐double negative T/NK‐lineage progenitor cells; however, the expression levels of tEGFR differed in six clones (Figure S3). Then, the progenitor cells were co–cultured with irradiated PBMC from the donor to generate and expand the NK/ILC‐like cells using phytohemagglutinin‐P (PHA‐P). Some features of these iPSC clone‐derived NK/ILC‐like cells were compared to determine the best clones for further study, and the G2 CAR‐transduced QHJI01s04 clone #22 was selected as the best iPSC clone for pre–clinical and clinical examinations because of their superior expansion capability and cytotoxicity of the iPSC clone‐derived CAR‐expressing NK/ILC‐like cells. (Figures [Link], [Link], [Link]).
Figure 2.
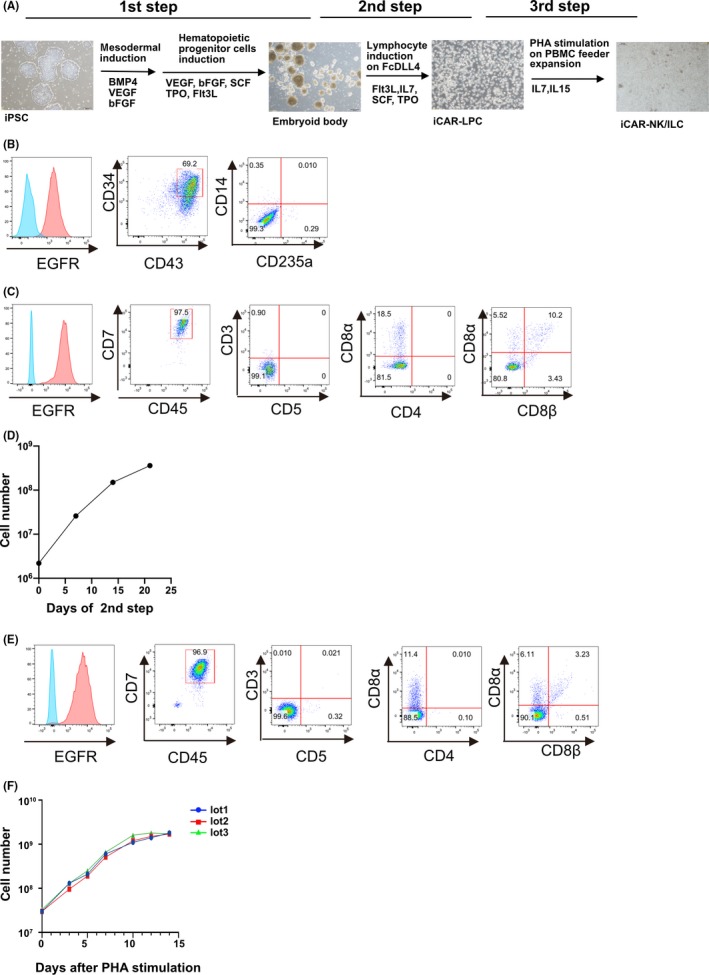
Clinically relevant manipulation of anti–GPC3 28bbz QHJI‐iPSC #22 to generate iCAR‐NK/ILC. A, Schematic for differentiation of anti–GPC3 28bbz QHJI‐iPSC #22 into iCAR‐NK/ILC. B, Flow‐cytometric analysis of differentiated cells at the end of the first step. C, Flow‐cytometric analysis of differentiated cells at the end of the second step. D, Cell number of differentiated cells during the second step. E, Flow‐cytometric analysis of differentiated cells at the end of the third step. F, Cell number of iCAR‐NK/ILC during the third step
3.3. Clinically relevant manipulation for iCAR‐NK/ILC generation from G2 CAR QHJI01s04 #22
Because of the success of clone #22, we created a master cell bank of the clone that would be suitable for use in non–clinical testing and clinical trials. Therefore, clone #22 was expanded and stored in a clinical cell processing facility for iPS cell‐based therapy (FiT) at Kyoto University.
Before moving to cell production for non–clinical studies, we changed some reagents and materials used in the differentiation protocol to comply with Japan’s regulatory standards for raw materials originating from living organisms (Table S1). Despite the changes, G2 CAR QHJI01s04 #22 successfully differentiated into CD7‐ and CD45‐double positive cells, hereafter called iCAR‐LPC, and expanded 164‐fold via CD34‐ and CD43‐double positive hematopoietic progenitor cells (Figure 2B–D). We further expanded these iCAR‐LPC on irradiated allogeneic PBMC feeders using PHA‐P. Under stimulation, almost all the cells maintained expression of both CD7 and CD45 throughout the expansion process (Figure 2E,F). The successful expansion profiles of iCAR‐NK/ILC from iCAR‐LPC in three trials using each donor PBMC is indicated in Table 1. In the majority of cases, the frozen iCAR‐NK/ILC cells maintained sufficient viability during shipment to a non–clinical test site, approximately 500 km from the production facility, and also maintained cell numbers and viability for 3 hours after thawing, when kept on ice, until injection into the test animals (Table 2). In addition, we thawed stored iCAR‐LPC and then tested their viability and expansion capacity to determine their stability in liquid nitrogen (Figures S7A‐C).
