

Abstract
Epstein‐Barr virus (EBV) is associated with particular forms of gastric cancer (GC). We previously showed that EBV infection into gastric epithelial cells induced aberrant DNA hypermethylation in promoter regions and silencing of tumor suppressor genes. We here undertook integrated analyses of transcriptome and epigenome alteration during EBV infection in gastric cells, to investigate activation of enhancer regions and related transcription factors (TFs) that could contribute to tumorigenesis. Formaldehyde‐assisted isolation of regulatory elements (FAIRE) sequencing (‐seq) data revealed 19 992 open chromatin regions in putative H3K4me1+ H3K4me3− enhancers in EBV‐infected MKN7 cells (MKN7_EB), with 10 260 regions showing increase of H3K27ac. Motif analysis showed candidate TFs, eg activating transcription factor 3 (ATF3), to possibly bind to these activated enhancers. ATF3 was considerably upregulated in MKN7_EB due to EBV factors including EBV‐determined nuclear antigen 1 (EBNA1), EBV‐encoded RNA 1, and latent membrane protein 2A. Expression of mutant EBNA1 decreased copy number of the EBV genome, resulting in relative downregulation of ATF3 expression. Epstein‐Barr virus was also infected into normal gastric epithelial cells, GES1, confirming upregulation of ATF3. Chromatin immunoprecipitation‐seq analysis on ATF3 binding sites and RNA‐seq analysis on ATF3 knocked‐down MKN7_EB revealed 96 genes targeted by ATF3‐activating enhancers, which are related with cancer hallmarks, eg evading growth suppressors. These 96 ATF3 target genes were significantly upregulated in MKN7_EB compared with MKN7 and significantly downregulated when ATF3 was knocked down in EBV‐positive GC cells SNU719 and NCC24. Knockdown of ATF3 in EBV‐infected MKN7, SNU719, and NCC24 cells all led to significant decrease of cellular growth through an increase of apoptotic cells. These indicate that enhancer activation though ATF3 might contribute to tumorigenesis of EBV‐positive GC.
Keywords: enhancer, epigenome, Epstein‐Barr virus, gastric cancer, transcription factor
Through integrated analyses on alterations of transcriptome, histone modification, and open chromatin status during Epstein‐Barr virus (EBV) infection in gastric cells, we here identified activating transcription factor 3 (ATF3) as a critical transcriptional activator with involvement in enhancer activation. We undertook screening of putative transcription factors binding to activated enhancer regions and identified ATF3 as a transcription factor that is upregulated by EBV infection and induces aberrant enhancer activation. Effect of ATF3 expression on cellular proliferation was confirmed, suggesting a tumorigenic role of aberrant enhancer activation by ATF3 upregulation, which could help our understanding of the tumorigenic mechanisms in this particular subtype of gastric cancer.

Abbreviations
- ATF3
activating transcription factor 3
- CDDP
cisplatin
- EBER
EBV‐encoded small RNA
- EBNA1
EBV‐determined nuclear antigen 1
- EBV
Epstein‐Barr virus
- FAIRE
formaldehyde‐assisted isolation of regulatory elements
- GC
gastric cancer
- GO
Gene Ontology
- GSEA
gene set enrichment analysis
- H3K27ac
histone H3 lysine 27 acetylation
- H3K4me1
histone H3 lysine 4 monomethylation
- H3K4me3
histone H3 lysine 4 trimethylation
- IL
interleukin
- IP
immunoprecipitation
- LMP2A
latent membrane protein 2A
- MKN7_EB
EBV‐infected MKN7
- MKN7_WT
MKN7 without EBV infection
- NES
normalized enrichment score
- qPCR
quantitative PCR
- seq
sequencing
- shATF3
shRNA against ATF3
- shNON
control nontarget shRNA
- TCGA
The Cancer Genome Atlas
- TF
transcription factor
- TGF‐β
transforming growth factor‐β
- TSG
tumor suppressor gene
1. INTRODUCTION
Gastric cancer is the second leading cause of cancer‐related deaths in the world, accounting for approximately 783 000 deaths in 2018. 1 The infectious agents, including the bacterium Helicobacter pylori 2 , 3 and EBV, 4 , 5 are the major pathogens of GC. As a human oncogenic virus, EBV has been identified in various malignant diseases, including endemic Burkitt lymphoma, nasopharyngeal carcinoma, approximately 50% of Hodgkin’s disease, and 7%‐16% of GC. 4 , 6 , 7 Due to the particular molecular subtypes in GC, EBV infection status provided us an independent classification that not only determined patient stratication, but revealed unique epigenetic phenotype and somatic genomic alterations. 8 Since the publication of the EBV genome sequence in 1984, 9 the molecular analysis of the virus allowed us to elucidate the mechanisms of action of the viral proteins that contribute to tumorigenesis in EBV+ GC. 10
A unique and characteristic feature of EBV is its capability to establish distinct latent gene expression patterns in vivo and in cultured cells (named latency type I, II, and III) in resting and proliferating cells. Epstein‐Barr virus‐positive GC, as well as Burkitt lymphoma, belong to latency I, in which the expression of viral latent genes is the most severely restricted to EBNA1, EBER, LMP2A, and BamHI A rightward transcripts. 11 Oncogenic functions of these viral transcripts have been studied to elucidate the tumorigenic mechanism of EBV‐associated cancer. Epstein‐Barr virus‐encoded small RNAs have been reported to contribute to tumorigenesis of GC through activation of growth factors such as insulin like growth factor 1. 12 Expression of LMP2A might also facilitate cell survival by inhibiting p53 activity and TGF‐β‐associated cellular apoptosis. 11 Another viral protein, EBNA1, has an important role to maintain episomal form of the EBV genome binding to the host chromosome during latent infection.
Although these functions of viral factors might partly contribute to tumorigenesis in EBV+ malignancies, other molecular aberrations, eg genomic and epigenomic aberrations, in host cells need to be elucidated. We have previously reported that EBV+ GC showed extensively high DNA methylation phenotype at gene promoter regions and abundant TSGs, eg p16, are repressed by the aberrant promoter hypermethylation. 13 , 14 We also found that in vitro EBV infection in gastric epithelial cells introduces extensive DNA hypermethylation resembling the hypermethylation phenotype of clinical EBV+ GC tissue samples. Although this unique DNA methylation phenotype was reportedly confirmed in EBV+ GC through comprehensive analyses of GC by TCGA, epigenetic aberrations have been considered important for EBV+ GC tumorigenesis in addition to genetic alteration, eg PIK3CA mutation and PD‐L1/2 overexpression. 8 , 15
Other than gene repression by aberrant DNA methylation, gene activation by aberrant enhancers is known as an epigenetic driver of many other types of cancer. 16 , 17 It is also reported that mutations in enhancers or enhancer‐binding TFs could cooperate with DNA methylation to aberrantly activate or repress neighboring genes and provoke cancer. 16 , 18 In EBV+ GC, we preliminarily reported aberrant activation and repression at enhancer regions as well as promoter regions; 19 , 20 however, the mechanisms to induce epigenetic activation during EBV infection in gastric epithelial cells is yet to be fully investigated.
