

Abstract
The signal transducer and activator of transcription 3 (STAT3) signaling pathway is a key mediator of cancer cell proliferation, survival and invasion. Aberrant STAT3 has been demonstrated in various malignant cancers. YHO‐1701 is a novel quinolinecarboxamide derivative generated from STX‐0119. Here, we examined the effect of YHO‐1701 on STAT3 and evaluated antitumor activity of YHO‐1701 as a single agent and in combination. YHO‐1701 inhibited STAT3‐SH2 binding to phospho‐Tyr peptide selectively and more potently than STX‐0119 in biochemical assays. Molecular docking studies with STAT3 suggested more stable interaction of YHO‐1701 with the SH2 domain. YHO‐1701 exhibited approximately 10‐fold stronger activity than STX‐0119 in abrogating the STAT3 signaling pathway of human oral cancer cell line SAS. YHO‐1701 also blocked multi‐step events by inhibiting STAT3 dimerization and suppressed STAT3 promoter activity. As expected, YHO‐1701 exerted strong antiproliferative activity against human cancer cell lines addicted to STAT3 signaling. Orally administered YHO‐1701 showed statistically significant antitumor effects with long exposure to high levels of YHO‐1701 at tumor sites in SAS xenograft models. Moreover, combination regimen with sorafenib led to significantly stronger antitumor activity. In addition, the suppression level of survivin (a downstream target) was superior for the combination as compared with monotherapy groups within tumor tissues. Thus, YHO‐1701 had a favorable specificity for STAT3 and pharmacokinetics after oral treatment; it also contributed to the enhanced antitumor activity of sorafenib. The evidence presented here provides justification using for this approach in future clinical settings.
Keywords: combination therapy, SH2 domain, signaling pathway, sorafenib, STAT3 inhibitor
YHO‐1701 is a novel small molecule STAT3 inhibitor with quinolinecarboxamide scaffold, which was designed to antagonize SH2‐dependent dimerization of STAT3. This compound inhibited proliferation of human cancer cell lines addicted to STAT3 signaling. YHO‐1701 showed a preferable PK profile with good oral bioavailability in mice and exhibited significant antitumor activity, especially in combination with a multi‐kinase inhibitor sorafenib in a SAS xenograft model.

1. INTRODUCTION
Signal transducer and activator of transcription 3 (STAT3) is a member of the STAT family proteins and has many biological functions. 1 When tyrosine kinases such as JAK are activated by the stimulation of various cytokines and growth factors, STAT3 Tyr705 is phosphorylated. Phospho‐STAT3 forms a dimer through the interaction of the SH2 domain with phospho‐Tyr motif, translocates into the nucleus, and binds to specific DNA sequences to activate the transcription of target genes. 2 Activated STAT3 promotes tumor proliferation and progression by regulating gene expression involved in not only the survival (eg, survivin, c‐Myc and Bcl‐2) and invasion (eg, matrix metalloproteinases) of cancer cells but also angiogenesis (eg, vascular endothelial growth factor) and immune escape (eg, interleukin [IL]‐6, IL‐10 and TGF‐β) in the tumor microenvironment. 3 STAT3 is constitutively activated in hematologic and solid tumors and activation remains transient in normal cells. 4 Thus, STAT3 is considered an attractive therapeutic target for cancers. 5 , 6
Inhibition of upstream tyrosine kinases (eg, JAK and Src) leads to downstream abrogation of STAT3 signaling. It can also block other STAT family members simultaneously, indicating the lack of specificity as a STAT3 inhibitor. 1 Although peptides, small molecules and natural products have been developed as direct STAT3 inhibitors, 2 , 5 , 7 some problems remain to be addressed before beginning clinical trials, including low cellular permeability, insufficient stability in vivo and antitumor activity. 8 , 9 , 10 , 11 Several STAT3 inhibitors have entered clinical development; however, given that drug candidates often drop out due to reasons such as unfavorable side effects and insufficient antitumor effects even at the clinical stage, 12 , 13 , 14 further development of new STAT3 inhibitors is still required. Although STAT3 inhibitors have mainly been developed as monotherapy to date, cancer cells can still utilize alternative salvage pathways such as the RAF/MEK/ERK and PI3K/AKT pathways that are also crucial in proliferation and survival in many cases. 15 Thus, we anticipated that blocking of the STAT3 pathway alone is insufficient to control tumor development and progression, and it is necessary to use STAT3 inhibitors in combination therapy. 6 , 14
We previously developed STX‐0119 as a STAT3 dimerization inhibitor using a virtual screening method, 16 which showed selective inhibition of STAT3 and desirable antitumor effects. 17 , 18 , 19 , 20 , 21 Nonetheless, as the STAT3 inhibitory activity may be improved further, we sought to develop a novel STAT3 inhibitor through structural optimization of STX‐0119 and recently identified a quinolinecarboxamide derivative YHO‐1701, which is predicted to exert greater oral bioavailability (BA) and binding activity to the STAT3‐SH2 domain compared with its lead compound STX‐0119. We characterized the ability of YHO‐1701 to inhibit STAT3 activation and evaluated whether YHO‐1701 could be a promising compound for patients. The results revealed that YHO‐1701 selectively targets STAT3 and exerts antitumor effects in vivo, and the combination of YHO‐1701 and the multi‐kinase inhibitor sorafenib exhibits stronger antitumor effects compared with each monotherapy.
2. MATERIALS AND METHODS
2.1. Human cancer cell lines
The human cancer cell lines are listed in Table S1. All cell lines were maintained according to the supplier’s instructions.
2.2. Reagents
YHO‐1701 was synthesized at the Center for Drug Discovery, University of Shizuoka (Shizuoka, Japan) or Yakult Honsha (Tokyo, Japan). STX‐0119 was synthesized at the Center for Drug Discovery, University of Shizuoka. Stattic was obtained from Cayman chemical. Sorafenib was purchased from Cayman Chemical for in vitro experiments or Bayer AG for in vivo experiments. In in vitro assays, these agents were dissolved in DMSO. In the in vivo antitumor study, YHO‐1701 and sorafenib were suspended in Tween80/propylene glycol/5% glucose (10:5:85) solution or Cremophor/ethanol/water (12.5:12.5:75) solution, respectively. Human IL‐6 was purchased from Cell Signaling Technologies.
