

Abstract
Sunitinib, a multitargeted receptor tyrosine kinase inhibitor including vascular endothelial growth factor, has been widely used as a first‐line treatment against metastatic renal cell carcinoma (mRCC). However, mRCC often acquires resistance to sunitinib, rendering it difficult to treat with this agent. Recently, Rapalink‐1, a drug that links rapamycin and the mTOR kinase inhibitor MLN0128, has been developed with excellent therapeutic effects against breast cancer cells carrying mTOR resistance mutations. The aim of the present study was to evaluate the in vitro and in vivo therapeutic efficacy of Rapalink‐1 against renal cell carcinoma (RCC) compared to temsirolimus, which is commonly used as a small molecule inhibitor of mTOR and is a derivative of rapamycin. In comparison with temsirolimus, Rapalink‐1 showed significantly greater effects against proliferation, migration, invasion and cFolony formation in sunitinib‐naïve RCC cells. Inhibition was achieved through suppression of the phosphorylation of substrates in the mTOR signal pathway, such as p70S6K, eukaryotic translation initiation factor 4E‐binding protein 1 (4EBP1) and AKT. In addition, Rapalink‐1 had greater tumor suppressive effects than temsirolimus against the sunitinib‐resistant 786‐o cell line (SU‐R 786‐o), which we had previously established, as well as 3 additional SU‐R cell lines established here. RNA sequencing showed that Rapalink‐1 suppressed not only the mTOR signaling pathway but also a part of the MAPK signaling pathway, the ErbB signaling pathway and ABC transporters that were associated with resistance to several drugs. Our study suggests the possibility of a new treatment option for patients with RCC that is either sunitinib‐sensitive or sunitinib‐resistant.
Keywords: mTOR inhibitor, Rapalink‐1, renal cell carcinoma, sunitinib resistance, temsirolimus
A new mTOR inhibitor, Rapalink‐1, suppresses the MAPK signaling pathway, the ErbB signaling pathway and ABC transporters.

1. INTRODUCTION
Renal cell carcinoma (RCC) is associated with more than 140 000 annual deaths worldwide, making it one of the most common cancers in the human kidney. 1 Molecularly targeted drugs that inhibit vascular endothelial growth factor (VEGF) or mammalian target of mTOR have been used for patients with advanced RCC. 2 , 3 Among them, sunitinib is one of the most common molecularly targeted drugs recommended as a first‐line therapy against advanced RCC. 3 , 4 However, sunitinib therapy does not have curative effects due to RCC’s acquisition of resistance. 4 Moreover, our previous study showed the occurrence of metabolic re‐programming in RCC cells, leading to sunitinib resistance. 5 In November 2015, anti–programmed death‐1 (PD‐1) antibodies were approved for the treatment of patients with sunitinib‐resistant RCC. However, in Tomita et al (2019), in advanced RCC, the complete and partial response rate to anti–PD‐1 antibodies was only 25%. 6 Therefore, it is necessary to consider more effective new treatment strategies for advanced RCC including cases of resistance to these drugs.
Temsirolimus and everolimus, termed rapalogs, have been used as first‐generation mTOR inhibitors for first‐line or second‐line therapy against advanced RCC. 7 mTOR is present in 2 complexes, mTORC1 and mTORC2. 8 , 9 The function of mTORC1 is mediated by phosphorylation of eukaryotic translation initiation factor 4E‐binding protein 1 (4EBP1) and p70S6K, promoting mRNA translation and cell proliferation. In contrast, mTORC2 provides phosphorylation of downstream targets such as the AGC family of protein kinases, including AKT, the activity of which is associated with cancer promotion. 8 , 10 , 11 Temsirolimus and everolimus block the substrates of mTORC1 but not those of mTORC2. 12 , 13 The inhibition of mTORC1 can remove negative feedback loops targeting AKT and increased AKT activity. 14 Therefore, the therapeutic effectiveness of mTORC1 inhibitors is inadequate. In contrast, mTOR kinase inhibitors (TORKi), second‐generation mTOR inhibitors, can block both mTORC1 and mTORC2 substrates. 15 , 16 In some preclinical studies, these inhibitors have shown promising anticancer effects. 17 , 18 , 19 Recently, it was reported that cancer patients who had not been treated with mTOR kinase inhibitors can carry a mutation in mTOR that renders them resistant to treatment with TORKi. 20 Therefore, a third‐generation mTOR inhibitor, Rapalink‐1, was developed that combined the high affinity of rapamycin for mTORC1 with the effective mTOR kinase inhibition of MLN0128 as a TORKi. 20 Excellent therapeutic effects with Rapalink‐1 have been achieved in glioblastoma and follicular lymphoma. 21 , 22 However, its anti–cancer efficacy has still not been extensively studied in RCC, including sunitinib‐resistant RCC (SU‐R‐RCC).
In this study, we evaluated the therapeutic efficacy of Rapalink‐1 against RCC cells in vitro and in vivo. In further analyses, we used 4 independent isolates of sunitinib‐resistant RCC cells: SUR‐786‐o (established by gavage feeding of sunitinib in a previous study), 5 and SU‐R‐A498, SU‐R‐ACHN and SU‐R‐caki1, which were established in the same way in this study. Moreover, we performed RNA sequencing to discover novel pathways underlying the effect of Rapalink‐1 in RCC cells.
2. MATERIALS AND METHODS
2.1. Renal cell carcinoma cell lines and culture
We used human RCC cell lines 786‐o, A498, ACHN, caki1 and caki2 that were obtained from the ATCC. The human RCC cell lines were cultured in RPMI 1640 medium supplemented with 10% FBS at 37°C in a humidified, 5% CO2 incubator. Murine SU‐R‐786‐o, SU‐R‐A498, SU‐R‐ACHN and SU‐R‐caki1 cell lines were established by performing gavage feeding of sunitinib (40 or 25 mg/kg, five times a week; Biorbyt) as previously reported. 5
2.2. Cell proliferation, cell migration and cell invasion assays
Temsirolimus (#ab141999, Abcam) and RapaLink‐1 (#A8764, APExBIO) were used as mTOR inhibitors. Cell proliferation was assessed using XTT assays (Roche Applied Science). Cell migration was evaluated with in vitro wound healing. Cell invasion was examined with modified Boyden chambers consisting of Transwell precoated Matrigel membrane filter inserts with 8‐μm pores in 24‐well tissue culture plates (BD Biosciences). The experimental procedures were conducted as previously described. 23 , 24
2.3. Colony formation assay
Cells (1000) were plated into a 10‐cm dish and cultured with temsirolimus or RapaLink‐1 for 7‐10 days to allow optimal colony formation, followed by staining with 0.04% crystal violet (Nacalai Tesque), as previously described. 24
2.4. Cell apoptosis and cell cycle assays
Cell cycle assays and cell apoptosis assays were performed by flow cytometry (CytoFLEX Analyzer; Beckman Coulter) using a FITC Annexin V Apoptosis Detection Kit (BD Biosciences) and a Cycletest PLUS DNA Reagent Kit (BD Biosciences) according to the manufacturer’s recommendations, as previously described. 24 , 25
2.5. Western blotting
Cell lysates were separated on NuPAGE 4%‐12% Bis‐tris gels (Invitrogen) and transferred to polyvinylidene difluoride membranes. Immunoblotting was performed with the following reagents from Cell Signaling Technology: anti–AKT antibodies (1:2000, #9272), anti–phospho‐AKT antibodies (1:1000, #9271), anti–4EBP1 antibodies (1:1000, #9452), anti–phospho‐4EBP1 antibodies (1:1000, #2855), anti–p70S6K antibodies (1:1000, #9202), anti–phospho‐p70S6K antibodies (1:1000, #9205) and anti–cleaved PARP antibodies (1:1000, #5625S). Specific complexes were visualized using an ECL detection system (GE Healthcare), as described previously. 23 , 24
2.6. Establishment of additional sunitinib‐resistant renal cell carcinoma cells in vivo
A mixture containing 100 µL A498 cells (4 × 106 cells), ACHN cells (5 × 106 cells) or caki1 (5 × 106 cells) and 100 µL Matrigel matrix (Corning) was injected subcutaneously into the flanks of female nude mice (BALB/c nu/nu, 6 to 8 weeks old) that had acquired resistance to sunitinib (40 mg/kg/mouse/d).
