

Abstract
The tumor suppressor protein p53 is often called “the genome guardian” and controls the cell cycle and the integrity of DNA, as well as other important cellular functions. Its main function is to trigger the process of apoptosis in tumor cells, and approximately 50% of all cancers are related to the inactivation of the p53 protein through mutations in the TP53 gene. Due to the association of mutant p53 with cancer therapy resistance, different forms of restoration of p53 have been subject of intense research in recent years. In this sense, this review focus on the main currently adopted approaches for activation and reactivation of p53 tumor suppressor function, focusing on the synthetic approaches that are involved in the development and preparation of such small molecules.
Keywords: tumor suppressor p53 protein, wild-type p53, mutant p53, MDM2
1. Introduction
Cancer is a multifactorial disease characterized by the dysregulated growth of cells; tumor cells replicate and spread in a process called metastasis, which is the cause of most of the deaths related to such disease. In this context, the presence of tumor suppressor genes, which act as regulators of cell proliferation, are essential in the control of cancer progression. One of these genes is TP53, which is activated when DNA damage takes place, promoting the transcription of the p53 protein (wild-type—WTp53), which is responsible for arresting the cell cycle, among other functions [1,2,3].
Since its discovery in 1979, the p53 tumor suppressor protein has been the subject of intense research and is considered an important and challenging target in cancer therapy. The increase in the level of wild-type p53 in the cell nucleus promotes cell repair, senescence and/or apoptosis through different mechanisms [4,5,6,7,8]. However, in approximately 50% of all cancers, there is an inactivation of the p53 function through somatic mutations in TP53 [9,10].
Due to the association of mutant p53 with cancer therapy resistance, different forms of reactivation of p53 have been subject of intense research in recent years. Some studies focus on the insertion of second-site suppressor mutations, which partially restore specific DNA binding and/or stabilize the folding structure of the protein. Others involve the use of synthetic peptides derived from the p53 C-terminus with the aim of restoring the specific DNA binding and transactivation function to mutant p53 and inducing p53-dependent apoptosis in tumor cells [9,10]. It is known that p53 controls the cell cycle and the integrity of DNA, among other important cellular functions, but in the cancer context, its main function is to trigger apoptosis in tumor cells [11]. Similar to the strategy of rescuing the wild-type function of mutant p53, the idea of trapping on-pathway oligomers from the p53 aggregation reaction seems reasonable to avoid the building of the complex assemblies observed in cancer cells [11]. A drawback still related to this approach is the sparse information regarding p53 oligomers as the building blocks of these cancer assemblages. Answering whether heterogeneous oligomers are on- or off-pathway intermediates and investigating those involved in cancer through different methods, such as cryo-EM and super resolution microscopy, will definitely point oncogenic mutants of p53 as potential targets for anticancer therapy [11].
Another promising strategy for the restoration of wild-type p53 function in tumors that have lost p53 tumor suppressor activity is the identification of natural or synthetic small organic molecules that can reactivate mutant p53 to its wild-type version. As a result, a large number of research groups have been focusing in the last few years on the synthesis of small molecules with p53-related activity and important advances have been accomplished thus far [12,13,14,15,16,17,18].
As mentioned before, TP53 is mutated in around 50% of human cancers. Other cases include different reasons for p53 activity impairment, such as the oncogene MDM2 overexpression, which leads to WTp53 inactivation. The MDM2 (Murine Double Minute 2 or HDM2) and MDMX (Murine Double Minute X, HDMX or MDM4) are important regulators of p53, but their overexpression may lead to the inactivation of p53. In this sense, numerous tumor suppressor approaches are related to p53-MDM2/MDMX, namely preventing the formation of p53-MDM2 complexes, preventing the p53 protein ubiquitination degradation and modifying p53 transcriptional active region to stabilize the p53 protein [19,20,21,22]. It is important to mention that comprehensive literature reviews on the rationale of using small molecules and peptide that function as MDMX inhibitors or as dual MDM2/MDMX inhibitors have been recently published [23,24].
Other approaches for the reactivation of p53 have also been successful, for instance the inhibition of the nuclear export factor CRM-1 [25]. Another viable approach for the activation of p53 is the inhibition of casein kinase 1A1 (CKIa) [26,27], as well as of the sirtuins SirT1 and SirT2, which are implicated in a series of essential cellular processes and disease conditions [28]. Additionally, considering that a significant fraction of mutant p53 is found in a self-aggregated amyloid-like state in cancer cells [29,30,31,32,33], and these destabilized oncogenic mutants in the aggregated state are formidable targets for stabilization by drugs [11,31,32,33]. With this aim in mind, several strategies were designed in order to recapitulate p53 function in cancer. Overall, they include the use of small molecule or peptide stabilizers of misfolded p53, zinc administration, gene therapy, metallochaperones, alkylating and DNA intercalators, [34] blockage of p53-MDM2 interaction, impaired reactive oxygen species (ROS) detoxification and other p53 regulators [17,18,33,35].
In this sense, this review will focus on the recent currently adopted methods for the activation and reactivation of the p53 tumor suppressor function with an emphasis on the synthetic approaches to obtain the small molecules used as reactivators. Although most of the mechanisms involved in such processes are still not completely known, this review will approach the reactivation of p53 via two main pathways: (1) The reactivation of mutant p53 via the covalent reaction with thiol in the cysteine residues and other approaches and (2) reactivation of wild-type p53 via the antagonists of the p53-MDM2/MDMX interactions. Lastly, additional approaches such as CRM1, CKIa, and SirT1/SirT2 inhibition will be discussed.
2. Targeting Mutant p53: Restoration of the p53 Function
2.1. Quinuclidinone Derivatives
It is already known that mutations in the central core domain of p53 can lead to the formation of a pocket in L1/S3, affecting specifically cysteine (Cys) residues Cys124, Cys135, Cys141, Cys182, and Cys277. In this sense, several small molecules can interact with this pocket and cause small conformational changes that can reactivate mutant p53, while others such as Michael acceptors may react via alkylation, causing permanent and significant changes to the structure of the protein, also restoring its DNA binding activity [36,37,38]. Although this alkylation approach can reportedly reactivate p53 function, the mechanism of this process is still not completely known. However, it has been reported that cysteines 124 and 277 are the main target for R175H mutant reactivation [39].
In view of these facts, the synthesis of low molecular weight molecules that can promote the alkylation of p53 and restore its activity has become particularly popular, especially since the 2000s. One of the pivotal studies in this field reported the discovery of PRIMA-1 (APR-017) through the screening of a database at the Developmental Therapeutics Program of the National Cancer Institute (NCI). Although other mechanisms may also take place, both PRIMA-1 and its methylated version (PRIMA-1MET, also known as APR-246) are both converted to methylene quinuclidinone (MQ), which is a Michael acceptor that reacts covalently with cysteine residues in p53 (WT or mutant), leading to the reactivation of mutant p53 into a WTp53-like conformation, to induce cell apoptosis [36,39,40,41,42]. Indeed, a phase Ib/II clinical trial is currently ongoing to evaluate the side effects and best dose of PRIMA-1 analog APR-246 when given along with azacitidine in treating patients with mutated TP53 mutant myeloid cancers, as well as a phase III study to compare the rate of complete response (CR) and duration of CR in patients with TP53-mutated Myelodysplastic Syndromes [43]. Recently, our group demonstrated that PRIMA-1 acts by inhibiting mutant p53 aggregation [44]. We were able to elucidate the molecular mechanism through which PRIMA-1 rescues amyloid aggregates of mutant p53 and thereby reduces the dominant negative (DN) and gain-of-function (GoF) effects, indicating that mutant p53 aggregation is an excellent target for the development of new anti-tumoral drugs [44].
The synthesis of PRIMA-1 is reported in patents, in which the quinuclidinone hydrochloride 1 is used as the starting material (Scheme 1). The synthesis of PRIMA-1 is achieved by the treatment of 1 with an excess of formaldehyde and potassium carbonate. As for PRIMA-1MET, it is also synthesized from 1, but in this case, 1 is treated with 1 equivalent of formaldehyde and potassium carbonate to form 2. The dehydration of 2 gives compound 3, which, upon treatment with sodium methoxide, affords 4 (not isolated), followed by a treatment with formaldehyde, which delivers PRIMA-1MET [45].
Scheme 1.

Synthetic routes for the preparation of PRIMA-1 and PRIMA-1MET.
Other types of α,β-unsaturated compounds such as maleimide derivatives can also be used to reactivate the ability of p53 to perform DNA binding and preserve its active conformation [46]. MIRA-1, for example, is a maleimide-type compound with low molecular weight that has been identified in screenings at the NCI data bank as a potential candidate for reactivating mutant p53-dependent cells in different human tumor cells, being more potent than PRIMA-1 in the induction of cell death [47]. Additionally, other structural analogues of MIRA-1 were tested in the inhibition of p53-dependent growths and showed similar activity (MIRA-2 and MIRA-3, Scheme 2). Interestingly, when the saturated analogues were evaluated, those did not present significant results, which clearly demonstrated that the unsaturated maleimide system is of utmost importance for the activity of such compounds [47]. Although very effective for p53 reactivation, MIRA-1 was shown to be highly toxic to different types of normal cells and demonstrated an acute toxicity which does not involve p53, inducing apoptosis in normal cells through a caspase-9-dependent pathway [48], which demonstrates the need for new derivatives aiming to reduce these toxic effects.
Scheme 2.

(a) Structure of the maleimide-derived MIRA compounds and (b) general synthetic route for their preparation.
2.2. Maleimide Derivatives
The synthesis of MIRA-type compounds has been known since the 1960s. It is based on maleimide as the building block, which is commercially available or may be easily obtained from the oxidation of pyrrole or by the acid-catalyzed cyclization of maleimide [49]. In this context, Tawney and co-workers explored the synthesis of several maleimide derivatives using fairly simple reactions and producing a huge diversity of compounds. Overall, the amine group of maleimide is reactive under basic conditions, and may undergo, for instance, methylation reactions with formaldehyde, producing the intermediate MIRA-2; this intermediate can easily react with phosphorus trichloride to produce N-chloromethylmaleimide 5 (80% yield), as shown in Scheme 2b. The reaction of 5 with anhydrides forms acetoxylmethylmaleimides (e.g., MIRA-1 and MIRA-3) [50,51].
2.3. α,β-Unsaturated Carbonyl Compounds
The positive results obtained with the compounds of the PRIMA and MIRA series stimulated the search for other small molecules with the ability to reactivate mutant p53 through covalent binding to its central domain to restore its tumor suppressor function. For instance, the open-chain version of the maleimide ring in the MIRA derivatives leads to α,β-unsaturated compounds capable of acting as Michael acceptors. With this perspective in mind, derivatives containing the 3-benzoylacrylic acid moiety have been synthesized and used as Michael acceptors. The 3-benzoylacrylic acid 6 and its fluorinated (E)-4-(4-fluorophenyl)-4-oxobut-2-enoic derivative 7 (Figure 1) raised the melting temperature of the core domain of WT p53 and the hotspot mutants R175H, Y220C, G245S, R249S, and R282 by 3 °C, which implicates on a thermodynamic stabilization of the mutant into a WTp53-like conformation [34]. Interestingly, in this work the authors were able to determine the order of reactivity of the different cysteine moieties in mutant p53 structure.
Figure 1.

Structure of 3-benzoylacrylic acid derivatives reactivators of p53.
Another class of compound that has shown promising results are N-alkylated and N,N-dialkylated 3,5-bis-(arylidene)piperidones, which are obtained from 4-piperidone [52]. Such molecules have been shown to have high activity against the A549 resistant human lung carcinoma cell line and are structurally related with the natural compound curcumin, which is the main component of the rhizome of Curcuma longa and has proved to be a powerful chemopreventive and anticancer agent, with several other reported biological activities. Further research in this area identified a series of synthetic curcumin analogs containing two arylidene-α-carbonyl units and a nitroxide group [53]. These compounds, which bear two structural antioxidant moieties, showed efficacy and selectivity in killing cancer cells A2780, MCF-7, and H9c2 by converting mutant p53 to a transcriptionally active WTp53-like form [54]. The central strategy was to combine the anticancer properties of curcumin derivatives and the antioxidant capacity of the nitroxide groups, which has the ability to reduce damage caused by ROS. Among the synthesized compounds, a very active one was identified and denominated as HO-3867. The synthetic route to HO-3867 involves a Claisen–Schmidt condensation between 4-piperidone hydrochloride 9 and p-fluorobenzaldehyde 8 giving 10, which leads to the product HO-3867 after alkylation with substituted pyrrol 11 (Scheme 3).
Scheme 3.

Synthesis of curcumin analogue HO-3867.
2.4. Pyrimidine, Quinazoline, and Quinoline Derivatives
Also considering that small molecules that act as Michael acceptors have the ability to react covalently with cysteine residues of mutant p53 without compromising its binding to DNA, Fersht et al. demonstrated that 5-chloro-2-(methylsulfonyl) pyrimidine-4-carboxylic acid, the so-called PK11000, increases the thermal stability of mutant p53 by reacting with cys182 or cys227 (Scheme 4a) [55,56]. Sulfonylpyrimidines are electrophilic compounds capable of reacting with nucleophiles, for example thiols, forming structures such as 13; indeed, K11000 has shown to be a potent and selective alkylating agent, and is currently being targeted in a preclinical investigation for the therapy of patients with triple negative breast cancer [57]. PK11000 is a commercially available compound and its synthesis can be accomplished via the reaction of dichlorooxobutenoic acid 14 with thiocompound 15 followed by oxidation of intermediate 16 with hydrogen peroxide (Scheme 4b) [58].
Scheme 4.

(a) Reaction of PK11000 with cysteines and (b) synthetic route for its preparation.
In a similar path, styryl quinazoline compound CP-31398, which contains a double bond conjugated between a benzene and a quinazoline ring, has also been able to stabilize the active conformation of mutant p53 [59]. This compound inhibits the growth of small human tumor xenografts in vivo and in tobacco-induced lung cancer. Recently, it was demonstrated that CP-31398 is a p53-modulating agent with potential to act as a chemopreventive agent for different cancer types [60,61,62,63]. The synthesis of CP-31398 was reported by Sutherland and co-workers in 2012 (Scheme 5); in this route, the condensation reaction between quinazolinone 17 and 4-methoxybenzaldehyde 18 in the presence of sodium acetate gives the styryl quinazoline 19, which after a chlorination/substitution reaction sequence leads to CP-31398 [64].
Scheme 5.

Synthesis of CP-31398.
Another very promising compound for the reactivation of p53 via covalent modification is 2-vinylquinazolin-4-(3H)-one, the so-called STIMA-1 (Scheme 6) [65]. Similarly, the activity of this compound is related to the exogenous olefin group that can act as a Michael acceptor. Zach and co-workers found that STIMA-1 is more potent than CP-31398 in suppressing the growth of mutant p53-containing tumor cells [65]. In this sense, there are two reported synthetic approaches towards STIMA-1 using anthranilic acid derivative 20 as starting material. In the method developed by Witt and Bergman, 3-chloropropionyl chloride 21 is used to prepare 2-(3-chloropropionylamino)-benzamide 22, which upon cyclization and dehydrohalogenation with sodium hydroxide in ethanol gives STIMA-1 (Scheme 6) [66]. Another approach was described recently by Abe and co-workers, and consists in a one-pot copper-catalyzed Ritter-type reaction with acrylonitrile 23 to achieve the same molecule (Scheme 6) [67].
Scheme 6.

Synthetic methods for the preparation of STIMA-1.
P53R3 is a quinazoline-type molecule which was originally identified using p53 DNA binding assays [68]. This compound restores sequence-specific DNA binding to several p53 hot spot mutants, but also improves the recruitment of wild-type p53 to target gene promoters. The synthesis of P53R3 was described back in 2007 in a patent by Mallams and co-workers (Scheme 7) [69]. 2,3-Dihydroquinazolinone 26 is obtained through the cyclization reaction between propargyl chloride 24 and ethyl 2-aminobenzoate 25. Next, 26 is reacted with piperazine to form intermediate 27, which gives 29 after treatment with 4,4’-(chloromethylene)bis(chlorobenzene) 28 in basic medium. The chlorination of 29 then leads to 30, which is converted to P53R3 after reaction with L-valine methyl ester 31.
Scheme 7.

Synthetic route towards compound P53R3.
In a similar path, SCH529074 is another quinazoline-type molecule which can reactivate mutant p53 and also bind to p53 DNA binding domain, interrupting HDM2-mediated ubiquitination of WTp53 [15,70]. SCH529074 synthesis can also be achieved using intermediate 27; the reaction of 27 with benzyl chloride gives 32, which is next chlorinated, furnishing 33 (Scheme 8) [69]. 33 is then reacted with N,N’-dimethylpropane-1,3-diamine, affording 34, which is next converted to 35 after a reduction step. Finally, the reaction of 35 with 4,4’-(chloromethylene)bis(chlorobenzene) 28 gives rise to the product.
Scheme 8.

Synthetic route towards compound SCH529074.
COTI-2, a thiosemicarbazone derivative, is another molecule which has showed promising activity in reactivating p53 (Scheme 9) [71]. Indeed, a phase 1 clinical trial in gynecological cancers is currently ongoing [72]. Although the mechanism of action of COTI-2 is not yet completely known, it has been already shown that this compound binds both to full length and DNA-binding domain of mutant p53, leading to the refolding of the mutant protein [73]. COTI-2 can be synthetized from the reaction of 1,1’-thiocarbonyldiimidazole 36 with N-(2-pyridyl) piperazine 37, giving 38 as intermediate. Next, 38 is reacted with hydrazine to furnish 39, which undergoes an addition reaction with 6,7-dihydro-5H-quinolin-8-one, giving the desired product [74].
Scheme 9.

