

Abstract
Hepatocellular carcinoma (HCC) is a common and particularly fatal form of cancer for which very few drugs are effective. The fibroblast growth factor 19 (FGF19) has been viewed as a driver of HCC development and a potential Ab target for developing novel HCC therapy. However, a previously developed anti‐FGF19 Ab disrupted FGF19’s normal regulatory function and caused severe bile‐acid‐related side‐effects despite of having potent antitumor effects in preclinical models. Here, we developed novel human Abs (G1A8 and HS29) that specifically target the N‐terminus of FGF19. Both Abs inhibited FGF19‐induced HCC cell proliferation in vitro and significantly suppressed HCC tumor growth in mouse models. Importantly, no bile‐acid‐related side effects were observed in preclinical cynomolgus monkeys. Fundamentally, our study demonstrates that it is possible to target FGF19 for anti‐HCC therapies without adversely affecting its normal bile acid regulatory function, and highlights the exciting promise of G1A8 or HS29 as potential therapy for HCC.
Keywords: antibody therapy, bile acid, drug development, FGF19, HCC
Fibroblast growth factor 19 (FGF19) is a driver in hepatocellular carcinoma (HCC); however, it exerts important physiological function in regulation of bile acid synthesis. Our study, for the first time, shows that selectively targeting a defined region of FGF19 by our newly developed Abs could provide an effective and safe novel treatment option for HCC.

1. INTRODUCTION
Liver cancer ranks as the third most common cause of cancer‐related deaths.1 Among all liver cancers, hepatocellular carcinoma (HCC) is the most common type; it accounts for approximately 90% of all liver cancer cases.2 Currently, two tyrosine kinase inhibitors (TKIs), sorafenib and lenvatinib, are the only systemic agents approved by the FDA as first‐line treatment of HCC. They can only extend patient survival by approximately 3 months.3, 4 Other drugs in second‐line therapy, including several TKIs and immune checkpoint inhibitors, show slight survival advantages and antitumor activities, have limited therapeutic efficacy.5 Thus, there is obviously a need for more effective therapeutic agents for HCC patients.
Several studies have identified that HCC tumors feature highly focal amplification of fibroblast growth factor 19 (FGF19),6, 7, 8, 9 a bile acid‐induced and ileum‐derived peptide growth factor that functions to regulate bile acid metabolism. FGF19 binds to its receptor, hepatocyte‐expressed FGF receptor 4 (FGFR4), and its co‐receptor, β‐klotho (KLB), to repress the hepatic transcription of a gene encoding cholesterol‐7‐alpha‐hydroxylase 1 (CYP7A1), an essential enzyme for bile acid biosynthesis.10, 11, 12, 13, 14 In addition to its bile‐acid‐regulatory function, both FGF19 and its cognate receptor FGFR4 are highly expressed in tumors compared to adjacent non‐tumorous tissues.15, 16 The high expression of these proteins promotes tumor progression; moreover, it is also associated with poor prognosis in HCC patients.16, 17 In transgenic mice, the overexpression of FGF19 caused hepatocellular dysplasia, neoplasia, and ultimately HCC,18 yet these outcomes were abolished in FGFR4 knockout mice,19 thus mechanistically confirming the tumorigenic activity of aberrant FGF19/FGFR4 signaling.
Although several selective FGFR4 small molecule inhibitors are under development for the treatment of HCC, each of these elevates bile acid synthesis and causes liver toxicity (either in preclinical animal models or in early human clinical trials).20, 21, 22, 23, 24 Another drug development effort directly targeting FGF19 for treating HCC was based on a neutralizing anti‐FGF19 Ab, 1A6. Treatment with 1A6 prevented transgenic mice overexpressing FGF19 from developing HCC, and such treatment also suppressed the growth of HCC xenografts in mice.6, 15 Unfortunately, however, in a toxicology study, treatment with the humanized 1A6 Ab to cynomolgus monkeys increased hepatic transcription of CYP7A1 and elevated bile acid synthesis, thus dramatically altering bile acid metabolism and causing severe dose‐limiting side effects.25 Considering both FGF19’s physiological function in regulating bile acid metabolism and its tumorigenic activity in driving the pathogenesis of HCC, it is unclear if a strategy that targets FGF19 can effectively treat HCC while being safe for patients.
Previous studies exploring the function of FGF19’s N‐terminus (NT) have established that a variant (M70) with NT substitutions and deletions, as well as a chimeric variant substituted with the 20 N‐terminal residues from FGF21 exhibit reduced ability to induce hepatocyte proliferation but retained their ability to suppress hepatic Cyp7a1 expression.26, 27, 28 Building on these insights, we surmised that the NT of FGF19 may be essential for its tumorigenic activity but may not be required for its physiological bile‐acid‐regulatory function. We further hypothesized that selectively targeting the NT of FGF19 with an Ab instead of a small molecule drug may be both effective and safe.
In this study, we first identified Abs that specifically bind to FGF19 in an NT‐dependent manner. We then demonstrated that one high‐affinity NT‐dependent Ab, G1A8, and its close variant Ab, HS29, effectively inhibit FGF19‐induced HCC cell proliferation in vitro and significantly suppress HCC tumor growth in cell line‐derived xenograft and patient‐derived xenograft (PDX) mouse models. Importantly, G1A8 did not affect FGF19‐mediated repression of mouse hepatic Cyp7a1 transcription. Moreover, G1A8 did not cause bile‐acid‐related side effects in cynomolgus monkeys. Collectively, our study demonstrates that selectively targeting the NT of FGF19 with an Ab can be both effective and safe, and the Abs we developed, G1A8 and HS29, show strong promise to be further developed into a safe and therapeutic agent for treating FGF19‐driven HCC.
2. MATERIALS AND METHODS
2.1. Cell lines
FreeStyle 293‐F cells were cultured according to the manufacturer’s instructions (Thermo Fisher Scientific). The human HCC Hep3B cells (ATCC) were cultured in DMEM supplemented with 10% FBS. The Hep3B‐Luc23 stable cell line expressing firefly luciferase was generated through lentivirus transduction.
2.2. Expression and purification of proteins
For FGF19 and its variants, including the NT deletion variant FGF19∆NT (residues Arg43‐Lys216), the coding sequences were cloned into an expression vector with a C‐terminal His‐Avi‐Tag. The vectors were transiently transfected alone or co‐transfected with a vector encoding BirA biotin‐protein ligase into 293‐F cells. Cell supernatants were collected at 3‐5 days after transfection, and proteins were purified using Ni‐NTA affinity chromatography (Qiagen). Human IgG1 Ab expression and purification were similar to procedures described previously.29
2.3. Screening of Ab library against FGF19
The synthetic NT‐peptide of FGF19 comprises residues Arg23‐Ile42 (corresponding to residues 1‐20 of FGF19 following signal peptide cleavage) with a biotin modification at its C‐terminus (CT). The NT‐peptide or the biotinylated FGF19 protein was captured on streptavidin‐conjugated magnetic M‐280 Dynabeads and then used for phage‐Ab library selection.29 After 2 rounds of selection, clones that bound to FGF19 with higher affinity than FGF19∆NT were screened out by ELISA for further characterization.