We assessed comparative time courses of tumor volumes of parental and SU‐R cells (n = 4 for each group) in nude mice after subcutaneous injection during sunitinib treatment (25 mg/kg/mouse/d). These experimental procedures were described in a previous report. 5
2.7. Xenograft analysis
A 100‐µL suspension containing 3 × 106 cells (SU‐R‐786‐o, SU‐R‐A498, SU‐R‐ACHN or SU‐R‐caki1) was mixed with 100‐µL Matrigel matrix (Corning). The suspensions were injected subcutaneously into the flanks of female nude mice (BALB/c nu/nu, 6 to 8 weeks old). After confirming that the tumors had engrafted, the mice were divided into 3 groups that were treated with daily i.p. injections of vehicle (20% DMSO, 40% PEG‐300 and 40% PBS) or temsirolimus (1.5 mg/kg, daily) or RapaLink‐1 (1.5 mg/kg every 5 days for 25 days). The dose was adjusted according to the weight of each mouse and the volume of injection did not exceed 100 μL. All animal experiments were approved by the animal care review board of Kagoshima University.
2.8. Immunohistochemistry
We performed immunohistochemistry with an UltraVision Detection System (Thermo Scientific) following the manufacturer’s protocol. The primary rabbit monoclonal antibodies against cleaved caspase‐3 (#9661S, Cell Signaling Technology) and cleaved PARP (#5625S, Cell Signaling Technology) were diluted 1:100. The apoptotic tumor cells were stained brown. Percentages of cleaved caspase‐3 and cleaved PARP positive cells were quantitated by counting 6 random microscopic fields. These experimental procedures were described in a previous report. 5
2.9. RNA sequencing analyses
Total RNA from 786‐o, A498 or SU‐R‐786‐o cell lines were subjected to RNA sequencing, which was performed by Eurofins Japan. mRNA profiles were generated by single‐read deep sequencing using Illumina HiSeq 2500/2000.
2.10. Statistical analysis
The relationships between two groups were analyzed using Mann‐Whitney U tests. The relationships between 3 variables and numerical values were analyzed using Bonferroni‐adjusted Mann‐Whitney U tests. All analyses were carried out using Expert StatView software, version 5.0.
3. RESULTS
3.1. Rapalink‐1 inhibited the activity of cell proliferation and induced apoptosis and cell cycle arrest in renal cell carcinoma cells
First, to identify the in vitro effects of the agents on cell viability, 786‐o and A498 cells were treated with 1‐1000 nmol/L of temsirolimus or Rapalink‐1 for 72 hours. Compared to mock, both temsirolimus and Rapalink‐1 decreased the viability of ccRCC cell lines (Figure S1A,B). Next, we investigated the effects of the same concentration of temsirolimus or Rapalink‐1 on viability. At 100 nmol/L, there were no significant effects of temsirolimus on cell viability, but Rapalink‐1 significantly reduced the viability of ccRCC cell lines (Figure S1C). Therefore, we continued to use this concentration.
To evaluate the effect of Rapalink‐1 on cell viability, 786‐o, A498, ACHN, caki1 and caki2 cells were treated with temsirolimus or Rapalink‐1 for 24‐96 hours. Both temsirolimus and Rapalink‐1 suppressed the proliferation of RCC cells over time and the effect of Rapalink‐1 was significantly greater than that of temsirolimus (Figure 1A). To investigate the mechanism of cell growth suppression, we assessed apoptosis in 786‐o and A498 cell lines. Temsirolimus induced apoptosis only in 786‐o cells. In contrast, Rapalink‐1 caused apoptosis in both RCC cell lines (Figure 1B). 26 In western blot analysis, the results showed that Rapalink‐1 increased the cleavage of PARP in RCC cells (Figure 1C). Rapamycin and rapalogs are known to arrest the cell cycle in the G1 phase. 27 , 28 In 786‐o and A498 lines, we found that Rapalink‐1 induced cell cycle arrest in G1 to a significantly greater extent than temsirolimus (Figure 1D).
FIGURE 1.
