

Abstract
The NF‐kappa B (NF‐κB) pathway plays a pivotal role in tumor progression and chemoresistance, and its inhibition has been shown to suppress tumor growth in a variety of preclinical models. Recently, we succeeded in synthesizing a water‐soluble injectable type of curcumin β‐D‐glucuronide (CMG), which is converted into a free‐form of curcumin by β‐glucuronidase in vivo. Herein, we aimed to clarify the efficacy, safety and pharmacokinetics of CMG in a xenograft mouse model. First, we confirmed that the presence of KRAS/TP53 mutations significantly increased the IC50 of oxaliplatin (L‐OHP) and NF‐κB activity in HCT116 cells in vitro. Then, we tested the efficacy of CMG in an HCT116 colon cancer xenograft mice model. CMG demonstrated superior anticancer effects compared to L‐OHP in an L‐OHP‐resistant xenograft model. With regard to safety, significant bodyweight loss, severe myelosuppression and AST/ALT elevation were observed in L‐OHP‐treated mice, whereas none of these toxicity was noted in CMG‐treated mice. The combination of CMG and L‐OHP exhibited additive effects in these xenograft models without increasing toxicity. Pharmacokinetic analysis revealed that high levels of free‐form curcumin were maintained in the tumor tissue after 48 hours following CMG administration, but it was not detected in other major organs, such as the heart, liver and spleen. Immunohistochemistry revealed reduced NF‐κB activity in the tumor tissue extracted from CMG‐treated mice compared with that from control mice. These results indicated that CMG could be a promising anticancer prodrug for treating colon cancer with minimal toxicity.
Keywords: colon cancer, NF‐κB, oxaliplatin, prodrug, β‐glucuronidase
Curcumin β‐D‐glucuronide exhibits anticancer effects in an oxaliplatin‐resistant colon cancer xenograft model with minimal toxicity.
![]()
1. INTRODUCTION
Colorectal cancer (CRC) is the third most common cause of cancer‐related death worldwide. 1 Systemic chemotherapy plays a pivotal role in treating CRC, and oxaliplatin (L‐OHP), a third‐generation diaminocyclohexane platinum compound, has been widely used as a key drug for both resectable and unresectable CRC. 2 , 3 , 4 Clinical data suggest that KRAS mutation, which occurs in approximately 40‐45% of CRC, 5 , 6 hampers the effects of L‐OHP. The survival benefit of CRC patients who received L‐OHP‐based chemotherapy as an adjuvant therapy was significantly poorer in the KRAS mutant group compared to the KRAS wild group. 7 , 8 The mechanisms of L‐OHP resistance are complex and could be affected by a variety of factors. 9 However, NF‐κB activation appears to play a pivotal role in L‐OHP resistance. Porras et al demonstrated that NF‐κB is activated in L‐OHP‐resistant colon cancer cell lines, and inhibition of the NF‐κB pathway using curcumin, a natural polyphenol molecule derived from turmeric (Curcumin longa), recovers L‐OHP‐sensitivity. 10
NF‐κB inhibition is one of the major pharmacologic properties of curcumin and has been demonstrated in a number of preclinical models. 11 , 12 , 13 , 14 For these reasons, curcumin could be a promising anticancer drug targeting the NF‐κB pathway and enhancing L‐OHP sensitivity. However, because curcumin is highly lipophilic, 15 its bioavailability is very poor, and this has been the major obstacle for its clinical application. Several researchers, including us, have tested the efficacy of orally administered curcumin in clinical trials and demonstrated several clinical benefits. 16 , 17 , 18 , 19 , 20 However, the reported curcumin blood levels after oral administration have been too low to derive the maximum anti–tumor effects of curcumin. 16 , 17 , 18 , 19 , 20 To overcome this problem, we recently succeeded in synthesizing a water‐soluble injectable type of curcumin β‐D‐glucuronide (curcumin monoglucuronide, CMG), which was the major metabolite after oral intake of curcumin. We also verified that intravenously administered synthetic CMG is converted into a free‐form of curcumin by β‐glucuronidase (Figure 1) and can achieve more than 1000 times higher plasma curcumin levels compared to orally administered conventional curcumin. 21
Figure 1.
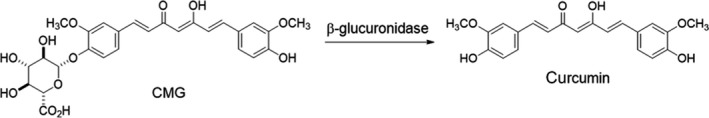
Curcumin β‐D‐glucuronide (CMG) as a water‐soluble prodrug that is converted into free‐form curcumin by β‐glucuronidase
In this study, we aimed to clarify the efficacy, safety and pharmacokinetics of CMG in a xenograft model using HCT116 cell lines with different mutation profiles of KRAS and TP53 genes.
2. MATERIALS AND METHODS
2.1. Chemicals
Oxaliplatin and curcumin were purchased from FUJIFILM Wako Pure Chemical. Three known NF‐κB inhibitors (Bay11‐7082; IκBα kinase inhibitor, TPCA1; IκB kinase inhibitor, MG132; proteasome inhibitor) were purchased from Abcam for Bay11‐7082 and TPCA1, or from Cayman Chemical for MG132, respectively. CMG was synthesized using a previous method (KNC Laboratories). 21
2.2. Cell culture
Three types of HCT116 colon cancer cell lines (KRAS mutant/TP53 wild parental cell line, KRASm/TP53w; KRAS mutant/TP53 homozygous knockout, KRASm/TP53‐; and KRAS mutant heterozygous knockout/TP53 wild, KRASw/TP53w) were purchased from the ATCC. RPMI1640 medium, FBS and penicillin/streptomycin solution were purchased from Sigma Aldrich. HCT116 cells were cultured once every 3 days in RPMI1640 medium with 10% FBS and 1% penicillin/streptomycin solution at 37°C in a 5% CO2 atmosphere.