2.3. Docking studies
The 3D structure of STAT3 homodimer was obtained from the protein data bank (code 1BG1). Using MOE 2018.01, 22 STAT3 was hydrogenated by the Protonate 3D module. After partial charges were assigned using an all‐atom force field combining Amber10 and extended Hueckel theory (EHT), 23 , 24 hydrogen atoms were minimized, followed by removing the DNA strands. The Alpha Site Finder module was used for definition of a ligand binding site targeting the SH2 domain of STAT3. 25 YHO‐1701 and STX‐0119 generated by the stochastic search method were docked on the respective binding sites, followed by the optimization of the Amber10: EHT force field. In AutoDock Vina 1.1.2, 26 water molecules within the 1BG1 were removed and polar hydrogens were added using AutoDock Tools. Docking runs were carried out using the standard parameters of the program, except for the parameters for setting grid box dimensions and the center. For both of the docking studies, a grid box the size of 25 Å × 25 Å × 25 Å was centered at coordinates 100.45 (x), 75.97 (y) and 68.79 (z) of the PDB structure.
2.4. STAT AlphaScreen
Bead‐based nonradioactive binding assays were performed as described in previous reports. 27 , 28 , 29 Biotinylated STAT were incubated for 90 minutes with YHO‐1701 or STX‐0119 and 5‐carboxyfluorescein (FITC)‐pTyr peptides and mixed with streptavidin‐coated donor beads and anti–FITC acceptor beads simultaneously before detection at 570 nm using EnVison Xcite (PerkinElmer). Phospho‐Tyr (pY) peptide probes used in this study were FITC‐GpYLPQTV (STAT3), FITC‐GpYDKPHVL (STAT1), FITC‐GpYKPFQDL (STAT6), FITC‐GpYLVLDKW (STAT5b) and FITC‐PSpYVNVQN (Grb2).
2.5. Western blot analysis
Cells were treated with compounds for 24 hours. Protein extraction and western blot analysis was performed as previously described. 30 Antibodies for phospho‐ERK1/2 (T202/Y204, CST #4370), ERK1/2 (CST #9102), phospho‐STAT3 (Y705, CST #9131) and STAT3 (CST #4904) were purchased from Cell Signaling Technologies. Antibodies for survivin (R&D AF886) and β‐actin (Sigma A5316) were obtained from R&D Systems and Sigma‐Aldrich, respectively.
2.6. Detection of dimer‐form STAT3
Cells were treated with compounds for 24 hours and then lysed in an ice‐cold isotonic buffer (20 mmol/L Tris [pH 7.0], 150 mmol/L NaCl, 6 mmol/L MgCl2, 0.8 mmol/L PMSF and 20% glycerol). The lysates were separated on native‐PAGE gels and immunoblotted with an anti–STAT3 antibody (CST #4904) as described earlier. 30
2.7. Immunocytochemistry for STAT3 localization
Serum‐starved cells were pretreated with 30 μM YHO‐1701 for 2 hours, followed by stimulation with 50 ng/mL of IL‐6 for 15 minutes. Cells were then fixed in 4% paraformaldehyde, incubated with primary anti–STAT3 antibody and Alexa Fluor 488‐conjugated secondary antibody, and counterstained with Hoechst 33342.
2.8. STAT3 DNA‐binding activity
Cells were treated as described in the previous section. Nuclear extracts were analyzed for STAT3 binding activity using the TransAM STAT3 Kits (Active Motif).
2.9. STAT3 transcription activity
STAT3 reporter HeLa stable cell line for the Luciferase reporter gene assay was obtained from Signosis. Cells were pretreated with YHO‐1701 for 2 hours, and 10 ng/mL oncostatin M was applied and incubated for 4 hours. Luciferase activity was measured using the Steady‐Glo Luciferase Assay System (Promega).
2.10. Caspase 3/7 activity
Cells were treated with YHO‐1701 and/or sorafenib for 24 hours. Caspase 3/7 activity was determined using Caspase‐Glo 3/7 Assays (Promega) as described previously. 31
2.11. Cell viability assay
Cells were grown in 96‐well plates for 24 hours and then treated with various concentrations of compounds. After 48 hours, the extent of cell proliferation was assessed by WST‐8 assay (Kishida chemical) as described previously. 30 Dose‐response curves were plotted to calculate 50% inhibitory concentration (IC50) values. To evaluate the effect of YHO‐1701 in combination with sorafenib, the combination index (CI) values were calculated using the Chou‐Talaly method with CalcuSyn software and plotted as a function of fraction affected (Fa, fraction of cell death induced by drug treatment). CI ≤ 0.9, 0.9‐1.1 and >1.1 represent synergism, additive effect and antagonism, respectively.
2.12. Evaluation of antitumor activity in vivo
Six‐week‐old male BALB/c nude mice and NOD.CB17‐Prkdcscid/J (NOD‐scid) were purchased from Japan SLC and Charles River Laboratories Japan, respectively. Tumor cells were inoculated subcutaneously into the right dorsal region of mice. When tumors became palpable (day 1), the mice were randomly allocated to the following 4 groups (n = 5): a vehicle group; a YHO‐1701 group (50 or 60 mg/kg); a sorafenib group (10 mg/kg); and a combination group. Treatment was started on day 1, and test compounds were administered orally with a 5‐day‐on/2‐day‐off ×4 cycle schedule. Tumor growth was monitored until day 29 by measuring two perpendicular diameters with a digital caliper (Mitutoyo), and tumor volume was calculated using the formula: tumor volume (mm3) = (long axis; mm) × (short axis; mm)2 × 0.5. On day 29, tumors were excised and weighed. Antitumor efficacy was expressed at day 29 as the percent tumor inhibition rate (% IR), calculated using the formula: IR (%) = (1 − mean tumor weight of the treated tumor/mean tumor weight of the vehicle group) × 100. The body weight was monitored twice a week to assess tolerability of this combination therapy. The relative body weight (RBW) at day n was calculated according to the formula: RBW = body weight on day n/body weight on day 1.