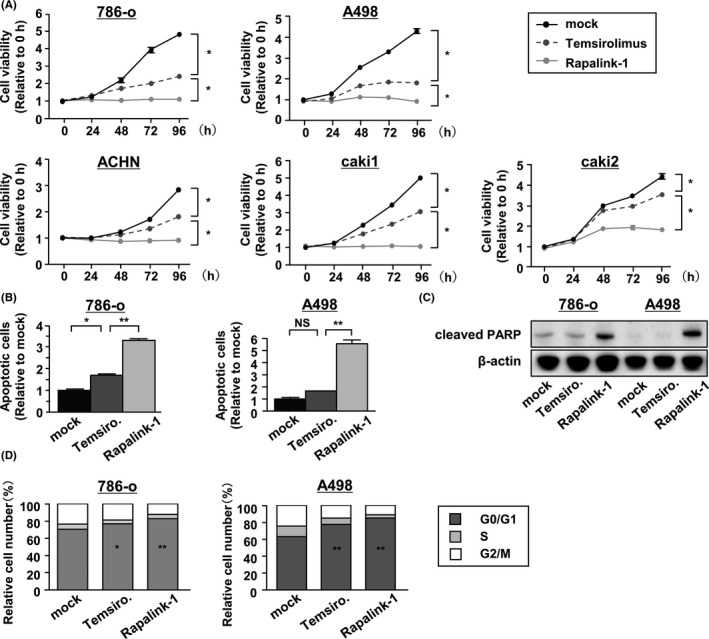
Rapalink‐1 suppressed renal cell carcinoma (RCC) cell proliferation by inducing apoptosis and cell cycle arrest. A, 786‐o, A498, ACHN and caki cell proliferation was determined by XTT assays during treatment with temsirolimus or Rapalink‐1 from 24 to 96 h. All experiments were performed in quadruplicate. *P < 0.0001. B, Apoptosis assays were carried out using flow cytometric analysis of 786‐o and A498 cells. All experiments were performed in triplicate. *P < 0.05; **P < 0.0001. C, Western blot analysis of apoptotic markers (cleaved PARP) in 786‐o and A498 cells. β‐actin was used as a loading control. D, Cell cycle assays were carried out using flow cytometric analyses of 786‐o and A498 cells. The bar charts represent the percentage of mock cells in G0/G1, S and G2/M phases. All experiments were performed in triplicate. *P < 0.01; **P < 0.001
3.2. Rapalink‐1 inhibited renal cell carcinoma cell migration, invasion and colony formation and suppressed PI3K/AKT/mTOR signaling to a greater extent than temsirolimus
Several reports suggested that the mTOR pathway plays important roles in the regulation of tumor cell motility, invasion and cancer metastasis. 29 , 30 We examined the inhibitory abilities of Rapalink‐1 in reducing cell migration, invasion and colony formation. The results showed that temsirolimus and Rapalink‐1 suppressed the abilities of 786‐o and A498 cells, and there were significant differences between the drugs (Figure 2A‐C).
FIGURE 2.
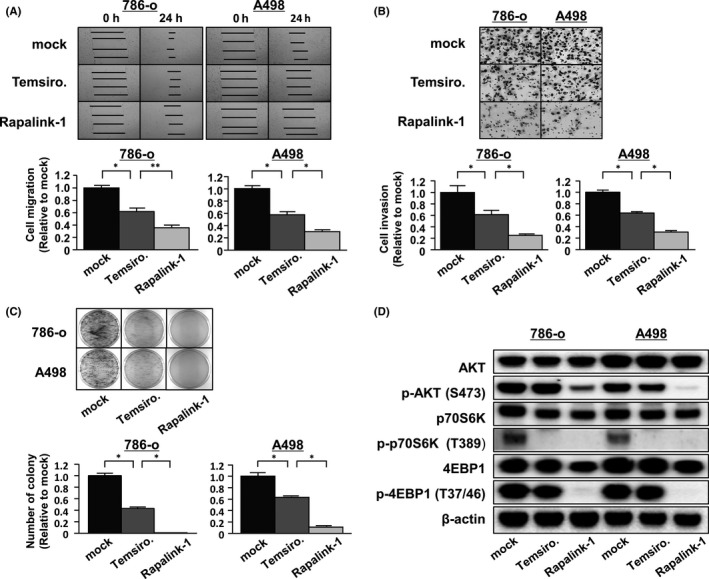
Rapalink‐1 suppressed cell migration, invasion and colony formation and blocked mTORC1 and mTORC2 in renal cell carcinoma (RCC) cells. A, B, Cell migration and invasion were determined by wound healing (A) and Matrigel invasion (B) assays of 786‐o and A498 cells, respectively. All experiments were performed in triplicate. *P < 0.0001; **P < 0.001. C, Colony formation by 786‐o and A498 cells was remarkably inhibited by Rapalink‐1 compared with temsirolimus. All experiments were performed in triplicate. *P < 0.0001. D, 786‐o and A498 cells were treated with temsirolimus or RapaLink‐1 at 100 nmol/L for 3 h and analyzed for PI3K/AKT/mTOR signaling by western blotting
Temsirolimus inhibits mTORC1, whereas Rapalink‐1 is a dual inhibitor of mTORC1/2. We examined these drugs’ abilities to inhibit the phosphorylation of proteins downstream from mTORC1/2. Using 2 RCC cell lines, 786‐o and A498, cultures were treated with these drugs and western blot analyses were performed. In 786‐o and A498 cells, the phosphorylation of Thr389 on p70S6K and Thr37/46 on 4EBP1 was inhibited by Rapalink‐1 (Figure 2D). Furthermore, the activity of mTORC2 kinase was prevented by Rapalink‐1, as shown by the inhibition of the phosphorylation of Ser473 on AKT. In contrast, temsirolimus inhibited the phosphorylation of Thr389 on p70S6K to approximately the same extent as Rapalink‐1 but did not inhibit Thr37/46 on 4EBP1. Rapamycin is a potent inhibitor of mTORC1 target p70S6K but is a relatively inefficient inhibitor of 4EBP1. 31 Rapamycin administration permits re‐phosphorylation of 4EBP1, whereas Rapalink‐1 impairs phosphorylation of 4EBP1 over time. 32 , 33 Unlike Rapalink‐1, the phosphorylation of Ser473 on AKT was not inhibited by temsirolimus. Therefore, Rapalink‐1 suppressed mTOR signaling efficiently compared to temsirolimus.
3.3. Rapalink‐1 showed similar effects on sunitinib‐resistant renal cell carcinoma cells in vitro and in vivo
Previously, a sunitinib‐resistant cell line, 786‐o cell (SU‐R‐786‐o), was established in our department. 5 Therefore, we examined whether Rapalink‐1 had effects on sunitinib‐resistant cells. To investigate the effects on cell viability, SU‐R‐786‐o cells were treated with temsirolimus or Rapalink‐1 for various lengths of time. Like parental 786‐o cells, Rapalink‐1 showed significantly greater cell growth suppression than did temsirolimus (Figure 3A).
FIGURE 3.