Table 1.
Profile of iCAR‐NK/ILC from iCAR‐LPC in three trials using different donor peripheral blood mononuclear cells
| Lot 1 | Lot 2 | Lot 3 | |
|---|---|---|---|
| End of culture | |||
| Total live cells | 1.8 × 109 | 1.7 × 109 | 1.7 × 109 |
| Live cells in a plate | 1.1 × 108 | 1.1 × 108 | 0.7 × 108 |
| Viability | 90% | 84% | 91% |
| Freezing | |||
| Total live cells | 1.6 × 109 | 1.3 × 109 | 2.5 × 109 |
| Viability | 92% | 93% | 86% |
| Thawing | |||
| Viability | 89% | 53% | 91% |
| Sterility test | |||
| Bacterial, Mycoplasma, Endotoxin | Negative | Negative | Negative |
Table 2.
Evaluation of viability and stability of iCAR‐NK/ILC during shipment
| Before transportation | After transportation | |||
|---|---|---|---|---|
| Viability (%) | Cell number/mL | Viability (%) | Cell number/mL | |
| Lot 1 | 92 | 550 000 | 92 | 550 000 |
| Lot 2 | 72 | 510 000 | 84 | 580 000 |
| Lot 3 | 98 | 600 000 | 93 | 510 000 |
3.4. CAR‐NK/ILC cells generated from G2 CAR iPSC #22 effectively suppress GPC3‐expressing tumor growth in vitro and in vivo
To investigate whether iCAR‐NK/ILC generated by a clinical‐grade manipulation protocol exerts enough tumor suppression on GPC3‐expressing cells in vitro, we performed a 51chromium‐releasing test using several cancer cell lines expressing GPC3 (SK‐Hep‐Vector and SK‐Hep‐GPC3). As was seen in previous reports, 8 iCAR‐NK/ILC killed not only the GPC3‐expressing cell line (SK‐Hep‐GPC3) but also the GPC3‐negative cell line (SK‐Hep‐Vector) (Figure 3A), possibly by using NK receptors, such as NKG2A, NKG2D, NKRP1, NKp30, NKp44 and DNAM1, which were expressed on iCAR‐NK/ILC (Figure 3B). The additional cytolysis observed against the GPC3‐positive target cells compared to negative cells was estimated to be due to CAR‐specific cytotoxicity. For that reason, the accumulative cytotoxicity of iCAR‐NK/ILC, which was composed of CAR‐dependent and CAR‐independent cytotoxicity, was estimated to be superior to the cytotoxicity of CAR‐transduced primary CD8 T cells (Figure S8). We measured the production of IFN‐γ, which is a major cytokine produced by NK cells. In a co–culture with GPC3‐positive tumor cells, iCAR‐NK/ILC cells produced significantly more IFN‐γ than cells cultured with GPC3‐negative tumor cells (Figure 3C,D).
Figure 3.