Synthetic route towards quinolinone-type compound COTI-2.
2.5. Pyrrol and Pyrazole and Derivatives
Recently, Fersht and co-workers identified a family of hybrid 1H-pyrrole-1H-pyrazole heterocycles from which compound PK7088 was highlighted, being biologically active in vitro against cancerous cells carrying the Y220C mutant p53 protein. Assays indicated that the PK7088 molecule increased the amount of WTp53 levels and restored its transcriptional functions and apoptosis [75]. This compound was identified in a library obtained by combinatorial chemistry, and was synthesized in four steps from 2-(4-fluorophenyl) acetonitrile; the key step was the synthesis of the pyrazole ring from 2-(4-fluorophenyl)-3-oxopropanenitrile via condensation with hydrazine. As for the construction of the pyrrole ring, it was achieved using the classic Paal-Knorr method from 40 (Scheme 10).
Scheme 10.

Synthetic route for the preparation of PK7088. MW = microwave irradiation.
Prodigiosin is a tripyrrole red pigment member of the prodiginine family; it is a secondary metabolite of the human pathogen Serratia marcescens [76]. This compound has been receiving growing attention owing to its reported antimicrobial, immunosuppressive, and anticancer properties. In 2014, El-Deiry and co-workers reported that prodigiosin is a promising p53 reactivator, restoring a deficient p53 signaling pathway and producing antitumor effects via a dual mechanism which involves p73 upregulation and disruption of the mutant p53/p73 complex. The first report on the total synthesis of prodigiosin dates back to 1962 [77], but other approaches have also been reported [76]. Tripathy, Lavallée and co-workers, for instance, reported the synthesis of fragment 42 via the reaction of 4-methoxy-3-pyrolin-2-one 41 with DMF in the presence of POBr3 followed by the Suzuki cross-coupling between 42 and boronic acid 43 (Scheme 11) [78]. Finally, fragment 43 is coupled with pyrrol 45 in acidic medium, giving prodigiosin as product.
Scheme 11.

Synthetic route towards prodigiosin.
2.6. Zinc Metallochaperones
Zinc plays a crucial role in the structure and properties of p53, since this protein binds to DNA through a zinc-stabilized structurally complex domain [79]. Considering these concepts, D’Orazi’s group investigated thoroughly the purpose of zinc in p53 reactivation in mutant p53-expressing cancer cells, as reported in 2011 [80]. The group observed that zinc partly induced the transition of mutant p53 into a functional conformation, being able to re-establish chemosensitivity in breast cancer cell lines expressing the R175H mutation, as well as in glioblastoma ones expressing the R273H mutation.
This study paved the path for a series of works involving a new class of compounds, the zinc metallochaperones (ZMCs), which have appeared as promising candidates for restoring p53 [81]. This new type of anti-cancer drug acts by targeting a precise set of zinc-binding p53 mutations. Carpizo’s group has very recently described the use of such types of compounds to treat BRCA1 deficient breast cancer and found very interesting results (Scheme 12) [82]. The compound ZMC1, which is commercially available, combined with olaparib, was very effective in inhibiting tumor growth, while its complexation with zinc (Zn-1) showed improved efficacy.
Scheme 12.

Preparation of zinc metallochaperone (ZMC)-type compound Zn-1 by Carpizo’s group.
Another interesting p53 reactivator is the case of a bifunctional ligand LH. The compound presents zinc metallochaperone features and strongly interacts with mutant p53. The simple insertion of an iodine atom to the compound structure (Figure 2) promotes inhibition of mutant p53 aggregation, restores zinc binding to mutant p53, and reactivates WTp53 transcriptional function. The effects were observed both in vitro and in tumoral cells. Also, the ligand presented minimal toxicity to non-cancerous organoids, showing a selective cytotoxicity to mutant p53 tumors [83].
Figure 2.
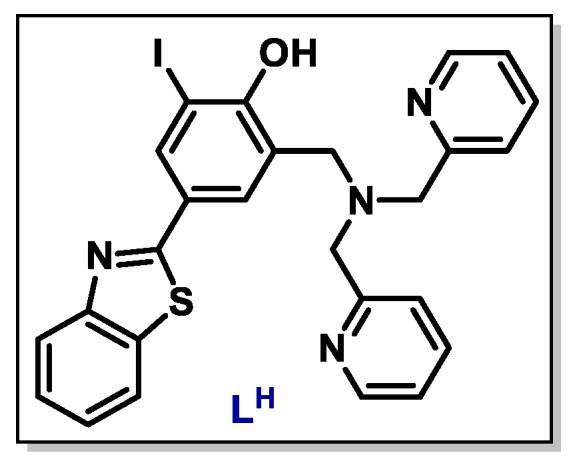
Chemical structure of the bifunctional ligand LH with p53 anti-aggregation effect.
2.7. Other Classes of Compounds for the Reactivation of Wild-Type p53
Inspired in natural products such as styryl lactones, which are known to present high cytotoxicity and are found in the Annonaceae plant gender, Kondaiah and Prasad’s group reported in 2013 the discovery of MPK-09, another promising compound that displays antitumor activity. Such molecule showed to be very selective and highly potent in the restoration of p53 functions of the mutants R175H, R249S, R273H, and R273C [84]. Although the mechanism through which MPK-09 reactivates p53 is not completely known, it is conceivable that its α,β-unsaturated dicarbonyl moiety may act as a Michael acceptor toward thiols (Scheme 13) [85].
Scheme 13.

Synthesis of styryl lactone-inspired compound MPK-09.
MPK-09 can be synthetized via a 5-step high yielding route from 2,2-dimethyl-1,3-dioxolane-4,5-dicarboxamide 46. Initially, 46 is converted into 47 after the reaction with a Grignard reagent, followed by a reduction step to afford 48. Next, an alkylation reaction gives 49, which is converted to 50 after the reaction with dimethyl methylphosphonate, and finally undergoes a Wittig-Horner olefination, finally giving MPK-09 as product.
PK5174 is also a noteworthy compound with good affinity for the Y220C mutant p53 that inhibits its aggregation by acting on specific points in the p53 core domain (Figure 3). Interestingly, in this compound the ethynyl group is a nonclassical bioisostere to iodine, forming halogen bonds within the protein pocket [86,87]. Chetomin (CTM), a natural product extracted from the fungus Chaetomium cochliodes, which has antibacterial and antifungal properties [88], restores the Hsp40-p53 axis in a R175H mutant model and p53 MDM2-dependent degradation (Figure 3) [89]. Thiosemicarbazones are also a very important class of compounds, with some of them having reached clinical trials stage. Compound NSC319726, for instance, has restored effectively the function of R175H mutant p53 with extensive apoptosis induction [90].
Figure 3.

Structure of p53 reactivators with varying features.
Additionally, through a virtual search based on a set of information on the pocket L1/S3 of the p53 protein structure, stictic acid (also known as NSC87511) was selected as a potential p53-reactivating compound by targeting cysteine 124 (Figure 3). Indeed, it has accomplished the reactivation of R175H mutant p53 of human osteosarcoma cells, being even more potent than PRIMA-1 [8]. Remarkably, its structure bears little resemblance to the conventional reactivators of p53 function. It is important to note that among all of the small molecules capable of restoring the function of p53, this natural product is the only one that does not bear a nitrogen atom. Resveratrol is another molecule that has shown to inhibit the aggregation of p53 mutants in vitro, in tumor cells and in xenotransplants implanted in a nude mouse model [91]. High doses of resveratrol (100 µM) were required to exert the anti-amyloid and antitumoral effects. There are great hopes that synthetic chemistry will lead into resveratrol analogs with higher potency that can be used as pharmaceuticals.
RETRA is another small-molecule which have shown to activate a series of p53- regulated genes, specifically suppressing mutant p53-bearing tumor cells in vitro and in mouse xenografts, as reported by reported by Chumakov’s group in 2008 (Figure 3) [92]. The treatment of mutant p53-expressing cancer cells with this compound has resulted in a considerable increase in the expression level of p73, as well as a discharge of p73 from the blocking complex with mutant p53. Importantly, RETRA has shown to be active against tumor cells expressing several p53 mutants, still not affecting normal cells. Later in 2015, Sonnemann and co-workers described the activity of RETRA against Ewing’s sarcoma cells and observed its activity was not dependent on their TP53 status, being effective against p53-deficient and p53 wild-type cell lines [93].
3. When p53 Is Inactivated by Oncogenes: Small Molecules Antagonists of p53-MDM2/MDMX
MDM2 (murine double minute 2), also known as E3 ubiquitin-protein ligase MDM2 or HDM2, is an important negative regulator of WTp53 in normal cells. In tumors, it acquires an oncogenic role due to its overexpression that leads to WTp53 inactivation. This biological target has been highly investigated to restore p53 activity, and also has the largest set of small molecule inhibitors. Moreover, MDM2 is the therapeutic target with the highest number of substances in clinical trials against various types of cancer in humans [94]. Thus, the disruption of the MDM2-p53 protein complex is a promising strategy for the treatment of various types of cancer via the restoration of WTp53 function. Indeed, a compilation of patents on small molecule MDM2 inhibitors has been recently published [24] and a critical discussion on the biological rationale of these strategies have also been conducted [23,95].
3.1. Cis-Imidazoline Derivatives
Back in 2004, 1,2,4,5-tetrasubstituted-4,5-cis-imidazolines, also known as Nutlins, were discovered via a high-throughput screening at Hoffman-La Roche. This family of compounds are potent and specific inhibitors of p53-MDM2, which have inspired the discovery of more selective MDM2 inhibitors [96,97,98]. In this family, Nutlin-2 (IC50 = 140 nM) and Nutlin-3a (IC50 = 90 nM) were the compounds with the highest affinity for the recombinant p53-MDM2 protein. Crystal structures of the complex of Nutlin-3 with p53-MDM2 revealed that the two 4-chlorophenyl rings interact with the residues Trp23 and Leu26 pockets and the isopropoxy group of the other benzene ring interacts in the Phe19 pocket (Figure 4). This type of interaction with three distinct pockets came to be known as the “three finger pharmacophore model”.
Figure 4.

Crystal structure of the complex of Nutlin-3 with p53-MDM2 (Murine Double Minute 2) available on PDB under code 5C5A (in the chemical structure, carbon atoms are drawn in pink, nitrogen in blue, oxygen in red and chlorine in green).
The synthesis of Nutlins was initially reported in patents by Hoffmann-La Roche, in which the authors describe their synthesis from benzil 51 as a racemic mixture (Scheme 14) [99].
Scheme 14.

First reported synthesis of Nutlin-3a.
Nutlin-3a can be obtained through the condensation of the substituted 1,2-diamine 52 with aromatic ester 53 in the presence of trimethylaluminum to give the imidazoline 54. Next, 54 is treated with phosgene in the presence of a base such as triethylamine to give the racemic carbamoyl chloride 55, which can be purified using chiral supercritical fluid chromatography (SFC). The coupling of the racemic carbamoyl chloride 55 with 2-oxopiperazine provides the Nutlin-3 as a racemic mixture; the enantiomers were then separated by SFC [100,101].
More recently, Johnston and co-workers reported the first enantioselective synthesis of (−)-Nutlin-3 [102]. The authors developed highly diastereo- and enantioselective aza-Henry additions, i.e., carbon–carbon bond-formation addition of aryl nitromethanes to imines, affording adducts that were used to prepare the cis-stilbene diamine key intermediate precursor (Scheme 15). The aza-Henry adduct 58 was obtained via the reaction of N-Boc-protected imine 56 with aryl nitroalkane 57 and chiral bisamidine catalyst BAM. Next, the reduction of the nitro group afforded amine 59, which was acylated with 2-isopropoxy-4-methoxybenzoic acid giving 60, and later submitted to standard TFA-promoted Boc removal for the obtainment of 61. Then, the monoamide 62 was achieved by a carbamoylation reaction with 2-oxopiperazine; finally, the Nutlin-3a compound was obtained after a Hendrickson’s dehydrative cyclization [103,104].
Scheme 15.

Johnston and co-workers synthetic route towards (−)-Nutlin-3.
Later on, the same group reported a further optimization in the synthetic route with the use of a novel monoamidine organocatalyst (MAM) that provided high enantioselectivity at a higher reaction temperature (−20 °C) employing low catalyst loadings. This additional refined approach led to the synthesis of (−)-Nutlin-3 in a 17 g batch scale with only three chromatographic purification steps [105].
More recently, Fantinatti and co-workers developed the synthesis of Nutlin-3 as a scalemic mixture in which the levorotatory isomer (−)-Nutlin-3 was obtained in 84:16 ratio (Scheme 16) [100]. Additionally, the synthesis of enantioenriched (+)-Nutlin-3 was discribed using catalyst 64, which was obtained using (1S,2S)-cyclohexanediamine as starting material (Scheme 16). The synthetic pathway in this case was initiated with 4-chlorobenzaldehyde 63, which leads to diamine 52 after the reaction with ammonium acetate; next, amide 61 is synthetized from 52 in the presence of 65 and catalyst 64. After two additional reaction steps, (+)-Nutlin-3 is obtained as a scalemic mixture. It is also worth mentioning that an array of biological tests showed that the activity of scalemic (−)-Nutlin-3 was comparable to that of the commercial eutomer in activating the p53 pathway [104].
Scheme 16.

Finatti and co-workers route to (−)-Nutlin 3.
Following a similar path, many new compounds were synthesized based on the structure of Nutlin-3a, with some of them displaying promising results. Vu and co-workers, for instance, reported a new member of the Nutlin family of MDM2 inhibitors, the so-called RG7112 (Scheme 17) [106,107,108]. This compound has advanced to phase I clinical trials in combination with cytarabine in patients with acute myelogenous leukemia, neoplasms and hematologic neoplasms [104]. Basically, as a strategy to optimize the structure of the original Nutlin compounds, the authors chose to preserve the most important features for binding to MDM2, e.g., the 4-chlorophenyl and isopropoxy groups, while exploring alterations in other sites of the molecule. Three main changes in the Nutlin structure were proposed: (a) methyl groups were placed at the imidazoline ring to prevent its oxidation; (b) a variety of polar groups were inserted into the amide side chain to explore their possible effects on binding and pharmacokinetic properties; and (c) a replacement of the 4-methoxy group for tert-butyl, owing to in vitro metabolism studies, which identified this as the most labile site in the molecule. The preparation of such compounds is exemplified in Scheme 17, which shows the synthesis of RG7112 [109]. Initially, the imidazoline 68 is obtained by the reaction of diamine 66 and benzoate ester 67 in the presence of trimethylaluminum. Next, the intermediate 69 is obtained by the phosgenation of 68. Then, the reaction of 69 with substituted piperazine 70 in the presence of triethylamine leads to racemic compound RG7112. The enantiomers are later separated by chiral chromatography.
Scheme 17.

Vu and co-workers route to RG7112.
Although compound RG7112 showed high affinity with the p53-MDM2 protein, the results of the phase I clinical trial of this compound alone or in combination with cytarabine in patients with liposarcoma showed limited reduction in tumor volume. Currently, a clinical trial with RG7112 combined with doxorubicin is ongoing [109].
In 2010, Domling’s group introduced a new process for predicting and preparing antagonists of the p53/HDM2 interaction [110]. This approach consisted on the use of multicomponent reactions (MCR) to synthesize new potential p53-HDM2 inhibitors starting from a fragment. Although no protein–protein interaction analyses were conducted, computational studies have indicated the potential of the new molecules, which imidazole PB11 being the most promising one. The synthesis of the compound PB11 was achieved starting with the Orru three-component reaction between p-chlorobenzaldehyde 63, isocyanoacetate 71, and cyclopropylmethylamine 72 followed by an amidation step (Scheme 18).
Scheme 18.

Synthesis of potential p53-HMD2 inhibitor PB11. TBD = 1,5,7-triazabicyclo[4.4.0]dec-5-ene.
3.2. Spiro-Oxindole, Indole, and Carbazole Derivatives
Since the discovery of the imidazolines or Nutlins family, many research groups have invested in the search for new nitrogen heterocycles that include benzodiazepines [111], indole-2-carboxylic acid derivatives [97] and pyrrolidines [112], among others. Wang and co-workers, for example, reported the design of a spiroindoline-3,3’-pyrrolidine series as potent, highly selective and orally active small-molecule inhibitors of the MDM2-p53 interaction [98,113]. The authors identified that the natural alkaloids spirotryprostatin A and Alstonisine displayed a steric hindrance that would keep them from fitting properly into the MDM2 cleft, but presented features in their structure that could be used as inspiration for designing new MDM2 inhibitors (Scheme 19).
Scheme 19.