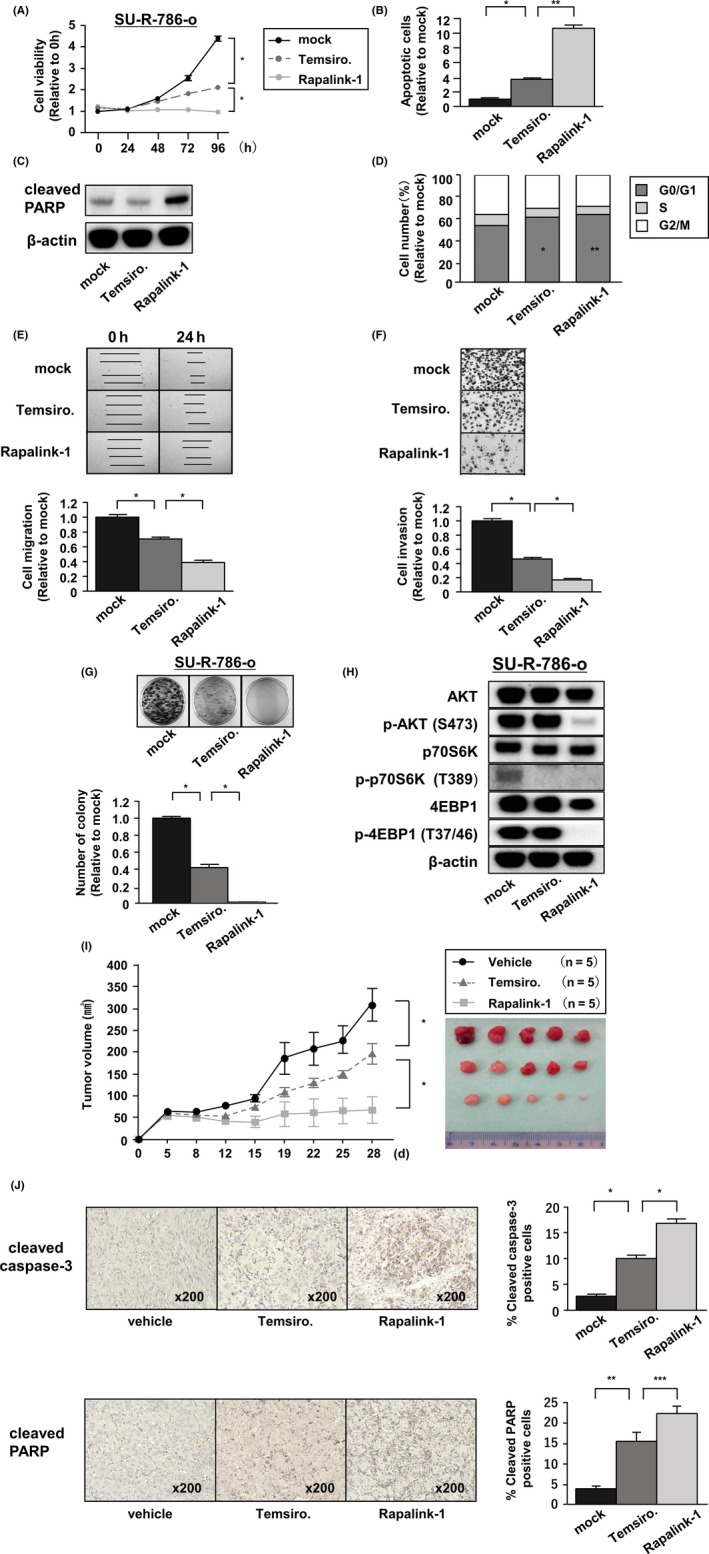
In sunitinib‐resistant renal cell carcinoma (RCC) cells, Rapalink‐1 had similar effects in vitro and vivo. A, Cell proliferation was determined by XTT assays during treatment with temsirolimus or Rapalink‐1 in SU‐R‐786‐o cells from 24 to 96 h. All experiments were performed in quadruplicate. *P < 0.0001. B, Apoptosis assays were carried out using flow cytometry. All experiments were performed in triplicate. *P < 0.001; **P < 0.0001. C, Western blot analysis of apoptotic markers (cleaved PARP). β‐actin was used as a loading control. D, Cell cycle assays were carried out using flow cytometry. The bar charts represent the percentage of mock cells in G0/G1, S or G2/M phases. All experiments were performed in triplicate. *P < 0.05; **P < 0.001. E, Cell migration was determined by wound healing assays. All experiments were performed in triplicate. *P < 0.0001. F, Cell invasion was determined by Matrigel invasion assays. All experiments were performed in triplicate. *P < 0.0001. G, Colony formation by SU‐R‐786‐o cells was remarkably inhibited by Rapalink‐1 compared with temsirolimus. All experiments were performed in triplicate. *P < 0.0001. H, SU‐R‐786‐o cells were treated with temsirolimus or RapaLink‐1 at 100 nmol/L for 3 h and analyzed for PI3K/AKT/mTOR signaling by western blotting. I, SU‐R‐786‐o tumor‐bearing nude mice (n = 5 per group) were given i.p. injections of vehicle (daily), temsirolimus (1.5 mg/kg, daily) or Rapalink‐1 (1.5 mg/kg, every 5 d). *P < 0.05. J, Tumors were extracted and immunostained with cleaved caspase‐3 and cleaved PARP antibodies. Percentages of cleaved caspase‐3 and cleaved PARP positive cells were quantitated by counting 6 random microscopic fields. *P < 0.0001; **P < 0.001; ***P < 0.05
Next, we evaluated the effects on apoptosis and the cell cycle. Rapalink‐1 induced apoptosis to a greater extent than did temsirolimus and increased cleaved‐PARP in western blot analysis (Figure 3B,C). Rapalink‐1 arrested the cell cycle in the G1 phase to an extent equal to or greater than temsirolimus treatment of SU‐R‐786‐o cells (Figure 3D). We examined the abilities of Rapalink‐1 to reduce cell migration, invasion and colony formation. The results showed that Rapalink‐1 had significantly greater ability to suppress the activities of SU‐R‐786‐o cells than did temsirolimus (Figure 3E‐G).
In western blot analysis, as observed with parental cells, Rapalink‐1 decreased the phosphorylation of Thr389 on p70S6K, Thr37/46 on 4EBP1 and Ser473 on AKT to greater extents than did temsirolimus (Figure 3H).
To evaluate the anti–tumor effects of Rapalink‐1 in vivo, nude mice were treated with Rapalink‐1. Control mice were treated with temsirolimus or vehicle. The results showed that temsirolimus decreased tumor volume by an average of 37%, whereas Rapalink‐1 reduced volumes by an average of 79% in SU‐R‐RCC cells (Figure 3I). We harvested tumors for immunostaining. Immunohistochemistry with cleaved caspase‐3 and cleaved PARP antibodies indicated that Rapalink‐1 significantly induced apoptosis compared to temsirolimus in vivo as well as in vitro. (Figure 3J).
3.4. Establishment of sunitinib‐resistant renal cell carcinoma cells
For more reliable verification of the effects of Rapalink‐1, we established new sunitinib‐resistant RCC cells. We subcutaneously injected A498, ACHN and caki1 cells into mice and started sunitinib treatment after tumor formation (Figure 4A). After tumors acquired resistance, tumors were extracted and harvested. Next, in xenograft assays, we confirmed that SU‐R‐A498, SU‐R‐ACHN and SU‐R‐caki1 cells showed resistance to sunitinib compared with parental cells (Figure 4B).
FIGURE 4.