2.3. MTS assay
HCT116 cells were plated in 48‐well plates at 4 × 105 cells per well. After 24 hours, the cells were treated with L‐OHP (0, 0.5, 1, 2, 4 and 10 μM in DMSO) or curcumin (0, 5, 10, 15, 20 and 25 μM in DMSO). Bay11‐7082 (0 and 5 μM in DMSO), TPCA1 (0 and 20 μM in DMSO) or MG132 (0 and 0.5 μM in DMSO) were also tested with L‐OHP (0 and 4 μM in DMSO) on KRASm/TP53‐ HCT116 cells. After 72 hours, cell viability was analyzed with the 3‐(4,5‐dimethylthiazol‐2‐yl)‐5‐(3‐ carboxymethoxyphenyl)‐2‐(4‐sulfophenyl)‐2H‐tetrazolium, inner salt) (MTS) assay using CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega).
2.4. NF‐κB activity assay
HCT116 cells were plated in 48‐well plates at 5 × 104 cells per well. After 1 day, the cells were cultured in RPMI1640 medium (1% FBS) for 24 hours. They were transfected with pGL4.32 luc2P/NF‐κB‐RE plasmid (Promega) and pSF‐PromMCS‐βGal containing CMV promoter (Boca Scientific). At 3 hours following gene transfection, the transfected cells were treated with various concentrations of curcumin in RPMI1640 medium (1% FBS) for 3 hours and then NF‐κB activity was measured using a PicaGene Luminescence Kit (Toyo Ink); β‐galactosidase activity was measured using a β‐Galactosidase Assay Kit (OZ Biosciences). Gene transfection into cells was performed using Lipofectamine 2000 Transfection Reagent (Invitrogen). The cell lysates were prepared with Reporter Lysis 5 × Buffer (Promega). NF‐κB activity was shown as luciferase/β‐galactosidase.
2.5. Assessment of efficacy and safety of curcumin β‐D‐glucuronide against KRASm/TP53w HCT116 xenograft mice model
All animal experiments were approved by the Jikei University Institutional Animal Care and Use Committee (Permission No. 2018‐004) and Nissei Bilis (Permission No. 1804‐13).
In BALB/c/AnNCr j‐nu/nude mice (7‐8 female mice per group, 6 weeks old, Charles River Laboratories Japan), 4 × 106 KRASm/TP53w HCT116 cells were subcutaneously transplanted. After 9 days following transplantation, they were grouped by stratified randomization with tumor volume (day 0). Saline (10 mL/kg) as a control, 90 mg/kg of CMG (equivalent to 61 mg/kg of curcumin) i.p. or i.v. and 500 mg/kg of synthetic curcumin orally were administered thrice a week for 3 weeks (days 0, 3, 5, 7, 10, 12, 14, 17 and 19). L‐OHP was administered at a dose of 8 mg/kg i.p. twice a week for 3 weeks (days 0, 3, 7, 10, 14 and 17). A combination of 8 mg/kg of L‐OHP (twice a week for 3 weeks) with 90 mg/kg of CMG (thrice a week for 3 weeks) was also tested. Tumor volume was assessed seven times, on days 0, 3, 7, 10, 14, 17 and 21, with the length, height and width of the tumor being measured using a pair of calipers. Tumor volume was calculated as length × width2 × 0.5 (mm3). Body weight was measured on days 0, 7, 14 and 21 using electronic scales (UW4200S, Shimadzu). Blood samples were collected and submitted for hematological and biochemical testing on day 21. Tumor tissue, the heart, the liver and the spleen were extracted from the mice after complete blood removal on day 21.
2.6. Assessment of efficacy and safety of curcumin β‐D‐glucuronide in KRASm/TP53‐ HCT116 xenograft mice model
In BALB/cSlc‐nu/nu mice (7 female mice per group, 6 weeks old, Japan SLC), 4 × 106 KRASm/TP53‐ HCT116 cells were subcutaneously transplanted. After 7 days following transplantation, with saline (10 mL/kg) as a control, 90 mg/kg of CMG were administered i.p. thrice a week for 3 weeks (days 0, 3, 5, 7, 10, 12, 14, 17 and 19). Eight mg/kg of L‐OHP alone or 8 mg/kg of L‐OHP (twice a week for 3 weeks) in combination with 90 mg/kg of CMG (thrice a week for 3 weeks) were administered i.p. twice a week for 3 weeks (days 0, 3, 7, 10, 14 and 17). Tumor volume and body weight were assessed as described above.
2.7. Pharmacokinetic analysis
Free‐form curcumin levels in serum, tumor tissue, heart, liver and spleen obtained from 90 mg/kg of CMG i.v.‐administered groups in the KRASm/TP53w HCT116 xenograft mice model on day 21 were measured.