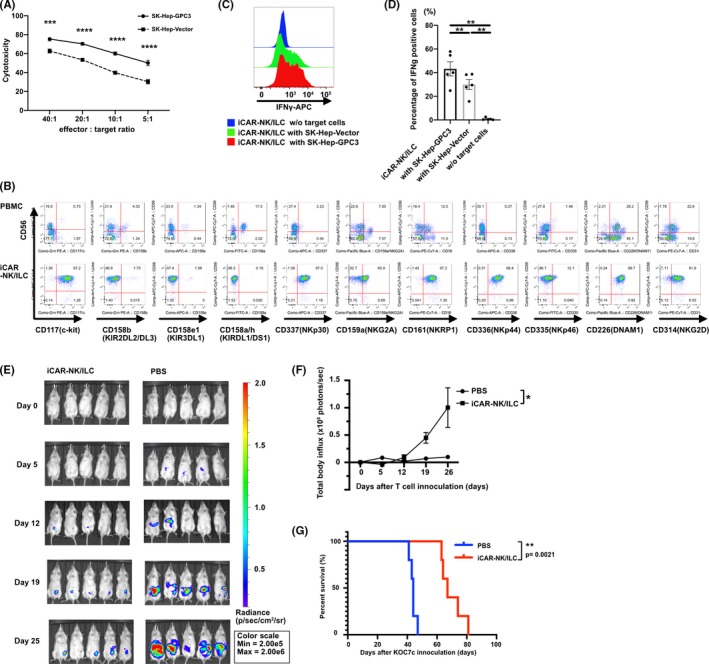
iCAR‐NK/ILC cells generated from anti–GPC3 28bbz QHJI‐iPSC #22 effectively suppress GPC3‐expressing tumor growth in vitro and in vivo. A, In vitro 51Cr‐release assay of iCAR‐NK/ILC co–cultured with SK‐Hep‐GPC3 or SK‐Hep‐Vector. n = 5 per point. B, Expression of NK‐related surface antigens was analyzed with flow cytometry. C, Intracellular IFN‐γ of iCAR‐NK/ILC was measured after co–culturing with SK‐Hep‐GPC3, SK‐Hep‐Vector or no target cells. D, Comparison of IFN‐γ production of iCAR‐NK/ILC to SK‐Hep‐GPC3, SK‐Hep‐Vector or no target cells. E, F, G, In vivo analysis of iCAR‐NK/ILC in NOD‐SCID IL2Rγcnull (NSG) xenograft model. A batch of 5 × 105 KOC7c were inoculated into the intraperitoneal cavity of NSG mice on day 0. Then, 5 × 106 iCAR‐NK/ILC was administered into the intraperitoneal cavity on days 3, 7, 10, 14, 17 and 21. Tumor burden was analyzed with in vivo imaging. E, Luciferase imaging of mice from each group. F, Summary of bioluminescence from each group. G, Kaplan‐Meier analysis of mouse survival. *p < .05, **p < .01, ***p < .001, ****p < .0001
To investigate whether iCAR‐NK/ILC suppress the growth of GPC3‐positive tumor cells in vivo, we administered iCAR‐NK/ILC into the peritoneal cavity of the intraperitoneal dissemination model for GPC3‐expressing ovarian cancer, KOC7c in the NOG mouse. Multiple injections of iCAR‐NK/ILC suppressed the intraperitoneal growth of KOC7c significantly better than PBS and prolonged the survival of the mice (Figure 3E‐G).
3.5. No evidence of ICAR‐NK/ILC‐mediated toxicity and tumorigenicity in non–clinical in vivo tests
The indicated therapeutic efficacy of iCAR‐NK/ILC by CAR‐dependent and CAR‐independent cytotoxicities raised concerns about the systemic toxicity of the product. In addition, there remained a potential risk of tumorigenicity of the final product due to malignant transformation or pluripotent stem cell contamination during cell manipulation. The risks must be adequately evaluated before moving to clinical trial.
We performed a cytokine‐independent cell proliferation assay to detect malignant transformation. In contrast to transformed cells, iCAR‐NK/ILC never proliferated in cultures without cytokines (Figure S9). We then evaluated the iPSC contamination in the final product. The mRNA of one of the pluripotency‐related genes, LIN28, of the final product was quantified by qPCR to detect undifferentiated iPS cell contamination (Figure S10). From the results, we estimated the possible contamination of the final product by iPSC to be less than 0.02%, which is below the minimum detection level of the assay. Based on the result and the planned number of cells for administration in clinical trials, we estimated the maximum number of contaminated iPS cells that will be injected into a patient. In the case of a sixfold injection of 3 × 106 cells/kg into a 60 kg patient, the estimated maximum possible number of contaminating iPS cells was calculated to be 3.6 × 105. To investigate whether the undifferentiated cells could form a teratoma in vivo, we administered 3.6 × 105 of G2 CAR‐transduced QHJI01s04 into the intraperitoneal cavity of 10 female NOG mice via the planned injection route for the clinical study. In parallel with the tumorigenicity evaluation of contaminated iPSC, we intraperitoneally administered 1 × 107 of iCAR‐NK/ILC cells into 10 female NOG mice to evaluate the risk of malignant transformation of the final product. During the 26‐week observation period, no tumorigenesis‐related symptoms were observed in the mice of either cohort. Then, on the 26th week after the injection, all mice in both cohorts were killed and evaluated by histological assay. Because iPS‐NK/ILC cells, iPS cells, transformed cells and teratoma were not detected in any specimen from the experiment, we confirmed that the clinical dose of iCAR‐NK/ILC cells did not contain tumorigenic cells.
