

Abstract
Nonalcoholic fatty liver disease is the leading cause of liver disease worldwide. It has expansive extrahepatic morbidity and mortality including increased rates of both cardiovascular disease and venous thromboembolism. Derangements in primary, secondary and tertiary hemostasis are found in nonalcoholic fatty liver disease independent of those ascribed to end-stage liver disease. The abnormalities across all stages of hemostasis explain the increased rates of clinically relevant thrombotic events, including pulmonary embolism, deep vein thrombosis and portal vein thrombosis, which on an epidemiologic basis appears to be independent of obesity and other traditional venous thromboembolic risk factors. However, given the complex interaction between obesity, body composition and nonalcoholic fatty liver disease and the potential for exercise to benefit all three, more research is needed to further define the role of each in contributing to the prohemostatic state of nonalcoholic fatty liver disease in order to improve patient oriented outcomes.
Keywords: Thrombosis, nonalcoholic steatohepatitis, liver transplantation, hypercoagulable, exercise
1. INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is the leading cause of chronic liver disease worldwide with prevalence rates exceeding 25 percent. [1] NAFLD is also the leading etiology of liver disease in the United States (US), where prevalence rates are even greater.[2] Often comorbid with obesity and metabolic syndrome, more than 70% of US adults are overweight or obese.[3] As a result, the healthcare burden of NAFLD is considerable with more than one hundred billion dollars in direct costs annually attributable to NAFLD.[7] According to the American Association for the Study of Liver Diseases guidelines, NAFLD is defined by 1) evidence of hepatic steatosis by imaging and 2) lack of secondary causes of hepatic fat accumulation including significant alcohol consumption (<21 drinks/week for men and <14 drinks/week for women).[115] NAFLD is an umbrella term for a spectrum of disease states that range from simple steatosis or nonalcoholic fatty liver (NAFL) to nonalcoholic steatohepatitis (NASH), which is characterized by inflammation and/or fibrosis. If uncorrected, NASH may progress to cirrhosis and end-stage liver disease. Currently, NASH is the second leading etiology for liver transplantation in the US after viral infection with chronic Hepatitis C [4,5]. In parallel with the worsening obesity epidemic, NASH cirrhosis is expected to become the most common reason for liver transplantation by 2025; it is already the leading indication for liver transplantation in women [6].
However, the impact of NAFLD is seen beyond that of chronic liver disease and progression to cirrhosis. Extrahepatic manifestations are common and include colorectal cancer, cardiovascular disease (CVD), endocrinopathies (e.g., diabetes, hypothyroidism, osteoporosis, polycystic ovarian syndrome iron overload, obstructive sleep apnea, psoriasis and venous thromboembolism (VTE). Hemostatic alterations in NAFLD affect lead to both CVD and VTE. In fact, CVD is the leading cause of death in patients with NAFLD, largely attributable to arterial thrombosis (e.g., myocardial infarction, cerebrovascular accident). Furthermore, multiple epidemiologic studies document increased rates of deep vein thrombosis (DVT), pulmonary embolism (PE) and portal vein thrombosis (PVT) in patients with NASH cirrhosis, independent of traditional risk factors.[15–17] While alterations in hemostasis have been described in advanced liver disease and cirrhosis, the focus of this systematic review is to highlight the abnormalities of hemostasis that are present across all types of NAFLD independent of those found in the presence of end-stage liver disease.
2. HEMOSTASIS AND CHRONIC LIVER DISEASE
Hemostasis involves a series of steps leading to the formation of a blood clot intended to avert hemorrhage following vascular injury. Hemostasis is divided into three phases (Fig. 1). Primary hemostasis is characterized by rapid formation of a platelet plug at the site of vascular injury [18]. Platelet activation and aggregation are mediated by von Willebrand factor (vWF)[19]. Secondary hemostasis refers to the generation and deposition of fibrin via the coagulation cascade. Secondary hemostasis is driven by the complex interaction of coagulation proteins or clotting factors that circulate in their inactive forms until activated by tissue factor [19]. Tertiary hemostasis or fibrinolysis, is critical for the dissolution of the fibrin clot and relies on plasminogen activation. Plasminogen, a proenzyme, generates plasmin by the action of the serine proteases tissue-type plasminogen activator (tPA) and urokinase-type plasminogen activator (uPA) on the surface of the fibrin clot or in the presence of the uPA receptor, respectively [20]. Dysregulation of fibrinolysis can lead to an increased risk of thrombosis or bleeding [21, 22].