2.8. Immunohistochemistry
The tumor tissues were extracted from the KRASm/TP53‐ HCT116 xenograft mice model on day 21. The samples were then fixed with formalin and embedded in paraffin. After heat‐induced antigen‐retrieval at 95℃ for 20 minutes, serial sections were incubated overnight at 4℃ with rabbit monoclonal anti–human p65 antibody (1:300 dilution; Abcam) or rabbit monoclonal anti–human Ikappa‐B (IκB) antibody (1:50 dilution; Cell Signaling Technology). Images were captured with NanoZoomer Digital Pathology 2 (Hamamatsu). A quantitative analysis of staining signals from 3 different images was performed using ImageJ software. 22
2.9. Statistical analysis
Data are shown as the mean ± standard deviation (SD). Significant difference was analyzed using Student's t test. P‐values < 0.05 were considered statistically significant.
3. RESULTS
3.1. KRAS mutation/TP53 loss attenuates sensitivity to L‐OHP
First, we tested whether KRAS/TP53 mutation status could affect sensitivity to L‐OHP using three HCT116 cell lines with/without KRAS/TP53 mutation. IC50 values of L‐OHP were 1.0/1.7/4.8 μM for KRASw/TP53w, KRASm/TP53w and KRASm/TP53‐ HCT116 cells, respectively (Figure 2A). In contrast, the IC50 values of curcumin were 19.6/20.0/17.4 μM for KRASw/TP53w, KRASm/TP53w and KRASm/TP53‐ HCT116 cells, respectively (Figure 2B). Thus, KRAS mutation or TP53 loss in HCT116 cells significantly increased the IC50 of L‐OHP but did not attenuate the anti–tumor effects of curcumin in vitro.
Figure 2.
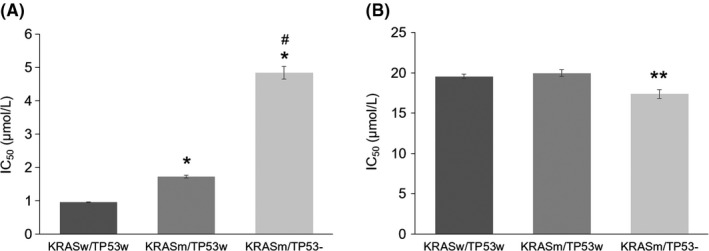
Anticancer effects of (A) L‐OHP and (B) curcumin on each HCT116 cell line in vitro. The IC50 values of L‐OHP and curcumin on each HCT116 cell line (KRASw/TP53w, KRASm/TP53w and KRASm/TP53‐) at 72 hours post−treatment. Data represent means ± SD (n = 3). *P < 0.01, as compared to KRASw/TP53w HCT116; #P < 0.01, as compared to KRASm/TP53w HCT116; **P < 0.01, as compared to KRASw/TP53w HCT116 and KRAS m/TP53w HCT116
3.2. KRAS mutation/TP53 loss increases NF‐κB activity
NF‐κB activity in the three HCT116 cell lines mentioned above was measured using luciferase assay. As shown in Figure 3A, NF‐κB activity was highest in KRASm/TP53‐ cells, followed by KRASm/TP53w and KRASw/TTP53w cells. As expected, curcumin suppressed NF‐κB activity in a dose‐dependent manner in KRASm/TP53‐ and KRASm/TP53w cells (Figure 3B,C).
Figure 3.
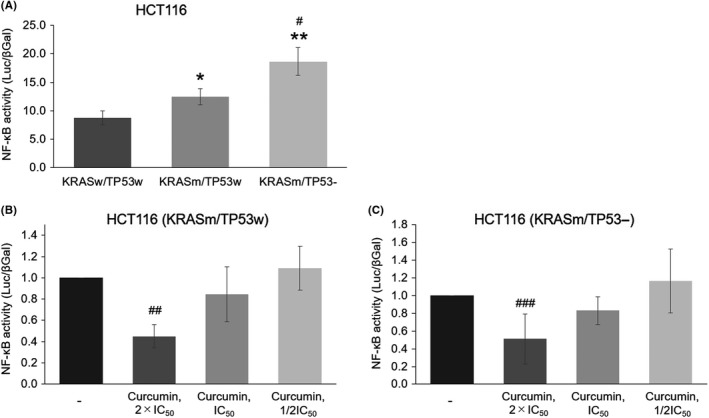
Inhibition of NF‐κB activity by curcumin in vitro. A, Baseline NF‐κB activity on each HCT116 cell line. *P < 0.05 and **P < 0.01, as compared to KRASw/TP53w cells; #P < 0.01, as compared to KRASm/TP53w. B, Dose‐dependent inhibition of NF‐κB activity by curcumin in KRASm/TP53w cells. The IC50 value of curcumin is 20.0 μM. ##P < 0.01, as compared to no treatment. C, Dose‐dependent inhibition of NF‐κB activity by curcumin in KRASm/TP53‐ cells. IC50 value of curcumin is 17.4 μM. ###P < 0.05, as compared to no treatment
3.3. Curcumin β‐D‐glucuronide and L‐OHP exhibit potent anti–tumor effects on KRASm/TP53w HCT116 xenograft model
First, we confirmed that there was no significant difference in the free‐form curcumin levels in the tumor tissue between the CMG i.v. and i.p. groups (Figure S1). Based on these data, the i.p. route was selected for our following in vivo experiments. As shown in Figure 4, the mean tumor volume of the control group increased over time from 28.6 ± 10.3 mm3 on day 0 to 344.0 ± 250.2 mm3 on day 21. The mean tumor volume of each treatment group on day 21 was 310.5 ± 221.6 mm3 in the conventional curcumin group, 213.4 ± 67.8 mm3 in the CMG group, 153.7 ± 112.7 mm3 in the L‐OHP group, and 107.8 ± 79.7 mm3 in the L‐OHP and CMG combination groups. Thus, both CMG and L‐OHP suppressed tumor growth by 38% and 55%, respectively, compared to the control group (Figure 4A). The combination of L‐OHP and CMG showed higher anti–tumor effects compared to CMG or L‐OHP monotherapy, and significantly suppressed tumor growth by 69% compared to the control group (P < 0.05).