Synthesis of spiro-oxindole compounds. MS = molecular sieves.
Considering that the p53-MDM2 crystal structure showed that Phe19, Trp23, and Leu26 hydrophobic residues in p53 interact with a hydrophobic cleft in MDM2, the authors designed small molecules that could block this interaction [112]. Specifically, the oxindole scaffold could mimic the Trp23 side chain of p53 by providing hydrogen-bonding and hydrophobic interactions with MDM2, while the spiropyrrolidone ring would provide a rigid system that could bear hydrophobic groups that could resemble the side chain of Phe10 and Leu26. In this sense, based on an early approach published in 2000 by Williams’ group for the synthesis of alkaloids [114], Wang’s group reported an approach having an asymmetric 1,3-dipolar reaction as the key step to synthetize spiro-oxindoles that was later used in the synthesis of a series of highly active MDM2 inhibitors [115]. In this sense, in 2005, Wang’s group reported the synthesis of a series of compounds (78a–f, Scheme 19), which had their activity evaluated in WTp53 LNCaP human prostate cancer cells. Remarkably, compound 78d proved to be a potent MDM2 inhibitor, being efficient in inhibiting cell growth with an IC50 value of 0.83 µM and also presenting low toxicity for normal cells [98].
Although compound 78d proved to be highly active and selective, it still had a low potency when compared to the most potent peptide-based inhibitors [98]. Considering this premise, Wang’s group conducted further structural optimizations and reached compound MI-63, which showed to be highly potent with a Ki of 3 nM, being 2000 times more active than the natural p53 peptide (Figure 5) [116]. Later on 2008, seeking to improve MI-63 PK profile, which was unsuitable for in vivo evaluation, the same group discovered a compound called MI-219 (Figure 5), which also contains a tert-butyl group in the spiropyrrolidone moiety but has a different substitution pattern in the amide group. MI-219 indeed has shown promising results, being highly active in p53 reactivation and presenting very low toxicity [117]. Another very interesting compound in this series was MI-147, which showed very low values of Ki, being a highly potent inhibitor; MI-147 was able to selectively activate p53 in SJSA-1 cell lines and to dose-dependently induce cell death, being also highly efficient in inhibiting tumor growth in the SJSA-1 xenograft model.
Figure 5.

Series of compounds containing the spiropyrrolidone scaffold that showed high activity to restore p53 via MDM2 inhibition.
Further structural optimization of MI-219 led to MI-888 (Scheme 20), in which the 1,2-diol moiety of the molecule was constrained by the insertion of a cyclobutyl ring, which would block this metabolic oxidative site [118]. In addition, the fluorine atom was removed from the oxindole ring, leading to an enhanced MDM2 binding affinity and improved pharmacokinetic profile. MI-888 showed to be stable in a variety of solvents, but when treated in the presence of MeOH or MeCN/H2O, formed four products that were later identified as stereoisomers [119]. In that way, the isomerization of MI-888 was further verified and the different stereoisomers had their p53-restoring activity evaluated. Interestingly, the different isomers presented markedly different activity as MDM2 inhibitors. An isomerization mechanism was proposed in which the trans-cis isomer is initially formed and undergoes a ring-opening reaction in the presence of MeCN/H2O, giving an imine intermediate 79. The intramolecular ring-closure reaction of imine 79 leads to the formation of the three remaining isomers (Scheme 20).
Scheme 20.

Isomerization of MI-888-type compounds in the presence of a acetonitrile/water solvent system. CAN = ceric ammonium nitrate.
It is known that MI-219 has a clear mechanism-based antitumor activity both in vitro and in vivo, however, high doses were required, whereas MI-888 failed to achieve complete tumor regression. Therefore, Wang and co-workers strived to further optimize the structure of such compounds and discovered MI-77301, also known as SAR405838 (Figure 4) [120]. Still presenting a structure similar to the one of pyrrolidones, MI-77301 displays a different stereochemistry in the quaternary carbon atom and changed halogen substitution patterns in both phenyl rings, as well as a conformationally-constrained cyclohexanol group (Figure 4) [115,121]. This compound stabilizes p53 and activates its pathway, resulting in the interruption of the cell proliferation, cell cycle arrest and apoptosis, and is currently in phase I clinical trials for cancer treatment [122,123,124,125]. Another clinical trial was initiated with cancer patients involving safety and efficacy of MI-77301 [126] and Pimasertib (AS-703026), which is a highly selective, potent, ATP non-competitive allosteric inhibitor of MEK1/2 [127].
Back in 2011, Graves and co-workers reported the synthesis of a new spiroindoline MDM2 inhibitor, RO8994, which was designed combining the structural features of RG7388 and the spiroindolinone core structure of MI-888 [128,129]. Indeed, RO8994 showed to be as active as RG7388 and more than MI-888; the compound showed marked tumor growth inhibition of WTp53, MDM2-amplified SJSA-1 osteosarcoma tumor xenograft model, displaying significant tumor growth inhibition (>60%) at 1.56 mg kg−1, tumor stasis at 3.125 mg kg−1 and regression at 6.25 mg kg−1. The synthetic route towards RO8994 developed by Graves’ group initiates with a cycloaddition reaction between 80 and 81 promoted by silver fluoride, giving compound 82a as intermediate (Scheme 21a). Next, 82a is isomerized to 82b in the presence of DBU via intermediate 83. Finally, the deprotection of the Boc group leads to 84, which after an amidation step, gives RO8994 as a racemic mixture. The desired enantiomer can be obtained via separation using chiral supercritical fluid chromatography (SFC). Some of the drawbacks of this synthetic pathway is the low overall yield (around 2%), which is a consequence of the formation of complex mixtures during the different steps and the need for SFC isolation of the enantiomers formed in the final step. Considering these factors, L. Shu and co-workers reported a new approach for the preparation of RO8994 (Scheme 21b). In this new approach, the cycloaddition reaction of 80 with 86 promoted by DBU gives 87 with high selectivity. Next, the hydrolysis of 87 affords acid intermediate rac-88, which undergoes a resolution step with N,N-dimethyl-((R)-1-phenylethyl)amine to give the enantiopure acid 88-salt. The desired product RO8994 is then formed after the amidation of 88 with ammonium hydroxide. This four-step procedure was able to generate enantiopure RO8994 with a 33% overall yield, and could be scaled up to produce up to multihundred grams of the product [130].
Scheme 21.

Synthesis of spiroindoline RO8994, a MDM2 inhibitor through (a) the original synthetic route and (b) an alternative route.
The strategy reported by L. Shu and co-workers was later used by Graves’ group to produce a new family of spiroindoline-type compounds containing bioisosteric replacements of the 6-chlorooxindole group in RO8994. This investigation led to the discovery of RO5353 and RO2468, which showed great potential for clinical development as highly potent, selective, and orally active p53-MDM2 inhibitors (Figure 6) [131].
Figure 6.

Structure of compounds RO2468 and RO5353, spiroindoline-type inhibitors of p53-MDM2. The pink color in the structures highlights the side chain of the bioisosteric replacements of the 6-chlorooxindole group in RO8994.
Later in 2015, Ivanenkov’s group reported the regioselective synthesis of a series of compounds containing the spiro-indolinone scaffold from the one-pot reaction of commercially available 2-thioxoimidazol-4-one 89, sarcosine 90 and isatin 91 (Scheme 22) [132]. Such molecules were evaluated on their ability induce apoptosis and block the proliferation of HepG2, Hek293, MCF-7, SiHa and HCT116 cell lines. Interestingly, the best activity was shown by compound 92a which showed a comparable performance to Nutlin-3 against MCF-7 cell lines, with an IC50 value of 4.88 ± 1.5 µM.
Scheme 22.

One-pot regioselective synthesis of spiro-indolinone scaffolds by Ivanenkov’s group.
Considering the high activity of spiropyrazoline oxindoles in the reactivation of WTp53, Santos’ group has recently described the synthesis of these scaffolds with a few structural modifications, such as the use of different substituents in the pyrazoline ring and their evaluation in the isogenic HCT116 cell lines pair, either in the presence or absence of TP53. The family of compounds 95 was synthetized through the 1,3-dipolar cycloaddition reaction between 3-methylene indoline-2-ones 93 and nitrile imines that were formed in situ via the dehydrohalogenation of hydrazonoyl chlorides 94. Among the 18 compounds tested, one showed promising results (R1 = F, R2 = Ph, R3 = tBu and R4 = Ph), against the cancer cell line expressing WT p53, with good activity and low cytotoxicity (Scheme 23) [133].
Scheme 23.

Synthesis of spiropyrazoline oxindole derivatives by the Santos’ group.
Very recently, two phase I clinical trials were initiated for the compound known as DS-3032b (Figure 7) aiming to study its safety, tolerability and pharmacokinetics in patients with hematological malignancies [134] and with advanced solid tumors or lymphomas [135]. DS-3032b appears to be a promising compound to treat hematological malignancies with MDM2 expression/amplification in leukemic blasts.
Figure 7.

Structure of DS-3032b, a MDM2 inhibitor.
Another interesting MDM2 inhibitor is MK-8242. Although its structure is not available to date, it has been evaluated singly [136] in clinical trials and in combination with cytarabine in adult participants with refractory or recurrent acute myelogenous leukemia (AML) [136]. This compound is also well tolerated by patients with liposarcoma and other advanced solid tumors by inhibiting the binding of the MDM2 protein to the transcriptional activation domain of the WTp53 [137,138,139,140,141].
Other types of molecules containing the indole/indoline moiety have also shown promising activity in inhibiting the MDM2-p53 interaction. Ji and Li’s group, for instance, reported the synthesis of a family of (E)-3-benzylideneindolin-2-one derivatives 99 from the alkylation of 2,4-dihydroxibenzaldehyde 96 with alkyl iodines, giving 97, followed by a Knoevenagel condensation with substituted indolinones 98 (Scheme 24) [142]. Among all of the synthetized compounds, 99a presented the highest binding affinity to MDM2, as well as the highest antiproliferative activity against HCT116 (WTp53). Another interesting feature of this compound was its Ki value (0.093 mM), which can be translated into a high ligand efficiency when the amount of non-hydrogen atoms in the molecule is considered [40,41].
Scheme 24.

Synthesis of functionalized of (E)-3-benzylideneindolin-2-one by Ji and Li’s group.
In 2006, Hardcastle, Lunec and co-workers reported the discovery of a series of indoline-type compounds as potent p53-MDM2 inhibitors by applying in silico screening and small library synthesis [143]. The compounds 105a and 105b showed promising biological activity results in SJSA cells, generating a dose-dependent increase of MDM2 and p21, which is related to the release of p53 transcriptional activity. The synthesis of the compounds was achieved starting from 2-benzoylbenzoic acid 100, which underwent intramolecular cyclization, giving 101 (Scheme 25). Next, an amidation step leads to indoline 102, which is then converted to indoline chloride 103. After the reaction of 103 with benzyl mercaptan, 104 is formed and converted to the desired indoline product 105 after reacting with the appropriate alcohol. Later in 2011, the same group reported that slight structural changes led to even more active indolinone scaffolds [144].
Scheme 25.

Synthetic route toward indoline scaffolds.
Other related compound is JNJ-26854165, also known as serdemetan, a tryptamine derivative which has showed pronounced p53 reactivating properties. In 2011, Deutsch and co-workers reported the action of JNJ-26854165 against several types of human cancer cell lines; although a mechanism of action was not proposed, the authors verified that this compound also has potential as a photosensitizer [145]. Also in 2010, Andreef and co-workers reported that JNJ-26854165 was able to induce p53-mediated apoptosis in acute leukemia cells, as well as S-phase delay and E2F1-mediated apoptosis in p53 mutant cells, acting along with 1-β-arabinofuranosyl-cytosine or doxorubicin in order to cause p53-mediated apoptosis [146]. Later in 2011, Zhuang and co-workers described a series of pharmacodynamic and pharmacokinetic analyses that were conducted in order to identify the minimal effective dose of JNJ-26854165 in patients with advanced stage/refractory solid tumors [147]. A synthetic route towards JNJ-26854165 was described back in 2006 by Lacrompe and co-workers [148]; pyridine-functionalized amide 106 and fluoronitrobenzene 107 were used as starting materials to give coupling product 108 in the presence of a copper catalyst (Scheme 26). Next, a deprotection step in basic medium leads to 109, which undergoes a Raney nickel-promoted reduction to furnish compound 110. Finally, the reduction of indole 111 followed by its reaction with compound 110 in the presence of polymer-supported cyanoborohydride (PSCBH) in acidic medium leads to product JNJ-26854165.
Scheme 26.

Synthetic route towards JNJ-26854165. PSCBH = polymer-supported cyanoborohydride.
In 2014, Buolamwini and Zhang’s group reported the activity of pyrido[b]indole SP-141 as a possible molecular-targeted chemotherapeutic agent [149]. SP-141 was able directly bind to MDM2, inhibit its expression and induce its autoubiquitination and proteasomal degradation. Importantly, this molecule has showed strong in vitro and in vivo antibreast cancer activity without evident host toxicity. The synthesis of SP-141 and several analogues was recently reported by Hiller and Mulcahy’s group, which evaluated such molecules as probes for the 5-hydroxytryptamine receptor (Scheme 27) [150]. The synthetic route starts with the Bischler-Napieralski reaction between indole 112 with triphosgene, giving intermediate 113. 113 is then oxidized with DDQ, leading to pyridone 114, which is next converted to the corresponding triflate 115. Finally, the palladium-catalyzed Suzuki cross-coupling between 115 and boronic acid 116 gives rise to SP-141.
Scheme 27.

Synthetic route towards SP-141.
3.3. Pyrrol/Pyrrolidine, Piperidine, and Morpholine Derivatives
Based on MI-219 and its analog MI-77301, Graves and co-workers reported in 2013 the discovery of a second-generation MDM2 inhibitor with superior potency and selectivity, called as RO5503781 (also known as RG7388, Scheme 28) [151,152].
Scheme 28.

Synthetic route towards of RG7388.
The key step in the synthesis of this compound is the formation of the pyrrolidine core through a 1,3-dipolar cycloaddition reaction between (Z)-α-cyanostilbene 119 (formed through the Knoevenagel condensation between 117 and 118) and the imine 122 (obtained from the reaction between amine 119 and aldehyde 120) promoted by AgF, giving substituted pyrrolidine intermediate 123. After a Boc-deprotection step using standard conditions, affording 124, and a subsequent amidation using HATU as coupling agent, the products were obtained as a racemic mixture. Next, a chiral SFC was used to separate the two enantiomers (Scheme 28). RG7388 has advanced to phase I clinical trials as a single agent or in combination with cytarabine in patients with acute myelogenous leukemia, neoplasms, and hematologic neoplasms [153].
Another class of compounds that has been successfully used as MDM2 inhibitors are piperidinones. Poppe and co-workers, for instance, reported in 2012 that a new class of piperidinones that are able to bind to the same three pockets as previously reported for the Nutlins, spiro-oxindoles and pyrrolidines, and also interacts in the N-terminal region of human MDM2, in particular Val14 and Thr16 [154]. Indeed, the synthesis and initial SAR of rigid cores capable of holding two aromatic rings in proximity had led Sun and co-workers closely before in the same year to the identification of a piperidinone derivative as a novel inhibitor of the MDM2-p53 protein–protein interaction [155]. Computational docking showed that the p-chloro substituted phenyl ring occupied the Trp23 pocket, while the m-chloro substituted one was in the Leu26 pocket and the cyclopropyl group occupied the Phe19 pocket. The carboxylic acid group at the 3-position of piperidinone also showed to have an additional interaction with the His96 residue of MDM2. In this sense, a series of optimizations led to a piperidinone derivative featuring a chiral tert-butyl 2-butanoate 126 which is 8-times more potent than 127 to MDM2 (Scheme 29a). The crystal structure of 126 showed that the two phenyl groups had a gauche-like orientation, which was less favored over the more stable anti-like conformation, as suggested by quantum mechanical calculations. Next, an extra methyl group was inserted at C3 along with further modification of the tert-butyl ester into a hydroxyl group, yielding AM-8553, which showed to be a highly potent, selective, and orally bioavailable inhibitor of MDM2-p53. The importance of the carboxylic acid moiety was confirmed by means of its removal from the molecule, which caused a 44-fold loss in binding affinity [156]. The same group also reported later the piperidone prototype AMG 232, which was created based on the structure of AM-8553 (Scheme 29a) [157].
Scheme 29.

(a) Examples of piperidinone derivatives and (b) synthetic route towards AMG 232.
The synthesis of AMG 232 was accomplished by initially reacting compound 127 with methyl methacrylate in the presence of a strong base, giving 128. The asymmetric reduction of 128 afforded 129, which next underwent an intramolecular cyclization step, furnishing lactone 130 (Scheme 29b). Lactone 130 was then alkylated with ally bromide to produce 131, following an amidation reaction with valinol, giving 132. After another intramolecular cyclization which leads to 133 and a thiol insertion/oxidation reaction, AMG 232 is obtained. AMG 232 is currently under clinical trials, in a phase 1b/2a study, for the evaluation of its activity in patients with metastatic melanoma in combination with trametinib and dabrafenib vs. trametinib combined with dabrafenib [158].
Rew and co-workers also reported later in 2014 the discovery of AM-7209, another piperidone that inhibits the MDM2-p53 interaction by inhibiting MDM2 [159]. This compound is structurally related to its predecessors, but the isopropylsulfonyl group was replaced by a butylsulfonyl group, and the isopropyl group for a cyclopropyl group in order to improve the interactions with the Phe19(p53) pocket in MDM2. Additionally, a m-fluoro group was inserted into the C6 phenyl ring, and the carboxylic acid was converted to a secondary amide. The structural changes led to a higher activity against the HCT-116 colorectal carcinoma xenograft model (SJSA-1 EdU IC50 = 1.6 nM). The synthesis of AM-7209 was accomplished using a fluorinated analogous of compound 131 (see the synthesis of 131 in Scheme 29) as a starting material (Scheme 30) [159]. After the conversion of 131 into amide 137 through the reaction with aminoalcohol 136, 137 undergoes an intramolecular cyclization step in the presence of p-toluenesulfonic anhydride, leading to lactam 138. 138 then furnishes bicyclic intermediate 139 after the reaction with p-toluenesulfonic acid, which next produces intermediate 140 in two steps. The carboxylic acid moiety in 140 is subsequently converted into an amide group via the reaction with 4-aminobenzoic acid 141 in the presence of EDC as a coupling agent, finally giving product AM-7209.
Scheme 30.