2.4. 31A3 Ab sublibrary construction and selection for affinity improvement
A 31A3 sublibrary (1.2 × 108) with random mutated complementarity‐determining regions (CDRs) was constructed using NNK degenerate codons.30 The sublibrary selection and screening were undertaken using a similar method as described above. To obtain high‐affinity hits, competitive elution with 31A3‐hIgG1 was used.
2.5. Enzyme‐linked immunosorbent assay
Antigens were captured on NeutrAvidin (Sigma‐Aldrich) coated 96‐well plates (MaxiSorp; Nunc). For direct‐binding ELISA, 3‐fold serially diluted testing Abs were added. For competition ELISA, 3‐fold serially diluted testing Abs were mixed with competitor FGFR4‐hFc. Binding was detected using an HRP‐conjugated goat anti‐hFc Ab.
2.6. Binding kinetic analysis by surface plasmon resonance
Kinetic analysis of human IgG1 Abs was carried out using a Biacore T200 system. Anti‐hFc Ab was immobilized on a CM5 sensor chip using an amine‐coupling kit (GE Healthcare). Antibodies were then captured on the sensor chip and followed by flowing FGF19 or FGF19 variants.
2.7. Cell proliferation assay
The Hep3B cells were treated with FGF19 or FGF19 variants at various concentrations in DMEM supplemented with 1% FBS. For evaluation of the inhibition activity against FGF19‐induced cell proliferation, 15 μg/mL Abs were added. Seventy‐two hours later, cell proliferation was measured using CCK‐8 (Dojindo Molecular Technologies).
2.8. Expression of Cyp7a1 in liver
C57BL/6 mice (5‐6 weeks old) were fasted overnight before i.p. injection of 2 μg FGF19, FGF19 variants, or FGF19 together with 60 μg anti‐FGF19 Abs. Mice livers were harvested 3 hours after i.p. injection for extracting total RNA and reverse transcribed into cDNA. Cyp7a1 mRNA expression level (relative to GAPDH) was evaluated by quantitative PCR with an ABI Fast 7500 instrument (Applied Biosystems).
2.9. Animal experiments
Mice (6‐8 weeks old NOD SCID and NSG) were s.c. injected with 5 × 106 Hep3B cells in the right flank. Based on similar mean tumor bioluminescence intensities or tumor volumes, mice were divided into groups and received i.p. injection of 10 mg/kg anti‐FGF19 or control Ab. Tumor bioluminescence intensities were measured using an IVIS Lumina III Imaging System (PerkinElmer). Tumor volume was measured with an electronic caliper and calculated using the formula 3.14 × L × W2/6, where L and W are the largest and smallest measured diameters, respectively. All animal experiments were carried out following the National Guidelines for Housing and Care of Laboratory Animals in China and under the approved Institutional Animal Care and Use Committee (IACUC) protocols at the National Institute of Biological Sciences. The study to evaluate antitumor efficacy of HS29 in PDXs in BALB/c nude mice was undertaken under the approved IACUC protocols at Crown Bioscience.
Safety assessment of G1A8 was carried out at JOINN Laboratories following approved IACUC protocols. Healthy cynomolgus monkeys (3‐4 years old) weighing approximately 3 kg were i.v. injected with G1A8. Blood samples were collected at various time points for pharmacokinetic analysis of G1A8. Liver, ileum, and kidney samples were collected at the end of the study for mRNA expression analysis of genes related to bile acid metabolism.
2.10. Crystallization and structural determination of the FGF19‐G1A8 complex
Full‐length FGF19 (residues 25‐216) and G1A8‐Fab were separately expressed in 293‐F cells and individually purified by Ni‐NTA chromatography (Qiagen). Subsequently, the FGF19‐G1A8 complex was purified by a Superdex S200 column (GE Healthcare), and then concentrated to 18 mg/mL for crystallization using the hanging‐drop vapor diffusion method. X‐ray diffraction data were collected at the Shanghai Synchrotron Radiation Facility.31 Data were processed in HKL2000 and XDS. The crystals were of the P212121 space group, and contained two copies of the FGF19‐G1A8 complex per asymmetric unit. The structure was determined by molecular replacement using Phaser in Phenix with the following structures as search models: FGF19 (PDB‐2P23) and a Fab 5F2 structure (PDB‐3KDM). The model was iteratively built in Coot and refined in PHENIX.
2.11. Statistical analysis
Ordinary one‐way ANOVA or unpaired Student’s t tests were used for comparisons between groups. Two‐way ANOVA and Turkey’s multiple comparison tests were used to assess continuous variables. Kaplan‐Meier survival analysis and log‐rank tests were used for survival analysis.
3. RESULTS
3.1. Generation of a fully human Ab G1A8 that targets NT of FGF19
To test whether the NT of FGF19 is essential for its tumorigenic activity but may not be required for its physiological bile‐acid‐regulatory function, we generated an FGF19 NT deletion variant (FGF19∆NT) and used a variety of assays to examine the functions of both full‐length FGF19 and FGF19∆NT. Compared to full‐length FGF19, the FGF19∆NT variant had significantly weaker binding affinity for FGFR4 (either in the presence or absence of its co‐receptor KLB), and exerted significantly reduced activity to induce tumor cell proliferation (Figure S1A,B). To compare the bile‐acid‐regulatory function between full‐length FGF19 and FGF19∆NT, we tested them in a mouse model in which exogenous FGF19 can exert its bile‐acid‐regulatory function through binding to the murine receptor FGFR4—which shares 90% amino acid identity with human FGFR4—to repress the murine hepatic transcription of Cyp7a1.28, 32 In this mouse model, we observed no difference between full‐length FGF19 and FGF19∆NT in repression of the hepatic Cyp7a1 gene expression level (Figure S1C). In light of these results, we further explored if selectively targeting the NT of FGF19 using an Ab could potentially inhibit its tumorigenic activity without deleteriously affecting its bile‐acid‐regulatory function.
To identify Abs targeting FGF19’s NT, we designed two selection strategies by using two different targets to select Abs from our large human Ab phage display library29: one was a synthetic NT‐peptide of FGF19, and the other one was the full‐length FGF19 (Figure 1A). After the library selections, we screened out positive clones for the full‐length FGF19, and then identified a number of Abs that bind to FGF19 in an NT‐dependent manner based on weak binding to FGF19∆NT as the screening criterion. Among the NT‐dependent Abs identified, the 31A3 Ab had relatively high binding affinity and was thus selected for further analysis (Figure 1A‐C).
Figure 1.