In the next series of experiments, SAS human oral tumor‐bearing nude mice were treated with YHO‐1701 and/or sorafenib as described above. On day 12, xenograft tumors were excised and snap‐frozen 6 hours following the last dose. For western blot analysis, tumor lysates were prepared as previously described. 30
2.13. Pharmacokinetic study
SAS xenograft mice were orally administered with YHO‐1701 suspended in 0.5% w/v methylcellulose 400cp solution at 80 mg/kg. Non–tumor‐bearing mice were intravenously administered YHO‐1701 dissolved with a mixture of polyethylene glycol 400, polyvinylpyrrolidone K30 and sterilized water (10.2:1.1:90 [w/w/w]) at 10 mg/kg. Their plasma and tumors were collected (n = 3), and the YHO‐1701 concentration in plasma and tumors was determined with LC‐MS/MS. Mean YHO‐1701 concentrations were calculated and used in pharmacokinetic (PK) analysis performed by Phoenix WinNonlin 6.4 (Pharsight Corporation) in a non–compartment model to calculate the terminal half‐life (T1/2), the area under the plasma and tumor concentration‐time curve from time 0 to infinity (AUC0‐inf), the total clearance (CLtot) and the distribution volume at steady state (Vdss). The oral BA of YHO‐1701 was calculated using the formula: BA (%) = AUC0‐inf of oral administration/Dose of oral administration × Dose of intravenous administration/AUC0‐inf of intravenous administration × 100.
All the animal studies were conducted at the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC)‐accredited facility in accordance with the Guidelines of the Yakult Central Institute and protocols approved by the Animal Experimental Committee of the Yakult Central Institute.
2.14. Statistical analysis
Data are presented as the mean ± standard deviation (SD). Statistical analyses were performed using SAS System Release 8.2 (SAS Preclinical Package, Version. 5.0, SAS Institute Japan). Differences were analyzed using the Tukey test. P‐values <0.05 were considered statistically significant.
3. RESULTS
3.1. Docking approach for the characterization of YHO‐1701 through comparison with STX‐0119
The phenyl group on the quinoline ring of STX‐0119 could effectively fit into the hydrophobic cleft around Ile634 and the side chain of Lys591 (Figure 1A). A hydrogen bond interaction with an amide moiety of STX‐0119 and Gln635 was also observed in accordance with our previous docking model. 16 The docked position of YHO‐1701 changed downward compared with STX‐0119, and, thus, YHO‐1701 could fit into the hydrophobic cleft more deeply and effectively (Figure 1B). Hydrogen bond interaction with the side chain of Ser636 was also observed, enabling YHO‐1701 to bind to STAT3 more strongly. These interactions were reflected in the final docking score and similar binding modes were also calculated from another program AutoDock Vina (Table S2). 32
Figure 1.

YHO‐1701 as a new signal transducer and activator of transcription 3 (STAT3) inhibitor acting on the SH2 domain. A, B, Docking model of STX‐0119 and YHO‐1701 to a cavity on the SH2 domain of STAT3 using MOE 2018.01. Visible by MOE, surface of the electrostatic map. pTyr (pY) peptide AAPpYLKTK, extracted from the crystal structure of STAT3 dimer, is colored green in the line representation. STX‐0119 highlighted in orange (A) and YHO‐1701 in yellow (B) are shown in space filling representation. C, Inhibitory activity of STX‐0119 and YHO‐1701 against STAT3‐SH2 binding to phosphotyrosine peptide in the AlphaScreen‐based assay. D, Expression levels of p‐STAT3 and survivin in SAS cells analyzed using western blotting after STX‐0119 or YHO‐1701 treatment for 24 h
3.2. YHO‐1701 inhibited STAT3 strongly and selectively
YHO‐1701 inhibited the binding of phospho‐Tyr peptide to the STAT3‐SH2 domain in a concentration‐dependent manner, and the inhibitory activity was approximately 10 times more potent than that of STX‐0119 (Figure 1C). We next compared the ability of YHO‐1701 and STX‐0119 to abrogate STAT3 signaling pathway in SAS oral cancer cells, which are known to produce IL‐6. 33 IL‐6 plays an essential role in activating STAT3 signaling in cancer cells. YHO‐1701 again showed 10 times stronger inhibitory effect than STX‐0119 on the expression of phospho‐STAT3 and the STAT3 downstream target, survivin (Figure 1D). Furthermore, to reveal the potential to block other STAT family members, we further examined the selectivity of YHO‐1701 for STAT3. YHO‐1701 exhibited a weaker inhibitory effect for STAT5 and STAT6 than for STAT3, and little or no effect toward STAT1 and Grb2, indicating higher specificity for STAT3 (Figure 2).
Figure 2.

Selective inhibition of signal transducer and activator of transcription 3 (STAT3) by YHO‐1701 in the AlphaScreen assay. Inhibitory activity of YHO‐1701 against SH2‐containing proteins STAT3, STAT1, STAT5, STAT6 and Grb2 in the AlphaScreen‐based assay. STAT were pre–incubated for 90 min with increasing concentration of YHO‐1701. The binding activity of phospho‐Tyr motif is shown as a percentage of DMSO treatment conditions (n = 3)
3.3. YHO‐1701 inhibited STAT3 dimerization and abrogated cellular events thereafter
To understand the effect of the YHO‐1701 against STAT3 function, we first analyzed the ability of YHO‐1701 to inhibit STAT3 dimerization in SAS cells and found that YHO‐1701 clearly inhibited the level of dimer formation of endogenous STAT3 compared with the commercially available STAT3 inhibitor stattic at 10 μM (Figure 3A). We next investigated the nuclear translocation of STAT3. As shown in Figure 3B, its nuclear translocation was only a few percent on serum‐starved SAS cells. However, when IL‐6 was added to the growth medium, this surged up to 80%, and treatment of the cells with YHO‐1701 before IL‐6 stimulation reduced the STAT3 nuclear translocation to approximately 15%. Moreover, after stimulation of SAS cells with IL‐6, binding of STAT3 to a specific DNA sequence increased approximately five times compared with that observed in untreated cells. However, it was clearly blocked by YHO‐1701 in a concentration‐dependent manner (Figure 3C). We also investigated the transcription of STAT3 after YHO‐1701 treatment in HeLa/STAT3‐Luc cells and found that YHO‐1701 inhibited the STAT3 promoter activity with oncostatin M stimulation (Figure 3D). Finally, we assessed the effects of YHO‐1701 on apoptosis. YHO‐1701 increased caspase 3/7 activity (Figure 3E) and sub–G1 cell population in flow cytometeric analysis with propidium iodide staining (data not shown), which are indicators of apoptosis in SAS cells. These results suggest that YHO‐1701 suppressed multi‐step events by inhibiting STAT3 dimerization.