Synthetic route towards compound AM-7209. EDC = 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide.
Another compound that has reached the clinical trials stage was CGM097, which was developed by Holzer and co-workers from Novartis and is being tested in patients with advanced solid tumors who have progressed despite standard therapy or for whom no standard therapy exists [160]. A Phase I dose escalation study of CGM097 in adult patients was also programed for selected patients with advanced solid tumors characterized by WTp53-dependent status [161,162]. The synthesis of CGM097 conducted using the convergent route is presented in Scheme 31.
Scheme 31.

Asymmetric synthesis of CGM097 published by Holzer’s group.
The synthetic route starts with compound 142, which initially undergoes an alkylation with iodopropane, giving 143. Next, a carbonylation reaction leads to 144, with a subsequent sulfonylation leading to 145. An asymmetric reduction of the imine using an organotin compound and a rhodium complex as catalyst then gives 146, which undergoes an intramolecular asymmetric cyclization step, furnishing fragment 147. The synthesis of the other moiety of the molecule starts with a reductive amination of aldehyde 148, giving 149. An alkylation of the amine group gives 150, which is submitted to a Boc deprotection step to furnish 151, which leads to 152 upon a nucleophilic substitution step with methyl 2-bromoacetate. Subsequently, the reductive amination of aldehyde 153 with compound 152 leads to the formation of 154, which is then submitted to an intramolecular cyclization reaction to produce 155. Finally, a coupling reaction of 155 with fragment 147 affords the desired compound CGM097.
Besides piperidones, structurally-related morpholine derivatives have also revealed to be effective and selective MDM2-p53 inhibitors [163,164]. In 2014, Gonzalez and co-workers described the discovery of AM-8735, a potent and selective MDM2 inhibitor (SJSA-1 EdU IC50 = 25 nM) that showed remarkable pharmacokinetic properties and in vivo efficacy, also displaying considerable improvements in hepatocyte stability when compared to the related piperidinone inhibitors. The synthetic route towards AM-8735 starts with the conversion of 4-chlorobenzyl bromide 156 to nitroalkane 157, which undergoes a Henry reaction with 3-chlorobenzaldehyde to furnish an amino alcohol, which subsequently undergoes an in situ protection to its corresponding silyl ether 158 (Scheme 32) [163]. Subsequently, the reduction of the nitro group in 158 leads to 159, which after a chiral resolution step delivers the desired amino alcohol 160 with a 97% enantiomeric excess (ee). Alternatively, 160 can be produced from commercially available amino acids in five steps (not shown in Scheme 32). Next, morpholine 161 is obtained through the reaction of aminoalcohol 160 with chloroacetyl chloride in basic medium. 161 then undergoes an alkylation reaction with bromide 162 to furnish 163 as a mixture of interconvertible diastereoisomers. After separation, 163b is reduced to alcohol 164 with superhydride and further converted to a thioether under Mitsunobu conditions. Finally, AM-8735 is obtained after an alkylation/oxidation sequence.
Scheme 32.

Synthetic route towards compound AM-8735.
3.4. Pyrimidine and Quinazoline Derivatives
In recent years, pyrimidines and quinazoline-type molecules have also been pointed as promising p53-MDM2 inhibitors. Back in 2005, Weisman and co-workers have identified a series of 7-nitro-5-deaza-flavin compounds, the so-called HDM2 ligase inhibitor 98 (HLI98) [165]. Interestingly, the HLI98 compounds were somewhat specific for HDM2/MDM2, but effects on HECT E3s (ubiquitin ligase) both in vitro and in cells, were also observed. Fischer and co-workers have reported the synthesis of the HLI98 compounds starting from 2,4,6-trichloropyrimidine 165, which was converted to 2-chlorouracil 166 via basic hydrolysis (Scheme 33) [166]. Next, the reaction of 166 with substituted anilines gives rise to 6-anilinouracils 167, which upon condensation with 2-halo- or 2-tosylaldehydes 168, furnish the products HLI98.
Scheme 33.

Synthetic route towards compound series HDM2 ligase inhibitor 98 (HLI98).
In 2009, high-throughput screening studies conducted by Allen and co-workers identified chromenotriazolopyrimidines as possible p53-MDM2 inhibitors [167]. In this case, rigid core of the compounds was believed to project the substituents into key p53 binding pockets of MDM2. Indeed, cellular assays proved the such compounds were able to to upregulate p53 protein and signaling, causing p53-dependent inhibition of proliferation and apoptosis. The synthesis of these compounds was achieved starting from substituted benzaldehyde 169 and 2′-hydroxyacetophenone 170, which reacted in basic medium giving chalcone derivative 171 (Scheme 34). 171 then undergoes a cyclization with 3-amino-1,2,4-triazole 172, leading to intermediate 173, which undergoes cyclization with benzaldehyde derivatives to furnish 174. Finally, the alkylation of 174 leads to 175 as a mixture of diastereoisomers.
Scheme 34.

Synthetic route towards chromenotriazolopyrimidines inhibitors of p53-MDM2.
HDM201 is another small molecule inhibitor of p53-MDM2 and is able to restore the normal function of WTp53, being inspired in the Nutlins family (Figure 8)[168]. This compound is under investigation in phase I clinical trials as an anticancer drug in patients with liposarcoma [169].
Figure 8.

Structure of the p53-MDM2 inhibitor HDM201.
In 2011, Lu, Lim and co-workers reported their findings on the development of pyrrolopyrimidines which act as R-helix mimetics able to interrupt R-helix-mediated protein–protein interactions, acting as dual MDMX/MDM2 inhibitors [170]. Importantly, the group developed a solid-phase method for the divergent synthesis of a wide library of pyrrolopyrimidines (Scheme 35). The synthetic route towards pyrrolopyrimidines 184 starts with the bromoacetylation of Rink amide MBHA resin 176, leading to 177. Then, the bromine group was substituted by amines, giving 178, which underwent the same procedures to give 179. Next, 179 is reacted with 4,6-dichloro-2-(methylthio)-pyrimidine-5-carbaldehyde 180, giving 181, which undergoes intramolecular cyclization in the presence of DBU, giving 182. The thioether group in 182 is then oxidized to the corresponding sulfone 183, which underwent substitution with several amines followed by the cleavage of the resin-bound compounds with trifluoroacetic acid (TFA), giving pyrrolopyrimidines 184 as products.
Scheme 35.

Solid-phase synthesis of R-helix mimetics pyrrolopyrimidines. DIAD = Diisopropyl azodicarboxylate, DIC = N,N’-Diisopropylcarbodiimide.
3.5. Pyrazole and Pyrrolone Derivatives
MDMX is another key regulator of the p53 pathways, and shares homology with MDM2 in its p53-binding domains [171,172]. However, MDMX is thought to regulate p53 via different mechanisms: while MDM2 mostly regulates the stability and subcellular localization of p53, MDMX directly regulates p53 transcription. Like MDM2, MDMX is genetically amplified in several types of cancers, and therefore, the inactivation of both MDM2 and MDMX is believed to be crucial to induce an effective p53 response in tumor cells that express wild-type p53. In this sense, in 2010 Dyer and co-workers reported the identification and characterization of the first small molecule inhibitor of MDMX, the pyrazole named SJ-172550 [173]. This compound was able to bind reversibly to the p53-binding pocket of MDMX, displacing p53 and effectively killing retinoblastoma cells in which the expression of MDMX was enlarged. Importantly, when combined with the MDM2 inhibitor nutlin-3a, SJ-172550 showed superior cytotoxicity in cells that expressed wild-type p53. The synthesis of SJ-172550 was outlined by Russel, Huber and co-workers (Scheme 36) [174]. Initially, 3-ethoxy-4-hydroxybenzaldehyde 185 is chlorinated to form 186, which is next submitted to a reaction with methyl 2-bromoacetate, giving methyl 2-(2-chloro-6-ethoxy-4-formylphenoxy)acetate 187 [175]. In parallel, phenyl hydrazine 188 is reacted with ethyl acetoacetate, forming pyrazolone 189. The base-catalyzed reaction of 189 with fragment 187 gives rise to the product SJ-172550.
Scheme 36.

Synthetic route towards compound SJ-172550.
In 2013, Sheng, Zhang and co-workers reported the synthesis of a set of pyrrolo[3,4-c]pyrazole derivatives 197 that were excellent simultaneous inhibitors of p53-MDM2 and NF-κB (five DNA-binding proteins that are often hyperactive in cancer and inflammatory processes) [176]. The developed molecules were able to efficiently inhibit tumor growth in the A549 xenograft model. Additionally, compound 197 displayed great oral bioavailability (72.9%). The synthesis of such derivatives starts with the generation of methyl pyruvates 192 from the Claisen condensation of diethyl oxalate 191 and acetophenone 190 (Scheme 37). Next, the reaction of 192 with 4-bromobenzaldehyde 193 and N-(3-aminopropyl)imidazole 194 leads to 195, which is then converted to pyrrolopyrazole 196 after reaction with hydrazine in acidic medium. Finally, the reaction of 196 with different haloalkanes gives rise to the desired products 197.
Scheme 37.

Synthetic route towards pyrrolopyrazole derivatives.
In a similar path, the same group described later a series of pyrrolones 199 which showed to be striking activity as MDM2/MDMX dual inhibitors, impeding the tumor growth in the A549 xenograft model (Scheme 38) [177]. The compounds could be synthetized in a similar fashion to that of pyrrolopyrazoles 197, but here intermediate 198 underwent a Mitsunobu reaction with a variety of alcohols to furnish the corresponding ether-pyrrolidone derivatives 199.
Scheme 38.

Synthetic route towards pyrrolone derivatives. DIAD = Diisopropyl azodicarboxylate.
3.6. Benzodiazepinone Derivatives
Benzodiazepinediones have also been studied with regards to their ability of reactivating p53 pathways. Using combinatorial chemistry techniques, Grasberger and co-workers identified compound DP222669 as antagonist of the HDM2-p53 interaction. The authors were also able to generate the X-ray crystal structure of the antagonists bound to HDM2, which revealed their R-helix mimetic properties. The synthesis of these compounds was achieved by using an Ugi multicomponent reaction with aldehydes 200, N-Boc protected carboxylic acids 201, amines 202 and isocyanide 203 to give intermediate 204. The intramolecular cyclization of 204 leads to the formation of benzodiazepinediones 205 (Scheme 39) [178].
Scheme 39.

Synthesis of benzodiazepinediones via a Ugi reaction/intramolecular cyclization sequence.
Later in 2011, Sheng, Zhang and co-workers described the synthesis of the thio-benzodiazepines and their biological activity of p53-MDM2 inhibitors [179]. Most compounds had nano/micromolar affinity towards MDM2, and some showed activity similar to that of nutlin-3a. Compound 211a (R = 4-CF3C6H4), in particular, exhibited a high antitumor activity against the U-2 OS human osteosarcoma cell line (IC50 = 1.06 mM). These molecules were synthetized through the Ugi multicomponent reaction of amine 206, benzaldehyde 63, isocyanide 208 and substituted 2-nitrobenzoic acids 207, giving bisamide 209 as intermediate (Scheme 40). Next, benzodiazepinone 210 is obtained after the reduction of 209 with iron powder followed by intramolecular cyclization, and is converted to 211 after a reductive amination step. Finally, benzodiazepinones 210 are converted to the corresponding thio-benzodiazepinones 211 after reacting with the Lawesson’s reagent.
Scheme 40.

Synthesis of thio-benzodiazepinedione derivatives.
3.7. Benzofuroxan and Furan Derivatives
In 2010, Yan’s group described the use of benzofuroxan derivative XI-006 (NSC207895) as an inhibitor of the MDMX expression in cancer cells; the treatment of MCF-7 cells with this compound activated p53, resulting in a higher expression of proapoptotic genes [180]. Interestingly, the compound also worked additively with nutlin-3a to activate p53. Later in 2015, Pishas and co-workers reported that XI-006 was able to induce p53-independent apoptosis in Ewing’s sarcoma [181]. The synthesis of XI-006 can be achieved starting with the base-catalyzed intramolecular cyclization of nitroaniline 213, giving 214 (Scheme 41) [182]. 214 then undergoes a nitration step, giving 215, which next is isomerized to 216 in acidic medium. The reaction of 216 with piperazine leads to 217, and a methylation step delivers the product XI-006.
Scheme 41.

Synthetic route towards XI-006.
Another valuable example is the compound called RITA (NSC 652287), which displays a very simple structure (Scheme 42). This compound induces both DNA-protein and DNA-DNA cross-links with no detectable DNA single-strand breaks, and has also been shown to inhibit the p53-MDM2 interaction as well as target p53 mutants R175H, R248W, R273H, and R280K, restoring transcriptional activity and inducing apoptosis [183,184]. The synthesis of RITA may be accomplished starting with the copper(I)-catalyzed homocoupling of terminal alkyne 218, giving 1,3-butadiyne 219, which next undergoes a cyclocondensation with water in basic medium, giving rise to the product [185] (Scheme 42).
Scheme 42.

Synthetic route towards RITA.
4. Small Molecules That Reactivate the p53 Protein through Other Pathways
Many compounds reported in the literature have shown the ability to restore the activity of p53 but apparently do not act through the aforementioned pathways. Leptomycin B (LMB), for instance, is a Streptomyces sp. metabolite that induces the transcriptional activity of a p53-dependent reporter plasmid [186]. Along with this LMB activity, an p53-dependent increase in the levels of the products of two p53-dependent genes, p21 and MDM2 were also observed [187]. Overall, LMB is believed to function mostly as an inhibitor of the export of proteins from the nucleus to the cytoplasm owing to its ability to interact with, and impair the function of the nuclear export factor CRM-1 [188] In this sense, LMB is a very effective inducer of the p53 response which does not act directly through the DNA damage pathway [189]. Indeed, LMB has showed to be effective against human lung adenocarcinoma cell lines, among others [190]. Other LMB derivatives have also showed to be efficient nuclear export inhibitors, having showed promising efficacy in multiple mouse xenograft models [191]. The total synthesis of leptomycin B was reported back in 1998 by Kobayashi and co-workers (Scheme 43) [192]. Initially, E-crotyl alcohol 220 undergoes a Sharpless oxidation reaction, giving epoxide 221. Next, a regioselective ring-opening leads to 222, which after a carboxylation/thermal treatment gives rise to lactone 223. Next, the lactone 223 is protected as isopropyl acetal 224, and after the deprotection of the benzyl group and Swern oxidation, aldehyde 225 is formed. 225 then undergoes a Wittig reaction with fragment 226, affording conjugated diene 227. Then, 227 is returned to its lactone form 228. In parallel, carboxylic acid 229 (formed from the ozonolysis of geraniol, not shown in Scheme 43) is condensed with a chiral oxazolidinone and submitted to a methylation step to give 230, which after removal of the chiral auxiliary and reduction, leads to aldehyde 231. Next, 231 undergoes an Evans aldol reaction, giving 232, which undergoes removal of the auxiliary group protection of the OH group, giving 233. A second aldol condensation leads to 234, which once again undergoes the removal of the chiral auxiliary and a reduction step, leading to 235. After the exchange of the terminal protecting group from MOM to TBS, fragment 236 is formed. Finally, the fragments 228 and 236 are combined, giving 237, which next undergoes removal of the alcohol protection group leading to 238, and an oxidation step gives leptomycin B.
Scheme 43.

Total synthesis of CRM1 inhibitor Leptomycin B.
Another efficient approach for the activation of p53 is the inhibition of casein kinase 1A1 (CKIa). Indeed, this method was successfully employed by several groups, with promising results in vivo in mouse models of intestinal cancer [26] and in cultured leukemia cells [193]. In 2018, Neriah’s group reported their findings on the use of a series of molecules, among them A51, in primary mouse acute myeloid leukemia cells [194]. Interestingly, the blocking of CKIa along with CDK7 and/or CDK9 synergistically stabilizes p53, depriving leukemia cells of survival and proliferation, maintaining SE-driven oncogenes and inducing apoptosis. The synthesis of A51 starts with 1-methylpyrazole 239, which undergoes deprotonation by a strong base and adds to cyclopropanecarboxaldehyde, giving 240 (Scheme 44). Next, 240 is dehydroxylated to produce 241, which next undergoes a bromination step giving 242. In sequence, 242 undergoes treatment with a strong base followed by the reaction with 2-isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane, giving 243, which is then is reacted with 2,4,5-trichloropyrimidine, giving substitution product 244. Finally, the reaction of with 244 with trans-diaminocyclohexane under microwave irradiation delivers the product A51.
Scheme 44.

Synthetic route towards casein kinase 1A1 (CKIa) inhibitor A51.
The human genome encompasses seven sirtuins (SirT1-7), and those are implicated in a series of essential cellular processes and disease conditions [195]. SirT1, especially, is involved in the regulation of the transcriptional function of p53, while SirT2 is involved in the direct deacetylation of α-tubulin and histone H4 [196,197,198,199]. Considering these facts, Lain and co-workers identified back in 2008 a small-molecule activator of p53 at low micromolar concentrations [200]. The authors were able to define the mechanism of action for this compound, which reversibly increased p53 and p21CIP/WAF1 protein levels and decreased cellular growth in several types of tumor cell lines. Indeed, tenovin-1 has shown similar potency in both mammalian cell-based assays and melanoma xenograft tumors. On the other hand, the low solubility of tenovin-1 in water led to the development of a molecule with higher water solubility, tenovin-6. Remarkably, tenovin-6 has also displayed a slightly higher potency in p53 activation [200]. The synthesis of such compounds can be accomplished by the initial conversion of p-tert-butylbenzoic acid 245 to the corresponding acyl chloride 246 (Scheme 45) [201]. Next, 246 is reacted with sodium thiocyanate and subsequently with phenylenediamine, giving thiourea 247. Finally, the reaction of 247 with the appropriate acyl chloride delivers the product.
Scheme 45.

