

Abstract

Cytochrome P450 46A1 (CYP46A1) is a central nervous system-specific enzyme, which catalyzes cholesterol 24-hydroxylation. Currently CYP46A1 is being evaluated in a clinical trial for activation by small doses of the anti-HIV drug efavirenz. Eight efavirenz-related compounds were investigated for CYP46A1 activation in vitro, induction of a CYP46A1 spectral response, spectral Kd values, interaction with the P450 allosteric sites, and a model of binding to the enzyme active site. We gained insight into structure–activity relationships of efavirenz for CYP46A1 activation and found that the investigated efavirenz primary metabolites are stronger and better activators of CYP46A1 than efavirenz. We also established that CYP46A1 is activated by racemates and that a conformational-selection mechanism is operative in CYP46A1. The results suggest structural modifications of efavirenz to further increase CYP46A1 activation without inhibition at high compound concentrations. It is possible that not only efavirenz but its metabolites activate CYP46A1 in vivo.
Introduction
CYP46A1 is an endoplasmic reticulum cytochrome P450 enzyme, which is expressed in the central nervous system, normally in the soma and dendrites of multiple neuron types.2−4 CYP46A1 converts cholesterol to 24-hydroxycholesterol (24HC), an oxysterol that rapidly diffuses across the blood–brain barrier to the systemic circulation and is delivered to the liver for further degradation to bile acids.2,5,6 Initially CYP46A1 was recognized for its key roles in brain cholesterol elimination and turnover.2,7,8 CYP46A1 was later discovered to be linked to other brain processes including memory and learning.9−12 CYP46A1 is now a focus for intensive investigations as a potential target for treatment of different brain disorders with both enzyme inhibition and activation having therapeutic potential.13 Animal studies suggest that increased CYP46A1 activity can ameliorate the manifestations of Alzheimer’s and Huntington’s diseases.14−18 Conversely, a decreased CYP46A1 activity is hypothesized to reduce seizure frequency and is now being tested in two clinical trials in subjects with Dravet, Lennox-Gastaut, and Duplication 15q syndromes as well as CDKL5 deficiency disorder.12,13 In addition to the brain, CYP46A1 is of importance for the retina and retinal blood vessels.19
This laboratory discovered that CYP46A1 could be activated pharmacologically in mice by (S)-efavirenz (EFV), an anti-HIV drug.20−22 Remarkably, the CYP46A1 activating EFV dose was very low (0.1 (mg/day)/kg body weight), at least 80 times lower than that (600 mg/day) given to HIV-positive individuals to keep their viral load low. When administered orally to normal (C57BL/6J) mice or a model of Alzheimer’s disease (5XFAD mice), this low EFV dose not only activated CYP46A1 in the brain but also elicited a compensatory increase in cholesterol biosynthesis and hence turnover.20−22 As a result, the steady-state levels of cholesterol in the brain remained unchanged in C57BL/6J mice and were normalized in 5XFAD mice. In the latter, EFV treatment also led to behavioral improvements with the effect on the total brain amyloid β load being the treatment paradigm-specific.21,22 However, when the EFV dose was increased to 0.22 (mg/day)/kg body weight, CYP46A1 activation was no longer observed and the higher 0.32 (mg/da)/kg body dose even inhibited the enzyme.20 Thus, the therapeutic window for CYP46A1 activation by EFV in mice appears to be quite narrow.
In vitro studies utilizing purified recombinant CYP46A1 revealed that the concentration dependence curves of EFV for the CYP46A1-mediated cholesterol 24-hydroxylation were bell-shaped with the enzyme activation at low compound concentrations and inhibition at high concentartions.20 Subsequent mechanistic investigations suggested an allosteric site for EFV on CYP46A1,20 which was then mapped experimentally to the cytosolic (proximal) surface of the P450 molecule.23 This site was close to the redox partner binding site but away from the CYP46A1 active site, a banana-shaped tunnel extending from the lipid-embedded distal CYP46A1 surface to the interior of the protein molecule.24 We proposed that at low concentrations EFV binds to the allosteric site and activates CYP46A1, whereas at high concentrations the drug likely binds to both the allosteric and active sites and inhibits CYP46A1 because of the competition with cholesterol for the active site.20 EFV was also found to increase the rate of the fast CYP46A1 reduction and the total amount of reduced P450.1 Furthermore, the analysis of the CYP46A1 crystal structure24 suggested that EFV could displace the water molecule in the allosteric site and thereby alter the hydrogen bond network that connects the allosteric and active sites and determines in part the hydration of the active site.23
Currently, we are involved in an ongoing clinical trial of EFV in patients with mild cognitive impairment due to Alzheimer’s disease (NCT03706885, ClinicalTrials.gov), which tests the 50 and 200 mg/day EFV doses. In parallel, we have a goal of generating a compound with a broader therapeutic window for CYP46A1 activation than EFV and preferably more potent to lower a possibility of CYP46A1 inhibition and hence side effects while perhaps enhancing the potentially beneficial drug effects. Therefore we initiated the current study to gain insights into the role of some of the EFV functionalities. We tested two types of EFV-related compounds for the effects on purified CYP46A1 in vitro: some of the primary EFV metabolites produced during hepatic drug clearance4,25−28 and EFV analogs with changes in the oxazinone and cyclopropyl rings.
Results
CYP46A1 Activation by (S)-EFV and Related Compounds
In mammals, (S)-EFV is mainly oxidized to (S)-8-hydroxyEFV (1), with the metabolism to (S)-7-hydroxyEFV (2) occurring to a lesser extent (Figure 1a). Both of these metabolites can be further hydroxylated to yield (S)-8,14-dihydroxyEFV (3) and (S)-7,8-dihydroxyEFV (4).4,25−28 (S)-EFV also has secondary metabolites produced as a result of glucuronidation and/or sulfation of the EFV oxidation products.25,29 Of the primary EFV metabolites, only (S)-1 is commercially available along with racemic (rac)-1, rac-2, and rac-3. Hence, all these compounds were tested in the enzyme assay using purified recombinant CYP46A1. Similar to CYP46A1 activation by (S)-EFV, the concentration dependence curves for (S)-1, rac-1, and rac-2 were bell-shaped, whereas that for rac-3 was essentially hyperbolic (Figure 1b). All of the tested metabolites had a maximal CYP46A1 activation higher than that with (S)-EFV (5.8-6.2-fold vs 4.1-fold, Table 1), which was observed at the same 20 μM metabolite concentration. (S)-1 and rac-1 had almost identical concentration dependence curves and along with rac-2 did not inhibit CYP46A1 at the 60–100 μM concentrations that were inhibitory for (S)-EFV. Thus, EFV hydroxylations at C-7, -8, and -14 render the drug properties that we sought when the study was initiated, namely, an increase in CYP46A1 activation at lower metabolite concentrations and a decrease in enzyme inhibition at higher compound concentrations. We then evaluated commercially available EFV analogs (Figure 1c): rac-5, which has a C atom instead of N-1; (S)-6, lacking the keto group; rac-7, which has a methyl group instead of the keto group; rac-8, which has a C atom instead of O-3; rac-9, which has an ethyl group instead of a cyclopropyl ring. All of these compounds, except rac-5, had bell-shaped concentration dependence curves, and CYP46A1 activation was either similar (rac-8) or lower than that with (S)-EFV(rac-5, (S)-6, rac-7, and rac-9). P450 inhibition was decreased for all compounds at their higher concentrations, and rac-5 as well as (S)-6 did not inhibit the enzyme at the concentrations tested. Thus, lack of O-3 (rac-8) did not significantly impair CYP46A1 activation by the derivative but decreased CYP46A1 inhibition at higher compound concentrations. Similarly, CYP46A1 inhibition was decreased by the replacement or elimination of EFV functionalities at positions 1 (rac-5), 2 ((S)-6), and 14 (rac-9), but the enzyme activation was less than that by (S)-EFV.
Figure 1.