Figure 4.
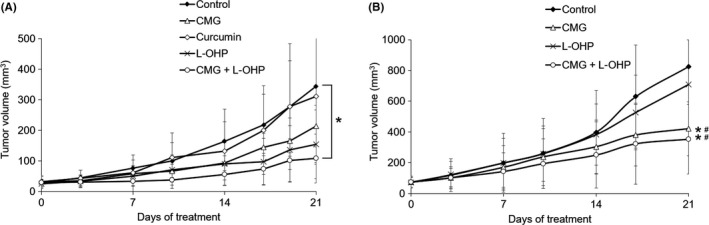
Efficacy of curcumin, curcumin β‐D‐glucuronide (CMG) and L‐OHP on the KRASm/TP53w or KRASm/TP53‐ HCT116 xenograft model. A, Tumor‐bearing mice with KRASm/TP53w HCT116 cells were treated with curcumin, CMG, L‐OHP, and the combination of CMG and L‐OHP, as described in the Materials and Methods. Data represent means ± SD (n = 5‐6). *P < 0.05, as compared to the control group on day 21. B, Tumor‐bearing mice with KRASm/TP53‐ HCT116 cells were treated with CMG, L‐OHP, and the combination of CMG and L‐OHP, as described in the Materials and Methods. Data represent means ± SD (n = 7). *P < 0.05, as compared to the control group on day 21, #P < 0.05, as compared to the L‐OHP group on day 21
3.4. Curcumin β‐D‐glucuronide exhibits superior anti–tumor effects on L‐OHP resistant KRASm/TP53‐ HCT116 xenograft model compared to L‐OHP
Next, we tested the efficacy of CMG using the xenograft model derived from KRASm/TP53‐ HCT116 cells, which demonstrated the highest NF‐κB activity and resistance to L‐OHP in vitro. As shown in Figure 4B, the mean tumor volume of the control group increased over time from 74.7 ± 38.8 mm3 on day 0 to 825.3 ± 404.1 mm3 on day 21. The mean tumor volume of each treatment group on day 21 was 708.0 ± 291.7 mm3 in the L‐OHP group, 420.7 ± 175.5 mm3 in the CMG group, and 353.0 ± 225.1 mm3 in the L‐OHP and CMG combination group. Thus, L‐OHP suppressed tumor growth by just 14%, whereas CMG suppressed tumor growth by 49% and this difference was statistically significant (P < 0.05). The combination of L‐OHP and CMG showed higher anti–tumor effects compared to monotherapy with each agent and significantly suppressed tumor growth by 57% compared to the control group (P < 0.05).
3.5. Safety profile is superior in curcumin β‐D‐glucuronide group compared to L‐OHP group
Compared to the control group, mean body weight was significantly lower on day 21 in L‐OHP‐treated mice (P < 0.01), but CMG or curcumin did not show a negative impact on body weight (Figure 5A). White blood cell count, hemoglobin and platelet count of L‐OHP‐administered groups significantly decreased compared to the control group (P < 0.01, Figure 5B). With regard to liver function, AST and ALT levels were elevated in the L‐OHP monotherapy group compared to the control group, reflecting possible L‐OHP‐induced liver damage, while CMG significantly reduced AST and ALT levels compared to the control group (P < 0.01 and P < 0.05, respectively) (Figure 5C). Interestingly, AST/ALT elevation was reduced in the combination L‐OHP and CMG group compared to the L‐OHP monotherapy group, indicating that CMG has hepato‐protective effects.
Figure 5.
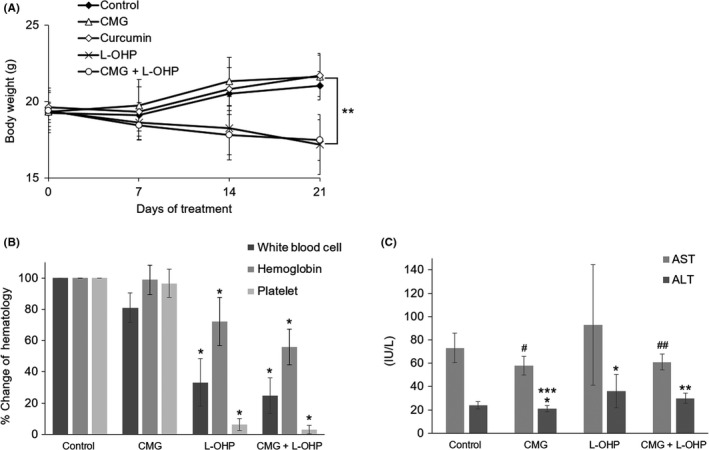
Body weight change, blood cell counts and AST/ALT levels on day 21 in KRASm/TP53w HCT116 xenograft model. A, Body weight change during curcumin, curcumin β‐D‐glucuronide (CMG) and L‐OHP treatment. **P < 0.01, as compared to the control, curcumin and CMG group on day 21. B, Blood cell counts on day 21. *P < 0.01, as compared to the control and CMG groups. C, AST/ALT levels on day 21 *P < 0.05, **P < 0.01, #P < 0.01 and ##P < 0.05, as compared to the control. ***P < 0.05, as compared to the L‐OHP group
3.6. High levels of free‐form curcumin are maintained in tumor tissue
The levels of free‐form curcumin in the serum, tumor tissue and major organs (heart, liver and spleen) on day 21 (after 48 hours following the final CMG administration on day 19) are shown in Figure 6. Free‐form curcumin levels were 15.2 ± 5.8 ng/mL and 136.9 ± 75.4 ng/g in the serum and tumor, respectively. In contrast, no free‐form curcumin was detected in the heart, liver and spleen.