Fig. (1).
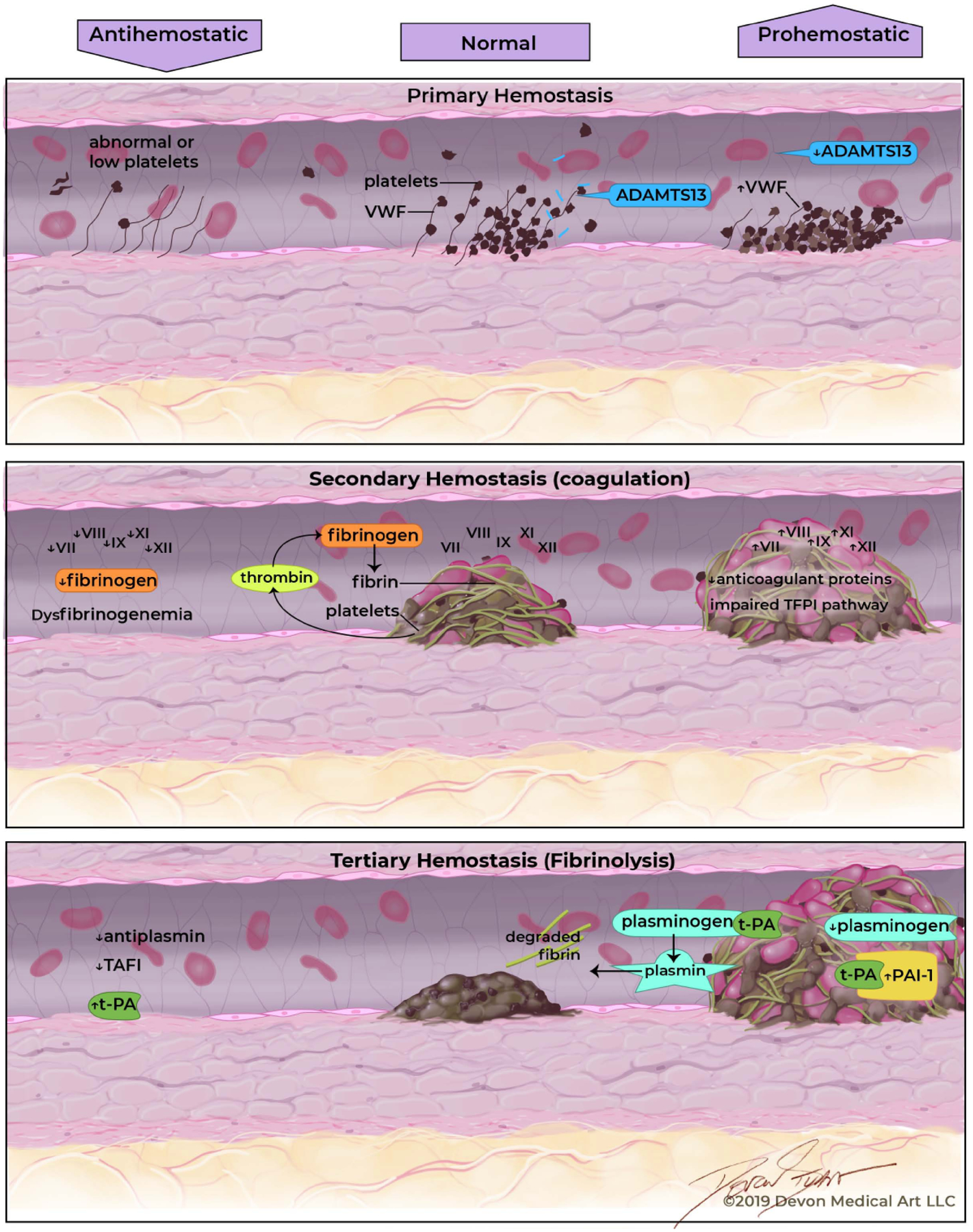
The precarious balance of hemostasis in patients with chronic liver disease.
In patients with cirrhosis, abnormalities exist within each phase of hemostasis that are both pro and antihemostatic. Thus, the hemostatic environment in cirrhosis is complicated and can often be tipped towards either bleeding or clotting. In order to discuss the abnormalities in hemostasis in less advanced forms of NAFLD, prohemostatic abnormalities that have been established in cirrhosis will be described as a model for comparison.