EFV metabolites and analogs. (a, c) Chemical structures of EFV, its metabolites, and analogs studied in the present work. Structural divergence from EFV is highlighted in either red font or circles. EFV metabolism by the cytochrome P450 enzymes is also shown and taken with permission from ref (4). (b, d) Dependence of CYP46A1 activity in vitro on the concentration of (S)-EFV or an EFV-related compound. CYP46A1 activity (the rate of cholesterol 24-hydroxylation) is presented as nanomoles of 24-hydroxycholesterol (24HC) formed per nmole of CYP46A1 per min. The range of the tested compound concentrations is based on our previous study,20 in which 100 μM (S)-EFV almost completely inhibited CYP46A1. The results are the mean ± SD of the measurements from the three independent experiments.
Table 1. Summary of the CYP46A1 Responses to the Studied EFV-Related Compounds.
| interacts via the allosteric site for |
||||||
|---|---|---|---|---|---|---|
| compd | substrate-free CYP46A1a | cholesterol-bound CYP46A1a | maximal activation (fold) | maximal activation in the presence of l-Glu (fold) | EFV | Glu |
| (S)-EFV | type 2; H | reverse type 1; S | 4.1 | 4.9 | + | – |
| (S)-1 | type 2; H | reverse type 1; S | 6.2 | 7.4 | + | + |
| rac-2 | type 2; H | reverse type 1; S | 5.8 | 6.8 | + | + |
| rac-5 | type 1; S | no spectral response | 1.9 | 2.8 | + | + |
| rac-3 | type 2; H | reverse type 1; H | 6.1 | 4.8 | – | + |
| (S)-6 | type 1; H | no spectral response | 2.5 | abolished | – | + |
| rac-7 | type 1; H | no spectral response | 2.6 | abolished | – | + |
| rac-8 | type 1; H | reverse type 1; S | 3.9 | 4.6 | + | – |
| rac-9 | type I; H | reverse type 1; S | 3.1 | 3.7 | + | – |
Presented is the spectral response type and fit: H, hyperbolic fit; S, sigmoidal fit.
Spectral Responses in CYP46A1 Induced by (S)-EFV and Related Compounds
CYP46A1 is a hemoprotein and, as with most P450s, has a water molecule coordinating its heme iron in the sixth axial position when in the resting, substrate-free state.24 When this water molecule is displaced and the heme iron is left uncoordinated, the CYP46A1 difference spectrum has a peak at ∼390 nm and a trough at ∼422 nm,20 a so-called type I spectral response, elicited in many cases by the P450 substrates.30−32 However, when the heme iron becomes coordinated with a nitrogen atom, a trough around ∼410 nm and a peak at ∼421–435 nm appear in the P450 difference spectrum,33 a so-called type 2 spectral response, often produced by P450 inhibitors.30−32 A third type of the spectral response for the P450 enzymes has also been described, reverse type I, which represents a mirror reflection of a type I difference spectrum (a trough at 380–390 nm and peak at 415–420 nm).32 This response is believed to reflect the formation of an Fe–O bond, where the oxygen donor atom is from either a water molecule or the added compound.32,34 Previously we reported that titrations with (S)-EFV induced a type 2 spectral response in substrate-free CYP46A1 (Figure 2), which could be fit (ΔA) to a hyperbolic curve.20 These data suggested that in the absense of cholesterol, EFV binds to the CYP46A1 active site and coordinates the heme iron with its N-1 atom. Similarly, type 2 spectral responses that fit to a hyperbolic curve were observed in substrate-free CYP46A1 upon the addition of (S)-1, rac-1, rac-2, and rac-3, an indication that the hydroxyl groups at C-7, C-8, and C-8,14 do not prevent EFV derivatives from binding to the CYP46A1 active site in an orientation that enables the formation of the N–Fe coordinate bond (Figure 2). Yet the apparent Kd values of the hydroxylated EFV metabolites were 2–5 times higher than that of (S)-EFV, suggesting that the affinity of these compounds for CYP46A1 was decreased (Figure 1b). Conversely, rac-5, (S)-6, rac-7, rac-8, and rac-9 elicited a type 1 spectral response in substrate-free CYP46A1 consistent with absence of N-1 (rac-5) as well as the hydrogen bond-forming functionalities ((S)-6, rac-7, rac-8) that may help EFV bind in the CYP46A1 active site in the orientation suitable for the ligation of the heme iron. In addition, testing of rac-9 revealed the importance of the EFV cyclopropyl ring for the drug coordination of the heme iron. As with type 2 ligands, the binding of type 1 ligands can be fit to a hyperbolic curve except in the case of rac-5, whose curve was sigmoidal and may underlie the sigmoidal-like curve in the concentration dependence enzyme assay (Figure 1d).
Figure 2.

Spectral responses induced in substrate-free CYP46A1 by EFV-related compounds. Data for (S)-EFV are shown in the center. ΔA is the amplitude of spectral changes in the P450 difference spectra (insets). The results are presented as the mean ± SD of the measurements in the three spectral titrations.
The allosteric response of CYP46A1 was only revealed when EFV was added to the P450 in the presence of the saturating (20 μM) cholesterol, i.e., substrate-bound enzyme.20 The spectral titration curve became sigmoidal, and the spectral response was of a reverse type 1 nature (Figure 3). On the basis of this and other data, we suggested EFV binding to an allosteric site and interpreted this response as an altered hydration of the CYP46A1 active site with the subsequent formation of the Fe–O coordinate bond.20,23 Similarly, sigmoidal curves and a reverse type 1-like spectral response were observed in CYP46A1 upon addition of (S)-1, rac-1, rac-2, rac-8, and rac-9, consistent with comparable (≤25%) or higher CYP46A1 activation by these compounds at lower concentrations and their bell-shape concentration dependence curves (Figure 1b,d). rac-3 also elicited a reverse type 1-like spectral response in cholesterol-bound CYP46A1, but its spectral binding was of a hyperbolic nature. Since this metabolite essentially did not inhibit CYP46A1 in the concentration dependence enzyme assay (Figure 1b), we suggest that this response could reflect preferential EFV binding only to the allosteric site. Lastly, rac-5, (S)-6, and rac-7 did not seem to elicit a quantifiable spectral response in cholesterol-bound CYP46A1 while still activating the P450 (Figure 2d), although to a lower extent than EFV (up to 2.6-fold vs 4.1-fold). These data suggest that compound interactions with the allosteric site(s) could be spectrally silent and that this spectral silence could be due to absence of the hydrogen-forming N-1 (rac-5) as well as the 2-keto group ((S)-6 and rac-7) in the tested derivatives. CYP46A1 spectral titrations with different EFV-related compounds are summarized in Table 1.
Figure 3.

Spectral responses induced in cholesterol-bound CYP46A1 by EFV-related compounds. Data for (S)-EFV are shown in the center. ΔA is the amplitude of spectral changes in the P450 difference spectra (insets). The results are presented as the mean ± SD of the measurements in the three spectral titrations.
Mapping of the Allosteric Site(s) for Compound Binding
We first conducted in silico docking of the (S)- and (R)-isomers of 3, the best CYP46A1 activator in vitro. Autodock, VINA, and Ledock were used, and none of the docking programs could fit these isomers to the allosteric site for (S)-EFV. The three programs did provide a consensus model for (S)-3 and (R)-3 binding to the so-called glutamate (Glu)-binding site, a region on the CYP46A1 proximal surface adjacent to the site EFV-binding (Figure 4a–c).1 Glu activates CYP46A1 in vitro either alone or synergistically with (S)-EFV,1 and we tested how CYP46A1 co-incubations with l-Glu and rac-3 or other compounds affected the enzyme activity under the conditions of the maximal P450 activation (Figure 4d). l-Glu did not synergistically activate CYP46A1 in the co-incubations with rac-3, (S)-6, or rac-7, with the P450 activation becoming either lower (rac-3) or even abolished ((S)-6 or rac-7) relative to the P450 activity in the presence of either activator. Conversely, co-incubations with l-Glu and (S)-1, rac-2, rac-5, rac-8, or rac-9 synergistically activated CYP46A1 in vitro. These data suggested that rac-3 and (S)-6 or rac-7 probably bind mainly to the allosteric site for Glu and compete with the neurotransmitter for this site, consistent with the computational predictions for (S)-3 and (R)-3. In contrast, other EFV-related compounds likely bind to both EFV- and Glu-binding sites. Accordingly we then tested co-incubations of (S)-EFV with rac-3, (S)-6, or rac-7 and found a synergistic CYP46A1 activation (Figure 4d). Thus we obtained support for the interaction of rac-3, (S)-6, and rac-7 mainly with the allosteric site for Glu.
