

Abstract
In a subgroup of Japanese patients in the ARCHER 1050 randomized phase 3 trial, we evaluated the efficacy and safety and determined the effects of dose modifications on adverse events (AE) and therapy management of first‐line oral dacomitinib 45 mg compared with oral gefitinib 250 mg, each once daily in 28‐d cycles, in patients with EGFR‐activating mutation–positive (EGFR‐positive; exon 19 deletion or exon 21 L858R substitution mutations) advanced non‐small cell lung cancer (NSCLC). The primary endpoint was progression‐free survival (PFS; RECIST, version 1.1, by blinded independent review). In 81 Japanese patients (40 dacomitinib, 41 gefitinib), PFS was longer with dacomitinib compared with gefitinib (hazard ratio [HR], 0.544 [95% confidence interval {CI}, 0.307‐0.961]; 2‐sided P = .0327; median 18.2 for dacomitinib [95% CI, 11.0‐31.3] mo, 9.3 [95% CI, 7.4‐14.7] mo for gefitinib). The most common Grade 3 AEs were dermatitis acneiform with dacomitinib (27.5%) and increased alanine aminotransferase with gefitinib (12.2%). A higher proportion of patients receiving dacomitinib (85.0%) compared with gefitinib (24.4%) had AEs leading to dose reduction. Incidence and severity of diarrhea, dermatitis acneiform, stomatitis and paronychia were generally reduced after dacomitinib dose reductions and dacomitinib treatment duration was generally longer in patients with a dose reduction in comparison with those without a dose reduction. Our results confirmed the efficacy and safety of first‐line dacomitinib in Japanese patients with EGFR‐positive advanced NSCLC.
Keywords: dacomitinib, epidermal growth factor receptor, Japanese, non‐small cell lung cancer, tyrosine kinase inhibitor
We evaluated the efficacy and safety of first‐line dacomitinib compared with gefitinib in a subgroup of Japanese patients with EGFR‐activating mutation–positive advanced non‐small cell lung cancer who were enrolled in the ARCHER 1050 randomized phase 3 trial. Results for the primary efficacy endpoint in the Japanese patients (n = 81) were consistent with the results in the overall ARCHER 1050 population; there was a clinically meaningful prolongation of median PFS by 8.9 mo favoring dacomitinib. The safety profile of dacomitinib was manageable by dose reduction or temporary dose interruption and no new safety signals were observed in the population of Japanese patients compared with the overall ARCHER 1050 study population.

Abbreviations
- AE
adverse event
- BIRC
blinded independent review committee
- CI
confidence interval
- DOR
duration of response
- EGFR
epidermal growth factor receptor
- HER
human epidural growth factor
- HR
hazard ratio
- ITT
intention‐to‐treat
- NSCLC
non‐small cell lung cancer
- ORR
objective response rate
- OS
overall survival
- PFS
progression‐free survival
- SAE
serious adverse event
- TKI
tyrosine kinase inhibitor
- TRAE
treatment‐related adverse event
1. INTRODUCTION
The second‐generation epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI) dacomitinib (VIZIMPRO®, Pfizer Oncology, New York, NY, USA) is characterized by irreversible inhibition of EGFR/human epidermal growth factor receptor 1 (HER1), HER2, and HER4 1 , 2 and had shown antitumor activity as first‐line therapy in patients with EGFR‐activating mutation–positive (EGFR‐positive) advanced non‐small cell lung cancer (NSCLC). This antitumor activity was confirmed in the international, multicenter, randomized, open‐label phase 3 ARCHER 1050 trial, which demonstrated that dacomitinib significantly improved progression‐free survival (PFS); the primary endpoint of the study; hazard ratio (HR), 0.59 (95% confidence interval [CI], 0.47‐0.74; P < .0001) compared with the first‐generation EGFR TKI gefitinib (IRESSA®, AstraZeneca Pharmaceuticals, Wilmington, DE, USA) when used as first‐line treatment in patients with EGFR‐positive advanced NSCLC. 3 Median PFS was 14.7 mo (95% CI, 11.1‐16.6 mo) in the dacomitinib arm and 9.2 mo (95% CI, 9.1‐11.0 mo) in the gefitinib arm. In the overall survival (OS) analysis of this trial, OS was significantly longer with dacomitinib than that with gefitinib (HR, 0.760 [95% CI, 0.582‐0.993]; 2‐sided P = .0438) with median OS of 34.1 mo (95% CI, 29.5‐37.7 mo) with dacomitinib and 26.8 mo (95% CI, 23.7‐32.1 mo) with gefitinib.4
Of particular interest, analysis of the effects of treatments administered after permanent discontinuation of dacomitinib indicated patients were able to receive subsequent systemic anti‐cancer therapies, including chemotherapy and other EGFR TKIs, in various sequences. Median OS in patients who received a third‐generation EGFR TKI as the first subsequent systemic anti‐cancer therapy appeared to be longer than that in patients who had received other subsequent systemic anti‐cancer therapy in the second line, although a small number of patients received third‐generation EGFR TKI. 4 In addition, analysis of PFS for patients who did and did not have a dose reduction to manage adverse events (AEs) found that PFS was longer in the patients who received a dose reduction than the patients who did not receive a dose reduction. 5
This report describes the subgroup analysis of efficacy and safety in Japanese patients randomized into the ARCHER 1050 trial. In addition, this report includes the analysis for dose modification of dacomitinib and subsequent systemic anti‐cancer therapies in Japanese patients, which will provide valuable information to oncologists on the appropriate use of dacomitinib in patients with EGFR‐positive NSCLC in Japan.
2. MATERIALS AND METHODS
2.1. Study design and treatments
Full details of the ARCHER 1050 trial design and methodology have been published. 3 Briefly, ARCHER 1050 was an international, multicenter, randomized, phase 3 trial designed to evaluate the efficacy and safety of dacomitinib compared with those of gefitinib as first‐line therapy in patients with EGFR‐activating mutation–positive advanced NSCLC. Of the 71 academic medical centers and universities in seven countries or regions (China, Hong Kong, Italy, Japan, Poland, South Korea, and Spain), 10 of the sites that participated in the study were in Japan. The trial is registered with ClinicalTrials.gov (NCT01774721).
Eligible patients were randomized 1:1 to receive oral dacomitinib 45 mg once daily or oral gefitinib 250 mg once daily, each in 28‐d cycles, until disease progression, initiation of new anti‐cancer therapy, discontinuation, or death. Dacomitinib dose reductions and interruptions and gefitinib dose reductions and interruptions were permitted per protocol. Dacomitinib dose reductions were permitted for Grade ≥3 treatment‐related AEs (TRAEs) or for Grade 2 TRAEs lasting >1 cycle. The first dacomitinib dose reduction was to 30 mg/d and the second dose was to 15 mg/d. No other dose reductions were permitted. Dose interruptions (<2 wk, or longer in consultation with the sponsor) were permitted per protocol. Because gefitinib was only available as a 250 mg dose, treatment was stopped for Grade 2 (intolerable only), Grade 3, or Grade 4 TRAEs, then resumed at 250 mg daily or at a reduced dose of 250 mg every other day at the investigator's discretion. The randomization was stratified by race (Japanese, Chinese, other East Asian, or non‐Asian) and EGFR mutation subtype (exon 19 deletion or exon 21 L858R substitution mutations).
2.2. Patients
Patients aged ≥18 y (≥20 y in Japan and South Korea) with newly diagnosed stage IIIB/IV or recurrent NSCLC harboring an EGFR‐activating mutation (exon 19 deletion ± T790M or exon 21 L858R substitution mutation ± T790M) and at least one target lesion that had not been irradiated and was measurable using RECIST, version 1.1 were eligible. Other inclusion criteria included ECOG performance status of 0 or 1; adequate renal, hepatic, and hematologic function; and availability of tumor specimens for central laboratory confirmation of EGFR‐activating mutation. Patients with mixed histology, central nervous system metastases, prior systemic anti‐cancer treatment for locally advanced or metastatic NSCLC were excluded. Prior treatment with EGFR TKIs was not allowed.
The trial protocol was approved by the institutional review board or ethics committee of each participating center and the trial was conducted in accordance with the International Conference on Harmonization Good Clinical Practice and the Declaration of Helsinki. All patients provided written informed consent to participate in the trial.