Figure 6.
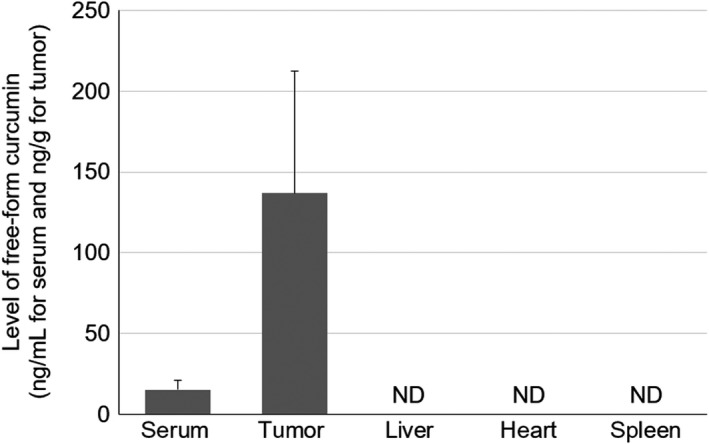
Levels of free‐form curcumin in the serum, tumor tissue and other major organs. Levels of free‐form curcumin were measured after 48 hours following intraperitoneal CMG administration. Data represent means ± SD (n = 6‐8). ND, not detected
3.7. NF‐κB activity is attenuated in curcumin β‐D‐glucuronide‐treated mice
We performed immunohistochemical analysis to test the NF‐κB and IκB expression levels in the tumor samples extracted from the KRASm/TP53‐ HCT116 xenograft mice model on day 21 (Figure 7A‐D). A quantitative analysis showed that NF‐κB staining signals were significantly lower in CMG‐treated mice compared with control mice (P < 0.05, Figure 7F). Furthermore, IκB staining signals were higher in CMG‐treated mice, although this difference did not reach statistical significance (Figure 7E). These data suggest that NF‐κB activity in the tumor tissue was suppressed in the CMG‐treated mice compared with the control mice.
Figure 7.
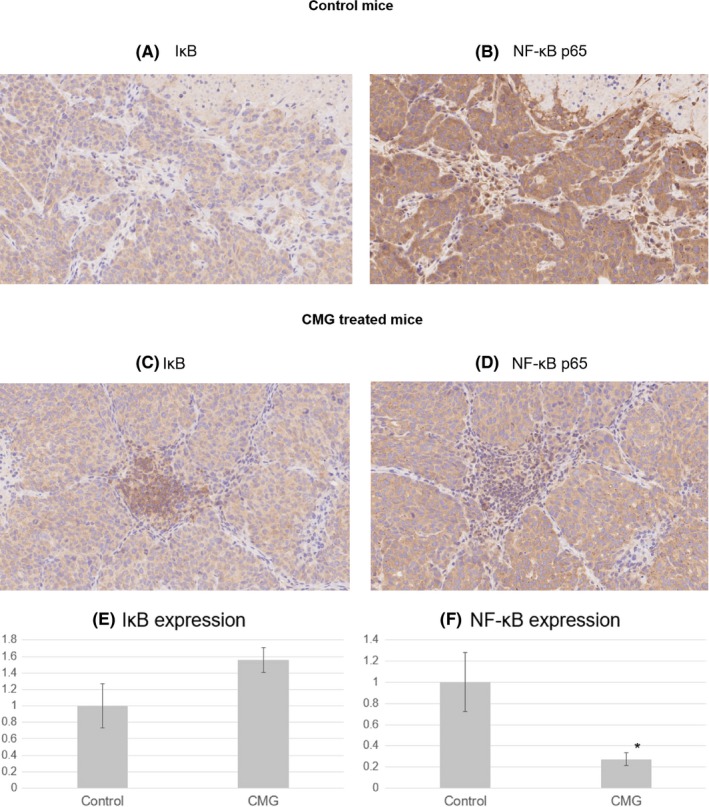
Immunohistochemical analysis of IκB and NF‐κB. Serial sections of the tumors were immunostained for IκB or NF‐κB (p65) antibodies. Representative images are shown (×400). A, IκB staining in control mice. B, NF‐κB staining in control mice. C, IκB staining in CMG‐treated mice. D, NF‐κB staining in CMG‐treated mice. E, A quantitative analysis of IκB staining signals. F, A quantitative analysis of NF‐κB staining signals. Data represent means ± SD (n = 3). *P < 0.05
4. DISCUSSION
In this study, we demonstrated that KRAS activation and TP53 inactivation could confer resistance to L‐OHP in HCT116 cells. The IC50 of L‐OHP for KRASm/TP53‐ HCT116 cells was more than five times higher than that of KRASw/TP53w cells (Figure 2A). NF‐κB activity assessed by luciferase assay was significantly higher in KRASm/TP53‐ HCT116 cells compared to other two cell lines (Figure 3). Because both KRAS and TP53 mutations reportedly upregulate NF‐κB activity and increase chemoresistance in other types of cancer, 23 , 24 , 25 , 26 these results were in line with those of previous reports. Curcumin, which is known to be a potent NF‐κB inhibitor, 11 , 12 suppressed HCT116 cell growth irrespective of the KRAS/TP53 mutation profile in vitro. As expected, NF‐κB activity was downregulated by curcumin in a dose‐dependent manner in vitro. In line with our current observations, Porras et al reported that NF‐κB activity is upregulated in acquired L‐OHP‐resistant CRC cells compared to parental cells and that treatment with curcumin recovered the sensitivity to L‐OHP. 10 Therefore, we tested whether known NF‐κB inhibitors could recover the efficacy of L‐OHP on KRASm/TP53‐ HCT116 cells. As shown in supplementary Figure S2, all of the tested NF‐κB inhibitors (Bay11‐7082, TPCA1 and MG132) suppressed cell growth rates. Among them, MG132 recovered the efficacy of L‐OHP to the same degree as that with curcumin. In contrast, Bay11‐7082 and TPCA1 did not recover the efficacy of L‐OHP. Unknown off‐target effects of these two compounds might have affected complicated results.