Figure 4.

Mapping of the CYP46A1 allosteric site for rac-3. (a–c) Computational models of (S)-EFV (black), l-Glu (yellow), (S)-3 (dark green), and (R)-3 (light green) binding to the CYP46A1 surface. Docking experiments were carried out on PDB code 2Q9G. Models for (S)-EFV and l-Glu are taken from ref (1), and those for (S)-3 and (R)-3 were generated in the present work. The heme group is in red. The nitrogen, oxygen, chlorine, and fluorine atoms are in blue, red, light green, and cyan, respectively. (d) Effect of l-Glu (100 μM), (S)-EFV (20 μM), or EFV analog (20 μM) on the maximal extent of CYP46A1 activation. CYP46A1 activity is presented as nanomoles of 24-hydroxycholesterol (24HC) formed per nmole of CYP46A1 per min. Control incubations with no activator, l-Glu only, and (S)-EFV only are shown as a white, yellow, and black bar, respectively. Incubations with an EFV-related compound only are shown as solid colored bars, and co-incubations with l-Glu and (S)-EFV are shown as dashed (yellow and black dashes, respectively) colored bars. The results are presented as the mean ± SD of the measurements from the three independent experiments.
To further map compound binding, we used the CYP46A1 mutants generated for this study as well as previously1,23 and assessed how the replacement of the amino acid residues specific for EFV binding site (R415A and F405A) and Glu binding site (K358R/L/A, F416A, and Y427F/A) as well as the loop region forming the common border between the two sites (K422A and R424A) affect maximal CYP46A1 activation by different EFV metabolites and derivatives. The P450 activation by rac-3, (S)-6, or rac-7 was not significantly affected by the smaller sized replacements within the EFV-binding site (R415A and F405A) but was largely reduced or abolished by the smaller sized replacements within the Glu-binding site (K358A, F416A, and Y427A) (Figure 5a). The effect of the alanine substitutions of the border-forming Lys-422 and Arg-424 was differential, and activation pattern of the CYP46A1 mutants by rac-8 and rac-9 was different with only EFV-site but not the Glu-site smaller sized mutations mainly affecting the P450 activation, evidence for the preferential EFV-site binding (Figure 5b). Lastly, the activation of all of the smaller sized CYP46A1 mutants was altered by (S)-1, rac-2, and rac-5, suggesting that these compounds bind to both EFV- and Glu-binding sites. Thus, depending on the structure, EFV-related compounds could bind either to both or mainly one of the allosteric sites and activate CYP46A1 in vitro with the extent of the P450 activation being compound-specific but not the allosteric site-specific (Table 1).
Figure 5.

Mapping of the CYP46A1 allosteric site(s) for binding of EFV-related compounds. (a, b) Maximal activity of wild type (WT) CYP46A1 and the P450 mutants in the absence (white bars) or presence of different EFV-related compounds (20 μM, colored bars). CYP46A1 activity is presented as nanomoles of 24-hydroxycholesterol (24HC) formed per nmole of CYP46A1 per min. The results are presented as the mean ± SD of the measurements from the three independent experiments. (c–e). Some of the amino acid residues forming EFV binding site (c) and Glu binding site (d and e) as well as the border between the sites (c–e). The positions of (S)-EFV, (S)-3, and (R)-3 within these sites are also shown. Docking experiments were carried out on PDB code 2Q9G.
Studies of a Model of (S)-EFV and rac-3 Binding to the CYP46A1 Active Site
These studies were carried out to gain insight into why there was no decrease in CYP46A1 activation and ultimately enzyme inhibition by rac-3 at higher metabolite concentrations as in the case with (S)-EFV (Figure 1b). Indeed, both (S)-EFV and rac-3 bind to the active site of substrate-free CYP46A1 and likely act as inhibitors by coordinating the P450 heme iron with the secondary amine group (Figure 2). Further, only a 2-fold difference in the apparent spectral Kd of rac-3 and (S)-EFV for substrate-free CYP46A1 is not large enough to predict that there would be no competition between rac-3 and cholesterol for the CYP46A1 active site as likely in the case with (S)-EFV. We first determined the binding rates of (S)-EFV and rac-3 for substrate-free CYP46A1 at different compound concentrations as diagnostic for a model of compound binding: conformation-selection vs induced-fit35,36 with the former discovered recently to be operative in several human P450 enzymes.37−40 Increasing compound concentrations increased the magnitude of the spectral changes with both (S)-EFV and rac-3 (Figure 6a–d) but clearly decreased the rates of binding (kobserved) (Figure 7). Notably, for rac-3, these rates were always lower (at least ∼7-fold) than those for (S)-EFV, and rac-3 showed essentially no spectral binding at 15–30 μM, the concentrations that still led to a measurable (S)-EFV binding. Similarly, when (S)-EFV and rac-3 were added to cholesterol-bound CYP46A1 (Figure 6e,f), the absorbance change for the type 2 spectral response was still observed for (S)-EFV at 50 μM but not rac-3 at 2 μM. A decrease in binding rates with increasing compound concentration indicates a conformation-selection model of binding.35,36 Collectively, the data obtained suggested that different CYP46A1 conformations are necessary for (S)-EFV and rac-3 binding to the P450 active site and that much less enzyme molecules exist in solution in the conformation favorable for rac-3, thus preventing it from efficient competition with cholesterol for the CYP46A1 active site.
Figure 6.

Binding of (S)-EFV and rac-3 to CYP46A1. The P450 (4 μM) was mixed with varying concentrations of a compound, and spectra were collected and analyzed. (a, b) Mixing CYP46A1 with low (2 μM) and (c, d) high (50 μM or 60 μM) compound concentrations. (e, f) Mixing of 4 μM P450 46A1·4 μM cholesterol complex with 50 μM or 2 μM compound. The fits shown are single exponentials. At least four individual traces were collected and averaged.
Figure 7.

Dependence of rates of binding to CYP46A1 as a function of compound concentration. Traces of Δ(A437 – A416) (e.g., Figure 6) were fit to single exponential plots. The error bars show the SD of the OLIS software fits. At least four separate reactions were averaged for each data point.
Next, we determined the apparent koff rates for cholesterol, (S)-EFV and rac-3 (Table 2, Figure S1). Release of cholesterol (koff) was very slow, 2 orders of magnitude slower than that of (S)- EFV and rac-3, perhaps a reason why the apparent Kd of cholesterol for CYP46A1 was also much lower than those of (S)-EFV and rac-3 (9 nM vs 5 μM). Accordingly the calculated kon rate of cholesterol was 1.7-fold lower than that for (S)-EFV but 1.9-fold higher than that for rac-3, another advantage of (S)-EFV relative to rac-3 in terms of binding to the CYP46A1 active site. Thus, not only the predominant enzyme conformation but also the steady state kon puts (S)-EFV at advantage,
Table 2. Compound Binding to CYP46A1a.
| compd | Kd for substrate-free CYP46A1 (M) | calculated konb (M–1 s–1) | experimental koff (s–1) |
|---|---|---|---|
| cholesterol | 9 × 10–9 c | 1.3 × 106 | 0.012 ± 0.002 |
| (S)-EFV | 5 × 10–6 d ± 0.4 × 10–6 | 2.2 × 106 | 11.2 ± 0.9 |
| rac-3 | 5 × 10–6 e ± 0.3 × 10–6 | 0.7 × 106 | 3.6 ± 0.2 |
Experimental data are presented as the mean ± SD of the measurements from the three independent experiments.
Values calculated
from Kd and koff assuming 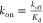 .
.
This value comes from an apparent Kd of 80 nM 20 after the correction for HPCD binding. See Experimental Section for details.
This is the Kd value for titrations at 24 °C 20 because the stopped-flow experiments were conducted at 23 °C.