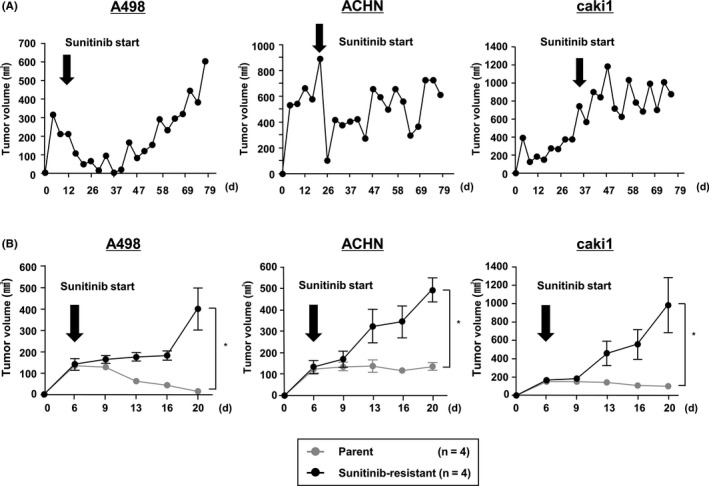
Establishment of additional sunitinib‐resistant clear cell renal cell carcinoma (ccRCC) cell lines (SU‐R‐A498, ACHN and caki1). A, As in our previous study, we established sunitinib‐resistant ccRCC cells. Time course of tumor volumes in nude mice after subcutaneous injection of parental A498, ACHN or caki1 cells, which acquired resistance to sunitinib treatment (40 mg/kg/mouse/d). B, Time course comparing tumor volumes of parental A498, ACHN, caki1 and each sunitinib‐resistant cell line in nude mice (n = 4 for each group) after subcutaneous injection under sunitinib treatment (25 mg/kg/mouse/d) *P < 0.05
3.5. Rapalink‐1 showed greater anti–tumor effects on several sunitinib‐resistant renal cell carcinoma cells than did temsirolimus in vitro and in vivo
To investigate the activity of the drugs on cell viability, newly established sunitinib‐resistant cells were treated with temsirolimus or Rapalink‐1 for various lengths of time. Rapalink‐1 suppressed cell proliferations to a significantly greater extent than did temsirolimus in all sunitinib‐resistant RCC cell lines (Figure 5A).
FIGURE 5.
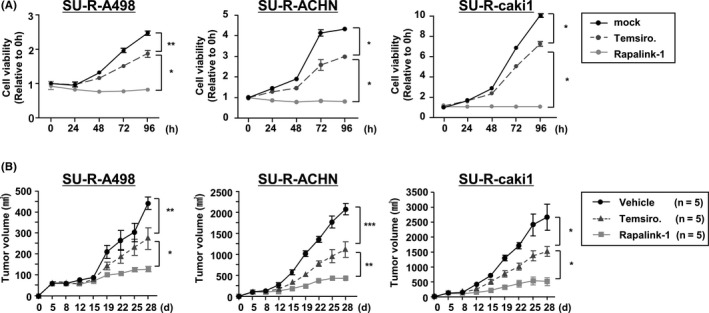
Rapalink‐1 suppressed cell proliferation in vitro and showed antitumor effects in sunitinib‐resistant renal cell carcinoma (RCC) cells. A, Cell proliferation was determined by XTT assays with temsirolimus or Rapalink‐1 in SU‐R‐A498, SU‐R‐ACHN and SU‐R‐caki1 cell lines from 24 to 96 h. All experiments were performed in triplicate. *P < 0.0001. B, Tumor‐bearing nude mice (n = 5 per group) using SU‐R‐A498, SU‐R‐ACHN or SU‐R‐caki1 cells were given i.p. injections of vehicle (daily), temsirolimus (1.5 mg/kg, daily) or Rapalink‐1 (1.5 mg/kg, every 5 d). *P < 0.05; **P < 0.01; ***P < 0.001
Next, to evaluate the anti–tumor effect of Rapalink‐1 in newly established sunitinib‐resistant cells in vivo, nude mice were treated with Rapalink‐1. Control mice were treated with temsirolimus or vehicle. In these 3 additional Sunitinib‐resistant cell lines, the results showed that temsirolimus decreased tumor volume by an average of 42%, while Rapalink‐1 reduced them by an average of 77%, showing that it had excellent antitumor effects (Figure 5B).
3.6. Rapalink‐1 suppressed mTOR signaling, some MAPK signaling, ErbB signaling and ABC transporters in sunitinib‐sensitive and sunitinib‐resistant renal cell carcinoma cells
We performed RNA sequencing of the 786‐o parental line, the A498 parental line and SU‐R‐786‐o cells. We identified 1726 genes that were downregulated by Rapalink‐1, less than half the number compared to temsirolimus and mock (Figure S2). We performed KEGG pathway analyses with these genes and identified multiple signals that were suppressed (Table 1). In particular, the MAPK signaling pathway, the ErbB signaling pathway and ATP‐binding cassette transporters were significantly suppressed (Table 1). Several reports have suggested that mTOR inhibitors cause feedback activation of MAPK signaling pathways, which serves as a major resistance factor. The combination of an mTOR inhibitor and MEK‐ERK inhibitor showed better antitumor effects than an mTOR inhibitor alone. 34 , 35 , 36 In addition, because PI3K binds directly to ERBB3 and ERBB4, the upregulation of the ERBB receptor activates the ERBB/PI3K/AKT signaling pathway. 37 Furthermore, the multi‐drug resistance of cancer cells is often attributable to the increased expression of ATP‐binding cassette (ABC) transporters that can remove various chemotherapeutic drugs from the cells. 38 Suppression of these pathways by Rapalink‐1 may contribute to its efficacy.
TABLE 1.