Figure 3.

Inhibition of signal transducer and activator of transcription 3 (STAT3) dimerization by YHO‐1701 and downstream events thereafter. A, SAS cells were treated with YHO‐1701 or stattic for 24 h and native‐PAGE analysis was performed using whole cell lysates to detect STAT3 dimerization. B, SAS cells were serum‐starved overnight and pretreated with YHO‐1701 for 2 h, followed by stimulation with interleukin‐6 for 15 min. Alexa Fluor 488‐labeled anti–mouse IgG is specific for STAT3 (green). Cells were counterstained with Hoechst 33342 to display the nuclei (blue). The fluorescent images were visualized and two images were merged to detect the localization of STAT3 in the nuclei. White arrowheads indicate clear examples of co–localization (bright blue signals). For quantification, cells were counted for nuclear translocation of STAT3 in each field. C, SAS cells were treated as in B. The ability of STAT3 in nuclear extracts to bind its corresponding consensus sequence was quantified. D, HeLa/STAT3‐Luc cells were treated with YHO‐1701 and oncostatin M, and STAT3 transcription activity was determined using the Steady‐Glo Luciferase Assay System. E, SAS cells were treated with YHO‐1701 at indicated doses for 24 h. The caspase 3/7 level was presented as the percentage of control cells. Data are representative of three independent experiments performed in triplicate
3.4. YHO‐1701 exerted antiproliferative activity against human cancer cell lines derived from various tumor types
YHO‐1701 was highly sensitive to most of the hematological cancer cell lines such as K562 (expressing BCR–ABL) and HEL 92.1.7 (expressing JAK2 V617F), where constitutive STAT3 activation is reported (Figure 4). 34 , 35 However, the antiproliferative activity of our compound against solid tumor cells differed greatly, and IC50 values <2 μM were observed in only 11 out of 28 cells tested. Among such sensitive cell lines, IC50 values for SAS, MDA‐MB‐468 and MKN45 were less than 1 μM where persistent STAT3 phosphorylation due to such factors as IL‐6 signaling or epigenetic silencing of suppressor of cytokine signaling‐1 (SOCS‐1) genes were reported, 33 , 36 , 37 and SAS cells, in particular, were the most sensitive to YHO‐1701, with an IC50 of 0.7 μM. In contrast, the insensitive group is comprised of cell lines such as SK‐MEL‐5 and SK‐MEL‐28 (BRAF V600E mutant), NCI‐H2228 (EML4‐ALK mutant) and HCC827 (EGFR exon 19 deficient), implying that their growth was addicted to not only the STAT3 pathway, but also other signaling pathways.
Figure 4.
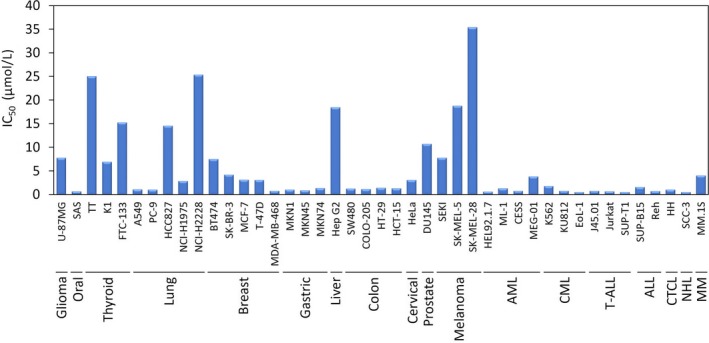
Antiproliferation activity of YHO‐1701 against a human cancer cell line panel. Cellular viability was measured by WST‐8 dye‐based assay after 48 h treatment with YHO‐1701. The percentage of viable cells was calculated, with 100% representing control cells treated with 0.1% DMSO alone, and the IC50 values were calculated by dose‐response curves. Data represent the mean of three independent experiments done in triplicate
3.5. YHO‐1701 synergized with sorafenib in downregulating STAT3/survivin axis and in inducing potent antiproliferative response
In anticipation of clinical development, we verified whether the synergism between YHO‐1701 and a clinically available molecular‐targeted agent was observed. At first, we screened for combination drugs that enhance the activity of YHO‐1701 using chemical libraries containing 270 kinase inhibitors, and selected sorafenib as a partner with a desirable combination effect with YHO‐1701 (data not shown).
For analysis of the drug combination, SAS cells were treated with YHO‐1701 and/or sorafenib at doses indicated in Figure 5A. The resulting dose‐response curves showed almost plateaus at high concentrations of sorafenib, and could not provide a satisfactory fit to calculate CI values. However, two out of the remaining three combinations resulted in synergy CI values of 0.47 and 0.65, showing greater efficiency in inhibiting cell proliferation.
Figure 5.