Discussion and Conclusion
The present work provides insight into the structure–activity relationships of (S)-EFV for CYP46A1 activation. Of the four functionalities tested (the N-1, 2-keto group, O-3, and cyclopropyl ring, Figure 1a), only one (the O-3) does not seem to be important for CYP46A1 activation as indicated by the concentration dependence curve for rac-8 (Figure 1d). This curve was similar to that of (S)-EFV, consistent with compound binding to the same allosteric site as EFF (Figure 5b). Yet interaction with the CYP46A1 the active site was different as indicted by a type 1 spectral response in substrate-free CYP46A1 (Figure 2). In addition, our studies revealed that introducing the hydroxyl group at C-7, C-8, or both C-8 and C-14 of EFV increased the maximal extent of CYP46A1 activation and eliminated the undesired CYP46A1 inhibition at higher compound concentrations (Figure 1b). All three hydroxylated compounds ((S)-1, rac-1, rac-2, and rac-3) appeared to bind similarly to the CYP46A1 active site (i.e., elicited a type 2 spectral response in substrate-free CYP46A1, Figure 2) and seemed to interact with either the allosteric site for Glu (rac-3) or both EFV and Glu ((S)-1 and rac-2). As with rac-8, a reverse type 1 spectral response induced in cholesterol-bound CYP46A1 was a common feature of the hydroxylated compounds. However, only in the case of rac-3 did this spectral response fit to a hyperbolic curve. It is thus possible that a reverse type 1 spectral response in cholesterol-bound CYP46A1 and a hyperbolic spectral fit are the features of a compound that does not inhibit the P450 at higher concentrations while activating CYP46A1 to the extent comparable with that of (S)-EFV (Table 1). Further studies are required to address this possibility.
Notably the apparent Kd values for substrate-free CYP46A1 (Figure 2) did not appear to be indicative of CYP46A1 inhibition in the concentration dependence curves, as was demonstrated by rac-1, rac-2, and rac-3 (Figure 1b). To clarify the reason for a weak predictive power of an apparent Kd value, we capitalized on a recent discovery in the P450 field showing that the P450-compound interactions are complex and that conformational-selection is a dominant feature of many of these interactions for human P450s 2C6, 2D6, 3A4, 4A11, 17A1, and 21A2.37−40 By conducting the stopped-flow experiments, we ascertained that (S)-EFV and rac-3 bind to substrate-free CYP46A1 in a mechanism involving conformational-selection, which is apparently more favorable to (S)-EFV than rac-3, a bulkier compound requiring more space in the P450 active site. In addition, a 3-fold lower kon of rac-3 than that of (S)-EFV when cholesterol release from the CYP46A1 active site (the koff) is extremely slow (Table 2) could be another reason for caution when considering the apparent Kd values.
An important finding of the present work was that CYP46A1 could be activated by racemic mixtures, which is in contrast to HIV reverse transcriptase, which is inhibited only by the (S)-isomer of EFV.41 CYP46A1 activation and response to (S)-1 and rac-1 were similar but not identical (Figures 1b, 2, 3), and molecular docking of (S)-3 and (R)-3 suggested the same binding site but a different binding mode (Figure 4b,c). Thus, at least in some cases, using a racemate as compared to an expensive (S)-isomer could be more advantageous (besides lower compound cost) for activating CYP46A1 in vivo. First, we could still see the benefits of the enzyme activation while possibly minimizing the negative, non-CYP46A1 related effects42−44 suggested by several cell culture studies, in which (S)-8-hydroxyEFV (as well as (S)-7-hydroxyEFV but to a lesser extent and not always) was shown to induce cell damage or death.45−47 However, these cell culture findings are not supported by a study in humans showing that neither plasma nor cerebrospinal fluid levels of (S)-8-hydroxyEFV are associated with central nervous system toxicity.29 Also, the metabolite concentrations in the cell culture studies were designed to mimic the 600 mg/day EFV dose, which is >80-fold higher (∼8.5 (mg/kg of body weight)/day for a 70 kg person) than that (0.1 (mg/kg body weight)/day) given to mice. Second, CYP2B6 metabolizes (S)-EFV 10 times more efficiently in vitro than (R)-EFV and does not generate the (R) isomers of 7-hydroxyEFV (2), 7,8-dihydroxyEFV (4), and 8,14-dihydroxyEFV (3) from (R)-EFV.48 Therefore the plasma half-life of racemic EFV or a hydroxylated EFV metabolite could be longer and hence requires a less frequent drug administration to activate CYP46A1. Lastly, potential toxic effects of the R-isomers of EFV metabolites have not been studied and may not be so detrimental as those suggested for the (S)-isomers.45−47 An in vivo investigation with racemic EFV and/or 8,14-dihydroxyEFV is required to evaluate potential benefits of the racemate use indicated by the current work in vitro.
Of immediate clinical significance is our finding that EFV hydroxylations at positions 7, 8, and 14, which represent initial steps in EFV clearance by the liver,4,25−28 enhance CYP46A1 activation at lower metabolite concentrations without inhibiting the enzyme at higher concentrations (Figure 1b). This finding raises a possibility that not only EFV but also its metabolites activate CYP46A1 in vivo. Indeed, all these compounds are present in the plasma of EFV-treated subjects: EFV at 2170–3350 ng/mL; 8-hydroxyEFV (1) at 315–2160 ng/mL; 7-hydroxyEFV (2) at 8.8–225 ng/mL; 8,14-dihydroxyEFV (3) at 10.2 ng/mL.4,49−54 Also, the total polar surface areas of 38.3 Å2 and 78.8 Å2 for EFV and 8,14-dihydroxyEFV, respectively, and the predicted log P values of 4.53 and 3.17, respectively (calculated using the Molinspiration property calculation service), are within the desirable range for good CNS availability (<90–120 and 2–5).55 Consistent with these properties, EFV (13.9–19.5 ng/mL), 8-hydroxyEFV (1, 3.1–5.3 ng/mL), 7-hydroxyEFV (2, 0.63 ng/mL), and 8,14-dihydroxyEFV (3, 0.38–0.44 ng/mL) were detected in the cerebrospinal fluid, an established marker for free compound levels in the brain.4,29,49−53 The detected compound levels in the cerebrospinal fluid were however low, probably because in the blood, EFV and EFV metabolites are ≥99.6% protein (albumin)-bound;4,50,51 hence only small drug amounts likely cross the blood–brain barrier.44 Then once in the brain, EFV seems to readily accumulate in the tissue due to high propensity for protein binding as indicated by the experiments showing that EFV brain levels could exceed those in the plasma 4.6 times within 1 h of an intraperitoneal administration.56 In addition, EFV metabolites in the brain could be produced locally because EFV-metabolizing CYP2B6, CYP1A1, and CYP2C8 are not only expressed in the liver but also in the brain.57−59 Thus, EFV metabolites could be present in the brain and activate CYP46A1 along with EFV, a possibility that would explain in part why only a small EFV dose (0.1 (mg /kg body weight)/day) is sufficient to activate CYP46A1 in mouse brain.
To conclude, structure–activity relationships of EFV for CYP46A1 activation were investigated and revealed that hydroxylations at C-7, -8, and -14 increase CYP46A1 activation at lower compound concentrations and decrease enzyme inhibition at higher compound concentrations. EFV N-1, 2-keto group, and the cyclopropyl ring were found to contribute to CYP46A1 activation, whereas the drug O-3 did not seem to be important. All of the investigated EFV metabolite and analogs decreased CYP46A1 inhibition at higher concentrations, possibly because their binding to the P450 active site involves the conformational selection mechanism. We also mapped the allosteric sites of compound binding on the CYP46A1 surface. The data obtained suggest the future direction of our work. This includes testing the following compounds, first in vitro and then, if justified, in vivo as potentially better CYP46A1 activators than (S)-EFV: (S)- and rac-7,8-dihydroxyEFV as well as (S)-8,14-dihydroxyEFV. Efforts are in progress for the generation of these compounds.