Synthetic route towards SirT1 and SirT2 inhibitors tenovin-1 and -6.
Another molecule that has shown to be a promising SirT1 inhibitor is inauhzin, as reported by Lu’s group [202]. This triazino[5,6-b]indol-phenothiazine-type molecule reactivates p53 by inhibiting SIRT1 activity and promoting p53-dependent apoptosis of cancer cells without provoking genotoxic stress. Structural changes in inauhzin have also led to analogues with promising p53 reactivating properties [203]. For the synthesis of inauhzin, 5H-[1,2,4]triazino[5,6-b]indole-3-thiol 250 is initially synthetized from commercially available isatin 248 and thiosemicarbazide 249 (Scheme 46) [203]. In parallel, bromide 254 is obtained from the reaction between 251 and 2-bromobutyryl bromide 252. Finally, fragments 250 and 254 undergo a reaction in basic medium to furnish inauhzin.
Scheme 46.

Synthetic route towards SirT1 inhibitor inauhzin.
Recently, El-Deiry and co-workers found that the imidazole-type molecule CB002 is a p53 pathway-restoring compound that induces tumor selective cell death via the induction of NOXA, a pro-apoptotic protein, being able of targeting mutant versions of p53 for MDM2-independent proteasomal degradation. Indeed, this compound was able to induce several p53 target genes in various mutant p53 cell lines without affecting their p53 protein turnover. The synthesis of CB002 was reported in 1994 by Kokel, which used 6-amino-1,3-dimethyluracil 255 as a starting material in the presence of (dichloromethylene)dimethylammonium chloride (Scheme 47) [204]. Next, the reaction of 256 with azidotrimethylsilane leads to 257, which then undergoes an addition-elimination step with aniline to give 258. Finally, 258 undergoes a base-catalyzed intramolecular step to furnish 8-anilinotheophylline CB002 as product.
Scheme 47.