Top 15 downregulated pathways in renal cell carcinoma cells treated by Rapalink‐1
|
KEGG ID |
Annotations | Number of genes | Corrected P‐value | Genes |
|---|---|---|---|---|
| 4010 | MAPK signaling pathway | 28 | 0.000812 | CACNA2D1, PPM1B, MEF2C, IL1R2, IL1R1, MAPT, FGFR3, TGFB3, RASA2, CACNA2D2, ARRB2, PLA2G6, MAPK10, MAPK8, EGF, MAP3K5, PAK1, MECOM, CACNA1G, NR4A1, MKNK1, MAPK14, RASGRF1, STK3, CACNB2, PRKACB, MAP3K8, PLA2G10 |
| 5152 | Tuberculosis | 21 | 0.000836 | CAMK2D, CIITA, IRAK4, CASP8, CREB1, SPHK2, ATP6V0A1, TGFB3, CASP9, MAPK10, MAPK8, FCGR2A, MAPK14, CLEC7A, CAMK2G, TLR1, CD209, JAK2, NFYC, RFX5, KSR1 |
| 4012 | ErbB signaling pathway | 13 | 0.002664 | CAMK2D, CIITA, IRAK4, CASP8, CREB1, SPHK2, ATP6V0A1, TGFB3, CASP9, MAPK10, MAPK8, FCGR2A, MAPK14, CLEC7A, CAMK2G, TLR1, CD209, JAK2, NFYC, RFX5, KSR1 |
| 2010 | ABC transporters | 9 | 0.002805 | CAMK2D, NRG1, NRG4, MAPK10, PIK3R3, MAPK8, EGF, PAK1, PTK2, PAK3, CAMK2G, STAT5A, CBLB |
| 5200 | Pathways in cancer | 29 | 0.003110 | AXIN2, PIAS2, CTNNA3, RUNX1T1, LAMA2, COL4A5, CASP8, FGFR3, RAD51, AR, TGFB3, CASP9, TCF7, MAPK10, PIK3R3, MAPK8, EGF, MECOM, WNT5B, CSF2RA, PTK2, TCF7L2, ZBTB16, TRAF3, PLD1, MLH1, DAPK2, STAT5A, CBLB |
| 4144 | Endocytosis | 20 | 0.004717 | STAMBP, SH3KBP1, DNM1, FGFR3, PIP5K1A, TGFB3, ARAP3, CXCR2, ARRB2, DNM3, GIT2, EGF, PRKCZ, ZFYVE16, IQSEC2, ARAP2, PLD1, LDLRAP1, PSD, CBLB |
| 4360 | Axon guidance | 15 | 0.007101 | ROBO1, LRRC4C, SEMA6D, NTNG1, EPHA6, SEMA4D, ABLIM1, UNC5B, PAK1, SEMA3E, PLXNC1, RGS3, PTK2, NFAT5, PAK3 |
| 4120 | Ubiquitin mediated proteolysis | 15 | 0.011088 |
ANAPC10, HERC3, KLHL13, PIAS2, DET1, UBA7, UBE3A, CDC27, ANAPC7, RHOBTB1, UBE2D4, FANCL, UBE2U, CBLB, TRIM37 |
| 5145 | Toxoplasmosis | 14 | 0.011838 | CIITA, IRAK4, LAMA2, CASP8, TGFB3, CASP9, PLA2G6, MAPK10, PIK3R3, MAPK8, MAPK14, JAK2, PDK1, PLA2G10 |
| 5412 | Arrhythmogenic right ventricular cardiomyopathy (ARVC) | 10 | 0.015495 | CACNA2D1, CTNNA3, LAMA2, SLC8A1, CACNA2D2, TCF7, DMD, TCF7L2, ITGB4, CACNB2 |
| 5210 | Colorectal cancer | 9 | 0.016725 | AXIN2, TGFB3, CASP9, TCF7, MAPK10, PIK3R3, MAPK8, TCF7L2, MLH1 |
| 5414 | Dilated cardiomyopathy | 11 | 0.017279 | CACNA2D1, LAMA2, TTN, SLC8A1, TGFB3, CACNA2D2, DMD, ITGB4, GNAS, CACNB2, PRKACB |
| 5213 | Endometrial cancer | 8 | 0.018332 | AXIN2, CTNNA3, CASP9, TCF7, PIK3R3, EGF, TCF7L2, MLH1 |
| 4666 | Fc gamma R‐mediated phagocytosis | 11 | 0.019509 | DNM1, SPHK2, PIP5K1A, PLA2G6, DNM3, PIK3R3, GSN, PRKCE, FCGR2A, PAK1, PLD1 |
| 130 | Ubiquinone and other terpenoid‐quinone biosynthesis | 3 | 0.028763 | COQ2, COQ6, COQ3 |
4. DISCUSSION
In various cancers, mTOR plays important roles in intracellular signaling pathways, including modulation of cell proliferation and angiogenesis. Therefore, temsirolimus and everolimus (termed “rapalogs”), which block the activity of mTORC1, are clinically useful for the treatment of RCC. 7 It is well known that VHL inactivation in RCC cells activates HIF‐regulated genes such as VEGF and PDGF. 39 , 40 Notably, HIF1α expression is regulated by both mTORC1 and mTORC2, whereas HIF2α is modulated only by mTORC2. 41 In RCC, tumor growth is driven by HIF2α. Thus, the inhibition of both mTORC1 and mTORC2 is highly effective for treatment. 42 , 43 Accordingly, TORKi, second‐generation mTOR inhibitors, have been developed, and a dual PI3K/mTOR inhibitor suppressed the level of HIF2a, whereas rapamycin could not. 44 A third‐generation mTOR inhibitor was developed that combined rapamycin with MLN0128 as the TORKi. 20 In glioblastoma, the FK506 binding protein 12 (FKBP12)‐rapamycin complex bound only to FK506 rapamycin binding (FRB) domain, whereas the FKBP12‐Rapalink‐1 complex bound to both FRB and the mTORC1 kinase domain. 21 Therefore, the dual binding ability of Rapalink‐1 showed a better antitumor effect. Furthermore, in follicular lymphoma, enhancer of zeste 2 polycomb repressive complex 2 subunit (EZH2) regulated mTORC1 via SESTRIN1 and Rapalink‐1 showed superior effects on EZH2 mutant cells. 22 Similarly, strong antitumor effects of Rapalink‐1 against RCC cells were observed in our experiments. Taken together, it appears necessary to suppress mTORC2 for RCC treatment.
Sunitinib, a tyrosine kinase inhibitor (TKI), is an anti–angiogenic agent that induces vascular destruction, hypoxia and tumor necrosis, and it has been approved as a first‐line treatment for advanced RCC. 4 However, sunitinib cannot eliminate tumors because resistance to sunitinib often occurs through various mechanisms. 45 In this study, we compared the therapeutic effects of RapaLink‐1 with temsirolimus in SUR‐RCC cells that were established in vivo. We showed that Rapalink‐1 inhibited proliferation, migration, invasion and colony formation in RCC cells, including SU‐R‐RCC cells, by inducing apoptosis and G1 arrest, and was more effective than temsirolimus. In addition, Rapalink‐1 blocked the phosphorylation downstream from both mTORC1 and mTORC2, including p70S6K, 4EBP1 and AKT. In contrast, temsirolimus completely inhibited the phosphorylation of p70S6K but phosphorylation of 4EBP1 was not significantly inhibited. Rapamycin potently inhibits long‐term p70S6K activity, but 4E‐BP1 restores phosphorylation within 6 hours and causes rapamycin resistance. 32 , 33 Therefore, temsirolimus could not sufficiently inhibit protein phosphorylation in PI3K/AKT/mTOR signaling, whereas Rapalink‐1 inhibited them. The same tumor‐inhibitory effect of Rapalink‐1 was demonstrated in vivo, where we used the sunitinib‐resistant 786‐o cell line 5 as well as 3 newly established SU‐R RCC cell lines (SU‐R‐A498, SU‐R‐ACHN and SU‐R‐caki1). In addition, Rapalink‐1 was superior to temsirolimus in terms of less frequency of administration (every 5 days vs daily).