Synergism between YHO‐1701 and sorafenib in SAS cells. A, The antiproliferative effect of YHO‐1701 and/or sorafenib was evaluated at 48 h of exposure. Dose‐response curves are shown in the upper panel. Combination index values are plotted as a function of fraction affected (Fa) in the lower panel. B, Phosphorylation/activation pattern of relevant molecules on the signal transducer and activator of transcription 3 (STAT3) pathway was analyzed at 24 h of exposure. C, Cells were treated with YHO‐1701 and/or sorafenib at indicated doses for 24 h. The caspase 3/7 level was presented as the percentage of control cells. Representative data are shown of at least three independent experiments
As the blockade of the STAT3/survivin axis is considered one of the major mechanisms of synergism, we next verified whether this combination could synergistically block this pathway. In contrast with the moderate suppression by single‐drug treatments, the combination treatments strongly and reproducibly inhibited phospho‐STAT3 and survivin (Figure 5B), suggesting that YHO‐1701 sensitized the SAS cells to sorafenib through downregulation of phospho‐STAT3 and its downstream target survivin.
In addition, we investigated the ability of this combination treatment to induce apoptosis in SAS cells. As a result, the combination treatment induced caspase 3/7 activity more effectively than treatment with either single agent (Figure 5C), suggesting that augmentation of apoptosis was one of the mechanisms of synergistic cell growth inhibition.
3.6. YHO‐1701 enhanced antitumor activity of sorafenib in an interleukin‐6 secreting SAS xenograft model
We prepared SAS xenograft tumors in nude mice. In this model, human IL‐6 and IL‐6 receptor were detected in plasma and tumor cell lysates, respectively (data not shown). At first, YHO‐1701 and sorafenib displayed significant antitumor responses compared with the vehicle group. The combined administration of YHO‐1701 and sorafenib resulted in significantly greater tumor growth inhibition compared with those of monotherapy groups. The combination regimen led to a 74.6% inhibition of SAS xenografts, which is suggestive of a favorable combination efficacy in vivo (Figure 6A,B). To further assess the systemic toxicity of mice receiving combination therapy, the body weight of mice was monitored. Our data showed that this combination had little or no effect on the mouse body weight throughout the experimental period (Figure 6C). Furthermore, no adverse effects in major organ weights and general conditions, including skin disorders, were induced by this therapeutically effective regimen. These results indicate that this combination is effective for the treatment of SAS tumors and does not lead to systemic toxicity. In addition, this combination effect was not limited to the SAS xenograft model, and a similar tendency was also found in both melanoma‐derived SEKI and cutaneous T‐cell lymphoma (CTCL)‐derived HH models, where constitutive activation of STAT3 was confirmed (Figures S1 and S2). 17 , 38 , 39
Figure 6.

In vivo characterization of orally administered YHO‐1701 in SAS xenograft model. The antitumor efficacy of YHO‐1701 in combination with sorafenib was explored (n = 5). YHO‐1701 (60 mg/kg) and sorafenib (10 mg/kg) were administered either alone or simultaneously. A, During the experimental period, the estimated tumor volume was calculated. B, Antitumor efficacy was expressed at day 29 as the tumor inhibition rate (IR) based on tumor weight. C, During the experimental period, the rate of change in body weight was calculated. Statistical significance was determined by Tukey test. *P < 0.05; ***P < 0.001 versus the vehicle group. †† P < 0.01; †† P < 0.001 versus the YHO‐1701 group. The independent experiments were performed twice with similar results. D, On day 12, xenograft tumors (n = 3) were excised 6 h following the last dose. The expression level of key molecules was detected by western blot
Furthermore, we assessed the key molecules in the STAT3 and RAF/MEK/ERK pathways by using tumor tissues at 6 hours following the final administration. YHO‐1701 monotherapy exhibited no effect on the phospho‐ERK, but moderate suppression on survivin, whereas sorafenib monotherapy clearly reduced both the phospho‐STAT3 and the phospho‐ERK, and moderately suppressed the level of survivin (Figure 6D), almost consistent with the in vitro findings for the phospho‐ERK (Figure 5B and Figure S3). As expected, the suppression level of survivin was greater in the combination therapy as compared to that with either single agent alone, supporting the fact that survivin is a downstream target oncogene in each pathway. Thus, the combination of YHO‐1701 and sorafenib suppresses tumor growth through the downregulation of STAT3 and ERK pathways.
3.7. Pharmacokinetics of YHO‐1701 in SAS xenograft model
YHO‐1701 concentration profiles in plasma and tumors are shown in Figure 7. The plasma concentration of YHO‐1701 after single intravenous administration decreased exponentially with a T1/2 of 2.0 hours. High AUC0‐inf (234 μg·h/mL), low CLtot (0.0428 L h−1 kg−1) and low Vdss (0.150 L/kg) indicated that YHO‐1701 is easily retained in the blood and has low distribution to tissues. In contrast, the level of YHO‐1701 gradually increased up to 8 hours after oral treatment and then gradually decreased, with a T1/2 of 6.6 hours, indicating that the gradual absorption of YHO‐1701 continued for a long time with the oral route of administration. In addition, the BA of YHO‐1701 was 46% following a single oral treatment. The tumor concentration of YHO‐1701 after oral administration also gradually increased up to 8 hours and then gradually decreased with a T1/2 of 9.0 hours. The AUC0‐inf ratio of tumor/plasma was 0.14, indicating low distribution to tumor tissues. Nevertheless, long exposure to high levels of YHO‐1701 was achieved at tumor sites owing to the high plasma concentration levels, allowing YHO‐1701 to show antitumor activity in vivo.
Figure 7.

YHO‐1701 concentration profiles in plasma and tumors after oral and intravenous administration. SAS tumor‐bearing mice were orally administered with YHO‐1701 suspension (80 mg/kg). They were killed at each time point and their plasma and tumors were collected (n = 3). Non–tumor‐bearing mice were intravenously administered with YHO‐1701 solution (10 mg/kg) and were bled over time from the saphenous vein (n = 3)
4. DISCUSSION
STX‐0119 blocks the binding of STAT3‐SH2 and exhibits desirable antitumor effects in xenograft models. 16 , 17 , 18 , 19 , 20 , 21 However, as STAT3 inhibitory activity could possibly be improved upon, we developed a novel STAT3 inhibitor by optimizing STX‐0119. Here, we characterized the ability of YHO‐1701 to inhibit STAT3 activation and evaluated whether YHO‐1701 can exert antiproliferative activities as a single agent and in combination with a clinically available molecular‐targeted agent, sorafenib.