Synthetic route towards CB002.
5. Conclusions and Outlook
In the last decades, a lot of groundbreaking work was conducted regarding the development of small molecules for p53 reactivation, and two main pathways have been especially highlighted: (1) Molecules that can react covalently with the thiol groups in the cysteine residues of mutant p53, which leads to conformational changes that can reactivate the protein and (2) molecules that are able to disrupt the MDM2-p53 protein complex, releasing the WTp53 protein to resume its function. In this sense, a lot of advanced studies were published and demonstrated that comprehending the structure of p53 is crucial for achieving its reactivation.
A recent breakthrough was the discovery that mutant p53 undergoes aggregation in a very similar way to that observed with other amyloid proteins [11,25,27], playing a crucial role in the development of cancer through loss of p53 function, a dominance-negative effect and gain of oncogenic function. Compounds and peptides that have been described to inhibit mutant p53 aggregation also lead to a decline on tumor proliferation and migration. Thus, the misfolded and aggregated states of mutant p53 have become highly promising targets for the development of novel therapeutic strategies against cancer [11].
On the other hand, although much has already been accomplished, there is still much to achieve, especially on the development of synthetic methods to reach the promising molecules. In addition, the use of computational approaches along with in vitro experiments will aid in the design of new drugs [205,206,207,208]. For instance, Lukman and coworkers have demonstrated by simulations how loop 1 of p53 DNA binding domain is the most dynamic segment among the DNA-contacting loops and how it can be used as a potential target [205].
The molecules tested so far have not yet reached the market and the effects can still be improved in terms of lower toxicity to normal cells, potency and affinity to targets. Also, many of the papers discussed in this review still employ outdated stoichiometric methods, toxic solvents and harsh reaction conditions, and such approaches can be easily substituted by greener ones. However, the growing awareness regarding environmental issues in the last few years have generated a change in the design of synthetic methods, which lead us to believe that this pressing change will come in due time.
Acknowledgments
F.P. Pauli, R.C.B. Ribeiro, T. de B. da Silva, C.G.S. Lima and G.D.S. Ferretti acknowledge CAPES, CNPq and FAPERJ for their fellowships.
Author Contributions
J.L.S., L.P.R., G.D.S.F., F.P.P., R.C.B.R., T.d.B.d.S., C.G.S.L., F.C.d.S., V.F.F. wrote and edited the text. All authors have read and agreed to the published version of the manuscript.
Funding
All authors thank FAPERJ for funding. J.L. Silva laboratory is supported by grants from CNPq, FINEP, CAPES and FAPERJ. Luciana Pereira Rangel also acknowledges the support by the Serrapilheira Institute (Grant Serra-1708-15204). J.L. Silva and Luciana Pereira Rangel thank Fundação do Câncer for funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Levine A.J., Oren M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer. 2009;9:749–758. doi: 10.1038/nrc2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lane D.P., Cheo C.F., Lain S. p53-based cancer therapy. Cold Spring Harb. Perspect. Biol. 2010;2:a001222. doi: 10.1101/cshperspect.a001222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lane D.P. p53, guardian of the genome. Nature. 1992;358:15–16. doi: 10.1038/358015a0. [DOI] [PubMed] [Google Scholar]
- 4.Powell E., Piwnica-Worms D., Piwnica-Worms H. Contribution of p53 to metastasis. Cancer Discov. 2014;4:405–414. doi: 10.1158/2159-8290.CD-13-0136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mowat M.R. p53 in tumor progression: Life, death, and everything. Adv. Cancer Res. 1998;74:25–48. doi: 10.1016/s0065-230x(08)60764-2. [DOI] [PubMed] [Google Scholar]
- 6.Pflaum J., Schlosser S., Müller M. p53 family and cellular stress responses in cancer. Front. Oncol. 2014;4:285. doi: 10.3389/fonc.2014.00285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gupta A., Shah K., Oza M.J., Behl T. Reactivation of p53 gene by MDM2 inhibitors: A novel therapy for cancer treatment. Biomed. Pharmacother. 2019;109:484–492. doi: 10.1016/j.biopha.2018.10.155. [DOI] [PubMed] [Google Scholar]
- 8.Wassman C.D., Baronio R., Demir O., Wallentine B.D., Chen C.K., Hall L.V., Salehi F., Lin D.W., Chung B.P., Hatfield G.W., et al. Computational identification of a transiently open L1/S3 pocket for reactivation of mutant p53. Nat. Commun. 2013;4:1407. doi: 10.1038/ncomms2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Levine A.J. p53, The cellular gatekeeper for growth and division. Cell. 1997;88:323–331. doi: 10.1016/S0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 10.Muller P.A.J., Vousden K.H. p53 mutations in cancer. Nat. Cell Biol. 2013;15:2–8. doi: 10.1038/ncb2641. [DOI] [PubMed] [Google Scholar]
- 11.Silva J.L., Cino E.A., Soares I.N., Ferreira V.F., de Oliveira G.A.P. Targeting the prion-like aggregation of mutant p53 to combat cancer. Acc. Chem. Res. 2018;51:181–190. doi: 10.1021/acs.accounts.7b00473. [DOI] [PubMed] [Google Scholar]
- 12.Wiman K.G. Pharmacological reactivation of mutant p53: From protein structure to the cancer patient. Oncogene. 2010;29:4245–4252. doi: 10.1038/onc.2010.188. [DOI] [PubMed] [Google Scholar]
- 13.Foster B.A., Coffey H.A., Morin M.J., Rastinejad F. Pharmacological rescue of mutant p53 conformation and function. Science. 1999;286:2507–2510. doi: 10.1126/science.286.5449.2507. [DOI] [PubMed] [Google Scholar]
- 14.Peng Y., Li C., Chen L., Sebti S., Chen J. Rescue of mutant p53 transcription function by ellipticine. Oncogene. 2003;22:4478–4487. doi: 10.1038/sj.onc.1206777. [DOI] [PubMed] [Google Scholar]
- 15.Demma M., Maxwel E., Ramos R., Liang L., Li C., Hesk D., Rossman R., Mallams A., Doll R., Liu M., et al. SCH529074, A small molecule activator of mutant p53, which binds p53 DNA binding domain (DBD), restores growth-suppressive function to mutant p53 and interrupts HDM2-mediated ubiquitination of wild type p53. J. Biol. Chem. 2010;285:10198–10212. doi: 10.1074/jbc.M109.083469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duffy M.J., Synnott N.C., Crown J. Mutant p53 as a target for cancer treatment. Eur. J. Cancer. 2017;83:258–265. doi: 10.1016/j.ejca.2017.06.023. [DOI] [PubMed] [Google Scholar]
- 17.Loh S.N. Follow the mutations: Toward class-specific, small-molecule reactivation of p53. Biomolecules. 2020;10:303. doi: 10.3390/biom10020303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lopes E.A., Gomes S., Saraiva L., Santos M.M.M. Small molecules targeting mutant P53: A promising approach for cancer treatment. Curr. Med. Chem. 2019;26:7323–7336. doi: 10.2174/0929867325666181116124308. [DOI] [PubMed] [Google Scholar]
- 19.Chène P. Inhibiting the p53-MDM2 interaction: Na importante target for cancer therapy. Nat. Rev. Cancer. 2003;3:102–109. doi: 10.1038/nrc991. [DOI] [PubMed] [Google Scholar]
- 20.Lemos A., Leão M., Soares J., Palmeira A., Pinto M., Saraiva L., Sousa M.E. Medicinal chemistry strategies to disrupt the p53–MDM2/MDMX interaction. Med. Res. Rev. 2016;36:789–844. doi: 10.1002/med.21393. [DOI] [PubMed] [Google Scholar]
- 21.Liu Y., Wang X., Wang G., Yang Y., Yuan Y., Ouyang L. The past, present and future of potential small-molecule drugs targeting p53-MDM2/MDMX for cancer therapy. Eur. J. Med. Chem. 2019;176:92–104. doi: 10.1016/j.ejmech.2019.05.018. [DOI] [PubMed] [Google Scholar]
- 22.Beloglazkina A., Zyk N., Majouga A., Beloglazkina E. Recent small-molecule inhibitors of the p53–MDM2 protein–protein interaction. Molecules. 2020;25:1211. doi: 10.3390/molecules25051211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Espadinha M., Barcherini V., Lopes E.A., Santos M.M.M. An update on MDMX and dual MDM2/X inhibitors. Curr. Top. Med. Chem. 2018;18:647–660. doi: 10.2174/1568026618666180604080119. [DOI] [PubMed] [Google Scholar]
- 24.Rusiecki R., Witkowski J., Joanna J.-A. MDM2-p53 interaction inhibitors: The current state-of-art and updated patent review (2010-Present) Recent Pat. Anticancer Drug Discov. 2019;14:324–369. doi: 10.2174/1574892814666191022163540. [DOI] [PubMed] [Google Scholar]
- 25.Kojima K., Kornblau S.M., Ruvolo V., Dilip A., Duvvuri S., Davis R.E., Zhang M., Wang Z., Coombes K.R., Zhang N., et al. Prognostic impact and targeting of CRM1 in acute myeloid leukemia. Blood. 2013;121:4166–4174. doi: 10.1182/blood-2012-08-447581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Elyada E., Pribluda A., Goldstein R.E., Morgenstern Y., Brachya G., Cojocaru G., Snir-Alkalay I., Burstain I., Haffner-Krausz R., Jung S., et al. CKIa ablation highlights a critical role for p53 in invasiveness control. Nature. 2011;470:409–413. doi: 10.1038/nature09673. [DOI] [PubMed] [Google Scholar]
- 27.Pribluda A., Elyada E., Wiener Z., Hamza H., Goldstein R.E., Biton M., Burstain I., Morgenstern Y., Brachya G., Billauer H., et al. A senescence-inflammatory switch from cancer-inhibitory to cancer-promoting mechanism. Cancer Cell. 2013;24:242–256. doi: 10.1016/j.ccr.2013.06.005. [DOI] [PubMed] [Google Scholar]
- 28.Yi J., Luo J. SIRT1 and p53, effect on cancer, senescence and beyond. Biochim. Biophys. Acta. 2010;1804:1684–1689. doi: 10.1016/j.bbapap.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ano Bom A.P.D., Rangel L.P., Costa D.C.F., de Oliveira G.A.P., Sanches D., Braga C.A., Gava L.M., Ramos C.H.I., Cepeda A.O.T., Stumbo A.C. Mutant p53 aggregates into prion-like amyloid oligomers and fibrils: Implications for cancer. J. Biol. Chem. 2012;287:28152–28162. doi: 10.1074/jbc.M112.340638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Levy C.B., Stumbo A.C., Ano Bom A.P.D., Portari E., Cordeiro Y., Silva J.L., De Moura-Gallo C.V. Co-localization of mutant p53 and amyloid-like protein aggregates in breast tumors. Int. J. Biochem. Cell Biol. 2011;43:60–64. doi: 10.1016/j.biocel.2010.10.017. [DOI] [PubMed] [Google Scholar]
- 31.Silva J.L., Gallo C.V., Costa D.C., Rangel L.P. Prion-like aggregation of mutant p53 in cancer. Trends Biochem. Sci. 2014;39:260–267. doi: 10.1016/j.tibs.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 32.Herrero A.B., Rojas E.A., Misiewicz-Krzeminska I., Krzeminski P., Gutiérrez N.C. Molecular mechanisms of p53 deregulation in cancer: An overview in multiple myeloma. Int. J. Mol. Sci. 2016;17:2003. doi: 10.3390/ijms17122003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Soragni A., Janzen D.M., Johnson L.M., Lindgren A.G., Thai-Quynh Nguyen A., Tiourin E., Soriaga A.B., Lu J., Jiang L., Faull K.F., et al. A designed inhibitor of p53 aggregation rescues p53 tumor suppression in ovarian carcinomas. Cancer Cell. 2016;29:90–103. doi: 10.1016/j.ccell.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaar J.L., Basse N., Joerger A.C., Stephens E., Rutherford T.J., Fersht A.R. Estabilization of mutant p53 via alkylation of cysteines and effects on DNA binding. Protein Sci. 2010;19:2267–2278. doi: 10.1002/pro.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schieber M., Chandel N.S. ROS Function in redox signaling and oxidative stress. Curr. Biol. 2014;24:R453–R462. doi: 10.1016/j.cub.2014.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu X., Narayanan S., Vazquez A., Carpizo D.R. Small molecule compounds targeting the p53 pathway: Are we finally making progress. Apoptosis. 2014;19:1055–1068. doi: 10.1007/s10495-014-0990-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Selivanova G. Wild type p53 reactivation: From lab bench to clinic. FEBS Lett. 2014;588:2628–2638. doi: 10.1016/j.febslet.2014.03.049. [DOI] [PubMed] [Google Scholar]
- 38.Selivanova G. Therapeutic targeting of p53 by small molecules. Semin. Cancer Biol. 2010;20:46–56. doi: 10.1016/j.semcancer.2010.02.006. [DOI] [PubMed] [Google Scholar]
- 39.Zhang Q., Bykov V.J.N., Wiman K.G., Zawacka-Pankau J. APR-246 reactivates mutant p53 by targeting cysteines 124 and 2772. Cell Death Dis. 2018;9:439. doi: 10.1038/s41419-018-0463-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bykov V.J.N., Issaeva N., Shlov A., Hultcrantz M., Pugacheva E., Chumakov P., Bergman J., Wiman K.G., Selivanova G. Restoration of the tumor supressor function to mutante p53 by a low-molecular-weight compound. Nat. Med. 2002;8:282–288. doi: 10.1038/nm0302-282. [DOI] [PubMed] [Google Scholar]
- 41.Reddy N.L., Hill J., Ye L., Fernandes P.B., Stout D.M. Identification and structure–activity relationship studies of 3-methylene-2-norbornanone as potent anti-proliferative agents presumably working through p53 mediated apoptosis. Bioorg. Med. Chem. Lett. 2004;14:5645–5649. doi: 10.1016/j.bmcl.2004.08.048. [DOI] [PubMed] [Google Scholar]
- 42.Parrales A., Iwakuma T. Targeting oncogenic mutant p53 for cancer therapy. Front. Oncol. 2015;5:288. doi: 10.3389/fonc.2015.00288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.The National Cancer Institute Website. [(accessed on 12 July 2019)]; Available online: https://www.cancer.gov/about-cancer/treatment/clinical-trials/intervention/prima-1-analog-apr-246?redirect=true.
- 44.Rangel L.P., Ferretti G.D.S., Costa C.L., Andrade S.M.M.V., Carvalho R.S., Costa D.C.F., Silva J.L. p53 reactivation with induction of massive apoptosis-1 (PRIMA-1) inhibits amyloid aggregation of mutant p53 in cancer cells. J. Biol. Chem. 2019;294:3670–3682. doi: 10.1074/jbc.RA118.004671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nielsen A.T. Systems with bridgehead nitrogen. b-ketols of the 1-azabicyclo[2.2.2]octane series. J. Org. Chem. 1996;31:1053–1059. doi: 10.1021/jo01342a016. [DOI] [Google Scholar]
- 46.Saha M.N., Chen Y., Chen G., Chang H. Small molecule MIRA-1 induces in vitro and in vivo anti-myeloma activity and synergizes with current anti-myeloma agents. Brit. J. Cancer. 2014;110:2224–2231. doi: 10.1038/bjc.2014.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bykov V.J.N., Issaeva N., Zache N., Shilov A., Hultcrantz N., Bergman J., Selivanova G., Wiman K.G. Reactivation of mutant p53 and induction of apoptosis in human tumor cells by maleimide analogs. J. Biol. Chem. 2005;280:30384–30391. doi: 10.1074/jbc.M501664200. [DOI] [PubMed] [Google Scholar]
- 48.Bou-Hanna C., Jarry A., Lode L., Schmitz I., Schulze-Osthoff K., Kury S., Bezieau S., Mosnier J.F., Laboisse C.L. Acute cytotoxicity of MIRA-1/NSC19630, a mutant p53-reactivating small molecule, against human normal and cancer cells via a caspase-9-dependent apoptosis. Cancer Lett. 2015;359:211–217. doi: 10.1016/j.canlet.2015.01.014. [DOI] [PubMed] [Google Scholar]
- 49.Ferreira V.F., Souza M.C.B.V., Cunha A.C., Perreira L.O.R., Ferreira M.L.G. Recent advances in the synthesis of pyrroles. Org. Prep. Proced. Int. 2001;33:411–454. doi: 10.1080/00304940109356613. [DOI] [Google Scholar]
- 50.Tawney P.O., Snyder R.H., Bryan C.E., Conger R.P., Dovell F.S., Kelly R.J., Stiteler C.H. The Chemistry of maleimide and its derivatives. I. N-Carbamylmaleimide. J. Org. Chem. 1960;25:56–60. doi: 10.1021/jo01071a017. [DOI] [Google Scholar]
- 51.Tawney P.O., Snyder R.H., Conger R.P., Leibbrand K.A., Stiteler C.H., Williams A.R. The chemistry of maleimide and its derivatives. II. Maleimide and N-methylolmaleimide. J. Org. Chem. 1961;26:15–21. doi: 10.1021/jo01060a004. [DOI] [Google Scholar]
- 52.Makarov M.V., Odinets I.L., Lyssenko K.A., Rybalkina E.Y., Kosilkin I.V., Antipin M.Y., Timofeeva T.V. N-Alkylated 3,5-bis(arylidene)-4-piperidones. Synthetic approaches, X-ray structure and anticancer activity. J. Heterocyclic Chem. 2008;45:729–736. doi: 10.1002/jhet.5570450315. [DOI] [Google Scholar]
- 53.Selvendiran K., Tong L., Bratasz A., Kuppusamy L.M., Ahmed S., Ravi Y., Trigg N.J., Rivera B.K., Kalai T., Hideg K., et al. Anticancer efficacy of a difluorodiarylidenyl piperidone (HO-3867) in human ovarian cancer cells and tumor xenografts. Mol. Cancer Ther. 2010;9:1169–1179. doi: 10.1158/1535-7163.MCT-09-1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Madan E., Parker T.M., Bauer M.R., Dhiman A., Pelham C.J., Nagane M., Kuppusamy M.L., Holmes M., Holmes T.R., Shaik K., et al. The curcumin analog HO-3867 selectively kills cancer cells by converting mutant p53 protein to transcriptionally active wildtype p53. J. Biol. Chem. 2018;293:4262–4276. doi: 10.1074/jbc.RA117.000950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fersht A.R., Bauer M.R. 2-Sulfonylpyrimidines. PCT/GB2016/052544. International Patent Application No. 2017 Feb 23
- 56.Bauera M.R., Joergera A.C., Fersht A.R. 2-Sulfonylpyrimidines: Mild alkylating agents with anticancer activity toward p53-compromised cells. Proc. Natl. Acad. Sci. USA. 2016;113:e5271–e5280. doi: 10.1073/pnas.1610421113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Synnott N.C., Bauer M.R., Madden S., Murray A., Klinger R., O’Donovan N., Duffy M.J. Mutant p53 as a therapeutic target for the treatment of triple-negative breast cancer: Preclinical investigation with the anti-p53 drug, PK11007. Cancer Lett. 2018;414:99–106. doi: 10.1016/j.canlet.2017.09.053. [DOI] [PubMed] [Google Scholar]
- 58.Liang Y.M., Luo S.J., Zhang Z.X., Ma Y.X. Efficient synthesis of a new pyrimidine derivative. Synth. Commun. 2002;32:153–157. doi: 10.1081/SCC-120001523. [DOI] [Google Scholar]
- 59.Jiang J.B., Hesson D.P., Dusak B.A., Dexter D.L., Kang G.J., Hamel E. Synthesis and biological evaluation of 2-styrylquinazolin-4(3H)-ones, a new class of antimitotic anticancer agents which inhibit tubulin polymerization. J. Med. Chem. 1990;33:1721–1728. doi: 10.1021/jm00168a029. [DOI] [PubMed] [Google Scholar]
- 60.Madka V., Zhang Y., Li Q., Mohammed A., Sindhwani P., Lightfoot S., Wu X.R., Kopelovich L., Rao C.V. p53-stabilizing agent CP-31398 prevents growth and invasion of urothelial cancer of the bladder in transgenic UPII-SV40T Mice. Neoplasia. 2013;15:966–974. doi: 10.1593/neo.13704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wischhusen J., Naumann U., Ohgaki H., Rastinejad F., Weller M. CP-31398, a novel p53-stabilizing agent, induces p53-dependent and p53-independent glioma cell death. Oncogene. 2003;22:8233–8245. doi: 10.1038/sj.onc.1207198. [DOI] [PubMed] [Google Scholar]
- 62.Tang X., Zhu Y., Han L., Kim A.L., Kopelovich L., Bickers D.R., Athar M. CP-31398 restores mutant p53 tumor suppressor function and inhibits UVB-induced skin carcinogenesis in mice. J. Clin. Invest. 2007;117:3753–3764. doi: 10.1172/JCI32481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rao C.V., Patlolla J.M., Qian L., Zhang Y., Brewer M., Mohammed A., Desai D., Amin S., Lightfoot S., Kopelovich L. Chemopreventive effects of the p53-modulating agents CP-31398 and PRIMA-1 in tobacco carcinogen–induced lung tumorigenesis in A/J mice. Neoplasia. 2013;15:1018–1027. doi: 10.1593/neo.131256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sutherland H.S., Hwang I.Y., Marshall E.S., Lindsay B.S., Denny W.A., Gilchrist C., Joseph W.R., Greenhalgh D., Richardson E., Kestell P., et al. Therapeutic reactivation of mutant p53 protein by quinazoline derivatives. Invest. New Drug. 2012;30:2035. doi: 10.1007/s10637-011-9744-z. [DOI] [PubMed] [Google Scholar]
- 65.Zache N., Lambert J.M.R., Rökaeus N., Shen J., Hainaut P., Bergman J., Wiman K.G., Bykov V.J.N. Mutant p53 targeting by the low molecular weight compound STIMA-1. Mol. Oncol. 2008;2:70–80. doi: 10.1016/j.molonc.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Witt A., Bergman J. Synthesis and reactions of some 2-vinyl-3H-quinazolin-4-ones. Tetrahedron. 2000;56:7245–7253. doi: 10.1016/S0040-4020(00)00595-0. [DOI] [Google Scholar]