2.3. Endpoints and assessments
The primary endpoint was PFS (time from randomization to date of disease progression according to RECIST, version 1.1 or death from any cause, whichever occurred first) according to a blinded independent review committee (BIRC). Secondary endpoints included investigator‐assessed PFS, objective response rate (ORR, best overall response of either complete response or partial response) determined by both BIRC and investigator assessment, DOR (time from first documented objective response to date of disease progression or death from any cause, whichever occurred first) determined by both BIRC and investigator assessment, OS (time from randomization to date of death from any cause), OS at 30 mo, pharmacokinetics (PK), and safety.
Tumor assessments by computed tomography or magnetic resonance imaging were conducted at screening, at the end of cycles 1 and 2, then at every other cycle until the end‐of‐treatment visit, and objective responses were measured using RECIST, version 1.1 by BIRC.
Safety assessments, including laboratory test abnormalities, concomitant medications, and AEs, were conducted on d 1 of each cycle, and AEs were assessed using the National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.0.
2.4. Statistical methods
The total sample size in this study was estimated to detect a 50% improvement in PFS that favored dacomitinib compared with gefitinib. The efficacy analyses were conducted for the Japanese subset in the intention‐to‐treat (ITT) population (all randomized patients) using data cutoff dates of July 29, 2016, for the primary efficacy analysis (PFS, ORR, DOR) and PK, and February 17, 2017, for the final OS analysis. A log‐rank test stratified by EGFR mutation status at randomization was used to assess PFS, and the two‐sided P value was calculated. However, as the study was not powered for the Japanese subset, all P values in this subset analysis were to be considered as nominal. A Cox proportional hazards model stratified by EGFR mutation status at randomization was used to calculate the HR and associated 95% CI for PFS. HRs and P values for PFS in a subgroup by EGFR mutation status at randomization, DOR, and OS were estimated from the unstratified Cox proportional hazards models and unstratified log‐rank tests, respectively. DOR was evaluated among the objective responders in the ITT population. OS at 30 mo was defined as the probability of a patient being alive at 30 mo from the date of random assignment. OS at 30 mo was estimated by using Kaplan‐Meier methods with a two‐sided 95% CI. The median survival time and two‐sided 95% CI for the median were provided by treatment arm. The ORR was compared between arms using Pearson’s chi‐square test.
The safety population comprised patients in the ITT population who received at least one dose of study drug. Medical Dictionary for Regulatory Activities, version 19.1 preferred terms were used to summarize AEs. The trial was monitored by an independent data and safety monitoring committee, who evaluated patient safety on a periodic basis and determined whether the study should be modified or terminated based on ongoing reviews of safety data. Statistical analyses were conducted using SAS, version 9.4.
In addition, frequency and severity of AEs of interest (diarrhea, dermatitis acneiform, stomatitis and paronychia) before and after dose reduction from 45 mg once daily were analyzed. Plasma steady‐state trough concentrations of dacomitinib were collected at d 1 of cycle 2, after at least 14 d of consecutive dacomitinib 45 mg once‐daily dosing. These concentrations were then used to descriptively compare the initial plasma exposure in patients who remained at 45 mg once daily for the duration of treatment, patients whose dose was reduced to 30 mg once daily as the lowest dose and patients whose dose was reduced to 15 mg once daily as the lowest dose. The patients who had available data of plasma steady‐state trough concentrations were included into the analysis. 6
3. RESULTS
3.1. Patient disposition
In total, 81 Japanese patients were randomly assigned to receive either dacomitinib or gefitinib; 40 patients were randomized to the dacomitinib arm and 41 patients were randomized to gefitinib. The disposition of these patients is shown in Figure 1. At the time of data cutoff for the primary analysis (July 29, 2016), study treatment was ongoing in 14 patients in the dacomitinib arm and six patients in the gefitinib arm.
Figure 1.
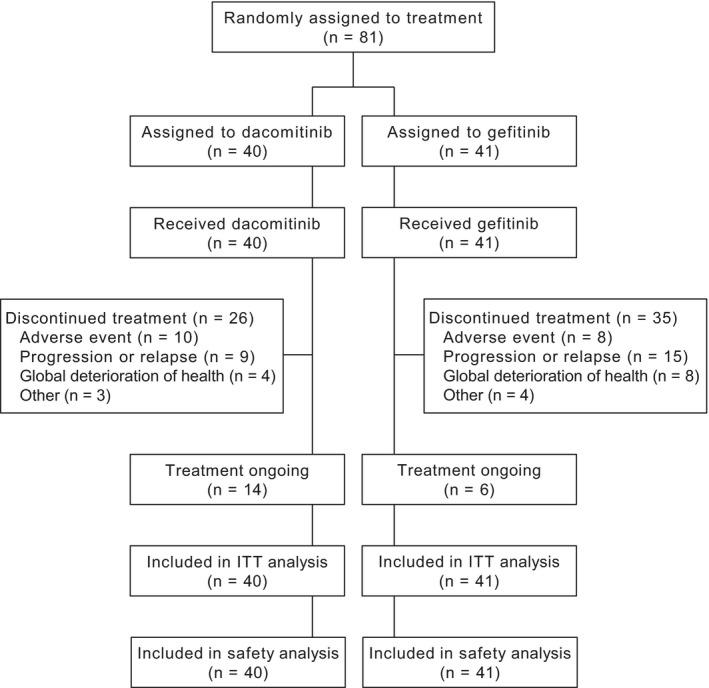
Disposition of Japanese subset in ARCHER 1050 (cutoff date: July 29, 2016). ITT, intention‐to‐treat
Patient demographics and disease characteristics of this Japanese population are shown in Table 1. The median age of patients was 66 y in the dacomitinib arm and 67 y in the gefitinib arm. The patient demographics and disease characteristics were generally balanced, however, a smaller proportion of patients in the dacomitinib arm (52.5%) than in the gefitinib arm (63.4%) were aged ≥65 y. The proportion of female patients in the dacomitinib arm was slightly higher (62.5%) than that in the gefitinib arm (51.2%). More patients in the dacomitinib arm (70.0%) than in the gefitinib arm (51.2%) had ECOG performance status of zero. Overall, the median body weight was 55.1 kg, 53.9 kg in the dacomitinib arm and 55.2 kg in the gefitinib arm. At randomization, approximately two‐thirds of patients in each treatment arm had EGFR gene mutations of exon 19 deletion, and the remainder had exon 21 L858R substitution mutation. The proportion of patients with smoking history was higher in the dacomitinib arm (52.5%) than that in the gefitinib arm (41.4%).
Table 1.
Patient demographics and disease characteristics (Japanese ITT population)
|
Dacomitinib n = 40 |
Gefitinib n = 41 |
|
|---|---|---|
| Age, median (range), y | 66 (39‐82) | 67 (49‐86) |
| 18‐64, n (%) | 19 (47.5) | 15 (36.6) |
| 65‐74 | 13 (32.5) | 18 (43.9) |
| ≥75 | 8 (20.0) | 8 (19.5) |
| Sex, n (%) | ||
| Male | 15 (37.5) | 20 (48.8) |
| Female | 25 (62.5) | 21 (51.2) |
| ECOG performance status, n (%) | ||
| 0 | 28 (70.0) | 21 (51.2) |
| 1 | 12 (30.0) | 20 (48.8) |
| Disease stage, n (%) | ||
| IIIB | 1 (2.5%) | 1 (2.4%) |
| IV | 38 (95%) | 40 (97.6%) |
| Unknown | 1 (2.5%) | 0 |
| Smoking status, n (%) | ||
| Never | 19 (47.5) | 24 (58.5) |
| Former | 20 (50.0) | 16 (39.0) |
| Current | 1 (2.5) | 1 (2.4) |
| EGFR mutation status, n (%) | ||
| Exon 19 deletion | 26 (65.0) | 26 (63.4) |
| Exon 21 L858R mutation | 14 (35.0) | 15 (36.6) |
Cutoff date: July 29, 2016.
Abbreviation: ITT, intention‐to‐treat.
The median duration of treatment and number of cycles administered was greater for dacomitinib than for gefitinib. The median duration of treatment was 74.9 wk (range, 2.1‐161.7 wk) in the dacomitinib arm and 51.7 wk (range, 3.9‐148.3 wk) in the gefitinib arm, and the median number of treatment cycles was 19.5 (range, 1‐41 cycles) for dacomitinib and 13.0 (range, 1‐38 cycles) for gefitinib.