Intraperitoneal injection‐related acute cytotoxicity of the product was then evaluated by injection of the maximum therapeutic dose of iPS‐NK/ILC cells into NOG mice, with a multiplying safety factor of 100. In total, 10 female mice were intraperitoneally injected with 6 × 107 iPS‐NK/ILC cells, divided into six injections over 3 weeks. The number of cells injected into the 20‐g mice corresponded to 3 × 109/kg for humans (ie, 1000‐fold higher than the therapeutic dose). The hematological and biochemistry assay data obtained during the observation period and the histological data from 42 organs obtained 4 days after the final injection are summarized in Figure 4A‐C. Some residual iCAR‐NK/ILC was observed in omentum, but there were no significant toxic effects compared to control mice, suggesting there was no risk of general or acute product toxicity, including severe GVHD. In addition, to estimate risks of unwanted scFv binding of the product to human cells, we performed a tissue cross‐reactivity assay with 20 specimens of normal adult human tissue. We observed no significant reactivity in normal human tissue, as suggested in various previous reports on GPC3 20 , 21 (data not shown).
Figure 4.
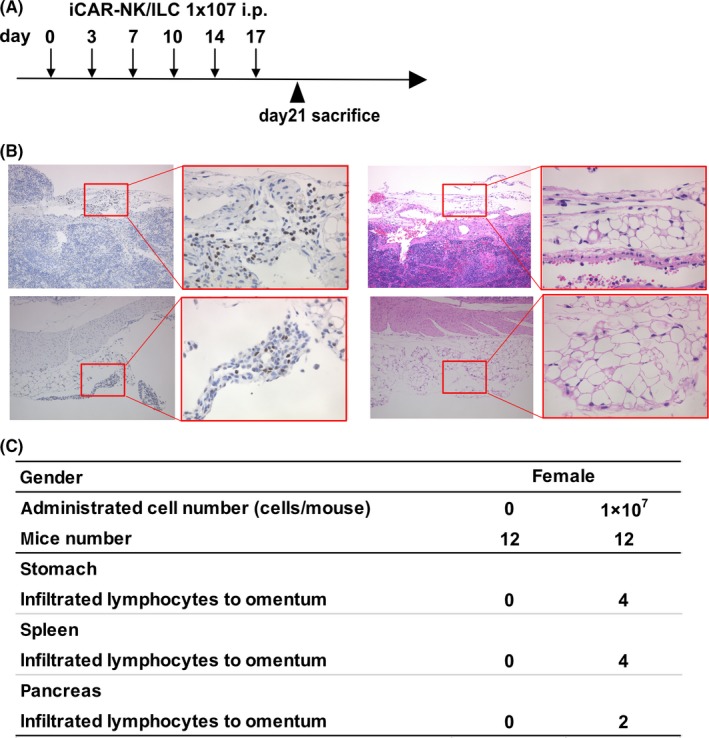
No evidence of ICAR‐NK/ILC‐mediated toxicity or tumorigenicity in non–clinical in vivo tests. A, Schematic of general toxicity test. B, Formalin‐fixed and paraffin‐embedded omentum sections stained with H&E for histologic analysis. Anti–human nuclear antibody staining was performed to distinguish human from mouse cells. C, Summary table of histological analyses
3.6. Differentiation of iCAR‐NK/ILC cells in a cell processing facility using clinically relevant materials and manipulation
For the final check before moving to the clinical study stage, a clinical dose of iCAR‐NK/ILC cells was manipulated in a cell processing facility of Kyoto University, according to normal processing flow and in‐process tests used (Figure 5). A batch of 2 × 105 of iPSC clone #22 cells was thawed and seeded onto six‐well plates, then expanded for EB formation from day 21. EB at day 35 was analyzed by FACS to estimate the HSC number in the culture. According to the flow cytometry results, 1.87 × 106 equivalent CD34+43+ cells (total cell number: 2.16 × 106, 86.8% CD34+43+) were seeded and cultured on DLL4‐coated plates for 21 days. On the last day of DLL4 culture, we confirmed that the differentiating cells had the iCAR‐LPC phenotype and we co–cultured them with irradiated healthy donor‐derived PBMC (an alternative only in pilot runs to patient‐derived PBMC) for expansion and final maturation to iCAR‐NK/ILC cells. Similar to our previous cell production experience for non–clinical tests, the estimated number (3.3 × 109) and quality of iCAR‐NK/ILC cells were successfully produced (Figure 5).
Figure 5.