- 67.Abe T., Kida K., Yamada K. A copper-catalyzed Ritter-type cascade via iminoketene for the synthesis of quinazolin-4(3H)-ones and diazocines. Chem. Commun. 2017;53:4362. doi: 10.1039/C7CC01406F. [DOI] [PubMed] [Google Scholar]
- 68.Weinmann L., Wischhusen J., Demma M.J., Naumann U., Roth P., DasMahapatra B., Weller M. A novel p53 rescue compound induces p53-dependent growth arrest and sensitises glioma cells to Apo2L/TRAIL-induced apoptosis. Cell Death Differ. 2008;15:718–729. doi: 10.1038/sj.cdd.4402301. [DOI] [PubMed] [Google Scholar]
- 69.Mallams A., Dasmahapatra B., Neustadt B.R., Demma M., Vaccaro H. Quinazoline derivatives useful in cancer treatment. PCT/US2006/027114. International Patent Application No. 2017 Jan 25
- 70.Nenkov M., Ma Y., Haase D., Zhou Z., Li Y., Petersen I., Lu G., Chen Y. Growth inhibitory role of the p53 activator SCH 529074 in non-small cell lung cancer cells expressing mutant p53. Oncol. Rep. 2020;43:2073–2082. doi: 10.3892/or.2020.7546. [DOI] [PubMed] [Google Scholar]
- 71.Salim K.Y., Vareki S.M., Danter W.R., Koropatnick J. COTI-2, a novel small molecule that is active against multiple human cancer cell lines in vitro and in vivo. Oncotarget. 2016;7:41363–41379. doi: 10.18632/oncotarget.9133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Salim K.Y., Vareki S.M., Danter W.R., Koropatnick J. COTI-2, a new anticancer drug currently under clinical investigation, targets mutant p53 and negatively modulates the PI3K/AKT/mTOR pathway. Eur. J. Cancer. 2016;69:S19. doi: 10.1016/S0959-8049(16)32638-7. [DOI] [Google Scholar]
- 73.Synnott N.C., O’Connell D., Crown J., Duffy M.J. COTI-2 reactivates mutant p53 and inhibits growth of triple-negative breast cancer cells. Breast Cancer Res. Treat. 2020;179:47–56. doi: 10.1007/s10549-019-05435-1. [DOI] [PubMed] [Google Scholar]
- 74.Danter W.R., Brown M., Lepifre F. Compounds and method for treatment of cancer. PCT/CA2008/000045. International Patent Application No. 2008 Jul 17
- 75.Liu X., Wilcken R., Joerger A.C., Chuckowree I.S., Amin J., Spencer J., Fersht A.R. Small molecule induced reactivation of mutant p53 in cancer cells. Nucleic Acids Res. 2013;41:6034–6044. doi: 10.1093/nar/gkt305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yip C.-H., Yarkoni O., Ajioka J., Wan K.-L., Nathan S. Recent advancements in high-level synthesis of the promising clinical drug, prodigiosin. Appl. Microbiol. Biot. 2019;103:1667–1680. doi: 10.1007/s00253-018-09611-z. [DOI] [PubMed] [Google Scholar]
- 77.Rapoport H., Holden K.G. The synthesis of prodigiosin. J. Am. Chem. Soc. 1962;84:635–642. doi: 10.1021/ja00863a026. [DOI] [Google Scholar]
- 78.Dairi K., Tripathy S., Attardo G., Lavallée J.-F. Two-step synthesis of the bipyrrole precursor of prodigiosin. Tet. Lett. 2006;47:2605–2606. doi: 10.1016/j.tetlet.2006.02.035. [DOI] [Google Scholar]
- 79.Hainaut P., Milner J. A structural role for metal ions in the “wild-type” conformation of the tumor suppressor protein p53. Cancer Res. 1993;53:1739–1742. [PubMed] [Google Scholar]
- 80.Puca R., Nardinocchi L., Porru M., Simon A.J., Rechavi G., Leonetti C., Givol D., D’Orazi G. Restoring p53 active conformation with zinc increases the response of mutant p53 tumor cells to anticancer drugs. Cell Cycle. 2011;10:1679–1689. doi: 10.4161/cc.10.10.15642. [DOI] [PubMed] [Google Scholar]
- 81.Blanden A.R., Yu X., Loh S.N., Levine A.J., Carpizo D.R. Reactivating mutant p53 using small molecules as zinc metallochaperones: Awakening a sleeping giant in cancer. Drug Discov. 2015;20:1391–1397. doi: 10.1016/j.drudis.2015.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Na B., Yu X., Withers T., Gilleran J., Yao M., Foo T.K., Chen C., Moore D., Lin Y., Kimball S.D., et al. Therapeutic targeting of BRCA1 and TP53 mutant breast cancer through mutant p53 reactivation. NJP Breast Cancer. 2019;5:14. doi: 10.1038/s41523-019-0110-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Miller J.J., Blanchet A., Orvain C., Nouchikian L., Reviriot Y., Clarke R.M., Martelino D., Wilson D., Gaiddon C., Storr T. Bifunctional ligand design for modulating mutant p53 aggregation in cancer. Chem. Sci. 2019;10:10802–10814. doi: 10.1039/C9SC04151F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Metri P.K., Naz S., Kondaiah P., Prasad K.R. MPK-09, a small molecule inspired from bioactive styryllactone restores the wild-type function of mutant p53. ACS Chem. Biol. 2013;8:1429–1434. doi: 10.1021/cb3005929. [DOI] [PubMed] [Google Scholar]
- 85.Mereyala H.B., Joe M. Cytotoxic activity of styryl lactones and their derivatives. Curr. Med. Chem. Anticancer Agents. 2001;1:293–300. doi: 10.2174/1568011013354606. [DOI] [PubMed] [Google Scholar]
- 86.Wilcken R., Wang G.Z., Boeckler F.M., Fersht A.R. Kinetic mechanism of p53 oncogenic mutant aggregation and its inhibition. Proc. Natl. Acad. Sci. USA. 2012;109:13584–13589. doi: 10.1073/pnas.1211550109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wilcken R., Zimmermann M.O., Bauer M.R., Rutherford T.J., Fersht A.R., Joerger A.C., Boeckler F.M. Experimental and theoretical evaluation of the ethynyl moiety as a halogen bioisostere. ACS Chem. Biol. 2015;10:2725–2732. doi: 10.1021/acschembio.5b00515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mazumder P.M., Sasmal D. Mycotoxins - limits and regulations. Anc. Sci. Life. 2001;20:1–19. [PMC free article] [PubMed] [Google Scholar]
- 89.Hiraki M., Hwang S.Y., Cao S., Ramadhar T.R., Byun S., Yoon K.W., Lee J.H., Chu K., Gurkar A.U., Kolev V., et al. Small molecule reactivation of mutant p53 to wt-like p53 through the p53-Hsp40 regulatory axis. Chem. Biol. 2015;22:1206–1216. doi: 10.1016/j.chembiol.2015.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yu X., Vazquez A., Levine A.J., Carpizo D.R. Allele specific p53 mutant reactivation. Cancer Cell. 2012;21:614–625. doi: 10.1016/j.ccr.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ferraz da Costa D.C., Campos N.P.C., Santos R.A., Guedes-da-Silva F.H., Martins-Dinis M.M.D.C., Zanphorlin L., Ramos C., Rangel L.P., Silva J.L. Resveratrol prevents p53 aggregation in vitro and in breast cancer cells. Oncotarget. 2018;9:29112–29122. doi: 10.18632/oncotarget.25631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kravchenko J.E., Ilyinskaya G.V., Komarov P.G., Agapova L.S., Kochetkov D.V., Strom E., Frolova E.I., Kovriga I., Gudkov A.V., Feinstein E., et al. Small-molecule RETRA suppresses mutant p53-bearing cancer cells through a p73-dependent salvage pathway. PNAS. 2008;105:6302–6307. doi: 10.1073/pnas.0802091105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sonnemann J., Grauel D., Blümel L., Hentschel J., Marx C., Blumrich A., Focke K., Becker S., Wittig S., Schinkel S., et al. RETRA exerts anticancer activity in Ewing’s sarcoma cells independent of their TP53 status. Eur. J. Cancer. 2015;51:841–851. doi: 10.1016/j.ejca.2015.02.016. [DOI] [PubMed] [Google Scholar]
- 94.Deb S.P., Deb S. Mutant p53 and MDM2 in Cancer. 1st ed. Springer; New York, NY, USA: 2014. [Google Scholar]
- 95.Ribeiro C.J., Rodrigues C.M., Moreira R., Santos M.M. Chemical variations on the p53 reactivation theme. Pharmaceuticals. 2016;9:25. doi: 10.3390/ph9020025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Vassilev L.T., Vu B.T., Graves B., Carvajal D., Podlaski F., Filipovic Z., Kong N., Kammlott U., Lukacs C., Klein C., et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 97.Neochoritis C.G., Wang K., Estrada-Ortiz N., Herdtweck E., Kubica K., Twarda A., Zak K.M., Holak T.A., Dömling A. 2,3´-Bis(1’H-indole) heterocycles: New p53/MDM2/MDMX antagonists. Bioorg. Med. Chem. Lett. 2015;25:5661–5666. doi: 10.1016/j.bmcl.2015.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ding K., Lu Y., Nikolovska-Coleska Z., Qiu S., Ding Y., Gao W., Stuckey J., Krajewski K., Roller P.P., Tomita Y., et al. Structure-based design of potent non-peptide MDM2 inhibitors. J. Am. Chem. Soc. 2005;127:10130–10131. doi: 10.1021/ja051147z. [DOI] [PubMed] [Google Scholar]
- 99.Kong N., Liu E.A., Vu B.T. Cis-imidazolines as MDM2 Inhibitors. PCT/EP2002/013905. International Patent Application No. 2003 Jun 26
- 100.Fantinati A., Bianco S., Cristofori V., Cavazzini A., Catani M., Zanirato V., Pacifico S., Rimondi E., Milani D., Voltan R., et al. Expeditious synthesis and biological characterization of enantio-enriched (-)-Nutlin-3. ChemistrySelect. 2017;2:8504–8508. doi: 10.1002/slct.201701059. [DOI] [Google Scholar]
- 101.Bartkovitz D.J., Cai J., Chu X.J., Li H., Lovey A.J., Vu B.T., Zhao C. Chiral Cis-Imidazolines. PCT/EP2008/063053. International Patent Application No. 2009 Apr 16;
- 102.Davis T.A., Johnston J.N. Catalytic, enantioselective synthesis of stilbene cis-diamines: A concise preparation of (-)-Nutlin-3, a potent p53/MDM2. Chem. Sci. 2011;2:1076–1079. doi: 10.1039/c1sc00061f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Davis T.A., Vilgelm A.E., Richmond A., Johnston J.N. Preparation of (−)-Nutlin-3 using enantioselective organocatalysis at decagram scale. J. Org. Chem. 2013;78:10605–10616. doi: 10.1021/jo401321a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.U.S. National Library of Medicine [(accessed on 12 July 2019)]; Available online: https://clinicaltrials.gov/ct2/show/NCT00623870.
- 105.Vara B.A., Mayasundari A., Tellis J.C., Danneman M.W., Arredondo V., Davis T.A., Min J., Finch K., Guy R.K., Johnston J.N. Organocatalytic, diastereo- and enantioselective synthesis of nonsymmetric cis-stilbene diamines: A platform for the preparation of single-enantiomer cis-imidazolines for protein−protein inhibition. J. Org. Chem. 2014;79:6913–6938. doi: 10.1021/jo501003r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Vu B., Wovkulich P., Pizzolato G., Lovey A., Ding Q., Jiang N., Liu J.J., Zhao C., Glenn K., Wen Y., et al. Discovery of RG7112: A small-molecule MDM2 inhibitor in clinical development. ACS Med. Chem. Lett. 2013;4:466–469. doi: 10.1021/ml4000657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tovar C., Graves B., Packman K., Filipovic Z., Higgins B., Xia M., Tardell C., Garrido R., Lee E., Kolinsky K., et al. MDM2 small-molecule antagonist RG7112 activates p53 signaling and regresses human tumors in preclinical cancer models. Cancer Res. 2013;73:2587–2597. doi: 10.1158/0008-5472.CAN-12-2807. [DOI] [PubMed] [Google Scholar]
- 108.Neochoritis C., Estrada-Ortiz N., Khoury K., Domling A. p53-MDM2 and MDMX Antagonists. Annu. Rep. Med. Chem. 2014;49:167–184. [Google Scholar]
- 109.Ray-Coquard I., Blay J.Y., Italiano A., Le Cesne A., Penel N., Zhi J., Heil F., Rueger R., Graves B., Ding M., et al. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: An exploratory proof-of-mechanism study. Lancet Oncol. 2012;13:1133–1140. doi: 10.1016/S1470-2045(12)70474-6. [DOI] [PubMed] [Google Scholar]
- 110.Czarna A., Beck B., Srivastava S., Popowicz G.M., Wolf S., Huang Y., Bista M., Holak T.A., Dömling A. Robust generation of lead compounds for protein–protein interactions by computational and MCR chemistry: p53/HDM2 antagonists. Angew. Chem. Int. Ed. Engl. 2010;49:5352–5356. doi: 10.1002/anie.201001343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Parks D.J., Lafrance L.V., Calvo R.R., Milkiewicz K.L., Gupta V., Lattanze J., Ramachandren K., Carver T.E., Petrella E.C., Cummings M.D., et al. 1,4-Benzodiazepine-2,5-diones as small molecule antagonists of the HDM2–p53 interaction: Discovery and SAR. Bioorg. Med. Chem. Lett. 2005;15:765–770. doi: 10.1016/j.bmcl.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 112.Hussie P.H., Gorina S., Marechal V., Elenbaas B., Moreau J., Levine A.J., Pavletich N.P. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science. 1996;274:948–953. doi: 10.1126/science.274.5289.948. [DOI] [PubMed] [Google Scholar]
- 113.Yu S., Qin D., Shangary S., Chen J., Wang G., Ding K., McEachern D., Qiu S., Nikolovska-Coleska Z., Miller R., et al. Potent and orally active small-molecule inhibitors of the MDM2−p53 interaction. J. Med. Chem. 2009;52:7970–7973. doi: 10.1021/jm901400z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sebahar P.R., Williams R.M. The asymmetric total synthesis of (+)-and (-)-spirotryprostatin B. J. Am. Chem. Soc. 2000;122:5666–5667. doi: 10.1021/ja001133n. [DOI] [Google Scholar]
- 115.Ding K., Wang G., Deschamps J.R., Parrish D.A., Wang S. Synthesis of spirooxindoles via asymmetric 1,3-dipolar cycloaddition. Tetrahedron Lett. 2005;46:5949–5951. doi: 10.1016/j.tetlet.2005.06.114. [DOI] [Google Scholar]
- 116.Ding K., Lu Y., Nikolovska-Coleska Z., Wang G., Qiu S., Shangary S., Gao W., Qin D., Stuckey J., Krajewski K., et al. Structure-based design of spiro-oxindoles as potent, specific small-molecule inhibitors of the MDM2-p53 interaction. J. Med. Chem. 2006;49:3432–3435. doi: 10.1021/jm051122a. [DOI] [PubMed] [Google Scholar]
- 117.Shangary S., Qin D., McEachern D., Liu M., Miller R.S., Qiu S., Nikolovska-Coleska Z., Ding K., Wang G., Chen J., et al. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc. Natl. Acad. Sci. USA. 2008;105:3393–3398. doi: 10.1073/pnas.0708917105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhao Y., Yu S., Sun W., Liu L., Lu J., McEachern D., Shargary S., Bernard D., Li X., Zhao T., et al. A potent small-molecule inhibitor of the MDM2−p53 interaction (MI-888) achieved complete and durable tumor regression in mice. J. Med. Chem. 2013;56:5553–5561. doi: 10.1021/jm4005708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhao Y., Liu L., Sun W., Lu J., McEachern D., Li X., Yu S., Bernard D., Ochsenbein P., Ferey V., et al. Diastereomeric spirooxindoles as highly potent and efficacious MDM2 inhibitors. J. Am. Chem. 2013;135:7223–7234. doi: 10.1021/ja3125417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wang S., Sun W., Zhao Y., McEachern D., Meaux I., Barrière C., Stuckey J., Meagher J., Bai L., Liu L., et al. SAR405838: An optimized inhibitor of MDM2-p53 interaction that induces complete and durable tumor regression. Cancer Res. 2014;74:5855–5865. doi: 10.1158/0008-5472.CAN-14-0799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zhao Y., Aguilar A., Bernard D., Wang S. Small-molecule inhibitors of the MDM2–p53 protein–protein interaction (MDM2 Inhibitors) in clinical trials for cancer treatment. J. Med. Chem. 2015;58:1038–1052. doi: 10.1021/jm501092z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Shangary S., Wang S. Targeting the MDM2-p53 interaction for cancer therapy. Clin. Cancer Res. 2008;14:5318–5324. doi: 10.1158/1078-0432.CCR-07-5136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bill K.L., Garnett J., Meaux I., Ma X., Creighton C.J., Bolshakov S., Barriere C., Debussche L., Lazar A.J., Prudner B.C., et al. SAR405838: A novel and potent inhibitor of the MDM2:p53 axis for the treatment of dedifferentiated liposarcoma. Clin. Cancer Res. 2016;22:1150–1160. doi: 10.1158/1078-0432.CCR-15-1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Jonge M., Weger V.A., Dickson M.A., Langenberg M., Le Cesne A., Wagner A.J., Hsu K., Zheng W., Macé S., Tuffal G., et al. A phase I study of SAR405838, a novel human double minute 2 (HDM2) antagonist, in patients with solid tumours. Eur. J. Cancer. 2017;76:144–151. doi: 10.1016/j.ejca.2017.02.005. [DOI] [PubMed] [Google Scholar]
- 125.U.S. National Library of Medicine [(accessed on 12 July 2019)]; Available online: https://clinicaltrials.gov/ct2/show/NCT01985191.
- 126.von Richter O., Massimini G., Scheible H., Udvaros I., Johne A. Pimasertib, a selective oral MEK1/2 inhibitor: Absolute bioavailability, mass balance, elimination route, and metabolite profile in cancer patients. Br. J. Clin. Pharmacol. 2016;82:1498–1508. doi: 10.1111/bcp.13078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bartkovitz D.J., Chu X.J., Ding Q., Graves B.J., Jiang N., Zhang J., Zhang Z. Spiroindolinone Pyrrolidines. PCT/EP2010/068353. International Patent Application No. 2011 Jun 9
- 128.Zhang Z., Ding Q., Liu J.J., Zhang J., Jiang N., Chu X.J., Bartkovitz D., Luk K.C., Janson C., Tovar C., et al. Discovery of potent and selective spiroindolinone MDM2 inhibitor, RO8994, for cancer therapy. Bioorg. Med. Chem. 2014;22:4001–4009. doi: 10.1016/j.bmc.2014.05.072. [DOI] [PubMed] [Google Scholar]
- 129.Shu L., Li Z., Gu C., Fishlock D. Synthesis of a Spiroindolinone Pyrrolidinecarboxamide MDM2 Antagonist. Org. Process Res. Dev. 2013;17:247–256. doi: 10.1021/op3003213. [DOI] [Google Scholar]
- 130.Zhang Z., Chu X.J., Liu J.J., Ding Q., Zhang J., Bartkovitz D., Jiang N., Karnachi P., So S.S., Tovar C., et al. Discovery of Potent and Orally Active p53-MDM2 Inhibitors RO5353 and RO2468 for Potential Clinical Development. ACS Med. Chem. Lett. 2014;5:124–127. doi: 10.1021/ml400359z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ivanenkov Y.A., Vasilevski S.V., Beloglazkina E.K., Kukushkin M.E., Machulkin A.E., Veselov M.S., Chufarova N.V., Chernyaginab E.S., Vanzcoo A.S., Zyk N.V., et al. Design, synthesis and biological evaluation of novel potent MDM2/p53 small-molecule inhibitors. Bioorg. Med. Chem. Lett. 2015;25:404–409. doi: 10.1016/j.bmcl.2014.09.070. [DOI] [PubMed] [Google Scholar]
- 132.Amaral J.D., Silva D., Rodrigues C.M.P., Solá S., Santos M.M.M. A novel small molecule p53 stabilizer for brain cell differentiation. Front. Chem. 2019;7:15. doi: 10.3389/fchem.2019.00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.U.S. National Library of Medicine [(accessed on 16 July 2019)]; Available online: https://clinicaltrials.gov/ct2/show/NCT02319369.
- 134.U.S. National Library of Medicine [(accessed on 16 July 2019)]; Available online: https://clinicaltrials.gov/ct2/show/NCT01877382.
- 135.U.S. National Library of Medicine [(accessed on 16 July 2019)]; Available online: https://clinicaltrials.gov/ct2/show/NCT01451437.
- 136.U.S. National Library of Medicine [(accessed on 16 July 2019)]; Available online: https://clinicaltrials.gov/show/NCT01463696.
- 137.Liao G., Yang D., Ma L., Li W., Hu L., Zeng L., Wu P., Duan L., Liu Z. The development of piperidinones as potent MDM2-P53 protein-protein interaction inhibitors for cancer therapy. Eur. J. Med. Chem. 2018;159:1–9. doi: 10.1016/j.ejmech.2018.09.044. [DOI] [PubMed] [Google Scholar]
- 138.Wagner A.J., Banerji U., Mahipal A., Somaiah N., Hirsch H., Fancourt C., Johnson-Levonas A.O., Lam R., Meister A.K., Russo G., et al. Phase I trial of the human double minute 2 inhibitor MK-8242 in patients with advanced solid tumors. J. Clin. Oncol. 2017;35:1304–1311. doi: 10.1200/JCO.2016.70.7117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Guerriero J.L., Sotayo A., Ponichtera H.E., Castrillon J.A., Pourzia A.L., Schad S., Johnson S.F., Carrasco R.D., Lazo S., Bronson R.T., et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature. 2017;543:428–432. doi: 10.1038/nature21409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ravandi F., Gojo I., Patnaik M.M., Minden M.D., Kantarjian H., Johnson-Levonas A.O., Fancourt C., Lam R., Jones M.B., Knox C.D., et al. A phase I trial of the human double minute 2 inhibitor (MK-8242) in patients with refractory/recurrent acute myelogenous leukemia (AML) Leuk. Res. 2016;48:92–100. doi: 10.1016/j.leukres.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zhang Q., Zeng S.X., Lu H. Targeting p53-MDM2-MDMX loop for cancer therapy. Subcell. Biochem. 2014;85:281–319. doi: 10.1007/978-94-017-9211-0_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zheng G.H., Shen J.J., Zhan Y.C., Yi H., Xue S.T., Wang Z., Ji X.Y., Li Z.R. Design, synthesis and in vitro and in vivo antitumour activity of 3-benzylideneindolin-2-one derivatives, a novel class of small-molecule inhibitors of the MDM2-p53 interaction. Eur. J. Med. Chem. 2014;81:277–288. doi: 10.1016/j.ejmech.2014.05.027. [DOI] [PubMed] [Google Scholar]