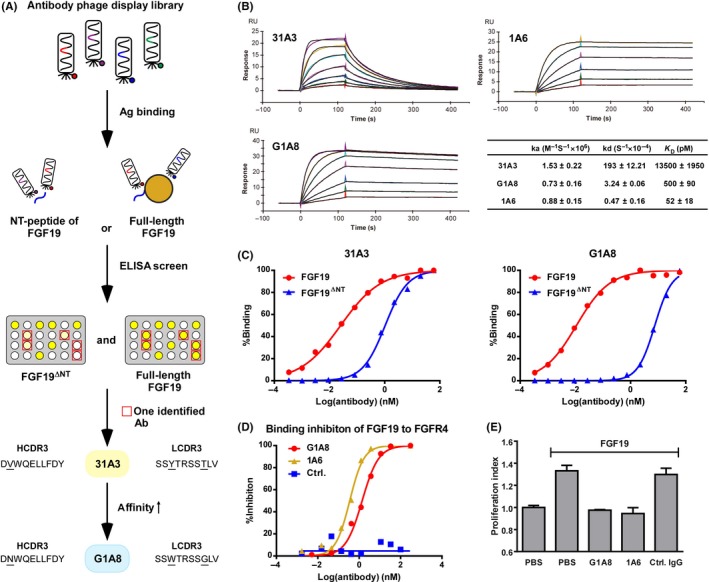
Generation and characterization of the G1A8 Ab. A, Schematic diagram illustrating the generation of Ab G1A8 using Ab phage display technology. Antibodies that bind to fibroblast growth factor 19 (FGF19) with higher affinity than to FGF19 N‐terminus (NT) deletion variant (FGF19∆NT) were identified and affinity matured. The amino acids of the complementarity‐determining regions 3 (CDR3) of one identified Ab, 31A3, and its affinity‐improved Ab G1A8 are shown; underlined amino acids indicate differences between the two Abs. HCDR3, heavy chain CDR3; LCDR3, light chain CDR3. B, Kinetic analysis of the binding of anti‐FGF19 Abs to FGF19 using surface plasmon resonance. Representative kinetic curves for each Ab are shown. All kinetic parameters shown represent the mean and SE of three independent experiments. C, The binding of 31A3 and G1A8 to FGF19 depends on the NT of FGF19. D, G1A8 competes with FGF receptor 4 (FGFR4) for binding to FGF19. Antibodies in their Fab form were mixed with 100 nM FGFR4‐Fc in the presence of 20 μg/mL heparin. The competition activity of Abs is shown as the percentage of inhibition for FGFR4 binding to FGF19. The control is an antihepatitis B virus preS1 Fab Ab 2H5‐A14.29 E, G1A8 inhibits FGF19‐induced hepatocellular carcinoma cell proliferation. Hep3B was cultured with 20 ng/mL FGF19 and 15 μg/mL Abs. Control IgG, rituximab
We next evaluated whether 31A3 can inhibit HCC cell proliferation as specifically induced by FGF19; we treated the human HCC cell line Hep3B with 31A3 in the presence of exogenous FGF19. Encouragingly, 31A3 inhibited FGF19‐induced proliferation of Hep3B cells (Figure S2A). Note that the antiproliferation effect of 31A3 was less pronounced than that of 1A6, the aforementioned anti‐FGF19 Ab that targets the CT region of FGF19.15 Partially explaining this observed difference, the FGF19‐binding affinity of 31A3 is much weaker than that of 1A6 (Figure 1B). Consistently, we found that 31A3 was not able to suppress tumor growth in a Hep3B xenograft mouse model (Figure S2B). These results collectively suggested that the FGF19‐binding affinity of 31A3 could be insufficiently strong to confer inhibition of tumor growth in vivo.
We subsequently improved the affinity of 31A3 through mutagenesis within 31A3’s two CDR3 regions that are known to confer particularly large impacts on binding were selected for mutagenesis. We constructed an Ab phage display sub‐library comprising 31A3‐derived Abs with randomized mutations within the two CDR3s. We then used 31A3‐hIgG1 as a competitor during this 31A3‐derived sub‐library selection to screen out binders with higher binding affinity to FGF19. This approach yielded a small panel of affinity‐improved Abs. The best performing hit from this panel, G1A8, having only three amino acid changes, exhibited a 27‐fold affinity increase and a 60‐fold slower dissociation rate, as well as substantially higher NT‐dependency over the parental Ab 31A3 (Figure 1A‐C). G1A8 also efficiently competed with the receptor FGFR4 for binding to FGF19, with an IC50 value of 1.39 nM (Figure 1D). Additionally, G1A8 exerted a strong antiproliferation effect on the growth of exogenous‐FGF19‐treated Hep3B cells (Figure 1E).
3.2. Structural characterization of the epitope of G1A8
To further understand the molecular basis of the G1A8‐FGF19 interactions and to precisely characterize the epitope of G1A8, we determined the structure of the Fab form of G1A8 in complex with FGF19 at 2.6 Å resolution by X‐ray crystallography. The structure was solved using a molecular replacement method and refined to a R work/R free of 0.216/0.278, with good geometry (Table S1). The residues 37‐172 of FGF19 showing well‐defined electron density were modeled. The complex structure shows an apparently perfect shape complementarity, burying a total surface area of 983 Å2.
In accordance with G1A8’s designed feature of specifically targeting the NT of FGF19 as described above, the majority of the residues comprising G1A8’s epitope are at the FGF19’s NT, which is situated above the cleft between the variable heavy chain (VH) and light chain (VL) (Figure 2A,B). The structure also revealed that residues 168‐172 of the FGF19 are in close proximity with the NT at the interaction interface of FGF19 and G1A8. The Leu169 residue and the NT together form hydrophobic interactions with Tyr51 and Pro57 from light chain CDR (LCDR) 2 (Figure 2A‐C). The electron density for the FGF19 CT (residues 173‐216) is invisible, likely due to its intrinsic flexibility.32, 33A stretch of roughly 8 residues (residues 38‐45) of the FGF19 NT engage extensive interactions with all six CDR loops from both the VH and VL of G1A8 through a mix of hydrophobic and polar contacts (Figure 2C and Table S2). The side chain of FGF19 NT residue Trp38 is anchored in a hydrophobic pocket formed by Ala33, Ser52, Ser57, Tyr59, Gln102, and Leu104 from heavy chain CDR (HCDR) 1‐3, while Arg45 of FGF19 on the other side engages salt bridge interactions with Glu103 and Asp52 from HCDR3 and LCDR2, respectively (Figure 2C). Indeed, an FGF19 Trp38Ala mutant had reduced binding affinity for G1A8, and Arg45Ala mutant completely lost the binding (Figure 2D), confirming the essentiality of these interactions. Arg45 also makes additional hydrogen bond interactions with Asn100 from HCDR3 of G1A8 (Figure 2C). G1A8’s parental Ab 31A3 has a Val at this same position (Figure 1A); this difference likely accounts for 31A3’s lower affinity than G1A8.
Figure 2.