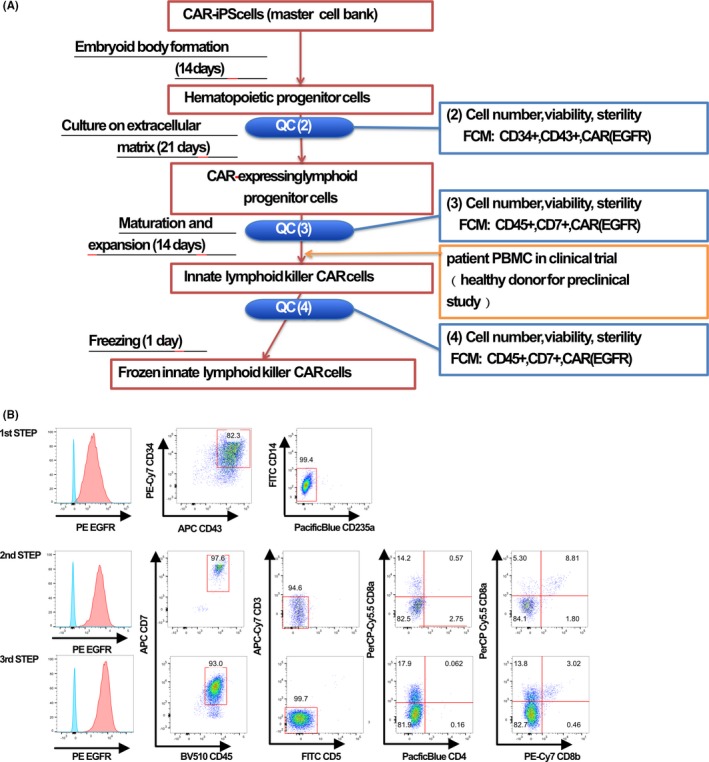
Differentiation of ICAR‐NK/ILC cells within cell processing facility using clinically relevant materials and manipulation. A, Cell processing chart of iCAR‐NK/ILC. B, Quality check of differentiated cells during cell processing
4. DISCUSSION
Since the first characterization of NK cells, immune cells have been continuously developed for clinical applications in cancer immunotherapy in autologous or allogeneic settings, and there are now an estimated 400 NK cell‐based immunotherapy clinical trials for various types of cancer being conducted worldwide. 22 Despite the numerous ongoing clinical studies involving NK cells and the vast repertoire of historical research reports, infused NK cells have shown short persistency, limited proliferation and inaccurate tumor‐targeting in patients, resulting in poor therapeutic efficacy. 23 , 24 , 25 , 26 , 27 However, recent advances in therapeutic technologies that support anti–tumor NK cell functions, such as Fc‐modified antibody‐dependent cellular cytotoxicity, 28 , 29 CAR modification, 4 , 30 efficient cytokine supply, 25 , 30 inhibitory receptor blockade 31 , 32 and use of allogeneic NK cells derived from cord blood with IL‐15 gene transduction, 33 have improved the efficiency of NK cell therapies. iPSC technology can provide further benefits to NK cell therapy development through increased speed, cell number and quality control of the allogeneic product, which may contribute to improved therapeutic efficacy.
iCAR‐NK/ILC cells will be repeatedly injected into the peritoneal cavity of a patient to treat GPC3‐expressing disseminated ovarian cancer in allogeneic settings in our planned clinical study. A strategy of peritumor local injection of CAR‐modified regenerated NK cells may overcome the natural disadvantages of NK cells, such as insufficiencies in tumor homing, target selectivity and continuance, while reducing the risk of adverse events, such as cytokine syndrome and CAR‐mediated unwanted on‐target and off‐target cytotoxicity. From that perspective, we can consider co–administering cytokines or transducing cytokine genes to iCAR‐NK/ILC for further enhancement of therapeutic efficacy. Even in a cytokine rich condition, theoretically, iCAR‐NK/ILC do not induce GVHD because they do not express alloreactive TCR.
In this translational study, we have demonstrated the feasibility of clinically differentiating the iCAR‐NK/ILC cells and have collated the non–clinical data for the cells, facilitating the transition into the clinical trial stage. The differentiation process for clinical‐grade iCAR‐NK/ILC cells was ascertained with regards to materials, methods and manipulation techniques, specifically to comply with Japanese regulations for regenerative medicine. In addition, we have set up a CAR‐transduced and CAR‐expanded iPSC clone #22 within an MCB, providing the starting material for GMP manufacturing, according to ICH‐Q5A guidelines. In addition, the method has allowed us to perform CAR transduction and clone selection of iPSC to establish a research cell bank under non–GMP conditions, allowing the regular research use of iPSC material, reducing labor costs and time without any negative safety impacts on the final product.
With regards to quality management, one advantage of using iPS cell‐based differentiated cells is that clonal manipulation of iPSC enables the very precise quality control of the product, especially at the genetic modification stage. In fact, previously, it was reported that retroviral insertion near protooncogenes of hematopoietic stem cells enhanced clonal leukemogenesis in immunodeficient patients. 34 In the case of CART, it was reported that TET2‐mutated CD19 CART cell clones became dominant after injection into a patient showing complete remission of leukemia. 35 Those clones with an in vivo proliferative advantage are usually hard to detect in polyclonally transduced cell therapy products prior to injection. Therefore, genomic characterization and safety tests, which are possible with the iPSC clone, contribute to a more precise risk evaluation of the final product.