In silico docking analysis clarified that YHO‐1701 forms a more energetically stable structure by utilizing the hydrophobic region of the STAT3‐SH2 domain that STX‐0119 does not utilize (Figure 1A,B and Table S2), which allows YHO‐1701 to bind to STAT3 more strongly. Consistent with this simulation, we found that YHO‐1701 exhibits approximately 10 times stronger activity in terms of inhibiting the binding of phospho‐Tyr peptide to the STAT3‐SH2 domain (Figure 1C), abrogating the STAT3 signaling pathway (Figure 1D) more than STX‐0119, which indicates that YHO‐1701 has the potential to be a more attractive STAT3 inhibitor than its predecessor STX‐0119.
We thought that targeting STAT3 specificity would be difficult because the STAT family has a highly homologous structure. 4 In fact, STAT3 inhibitors such as stattic, C188‐9 and OPB‐31121 have been reported to inhibit STAT1 and STAT5, in addition to STAT3. 9 , 11 , 40 However, we showed that YHO‐1701 has a higher selectivity for STAT3 than for STAT1, 5 and 6 (Figure 2). Given that the blockage of STAT1 may act in reverse to STAT3 inhibition on inducing apoptosis of cancer cells and may also cause side effects such as infections, 1 , 5 this favorable selectivity for STAT3 will make YHO‐1701 an attractive drug candidate for cancer therapy. Furthermore, YHO‐1701 inhibited multi‐step events by abrogating STAT3 dimerization (Figure 3). As assumed, YHO‐1701 exerted strong antiproliferative activity, especially against cancer cell lines where addiction to enhanced STAT3 signaling has been reported, 17 , 34 , 35 supporting the STAT3‐dependent antiproliferative activity of YHO‐1701 (Figure 4). Moreover, YHO‐1701 suppressed the STAT3 promoter activity, suggesting that the blockade of STAT3 signaling is the crucial event behind antiproliferative activity of YHO‐1701. Although YHO‐1701 was designed as the STAT3 dimerization inhibitor by blocking the SH2 domain, it also reduced phospho‐STAT3 in SAS cells in vitro (Figures 1D and 5B). The underlying mechanism responsible for this has not yet been elucidated; however, considering that the STAT3‐SH2 domain is required for both tyrosine phosphorylation and dimerization of STAT3, 34 , 41 , 42 YHO‐1701 may abrogate the interaction of a STAT3 monomer with cytokine receptors.
Unexpectedly, YHO‐1701 did not reduce phospho‐STAT3 in tumor tissues under the current experimental conditions (Figure 6D). However, we believe that inhibition of STAT3 phosphorylation is not essential for YHO‐1701 to block STAT3 signaling. The difference of phospho‐STAT3 levels in vitro and in vivo after YHO‐1701 treatment might be explained, at least in part, by the assay system of cells growing in a monolayer in vitro and the presence of different kinds of cells and concentrations of growth factors and cytokines in vivo. Although YHO‐1701 did not reduce STAT3 phosphorylation, it inhibited STAT3 dimerization (Figure S4) and suppressed the expression of the STAT3 downstream target, survivin, in tumor tissues (Figure 6D). Hence, we believe that YHO‐1701 exerted antitumor effects through the suppression of STAT3 dimerization. Here, the cell line SAS was the most sensitive to our compound among the solid tumor cell lines tested. The IL‐6 signaling is known as a dominant activated signal pathway in the cell line. 33 Consistent with this fact, orally administered YHO‐1701 showed significant antitumor effect and inhibition of survivin in the SAS xenograft model. On the basis of PK profiles, we speculate that long‐term exposure to a high level of YHO‐1701 at the tumor site was an indispensable event behind in vivo antitumor activity. Although YHO‐1701 exerted a higher antiproliferation activity against SAS cells than SEKI cells in vitro (Figure 4), it exhibited similar effects in both xenograft models in vivo (Figure 6 and Figure S1). Therefore, the antitumor effect of YHO‐1701 in vivo may have been achieved not only by directly acting on cancer cells but also by regulating the tumor microenvironment. 1 , 2 , 3
Simultaneous inhibition of MEK and STAT3 pathways shows favorable combined effects 43 ; therefore, we hypothesized that the inhibition of the STAT3 pathway potentiates the antitumor effect achieved by inhibition of the RAF/MEK/ERK pathway. Here, we selected sorafenib, a multi‐kinase inhibitor, as a combination partner, which can suppress ERK by blocking RAF, and additionally inhibit STAT3. 44 , 45 As expected, this combination therapy led to a significantly greater antitumor effect to those of monotherapy groups in xenograft models without increasing systemic toxicity (Figure 6, Figures S1 and S2). Furthermore, the suppression level of survivin (downstream target oncogene in the ERK and STAT3 pathways) in the tumor site appeared superior for the combination than either single agent alone, supporting the importance of dual inhibition of ERK and STAT3 signaling pathways. Considering that the antitumor efficacy of sorafenib as a monotherapy is not sufficient in clinical settings and some combination therapies are being tested, 46 , 47 , 48 this combination may provide a therapeutic advantage for cancer patients. The abovementioned findings show that YHO‐1701 is an attractive STAT3 inhibitor and that motivated us to conduct further testing of drug combinations with other standard‐of‐care agents, and such research is currently being pursued.
In conclusion, we identified a novel quinolinecarboxamide derivative, YHO‐1701, as the orally available inhibitor of STAT3‐SH2. Although it is meaningful to validate our findings using clinical specimens, the preclinical evidence presented here reveals a promising approach for the treatment of cancer patients in the future.