To identify factors that can induce aberrant enhancer activation during EBV infection, we undertook comprehensive analyses of histone modification and open chromatin regions to predict their putative binding elements. We identified that ATF3, which is activated by various environmental stresses and reportedly has dual functions as a TSG or an oncogene in several types of cancer, 21 , 22 promoted proliferation in EBV+ gastric cells. This investigation could provide insights into the epigenomic activations in enhancer regions, and thus tumorigenesis, by EBV infection in gastric epithelial cells.
2. MATERIAL AND METHODS
2.1. Cell culture
The SNU719 EBV+ GC cell line was obtained from the Korean Cell Line Bank, and NCC24 from Creative Bioarray. The MKN7 EBV− GC cell line (MKN7_WT), which shows a low DNA methylation epigenotype, 13 was purchased from Riken BioResource Center Cell Bank. GES1 is a normal fetal gastric epithelial cell line immortalized with SV40 (Beijing Institute for Cancer Research). 23 Cells were cultured in RPMI‐1640 medium supplemented with 10% FBS and penicillin/streptomycin. MKN7 and GES1 cells were infected with recombinant EBV using the Akata system to establish EBV‐infected MKN7 cells (MKN7_EB) as previously reported. 13 , 24 The study design was approved by the institutional review board of Chiba University.
2.2. Chromatin immunoprecipitation‐seq and FAIRE‐seq analysis
Chromatin immunoprecipitation assays for histone modifications and FAIRE assays for open chromatin regions were carried out as previously reported. 25 For ChIP assays for ATF3 binding regions, cells were treated with 30 μM CDDP for 48 hours to induce ATF3 expression. Approximately 1 × 107 cells were cross‐linked with 1% formaldehyde for 10 minutes at room temperature and formaldehyde was quenched by addition of 2.5 M glycine to a final concentration of 0.125 M. Cross‐linked chromatin was sonicated to a size of 0.2‐1 kb using an ultrasonic disruptor (Branson Digital Sonifier). A total of 15 μg anti‐ATF3 Ab (sc‐188 X, rabbit mAb; Santa Cruz Biotechnology) and 20 μL Protein G Sepharose beads were mixed in IP dilution buffer and incubated for 6 hours at 4°C. After washing with IP dilution buffer, Ab‐binding beads were added to the sonicated chromatin sample and incubated overnight at 4°C. Beads were washed and chromatin was eluted, followed by reversal of the cross‐linking and DNA purification. Chromatin‐immunoprecipitated DNA was dissolved in EB buffer (Qiagen). Libraries were constructed by using NEBNext ChIP‐seq Library Prep Reagent Set for Illumina (NEB) according to the manufacturer’s instructions. The sequencing libraries were quantified by Bioanalyzer (Agilent) and sequenced at a concentration of 4 pM on an Illumina Hiseq (Illumina).
Sequenced reads in ChIP‐seq and FAIRE‐seq experiments were mapped to the UCSC human genome (hg19) using bowtie 2. Duplicated reads were removed with Picard tools. Peak calling and motif analyses were carried out by using HOMER software (bib26://homer.salk.edu/homer/index.html), 26 which was also used to count reads for each signal. Peak annotation to the nearest genes was undertaken by using bedtools. Peak heatmaps were produced with the use of HOMER and TreeView for enrichment calculation and visualization.
2.3. RNA sequencing
RNA was extracted by using the RNeasy Mini Kit (Qiagen) following the manufacturer’s protocol and treated with DNaseI (Qiagen). Libraries for RNA‐seq were prepared using the TruSeq Stranded mRNA Sample Prep Kit (Illumina), following the manufacturer’s protocol. Deep sequencing was carried out on the Illumina HiSeq 1500 or NextSeq 500 platform using the TruSeq Rapid SBS Kit (Illumina) in 50‐base single‐end mode according to the manufacturer’s protocol. Sequenced reads from the RNA‐seq experiment were aligned by using HISAT2, and Cufflinks was used for transcript assembly. Gene expression levels were expressed as fragments per kilobase of exon per million mapped sequence reads.
2.4. Cancer hallmark analysis
The ATF3 target genes identified were compared with GO categories related with cancer hallmarks. 27 The GO categories used for the analysis of cancer hallmarks are shown in Table S1.
2.5. Knockdown by shRNA
To knock down ATF3, double‐stranded oligonucleotide DNA encoding shRNA against ATF3 was cloned into the pLKO.1 vector between EcoRI and AgeI sites, as previously described. 28 Oligonucleotide sequences to ligate in the vector for construction of shATF3 and shNON are as follows, where 21‐mer target sequences are underlined: shATF3, CCGG GCTGAACTGAAGGCTCAGATT CTCGAG AATCTGAGCCTTCAGTTCAGC TTTTTG and AATTCAAAAA GCTGAACTGAAGGCTCAGATT CTCGAG AATCTGAGCCTTCAGTTCAGC; and shNON CCGG CAACAAGATGAAGAGCACCAA CTCGAG TTGGTGCTCTTCATCTTGTTG TTTTT and AATTCAAAAA CAACAAGATGAAGAGCACCAA CTCGAG TTGGTGCTCTTCATCTTGTTG. Viral packaging for shRNA retrovirus vectors was undertaken using 293T cells and FuGENE 6 (Promega), and medium containing the virus was collected 48 hours after transfection.
2.6. Overexpression of latent EBV factors
Latent viral factors, BARF, EBER1, EBNA1, and LMP2A, were previously constructed in pcDNA3 vector. 13 Each vector or mock vector was introduced in MKN7 cells using FuGene6, and gene expression was analyzed by RNA‐seq as above.
2.7. Overexpression of mutant EBNA1
Mutant ENBA1 to act as a dominant‐negative inhibitor of EBNA1 was constructed according to the previous report. 29 Mutant EBNA1 fused by mCherry fluorescent protein at the C‐terminal region (mtEBNA1‐mCherry) was inserted into the modified pcDNA5 vector. 28 Mock vector with only mCherry inserted was also constructed and used as a control. Vectors with mtEBNA1‐mCherry or mCherry were transfected in MKN7_EB cells by Lipofectamine 3000. On day 7, strongly red‐colored cells caused by mCherry were sorted by a cell sorter HS800 (Sony). Decrease of EBV copy numbers was examined by qPCR.
2.8. Real‐time qPCR
Real‐time qPCR was undertaken using SYBR Green and CFX96 Touch Real‐Time PCR (Bio‐Rad Laboratories). The quantity of genomic DNA in a sample was estimated by comparisons with standard samples that contained 101 to 106 gene copies, as previously described. 13 The quantity of EBV genome was normalized to that of the HBB region in the human genome. Primers for the EBV genome are TCGTACATCT CCAGGCGACA CT and CATACAACCA CTGGCGATCC CC. Primers for HBB are CAGGGTGAGG TCTAAGTGAT GA and TTGAAGTCCA ACTCCTAAGC CA.