Recently, we have succeeded in synthesizing CMG. 21 CMG itself has no anticancer effects in vitro; 27 however, we found that it is converted to a free‐form curcumin by β‐glucuronidase after intravenous injection (Figure 1) and previously showed that it could achieve >1000 times higher plasma curcumin levels compared to those of orally administered curcumin. 21 Herein, we aimed to clarify the efficacy, safety and pharmacokinetics of CMG using KRASm/TP53w and KRASm/TP53‐ HCT116 xenograft models. In the KRASm/TP53w HCT116 xenograft model, CMG and L‐OHP showed higher anti–tumor effects compared to orally administered curcumin, although these differences did not reach statistical significance; however, the combination of L‐OHP and CMG significantly suppressed tumor growth compared to the control group (Figure 4A). In contrast, the KRASm/TP53‐ HCT116 xenograft model showed a higher tumor growth rate than the KRASm/TP53w HCT116 xenograft model, which was consistent with the in vitro data showing that KRASm/TP53‐ cells have the highest IC50 of L‐OHP and NF‐κB activity. The mean tumor volume of the control group on day 21 was 2.4 times larger than that of the KRASm/TP53w HCT116 xenograft model. In the KRASm/TP53‐ HCT116 xenograft model, CMG showed significantly superior anti–tumor effects compared to L‐OHP monotherapy (Figure 4B). In both the KRASm/TP53w and KRASm/TP53‐ HCT116 xenograft models, the combination of L‐OHP with CMG groups showed additive anti–tumor effects without increasing toxicity. These results suggest that CMG monotherapy and combination of L‐OHP with CMG could be promising treatments for L‐OHP‐resistant CRC. Supporting our hypothesis, Howells et al tested the efficacy of conventional curcumin in combination with FOLFOX in a randomized phase IIa trial for patients with advanced CRC and reported that combination of FOLFOX with 2 g daily oral curcumin improved overall survival compared to FOLFOX alone. 28 However, because only 27 patients were enrolled and their baseline characteristics appeared to be different between two arms, these results need to be interpreted with caution and further studies are warranted.
With regard to safety, CMG did not cause any serious toxicity. In L‐OHP‐treated mice, significant body weight loss and severe myelosuppression, and AST/ALT elevation were observed, whereas none of these adverse events were observed in CMG‐treated mice (Figure 5A‐C). Liver damage, referred to as sinusoidal obstruction syndrome, is one of the major adverse events associated with L‐OHP treatment and is observed in up to 50% of patients. 29 , 30 Interestingly, CMG reversed L‐OHP‐induced AST/ALT elevation, indicating that CMG has hepato‐protective effects (Figure 5C). Further studies are warranted to clarify the underlying mechanisms.
β‐glucuronidase, which is a key enzyme that hydrolyzes glucuronide prodrugs, such as CMG, 31 , 32 , 33 could be attributable to the higher deposition of free‐form curcumin in the tumor tissue (Figure 6). β‐glucuronidase is secreted extracellularly in necrotic lesions by monocytes/granulocytes in tumor tissues, while it is localized to lysosomes in normal tissues. 34 In line with our observation, Murdter et al tested the pharmacokinetics of glucuronyl‐spacer‐doxorubicin prodrugs, which are converted to active doxorubicin by β‐glucuronidase, and reported that deposition of doxorubicin in tumor tissue was increased when administering glucuronyl‐spacer‐doxorubicin prodrugs compared to standard doxorubicin in xenograft models. 35
Our current study has the following limitation. We have demonstrated that NF‐κB activity was suppressed by free‐form curcumin in vitro and by CMG in vivo; however, because free‐form curcumin can affect a variety of molecules, we could not exclude the involvement of mechanisms other than NF‐κB inhibition in our experimental models. A recent study has clarified that curcumin perturbs 26S proteasome activity through direct inhibition of dual‐specificity tyrosine phosphorylation‐regulated kinase 2 (DYRK2), leading to tumor regression in the mice model. 36 Because IκB degradation by proteasome is necessary for NF‐κB activation, DYRK2‐dependent proteasome inhibition by free‐form curcumin, which is converted from CMG in vivo, could lead to NF‐κB inactivation. We have confirmed that curcumin suppresses proteasome activity in vitro and are now investigating whether DYRK2 is involved in the underlying mechanisms in our models.