Experimental Section
Materials
(S)-EFV ((S)-6-chloro-4-(cyclopropylethynyl)-4-(trifluoromethyl)-1,4-dihydro-2H-benzo[d][1,3]oxazin-2-one) and all EFV metabolites, as well as analogs, were purchased from Toronto Research Chemicals (Toronto, ON, Canada) and were >95% pure according to the manufacturer’s certificates of analysis (Figures S2–S11). 1H NMR and 19F NMR spectra of these compounds conformed to their structures. l-Glutamate was from MilliporeSigma (St. Louis, MO), and a 2 mM stock solution was prepared in water. Cholesterol was obtained from Steraloids (Newport, RI), and 24-hydroxy-[25,26,26,26,27,27,27-2H7]-cholesterol was from Medical Isotopes (Pelham, NH). Cholesterol was added from a 1 mM stock in 4.5%, w/v, aqueous 2-hydroxypropyl-β-cyclodextrin (HPCD), and deuterated 24-hydroxycholesterol was added from a 0.1 mM stock in methanol. Human truncated Δ(2–50)CYP46A1 with a four-histidine tag on the C terminus and rat cytochrome P450 oxidoreductase were expressed in Escherichia coli and purified as described.60,61 The F405A Δ(2–50)CYP46A1 mutant was generated by using an in vitro QuikChange site-directed mutagenesis kit (Stratagene, San Diego, CA) according to the instructions. The correct generation of the desired mutation and absence of undesired mutations were confirmed by nucleotide sequencing of the entire CYP46A1 coding region as well as by the restriction analysis.
Enzyme Assays
Incubations for the concentration dependence curves were carried out in 1 mL of 50 mM KPi buffer (pH 7.2) containing 100 mM NaCl, 40 μg/mL l-α-1,2-dilauroyl-sn-glycero-3-phosphocholine, 0.5 μM purified Δ(2-50)CYP46A1, 1.0 μM cytochrome P450 oxidoreductase, 40 μM cholesterol, varying concentrations of a test compound (0–100 μM added from 0.5 to 5 mM methanol stocks to keep the 2% final methanol concentration), 2 units of catalase, and an NADPH-regenerating system (1 mM NADPH, 10 mM glucose 6-phosphate, and 2 units of glucose 6-phosphate dehydrogenase). Enzymatic reactions proceeded for 30 min at 37 °C, followed by sterol extraction with 5 mL of dichloromethane containing 1 nmol of 24-hydroxy-[25,26,26,26,27,27,27-2H7]-cholesterol, which served as an internal standard. Sterol extracts were then processed and analyzed by gas chromatography–mass spectrometry as described.62 Incubations to determine for the maximal extent of CYP46A1 activation and the activation of the CY46A1 mutants were carried out similarly, except a fixed concentration (20 μM) of a test compound (added from a 5 mM stock in methanol) was used. Co-incubations with l-Glu were with 100 μM neurotransmitter.
Spectral Titrations
Spectral titrations were carried out at 18 °C as described,63 i.e., in 1 mL of 50 mM KPi buffer (pH 7.2) containing 100 mM NaCl and 0.4–0.5 μM Δ(2-50)CYP46A1, either substrate free or substrate-bound, i.e., in the presence of 20 μM cholesterol. Stock solutions of a 0.5–5 mM test compound in methanol or 0.5 mM cholesterol in 4.5% (w/v) aqueous HPCD were used. Data were fit by the GraphPad Prism software to either of the following equations:
or an allosteric sigmoidal curve, in which [E] is the enzyme concentration, ΔA is the spectral response at different compound concentrations [L], and ΔAmax is the maximal amplitude of the spectral response.
Computational Dockings
The structures of (S)-3 and (R)-3 were optimized with density functional theory calculations at the B3LYP/6-31+G* level of theory using Gaussian 16.64 These architectures were then used as the input for docking experiments, using Autodock 4.2.6,65 Autodock VINA,66 and Ledock.67 Input files were generated using Autodock Tools 1.5.668 and converted using OpenBabel 2.3.1.69 Docking experiments were carried out on PDB code 2Q9G,24 with a search box centered on the l-Glu binding pocket. The search box was similarly sized for all docking approaches (Autodock, 74 × 74 × 74, grid spacing 0.375 Å; Autodock VINA, 28 Å × 28 Å × 28 Å; Ledock, 28 Å × 28 Å × 28 Å). Visualization of docked compounds on the protein surface was carried out using PyMol.
Stopped-Flow Kinetics
Measurements were made in an OLIS RSM-1000 stopped-flow spectrophotometer (On-Line Instrument Systems, Bogart, GA) in the rapid-scanning mode, using a 20 mm × 4 mm cell, 16 mm × 0.2 mm Scandisk, 1.24 mm slits (8 nm bandpass), and 600 lines/500 nm gratings, operating at 23 °C. When the collection period was ≤4 s, data were collected at 1000 scans/s; when the collection time was >4 s, 62 scans/s were collected in the signal average mode. The wavelength range used was 330–570 nm. Measurements typically involved mixing one syringe containing 4 μM CYP46A1 in 50 mM KPi (pH 7.2), supplemented 100 mM NaCl, and another syringe containing a varying compound concentration in an equal volume of the same buffer. When CYP46A1 was premixed with equimolar cholesterol, the latter was added from a 2 mM stock in 4.5% (w/v) 2-hydroxypropyl-β-cyclodextrin. The data were saved as OLIS (.ols), and Excel files and converted to ΔAmax – ΔAmin plots. The resulting Excel files were corrected to ΔAt = 0 > 0, saved as txt files, and imported into KinTek Explorer software version 8.0 (KinTek, Snowshoe, PA).70 Analyses utilized an Apple iMac OSX 10.13.6 system.
The apparent rates of release (koff) of cholesterol, (S)-EFV,
and rac-3 from CYP46A1 were estimated
by mixing a prebound complex with compound under study (4 μM
CYP46A1 and 4 μM compound) with an excess of the inhibitor clotrimazole
(40 μM), which binds tightly and has a different spectral complex.33,71 An increase in the Δ(A437-A416) absorbance was measured, and
data from 8 to 12 plots were averaged. KinTek Explorer was used for
data fitting. Since 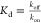 , the kon rates
were then calculated from the compound Kd values for substrate-free CYP46A1 (Figure 2). Cholesterol binds to both HPCD and CYP46A1;
therefore the true Kd of cholesterol for
CYP46A1 was calculated (9 nM) using KinTek Explorer and an apparent Kd of cholesterol for CYP46A1 (0.08 μM 20), the Kd of cholesterol
for HPCD in 50 mM KPi buffer (pH 7.2) containing 100 mM
NaCl at 24 °C (540 μM, determined in the present work as
described72), and the HPCD concentration
(3.1 mM).
, the kon rates
were then calculated from the compound Kd values for substrate-free CYP46A1 (Figure 2). Cholesterol binds to both HPCD and CYP46A1;
therefore the true Kd of cholesterol for
CYP46A1 was calculated (9 nM) using KinTek Explorer and an apparent Kd of cholesterol for CYP46A1 (0.08 μM 20), the Kd of cholesterol
for HPCD in 50 mM KPi buffer (pH 7.2) containing 100 mM
NaCl at 24 °C (540 μM, determined in the present work as
described72), and the HPCD concentration
(3.1 mM).
Acknowledgments
This work was supported in part by United States Public Health Service Grants R01 AG067552 (to I.A.P), P30 Core Grant EY011373, and Grant R01 GM118122 (to F.P.G.). The authors thank John Denker from the Molecular Biology and Genotyping Core of the Visual Sciences Research Center Core Facilities (supported by National Institutes of Health Grant P30 EY11373) for the generation of the CYP46A1 mutants.
Glossary
Abbreviations Used
- 24HC
24-hydroxycholesterol
- CYP
cytochrome P450
- EFV
efavirenz
- Glu
glutamate
- HPCD
2-hydroxypropyl-β-cyclodextrin
- rac
racemic
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.9b01383.
Author Contributions
N.M., P.V., C.J.W., and F.P.G. conducted experiments. N.M., P.V., F.P.G., and I.A.P. analyzed the data. N.M., P.V., F.P.G., and I.A.P wrote or contributed to the manuscript writing.
The authors declare no competing financial interest.