2.9. Real‐time qPCR
cDNA was prepared from 1 μg total RNA using SuperScript III Reverse Transcriptase (Thermo Fisher). Real‐time qPCR was undertaken as above, using cDNA as a template, and the quantity of mRNA for each gene in a sample was estimated by comparisons with standard samples that contained 101 to 106 gene copies. 13 The expression level of ATF3 was normalized to that of B2M. The PCR primers for ATF3 are GGATGTCCTC TGCGCTGGAA TC and TCCTTTCATC TTCTTCAGGG GC. Primers for B2M are GATGAGTATG CCTGCCGTGT and CTGCTTACAT GTCTCGATCC CA.
2.10. Western blot analysis
Cells were lysed in a lysis buffer containing 62.5 mM Tris‐HCl (pH6.8), 2.3% SDS, 10% glycerol, 0.2% bromophenol blue for whole cell or 10 mM Tris‐HCl (pH 7.4), 10 mM NaCl, 2.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 0.5% NP40, and proteinase inhibitor cocktails (Roche) for cytoplasm and nuclear fraction. The following primary Abs were used: anti‐ATF3 (sc‐188 X; Santa Cruz Biotechnology for whole cell; or #33593, rabbit mAb; Cell Signaling Technology for cytoplasm and nuclear fraction), anti‐Actin (MA5‐11869, mouse mAb; Invitrogen), anti‐Lamin A (NA934V, rabbit polyclonal; GE Healthcare), and anti‐α‐tubulin (sc‐5286, mouse mAb; Santa Cruz Biotechnology). Protein‐blotted membranes were incubated with Abs using Can Get Signal Immunoreaction Enhancer Solution (Toyobo) at 4°C overnight for the primary Abs, and at room temperature for 1 hour for secondary Abs, followed by visualization using the ECL prime system (GE Healthcare). The protein signals were detected using a Luminescent Image Analyzer LAS‐3000 (Fujifilm).
2.11. Cell growth assay
Cells were seeded into 6 wells of 96‐well plates at 2000 cells per well in 200 μL culture medium. After an appropriate period of culturing, WST‐8 reagent (Dojindo) was added to each well, followed by incubation for 4 hours at 37°C in 5% CO2. Absorbance at 450 nm was measured using a SpectraMax PLUS 384 microplate reader (Molecular Devices).
2.12. Caspase 3/7 apoptosis staining
Analysis of apoptosis staining was undertaken using CellEvent Caspase‐3/7 Green Detection Reagent (Caspase 3/7) (Invitrogen). The apoptotic cells were imaged using a BZ‐X710 microscope (Keyence). Imaged data were processed and quantified using ImageJ. 30
2.13. Statistical analysis
The P values for expression analysis were obtained using paired t test to evaluate differences between 2 groups, with P less than .05 considered statistically significant. Gene Ontology analysis was undertaken by using Metascape (http://metascape.org/gp/index.html#/main/step1). 31 Gene set enrichment analysis was undertaken using GSEA software version 4.0.3 (http://software.broadinstitute.org/gsea/index.jsp). 32
3. RESULTS
3.1. Detection of activated enhancers in EBV‐infected MKN7 cells
We undertook a FAIRE‐seq analysis in MKN7_EB to detect open chromatin regions in gene promoters and enhancers. Promoter regions were defined by H3K4me3 peaks, and enhancer regions were defined by H3K4me1 peaks without H3K4me3 signal (Figure 1A,B). We identified a total of 52 850 FAIRE peaks in MKN7_EB, including 13 209 peaks (25%) in promoters and 19 992 peaks (38%) in enhancers. While the majority of FAIRE peaks were distributed to enhancer regions, 10 260 (51%) of those showed an increase of H3K27ac signals and thus were activated, 4523 (23%) showed no change in H3K27ac signals, and 5209 (26%) showed a decrease of H3K27ac signals and thus were inactivated (Figure 1C).
Figure 1.
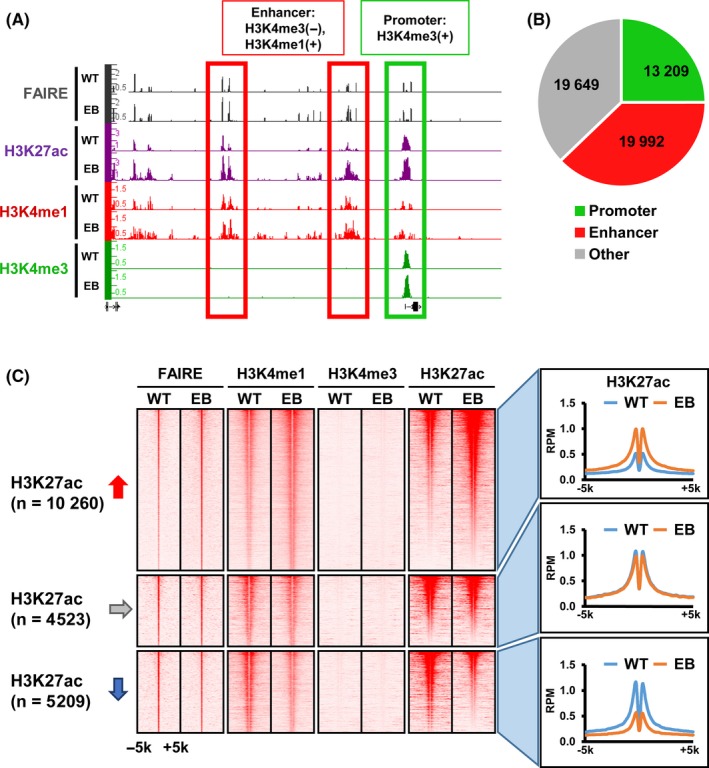
Detection of activated enhancers in Epstein‐Barr virus (EBV)‐infected MKN7 cells, MKN7_EB WT, MKN7_WT EB, and MKN7_EB. A, Representative formaldehyde‐assisted isolation of regulatory elements (FAIRE) signals on promoter and enhancer regions. Promoter and enhancer regions were defined as H3K4me3 peaks, and as H3K4me1 peaks without H3K4me3 signal, respectively. B, Distribution of FAIRE peaks. Among 52 850 FAIRE peaks in MKN7_EB, 13 209 and 19 992 peaks were detected in promoter regions and enhancer regions, respectively. C, Alteration of H3K27ac signals around 19 992 FAIRE peaks on enhancer regions. Heatmaps represent read densities of FAIRE, H3K4me1, H3K4me3 and H3K27ac within the ±5‐kb region from the FAIRE peak center (left). Histograms indicate the average read per million (RPM) reads of H3K27ac within ±5‐kb regions from the FAIRE peak center (right). The 10 260 and 5209 regions showed increase and decrease of the H3K27ac signals, respectively
Using the top 3000 regions showing higher H3K27ac signals among 10 260 activated enhancers, motif analysis was used to search for candidate TFs binding to these regions (Figure 2A). The top 5 significant motifs (P values ranging 1 × 10−55 to 1 × 10‐819) were detected, and candidate TFs were identified to match those significant motifs. Expression levels of genes near the motifs were analyzed by RNA‐seq and compared between MKN7_WT and MKN7_EB (Figure 2B). Significant upregulation of these neighboring genes was confirmed in MKN7_EB, suggesting that the activated enhancers could function in upregulation of the nearby genes. Among the candidate TFs, ATF3, TEAD2, and RUNX3 were upregulated more than 2‐fold in MKN7_EB in mRNA levels (Figure 2C). When EBV was infected in GES1 normal gastric epithelial cells, upregulation of ATF3 was confirmed, but TEAD2 and RUNX3 were not upregulated (Figure 2D). We therefore focused on ATF3, and upregulation of ATF3 in MKN7_EB was detected in not only mRNA levels but also protein levels (Figure 2E).