3.2. Efficacy
Consistent with the overall study results, 3 in the Japanese population, there was a clinically meaningful prolongation of PFS as assessed by BIRC with dacomitinib in comparison with that with gefitinib (stratified HR, 0.544 [95% CI, 0.307‐0.961]; 2‐sided P = .0327; median PFS, 18.2 mo [95% CI, 11.0‐31.3 mo] for dacomitinib vs 9.3 mo [95% CI, 7.4‐14.7 mo] for gefitinib; Figure 2). PFS results by investigator assessment, stratified HR, 0.626 (95% CI, 0.367‐1.069); 2‐sided P = .0830; median PFS of 18.3 mo (95% CI, 14.6‐22.1) with dacomitinib and 10.2 mo with gefitinib (95% CI, 7.3‐16.9) were consistent with the BIRC findings.
Figure 2.
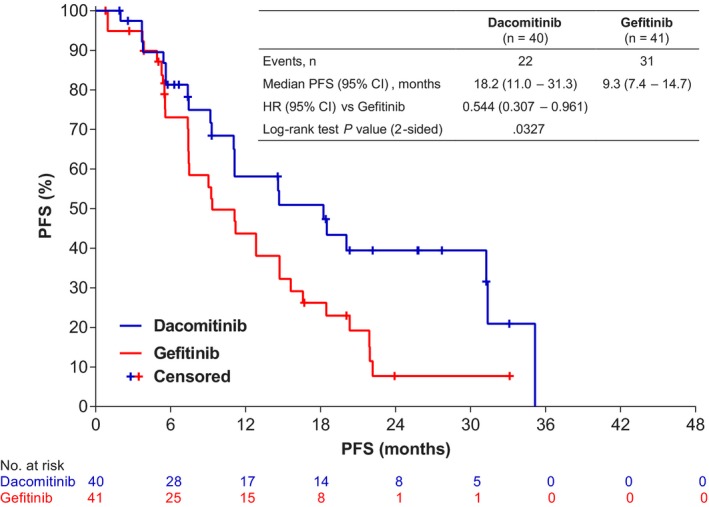
PFS of dacomitinib compared with gefitinib by blinded independent review committee (Japanese ITT population; cutoff date: July 29, 2016). The HR and associated CI was obtained from a stratified Cox Regression and the P‐value was based on a stratified log‐rank test with EGFR mutation status at randomization as the stratification factor. CI, confidence interval; HR, hazard ratio; ITT, intention‐to‐treat; PFS, progression‐free survival
Dacomitinib also improved PFS in patients with both exon 19 deletion (unstratified HR, 0.714 [95% CI, 0.360‐1.413]; 2‐sided P = .3295) and exon 21 L858R substitution mutation (unstratified HR, 0.302 [95% CI, 0.104‐0.875]; 2‐sided P = .0190) compared with gefitinib (Figure S1), although patient numbers were small in each arm.
There was no significant difference in ORR by BIRC between treatment arms (75.0% [95% CI, 58.8%‐87.3%] for dacomitinib vs 75.6% [95% CI, 59.7%‐87.6%] for gefitinib; 2‐sided P = .9493; Table 2). There was one complete response in each treatment arm. However, median DOR was longer in the dacomitinib arm than that in the gefitinib arm (median DOR, 17.5 mo [95% CI, 10.2‐34.3 mo] compared with 8.3 mo [95% CI, 5.6‐12.9 mo]; unstratified HR, 0.435 [95% CI, 0.224‐0.843]; 2‐sided P = .0112). The ORR and DOR results by investigator assessment were consistent with the results by BIRC.
Table 2.
Best overall response (Japanese ITT population) a
|
Dacomitinib n = 40 |
Gefitinib n = 41 |
|
|---|---|---|
| Type of response, n (%) | ||
| Complete response | 1 (2.5) | 1 (2.4) |
| Partial response | 29 (72.5) | 30 (73.2) |
| Stable disease | 9 (22.5) | 5 (12.2) |
| Progressive disease | 1 (2.5) | 2 (4.9) |
| Not evaluable | 0 (0.0) | 3 (7.3) |
| Objective response rate (95% CI) | 75.0 (58.8‐87.3) | 75.6 (59.7‐87.6) |
| 2‐sided P b | 0.9493 | |
| Duration of response in responders, mo | ||
| Median (95% CI) | 17.5 (10.2‐34.3) | 8.3 (5.6‐12.9) |
| HR (95% CI) | 0.435 (0.224‐0.843) | |
| 2‐sided P c | 0.0112 | |
Cutoff date: July 29, 2016.
Abbreviations: CI, confidence interval; HR, hazard ratio; ITT, intention‐to‐treat.
By blinded independent review committee.
P (2‐sided) is from Pearson’s chi‐square test.
P (2‐sided) is from log‐rank test.
The maximum change in tumor size from baseline by best overall response based on BIRC is shown in Figure 3. In both treatment arms, most patients had a reduction in tumor size of >30%, although reductions in tumor size were greater in the dacomitinib arm than reductions in the gefitinib arm.
Figure 3.
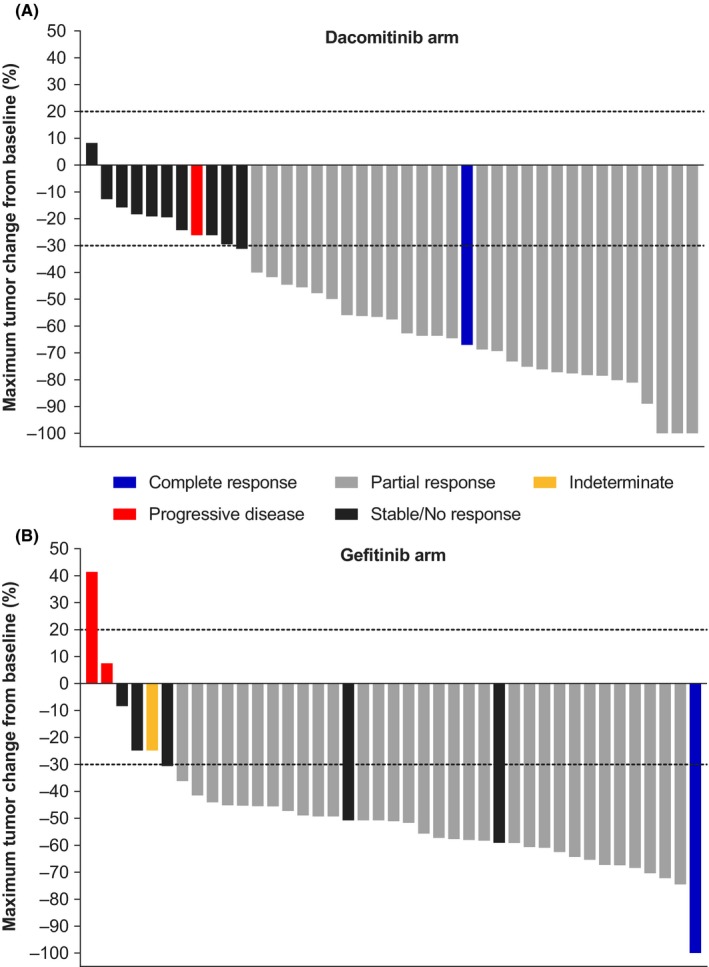
Maximum tumor change from baseline by best overall response based on blinded independent review committee (Japanese ITT population; cutoff date: July 29, 2016). A, dacomitinib arm. B, gefitinib arm. Dashed line at ≥20% increase shows cutoff for progressive disease and at ≥30% reduction shows cutoff for partial response. ITT, intention‐to‐treat. Indeterminate was defined as progression not documented within 12 wk after start of treatment date and where none of the other categories (complete response, partial response, stable disease, or progressive disease) was applicable
The OS data in the Japanese population were immature at the time of the final OS data cutoff (February 17, 2017) and median OS was not reached in either treatment arm. Deaths occurred in 13 patients (32.5%) in the dacomitinib arm and in 13 patients (31.7%) in the gefitinib arm (unstratified HR, 1.003 [95% CI, 0.465‐2.165]; 2‐sided P = .9941). The OS rates at 30 mo were 72.5% (95% CI, 54.0%‐84.5%) in the dacomitinib arm and 69.1% (95% CI, 51.9%‐81.2%) in the gefitinib arm.