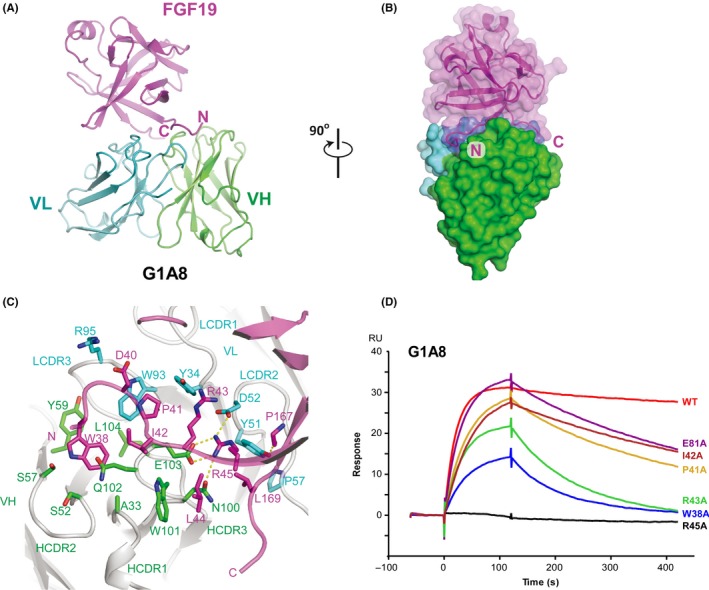
Structural analysis of the fibroblast growth factor 19 (FGF19)‐G1A8 complex. A, Ribbon representation of FGF19‐G1A8 in orthogonal views. FGF19 is shown in magenta; the variable heavy chain (VH) and variable light chain (VL) of G1A8 are in green and cyan, respectively. C, C‐terminus; N, N‐terminus. B, Surface view of the FGF19‐G1A8 complex. C, Detailed view of the G1A8‐FGF19 interface. Dashed lines represent hydrogen bonds. LCDR3, light chain complementarity‐determining region; HCDR3, heavy chain complementarity‐determining region. D, Binding activity of G1A8 to WT FGF19 and FGF19 alanine mutants as analyzed using surface plasmon resonance
3.3. Antitumor activity of G1A8 in xenograft mouse models
As G1A8 significantly inhibited FGF19‐induced proliferation of tumor cells in vitro (Figure 1E), we further evaluated its antitumor activity using xenograft mouse models (Figure 3). We first established a Hep3B‐Luc23 cell line that stably expresses luciferase and thus enabled measurement of tumor growth using in vivo bioluminescence imaging. Mice were s.c. injected with these Hep3B‐Luc23 cells, and then were divided into three groups with similar mean tumor bioluminescence intensities prior to receiving Ab treatment (10 mg/kg) twice each week for 4 weeks. The Ab treatment started at day 4 after tumor cell implantation, an early stage of tumor development. Monitoring of tumor growth over time by measuring tumor volumes and bioluminescence intensities showed that G1A8 and 1A6 each significantly suppressed tumor growth compared to the isotype control Ab; there was no significant difference between the two Abs (Figure 3A‐C).
Figure 3.
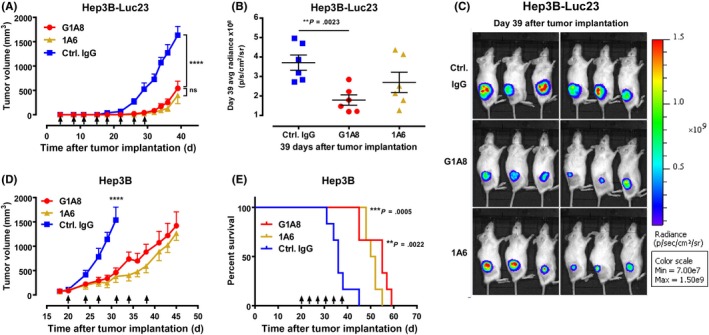
Antitumor activity of antifibroblast growth factor 19 (FGF19) Abs in hepatocellular carcinoma xenograft mouse models. A‐C, G1A8 inhibited tumor growth in a cell line‐derived xenograft model established using luciferized Hep3B‐Luc23 cells in NOD SCID mice (n = 6/group). D, E, G1A8 inhibited tumor growth in a xenograft model established using WT Hep3B cells in NSG mice. Tumor volumes and survival curves are shown. Time points for Ab treatment are marked by arrows. Control (Ctrl.) IgG, rituximab
We also evaluated G1A8’s antitumor efficacy against xenograft tumors formed from transplanted WT Hep3B cells. Antibody treatment was started when tumor volumes reached approximately 100 mm3 in size at day 20 after tumor cell implantation, a relatively late stage of tumor development. Hep3B‐bearing mice were divided into three groups with similar mean tumor volumes and received Ab treatment (10 mg/kg) twice each week for 3 weeks. As with the aforementioned results for Hep3B‐Luc23 xenografts, both G1A8 and 1A6 significantly inhibited Hep3B tumor progression (Figure 3D); moreover, both Abs significantly prolonged survival of mice (Figure 3E).
3.4. Safety assessment of G1A8 in mouse and cynomolgus monkey
As G1A8 cannot bind to murine FGF15, the aforementioned mouse tumor xenograft models are not suitable for assessing the safety profiles of G1A8 treatment; specifically, FGF15 is the murine ortholog of human FGF19 and shares only 49% amino acid identity with FGF19.34 Nevertheless, recalling that human FGF19 can exert its bile‐acid‐regulatory function in mouse via murine FGFR4 to repress hepatic Cyp7a1 transcription28, 32 (Figure S1C), we were able to assess whether G1A8 affects hepatic Cyp7a1 transcription in this mouse model. Encouragingly, and unlike 1A6, G1A8 did not affect FGF19‐induced repression of hepatic Cyp7a1 transcription (Figure S3A), indicating that G1A8 does not apparently interfere with FGF19’s bile‐acid‐regulatory function.
Pai et al.25 showed that treatment with the humanized anti‐FGF19 Ab (1A6) in cynomolgus monkeys caused disrupted bile acid metabolism by interrupting FGF19’s function and thereby increasing CYP7A1 expression; this was accompanied by clinical manifestations including reduced body weight, low food consumption, and severe diarrhea, and ultimately resulted in unscheduled euthanasia of all animals after one dose in the 10 and 30 mg/kg treatment groups.25 We first verified that G1A8 binds cynomolgus FGF19 and human FGF19 with similar binding affinities (Figures 1B and S3B). We then assessed whether G1A8 impairs the bile‐acid‐regulatory function of FGF19 in cynomolgus monkeys. Four cynomolgus monkeys were randomized into two groups and received control saline or 10 mg/kg G1A8 at day 1 and 30 mg/kg G1A8 at day 16. Each group included one male and one female monkey. Blood samples were collected throughout the duration of the study (Figure 4A).
Figure 4.