Figure 2.
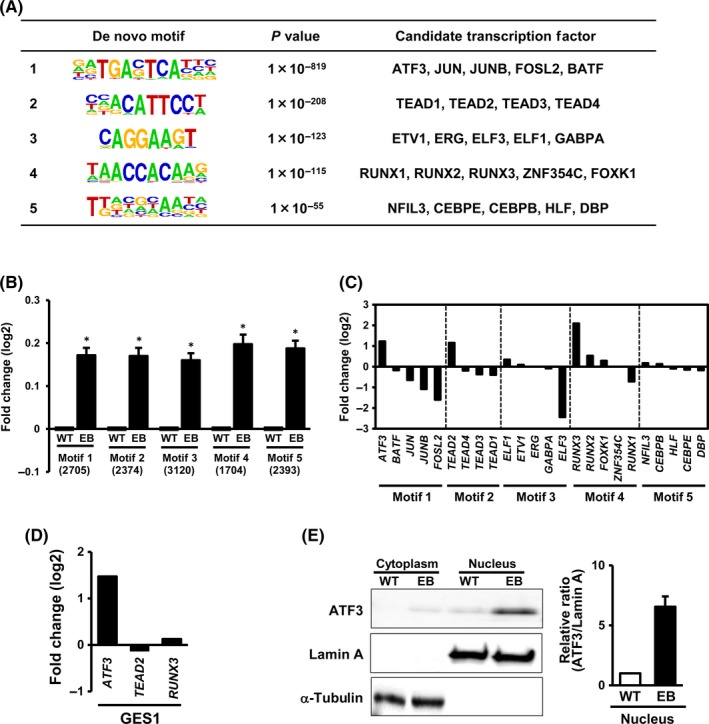
Detection of activating transcription factor 3 (ATF3) as a candidate TF. A, Motif analysis for activated enhancer regions. Among 10 260 activated enhancer regions, the top 3000 regions with highly expressed H3K27ac signal were extracted and subjected to de novo motif analysis. Top 5 significant motifs and candidate TFs, ATF3, JUN, JUNB, FOSL2, and BATF, are shown. B, Expression of genes neighboring the top 5 motifs. Neighboring genes were significantly upregulated in Epstein‐Barr virus (EBV)‐infected MKN7 cells (MKN7_EB). C, Relative expression levels of candidate TFs in MKN7_EB. While ATF3, TEAD2, and RUNX3 were clearly upregulated in MKN7_EB, other candidate TFs were not. D, Expression of genes in EBV‐infected GES1 cells. ATF3 was confirmed to be upregulated after EBV infection, whereas TEAD2 and RUNX3 were not. E, ATF3 protein level in cytoplasm and nucleus. Western blot showed ATF3 localized in nucleus, and expressed at higher levels in MKN7_EB cells. Open and closed boxes indicate relative ratio of the intensity of ATF3 to that of lamin A in the nucleus in MKN7_WT and MKN7_EB cells, respectively (n = 3). Values are shown as the mean ± SD
To gain insight into the viral factors that might upregulate ATF3 expression in host cells, viral factors including BARF, EBER1, EBNA1, and LMP2A, which are known to be expressed in latent infection of EBV, were overexpressed in MKN7 cells, and RNA‐seq was carried out. Activating transcription factor 3 was found to be upregulated the most by EBNA1 overexpression, and relative upregulation by more than 1.5‐fold was also detected by EBER1or LMP2A overexpression (Figure 3A). Previous ChIP‐seq analysis for EBNA1 target regions 33 , 34 showed that EBNA1 can bind to an enhancer region upstream of ATF3. When mutant EBNA1 was overexpressed in MKN7_EB, the copy number of the EBV genome was confirmed to be decreased, resulting in a considerable decrease of ATF3 expression level (Figure 3C). These results indicate that ATF3 could be upregulated, at least partly, by expression of EBV factors, eg EBNA1.
Figure 3.

Upregulation of activating transcription factor 3 (ATF3) by Epstein‐Barr virus (EBV) factors. A, Relative expression of ATF3 in conditions of overexpression of EBV factors. Each EBV factor, known to express in latent infection in EBV+ gastric cancer, was overexpressed in MKN7 cells and expression level of ATF3 was analyzed by RNA sequencing (RNA‐seq). ATF3 was most upregulated by overexpression of EBV‐determined nuclear antigen 1 (EBNA1), and >1.5‐fold by EBV‐encoded small RNA 1 (EBER1) and latent membrane protein 2A (LMP2A). B, EBNA1 target region for ATF3. Previous ChIP‐seq data for EBNA1 binding regions showed that EBNA1 could bind to an enhancer region upstream of ATF3. C, EBNA1 inhibitor. Overexpression of mutant EBNA1, known as EBNA1 inhibitor, in MKN7_EB cells resulted in decrease of the copy number of EBV genome and ATF3 expression
3.2. Identification of possible ATF3 binding sites by ChIP‐seq
To identify target regions of ATF3 genome‐wide, ChIP‐seq using anti‐ATF3 Ab was used against ATF3 binding sites (Figure 4). As ATF3 overexpression using pcDNA5 expression vector in MKN7 led to more than 100‐fold upregulation of ATF3 and cell death at 36 hours, ATF3 upregulation was induced in MKN7 cells by exposure to 30 μM CDDP instead. As ATF3 expression was increased in a time‐dependent manner up to 48 hours (Figure 4A), ChIP‐seq analysis of possible ATF3 binding sites was carried out using MKN7 cells at 48 hours after CDDP exposure (Figure 4B,C). Among a total of 3345 ATF3 peaks defined by ChIP‐seq analysis, 1339 peaks were distributed to enhancer regions, and 590 of those showed increase of H3K27ac signals and thus were activated (Figure 4D). Expression levels of 659 genes neighboring ATF3 peaks on the 590 activated enhancers were significantly increased (P = 1.5 × 10‐6).
Figure 4.