The majority of patients who discontinued dacomitinib therapy received subsequent systemic anti‐cancer therapy and their total time on treatment was extended, as shown in Figure 4. The kinds of subsequent systemic anti‐cancer therapy drugs and sequences of administration in the dacomitinib and gefitinib arms varied. Of 31 patients in the dacomitinib arm who received subsequent systemic anti‐cancer therapy, osimertinib was a subsequent systemic anti‐cancer therapy in nine of those patients (Table S1). The proportion of patients who received any subsequent systemic anti‐cancer therapy was 77.5% in the dacomitinib arm and 85.4% in the gefitinib arm; 20.0% of patients receiving dacomitinib and 24.4% of patients receiving gefitinib had > 3 subsequent systemic anti‐cancer therapies (Table 3).
Figure 4.
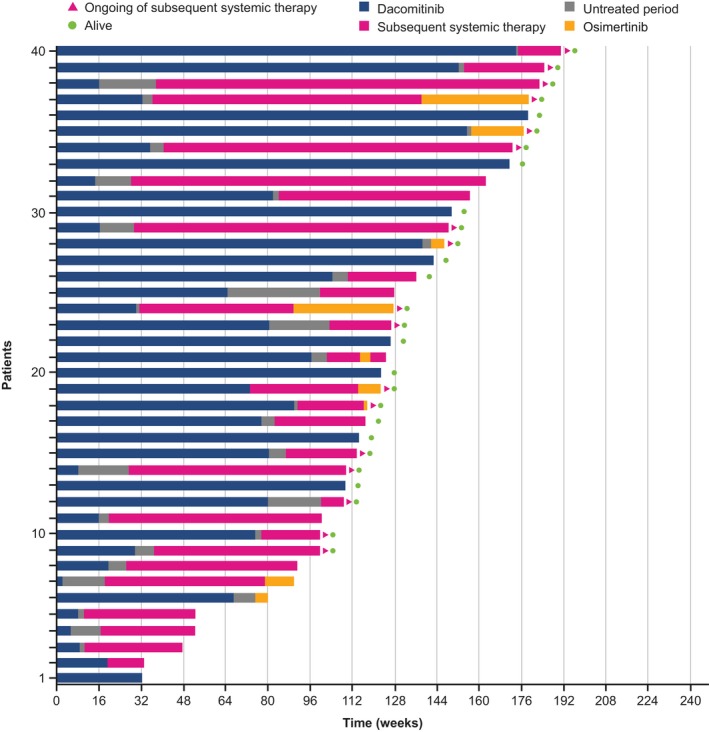
Total duration of treatment by dacomitinib and subsequent systemic anti‐cancer therapy (Japanese safety population; cutoff date: February 17, 2017). The duration of treatment with dacomitinib is shown in blue, with subsequent systemic anti‐cancer therapy (including the interval between subsequent systemic anti‐cancer therapies) in red, with osimertinib in yellow. The period between dacomitinib treatment and subsequent systemic anti‐cancer therapy is shown in gray. Green dots indicate patients who remained alive at the time of data cutoff. Red arrowheads indicate patients who are ongoing the subsequent systemic therapy at the time of data cutoff
Table 3.
Subsequent systemic anti‐cancer therapy (Japanese ITT population)
|
Dacomitinib n = 40 |
Gefitinib n = 41 |
|
|---|---|---|
| Number of SSTs, n (%) | ||
| Any | 31 (77.5) | 35 (85.4) |
| 1 | 8 (20.0) | 13 (31.7) |
| 2 | 9 (22.5) | 5 (12.2) |
| 3 | 6 (15.0) | 7 (17.1) |
| >3 | 8 (20.0) | 10 (24.4) |
| Number of SSTs per patient | ||
| Number of patients with any SST, n | 31 | 35 |
| Mean | 2.7 | 2.8 |
| Median (range) | 2.0 (1‐7) | 2.0 (1‐8) |
| Time from last dose of study treatment to first SST, wk | ||
| Number of patients with any SST | 31 | 35 |
| Mean | 8.4 | 6.8 |
| Median (range) | 5.6 (0.3‐35.4) | 3.3 (0.3‐59.0) |
Cutoff date: February 17, 2017.
Abbreviation: SST, subsequent systemic anti‐cancer therapy.
3.3. Safety
All 81 Japanese patients received study treatment, and all reported AEs of any cause. There were no notable differences in the proportion of patients with serious adverse events (SAEs) of any cause and Grade 3 AEs between the dacomitinib and gefitinib arms, and no new safety signals were identified in Japanese patients compared with the overall study population (Table 4). No Grade 4 or 5 AEs were observed with dacomitinib, whereas three Grade 4 AEs (suicide attempt [n = 1] and hepatic enzyme increased [n = 2]) and one Grade 5 AE (disease progression) occurred with gefitinib.
Table 4.
Adverse events of any cause (Japanese safety population)
|
Dacomitinib n = 40 |
Gefitinib n = 41 |
|
|---|---|---|
| Patients with any adverse event, n (%) | 40 (100) | 41 (100) |
| Serious adverse events, n (%) | 6 (15.0) | 7 (17.1) |
| Grade 3 adverse events, n (%) | 19 (47.5) | 18 (43.9) |
| Grade 4 adverse events, n (%) | 0 (0.0) | 3 (7.3) |
| Grade 5 adverse events, n (%) | 0 (0.0) | 1 (2.4) |
| Adverse event leading to treatment discontinuation, n (%) | 10 (25.0) | 8 (19.5) |
| Adverse event leading to dose reduction, n (%) | 34 (85.0) | 10 (24.4) |
| Adverse event leading to dose interruption, n (%) | 27 (67.5) | 18 (43.9) |
Cutoff date: July 29, 2016.
In the dacomitinib arm, AEs of any cause were primarily gastrointestinal, skin, and nail, respiratory, and/or systemic symptoms. The most commonly reported Grade 3 AEs were dermatitis acneiform (27.5%) and paronychia (22.5%) with dacomitinib and increased alanine aminotransferase (12.2%) with gefitinib (Table 5).
Table 5.
Treatment‐emergent adverse events from any cause occurring in > 15% of patients in either treatment arm (Japanese safety population)
| Adverse event a |
Dacomitinib, n (%) n = 40 |
Gefitinib, n (%) n = 41 |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Any grade | Grade 1 | Grade 2 | Grade 3 | Grade 4 | Grade 5 | Any grade | Grade 1 | Grade 2 | Grade 3 | Grade 4 | Grade 5 | |
| Any adverse event | 40 (100.0) | 0 | 21 (52.5) | 19 (47.5) | 0 | 0 | 41 (100.0) | 2 (4.9) | 20 (48.8) | 15 (36.6) | 3 (7.3) | 1 (2.4) |
| Dermatitis acneiform | 39 (97.5) | 11 (27.5) | 17 (42.5) | 11 (27.5) | 0 | 0 | 31 (75.6) | 19 (46.3) | 12 (29.3) | 0 | 0 | 0 |
| Diarrhea | 39 (97.5) | 15 (37.5) | 19 (47.5) | 5 (12.5) | 0 | 0 | 30 (73.2) | 25 (61.0) | 4 (9.8) | 1 (2.4) | 0 | 0 |
| Paronychia | 38 (95.0) | 2 (5.0) | 27 (67.5) | 9 (22.5) | 0 | 0 | 18 (43.9) | 10 (24.4) | 6 (14.6) | 2 (4.9) | 0 | 0 |
| Stomatitis | 32 (80.0) | 16 (40.0) | 15 (37.5) | 1 (2.5) | 0 | 0 | 16 (39.0) | 11 (26.8) | 5 (12.2) | 0 | 0 | 0 |
| Dry skin | 24 (60.0) | 11 (27.5) | 11 (27.5) | 2 (5.0) | 0 | 0 | 24 (58.5) | 21 (51.2) | 3 (7.3) | 0 | 0 | 0 |
| Pruritus | 17 (42.5) | 10 (25.0) | 7 (17.5) | 0 | 0 | 0 | 9 (22.0) | 6 (14.6) | 2 (4.9) | 1 (2.4) | 0 | 0 |
| Decreased appetite | 15 (37.5) | 9 (22.5) | 5 (12.5) | 1 (2.5) | 0 | 0 | 16 (39.0) | 13 (31.7) | 2 (4.9) | 1 (2.4) | 0 | 0 |
| Dysgeusia | 14 (35.0) | 9 (22.5) | 5 (12.5) | 0 | 0 | 0 | 6 (14.6) | 5 (12.2) | 1 (2.4) | 0 | 0 | 0 |
| Weight decreased | 11 (27.5) | 5 (12.5) | 6 (15.0) | 0 | 0 | 0 | 7 (17.1) | 5 (12.2) | 2 (4.9) | 0 | 0 | 0 |
| AST increased | 10 (25.0) | 10 (25.0) | 0 | 0 | 0 | 0 | 20 (48.8) | 16 (39.0) | 3 (7.3) | 1 (2.4) | 0 | 0 |
| Nausea | 10 (25.0) | 7 (17.5) | 2 (5.0) | 1 (2.5) | 0 | 0 | 9 (22.0) | 8 (19.5) | 1 (2.4) | 0 | 0 | 0 |
| Nasopharyngitis | 9 (22.5) | 7 (17.5) | 2 (5.0) | 0 | 0 | 0 | 8 (19.5) | 7 (17.1) | 1 (2.4) | 0 | 0 | 0 |
| ALT increased | 8 (20.0) | 8 (20.0) | 0 | 0 | 0 | 0 | 21 (51.2) | 12 (29.3) | 4 (9.8) | 5 (12.2) | 0 | 0 |
| Cheilitis | 8 (20.0) | 3 (7.5) | 5 (12.5) | 0 | 0 | 0 | 4 (9.8) | 4 (9.8) | 0 | 0 | 0 | 0 |