We conducted RNA sequencing of SUR‐cells to evaluate the mechanisms associated with the tumor‐suppressive effects of Rapalink‐1. The data showed that Rapalink‐1 suppressed a portion of the MAPK signaling pathway, whereas temsirolimus did not. Interestingly, long‐term treatment with sunitinib induces epigenetic silencing of the PTEN gene, 46 a negative regulator of the PI3K/AKT/mTOR signaling pathway. Moreover, there is an inverse correlation between PTEN expression and sunitinib resistance in RCC cells. 47 In addition, the expression of IL‐8 stimulates VEGF expression via the MAPK pathway and the PI3K/AKT/mTOR pathway in sunitinib‐resistant RCC cells. Suppression of IL‐8 inhibition is tumor‐suppressive. 48 Therefore, the tumor suppressive effects of Rapalink‐1 against sunitinib‐resistant RCC might occur through inhibition of the PI3K/AKT/mTOR pathway. The RNA sequencing analyses also indicated that the ErbB signaling pathway and ATP‐binding cassette transporters were suppressed by Rapalink‐1 in SUR‐cells. Note also that upregulation of the ErbB receptor activates the ErbB/PI3K/AKT signaling pathway, 37 and that ATP‐binding cassette (ABC) transporters contribute to drug resistance. 38 , 49 Thus, the suppression of these pathways by Rapalink‐1 may enhance its tumor‐suppressive effects. Further studies are needed to clarify the genetic or epigenetic mechanisms associated with Rapalink‐1 in the setting of drug resistance.
In conclusion, Rapalink‐1 had better antitumor effects than did temsirolimus in the treatment of sunitinib‐sensitive and sunitinib‐resistant RCC cells in vitro and vivo. We also found that Rapalink‐1 significantly inhibited not only PI3K/AKT/mTOR signaling but also ErbB signaling and ABC transporters. To the best of our knowledge, this is the first paper suggesting that Rapalink‐1 is a new option for the treatment of RCC patients who have acquired resistance to traditional molecularly targeted drugs. Early clinical trials with Rapalink‐1 for the treatment of RCC are expected.
DISCLOSURE
The authors declare no conflicts of interest.
Supporting information
Figure S1
Figure S2
ACKNOWLEDGMENTS
This study was supported by KAKENHI (KIBAN‐B) 16H05464 and 17H04332, KAKENHI (KIBAN‐C) 16K11015, KAKENHI (WAKATE‐B) 17K16799, the Shinnihon Foundation of Advanced Medical Treatment Research, the Takeda Science Foundation and the Foundation for Promotion of Cancer Research in Japan. We thank Ms Keiko Yoshitomi, of the Department of Urology, Graduate School of Medical and Dental Sciences, Kagoshima University (Kagoshima, Japan), for excellent laboratory assistance.
Kuroshima K, Yoshino H, Okamura S, et al. Potential new therapy of Rapalink‐1, a new generation mammalian target of rapamycin inhibitor, against sunitinib‐resistant renal cell carcinoma. Cancer Sci. 2020;111:1607–1618. 10.1111/cas.14395
REFERENCES
- 1. Capitanio U, Montorsi F. Renal cancer. Lancet (London, England). 2016;387:894‐906. [DOI] [PubMed] [Google Scholar]
- 2. Hutson TE, Figlin RA, Kuhn JG, Motzer RJ. Targeted therapies for metastatic renal cell carcinoma: an overview of toxicity and dosing strategies. Oncologist. 2008;13:1084‐1096. [DOI] [PubMed] [Google Scholar]
- 3. Margulis V, Master VA, Cost NG, et al. International consultation on urologic diseases and the European Association of Urology international consultation on locally advanced renal cell carcinoma. Eur Urol. 2011;60:673‐683. [DOI] [PubMed] [Google Scholar]
- 4. Garcia‐Donas J, Leandro‐García LJ, González del Alba A, et al. Prospective study assessing hypoxia‐related proteins as markers for the outcome of treatment with sunitinib in advanced clear‐cell renal cell carcinoma. Ann Oncol. 2013;24:2409‐2414. [DOI] [PubMed] [Google Scholar]
- 5. Yoshino H, Nohata N, Miyamoto K, et al. PHGDH as a key enzyme for serine biosynthesis in HIF2alpha‐targeting therapy for renal cell carcinoma. Can Res. 2017;77:6321‐6329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tomita Y, Fukasawa S, Shinohara N, et al. Nivolumab versus everolimus in advanced renal cell carcinoma: Japanese subgroup 3‐year follow‐up analysis from the Phase III CheckMate 025 study. Jpn J Clin Oncol. 2019;49:506‐514. [DOI] [PubMed] [Google Scholar]
- 7. Furge KA, MacKeigan JP, Teh BT. Kinase targets in renal‐cell carcinomas: reassessing the old and discovering the new. Lancet Oncol. 2010;11:571‐578. [DOI] [PubMed] [Google Scholar]
- 8. Foster KG, Fingar DC. Mammalian target of rapamycin (mTOR): conducting the cellular signaling symphony. J Biol Chem. 2010;285:14071‐14077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9‐22. [DOI] [PubMed] [Google Scholar]
- 10. Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4:648‐657. [DOI] [PubMed] [Google Scholar]
- 11. Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol. 2007;9:316‐323. [DOI] [PubMed] [Google Scholar]
- 12. Rini BI. Temsirolimus, an inhibitor of mammalian target of rapamycin. Clin Cancer Res. 2008;14:1286‐1290. [DOI] [PubMed] [Google Scholar]
- 13. Pal SK, Quinn DI. Differentiating mTOR inhibitors in renal cell carcinoma. Cancer Treat Rev. 2013;39:709‐719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Carracedo A, Pandolfi PP. The PTEN‐PI3K pathway: of feedbacks and cross‐talks. Oncogene. 2008;27:5527‐5541. [DOI] [PubMed] [Google Scholar]
- 15. Sun SY. mTOR kinase inhibitors as potential cancer therapeutic drugs. Cancer Lett. 2013;340:1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhou HY, Huang SL. Current development of the second generation of mTOR inhibitors as anticancer agents. Chin J Cancer. 2012;31:8‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang H, Berel D, Wang Y, et al. A comparison of Ku0063794, a dual mTORC1 and mTORC2 inhibitor, and temsirolimus in preclinical renal cell carcinoma models. PLoS ONE. 2013;8:e54918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pike KG, Malagu K, Hummersone MG, et al. Optimization of potent and selective dual mTORC1 and mTORC2 inhibitors: the discovery of AZD8055 and AZD2014. Bioorg Med Chem Lett. 2013;23:1212‐1216. [DOI] [PubMed] [Google Scholar]
- 19. Li Q, Song XM, Ji YY, Jiang H, Xu LG. The dual mTORC1 and mTORC2 inhibitor AZD8055 inhibits head and neck squamous cell carcinoma cell growth in vivo and in vitro. Biochem Biophys Res Comm. 2013;440:701‐706. [DOI] [PubMed] [Google Scholar]