In summary, we have demonstrated that CMG could exert significant anti–tumor effects against the L‐OHP‐resistant CRC xenograft model with minimal toxicity. Pharmacokinetic analysis showed that CMG had a better distribution in the tumor tissue compared to that in blood or other major organs. Taken together, our data indicate that CMG could be a novel anticancer prodrug with a high safety profile, and we are continuing to develop this promising compound in combination with L‐OHP for patients with CRC.
DISCLOSURE
TH is a chief executive officer of Therabiopharma. AI is a senior vice president of Therabiopharma. HU and AK are employees of Therabiopharma. MK and HK own equity and are scientific consultants for Therabiopharma.
Supporting information
Fig S1
Fig S2
ACKNOWLEDGMENTS
This work was supported in part by a Grant‐in Aid for Scientific Research on Innovative Areas “Frontier Research on Chemical Communications” (No. 17H06401 to HK) from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT), Japan.
Ozawa‐Umeta H, Kishimoto A, Imaizumi A, et al. Curcumin β‐D‐glucuronide exhibits anti–tumor effects on oxaliplatin‐resistant colon cancer with less toxicity in vivo. Cancer Sci. 2020;111:1785–1793. 10.1111/cas.14383
Contributor Information
Hideaki Kakeya, Email: scseigyo-hisyo@pharm.kyoto-u.ac.jp.
Masashi Kanai, Email: kanai@kuhp.kyoto-u.ac.jp.
REFERENCES
- 1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394‐424. [DOI] [PubMed] [Google Scholar]
- 2. de Gramont A, Figer A, Seymour M, et al. Leucovorin and fluorouracil with or without oxaliplatin as first‐line treatment in advanced colorectal cancer. J Clin Oncol. 2000;18:2938‐2947. [DOI] [PubMed] [Google Scholar]
- 3. Andre T, Boni C, Mounedji‐Boudiaf L, et al. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N Engl J Med. 2004;350:2343‐2351. [DOI] [PubMed] [Google Scholar]
- 4. Grothey A, Sobrero AF, Shields AF, et al. Duration of adjuvant chemotherapy for stage III colon cancer. N Engl J Med. 2018;378:1177‐1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Van Cutsem E, Kohne CH, Lang I, et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first‐line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J Clin Oncol. 2011;29:2011‐2019. [DOI] [PubMed] [Google Scholar]
- 6. Cancer Genome Atlas N . Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330‐337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lee DW, Kim KJ, Han SW, et al. KRAS mutation is associated with worse prognosis in stage III or high‐risk stage II colon cancer patients treated with adjuvant FOLFOX. Ann Surg Oncol. 2015;22:187‐194. [DOI] [PubMed] [Google Scholar]
- 8. Taieb J, Zaanan A, Le Malicot K, et al. Prognostic effect of BRAF and KRAS mutations in patients with stage III colon cancer treated with leucovorin, fluorouracil, and oxaliplatin with or without cetuximab: A post hoc analysis of the PETACC‐8 trial. JAMA Oncol. 2016;2:643‐653. [DOI] [PubMed] [Google Scholar]
- 9. Martinez‐Balibrea E, Martinez‐Cardus A, Gines A, et al. Tumor‐related molecular mechanisms of oxaliplatin resistance. Mol Cancer Ther. 2015;14:1767‐1776. [DOI] [PubMed] [Google Scholar]
- 10. Ruiz de Porras V, Bystrup S, Martinez‐Cardus A, et al. Curcumin mediates oxaliplatin‐acquired resistance reversion in colorectal cancer cell lines through modulation of CXC‐Chemokine/NF‐kappaB signalling pathway. Sci Rep. 2016;6:24675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Singh S, Aggarwal BB. Activation of transcription factor NF‐kappa B is suppressed by curcumin (diferuloylmethane). [corrected] J Biol Chem. 1995;270:24995‐25000. [DOI] [PubMed] [Google Scholar]
- 12. Bharti AC, Donato N, Singh S, Aggarwal BB. Curcumin (diferuloylmethane) down‐regulates the constitutive activation of nuclear factor‐kappa B and IkappaBalpha kinase in human multiple myeloma cells, leading to suppression of proliferation and induction of apoptosis. Blood. 2003;101:1053‐1062. [DOI] [PubMed] [Google Scholar]
- 13. Kunnumakkara AB, Guha S, Krishnan S, Diagaradjane P, Gelovani J, Aggarwal BB. Curcumin potentiates antitumor activity of gemcitabine in an orthotopic model of pancreatic cancer through suppression of proliferation, angiogenesis, and inhibition of nuclear factor‐kappaB‐regulated gene products. Cancer Res. 2007;67:3853‐3861. [DOI] [PubMed] [Google Scholar]
- 14. Shakibaei M, Mobasheri A, Lueders C, Busch F, Shayan P, Goel A. Curcumin enhances the effect of chemotherapy against colorectal cancer cells by inhibition of NF‐kappaB and Src protein kinase signaling pathways. PLoS ONE. 2013;8:e57218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Solubility THH. chemical and photochemical stability of curcumin in surfactant solutions. Studies of curcumin and curcuminoids, XXVIII. Pharmazie. 2002;57:820‐824. [PubMed] [Google Scholar]