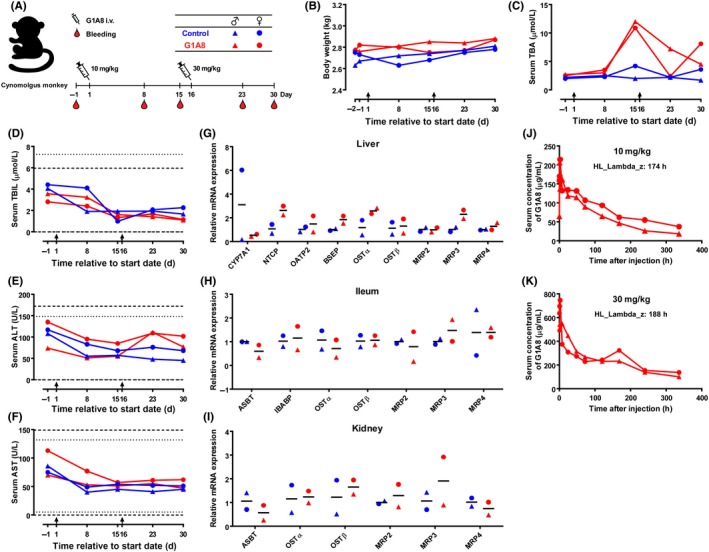
Safety assessment of G1A8 in cynomolgus monkeys. A, Schematic diagram illustrating the timeline of G1A8 treatment and sampling from animals. Four cynomolgus monkeys were divided into two groups receiving i.v. of control saline or G1A8. Each group consists of one male and one female monkey. B, Body weight of cynomolgus monkeys. C‐F, Blood biochemistry: serum total bile acid (TBA), total bilirubin (TBIL), alanine transaminase (ALT), and aspartate transaminase (AST) at various time points. Arrows indicate treatment days. Dashed lines and dotted lines indicate normal reference intervals of biochemical parameters in the male and female cynomolgus monkeys, respectively. G‐I, Expression of genes related to bile acid metabolism. Tissue samples were harvested at the end of the study. Expression of selected genes in liver, ileum, and kidney were analyzed by quantitative PCR. J, K, Pharmacokinetic profile of G1A8. Serum concentrations of G1A8 following treatment with 10 mg/kg G1A8 on day 1 or 30 mg/kg G1A8 on day 16 are shown
Encouragingly, all of the monkeys finished two full courses of G1A8 treatment, and none of them exhibited any of the clinical side effects reported for 1A6, such as reduced body weight, low food consumption, or diarrhea (Figure 4B). The previous study of 1A6 treatment (a single dose) in monkeys reported a marked increase in serum total bile acid (TBA) levels, alanine transaminase (ALT), and aspartate transaminase (AST).25 Although monkeys in the G1A8 treatment group initially showed a slight increase in serum TBA concentration at day 15 (first testing time point) after G1A8 (10 mg/kg) treatment, no further increase was observed following the second injection of a higher dose of 30 mg/kg at day 16; rather, a decrease was observed (Figure 4C). This suggests that the slight increase in the serum TBA level observed at day 15 can likely be explained by normal physiological variations in the animals. The serum levels of total bilirubin, ALT, and AST showed no significant differences in monkeys treated with either control saline or G1A8, suggesting that G1A8 does not cause liver damage (Figure 4D‐F).
We also collected monkey tissue samples from the organs (liver, ileum, and kidney) that are responsible for bile acid recycling to evaluate the expression levels of multiple genes known to impact bile acid metabolism (Figure 4G,H). In the liver, no increase in CYP7A1 gene expression was found in animals treated with G1A8, and the expression of other genes known to encode bile acid transporter proteins did not show any significant increase compared to control animals (Figure 4G). In the kidney and ileum, the expression of bile acid transporter genes did not differ between the G1A8 and control groups (Figure 4H,I), indicating G1A8 does not disrupt the recycling and metabolism of bile acid. Pathological examination revealed no abnormalities in any animal. The G1A8 serum concentration‐time profiles for the two different dosages revealed similar terminal half‐life durations for G1A8 of 174 and 188 hours in cynomolgus monkeys dosed with 10 and 30 mg/kg, respectively (Figure 4J,K). Taken together, these pilot safety assessment experiments in cynomolgus monkey did not detect any bile‐acid‐related toxicity in response to G1A8, thus highlighting that G1A8 treatment in a therapeutic setting is unlikely to cause significant malabsorption of bile acid or other bile‐acid‐related side effects.
3.5. Fibroblast growth factor‐19‐dependent antitumor activity of G1A8’s close derivative HS29 in PDX models
We subsequently used Ab engineering to further improve the physicochemical properties of G1A8 and eventually obtained an Ab, HS29, with only two amino acid substitutions in the HCDR3 region of G1A8 that had highly favorable physicochemical properties, eg, improved thermal stability and expression yield. Importantly, HS29 retains G1A8’s binding epitope, high binding affinity, competition activity against FGF19 binding to FGFR4 in the presence or absence of co‐receptor KLB, and strong antiproliferation activity (Figure S4). In addition, G1A8 and HS29 did not show apoptotic or cytotoxic effects (Figure S5).
We next examined HS29’s antitumor activity in human HCC PDX models, including an FGF19‐expressing HCC model and an FGF19‐negative HCC model (Figure 5A,B). Treatment with HS29 started when the mean tumor volume reached approximately 150 mm3. Mice bearing FGF19‐expressing tumors showed arrested tumor growth after HS29 treatment (Figure 5A). Whereas the HS29‐treated mice maintained steady weights and remained healthy throughout the experiment, mice in the control group showed significant weight loss as tumors continued to grow, and two control mice died prior to the end of the study because of rapid tumor progression (Figure 5C). Notably, no response to HS29 treatment was observed for the PDX model that lacked FGF19 expression (Figure 5B). At the end of the study, FGF19 mRNA expression in tumor samples was examined by quantitative PCR and confirmed that the FGF19‐expressing tumor had strong FGF19 mRNA expression (more than 20‐fold higher than the Hep3B cell line) whereas the FGF19‐negative tumor had barely detectable FGF19 mRNA levels (Figure 5D). Collectively, these results confirmed that the antitumor effects of HS29 are FGF19‐dependent.
Figure 5.