In this study, we discovered that the anti–tumor efficacy of our product was due to both CAR‐dependent and CAR‐independent cytotoxicities. The modest contribution of G2‐CAR to the cytotoxicity was partially explained by the choice of signaling molecules in the CAR. Originally, a third generation anti–GPC3‐CAR 28bbz, containing the CD8a transmembrane domain and signaling domains from CD28, 4‐1BB and CD3z, was designed for primary T cells. It was recently reported that iPSC‐derived NK cells armed with an optimized anti–mesothelin CAR, consisting of NK cell receptor components such as the NKG2D transmembrane domain, the signaling domain from 2B4 and CD3z, inhibited target molecule‐expressing tumor growth significantly longer than third generation anti–mesothelin CAR‐armed iPS‐NK cells in vitro and in vivo. 9 The results suggested that the optimum choice of CAR for NK cells may also have improved the CAR‐mediated cytotoxicity for iPSC‐derived NK/ILC cells in this study. Although there are no available commercial products that adhere to Japanese standards for biological ingredients, we have observed the feasibility of using proinflammatory cytokines, such as IL‐12 and IL‐18, to increase the proliferative and cytotoxic potential of iPSC‐derived NK/ILC cells (data not shown). From these observations, we speculated that the addition of proinflammatory cytokines in the expansion process of the final product may enhance its anti–tumor functions; however, their use in clinical trials requires IL‐12 and IL‐18 that fulfil Japanese standards for biological ingredients.
A frequent problem in the field of cell therapy is the selection of adequate assays to test the final product. In fact, the experience accumulated during the development of anti–CD19 CART therapy has shown that cytotoxicity or cytokine production tests against CD19‐expressing leukemia cell lines in vitro never correctly predict the therapeutic efficacy of the product. 36 Therefore, we plan to put some functional assays as reference tests, and put only a few assays for standard tests such as sterality, cell number, cell viability, cell phenotyping by flowcytometry, and IFN‐g secretion, until reliability of the reference assays is confirmed to predict the in vivo reaction of the immune cells (Table 3).
Table 3.
Standard and reference tests for the final product
| Test items | Method | |
|---|---|---|
| Standard test | Sterilization test | BacT/ALERT and Japanese Pharmacopoeia |
| Mycoplasma test | PCR | |
| Bacterial endotoxin test | Japanese Pharmacopoeia | |
| Purity of ILC | Flow cytometry | |
| Purity of EGFR positive cells | Flow cytometry | |
| IFN‐γ production ability | Flow cytometry or ELISA | |
| Cell number | Trypan blue stain | |
| Cell viability | Trypan blue stain or flow cytometry | |
| Reference test | Cell growth rate | Co–culture with GPC3 positive cells trypan blue stain or Thymidine incorporation assay |
| Cytotoxicity in vitro | 51Cr releasing test | |
| Expression of undifferentiated cell marker | PCR (Lin28) |
In summary, we have reported a reliable method for the production of anti–GPC3 CAR‐expressing iPSC‐derived NK/ILC cells using clinically relevant materials and manipulations. In addition, we have confirmed various aspects of the therapeutic quality and safety of the final product. Our study has not only demonstrated the feasibility of the product (ie, anti–GPC3 CAR‐expressing iPSC‐derived NK/ILC cells) but has also provided a platform for the production and quality control of iPSC‐derived NK/ILC cells expressing various types of CAR.
DISCLOSURE
Shin Kaneko is a founder, shareholder and chief scientific officer at Thyas and received research funding from Takeda Pharmaceutical, Kirin Holdings, Terumo, Tosoh, Sumitomo Chemical and Thyas. Tetsuya Nakatsura is a shareholder at Killer T Save You. Koji Tamada is a shareholder of Noile‐Immune Biotech and receives consulting fees and research funding from Noile‐Immune Biotech. Yutaka Yasui is an employee of Thyas. The remaining authors declare no competing financial interests.
Supporting information
Fig S1
Fig S2
Fig S3
Fig S4
Fig S5
Fig S6
Fig S7
Fig S8
Fig S9
Fig S10
Table S1
ACKNOWLEDGMENTS
This work was supported in part by Grants‐in‐Aid 15H04655, 25293226 and 26293357 from: the Ministry of Education, Culture, Sports, Science and Technology Japan; the Project for Development of Innovative Research on Cancer Therapeutics, Practical Research for Innovative Cancer Control, and Core Center for iPS Cell Research from Japan Agency for Medical Research and Development; and the National Cancer Center Research Fund. We thank Prof Shinya Yamanaka (Kyoto University) for providing critical advice for our research work, Drs Izumi Ihara, Hiroaki Suzuki, Hiroki Maruyama, Munenori Nanao (KIRIN), Youichi Higuchi (Terumo), Yuta Mishima, Wang Bo and Hisashi Yano (Kyoto University), Mr Shuichi Kitayama, (Kyoto University), Mses Yoshie Tanikawa (KIRIN), Reiko Saikawa (Thyas), Yuuhi Kumagiri, Sayaka Okamoto, Kaede Makino, Sanae Kamibayashi, Eri Imai, Hitomi Takakubo and Katsura Noda (Kyoto University) for technical assistance, and Drs Toshihiko Doi, Kenichi Harano, Junichiro Yuda, Yukiko Ishiguro (National Cancer Center) for clinical development planning. The entire study was conducted in accordance with the Declaration of Helsinki and was permitted by the institutional ethical board and ACUC of Kyoto University.