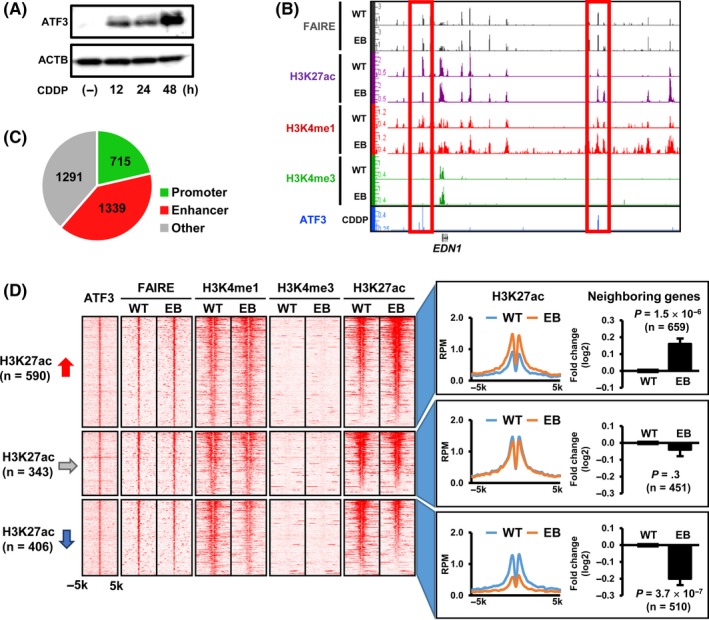
Identification of possible activating transcription factor 3 (ATF3) binding sites. A, ATF3 induction in MKN7 cells by exposure to cisplatin (CDDP). To analyze the possible ATF3 binding site in MKN7 cells, ATF3 was induced by exposure to 30 μM CDDP for 48 h. B, Representative ATF3 signals. Possible ATF3 binding sites were analyzed by ChIP sequencing (ChIP‐seq) in ATF3‐induced MKN7 cells at 48 h after CDDP treatment. C, Distribution of ATF3 peaks. Among 3345 ATF3 peaks detected, 715 and 1339 peaks were distributed to promoter and enhancer regions, respectively. D, Heatmaps of read densities in ATF3‐bound enhancer regions. Read densities of ChIP‐seq for ATF3, H3K4me1, H3K4me3, and H3K27ac, and those for formaldehyde‐assisted isolation of regulatory elements (FAIRE)‐seq within ±5‐kb regions from the ATF3 peak center, are shown (left). Histogram indicated the average read per million (RPM) reads of H3K27ac (right). The 590 and 406 regions showed increase and decrease of the H3K27ac signals, respectively. Expression levels of 659 and 510 genes neighboring ATF3 peaks in the 590 activated or 406 inactivated enhancer regions were significantly increased (P = 1.5 × 10−6), or decreased (P = 3.7 × 10−7), respectively. Values are shown as the mean ± SEM
3.3. Identification of ATF3 target genes
As the possible ATF3 binding sites might include regions targeted specifically in the condition of CDDP exposure, ATF3 was knocked down in MKN7_EB cells by a shATF3 to screen genes targeted by ATF3 in EBV‐infected cells. Repression of ATF3 was confirmed by western blot analysis (Figure 5A). Gene expression alteration by knockdown of ATF3 was analyzed by RNA‐seq. Among the 659 genes neighboring possible ATF3 binding sites at active enhancer regions, we identified 96 genes downregulated by ATF3 knockdown in MKN7_EB cells, namely ATF3 target genes (Figure 5B). By GSEA, it was confirmed that these ATF3 target genes were shown to be significantly enriched in highly expressed genes in MKN7_EB compared with MKN7_WT (NES = 1.88, P < .001) (Figure 5C). The top 30 ATF3 target genes that showed high expression in MKN7_EB cells in the GSEA were found to be considerably related with cancer hallmarks, such as evading growth suppressors, sustaining proliferative signaling, and resisting cell death (Figure 5D).
Figure 5.
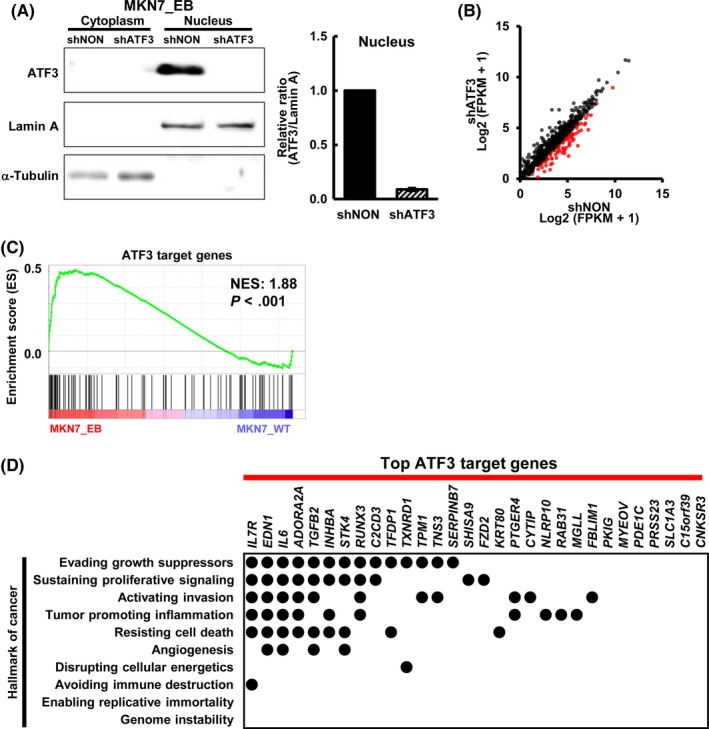
Identification of activating transcription factor 3 (ATF3) target genes. A, Knockdown of ATF3 in Epstein‐Barr virus (EBV)‐infected MKN7 (MKN7_EB) cells. ATF3 was knocked down by shRNA lentivirus targeting ATF3 (shATF3). Western blot analysis showed marked decrease of ATF3 expression in the nucleus. Right, comparison of ratios of the intensity of ATF3 to that of lamin A in nucleus (n = 3). Values are shown as the mean ± SD. B, RNA sequencing analysis of gene expression alterations by knockdown of ATF3. Among the 659 genes neighboring possible peaks bound by ATF3, 96 genes were downregulated in MKN7_EB when ATF3 was knocked down (red dots). C, Gene set enrichment analysis (GSEA) for the 96 ATF3 target genes. The 96 ATF3 target genes were significantly enriched in the genes highly expressed in MKN7_EB (normalized enrichment score [NES] = 1.88, P < .001). D, Relationship with cancer hallmarks. Among the 96 ATF3 target genes, the top 30 genes showing higher expression in MKN7_EB in GSEA were extracted. They were frequently related with cancer hallmarks, such as evading growth suppressors, sustaining proliferative signaling, and resisting cell death. FPKM, fragments per kilobase of exon per million mapped sequence reads; shNON, control nontarget shRNA
3.4. Knockdown of ATF3 in EBV+ GC cell lines NCC24 and SNU719
Additionally, ATF3 was knocked down by shATF3 in 2 EBV+ GC cell lines NCC24 and SNU719, and repression of ATF3 was confirmed by western blot (Figure 6). RNA sequencing was used to analyze gene expression alteration. Gene set enrichment analysis showed that the 96 ATF3 target genes were significantly enriched in genes downregulated by knockdown of ATF3, in both NCC24 and SNU719 cells, compared with shNON control cells (Figure 6A‐D). We analyzed the cellular growth of ATF3 knocked‐down cells by WST‐8 assay (Figure 6E). Significant repression of cellular growth was detected in MKN7_EB, NCC24, and SNU719 cells when ATF3 was knocked down. Caspase 3/7 activity was analyzed to detect apoptotic cells, and apoptosis was observed significantly frequently in MKN7_EB, NCC24, and SNU719 cells when ATF3 was knocked down (Figure 6F).