DISCLOSURE
Fukiko Nishisaka, Keisuke Taniguchi, Momomi Tsugane, Genya Hirata, Akimitsu Takagi, Naoyuki Asakawa, Akinobu Kurita, Hiroyuki Takahashi and Yoshiyuki Shishido are employees of Yakult Honsha, Tokyo, Japan.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Dr Takeshi Matsuzaki, Dr Ryuta Yamazaki and Mr Hiroshi Kodaira for their helpful advice. We are also grateful to Mr Toshio Sasai, Mr Hiroaki Konishi, Mr Takahiro Iijima and Mr Satoru Ishii for their discussions and comments. We thank Ms Tomomi Okamura, Ms Kyoko Taguchi and Ms Nao Miyoshi for their excellent technical support. This work was partially supported by a JSPS Grant‐in Aid for Scientific Research (26430166).
Nishisaka F, Taniguchi K, Tsugane M, et al. Antitumor activity of a novel oral signal transducer and activator of transcription 3 inhibitor YHO‐1701. Cancer Sci. 2020;111:1774–1784. 10.1111/cas.14369
Contributor Information
Fukiko Nishisaka, Email: fukiko-nishisaka@yakult.co.jp.
Akira Asai, Email: aasai@u-shizuoka-ken.ac.jp.
REFERENCES
- 1. Shahmarvand N, Nagy A, Shahryan J, Ohgami RS. Mutations in the signal transducer and activator of transcription family of genes in cancer. Cancer Sci. 2018;109:926‐933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Furtek SL, Backos DS, Matheson CJ, Reigan P. Strategies and approaches of targeting STAT3 for cancer treatment. ACS Chem Biol. 2016;11:308‐318. [DOI] [PubMed] [Google Scholar]
- 3. Wang Y, Shen Y, Wang S, Shen Q, Zhou X. The role of STAT3 in leading the crosstalk between human cancers and the immune system. Cancer Lett. 2018;415:117‐128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lavecchia A, Giovanni CD, Novellino E. STAT‐3 inhibitors: state of the art and new horizons for cancer treatment. Curr Med Chem. 2011;18:2359‐2375. [DOI] [PubMed] [Google Scholar]
- 5. Miklossy G, Hilliard TS, Turkson J. Therapeutic modulators of STAT signaling for human diseases. Nat Rev Drug Discov. 2013;12:611‐629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Beebe JD, Liu JY, Zhang JT. Two decades of research in discovery of anticancer drugs targeting STAT3, how close are we? Pharmacol Therapeut. 2018;191:74‐91. [DOI] [PubMed] [Google Scholar]
- 7. Yue P, Turkson J. Targeting STAT3 in cancer: how successful are we? Expert Opin. Investig. Drugs. 2009;18:45‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen H, Yang Z, Ding C, et al. Fragment‐based drug design and identification of HJC0123, a novel orally bioavailable STAT3 inhibitor for cancer therapy. Eur J Med Chem. 2013;62:498‐507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kolosenko I, Yu Y, Busker S, et al. Identification of novel small molecules that inhibit STAT3‐deoendent transcription and function. PLoS ONE. 2017;7:e0178844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhao C, Wang W, Yu W, et al. A novel small molecule STAT3 inhibitor, LY5, inhibits cell viability, colony formation, and migration of colon and liver cancer cells. Oncotarget. 2016;7:12917‐12926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bharadwaj U, Eckols TK, Xu X, et al. Small‐molecule inhibition of STAT3 in radioresistant head and neck squamous cell carscinoma. Oncotarget. 2016;7:26307‐26330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bendell JC, Hong DS, Burris HA, et al. Phase 1, open‐label, dose‐escalation, and pharmacokinetic study of STAT3 inhibitor OPB‐31121 in subjects with advanced solid tumors. Cancer Chemother Pharmacol. 2014;74:125‐130. [DOI] [PubMed] [Google Scholar]
- 13. Ogura M, Uchida T, Terui Y, et al. Phase I study of OPB‐51602, an oral inhibitor of signal transducer and activator of transcription 3, in patients with relapsed/refractory hematological malignancies. Cancer Sci. 2015;106:896‐901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wong ALA, Hirpara JL, Pervaiz S, et al. Do STAT3 inhibitors have potential in the future for cancer therapy? Expert Opin Invest Drugs. 2017;26:883‐887. [DOI] [PubMed] [Google Scholar]
- 15. Li F, Zhao C, Wang L. Molecular‐targeted agents combination therapy for cancer: developments and potentials. Int J Cancer. 2014;134:1257‐1269. [DOI] [PubMed] [Google Scholar]
- 16. Matsuno K, Masuda Y, Uehara Y, et al. Identification of a new series of STAT3 inhibitors by virtual screening. ACS Med Chem Lett. 2010;1:371‐375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ashizawa T, Miyata H, Ishii H, et al. Antitumor activity of a novel small molecule STAT3 inhibitor against a human lymphoma cell line with high STAT3 activation. Int J Oncol. 2011;38:1245‐1252. [DOI] [PubMed] [Google Scholar]
- 18. Ashizawa T, Akiyama Y, Miyata H, et al. Effect of the STAT3 inhibitor STX‐0119 on the proliferation of a temozolomide‐resistant glioblastoma cell line. Int J Oncol. 2014;45:411‐418. [DOI] [PubMed] [Google Scholar]
- 19. Ashizawa T, Miyata H, Iizuka A, et al. Effect of the STAT3 inhibitor STX‐0119 on the proliferation of cancer stemlike cells derived from recurrent glioblastoma. Int J Oncol. 2013;43:219‐227. [DOI] [PubMed] [Google Scholar]
- 20. Akiyama Y, Nonomura C, Ashizawa T, et al. The anti–tumor activity of the STAT3 inhibitor STX‐0119 occurs via promotion of tumor‐infiltrating lymphocyte accumulation in temozolomideresistant glioblastoma cell line. Immunol Lett. 2017;190:20‐25. [DOI] [PubMed] [Google Scholar]
- 21. Miyata H, Ashizawa T, Iizuka A, et al. Combination of a STAT3 inhibitor and an mTOR inhibitor against a temozolomide‐resistant glioblastoma cell line. Cancer Genom Proteom. 2017;14:83‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Molecular Operating Environment (MOE), 2018.01; Chemical Computing Group ULC, 1010 Sherbrooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2018.