- 20. Rodrik‐Outmezguine VS, Okaniwa M, Yao Z, et al. Overcoming mTOR resistance mutations with a new‐generation mTOR inhibitor. Nature. 2016;534:272‐276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fan QiWen, Aksoy O, Wong RA, et al. A Kinase Inhibitor Targeted to mTORC1 Drives Regression in Glioblastoma. Cancer Cell. 2017;31:424‐435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Oricchio E, Katanayeva N, Donaldson MC, et al. Genetic and epigenetic inactivation of SESTRIN1 controls mTORC1 and response to EZH2 inhibition in follicular lymphoma. Sci Transl Med. 2017;9:pii: eaak9969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chiyomaru T, Yamamura S, Fukuhara S, et al. Genistein inhibits prostate cancer cell growth by targeting miR‐34a and oncogenic HOTAIR. PLoS ONE. 2013;8:e70372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Osako Y, Yoshino H, Sakaguchi T, et al. Potential tumorsuppressive role of microRNA99a3p in sunitinibresistant renal cell carcinoma cells through the regulation of RRM2. Int J Oncol. 2019;54:1759‐1770. [DOI] [PubMed] [Google Scholar]
- 25. Sakaguchi T, Yoshino H, Sugita S, et al. Bromodomain protein BRD4 inhibitor JQ1 regulates potential prognostic molecules in advanced renal cell carcinoma. Oncotarget. 2018;9:23003‐23017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hosoi H, Dilling MB, Shikata T, et al. Rapamycin causes poorly reversible inhibition of mTOR and induces p53‐independent apoptosis in human rhabdomyosarcoma cells. Can Res. 1999;59:886‐894. [PubMed] [Google Scholar]
- 27. Fingar DC, Richardson CJ, Tee AR, Cheatham L, Tsou C, Blenis J. mTOR controls cell cycle progression through its cell growth effectors S6K1 and 4E‐BP1/eukaryotic translation initiation factor 4E. Mol Cell Biol. 2004;24:200‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hashemolhosseini S, Nagamine Y, Morley SJ, Desrivieres S, Mercep L, Ferrari S. Rapamycin inhibition of the G1 to S transition is mediated by effects on cyclin D1 mRNA and protein stability. J Biol Chem. 1998;273:14424‐14429. [DOI] [PubMed] [Google Scholar]
- 29. Chen J‐S, Wang Q, Fu X‐H, et al. Involvement of PI3K/PTEN/AKT/mTOR pathway in invasion and metastasis in hepatocellular carcinoma: association with MMP‐9. Hepatol Res. 2009;39:177‐186. [DOI] [PubMed] [Google Scholar]
- 30. Busch S, Renaud SJ, Schleussner E, Graham CH, Markert UR. mTOR mediates human trophoblast invasion through regulation of matrix‐remodeling enzymes and is associated with serine phosphorylation of STAT3. Exp Cell Res. 2009;315:1724‐1733. [DOI] [PubMed] [Google Scholar]
- 31. Baretic D, Williams RL. The structural basis for mTOR function. Semin Cell Dev Biol. 2014;36:91‐101. [DOI] [PubMed] [Google Scholar]
- 32. Choo AY, Yoon SO, Kim SG, Roux PP, Blenis J. Rapamycin differentially inhibits S6Ks and 4E‐BP1 to mediate cell‐type‐specific repression of mRNA translation. Proc Natl Acad Sci USA. 2008;105:17414‐17419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fan QW, Nicolaides TP, Weiss WA. Inhibiting 4EBP1 in Glioblastoma. Clin Cancer Res. 2018;24:14‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Carracedo A, Ma LI, Teruya‐Feldstein J, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K‐dependent feedback loop in human cancer. J Clin Investig. 2008;118:3065‐3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dogan Turacli I, Ozkan AC, Ekmekci A. The comparison between dual inhibition of mTOR with MAPK and PI3K signaling pathways in KRAS mutant NSCLC cell lines. Tumour Biol. 2015;36:9339‐9345. [DOI] [PubMed] [Google Scholar]
- 36. Xiong Z, Zang Y, Zhong S, et al. The preclinical assessment of XL388, a mTOR kinase inhibitor, as a promising anti–renal cell carcinoma agent. Oncotarget. 2017;8:30151‐30161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hynes NE, MacDonald G. ErbB receptors and signaling pathways in cancer. Curr Opin Cell Biol. 2009;21:177‐184. [DOI] [PubMed] [Google Scholar]
- 38. Fletcher JI, Haber M, Henderson MJ, Norris MD. ABC transporters in cancer: more than just drug efflux pumps. Nat Rev Cancer. 2010;10:147‐156. [DOI] [PubMed] [Google Scholar]
- 39. Gordan JD, Simon MC. Hypoxia‐inducible factors: central regulators of the tumor phenotype. Curr Opin Genet Dev. 2007;17:71‐77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Linehan WM, Srinivasan R, Schmidt LS. The genetic basis of kidney cancer: a metabolic disease. Nature Rev Urol. 2010;7:277‐285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Toschi A, Lee E, Gadir N, Ohh M, Foster DA. Differential dependence of hypoxia‐inducible factors 1 alpha and 2 alpha on mTORC1 and mTORC2. J Biol Chem. 2008;283:34495‐34499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kondo K, Kim WY, Lechpammer M, Kaelin WG Jr. Inhibition of HIF2alpha is sufficient to suppress pVHL‐defective tumor growth. PLoS Biol. 2003;1:E83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Maranchie JK, Vasselli JR, Riss J, Bonifacino JS, Linehan WM, Klausner RD. The contribution of VHL substrate binding and HIF1‐alpha to the phenotype of VHL loss in renal cell carcinoma. Cancer Cell. 2002;1:247‐255. [DOI] [PubMed] [Google Scholar]
- 44. Cho DC, Cohen MB, Panka DJ, et al. The efficacy of the novel dual PI3‐kinase/mTOR inhibitor NVP‐BEZ235 compared with rapamycin in renal cell carcinoma. Clin Cancer Res. 2010;16:3628‐3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Joosten SC, Hamming L, Soetekouw PM, et al. Resistance to sunitinib in renal cell carcinoma: From molecular mechanisms to predictive markers and future perspectives. Biochem Biophys Acta. 2015;1855:1‐16. [DOI] [PubMed] [Google Scholar]
- 46. Yang J, Ikezoe T, Nishioka C, et al. Long‐term exposure of gastrointestinal stromal tumor cells to sunitinib induces epigenetic silencing of the PTEN gene. Int J Cancer. 2012;130:959‐966. [DOI] [PubMed] [Google Scholar]
- 47. Makhov PB, Golovine K, Kutikov A, et al. Modulation of Akt/mTOR signaling overcomes sunitinib resistance in renal and prostate cancer cells. Mol Cancer Ther. 2012;11:1510‐1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Martin D, Galisteo R, Gutkind JS. CXCL8/IL8 stimulates vascular endothelial growth factor (VEGF) expression and the autocrine activation of VEGFR2 in endothelial cells by activating NFkappaB through the CBM (Carma3/Bcl10/Malt1) complex. J Biol Chem. 2009;284:6038‐6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Choi CH. ABC transporters as multidrug resistance mechanisms and the development of chemosensitizers for their reversal. Cancer Cell Int. 2005;5:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2