Supplementary Material
References
- Mast N.; Anderson K. W.; Johnson K. M.; Phan T. T. N.; Guengerich F. P.; Pikuleva I. A. In vitro cytochrome P450 46A1 (CYP46A1) activation by neuroactive compounds. J. Biol. Chem. 2017, 292, 12934–12946. 10.1074/jbc.M117.794909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund E. G.; Guileyardo J. M.; Russell D. W. cDNA cloning of cholesterol 24-hydroxylase, a mediator of cholesterol homeostasis in the brain. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 7238–7243. 10.1073/pnas.96.13.7238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez D. M.; Andersson S.; Russell D. W. Neuronal expression and subcellular localization of cholesterol 24-hydroxylase in the mouse brain. J. Comp. Neurol. 2008, 507, 1676–1693. 10.1002/cne.21605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avery L. B.; VanAusdall J. L.; Hendrix C. W.; Bumpus N. N. Compartmentalization and antiviral effect of efavirenz metabolites in blood plasma, seminal plasma, and cerebrospinal fluid. Drug Metab. Dispos. 2013, 41, 422–429. 10.1124/dmd.112.049601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretillon L.; Lutjohann D.; Stahle L.; Widhe T.; Bindl L.; Eggertsen G.; Diczfalusy U.; Björkhem I. Plasma levels of 24S-hydroxycholesterol reflect the balance between cerebral production and hepatic metabolism and are inversely related to body surface. J. Lipid Res. 2000, 41, 840–845. [PubMed] [Google Scholar]
- Meaney S.; Bodin K.; Diczfalusy U.; Björkhem I. On the rate of translocation in vitro and kinetics in vivo of the major oxysterols in human circulation: critical importance of the position of the oxygen function. J. Lipid Res. 2002, 43, 2130–2135. 10.1194/jlr.M200293-JLR200. [DOI] [PubMed] [Google Scholar]
- Lutjohann D.; Breuer O.; Ahlborg G.; Nennesmo I.; Siden A.; Diczfalusy U.; Björkhem I. Cholesterol homeostasis in human brain: evidence for an age-dependent flux of 24S-hydroxycholesterol from the brain into the circulation. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 9799–9804. 10.1073/pnas.93.18.9799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund E. G.; Xie C.; Kotti T.; Turley S. D.; Dietschy J. M.; Russell D. W. Knockout of the cholesterol 24-hydroxylase gene in mice reveals a brain-specific mechanism of cholesterol turnover. J. Biol. Chem. 2003, 278, 22980–22988. 10.1074/jbc.M303415200. [DOI] [PubMed] [Google Scholar]
- Kotti T. J.; Ramirez D. M.; Pfeiffer B. E.; Huber K. M.; Russell D. W. Brain cholesterol turnover required for geranylgeraniol production and learning in mice. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 3869–3874. 10.1073/pnas.0600316103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maioli S.; Bavner A.; Ali Z.; Heverin M.; Ismail M. A.; Puerta E.; Olin M.; Saeed A.; Shafaati M.; Parini P.; Cedazo-Minguez A.; Björkhem I. Is it possible to improve memory function by upregulation of the cholesterol 24S-hydroxylase (CYP46A1) in the brain?. PLoS One 2013, 8, e68534. 10.1371/journal.pone.0068534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M. Y.; Izumi Y.; Benz A.; Zorumski C. F.; Mennerick S. Endogenous 24S-hydroxycholesterol modulates NMDAR-mediated function in hippocampal slices. J. Neurophysiol. 2016, 115, 1263–1272. 10.1152/jn.00890.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M. Y.; Linsenbardt A. J.; Emnett C. M.; Eisenman L. N.; Izumi Y.; Zorumski C. F.; Mennerick S. 24(S)-Hydroxycholesterol as a modulator of neuronal signaling and survival. Neuroscientist 2016, 22, 132–144. 10.1177/1073858414568122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrov A. M.; Pikuleva I. A. Cholesterol 24-hydroxylation by CYP46A1: benefits of modulation for brain diseases. Neurotherapeutics 2019, 16, 635–648. 10.1007/s13311-019-00731-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudry E.; Van Dam D.; Kulik W.; De Deyn P. P.; Stet F. S.; Ahouansou O.; Benraiss A.; Delacourte A.; Bougneres P.; Aubourg P.; Cartier N. Adeno-associated virus gene therapy with cholesterol 24-hydroxylase reduces the amyloid pathology before or after the onset of amyloid plaques in mouse models of Alzheimer’s disease. Mol. Ther. 2010, 18, 44–53. 10.1038/mt.2009.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burlot M. A.; Braudeau J.; Michaelsen-Preusse K.; Potier B.; Ayciriex S.; Varin J.; Gautier B.; Djelti F.; Audrain M.; Dauphinot L.; Fernandez-Gomez F. J.; Caillierez R.; Laprevote O.; Bieche I.; Auzeil N.; Potier M. C.; Dutar P.; Korte M.; Buee L.; Blum D.; Cartier N. Cholesterol 24-hydroxylase defect is implicated in memory impairments associated with Alzheimer-like Tau pathology. Hum. Mol. Genet. 2015, 24, 5965–5976. 10.1093/hmg/ddv268. [DOI] [PubMed] [Google Scholar]
- Djelti F.; Braudeau J.; Hudry E.; Dhenain M.; Varin J.; Bieche I.; Marquer C.; Chali F.; Ayciriex S.; Auzeil N.; Alves S.; Langui D.; Potier M. C.; Laprevote O.; Vidaud M.; Duyckaerts C.; Miles R.; Aubourg P.; Cartier N. CYP46A1 inhibition, brain cholesterol accumulation and neurodegeneration pave the way for Alzheimer’s disease. Brain 2015, 138, 2383–2398. 10.1093/brain/awv166. [DOI] [PubMed] [Google Scholar]
- Chali F.; Djelti F.; Eugene E.; Valderrama M.; Marquer C.; Aubourg P.; Duykaerts C.; Miles R.; Cartier N.; Navarro V. Inhibiting cholesterol degradation induces neuronal sclerosis and epileptic activity in mouse hippocampus. Eur. J. Neurosci. 2015, 41, 1345–1355. 10.1111/ejn.12911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boussicault L.; Alves S.; Lamaziere A.; Planques A.; Heck N.; Moumne L.; Despres G.; Bolte S.; Hu A.; Pages C.; Galvan L.; Piguet F.; Aubourg P.; Cartier N.; Caboche J.; Betuing S. CYP46A1, the rate-limiting enzyme for cholesterol degradation, is neuroprotective in Huntington’s disease. Brain 2016, 139, 953–970. 10.1093/brain/awv384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saadane A.; Mast N.; Trichonas G.; Chakraborty D.; Hammer S.; Busik J. V.; Grant M. B.; Pikuleva I. A. Retinal vascular abnormalities and microglia activation in mice with deficiency in cytochrome P450 46A1-mediated cholesterol removal. Am. J. Pathol. 2019, 189, 405–425. 10.1016/j.ajpath.2018.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mast N.; Li Y.; Linger M.; Clark M.; Wiseman J.; Pikuleva I. A. Pharmacologic stimulation of cytochrome P450 46A1 and cerebral cholesterol turnover in mice. J. Biol. Chem. 2014, 289, 3529–3538. 10.1074/jbc.M113.532846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mast N.; Saadane A.; Valencia-Olvera A.; Constans J.; Maxfield E.; Arakawa H.; Li Y.; Landreth G.; Pikuleva I. A. Cholesterol-metabolizing enzyme cytochrome P450 46A1 as a pharmacologic target for Alzheimer’s disease. Neuropharmacology 2017, 123, 465–476. 10.1016/j.neuropharm.2017.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrov A. M.; Lam M.; Mast N.; Moon J.; Li Y.; Maxfield E.; Pikuleva I. A. CYP46A1 activation by efavirenz leads to behavioral Improvement without significant changes in amyloid plaque load in the brain of 5XFAD mice. Neurotherapeutics 2019, 16, 710–724. 