| Epistaxis | 8 (20.0) | 8 (20.0) | 0 | 0 | 0 | 0 | 1 (2.4) | 1 (2.4) | 0 | 0 | 0 | 0 |
| Angular cheilitis | 7 (17.5) | 5 (12.5) | 2 (5.0) | 0 | 0 | 0 | 2 (4.9) | 2 (4.9) | 0 | 0 | 0 | 0 |
| Constipation | 7 (17.5) | 5 (12.5) | 2 (5.0) | 0 | 0 | 0 | 5 (12.2) | 3 (7.3) | 2 (4.9) | 0 | 0 | 0 |
| Fatigue | 7 (17.5) | 6 (15.0) | 1 (2.5) | 0 | 0 | 0 | 6 (14.6) | 5 (12.2) | 1 (2.4) | 0 | 0 | 0 |
| Palmar‐plantar erythrodysesthesia syndrome | 7 (17.5) | 3 (7.5) | 4 (10.0) | 0 | 0 | 0 | 3 (7.3) | 2 (4.9) | 1 (2.4) | 0 | 0 | 0 |
| Maculopapular rash | 3 (7.5) | 0 | 1 (2.5) | 2 (5.0) | 0 | 0 | 7 (17.1) | 5 (12.2) | 1 (2.4) | 1 (2.4) | 0 | 0 |
Cutoff date: July 29, 2016.
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase.
Adverse events are listed in descending order of frequency in the dacomitinib arm. Patients are counted only once, at the maximum Common Terminology Criteria for Adverse Events grade reported for each preferred term.
Interstitial lung disease or pneumonitis was reported in two patients (5.0%; 1 Grade 1 AE and one Grade 2 AE) in the dacomitinib arm and one patient (2.4%; 1 Grade 1 AE) in the gefitinib arm. However, the sample size did not permit conclusions to be drawn about the incidence of interstitial lung disease AEs. In the dacomitinib arm, there were no deaths reported as Grade 5 AEs; in the gefitinib arm, death reported as a Grade 5 AE occurred in one patient (2.4%, not related to the study drug). SAEs of any cause occurred in six (15.0%) patients in the dacomitinib arm and seven (17.1%) patients in the gefitinib arm. The most common SAE in the dacomitinib arm was diarrhea occurring in two patients (5%) and the most common SAE in the gefitinib arm was hepatic enzyme increases occurring in two patients (4.9%).
3.4. Dose modification
Dose modification was more frequent with dacomitinib than with gefitinib. The proportion of Japanese patients with AEs leading to dose reductions (85.0% and 24.4%, respectively) or dosing interruptions (67.5% and 43.9%, respectively) was higher with dacomitinib than with gefitinib (Table 4). AEs leading to permanent treatment discontinuation occurred in 25.0% of patients receiving dacomitinib and 19.5% of patients receiving gefitinib (Table 4). The lowest dacomitinib dose on study was 30 mg once daily for 27.5% of patients and 15 mg once daily for 57.5% of patients (Table 6). A higher proportion of patients in the Japanese population in both treatment arms had AEs leading to a dose reduction or dosing interruption than that in the overall population. 3 The most common AEs leading to a dose reduction or dosing interruption were paronychia (55%), dermatitis acneiform (55%), and diarrhea (25%) in the dacomitinib arm and alanine aminotransferase increased (12.2%), abnormal hepatic function (7.3%), dermatitis acneiform (7.3%), and paronychia (7.3%) in the gefitinib arm.
Table 6.
Dose reductions (Japanese safety population)
|
Dacomitinib n = 40 |
Gefitinib n = 41 |
|
|---|---|---|
| No dose reduction, n (%) | 6 (15.0) | 31 (75.6) |
| Lowest dose on study (dacomitinib: reduction to 30 mg/d; gefitinib: reduction to 250 mg every 2 d), n (%) | 11 (27.5) | 6 (14.6) |
| Lowest dose on study (dacomitinib: reduction to 15 mg/d; gefitinib: >1 dosing change a ), n (%) | 23 (57.5) | 4 (9.8) |
| Any dose reduction, n (%) | 34 (85.0) | 10 (24.4) |
Cutoff date: July 29, 2016.
Additional dose reduction(s) for gefitinib followed re‐escalation of dose to 250 mg once daily after dose reduction.
Reasons for permanent treatment discontinuation in the dacomitinib arm among the six patients who did not have a dose reduction were as follows: AEs (n = 3; 1 case for each of interstitial lung disease, dermatitis acneiform, and infection), objective progression or relapse (n = 2), and global deterioration of health (n = 1). Five of these patients received subsequent systemic anti‐cancer therapy.
In general, the incidence and severity of AEs decreased following dose reductions. The incidence of Grade 2 diarrhea and stomatitis decreased in the dacomitinib arm after the first dose reduction while there was an accompanying increase in the incidence of Grade 1 diarrhea and stomatitis, reflecting a reduction in the severity of these AEs after the first dose reduction; for dermatitis acneiform and paronychia, the incidence of Grade 3 AEs was decreased, while the incidence of Grade 2 AEs was increased after the dose reduction (Figure 5). Dacomitinib treatment duration was generally longer in patients who received a dose reduction than that in those who did not receive a dose reduction (Figure 6). The median PFS was 18.5 mo (95% CI: 11.0‐31.3) in those patients who received a dose reduction (n = 34) and 18.2 mo (95% CI: 11.0‐31.3) in all patients (n = 40) (Figure S2). In addition, after the first treatment cycle, geometric mean (geometric coefficient of variation; gCV%) of plasma steady‐state trough concentrations at cycle 2, d 1 was 67 ng/ml (26.9%) for patients without any dose reduction (n = 4), 81 ng/mL (38.8%) for patients with 30 mg once a day as the lowest dose (n = 9), and 78.1 ng/mL (39.0%) for patients with 15 mg once a day as the lowest dose (n = 16), respectively (Figure 7).
Figure 5.
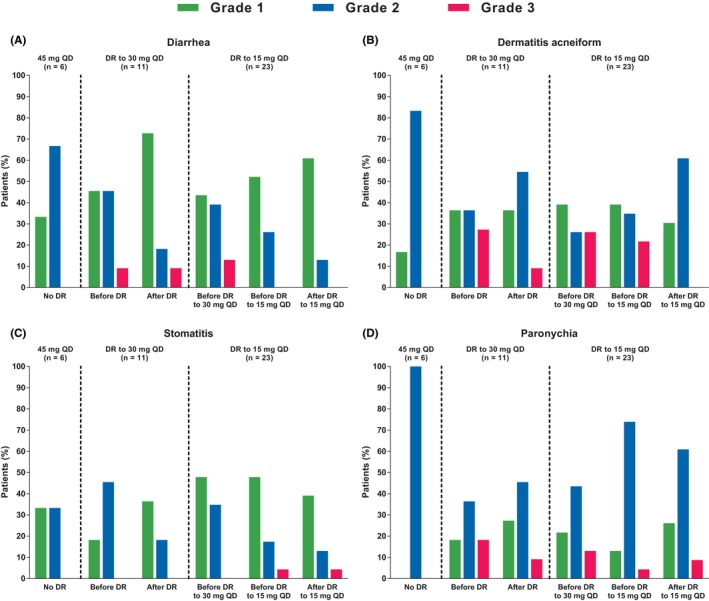
Incidences and severity of adverse events of interest before and after dacomitinib dose reduction (Japanese safety population; cutoff date: July 29, 2016). (A, diarrhea; B, dermatitis acneiform; C, stomatitis; D, paronychia. QD, once daily). The incidences and severity of the adverse events are summarized in patients who did or did not undergo dose reductions because of adverse events. The frequencies of adverse events in the interval before the dose reductions and in the interval after dose reductions are indicated. For diarrhea, dermatitis acneiform, stomatitis, or paronychia, there were no Grade 4 adverse events requiring dose reductions
Figure 6.