Figure 6.
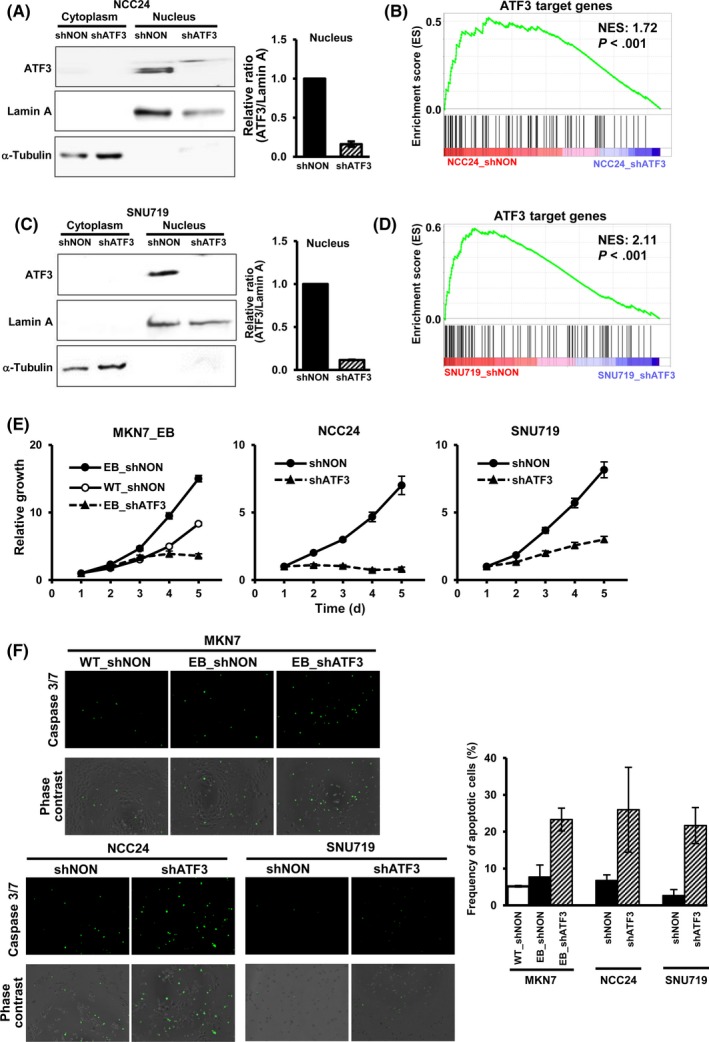
Knockdown of activating transcription factor 3 (ATF3) and repression of cellular growth. A, Knockdown of ATF3 in NCC24 cells. ATF3 was knocked down in Epstein‐Barr virus (EBV)+ gastric cancer (GC) cell line NCC24 by shRNA lentivirus targeting ATF3 (shATF3). Right, ratio of the intensity of ATF3 to that of lamin A in nucleus (n = 3). Values are shown as the mean ± SD. B, Gene set enrichment analysis (GSEA) for the 96 ATF3 target genes in NCC24 cells. ATF3 target genes were significantly downregulated in NCC24 cells with ATF3 knockdown. C, Knockdown of ATF3 in EBV+ GC cell line SNU719. D, GSEA of the 96 ATF3 target genes in SNU719 cells. E, Repression of cellular growth of MKN7_EB, NCC24, and SNU719. Cellular growth was measured by WST‐8 assay for 5 days (n = 6), and relative growth is shown. Knockdown of ATF3 led to decrease of cellular growth in all the EBV+ cells analyzed. F, Caspase 3/7 assay. Apoptotic cells were counted by caspase 3/7 activity (green). Frequency of apoptotic cells was increased by knockdown of ATF3 in all the EBV+ cells analyzed. Values are shown as the mean ± SD (n = 3). NES, normalized enrichment score; shNON, control nontarget shRNA
4. DISCUSSION
Through integrated analyses on alterations of transcriptome, histone modification, and open chromatin status, we here identified ATF3 as a critical transcriptional activator after EBV infection in gastric cells. We undertook screening of putative TF binding to activated enhancer regions that are predicted by motif analysis and refined by gene expression analysis. ATF3 was identified as a candidate TF that is upregulated by EBV infection and induces aberrant enhancer activation. Expression of viral factors might be the cause of upregulated ATF3 expression, at least partly, and the effect of ATF3 expression on cellular proliferation was confirmed, suggesting a tumorigenic role of aberrant enhancer activation by ATF3 upregulation.
ATF3 is known to play different roles depending on various interacting partners. It is activated by a variety of environmental stress signals and is associated with the pathogenesis of various diseases including cancer, cardiac hypertrophy, and infection. 35 In cancer, reciprocal functions of ATF3 as either an oncogene or a TSG have been reported, depending on the condition of the cell. It reportedly promotes apoptosis of human cancer cells, such as T‐cell lymphoma, multiple myeloma, blood, lung, prostate, and colon cancer cells, 36 , 37 , 38 , 39 and overexpression of ATF3 reportedly promotes the metastasis of prostate and breast cancer. 40 , 41 As an oncogene, TGF‐β, p53, and Wnt/β‐catenin signaling pathways are the known factors to activate ATF3, leading to the expression of downstream factors, eg MMP13, Snail, Slug, and Twist, which are key regulators of epithelial‐mesenchymal transition and metastasis. 42 ATF3 was also reported to promote Runx2 expression in metastatic breast cancer cells, aiding bone metastasis of tumor cells. 43 In this study, ATF3 was found to be upregulated by EBV infection through expression of EBV factors, eg EBNA1, EBER1, and LMP2A, and downregulation of ATF3 was confirmed when the copy number of the EBV genome was decreased in MKN7_EB due to overexpression of mutant EBNA1. Knockdown experiments of ATF3 showed that ATF3 has an oncogenic role to promote proliferation in EBV‐infected gastric cells. Downstream targets of ATF3 were also shown to correlate with hallmarks of cancer, which might contribute to EBV+ GC progression.