In patients with cirrhosis, prohemostatic alterations in primary hemostasis involve vWF, ADAMTS13 (A Disintegrin and Metalloproteinase with a ThromboSpondin type 1 motif, member 13), as well as platelet count and function. The changes which promote hemostasis are elevated levels of vWF and low levels of ADAMTS13. The hepatic stellate cells generate ADAMTS13 which cleaves vWF. In chronic liver disease, hepatic stellate cells are damaged resulting in lower levels of ADAMTS13. Decreased plasma ADAMTS13 activity may serve as a prognostic indicator for patients with liver cirrhosis. The severity of deficiency of ADAMTS13 activity (ADAMTS13:AC) has been used to estimate survival rates in patients with liver cirrhosis. Diminishing survival rates correlated with the degree of ADAMTS13:AC deficiency and may be a useful adjunct alongside well-established predictors including the Child Turcotte-Pugh Score and Model for End-Stage Liver Disease score [25]. While alterations in levels of vWF and ADAMTS13 promote hemostasis, thrombocytopenia acts as a driving factor in direct opposition.
In secondary hemostasis, dysregulation of the coagulation cascade is a consequence of the liver failing to synthesize coagulation factors [26]. While the synthesis of most clotting factors is reduced, an elevation in plasma Factor VIII is seen in chronic liver disease. This is in part due to increased levels of vWF as together, vWF and Factor VIII circulate as a noncovalent complex [27,28].. Both procoagulant and anticoagulant factors are affected in cirrhosis and while a new equilibrium may be established, a delicate balance exists between pro and anticoagulant factors. Drivers that promote secondary hemostasis include low levels of anticoagulant protein C, protein S, and antithrombin [29–31]..In contrast, low levels of procoagulants fibrinogen and Factors II, V, VII, IX, X, XI are found in cirrhosis. Low levels of these procoagulant factors oppose the effects of hemostasis. Furthermore, not only are the quantity of factors affected, but there are also qualitative defects in these coagulation factors, especially with vitamin K dependent factors [32].
In the last stage of liver disease, alterations in tertiary hemostasis or fibrinolysis are also common. As seen in secondary hemostasis, the major components of tertiary hemostasis involved in fibrinolysis are a product of liver synthesis [33]. Fibrinolysis occurs along the fibrin surface and is mediated by tPA and uPA, serine proteases found on endothelial cells. tPA and uPA bind to plasminogen, a zymogen that is then activated into plasmin, the major driver of the breakdown of fibrin into fibrin degradation products. Regulation of these activators is mediated by plasmin inhibitor as well as plasminogen activator inhibitors. The principal inhibitor at the level of endothelial cell is plasminogen activator inhibitor (PAI)-1, which is produced by several sources including endothelial cells and adipose tissue [21]. The prohemostatic imbalance in cirrhosis is partially steered by low plasminogen levels and elevated levels of PAI-1 [35–37] While the antihemostatic balance is propelled by elevated levels of tPA, low levels of thrombin activatable fibrinolysis inhibitor and plasmin inhibitor also contribute to the imbalance [38–41]. Plasma levels of tPA are elevated due to both increased secretion from endothelial cells and also reduced clearance by the diseased liver [42].
3. THE PROCOAGULANT IMBALANCE IN NAFLD
Independent of the hemostatic imbalance of cirrhosis, there is a growing body of literature stating that NAFLD is a prohemostatic state (Fig. 2). Over time, hepatic steatosis leads to chronic inflammation which causes changes in normal hemostasis [36]. In fact, there is a step-wise progression in hemostatic abnormality with the lowest aberration seen in NAFL and the greatest in NASH cirrhosis. Similar to the alterations found in cirrhosis, there are changes in all three stages of hemostasis in NAFLD.
Fig. (2).