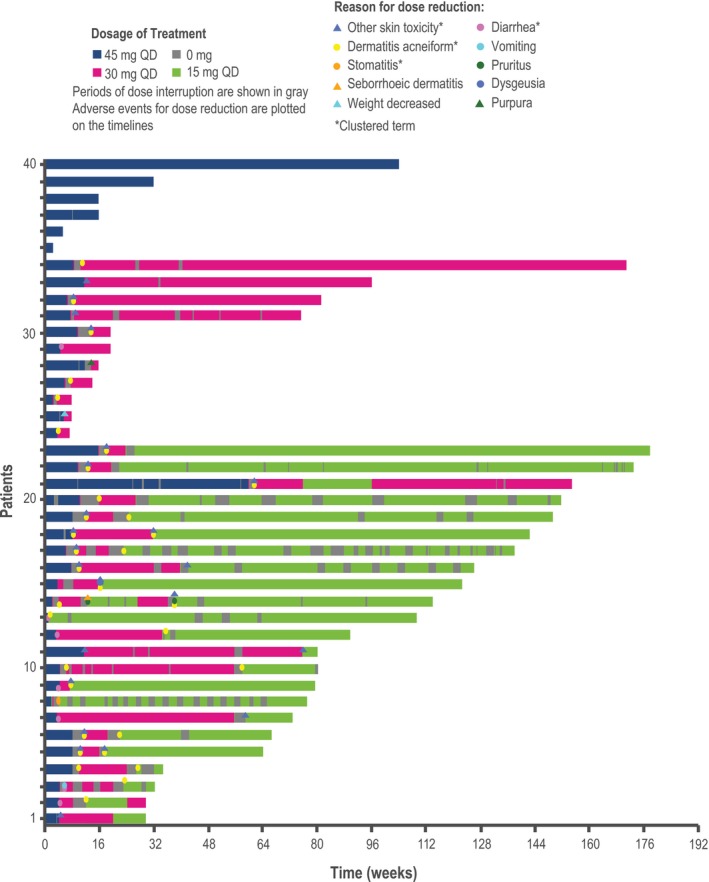
Treatment duration and dacomitinib dose adjustments (Japanese safety population; cutoff date: February 17, 2017). QD, once daily. The duration of treatment with 45 mg QD is shown in blue, with 30 mg QD in red, and with 15 mg QD in green. The period of dose interruption is shown in gray. Adverse events leading to dose reduction are plotted on the timing. The other skin toxicity cluster term includes dry skin, nail disorder, palmar‐plantar erythrodysesthesia syndrome, paronychia, skin fissures, skin ulcer, or xerosis; the diarrhea cluster term includes acute prerenal failure, azotemia, dehydration, diarrhea, blood urea nitrogen/creatinine ratio increased, electrolyte imbalance, hypovolemia, or prerenal failure; the dermatitis acneiform cluster term includes any preferred term within the high level term acnes, drug eruption, rash, rash erythematous, rash generalized, rash maculopapular, or rash pruritic; the stomatitis cluster term includes any preferred term within the high level term stomatitis and ulceration, cheilitis, oral pain, or oropharyngeal discomfort, oropharyngeal pain or mucosal inflammation
Figure 7.
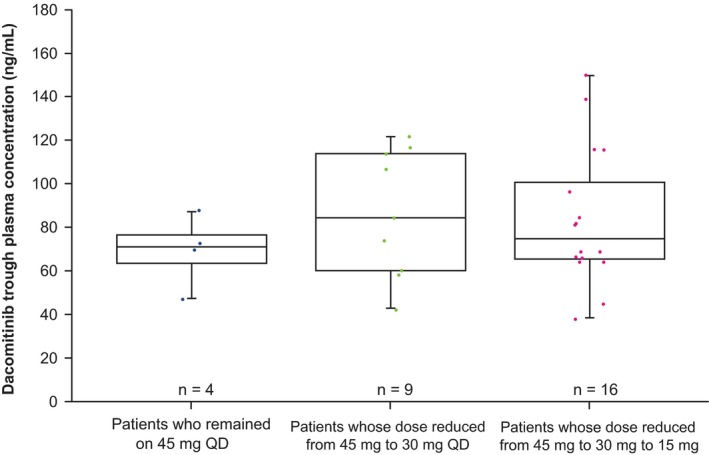
Comparison of dacomitinib initial exposure after the first cycle of treatment by lowest dose on study arm (Japanese safety population; cutoff date: July 26, 2016). Horizontal bars within each box represent the median. The top and bottom borders of each box represent the 75th and 25th percentiles. The error bars above and below each box represent the 90th and 10th percentiles. QD, once daily
4. DISCUSSION
Consistent with the overall study results from ARCHER 1050, 3 dacomitinib improved PFS as assessed by BIRC compared with gefitinib when administered as first‐line treatment in Japanese patients with EGFR‐positive advanced NSCLC. The prolongation in median PFS of 8.9 mo in favor of dacomitinib was clinically meaningful. Dacomitinib also was efficacious in patients with either EGFR exon 19 deletion or exon 21 L858R substitution mutation, with a greater improvement in those with the exon 21 L858R substitution mutation. Although patient numbers were limited in these subgroups, these results are also consistent with what was reported for the overall population. 3 The median OS was immature in both treatment arms as there were a limited number of OS events. Although there was no significant difference in ORR between treatment arms, DOR was longer in the dacomitinib arm than that in the gefitinib arm. The greater clinical benefit observed in the dacomitinib arm compared to the gefitinib may have been related to the fact that gefitinib is a first‐generation EGFR TKI that selectively targets EGFR and is reversible, whereas dacomitinib is a second‐generation EGFR TKI that is irreversible and is an inhibitor of EGFR/HER1, HER2, and HER4. 3 The median PFS by investigator assessment with dacomitinib was 16.6 mo in ARCHER 1050.3 The median PFS by investigator assessment reported with afatinib 7 (another second‐generation irreversible inhibitor of EGFR TKI) was 11.1 mo in patients with NSCLC that had EGFR mutations. In the FLAURA study of first‐line therapy in patients with EGFR‐positive NSCLC, the investigator‐assessed median PFS with osimertinib (a third‐generation EGFR TKI with activity against the T790M resistance mutation) 8 , 9 was 18.9 mo.