- 23. Case DA, Darden TA, Cheatham TE III, et al. AMBER 10. San Francisco, CA: University of California; 2008. [Google Scholar]
- 24. Gerber PR, Müller K. MAB, a generally applicable molecular force field for structure modelling in medicinal chemistry. J Comput Aided Mol Des. 1995;9:251‐268. [DOI] [PubMed] [Google Scholar]
- 25. Soga S, Shirai H, Kobori M, Hirayama N. Use of amino acid composition to predict ligand‐binding site. J. Chem. Inf. Model. 2007;47:400‐406. [DOI] [PubMed] [Google Scholar]
- 26. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem. 2010;31:455‐461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Uehara Y, Mochizuki M, Matsuno K, Haino T, Asai A. Novel high‐throughput screening system for identifying STAT3‐SH2 antagonist. Biochem Biophys Res Commun. 2009;380:627‐631. [DOI] [PubMed] [Google Scholar]
- 28. Takakuma K, Oga N, Uehara Y, Takahashi S, Miyoshi N, Asai A. Novel multiplexed assay for identifying SH2 domain antagonists of STAT family proteins. PLoS ONE. 2013;8:e71646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hato SV, Figdor CG, Takahashi S, et al. Direct inhibition of STAT signaling by platinum drugs contributes to their anti–cancer activity. Oncotarget. 2017;8:54434‐54443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Haibara H, Yamazaki R, Nishiyama Y, et al. YPC‐21661 and YPC‐22026, novel small molecules, inhibit ZNF143 activity in vitro and in vivo . Cancer Sci. 2017;108:1042‐1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Konishi H, Takagi A, Takahashi H, Ishii S, Inutake Y, Matsuzaki T. Acetylation of α‐tubulin by a histone deacetylase inhibitor, resminostat, leads synergistic antitumor effect with docetaxel in non–small cell lung cancer models. Int J Cancer Clin Res. 2017;4:077. [Google Scholar]
- 32. Corbeil CR, Williams CI, Labute P. Variability in docking success rates due to dataset preparation. J Comput Aided Mol Des. 2012;26:775‐786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shinriki S, Jono H, Ota K, et al. Humanized anti–interleukin‐6 receptor antibody suppresses tumor angiogenesis and in vivo growth of human oral squamous cell carcinoma. Clin Cancer Res. 2009;15:5426‐5434. [DOI] [PubMed] [Google Scholar]
- 34. Hayakawa F, Sugimoto K, Harada Y, et al. A novel STAT inhibitor, OPB‐31121, has a significant antitumor effect on leukemia with STAT‐addictive oncokinases. Blood Cancer J. 2013;3:e166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fiskus W, Verstovsek S, Manshouri T, et al. Heat shock protein 90 inhibitor is synergistic with JAK2 inhibitor and overcomes resistance to JAK2‐TKI in human myeloproliferative neoplasm cells. Clin Cancer Res. 2011;17:7347‐7358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Berishaj M, Gao SP, Ahmed S, et al. Stat3 is throsine‐phosphorylated through the interleukin‐6/glycoprotein 130/Janus kinase pathway in breast cancer. Breast Cancer Res. 2007;9:R32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Souma Y, Nishida T, Serada S, et al. Antiproliferative effect of SOCS‐1 throught the suppression of STAT3 and p38 MAPK activation in gastric cancer cells. Int J Cancer. 2012;131:1287‐1296. [DOI] [PubMed] [Google Scholar]
- 38. Tomida M, Saito T. The human hepatocyte growth factor (HGF) gene is transcriptionally activated by leukemia inhibitory factor through the Stat binding element. Oncogene. 2004;23:679‐686. [DOI] [PubMed] [Google Scholar]
- 39. Zhang C, Li B, Zhang X, Hazarika P, Aggarwal BB, Duvic M. Curcumin selectively induces apoptosis in cutaneous T‐cell lymphoma cell lines and patients’ PBMCs: potential role for STAT‐3 and NF‐κB signaling. J Invest Dermatol. 2010;130:2110‐2119. [DOI] [PubMed] [Google Scholar]
- 40. Kim MJ, Nam HG, Kim HP, et al. OPB‐31121, a novel small molecular inhibitor, disrupts the JAK2/STAT3 pathway and exhibits an antitumor activity in gastric cancer cells. Cancer Lett. 2013;335:145‐152. [DOI] [PubMed] [Google Scholar]
- 41. Siddiquee K, Zhang S, Guida WC, et al. Selective chemical probe inhibitor of Stat3, identified through structure‐based virtual screening, induces antitumor activity. Proc Natl Acad Sci USA. 2007;104:7391‐7396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang X, Yue P, Fletcher S, et al. A novel small‐molecule disrupts Stat3 SH2 domain‐phosphotyrosine interactions and Stat3‐dependent tumor processes. Biochem Pharmacol. 2010;79:1398‐1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhao C, Xiao H, Wu X, et al. Rational combination of MEK inhibitor and the STAT3 pathway modulator for the therapy in K‐Ras mutated pancreatic and colon cancer cells. Oncotarget. 2015;6:14472‐14487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wilhelm SM, Carter C, Tang L, et al. BAY 43–9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinase involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099‐7109. [DOI] [PubMed] [Google Scholar]
- 45. Chao TI, Tai WT, Hung MH, et al. A combination of sorafenib and SC‐43 is a synergistic SHP‐1 agonist duo to advance hepatocellular carcinoma therapy. Cancer Lett. 2016;371:205‐213. [DOI] [PubMed] [Google Scholar]
- 46. Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378‐390. [DOI] [PubMed] [Google Scholar]
- 47. Liangtao Y, Mayerle J, Ziesch A, Reiter FP, Gerbes AL, DeToni EN. The PI3K inhibitor copanlisib synergizes with sorafenib to induce cell death in hepatocellular carcinoma. Cell Death Discov. 2019;5:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Singh AR, Joshi S, Burgoyre AM, et al. Single agent and synergistic activity of the "first‐in‐class" dual PI3K/BRD4 inhibitor SF1126 with Sorafenib in hepatocellular carcinoma. Mol Cancer Ther. 2016;15:2553‐2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material