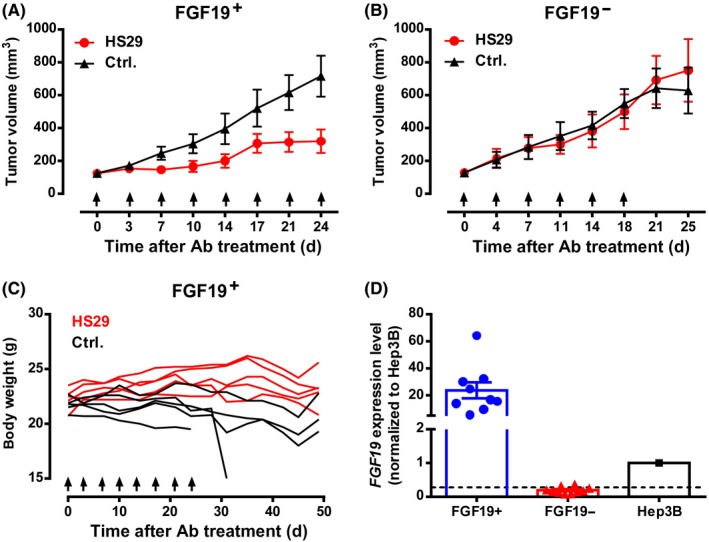
Evaluation of antitumor activity of HS29 in hepatocellular carcinoma patient‐derived xenograft (PDX) models. A‐C, Tumor volumes or body weights of BALB/c nude mice bearing fibroblast growth factor‐19 (FGF19)+ or FGF19− PDX. Mice were divided into groups (n = 5/group) with similar mean tumor volume and received 10 mg/kg HS29 i.p. or no treatment as control (Ctrl.). D, FGF19 mRNA expression in tumor samples. Each data point represents one mouse tumor sample. The level of FGF19 mRNA expression in Hep3B was used as a reference. The dashed line indicates the reliable detection limit of the assay
4. DISCUSSION
Drug development for HCC therapy has been disappointing to date. The multikinase inhibitors sorafenib and lenvatinib are the only two FDA approved first‐line treatments for HCC. However, sorafenib lacks efficient inhibition activity against FGFR4,35 and lenvatinib shows much more potent inhibition activity for vascular endothelial growth factor receptors than for FGFRs.36, 37 It is therefore reasonable to surmise that these agents might be ineffective for the treatment of patients with aberrant FGF19‐FGFR4 signaling axis. Increased FGF19 expression was found in HCC patients, and as many as 48% of liver tumors were positive for FGF19 expression.15, 17 Moreover, after surgical resection of liver tumors, HCC patients still have higher serum FGF19 levels than healthy individuals,16 and most HCC patients experience disease recurrence even after tumor resection, suggesting that a supra‐physiological level of FGF19 might induce excessive cell proliferation and drive HCC development. Hence, a therapeutic strategy specifically targeting FGF19 that could inhibit the excessive cell proliferation induced by FGF19 during HCC development may also protect patients from HCC recurrence.
In this study, we generated high‐affinity NT‐targeting Abs (G1A8 and HS29) by using extensive library screening and Ab engineering, and demonstrated their strong antitumor efficacy in xenograft mouse models and their safety in cynomolgus monkeys. Although we only included four cynomolgus monkeys in the safety assessment study of G1A8 in our preclinical pilot study, it is notable that none of the G1A8‐treated monkeys manifested any bile‐acid‐related toxicity or clinical symptoms. To more reliably evaluate G1A8’s potential side‐effects, a safety study that includes larger numbers of animals, as well as higher doses and long‐term treatment with G1A8 or its close engineered derivative HS29 under good laboratory practice conditions will be needed. In such a safety study, detailed analysis of bile acid composition after G1A8 or HS29 treatment will also help to more comprehensively evaluate bile acid metabolism in response to these Ab therapies.
It is noteworthy that it is very challenging to generate high‐affinity Abs specifically targeting the NT of FGF19. The NT (residues 23‐39, corresponding to residues 1‐17 of FGF19 following signal peptide cleavage) is intrinsically flexible as revealed by two previously determined FGF19 structures.32, 33 Consistently, in our FGF19/G1A8 complex structure, these NT residues are also disordered; only residues 38‐45 that engage extensive interactions with the six CDR loops of G1A8 show well‐ordered electron density. Thus, both the intrinsic flexibility of the NT and the limited number of targetable epitope residues at the NT contribute to the challenges of obtaining NT‐selectively targeting Abs.
The FGF family members have a homologous core region formed by 12 antiparallel β‐strands flanked by divergent NT and CT.38 The primary sequence variations among the NT and CT of various FGFs account for their different biological activities.39, 40 FGF19 belongs to the endocrine FGF19 subfamily that also includes FGF21 and FGF23. The crystal structure of FGF19’s CT tail bound to soluble KLB has been determined,41 whereas the structure of the ternary complex of FGF19‐FGFR4‐KLB is yet to be solved. A recently solved crystal structure of FGF23 in complex with its receptor FGFR1c and co‐receptor α‐klotho (KLA) revealed that KLA tethers FGFR1c and FGF23 to form a stable ternary complex. In the complex, the globular NT region and the rod‐like CT region of FGF23 are critical for binding to FGFR1c and KLA, respectively. Heparin sulfate (HS) is required for the dimerization of the 1:1:1 FGF23‐KLA‐FGFR1c complex; the FGF23 signaling through FGFR1c depends on both KLA binding and HS‐mediated dimerization.42 It remains to be determined whether FGF19, KLB, and FGFR4 form a ternary and/or quaternary complex in a similar manner. As FGFR4 is the essential and indispensable receptor for mediating both FGF19’s physiological function in regulating bile acid metabolism and its tumorigenic activity in driving the pathogenesis of HCC,43, 44, 45 it is therefore especially important to uncover the structure‐function relationships that govern FGF19’s tumorigenic and bile‐acid‐regulatory activities.
Nonetheless, our current understanding is that the CT of FGF19 determines FGF19’s binding specificity towards co‐receptor KLB,41, 46, 47, 48 and thus FGF19’s endocrine actions primarily function in the liver—the only tissue that highly expresses both FGFR4 and KLB; the NT of FGF19 may be essential for its tumorigenic activity but may not be required for its physiological bile‐acid‐regulatory function (Figure S1).26, 27, 28, 45, 49 It appears that the NT 38‐42 residue region of FGF19 (the critical binding site for G1A8 and HS29) can contribute to FGFR4 activation with respect to hepatocyte proliferation, which is independent of the binding between KLB and FGF19’s CT.48, 49 Moreover, recall that the aforementioned FGF19 NT variant, M70, exerts different biological activities—in bile acid regulation and tumorigenicity—but exhibits no difference in its binding affinity for its receptor FGFR4.28 Specifically, M70 does not induce tumor formation owing to its inability to activate the STAT3 pathway downstream of FGFR4 compared to FGF19, but retains the ability to activate the FGFR4‐dependent signaling pathways of bile acid regulation.27, 28
It is intriguing how our NT‐targeting Abs (GA18 and HS29) exert antitumor growth activities without causing obvious side effects related to FGF19’s bile‐acid‐regulatory function. The unique features of our NT‐targeting Abs likely result from their selective interruption of the FGF19‐FGFR4 interactions that is required for tumorigenesis. Building on the aforementioned structural and functional findings, we surmised that the interaction of FGF19’s CT with KLB plays the most critical role in tethering receptors and implementing all of FGF19’s activities; whereas the globular NT interacts with FGFR4 and may modulate the receptor dimerization, hence regulates downstream receptor signaling strength. Thus, Abs (ie, 1A6) targeting a region proximal to FGF19’s CT may lead to a complete loss of FGF19’s activities, including its physiological activity. Antibodies (ie, G1A8 and HS29) targeting the FGF19‐NT may only reduce receptor signaling strength to a level that can still maintain FGF19’s physiological activity in bile acid metabolism (Figure S6).
In summary, we developed Abs that open the door for further exploitation of the widely‐appreciated potential benefits of FGF19‐targeted therapy for HCC patients. More generally, our study is a clear example of how an Ab can successfully disrupt a cancer‐related target that was previously thought to be undruggable. Importantly, aberrant FGF19‐FGFR4 signaling has also been associated with the pathogenesis of many other cancers, including breast cancer,50, 51, 52 prostate cancer,53, 54 colon cancer,15, 55 and lung cancer,15, 56, 57 suggesting that blockade of FGF19 signaling using G1A8 or HS29 Ab could be a useful strategy for developing treatments of many types of cancer.