10.1007/s13311-019-00737-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson K. W.; Mast N.; Hudgens J. W.; Lin J. B.; Turko I. V.; Pikuleva I. A. Mapping of the allosteric site in cholesterol hydroxylase CYP46A1 for efavirenz, a drug that stimulates enzyme activity. J. Biol. Chem. 2016, 291, 11876–11886. 10.1074/jbc.M116.723577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mast N.; White M. A.; Björkhem I.; Johnson E. F.; Stout C. D.; Pikuleva I. A. Crystal structures of substrate-bound and substrate-free cytochrome P450 46A1, the principal cholesterol hydroxylase in the brain. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 9546–9551. 10.1073/pnas.0803717105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutlib A. E.; Chen H.; Nemeth G. A.; Markwalder J. A.; Seitz S. P.; Gan L. S.; Christ D. D. Identification and characterization of efavirenz metabolites by liquid chromatography/mass spectrometry and high field NMR: species differences in the metabolism of efavirenz. Drug Metab. Dispos. 1999, 27, 1319–1333. [PubMed] [Google Scholar]
- Ward B. A.; Gorski J. C.; Jones D. R.; Hall S. D.; Flockhart D. A.; Desta Z. The cytochrome P450 2B6 (CYP2B6) is the main catalyst of efavirenz primary and secondary metabolism: implication for HIV/AIDS therapy and utility of efavirenz as a substrate marker of CYP2B6 catalytic activity. J. Pharmacol. Exp. Ther. 2003, 306, 287–300. 10.1124/jpet.103.049601. [DOI] [PubMed] [Google Scholar]
- Bumpus N. N.; Kent U. M.; Hollenberg P. F. Metabolism of efavirenz and 8-hydroxyefavirenz by P450 2B6 leads to inactivation by two distinct mechanisms. J. Pharmacol. Exp. Ther. 2006, 318, 345–351. 10.1124/jpet.106.102525. [DOI] [PubMed] [Google Scholar]
- Ogburn E. T.; Jones D. R.; Masters A. R.; Xu C.; Guo Y.; Desta Z. Efavirenz primary and secondary metabolism in vitro and in vivo: identification of novel metabolic pathways and cytochrome P450 2A6 as the principal catalyst of efavirenz 7-hydroxylation. Drug Metab. Dispos. 2010, 38, 1218–1229. 10.1124/dmd.109.031393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aouri M.; Barcelo C.; Ternon B.; Cavassini M.; Anagnostopoulos A.; Yerly S.; Hugues H.; Vernazza P.; Gunthard H. F.; Buclin T.; Telenti A.; Rotger M.; Decosterd L. A. In vivo profiling and distribution of known and novel phase I and phase II metabolites of efavirenz in plasma, urine, and cerebrospinal fluid. Drug Metab. Dispos. 2016, 44, 151–161. 10.1124/dmd.115.065839. [DOI] [PubMed] [Google Scholar]
- Remmer H.; Schenkman J.; Estabrook R. W.; Sasame H.; Gillette J.; Narasimhulu S.; Cooper D. Y.; Rosenthal O. Drug interaction with hepatic microsomal cytochrome. Mol. Pharmacol. 1966, 2, 187–190. [PubMed] [Google Scholar]
- Schenkman J. B.; Remmer H.; Estabrook R. W. Spectral studies of drug interaction with hepatic microsomal cytochrome. Mol. Pharmacol. 1967, 3, 113–123. [PubMed] [Google Scholar]
- Mailman R. B.; Kulkarni A. P.; Baker R. C.; Hodgson E. Cytochrome P-450 difference spectra: effect of chemical structure on Type II spectra in mouse hepatic microsomes. Drug Metab. Dispos. 1974, 2, 301–308. [PubMed] [Google Scholar]
- Mast N.; Charvet C.; Pikuleva I. A.; Stout C. D. Structural basis of drug binding to CYP46A1, an enzyme that controls cholesterol turnover in the brain. J. Biol. Chem. 2010, 285, 31783–31795. 10.1074/jbc.M110.143313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumaki K.; Sato M.; Kon H.; Nebert D. W. Correlation of Type I, Type II, and Reverse Type I difference spectra with absolute changes in spin state of hepatic microsomal cytochrome P-450 iron from five mammalian species. J. Biol. Chem. 1978, 253, 1048–1058. [PubMed] [Google Scholar]
- Vogt A. D.; Di Cera E. Conformational selection or induced fit? A critical appraisal of the kinetic mechanism. Biochemistry 2012, 51, 5894–5902. 10.1021/bi3006913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianni S.; Dogan J.; Jemth P. Distinguishing induced fit from conformational selection. Biophys. Chem. 2014, 189, 33–39s. 10.1016/j.bpc.2014.03.003. [DOI] [PubMed] [Google Scholar]
- Gonzalez E.; Guengerich F. P. Kinetic processivity of the two-step oxidations of progesterone and pregnenolone to androgens by human cytochrome P450 17A1. J. Biol. Chem. 2017, 292, 13168–13185. 10.1074/jbc.M117.794917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guengerich F. P.; Wilkey C. J.; Glass S. M.; Reddish M. J. Conformational selection dominates binding of steroids to human cytochrome P450 17A1. J. Biol. Chem. 2019, 294, 10028–10041. 10.1074/jbc.RA119.008860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guengerich F. P.; Wilkey C. J.; Phan T. T. N. Human cytochrome P450 enzymes bind drugs and other substrates mainly through conformational-selection modes. J. Biol. Chem. 2019, 294, 10928–10941. 10.1074/jbc.RA119.009305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddish M. J.; Guengerich F. P. Human cytochrome P450 11B2 produces aldosterone by a processive mechanism due to the lactol form of the intermediate 18-hydroxycorticosterone. J. Biol. Chem. 2019, 294, 12975–12991. 10.1074/jbc.RA119.009830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young S. D.; Britcher S. F.; Tran L. O.; Payne L. S.; Lumma W. C.; Lyle T. A.; Huff J. R.; Anderson P. S.; Olsen D. B.; Carroll S. S.; et al. L-743, 726 (DMP-266): a novel, highly potent nonnucleoside inhibitor of the human immunodeficiency virus type 1 reverse transcriptase. Antimicrob. Agents Chemother. 1995, 39, 2602–2605. 10.1128/AAC.39.12.2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostolova N.; Blas-Garcia A.; Galindo M. J.; Esplugues J. V. Efavirenz: What is known about the cellular mechanisms responsible for its adverse effects. Eur. J. Pharmacol. 2017, 812, 163–173. 10.1016/j.ejphar.2017.07.016. [DOI] [PubMed] [Google Scholar]
- Apostolova N.; Funes H. A.; Blas-Garcia A.; Galindo M. J.; Alvarez A.; Esplugues J. V. Efavirenz and the CNS: what we already know and questions that need to be answered. J. Antimicrob. Chemother. 2015, 70, 2693–2708. 10.1093/jac/dkv183. [DOI] [PubMed] [Google Scholar]
- Dalwadi D. A.; Ozuna L.; Harvey B. H.; Viljoen M.; Schetz J. A. Adverse neuropsychiatric events and recreational use of efavirenz and other HIV-1 antiretroviral drugs. Pharmacol. Rev. 2018, 70, 684–711. 10.1124/pr.117.013706. [DOI] [PubMed] [Google Scholar]
- Bumpus N. N. Efavirenz and 8-hydroxyefavirenz induce cell death via a JNK- and BimEL-dependent mechanism in primary human hepatocytes. Toxicol. Appl. Pharmacol. 2011, 257, 227–234. 10.1016/j.taap.2011.09.008. [DOI] [PubMed] [Google Scholar]
- Tovar-Y-Romo L. B.; Bumpus N. N.; Pomerantz D.; Avery L. B.; Sacktor N.; McArthur J. C.; Haughey N. J. Dendritic spine injury induced by the 8-hydroxy metabolite of efavirenz. J. Pharmacol. Exp. Ther. 2012, 343, 696–703. 10.1124/jpet.112.195701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandmann M.; Nehls U.; Dringen R. 8-Hydroxy-efavirenz, the primary metabolite of the antiretroviral drug Efavirenz, stimulates the glycolytic flux in cultured rat astrocytes. Neurochem. Res. 2013, 38, 2524–2534. 10.1007/s11064-013-1165-2. [DOI] [PubMed] [Google Scholar]