The median PFS by blinded independent central review (BICR) with dacomitinib was 18.2 mo in the Japanese population of ARCHER 1050. Nevertheless, comparisons of efficacy across trials are limited by differences in trial methodology and disease characteristics. For example, ARCHER 1050 excluded patients with central nervous system metastases, 4 whereas the LUX‐Lung 3 and FLAURA trials included patients with these metastases. 7 , 9
Although patient numbers were small in each arm, in the Japanese subgroup of ARCHER 1050 dacomitinib improved PFS in patients with either exon 19 deletion and with exon 21 L858R substitution compared with gefitinib. In the afatinib LUX‐Lung 3, including the subgroup analysis of Japanese patients, and LUX‐Lung 6 trials, but not LUX‐Lung 7, PFS improvement was greater in patients with an exon 19 deletion than in those with an exon 21 L858R substitution mutation. 7 , 10 , 11 , 12
In the present analysis, the safety profile of dacomitinib was manageable, and no new safety signals were observed in this population of Japanese patients compared with the overall study population. The most frequently reported AEs related to dacomitinib in the Japanese patients were skin and gastrointestinal disorders, consistent with the known safety profile of dacomitinib and other first‐ and second‐generation EGFR TKIs. 3 , 13 , 14 Most Japanese patients in the dacomitinib arm had dose reductions or dosing interruptions, primarily due to AEs. A higher proportion of patients in the Japanese subgroup in both treatment arms had AEs leading to dose reduction or dosing interruptions than that in the overall population, 3 possibly related to the lower median body weight, 55.1 vs 59.9 kg, respectively, and higher initial dacomitinib exposure in the Japanese patients compared to that of the overall population. 6 At both dacomitinib dose reduction levels (30 mg and 15 mg daily), median weights, as well as upper and lower boundaries for weight, were lower than for patients who remained on the initial 45 mg daily dose of dacomitinib. 6
Following dose reductions, the severity of common AEs associated with dacomitinib (ie, diarrhea, stomatitis, dermatitis acneiform, and paronychia) was reduced in the Japanese population (Figure 5), consistent with the overall population which has been previously reported. 5 , 6 As seen with the overall ARCHER 1050 population, following an initial dacomitinib dose of 45 mg once daily, dose reductions based on the tolerability in each patient can be implemented to decrease severity of toxicities, improve tolerability, and allow for prolongation of the treatment duration, thereby allowing patients an opportunity to receive the greatest benefit from therapy. As similarly described for afatinib (LUX‐Lung 3), these findings suggest that emergence of treatment‐related AEs associated with dacomitinib, and subsequent dose modifications, are potential predictive markers for treatment duration. 7
For the overall population, although PFS results by lowest dose received were not based on criteria present at randomization, median PFS was similar between all dacomitinib‐treated patients and those with dose reduction. The benefit of PFS was maintained in patients who received dose reductions. 6 For the Japanese population, although the sample size was limited, the results were similar to the overall population.
In the overall population, patients who remained at a dacomitinib dose of 45 mg once a day tended to have the lowest initial dacomitinib exposure, compared with those patients who had their dose reduced to 30 mg or 15 mg once a day as the lowest dose. 6 In the Japanese subgroup, after the first cycle of treatment, patients who did not have any dose reductions from the initial dacomitinib dose of 45 mg once a day appeared to have slightly lower initial exposures than patients who had dacomitinib dose reductions, although the patient numbers in each treatment arm were limited (Figure 7). This relationship between dose reduction and drug exposure has been reported with afatinib. 7 The management of AEs by means of dose reduction in the afatinib LUX‐Lung 6 trial allowed more patients to continue receiving afatinib and to optimally benefit from therapy. 12 Furthermore, in a real‐world observational study of afatinib in patients with EGFR‐positive advanced NSCLC including Japanese patients, the severity of AEs associated with afatinib was reduced as a result of dose modifications without loss of effectiveness. 15 By following established AE management protocols for second‐generation EGFR TKIs such as afatinib 16 and dacomitinib,5 treatment may be optimized in clinical practice in Japan.
The overall treatment of patients with EGFR‐positive advanced NSCLC treated with EGFR TKIs may be prolonged by the appropriate use of subsequent systemic anti‐cancer therapies. Most patients in this Japanese population from the ARCHER 1050 trial received ≥1 subsequent systemic anti‐cancer therapy, including chemotherapy and other EGFR TKIs (including osimertinib). The numbers and sequences of different subsequent systemic anti‐cancer therapies administered to Japanese patients in this study varied considerably in both the dacomitinib and gefitinib arms; however, the proportion of osimertinib use as subsequent therapy was similar (9.7% of dacomitinib patients and 11.1% of gefitinib patients) in both arms.
The factor that may have contributed to improvement in OS is the impact of subsequent therapy after discontinuation of study drugs. 4 Patients who received third‐generation EGFR TKIs as subsequent therapy appeared to have longer survival than patients who received chemotherapy. 4 In the case of Japanese patients, the OS is immature and the number of patients who had access to osimertinib was very limited because it was not widely available until 1 y after study enrollment had closed. The importance of sequential therapy is noted in treatment guidelines for EGFR‐positive NSCLC, which recommend osimertinib (if not previously received), chemotherapy, or other targeted agents according to the patient's disease characteristics. 17 The benefit of sequential therapies for EGFR‐positive NSCLC was shown in the multinational, retrospective, observational, GioTag study, in which treatment with a second‐generation EGFR TKI (afatinib) followed by osimertinib prolonged time on treatment and clinical benefit was reported in patients with EGFR‐positive NSCLC, suggesting that use of osimertinib as subsequent systemic anti‐cancer therapy after first‐line second‐generation EGFR TKI was an effective treatment sequence. 18
Finally, consistent with overall study results, dacomitinib improved median PFS and median DOR vs gefitinib in first‐line treatment of Japanese patients. Dacomitinib dose modifications based on tolerability in the Japanese population were more frequent compared with the overall population and are a key management strategy for extending the duration of treatment with dacomitinib. Based on these results, dacomitinib should be considered a first‐line treatment option in Japanese patients with EGFR‐positive NSCLC.
DISCLOSURE
MN has received honoraria from AstraZeneca, Boehringer Ingelheim, Bristol‐Myers Squibb Company, Chugai Pharmaceutical Co., Ltd., Eli Lilly and Company, Merck, Sharpe & Dohme (MSD), Novartis International AG, Pfizer Inc., Taiho Pharmaceutical, and Ono Pharmaceutical Co., Ltd. He has served as a consultant for AstraZeneca, Boehringer Ingelheim, Bristol‐Myers Squibb, Chugai Pharmaceutical, Daiichi Sankyo Healthcare, Eli Lilly, Merck Serono, MSD, Novartis, Ono Pharmaceutical, Pfizer, and Taiho Pharmaceutical, and received research funding from Astellas, AstraZeneca, Bristol‐Myers Squibb, Chugai Pharmaceutical, Eli Lilly, MSD, Novartis, Ono Pharmaceutical, Pfizer, Taiho Pharmaceutical. TK has received honoraria from AstraZeneca, Boehringer Ingelheim, Chugai, Eli Lilly, MSD, and Ono Pharmaceutical and research funding from AbbVie, AstraZeneca, Bristol‐Myers Squibb, Chugai, Eli Lilly, Kyorin Pharmaceutical Co., Ltd., Kyowa Kirin, Co., Ltd., Merck Serono, MSD, Novartis, Ono Pharmaceutical, Pfizer, Regeneron Pharmaceuticals, Inc., and Taiho. SN has received honoraria from AstraZeneca, Chugai, and Pfizer, and research funding from AstraZeneca. NY has received payments for serving as an advisor to Boehringer Ingelheim, Cimic Group Limited, Eisai Co., Ltd., Otsuka Pharmaceutical Co., Ltd., and Takeda Pharmaceutical Company Ltd. He has also received honoraria from AstraZeneca, Bristol‐Myers Squibb, Chugai, Eli Lilly, Ono Pharmaceutical, and Pfizer, and research funding from AbbVie, Astellas, Bayer, Boehringer Ingelheim, Bristol‐Myers Squibb, Chugai, Daiichi Sankyo, Eisai, Janssen Pharma, Kyowa Hakko Kirin Co., Ltd., Merck, MSD, Novartis, Ono Pharmaceutical, Pfizer, Taiho, and Takeda. TT has received honoraria from AstraZeneca KK, Chugai Pharmaceutical Co., Ltd., and Eli Lilly Japan KK, and has received research funding from AstraZeneca KK, Chugai Pharmaceutical Co., Ltd., Eli Lilly Japan K.K., Ono, Pfizer Japan Inc., and MSD KK. NN has received honoraria from AstraZeneca, Chugai, Eli Lilly, Kyowa Hakko Kirin, MSD, Pfizer Inc., and Taiho Pharmaceutical. HK has received lecture fees from AstraZeneca and Pfizer. YF has received research funding from Chugai, Eli Lilly, Pfizer, and Taiho. KW is an employee of Pfizer, Inc. MY is an employee of Pfizer, Inc. MI is an employee of Pfizer, Inc. SW is an employee of Pfizer, Inc. FT has nothing to disclose. KN has received payments for serving as an advisor to Astellas Pharma Inc., Eli Lilly Japan KK, Ono, and Takeda. He has also received honoraria from Astellas Pharma Inc., AstraZeneca KK, AYUMI Pharmaceutical Corporation, Bristol‐Myers Squibb, CareNet, Inc., Chugai Pharmaceutical, Clinical Trial Co., Ltd., Daiichi Sankyo Co., Ltd., Eli Lilly Japan KK, Hisamitsu Pharmaceutical Co., Inc., KYORIN Pharmaceutical Co., Ltd., Medical Review Co., Ltd., MEDICUS SHUPPAN, Publishers Co., Ltd., MSD KK, NANZANDO Co., Ltd., Nichi‐Iko Pharmaceutical Co., Ltd., Nikkei Business Publications, Inc., Nippon Boehringer Ingelheim Co., Ltd., Novartis Pharma KK, Ono, Pfizer Japan Inc., Reno Medical KK/SymBio Pharmaceuticals Ltd., Taiho Pharmaceutical Co., Ltd., Takeda Pharmaceutical Co., Ltd., Thermo Fisher Scientific KK, YODOSHA Co., Ltd. and YOMIURI TELECASTING CORPORATION, and research funding from A2 Healthcare Corp., AbbVie, Astellas Pharma, AstraZeneca KK, Bayer Yakuhin, Ltd., Bristol‐Myers Squibb, Chugai, CMIC Shift Zero KK, Covance Inc., Daiichi Sankyo, Eisai, Eli Lilly Japan KK, EP‐CRSU Co., Ltd., EPS Corporation, EPS International Co., Ltd., GlaxoSmithKline KK, Gritstone Oncology, ICON Japan KK, inVentiv Health Japan, IQVIA Services JAPAN KK, Kissei Pharmaceutical Co., Ltd., Kyowa Hakko Kirin, Linical Co., Ltd., Merck Serono, MSD KK, Nippon Boehringer Ingelheim, Novartis Pharma KK, Ono, Otsuka Pharmaceutical Co., Ltd., PAREXEL International, Pfizer Japan, Quintiles Inc., SymBio Pharmaceuticals Ltd., Taiho, Takeda, and Yakult Honsha Co., Ltd.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
This study was sponsored by SFJ Pharmaceuticals® and Pfizer, Inc. The authors thank Hironori Kikkawa and Koichi Matsumura of Pfizer Japan, Inc., for their contributions to concept preparation, project management, and manuscript preparation. Medical writing support was provided by Michelle Daniels, in Science Communications, Springer Healthcare (Philadelphia, PA, USA) and funded by Pfizer.