DISCLOSURE
The authors have no conflict of interest.
Supporting information
Suppinfo
ACKNOWLEDGMENTS
This work was supported by the Beijing Municipal Science and Technology Commission, and Beijing Key Laboratory of Pathogen Invasion and Immune Defense (Z171100002217064 to JS). This work was also supported by the National Natural Science Foundation of China (NSFC81525018 to WL). We thank W. Chen, X. Huang, in the Sui laboratory for their technical assistance, and Z. Gao and F. Mao in Dr Wenhui Li’s laboratory at NIBS for their helpful discussion. We would also like to thank the NIBS Biological Resource Center for DNA sequencing, the NIBS Animal Facility for their help in animal handling and care, and the staff at beamline BL19U of the Shanghai Synchrotron Radiation Facility for assistance in collection of X‐ray diffraction data of crystals.
Liu H, Zheng S, Hou X, et al. Novel Abs targeting the N‐terminus of fibroblast growth factor 19 inhibit hepatocellular carcinoma growth without bile‐acid‐related side‐effects. Cancer Sci. 2020;111:1750–1760. 10.1111/cas.14353
REFERENCES
- 1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394‐424. [DOI] [PubMed] [Google Scholar]
- 2. Llovet JM, Zucman‐Rossi J, Pikarsky E, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2016;2:16018. [DOI] [PubMed] [Google Scholar]
- 3. Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378‐390. [DOI] [PubMed] [Google Scholar]
- 4. Kudo M, Finn RS, Qin S, et al. Lenvatinib versus sorafenib in first‐line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non‐inferiority trial. Lancet. 2018;391:1163‐1173. [DOI] [PubMed] [Google Scholar]
- 5. Kudo M. Targeted and immune therapies for hepatocellular carcinoma: predictions for 2019 and beyond. World J Gastroenterol. 2019;25:789‐807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sawey ET, Chanrion M, Cai C, et al. Identification of a therapeutic strategy targeting amplified FGF19 in liver cancer by oncogenomic screening. Cancer Cell. 2011;19:347‐358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang K, Lim HY, Shi S, et al. Genomic landscape of copy number aberrations enables the identification of oncogenic drivers in hepatocellular carcinoma. Hepatology. 2013;58:706‐717. [DOI] [PubMed] [Google Scholar]
- 8. Ahn SM, Jang SJ, Shim JH, et al. Genomic portrait of resectable hepatocellular carcinomas: implications of RB1 and FGF19 aberrations for patient stratification. Hepatology. 2014;60:1972‐1982. [DOI] [PubMed] [Google Scholar]
- 9. Schulze K, Imbeaud S, Letouze E, et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet. 2015;47:505‐511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xie MH, Holcomb I, Deuel B, et al. FGF‐19, a novel fibroblast growth factor with unique specificity for FGFR4. Cytokine. 1999;11:729‐735. [DOI] [PubMed] [Google Scholar]
- 11. Holt JA, Luo G, Billin AN, et al. Definition of a novel growth factor‐dependent signal cascade for the suppression of bile acid biosynthesis. Genes Dev. 2003;17:1581‐1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kurosu H, Choi M, Ogawa Y, et al. Tissue‐specific expression of betaKlotho and fibroblast growth factor (FGF) receptor isoforms determines metabolic activity of FGF19 and FGF21. J Biol Chem. 2007;282:26687‐26695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lin BC, Wang M, Blackmore C, Desnoyers LR. Liver‐specific activities of FGF19 require Klotho beta. J Biol Chem. 2007;282:27277‐27284. [DOI] [PubMed] [Google Scholar]
- 14. Jones SA. Physiology of FGF15/19 In: Kuro‐o M, ed. Endocrine FGFs and Klothos. New York, NY: Springer US; 2012:171‐182. [Google Scholar]
- 15. Desnoyers LR, Pai R, Ferrando RE, et al. Targeting FGF19 inhibits tumor growth in colon cancer xenograft and FGF19 transgenic hepatocellular carcinoma models. Oncogene. 2008;27:85‐97. [DOI] [PubMed] [Google Scholar]
- 16. Miura S, Mitsuhashi N, Shimizu H, et al. Fibroblast growth factor 19 expression correlates with tumor progression and poorer prognosis of hepatocellular carcinoma. BMC Cancer. 2012;12:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hyeon J, Ahn S, Lee JJ, Song DH, Park CK. Expression of fibroblast growth factor 19 is associated with recurrence and poor prognosis of hepatocellular carcinoma. Dig Dis Sci. 2013;58:1916‐1922. [DOI] [PubMed] [Google Scholar]
- 18. Nicholes K, Guillet S, Tomlinson E, et al. A mouse model of hepatocellular carcinoma: ectopic expression of fibroblast growth factor 19 in skeletal muscle of transgenic mice. Am J Pathol. 2002;160:2295‐2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. French DM, Lin BC, Wang M, et al. Targeting FGFR4 inhibits hepatocellular carcinoma in preclinical mouse models. PLoS ONE. 2012;7:e36713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hagel M, Miduturu C, Sheets M, et al. First selective small molecule inhibitor of FGFR4 for the treatment of hepatocellular carcinomas with an activated FGFR4 signaling pathway. Cancer Discov. 2015;5:424‐437. [DOI] [PubMed] [Google Scholar]
- 21. Kim R, Sharma S, Meyer T, et al. First‐in‐human study of BLU‐554, a potent, highly‐selective FGFR4 inhibitor designed for hepatocellular carcinoma (HCC) with FGFR4 pathway activation. Eur J Cancer. 2016;69:S41. [Google Scholar]
- 22. Chan SL, Yen C‐J, Schuler M, et al. Abstract CT106: Ph I/II study of FGF401 in adult pts with HCC or solid tumors characterized by FGFR4/KLB expression. Can Res. 2017;77:CT106. [Google Scholar]
- 23. Joshi JJ, Coffey H, Corcoran E, et al. H3B–6527 is a potent and selective inhibitor of FGFR4 in FGF19‐driven hepatocellular carcinoma. Cancer Res. 2017;77:6999‐7013. [DOI] [PubMed] [Google Scholar]
- 24. Ruggeri B, Stubbs M, Yang Y‐O, et al. Abstract 1234: the novel FGFR4‐selective inhibitor INCB062079 is efficacious in models of hepatocellular carcinoma harboring FGF19 amplification. Can Res. 2017;77:1234‐1234. [Google Scholar]
- 25. Pai R, French D, Ma N, et al. Antibody‐mediated inhibition of fibroblast growth factor 19 results in increased bile acids synthesis and ileal malabsorption of bile acids in cynomolgus monkeys. Toxicol Sci. 2012;126:446‐456. [DOI] [PubMed] [Google Scholar]
- 26. Wu AL, Coulter S, Liddle C, et al. FGF19 regulates cell proliferation, glucose and bile acid metabolism via FGFR4‐dependent and independent pathways. PLoS ONE. 2011;6:e17868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Luo J, Ko B, Elliott M, et al. A nontumorigenic variant of FGF19 treats cholestatic liver diseases. Sci Transl Med. 2014;6:247ra100. [DOI] [PubMed] [Google Scholar]