- 16. Cheng AL, Hsu CH, Lin JK, et al. Phase I clinical trial of curcumin, a chemopreventive agent, in patients with high‐risk or pre‐malignant lesions. Anticancer Res. 2001;21:2895‐2900. [PubMed] [Google Scholar]
- 17. Dhillon N, Aggarwal BB, Newman RA, et al. Phase II trial of curcumin in patients with advanced pancreatic cancer. Clin Cancer Res. 2008;14:4491‐4499. [DOI] [PubMed] [Google Scholar]
- 18. Kanai M, Yoshimura K, Asada M, et al. A phase I/II study of gemcitabine‐based chemotherapy plus curcumin for patients with gemcitabine‐resistant pancreatic cancer. Cancer Chemother Pharmacol. 2011;68:157‐164. [DOI] [PubMed] [Google Scholar]
- 19. Kanai M, Otsuka Y, Otsuka K, et al. A phase I study investigating the safety and pharmacokinetics of highly bioavailable curcumin (Theracurmin) in cancer patients. Cancer Chemother Pharmacol. 2013;71:1521‐1530. [DOI] [PubMed] [Google Scholar]
- 20. Sharma RA, Euden SA, Platton SL, et al. Phase I clinical trial of oral curcumin: biomarkers of systemic activity and compliance. Clin Cancer Res. 2004;10:6847‐6854. [DOI] [PubMed] [Google Scholar]
- 21. Ozawa H, Imaizumi A, Sumi Y, et al. Curcumin beta‐D‐glucuronide plays an important role to keep high levels of free‐form curcumin in the blood. Biol Pharm Bull. 2017;40:1515‐1524. [DOI] [PubMed] [Google Scholar]
- 22. Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671‐675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ling J, Kang Y, Zhao R, et al. KrasG12D‐induced IKK2/beta/NF‐kappaB activation by IL‐1alpha and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:105‐120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Meylan E, Dooley AL, Feldser DM, et al. Requirement for NF‐kappaB signalling in a mouse model of lung adenocarcinoma. Nature. 2009;462:104‐107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Weisz L, Damalas A, Liontos M, et al. Mutant p53 enhances nuclear factor kappaB activation by tumor necrosis factor alpha in cancer cells. Cancer Res. 2007;67:2396‐2401. [DOI] [PubMed] [Google Scholar]
- 26. Yang L, Zhou Y, Li Y, et al. Mutations of p53 and KRAS activate NF‐kappaB to promote chemoresistance and tumorigenesis via dysregulation of cell cycle and suppression of apoptosis in lung cancer cells. Cancer Lett. 2015;357:520‐526. [DOI] [PubMed] [Google Scholar]
- 27. Pal A, Sung B, Bhanu Prasad BA, et al. Curcumin glucuronides: assessing the proliferative activity against human cell lines. Bioorg Med Chem. 2014;22:435‐439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Howells LM, Iwuji COO, Irving GRB, et al. Curcumin combined with FOLFOX chemotherapy is safe and tolerable in patients with metastatic colorectal cancer in a randomized phase IIa trial. J Nutr. 2019;149:1133‐1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rubbia‐Brandt L, Audard V, Sartoretti P, et al. Severe hepatic sinusoidal obstruction associated with oxaliplatin‐based chemotherapy in patients with metastatic colorectal cancer. Ann Oncol. 2004;15:460‐466. [DOI] [PubMed] [Google Scholar]
- 30. Tamandl D, Klinger M, Eipeldauer S, et al. Sinusoidal obstruction syndrome impairs long‐term outcome of colorectal liver metastases treated with resection after neoadjuvant chemotherapy. Ann Surg Oncol. 2011;18:421‐430. [DOI] [PubMed] [Google Scholar]
- 31. Sperker B, Backman JT, Kroemer HK. The role of beta‐glucuronidase in drug disposition and drug targeting in humans. Clin Pharmacokinet. 1997;33:18‐31. [DOI] [PubMed] [Google Scholar]
- 32. Tranoy‐Opalinski I, Legigan T, Barat R, et al. β‐Glucuronidase‐responsive prodrugs for selective cancer chemotherapy: an update. Eur J Med Chem. 2014;74:302‐313. [DOI] [PubMed] [Google Scholar]
- 33. Renoux B, Raes F, Legigan T, et al. Targeting the tumour microenvironment with an enzyme‐responsive drug delivery system for the efficient therapy of breast and pancreatic cancers. Chem Sci. 2017;8:3427‐3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bosslet K, Straub R, Blumrich M, et al. Elucidation of the mechanism enabling tumor selective prodrug monotherapy. Cancer Res. 1998;58:1195‐1201. [PubMed] [Google Scholar]
- 35. Murdter TE, Sperker B, Kivisto KT, et al. Enhanced uptake of doxorubicin into bronchial carcinoma: beta‐glucuronidase mediates release of doxorubicin from a glucuronide prodrug (HMR 1826) at the tumor site. Cancer Res. 1997;57:2440‐2445. [PubMed] [Google Scholar]
- 36. Banerjee S, Ji C, Mayfield JE, et al. Ancient drug curcumin impedes 26S proteasome activity by direct inhibition of dual‐specificity tyrosine‐regulated kinase 2. Proc Natl Acad Sci U S A. 2018;115:8155‐8160. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2