The sequence‐specific DNA binding protein EBNA1 binds with high affinity to 3 well‐characterized sites in the viral genome that are important for maintenance of the episomal viral genome, DNA replication, and viral gene regulation. Several papers reported that there exist numerous EBNA1 binding sites in the host genome and that EBNA1 binds to these sites to activate the expression of neighboring host genes. 33 , 34 It has been reported that EBNA1 activates many other host genes directly, which can function in various pathways related to cancer proliferation, including several cytokine pathways (IL‐18, IL‐6, IL‐12, and IL‐2), the p38 MAPK and MAP3K1 network in B cells, 33 , 34 the activator protein 1 (AP1) transcription factor pathway in nasopharyngeal carcinoma cells, 44 and signal transducer and activator of transcription 1 (STAT1) in carcinoma and B‐lymphoma cells, 45 , 46 although ATF3 had not been identified as an EBNA1 downstream target. This study showed that EBNA1 might be an EBV factor to potentially induce upregulation of ATF3, and thus promote proliferation in GC cells; mechanisms to activate ATF3 expression, eg function analysis of EBNA1 binding site nearby ATF3 as an enhancer, should be further investigated.
Previous comprehensive analyses of GC by us or TCGA revealed that EBV+ GC shows a unique DNA hypermethylation phenotype that is the most extensive hypermethylation among all the human malignancies. 8 , 13 Distinct mutations of genes, eg PIK3CA and ARID1A, are also observed in EBV+ GC. 8 Most of these aberrations are related with repression of tumor suppressive functions in gastric epithelial cells, and viral proteins also mainly function to repress tumor suppressive functions, except EBERs that reportedly promote tumor progression through activation of growth factors. Recently, aberrant enhancer activation has been reported in many types of cancer to promote oncogene activation and is reportedly caused by genomic aberrations such as amplification or translocation, or activation of oncogenic master regulators in various cancers. Whereas enhancer activation in EBV+ GC has been reported in only a few published works, we here found that ATF3 is upregulated and contributes to enhancer activation in EBV‐infected gastric cells.
In summary, we undertook integrated analyses on alterations of the transcriptome and epigenome during EBV infection in gastric epithelial cells and identified upregulation of ATF3 that could contribute to aberrant enhancer activation in EBV+ GC and enhance proliferation of gastric cells by EBV infection.
CONFLICT OF INTEREST
Yuta Asakawa is hired by Otsuka Pharmaceutical Co., Ltd. The authors have no other conflicts of interest to disclose.
Supporting information
Table S1
ACKNOWLEDGMENTS
We thank Haruka Maruyama for technical assistance. This study was funded by: 19ck0106263h0003 (to AK), and 19cm0106510h0004 (to AK) from the Japan Agency for Medical Research and Development (AMED); Grants‐in‐Aid for Scientific Research (KAKENHI) from the Japan Society for the Promotion of Science (16H05412 and 19H03726 to AK); and a grant from Global and Prominent Research, Chiba University to AK.
Asakawa Y, Okabe A, Fukuyo M, et al. Epstein‐Barr virus‐positive gastric cancer involves enhancer activation through activating transcription factor 3. Cancer Sci. 2020;111:1818–1828. 10.1111/cas.14370
DATA AVAILABILITY STATEMENT
NGS data generated in this study have been deposited in Gene Expression Omnibus under accession # http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE141385 (GSM4202156 ‐ GSM4202165, GSM4308257, GSM4308258). Previously deposited NGS data (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE97837 and http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE97838) that were used in this study are also available in Gene Expression Omnibus. The authors declare that all other data are available within the article or associated supplementary information files, or available from the authors on request.
REFERENCES
- 1. Ferlay J, Colombet M, Soerjomataram I, et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer. 2019;144:1941‐1953. [DOI] [PubMed] [Google Scholar]
- 2. Amieva M, Peek RM. Pathobiology of Helicobacter pylori‐Induced Gastric Cancer. Gastroenterology. 2016;150:64‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Uemura N, Okamoto S, Yamamoto S, et al. Helicobacter pylori infection and the development of gastric cancer. N Engl J Med. 2001;345:784‐789. [DOI] [PubMed] [Google Scholar]
- 4. Shibata D, Weiss LM. Epstein‐Barr virus‐associated gastric adenocarcinoma. Am J Pathol. 1992;140:769‐774. [PMC free article] [PubMed] [Google Scholar]
- 5. Burke AP, Yen TS, Shekitka KM, Sobin LH. Lymphoepithelial carcinoma of the stomach with Epstein‐Barr virus demonstrated by polymerase chain reaction. Mod Pathol. 1990;3:377‐380. [PubMed] [Google Scholar]
- 6. Murphy G, Pfeiffer R, Camargo MC, Rabkin CS. Meta‐analysis shows that prevalence of Epstein‐Barr virus‐positive gastric cancer differs based on sex and anatomic location. Gastroenterology. 2009;137:824‐833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cohen JI, Fauci AS, Varmus H, Nabel GJ. Epstein‐Barr virus: an important vaccine target for cancer prevention. Sci Transl Med. 2011;3:107fs107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cancer T, Atlas G, Bass AJ, et al. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Baer R, Bankier AT, Biggin MD, et al. DNA sequence and expression of the B95–8 Epstein‐Barr virus genome. Nature. 1984;310:207‐211. [DOI] [PubMed] [Google Scholar]
- 10. Young LS, Yap LF, Murray PG. Epstein‐Barr virus: more than 50 years old and still providing surprises. Nat Rev Cancer. 2016;16:789‐802. [DOI] [PubMed] [Google Scholar]
- 11. Fukayama M, Hino R, Uozaki H. Epstein‐Barr virus and gastric carcinoma: virus‐host interactions leading to carcinoma. Cancer Sci. 2008;99:1726‐1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Iwakiri D, Eizuru Y, Tokunaga M, Takada K. Autocrine growth of Epstein‐Barr virus‐positive gastric carcinoma cells mediated by an Epstein‐Barr virus‐encoded small RNA. Cancer Res. 2003;63:7062. [PubMed] [Google Scholar]
- 13. Matsusaka K, Kaneda A, Nagae G, et al. Classification of Epstein‐Barr virus‐positive gastric cancers by definition of DNA methylation epigenotypes. Cancer Res. 2011;71:7187‐7197. [DOI] [PubMed] [Google Scholar]
- 14. Matsusaka K, Funata S, Fukuyo M, et al. Epstein‐Barr virus infection induces genome‐wide de novo DNA methylation in non‐neoplastic gastric epithelial cells. J Pathol. 2017;242:391‐399. [DOI] [PubMed] [Google Scholar]
- 15. Saito R, Abe H, Kunita A, Yamashita H, Seto Y, Fukayama M. Overexpression and gene amplification of PD‐L1 in cancer cells and PD‐L1+ immune cells in Epstein‐Barr virus‐associated gastric cancer: the prognostic implications. Mod Pathol. 2017;30:427‐439. [DOI] [PubMed] [Google Scholar]