- Wang P. F.; Neiner A.; Kharasch E. D. Efavirenz metabolism: influence of polymorphic CYP2B6 variants and stereochemistry. Drug Metab. Dispos. 2019, 47, 1195–1205. 10.1124/dmd.119.086348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best B. M.; Koopmans P. P.; Letendre S. L.; Capparelli E. V.; Rossi S. S.; Clifford D. B.; Collier A. C.; Gelman B. B.; Mbeo G.; McCutchan J. A.; Simpson D. M.; Haubrich R.; Ellis R.; Grant I.; Efavirenz concentrations in CSF exceed IC50 for wild-type HIV. J. Antimicrob. Chemother. 2011, 66, 354–357. 10.1093/jac/dkq434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avery L. B.; Bakshi R. P.; Cao Y. J.; Hendrix C. W. The male genital tract is not a pharmacological sanctuary from efavirenz. Clin. Pharmacol. Ther. 2011, 90, 151–156. 10.1038/clpt.2011.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avery L. B.; Sacktor N.; McArthur J. C.; Hendrix C. W. Protein-free efavirenz concentrations in cerebrospinal fluid and blood plasma are equivalent: applying the law of mass action to predict protein-free drug concentration. Antimicrob. Agents Chemother. 2013, 57, 1409–1414. 10.1128/AAC.02329-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winston A.; Amin J.; Clarke A.; Else L.; Amara A.; Owen A.; Barber T.; Jessen H.; Avihingsanon A.; Chetchotisakd P.; Khoo S.; Cooper D. A.; Emery S.; Puls R.; Cerebrospinal fluid exposure of efavirenz and its major metabolites when dosed at 400 mg and 600 mg once daily: a randomized controlled trial. Clin. Infect. Dis. 2015, 60, 1026–1032. 10.1093/cid/ciu976. [DOI] [PubMed] [Google Scholar]
- Nightingale S.; Chau T. T.; Fisher M.; Nelson M.; Winston A.; Else L.; Carr D. F.; Taylor S.; Ustianowski A.; Back D.; Pirmohamed M.; Solomon T.; Farrar J.; Torok M. E.; Khoo S. Efavirenz and metabolites in cerebrospinal fluid: relationship with CYP2B6 c.516G—>T genotype and perturbed blood-brain barrier due to tuberculous meningitis. Antimicrob. Agents Chemother. 2016, 60, 4511–4518. 10.1128/AAC.00280-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngaimisi E.; Mugusi S.; Minzi O. M.; Sasi P.; Riedel K. D.; Suda A.; Ueda N.; Janabi M.; Mugusi F.; Haefeli W. E.; Burhenne J.; Aklillu E. Long-term efavirenz autoinduction and its effect on plasma exposure in HIV patients. Clin. Pharmacol. Ther. 2010, 88, 676–684. 10.1038/clpt.2010.172. [DOI] [PubMed] [Google Scholar]
- Hitchcock S. A.; Pennington L. D. Structure-brain exposure relationships. J. Med. Chem. 2006, 49, 7559–7583. 10.1021/jm060642i. [DOI] [PubMed] [Google Scholar]
- Dirson G.; Fernandez C.; Hindlet P.; Roux F.; German-Fattal M.; Gimenez F.; Farinotti R. Efavirenz does not interact with the ABCB1 transporter at the blood-brain barrier. Pharm. Res. 2006, 23, 1525–1532. 10.1007/s11095-006-0279-5. [DOI] [PubMed] [Google Scholar]
- Miksys S. L.; Tyndale R. F. Drug-metabolizing cytochrome P450s in the brain. J. Psychiatry Neurosci. 2002, 27, 406–415. [PMC free article] [PubMed] [Google Scholar]
- McFadyen M. C. E.; Melvin W. T.; Murray G. I. Regional distribution of individual forms of cytochrome P450 mRNA in normal adult human brain. Biochem. Pharmacol. 1998, 55, 825–830. 10.1016/S0006-2952(97)00516-9. [DOI] [PubMed] [Google Scholar]
- Klose T. S.; Blaisdell J. A.; Goldstein J. A. Gene structure of CYP2C8 and extrahepatic distribution of the human CYP2Cs. J. Biochem. Mol. Toxicol. 1999, 13, 289–295. . [DOI] [PubMed] [Google Scholar]
- White M. A.; Mast N.; Björkhem I.; Johnson E. F.; Stout C. D.; Pikuleva I. A. Use of complementary cation and anion heavy-atom salt derivatives to solve the structure of cytochrome P450 46A1. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2008, 64, 487–495. 10.1107/S0907444908004046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna I. H.; Teiber J. F.; Kokones K. L.; Hollenberg P. F. Role of the alanine at position 363 of cytochrome P450 2B2 in influencing the NADPH- and hydroperoxide-supported activities. Arch. Biochem. Biophys. 1998, 350, 324–332. 10.1006/abbi.1997.0534. [DOI] [PubMed] [Google Scholar]
- Mast N.; Reem R.; Bederman I.; Huang S.; DiPatre P. L.; Björkhem I.; Pikuleva I. A. Cholestenoic acid is an important elimination product of cholesterol in the retina: comparison of retinal cholesterol metabolism with that in the brain. Invest. Ophthalmol. Visual Sci. 2011, 52, 594–603. 10.1167/iovs.10-6021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mast N.; Zheng W.; Stout C. D.; Pikuleva I. A. Binding of a cyano- and fluoro-containing drug bicalutamide to cytochrome P450 46A1: unusual features and spectral response. J. Biol. Chem. 2013, 288, 4613–4624. 10.1074/jbc.M112.438754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.; Nakatsuji H.; Li X.; Caricato M.; Marenich A. V.; Bloino J.; Janesko B. G.; Gomperts R.; Mennucci B.; Hratchian H. P.; Ortiz J. V.; Izmaylov A. F.; Sonnenberg J. L.; Williams; Ding F.; Lipparini F.; Egidi F.; Goings J.; Peng B.; Petrone A.; Henderson T.; Ranasinghe D.; Zakrzewski V. G.; Gao J.; Rega N.; Zheng G.; Liang W.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Throssell K.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M. J.; Heyd J. J.; Brothers E. N.; Kudin K. N.; Staroverov V. N.; Keith T. A.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A. P.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Millam J. M.; Klene M.; Adamo C.; Cammi R.; Ochterski J. W.; Martin R. L.; Morokuma K.; Farkas O.; Foresman J. B.; Fox D. J.. Gaussian 16, revision A.03; Gaussian, Inc.: Wallingford, CT, 2016.
- Morris G. M.; Huey R.; Lindstrom W.; Sanner M. F.; Belew R. K.; Goodsell D. S.; Olson A. J. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N.; Zhao H. Enriching screening libraries with bioactive fragment space. Bioorg. Med. Chem. Lett. 2016, 26, 3594–3597. 10.1016/j.bmcl.2016.06.013. [DOI] [PubMed] [Google Scholar]
- Sanner M. F. Python: a programming language for software integration and development. J. Mol. Graphics Modell. 1999, 17, 57–61. [PubMed] [Google Scholar]
- O’Boyle N. M.; Banck M.; James C. A.; Morley C.; Vandermeersch T.; Hutchison G. R. Open Babel: An open chemical toolbox. J. Cheminf. 2011, 3, 33. 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson K. A.; Simpson Z. B.; Blom T. Global kinetic explorer: a new computer program for dynamic simulation and fitting of kinetic data. Anal. Biochem. 2009, 387, 20–29. 10.1016/j.ab.2008.12.024. [DOI] [PubMed] [Google Scholar]
- Mast N.; Zheng W.; Stout C. D.; Pikuleva I. A. Antifungal azoles: structural insights into undesired tight binding to cholesterol-metabolizing CYP46A1. Mol. Pharmacol. 2013, 84, 86–94. 10.1124/mol.113.085902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mast N.; Graham S. E.; Andersson U.; Björkhem I.; Hill C.; Peterson J.; Pikuleva I. A. Cholesterol binding to cytochrome P450 7A1, a key enzyme in bile acid biosynthrsis. Biochemistry 2005, 44, 3259–3271. 10.1021/bi047566a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.