- 143.Hardcastle I.R., Ahmed S.U., Atkins H., Farnie G., Golding B.T., Griffin R.J., Guyenne S., Hutton C., Kallblad P., Kemp S.J., et al. Small-molecule inhibitors of the MDM2-p53 protein–protein interaction based on an isoindolinone scaffold. J. Med. Chem. 2006;49:6209–6221. doi: 10.1021/jm0601194. [DOI] [PubMed] [Google Scholar]
- 144.Hardcastle I.R., Liu J., Valeur E., Watson A., Ahmed S.U., Blackburn T.J., Bennaceur K., Clegg W., Drummond C., Endicott J.A., et al. Isoindolinone inhibitors of the Murine Double Minute 2 (MDM2)-p53 protein–protein interaction: Structure-activity studies leading to improved potency. J. Med. Chem. 2011;54:1233–1243. doi: 10.1021/jm1011929. [DOI] [PubMed] [Google Scholar]
- 145.Chargari C., Leteur C., Angevin E., Bashir T., Schoentjes B., Arts J., Janicot M., Bourhis J., Deutsch E. Preclinical assessment of JNJ-26854165 (Serdemetan1), a novel tryptamine compound with radiosensitizing activity in vitro and in tumor xenografts. Cancer Lett. 2011;312:209–218. doi: 10.1016/j.canlet.2011.08.011. [DOI] [PubMed] [Google Scholar]
- 146.Kojima K., Burks J.K., Arts J., Andreeff M. The novel tryptamine derivative JNJ- 26854165 induces wild-type p53- and E2F1-mediated apoptosis in acute myeloid and lymphoid leukemias. Mol. Cancer Ther. 2010;9:2545–2557. doi: 10.1158/1535-7163.MCT-10-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Tabernero J., Dirix L., Schoffski P., Cervantes A., Lopez-Martin J.A., Capdevila J., van Beijsterveldt L., Platero S., Hall B., Yuan Z., et al. A phase I first-in-human pharmacokinetic and pharmacodynamic study of serdemetan in patients with advanced solid tumors. Clin. Cancer Res. 2011;17:6313–6321. doi: 10.1158/1078-0432.CCR-11-1101. [DOI] [PubMed] [Google Scholar]
- 148.Lacrampe J.F.A., Meyer C., Ligny Y.A.E., Csoka I.C.F., Van Hijfte L., Arts J., Schoentjes B., Wermuth C.G., Giethlen B., Contreras J.-M., et al. Inhibitors of the interaction between MDM2 and P53. PCT/EP2005/054604. International Patent Application No. 2006 Mar 30
- 149.Wang W., Qin J.-J., Voruganti S., Srivenugopal K.S., Nag S., Patil S., Sharma H., Wang M.-H., Wang H., Buolamwini J.K., et al. The pyrido[b]indole MDM2 inhibitor SP-141 exerts potent therapeutic effects in breast cancer models. Nat. Commun. 2014;5:5086. doi: 10.1038/ncomms6086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Foley C.A., Al-Issa Y.A., Hiller K.P., Mulcahy S.P. Synthesis and structure-activity relationships of 1-aryl-β-carbolines as affinity probes for the 5-hydroxytryptamine receptor. ACS Omega. 2019;4:9807–9812. doi: 10.1021/acsomega.9b01111. [DOI] [Google Scholar]
- 151.Ding Q., Zhang Z., Liu J.J., Jiang N., Zhang J., Ross T.M., Chu X.J., Bartkovitz D., Podlaski F., Janson C., et al. Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J. Med. Chem. 2013;56:5979–5983. doi: 10.1021/jm400487c. [DOI] [PubMed] [Google Scholar]
- 152.Rimmler G., Alker A., Bosco M., Diodone R., Fishlock D., Hildbrand S., Kuhn B., Moessner C., Peters C., Rege P.D., et al. Practical synthesis of MDM2 antagonist RG7388. Part 2: Development of the Cu(I) catalyzed [3 + 2] asymmetric cycloaddition process for the manufacture of idasanutlin. Org. Process Res. Dev. 2016;20:2050–2056. doi: 10.1021/acs.oprd.6b00319. [DOI] [Google Scholar]
- 153.U.S. National Library of Medicine [(accessed on 16 July 2019)]; Available online: https://clinicaltrials.gov/ct2/show/study/NCT01773408.
- 154.Michelsen K., Jordan J.B., Long A.M., Lewis J.K., Yang E., Rew Y., Zhou J., Yakowec P., Schnier P.D., Huang X., et al. Ordering of the N-terminus of human MDM2 by small molecule inhibitors. J. Am. Chem. Soc. 2012;134:17059–17067. doi: 10.1021/ja305839b. [DOI] [PubMed] [Google Scholar]
- 155.Rew Y., Sun D., Turiso F.G.L., Bartberger M.D., Beck H.P., Canon J., Chen A., Chow D., Deignan J., Fox B.M., et al. Structure-based design of novel inhibitors of the MDM2−p53 interaction. J. Med. Chem. 2012;55:4936–4954. doi: 10.1021/jm300354j. [DOI] [PubMed] [Google Scholar]
- 156.Yu M., Wang Y., Zhu J., Bartberger M.D., Canon J., Chen A., Chow D., Eksterowicz J., Fox B., Fu J., et al. Discovery of potent and simplified piperidinone-based inhibitors of the MDM2–p53 interaction. ACS Med. Chem. Lett. 2014;5:894–899. doi: 10.1021/ml500142b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Sun D., Li Z., Rew Y., Gribble M., Bartberger M.D., Beck H.P., Canon J., Chen A., Chen X., Chow D., et al. Discovery of AMG 232, a potent, selective, and orally bioavailable MDM2−p53 inhibitor in clinical development. J. Med. Chem. 2014;57:1454–1472. doi: 10.1021/jm401753e. [DOI] [PubMed] [Google Scholar]
- 158.U.S. National Library of Medicine [(accessed on 16 July 2019)]; Available online: https://clinicaltrials.gov/ct2/show/NCT02110355.
- 159.Rew Y., Sun D., Yan X., Beck H.P., Canon J., Chen A., Duquette J., Eksterowicz J., Fox B.M., Fu J., et al. Discovery of AM-7209, a Potent and Selective 4.Amidobenzoic Acid Inhibitor of the MDM2.p53 Interaction. J. Med. Chem. 2014;57:10499–10511. doi: 10.1021/jm501550p. [DOI] [PubMed] [Google Scholar]
- 160.Holzer P., Masuya K., Furet P., Kallen J., Valat-Stachyra T.S., Ferretti S., Berghausen J., Bouisset-Leonard M., Buschmann N., Pissot-Soldermann C., et al. Discovery of a dihydroisoquinolinone derivative (NVP-CGM097) – a highly potent and selective MDM2 inhibitor undergoing phase 1 clinical trials I p53wt tumors. J. Med. Chem. 2015;58:6348–6358. doi: 10.1021/acs.jmedchem.5b00810. [DOI] [PubMed] [Google Scholar]
- 161.U.S. National Library of Medicine [(accessed on 16 July 2019)]; Available online: https://clinicaltrials.gov/ct2/show/NCT01760525.
- 162.Townsend E.C., De Souza T., Murakami M.A., Montero J., Stevenson K., Christie A.L., Christodolou A.N., Vojinovic U., Kopp N., Barzaghi-Rinaudo P., et al. The MDM2 inhibitor NVP-CGM097 is highly active in a randomized preclinical trial of B-cell acute lymphoblastic leukemia patient derived xenografts. Blood. 2015;126:797. doi: 10.1182/blood.V126.23.797.797. [DOI] [Google Scholar]
- 163.de Turiso F.G.-L., Sun D., Rew Y., Bartberger M.D., Beck H.P., Canon J., Chen A., Chow D., Correll T.L., Huang X., et al. Rational design and binding mode duality of MDM2–p53 inhibitors. Med. Chem. 2013;56:4053–4070. doi: 10.1021/jm400293z. [DOI] [PubMed] [Google Scholar]
- 164.Gonzalez A.Z., Eksterowicz J., Bartberger M.D., Beck H.P., Canon J., Chen A., Chow D., Duquette J., Fox B.M., Fu J., et al. Selective and potent morpholinone inhibitors of the MDM2-p53 protein−protein interaction. J. Med. Chem. 2014;57:2472–2488. doi: 10.1021/jm401767k. [DOI] [PubMed] [Google Scholar]
- 165.Yang Y., Ludwig R.L., Jensen J.P., Pierre S.A., Medaglia M.V., Davydov I.V., Safiran Y.J., Oberoi P., Kenten J.H., Phillips A.C., et al. Small molecule inhibitors of HDM2 ubiquitin ligase activity stabilize and activate p53 in cells. Cancer Cell. 2005;7:547–559. doi: 10.1016/j.ccr.2005.04.029. [DOI] [PubMed] [Google Scholar]
- 166.Dickens M.P., Roxburgh P., Hock A., Mezna M., Kellam B., Vousden K.H., Fischer P.M. 5-Deazaflavin derivatives as inhibitors of p53 ubiquitination by HDM2. Bioorg. Med. Chem. 2013;21:6868–6877. doi: 10.1016/j.bmc.2013.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Allen J.G., Bourbeau M.P., Wohlhieter G.E., Bartberger M.D., Michelsen K., Hungate R., Gadwood R.C., Gaston R.D., Evans B., Mann L.W., et al. Discovery and optimization of chromenotriazolopyrimidines as potent inhibitors of the Mouse Double Minute 2-tumor protein 53 protein–protein interaction. J. Med. Chem. 2009;52:7044–7053. doi: 10.1021/jm900681h. [DOI] [PubMed] [Google Scholar]
- 168.Furet P., Masuya K., Kallen J., Stachyra-Valat T., Ruetz S., Guagnano V., Holzer P., Mah R., Stutz S., Vaupel A., et al. Discovery of a novel class of highly potent inhibitors of the p53-MDM2 interaction by structure-based design starting from a conformational argument. Bioorg. Med. Chem. Lett. 2016;26:4837–4841. doi: 10.1016/j.bmcl.2016.08.010. [DOI] [PubMed] [Google Scholar]
- 169.U.S. National Library of Medicine [(accessed on 16 July 2019)]; Available online: https://clinicaltrials.gov/ct2/show/NCT02343172.
- 170.Lee J.H., Zhang Q., Jo S., Chai S.C., Oh M., Im W., Lu H., Lim H.-S. Novel pyrrolopyrimidine-based R-helix mimetics: Cell-permeable inhibitors of protein–protein interactions. J. Am. Chem. Soc. 2011;133:676–679. doi: 10.1021/ja108230s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Danovi D., Meulmeester E., Pasini D., Migliorini D., Capra M., Frenk R., de Graaf P., Francoz S., Gasparini P., Gobbi A., et al. Amplification of Mdmx (or Mdm4) directly contributes to tumor formation by inhibiting p53 tumor suppressor activity. Mol. Cell. Biol. 2004;24:5835–5843. doi: 10.1128/MCB.24.13.5835-5843.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Bartel F., Schulz J., Bohnke A., Blumke K., Kappler M., Bache M., Schmidt H., Wurl P., Taubert H., Hauptmann S. Significance of HDMX-S (or MDM4) mRNA splice variant overexpression and HDMX gene amplification on primary soft tissue sarcoma prognosis. Int. J. Cancer. 2005;117:469–475. doi: 10.1002/ijc.21206. [DOI] [PubMed] [Google Scholar]
- 173.Reed D., Shen Y., Shelat A.A., Arnold L.A., Ferreira A.M., Zhu F., Mills N., Smithson D.C., Regni C.A., Bashford D., et al. Identification and characterization of the first small molecule inhibitor of MDMX. J. Biol. Chem. 2010;285:10786–10796. doi: 10.1074/jbc.M109.056747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Stefaniak J., Lewis A.M., Conole D., Galan S.R.G., Bataille C.J.R., Wynne G.M., Castaldi M.P., Lundbäck T., Russell A.J., Huber K.V.M. chemical instability and promiscuity of arylmethylidenepyrazolinone-based MDMX Inhibitors. ACS Chem. Biol. 2018;13:2849–2854. doi: 10.1021/acschembio.8b00665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Noushini S., Alipour E., Emami S., Safavi M., Ardestani S.K., Gohari A.R., Shafiee A., Foroumadi A. Synthesis and cytotoxic properties of novel (E)-3-benzylidene-7-methoxychroman-4-one derivatives. Daru. 2013;21:31. doi: 10.1186/2008-2231-21-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Zhuang C., Miao Z., Wu Y., Guo Z., Li J., Yao J., Xing C., Sheng C., Zhang W. Double-edged swords as cancer therapeutics: Novel, orally active, small molecules simultaneously inhibit p53-MDM2 interaction and the NF-kB pathway. J. Med. Chem. 2014;57:567–577. doi: 10.1021/jm401800k. [DOI] [PubMed] [Google Scholar]
- 177.Zhuang C., Miao Z., Zhu L., Dong G., Guo Z., Wang S., Zhang Y., Wu Y., Yao J., Sheng C., et al. Discovery, Synthesis, and biological evaluation of orally active pyrrolidone derivatives as novel inhibitors of p53-MDM2 protein–protein interaction. J. Med. Chem. 2012;55:9630–9642. doi: 10.1021/jm300969t. [DOI] [PubMed] [Google Scholar]
- 178.Grasberger B.L., Lu T., Schubert C., Parks D.J., Carver T.E., Koblish H.K., Cummings M.D., LaFrance L.V., Milkiewicz K.L., Calvo R.R., et al. Discovery and cocrystal structure of benzodiazepinedione HDM2 antagonists that activate p53 in cells. J. Med. Chem. 2005;48:909–912. doi: 10.1021/jm049137g. [DOI] [PubMed] [Google Scholar]
- 179.Zhuang C., Miao Z., Zhu L., Zhang Y., Guo Z., Yao J., Dong G., Wang S., Liu Y., Chen H., et al. Synthesis and biological evaluation of thio-benzodiazepines as novel small molecule inhibitors of the p53-MDM2 protein–protein interaction. Eur. J. Med. Chem. 2011;46:5654–5661. doi: 10.1016/j.ejmech.2011.09.043. [DOI] [PubMed] [Google Scholar]
- 180.Wang H., Ma X., Ren S., Buolamwini J.K., Yan C. A small-molecule inhibitor of MDMX activates p53 and induces apoptosis. Mol. Cancer Ther. 2011;10:69–79. doi: 10.1158/1535-7163.MCT-10-0581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Pishas K.I., Adwal A., Neuhaus S.J., Clayer M.T., Farshid G., Staudacher A.H., Callen D.F. XI-006 induces potent p53-independent apoptosis in Ewing sarcoma. Sci. Rep. 2015;5:11465. doi: 10.1038/srep11465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Qiao C., Yonghao Y., Wang L. Application of benzofuroxan derivative to resisting plant pathogenic fungi. 201310003647.5. Chinese Patent Application No. 2013 Apr 3
- 183.Issaeva N., Bozko P., Engel M., Protopopova M., Verhoef1 L.G.G.C., Masucci M., Pramanik A., Selivanova G. Small molecule RITA binds to p53, blocks p53–HDM-2 interaction and activates p53 function in tumors. Nat. Med. 2004;10:1321–1328. doi: 10.1038/nm1146. [DOI] [PubMed] [Google Scholar]
- 184.Burmakin K., Shi Y., Hedström E., Kogner P., Selivanova G. Dual targeting of wild-type and mutant p53 by small molecule RITA results in the inhibition of N-Myc and key survival oncogenes and kills neuroblastoma cells in vivo and in vitro. Clin. Cancer Res. 2013;19:5092–5103. doi: 10.1158/1078-0432.CCR-12-2211. [DOI] [PubMed] [Google Scholar]
- 185.Jiang J., Ding C., Li L., Gao C., Jiang Y., Tan C., Hua R. Synthesis and antiproliferative activity of RITA and its analogs. Tet. Lett. 2014;55:6635–6638. doi: 10.1016/j.tetlet.2014.10.074. [DOI] [Google Scholar]
- 186.Lain S., Midgley C., Sparks A., Lane E.B., Lane D.P. An inhibitor of nuclear export activates the p53 response and induces the localization of HDM2 and p53 to U1A-positive nuclear bodies associated with the PODs. Exp. Cell. Res. 1999;248:457–472. doi: 10.1006/excr.1999.4433. [DOI] [PubMed] [Google Scholar]
- 187.Freedman D.A., Levine A.J. Nuclear export is required for degradation of endogenous p53 by MDM2 and human papillomavirus E6. Mol. Cell. Biol. 1998;18:7288–7293. doi: 10.1128/MCB.18.12.7288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Kudo N., Matsumori N., Taoka H., Fujiwara D., Schreiner E.P., Wolff B., Yoshida M., Horinouchi S. Leptomycin B inactivates CRM1/exportin 1 by covalent modification at a cysteine residue in the central conserved region. Proc. Natl. Acad. Sci. USA. 1999;96:9112–9117. doi: 10.1073/pnas.96.16.9112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Lain S., Xirodimas D., Lane D.P. Accumulating active p53 in the nucleus by inhibition of nuclear export: A novel strategy to promote the p53 tumor suppressor function. Exp. Cell. Res. 1999;253:315–324. doi: 10.1006/excr.1999.4672. [DOI] [PubMed] [Google Scholar]
- 190.Shao C., Lu C., Chen L., Koty P.P., Cobos E., Gao W. p53-dependent anticancer effects of leptomycin B on lung adenocarcinoma. Cancer Chemother. Pharmacol. 2011;67:1369–1380. doi: 10.1007/s00280-010-1434-6. [DOI] [PubMed] [Google Scholar]
- 191.Mutka S.C., Yang W.Q., Dong S.D., Ward S.L., Craig D.A., Timmermans P.B.M.W.M., Murli S. Identification of nuclear export inhibitors with potent anticancer activity in vivo. Cancer Res. 2009;69:510–517. doi: 10.1158/0008-5472.CAN-08-0858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Kobayashi M., Wang W., Tsutsui Y., Sugimoto M., Murakami N. Absolute stereostructure and total synthesis of leptomycin B. Tetrahedron Lett. 1998;39:8291–8294. doi: 10.1016/S0040-4039(98)01809-7. [DOI] [Google Scholar]
- 193.Jaras M., Miller P.G., Chu L.P., Puram R.V., Fink E.C., Schneider R.K., Al- Shahrour F., Peña P., Breyfogle L.J., Hartwell K.A., et al. Csnk1a1 inhibition has p53-dependent therapeutic efficacy in acute myeloid leukemia. J. Exp. Med. 2014;211:605–612. doi: 10.1084/jem.20131033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Minzel W., Venkatachalam A., Fink A., Hung E., Brachya G., Burstain I., Shaham M., Rivlin A., Omer I., Zinger A., et al. Small molecules co-targeting CKIa and the transcriptional kinases CDK7/9 control AML in preclinical models. Cell. 2018;175:171–185. doi: 10.1016/j.cell.2018.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Haigis M.C., Sinclair D.A. Mammalian sirtuins: Biological insights and disease relevance. Annu. Rev. Pathol. 2010;5:253–295. doi: 10.1146/annurev.pathol.4.110807.092250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Van Leeuwen I., Lain S. Sirtuins and p53. Adv. Cancer Res. 2009;102:171–195. doi: 10.1016/S0065-230X(09)02005-3. [DOI] [PubMed] [Google Scholar]
- 197.Luo J., Nikolaev A.Y., Imai S., Chen D., Su F., Shiloh A., Guarente L., Gu W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107:137–148. doi: 10.1016/S0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- 198.Langley E., Pearson M., Faretta M., Bauer U.M., Frye R.A., Minucci S., Pelicci P.G., Kouzarides T. Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. EMBO J. 2002;21:2383–2396. doi: 10.1093/emboj/21.10.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Vaziri H., Dessain S.K., Ng Eaton E., Imai S.I., Frye R.A., Pandita T.K., Guarente L., Weinberg R.A. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/S0092-8674(01)00527-X. [DOI] [PubMed] [Google Scholar]
- 200.Lain S., Hollick J.J., Campbell J., Staples O.D., Higgins M., Aoubala M., McCarthy A.R., Appleyard V., Murray K.E., Baker L., et al. Discovery, in vivo activity, and mechanism of action of a small-molecule p53 activator. Cancer Cell. 2008;13:454–463. doi: 10.1016/j.ccr.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.McCarthy A.R., Pirrie L., Hollick J.J., Ronseaux S., Campbell J., Higgins M., Staples O.D., Tran F., Slawin A.M.Z., Lain S., et al. Synthesis and biological characterisation of sirtuin inhibitors based on the tenovins. Bioorg. Med. Chem. 2012;20:1779–1793. doi: 10.1016/j.bmc.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 202.Zhang Q., Zeng S.X., Zhang Y., Zhang Y., Ding D., Ye Q., Meroueh S.O., Lu H. A small molecule Inauhzin inhibits SIRT1 activity and suppresses tumour growth through activation of p53. EMBO Mol. Med. 2012;4:298–312. doi: 10.1002/emmm.201100211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Zhang Q., Ding D., Zeng S.X., Ye Q.-Z., Lu H. Structure and activity analysis of inauhzin analogs as novel antitumor compounds that induce p53 and inhibit cell growth. PLoS ONE. 2012;7:e46294. doi: 10.1371/journal.pone.0046294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Kokel B. A new entry to the imidazo[4,5-d] pyrimidine system. The reaction of 1,3-dimethyl-6-aminouracil with N,N-dimethyldichloromethyleniminium chloride (phosgeniminium chloride) and trimethylsilyl azide, a novel and convenient “one pot” synthesis of 8-N-arylaminotheophyllines (2-N-arylamino-4,6-dimethylimidazo[4,5-d]pyrimidine-(4H,6H)-5,7-diones), starting from 1,3-dimethyl-6-aminouracil. Heterocycl. Chem. 1994;31:1185. [Google Scholar]
- 205.Lukman S., Lane D.P., Verma C.S. Mapping the structural and dynamical features of multiple p53 DNA binding domains: Insights into loop 1 intrinsic dynamics. PLoS ONE. 2013;8:e80221. doi: 10.1371/journal.pone.0080221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Cino E.A., Soares I.N., Pedrote M.M., de Oliveira G.A., Silva J.L. Aggregation tendencies in the p53 family are modulated by backbone hydrogen bonds. Sci. Rep. 2016;6:32535. doi: 10.1038/srep32535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Lima I., Navalkar A., Maji S.K., Silva J.L., de Oliveira G.A.P., Cino E.A. Biophysical characterization of p53 core domain aggregates. Biochem. J. 2020;477:111–120. doi: 10.1042/BCJ20190778. [DOI] [PubMed] [Google Scholar]
- 208.de Oliveira G.A.P., Petronilho E.C., Pedrote M.M., Marques M.A., Vieira T.C.R.G., Cino E.A., Silva J.L. The status of p53 oligomeric and aggregation states in cancer. Biomolecules. 2020;10:548. doi: 10.3390/biom10040548. [DOI] [PMC free article] [PubMed] [Google Scholar]