Ueda T, Kumagai A, Iriguchi S, et al. Non–clinical efficacy, safety and stable clinical cell processing of induced pluripotent stem cell‐derived anti–glypican‐3 chimeric antigen receptor‐expressing natural killer/innate lymphoid cells. Cancer Sci. 2020;111:1478–1490. 10.1111/cas.14374
REFERENCES
- 1. Kiessling R, Klein E, Wigzell H. Natural killer cells in the mouse. Eur J Immunol. 1975;5:112‐117. [DOI] [PubMed] [Google Scholar]
- 2. Pross HF, Jondal M. Cytotoxic lymphocytes from normal donors. A functional marker of human non–T lymphocytes. Clin Exp Immunol. 1975;21:226‐235. [PMC free article] [PubMed] [Google Scholar]
- 3. Spits H, Artis D, Colonna M, et al. Innate lymphoid cells‐a proposal for uniform nomenclature. Nat Rev Immunol. 2013;13:145‐149. [DOI] [PubMed] [Google Scholar]
- 4. Chang YH, Connolly J, Shimasaki N, Mimura K, Kono K, Campana D. A chimeric receptor with NKG2D specificity enhances natural killer cell activation and killing of tumor cells. Cancer Res. 2013;73:1777‐1786. [DOI] [PubMed] [Google Scholar]
- 5. Hermanson DL, Kaufman DS. Utilizing chimeric antigen receptors to direct natural killer cell activity. Front Immunol. 2015;6:1‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang C, Oberoi P, Oelsner S, et al. Chimeric antigen receptor‐engineered NK‐92 cells: an off‐the‐shelf cellular therapeutic for targeted elimination of cancer cells and induction of protective antitumor immunity. Front Immunol. 2017;8:533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kitayama S, Zhang R, Liu T‐Y, et al. Cellular adjuvant properties, direct cytotoxicity of re‐differentiated Vα24 invariant NKT‐like cells from human induced pluripotent stem cells. Stem Cell Rep. 2016;6:213‐227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ueda N, Uemura Y, Zhang R, et al. Generation of TCR‐expressing innate lymphoid‐like helper cells that induce cytotoxic T cell‐mediated anti–leukemic cell response. Stem Cell Rep. 2018;10:1935‐1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li Y, Hermanson DL, Moriarity BS, Kaufman DS. Human iPSC‐derived natural killer cells engineered with chimeric antigen receptors enhance anti–tumor activity. Cell Stem Cell. 2018; 23:181‐192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nishimura T, Kaneko S, Kawana‐Tachikawa A, et al. Generation of rejuvenated antigen‐specific T cells by reprogramming to pluripotency and redifferentiation. Cell Stem Cell. 2013;12:114‐126. [DOI] [PubMed] [Google Scholar]
- 11. Minagawa A, Yoshikawa T, Yasukawa M, et al. Enhancing T cell receptor stability in rejuvenated iPSC‐derived T cells improves their use in cancer immunotherapy. Cell Stem Cell. 2018; 23:850‐858. [DOI] [PubMed] [Google Scholar]
- 12. Xu H, Wang B, Ono M, et al. Targeted disruption of HLA genes via CRISPR‐Cas9 generates iPSCs with enhanced immune compatibility. Cell Stem Cell. 2019;24:566‐578.e7. [DOI] [PubMed] [Google Scholar]
- 13. Iriguchi S, Kaneko S. Toward the development of true “off‐the‐shelf” synthetic T‐cell immunotherapy. Cancer Sci. 2019;110:16‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Croze RH, Buchholz DE, Radeke MJ, et al. ROCK inhibition extends passage of pluripotent stem cell‐derived retinal pigmented epithelium. Stem Cells Transl Med. 2014;3:1066‐1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hermanson DL, et al. Induced pluripotent stem cell‐derived natural killer cells for treatment of ovarian cancer. Stem Cells. 2016;34:93‐101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhai B, Shi D, Gao H, et al. A phase I study of anti–GPC3 chimeric antigen receptor modified T cells (GPC3 CAR‐T) in Chinese patients with refractory or relapsed GPC3+ hepatocellular carcinoma (r/r GPC3+ HCC). J Clin Oncol. 2017;35:3049. [Google Scholar]
- 17. Suzuki S, Yoshikawa T, Hirosawa T, et al. Glypican‐3 could be an effective target for immunotherapy combined with chemotherapy against ovarian clear cell carcinoma. Cancer Sci. 2011;102:1622‐1629. [DOI] [PubMed] [Google Scholar]