- 16. Herz HM, Hu D, Shilatifard A. Enhancer malfunction in cancer. Mol Cell. 2014;53:859‐866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bradner JE, Hnisz D, Young RA. Transcriptional addiction in cancer. Cell. 2017;168:629‐643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Aran D, Hellman A. DNA methylation of transcriptional enhancers and cancer predisposition. Cell. 2013;154:11‐13. [DOI] [PubMed] [Google Scholar]
- 19. Okabe A, Funata S, Matsusaka K, et al. Regulation of tumour related genes by dynamic epigenetic alteration at enhancer regions in gastric epithelial cells infected by Epstein‐Barr virus. Sci Rep. 2017;7:7924‐7924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Funata S, Matsusaka K, Yamanaka R, et al. Histone modification alteration coordinated with acquisition of promoter DNA methylation during Epstein‐Barr virus infection. Oncotarget. 2017;8:55265‐55279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li X, Zang S, Cheng H, Li J, Huang A. Overexpression of activating transcription factor 3 exerts suppressive effects in HepG2 cells. Mol Med Rep. 2019;19:869‐876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rohini M, Haritha Menon A, Selvamurugan N. Role of activating transcription factor 3 and its interacting proteins under physiological and pathological conditions. Int J Biol Macromol. 2018;120:310‐317. [DOI] [PubMed] [Google Scholar]
- 23. Ke Y, Ning T, Wang B. Establishment and characterization of a SV40 transformed human fetal gastric epithelial cell line‐GES‐1. Zhonghua Zhong Liu Za Zhi. 1994;16:7‐10. [PubMed] [Google Scholar]
- 24. Imai S, Nishikawa JUN, Takada K. Cell‐to‐cell contact as an efficient mode of Epstein‐Barr virus infection of diverse human epithelial cells. J Virol. 1998;72:4371‐4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kaneda A, Fujita T, Anai M, et al. Activation of Bmp2‐Smad1 signal and its regulation by coordinated alteration of H3K27 trimethylation in Ras‐induced senescence. PLoS Genet. 2011;7:e1002359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Heinz S, Benner C, Spann N, et al. Simple combinations of lineage‐determining transcription factors prime cis‐regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576‐589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646‐674. [DOI] [PubMed] [Google Scholar]
- 28. Namba‐Fukuyo H, Funata S, Matsusaka K, et al. TET2 functions as a resistance factor against DNA methylation acquisition during Epstein‐Barr virus infection. Oncotarget. 2016;7:81512‐81526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kirchmaier AL, Sugden B. Dominant‐negative inhibitors of EBNA‐1 of Epstein‐Barr virus. J Virol. 1997;71:1766‐1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671‐675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tripathi S, Pohl MO, Zhou Y, et al. Meta‐ and orthogonal integration of influenza "oMICs" data defines a role for UBR4 in virus budding. Cell Host Microbe. 2015;18:723‐735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545‐15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tempera I, Leo AD, Kossenkov AV, et al. Identification of MEF2B, EBF1, and IL6R as direct gene targets of Epstein‐Barr virus (EBV) nuclear antigen 1 critical for EBV‐infected B‐lymphocyte survival. J Virol. 2016;90:345‐355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lu F, Wikramasinghe P, Norseen J, et al. Genome‐wide analysis of host‐chromosome binding sites for Epstein‐Barr Virus Nuclear Antigen 1 (EBNA1). Virol J. 2010;7:1‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhao J, Li X, Guo M, Yu J, Yan C. The common stress responsive transcription factor ATF3 binds genomic sites enriched with p300 and H3K27ac for transcriptional regulation. BMC Genomics. 2016;17:1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chüeh AC, Tse JWT, Dickinson M, et al. ATF3 repression of BCL‐X<sub>L</sub> determines apoptotic sensitivity to HDAC inhibitors across tumor types. Clin Cancer Res. 2017;23:5573‐5584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang Z, Xu D, Ding HF, et al. Loss of ATF3 promotes Akt activation and prostate cancer development in a Pten knockout mouse model. Oncogene. 2015;34:4975‐4984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Joo JH, Ueda E, Bortner CD, Yang XP, Liao G, Jetten AM. Farnesol activates the intrinsic pathway of apoptosis and the ATF4‐ATF3‐CHOP cascade of ER stress in human T lymphoblastic leukemia Molt4 cells. Biochem Pharmacol. 2015;97:256‐268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jan YH, Tsai HY, Yang CJ, et al. Adenylate kinase‐4 is a marker of poor clinical outcomes that promotes metastasis of lung cancer by downregulating the transcription factor ATF3. Cancer Res. 2012;72:5119‐5129. [DOI] [PubMed] [Google Scholar]
- 40. Wolford CC, McConoughey SJ, Jalgaonkar SP, et al. Transcription factor ATF3 links host adaptive response to breast cancer metastasis. J Clin Invest. 2013;123:2893‐2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bandyopadhyay S, Wang Y, Zhan R, et al. The tumor metastasis suppressor gene <em>Drg‐1</em> down‐regulates the expression of activating transcription factor 3 in prostate cancer. Cancer Res. 2006;66:11983‐11990. [DOI] [PubMed] [Google Scholar]
- 42. Yin X, Wolford CC, Chang YS, et al. ATF3, an adaptive‐response gene, enhances TGF{beta} signaling and cancer‐initiating cell features in breast cancer cells. J Cell Sci. 2010;123:3558‐3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gokulnath M, Partridge NC, Selvamurugan N. Runx2, a target gene for activating transcription factor‐3 in human breast cancer cells. Tumour Biol. 2015;36:1923‐1931. [DOI] [PubMed] [Google Scholar]
- 44. O'Neil JD, Owen TJ, Wood VHJ, et al. Epstein‐Barr virus‐encoded EBNA1 modulates the AP‐1 transcription factor pathway in nasopharyngeal carcinoma cells and enhances angiogenesis in vitro. J Gen Virol. 2008;89:2833‐2842. [DOI] [PubMed] [Google Scholar]
- 45. AlQarni S, Al‐Sheikh Y, Campbell D, et al. Lymphomas driven by Epstein‐Barr virus nuclear antigen‐1 (EBNA1) are dependant upon Mdm2. Oncogene. 2018;37:3998‐4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wood VHJ, O'Neil JD, Wei W, Stewart SE, Dawson CW, Young LS. Epstein‐Barr virus‐encoded EBNA1 regulates cellular gene transcription and modulates the STAT1 and TGFβ signaling pathways. Oncogene. 2007;26:4135‐4147. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Data Availability Statement
NGS data generated in this study have been deposited in Gene Expression Omnibus under accession # http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE141385 (GSM4202156 ‐ GSM4202165, GSM4308257, GSM4308258). Previously deposited NGS data (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE97837 and http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE97838) that were used in this study are also available in Gene Expression Omnibus. The authors declare that all other data are available within the article or associated supplementary information files, or available from the authors on request.