Nishio M, Kato T, Niho S, et al. Safety and efficacy of first‐line dacomitinib in Japanese patients with advanced non‐small cell lung cancer. Cancer Sci. 2020;111:1724–1738. 10.1111/cas.14384
ClinicalTrials.gov identifier: NCT01774721
DATA AVAILABILITY STATEMENT
Upon request, and subject to certain criteria, conditions, and exceptions (see https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information), Pfizer will provide access to individual deidentified participant data from Pfizer‐sponsored global interventional clinical studies conducted for medicines, vaccines, and medical devices: (1) for indications that have been approved in the United States and/or European Union or (2) in programs that have been terminated (ie, development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data may be requested from Pfizer trials 24 mo after study completion. The deidentified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.
REFERENCES
- 1. Engelman JA, Zejnullahu K, Gale CM, et al. PF00299804, an irreversible pan‐ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res. 2007;67:11924‐11932. [DOI] [PubMed] [Google Scholar]
- 2. Gonzales AJ, Hook KE, Althaus IW, et al. Antitumor activity and pharmacokinetic properties of PF‐00299804, a second‐generation irreversible pan‐erbB receptor tyrosine kinase inhibitor. Mol Cancer Ther. 2008;7:1880‐1889. [DOI] [PubMed] [Google Scholar]
- 3. Wu YL, Cheng Y, Zhou X, et al. Dacomitinib versus gefitinib as first‐line treatment for patients with EGFR‐mutation‐positive non‐small‐cell lung cancer (ARCHER 1050): a randomised, open‐label, phase 3 trial. Lancet Oncol. 2017;18:1454‐1466. [DOI] [PubMed] [Google Scholar]
- 4. Mok TS, Cheng Y, Zhou X, et al. Improvement in overall survival in a randomized study that compared dacomitinib with gefitinib in patients with advanced non‐small‐cell lung cancer and EGFR‐activating mutations. J Clin Oncol. 2018;36:2244‐2250. [DOI] [PubMed] [Google Scholar]
- 5. Zhou Q, Wu YL, Corral J, et al. Management of common adverse events related to first‐line dacomitinib use in EGFR mutation‐positive non‐small‐cell lung cancer: a pooled safety analysis. Future Oncol. 2019. [DOI] [PubMed] [Google Scholar]
- 6. Corral J, Mok TS, Nakagawa K, et al. Effects of dose modifications on the safety and efficacy of dacomitinib for EGFR mutation‐positive non‐small‐cell lung cancer. Future Oncol. 2019;15:2795‐2805. [DOI] [PubMed] [Google Scholar]
- 7. Sequist LV, Yang JC, Yamamoto N, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013;31:3327‐3334. [DOI] [PubMed] [Google Scholar]
- 8. Cross DA, Ashton SE, Ghiorghiu S, et al. AZD9291, an irreversible EGFR TKI, overcomes T790M‐mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014;4:1046‐1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Soria JC, Ohe Y, Vansteenkiste J, et al. Osimertinib in Untreated EGFR‐Mutated Advanced Non‐Small‐Cell Lung Cancer. N Engl J Med. 2018;378:113‐125. [DOI] [PubMed] [Google Scholar]
- 10. Kato T, Yoshioka H, Okamoto I, et al. Afatinib versus cisplatin plus pemetrexed in Japanese patients with advanced non‐small cell lung cancer harboring activating EGFR mutations: Subgroup analysis of LUX‐Lung 3. Cancer Sci. 2015;106:1202‐1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Park K, Tan EH, O'Byrne K, et al. Afatinib versus gefitinib as first‐line treatment of patients with EGFR mutation‐positive non‐small‐cell lung cancer (LUX‐Lung 7): a phase 2B, open‐label, randomised controlled trial. Lancet Oncol. 2016;17:577‐589. [DOI] [PubMed] [Google Scholar]
- 12. Wu YL, Zhou C, Hu CP, et al. Afatinib versus cisplatin plus gemcitabine for first‐line treatment of Asian patients with advanced non‐small‐cell lung cancer harbouring EGFR mutations (LUX‐Lung 6): an open‐label, randomised phase 3 trial. Lancet Oncol. 2014;15:213‐222. [DOI] [PubMed] [Google Scholar]
- 13. Janne PA, Ou SH, Kim DW, et al. Dacomitinib as first‐line treatment in patients with clinically or molecularly selected advanced non‐small‐cell lung cancer: a multicentre, open‐label, phase 2 trial. Lancet Oncol. 2014;15:1433‐1441. [DOI] [PubMed] [Google Scholar]
- 14. Lacouture ME, Keefe DM, Sonis S, et al. A phase II study (ARCHER 1042) to evaluate prophylactic treatment of dacomitinib‐induced dermatologic and gastrointestinal adverse events in advanced non‐small‐cell lung cancer. Ann Oncol. 2016;27:1712‐1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Halmos B, Tan E‐H, Soo RA, et al. Impact of afatinib dose modification on safety and effectiveness in patients with EGFR mutation‐positive advanced NSCLC: Results from a global real‐world study (RealGiDo). Lung Cancer. 2019;127:103‐111. [DOI] [PubMed] [Google Scholar]
- 16. Edwards RL, Andan C, Lalla RV, Lacouture ME, O'Brien D, Sequist LV. Afatinib therapy: practical management of adverse events with an oral agent for non‐small cell lung cancer treatment. Clin J Oncol Nurs. 2018;22:542‐548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wu Y‐L, Planchard D, Lu S, et al. Pan‐Asian adapted Clinical Practice Guidelines for the management of patients with metastatic non‐small‐cell lung cancer: a CSCO–ESMO initiative endorsed by JSMO, KSMO, MOS. SSO and TOS. Ann Oncol. 2018;30:171‐210. [DOI] [PubMed] [Google Scholar]
- 18. Hochmair MJ, Morabito A, Hao D, et al. Sequential treatment with afatinib and osimertinib in patients with EGFR mutation‐positive non‐small‐cell lung cancer: an observational study. Future Oncology. 2018;14:2861‐2874. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
Upon request, and subject to certain criteria, conditions, and exceptions (see https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information), Pfizer will provide access to individual deidentified participant data from Pfizer‐sponsored global interventional clinical studies conducted for medicines, vaccines, and medical devices: (1) for indications that have been approved in the United States and/or European Union or (2) in programs that have been terminated (ie, development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data may be requested from Pfizer trials 24 mo after study completion. The deidentified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.