- 18. Tamada K, Geng D, Sakoda Y, et al. Redirecting gene‐modified T cells toward various cancer types using tagged antibodies. Clin Cancer Res. 2012;18:6436‐6445. [DOI] [PubMed] [Google Scholar]
- 19. Adachi K, Kano Y, Nagai T, et al. IL‐7 and CCL19 expression in CAR‐T cells improves immune cell infiltration and CAR‐T cell survival in the tumor. Nat Biotechnol. 2018;36:346‐351. [DOI] [PubMed] [Google Scholar]
- 20. Ho M, Kim H. Glypican‐3: a new target for cancer immunotherapy. Eur J Cancer. 2011;47:333‐338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gao H, Li K, Tu H, et al. Development of T cells redirected to glypican‐3 for the treatment of hepatocellular carcinoma. Clin Cancer Res. 2014;20:6418‐6428. [DOI] [PubMed] [Google Scholar]
- 22. Dianat‐Moghadam H, Rokni M, Marofi F, Panahi Y, Yousefi M. Natural killer cell–based immunotherapy: from transplantation toward targeting cancer stem cells. J Cell Physiol. 2018;234:259‐273. [DOI] [PubMed] [Google Scholar]
- 23. Rezvani K, Rouce RH. The application of natural killer cell immunotherapy for the treatment of cancer. Front Immunol 2015;6:578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang Y, Wallace DL, de Lara CM, et al. In vivo kinetics of human natural killer cells: the effects of ageing and acute and chronic viral infection. Immunology. 2007;121:258‐265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bachanova V, Cooley S, Defor TE, et al. Clearance of acute myeloid leukemia by haploidentical natural killer cells is improved using IL‐2 diphtheria toxin fusion protein. Blood. 2014;123:3855‐3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Miller JS, Soignier Y, Panoskaltsis‐Mortari A, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood. 2005;105:3051‐3057. [DOI] [PubMed] [Google Scholar]
- 27. Romee R, Rosario M, Berrien‐Elliott MM, et al. Cytokine‐induced memory‐like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med. 2016;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Raab S, Steinbacher J, Schmiedel BJ, et al. Fc‐Optimized NKG2D–Fc constructs induce NK Cell antibody‐dependent cellular cytotoxicity against breast cancer cells independently of HER2/neu expression status. J Immunol. 2014;193:4261‐4272. [DOI] [PubMed] [Google Scholar]
- 29. Snyder KM, Hullsiek R, Mishra HK, et al. Expression of a recombinant high affinity IgG Fc receptor by engineered NK cells as a docking platform for therapeutic mAbs to target cancer cells. Front Immunol. 2018;9:2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mehta RS, Rezvani K. Chimeric antigen receptor expressing natural killer cells for the immunotherapy of cancer. Front Immunol. 2018;9:1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hsu J,Hodgins J, Marathe M, et al. Contribution of NK cells to immunotherapy mediated by PD‐1/PD‐L1 blockade. J Clin Invest. 2018;128:4654‐4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. André P, Denis C, Soulas C, et al. Anti–NKG2A mAb is a checkpoint inhibitor that promotes anti–tumor immunity by unleashing both T and NK cells. Cell. 2018;175:1731‐1743.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu E, Dotti G, Shaim H, et al. Cord blood NK cells engineered to express IL‐15 and a CD19− targeted CAR show long‐term persistence and potent anti–tumor activity. Leukemia. 2018;32:520‐531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hacein‐Bey‐Abina S, Von Kalle C , Schmidt M, et al. LMO2‐associated clonal T cell proliferation in two patients after gene therapy for SCID‐X1. Science. 2003;302:415‐419. [DOI] [PubMed] [Google Scholar]
- 35. Fraietta JA, Nobles CL, Sammons MA, et al. Disruption of TET2 promotes the therapeutic efficacy of CD19‐targeted T cells. Nature. 2018;558:307‐312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li Y, Huo Y, Yu L, Wang J. Quality control and nonclinical research on CAR‐T cell products: general principles and key issues. Engineering. 2019;5:122‐131. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Fig S4
Fig S5
Fig S6
Fig S7
Fig S8
Fig S9
Fig S10
Table S1