- 28. Zhou M, Wang X, Phung V, et al. Separating tumorigenicity from bile acid regulatory activity for endocrine hormone FGF19. Cancer Res. 2014;74:3306‐3316. [DOI] [PubMed] [Google Scholar]
- 29. Li D, He W, Liu X, et al. A potent human neutralizing antibody Fc‐dependently reduces established HBV infections. eLife. 2017;6:e26738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sui J, Aird DR, Tamin A, et al. Broadening of neutralization activity to directly block a dominant antibody‐driven SARS‐coronavirus evolution pathway. PLoS Pathog. 2008;4:e1000197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang Q‐S, Zhang K‐H, Cui Y, et al. Upgrade of macromolecular crystallography beamline BL17U1 at SSRF. Nucl Sci Tech. 2018;29:68. [Google Scholar]
- 32. Goetz R, Beenken A, Ibrahimi OA, et al. Molecular insights into the klotho‐dependent, endocrine mode of action of fibroblast growth factor 19 subfamily members. Mol Cell Biol. 2007;27:3417‐3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Harmer NJ, Pellegrini L, Chirgadze D, Fernandez‐Recio J, Blundell TL. The crystal structure of fibroblast growth factor (FGF) 19 reveals novel features of the FGF family and offers a structural basis for its unusual receptor affinity. Biochemistry. 2004;43:629‐640. [DOI] [PubMed] [Google Scholar]
- 34. Wright TJ, Ladher R, McWhirter J, Murre C, Schoenwolf GC, Mansour SL. Mouse FGF15 is the ortholog of human and chick FGF19, but is not uniquely required for otic induction. Dev Biol. 2004;269:264‐275. [DOI] [PubMed] [Google Scholar]
- 35. Davis MI, Hunt JP, Herrgard S, et al. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol. 2011;29:1046‐1051. [DOI] [PubMed] [Google Scholar]
- 36. Yamamoto Y, Matsui J, Matsushima T, et al. Lenvatinib, an angiogenesis inhibitor targeting VEGFR/FGFR, shows broad antitumor activity in human tumor xenograft models associated with microvessel density and pericyte coverage. Vasc Cell. 2014;6:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hussein Z, Mizuo H, Hayato S, Namiki M, Shumaker R. Clinical pharmacokinetic and pharmacodynamic profile of lenvatinib, an orally active, small‐molecule, multitargeted tyrosine kinase inhibitor. Eur J Drug Metab Pharmacokinet. 2017;42:903‐914. [DOI] [PubMed] [Google Scholar]
- 38. Goetz R, Mohammadi M. Exploring mechanisms of FGF signalling through the lens of structural biology. Nat Rev Mol Cell Biol. 2013;14:166‐180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Beenken A, Mohammadi M. The FGF family: biology, pathophysiology and therapy. Nat Rev Drug Discov. 2009;8:235‐253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Itoh N, Ornitz DM. Fibroblast growth factors: from molecular evolution to roles in development, metabolism and disease. J Biochem. 2011;149:121‐130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kuzina ES, Ung PM, Mohanty J, et al. Structures of ligand‐occupied beta‐Klotho complexes reveal a molecular mechanism underlying endocrine FGF specificity and activity. Proc Natl Acad Sci USA. 2019;116:7819‐7824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen G, Liu Y, Goetz R, et al. alpha‐Klotho is a non‐enzymatic molecular scaffold for FGF23 hormone signalling. Nature. 2018;553:461‐466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Inagaki T, Choi M, Moschetta A, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005;2:217‐225. [DOI] [PubMed] [Google Scholar]
- 44. Yu C, Wang F, Kan M, et al. Elevated cholesterol metabolism and bile acid synthesis in mice lacking membrane tyrosine kinase receptor FGFR4. J Biol Chem. 2000;275:15482‐15489. [DOI] [PubMed] [Google Scholar]
- 45. Wu X, Ge H, Lemon B, et al. FGF19‐induced hepatocyte proliferation is mediated through FGFR4 activation. J Biol Chem. 2010;285:5165‐5170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wu X, Lemon B, Li X, et al. C‐terminal tail of FGF19 determines its specificity toward Klotho co‐receptors. J Biol Chem. 2008;283:33304‐33309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Goetz R, Ohnishi M, Kir S, et al. Conversion of a paracrine fibroblast growth factor into an endocrine fibroblast growth factor. J Biol Chem. 2012;287:29134‐29146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lee S, Choi J, Mohanty J, et al. Structures of beta‐klotho reveal a 'zip code'‐like mechanism for endocrine FGF signalling. Nature. 2018;553:501‐505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kuro‐o M. Understanding the structure‐fuction relationship between FGF19 and its miogenic and metabolic activities In: Kuro‐o M, ed. Endocrine FGFs and Klothos. New York, NY: Springer US; 2012:25‐40. [Google Scholar]
- 50. Tiong KH, Tan BS, Choo HL, et al. Fibroblast growth factor receptor 4 (FGFR4) and fibroblast growth factor 19 (FGF19) autocrine enhance breast cancer cells survival. Oncotarget. 2016;7:57633‐57650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhao X, Xu F, Dominguez NP, et al. FGFR4 provides the conduit to facilitate FGF19 signaling in breast cancer progression. Mol Carcinog. 2018;57:1616‐1625. [DOI] [PubMed] [Google Scholar]
- 52. Buhmeida A, Dallol A, Merdad A, et al. High fibroblast growth factor 19 (FGF19) expression predicts worse prognosis in invasive ductal carcinoma of breast. Tumour Biol. 2014;35:2817‐2824. [DOI] [PubMed] [Google Scholar]
- 53. Feng S, Dakhova O, Creighton CJ, Ittmann M. Endocrine fibroblast growth factor FGF19 promotes prostate cancer progression. Cancer Res. 2013;73:2551‐2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nagamatsu H, Teishima J, Goto K, et al. FGF19 promotes progression of prostate cancer. Prostate. 2015;75:1092‐1101. [DOI] [PubMed] [Google Scholar]
- 55. Pai R, Dunlap D, Qing J, Mohtashemi I, Hotzel K, French DM. Inhibition of fibroblast growth factor 19 reduces tumor growth by modulating beta‐catenin signaling. Cancer Res. 2008;68:5086‐5095. [DOI] [PubMed] [Google Scholar]
- 56. Tan Q, Li F, Wang G, et al. Identification of FGF19 as a prognostic marker and potential driver gene of lung squamous cell carcinomas in Chinese smoking patients. Oncotarget. 2016;7:18394‐18402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhang X, Kong M, Zhang Z, et al. FGF19 genetic amplification as a potential therapeutic target in lung squamous cell carcinomas. Thorac Cancer. 2017;8:655‐665. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Suppinfo