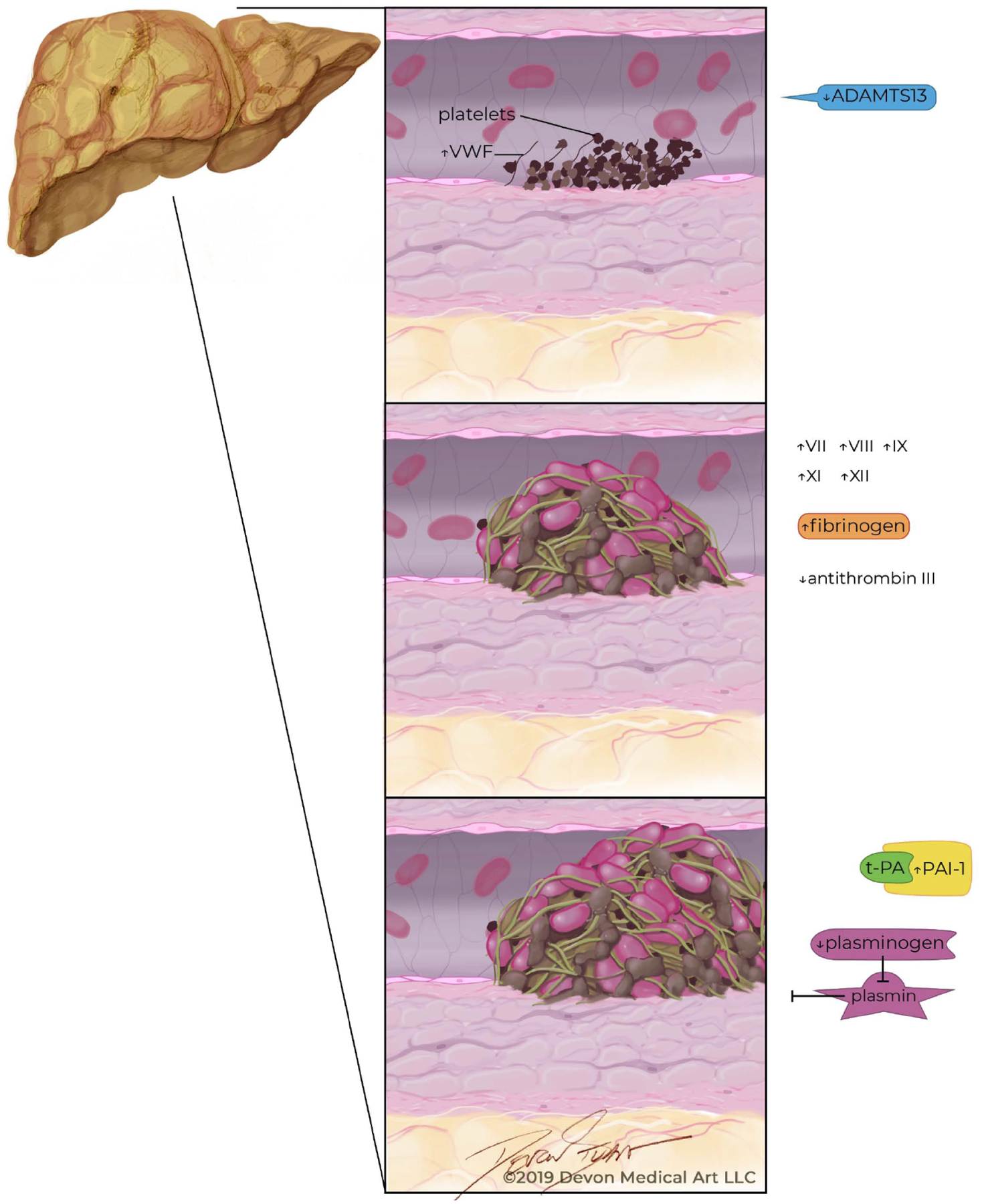
Abnormalities across all three phases of hemostasis in patients with nonalcoholic fatty liver disease.
4. PRIMARY HEMOSTASIS
Patients with NAFLD have abnormal primary hemostasis due to aberrations in both platelet recruitment and function. Mean platelet volume (MPV), a marker of platelet function, is associated with platelet dysfunction. In patients with increased MPV, greater amounts of platelet activation and aggregation are observed [47–52]. MPV is greater in patients with NAFLD in response to chronic inflammatory mediators and worsening insulin resistance [53]. Moreover, increased MPV is associated with the development of CVD, such as coronary artery disease. MPV is also a marker for unprovoked VTE. Collectively, these findings suggest a role for platelet dysfunction in thrombosis generation [54, 56]. Further investigation to elucidate the role of platelets in the NAFLD state would be of significant clinical importance, especially given the widespread use of aspirin for comorbid CVD. While the role of antiplatelet agents, such as aspirin, is well established both for primary prevention and treatment of CVD in patients with NAFLD, whether or not antiplatelet agents reduce the risk of VTE remains unknown and offers an intriguing avenue for future study as the fibrin rich venous clot still requires activated platelets for thrombosis formation.
Increased levels of vWF are also found in NAFLD [36]. However, its’ clinical significance remains unclear as alterations in vWF correlate more strongly with characteristics of metabolic syndrome including increased body mass index (BMI) and visceral adipose tissue rather than histologic features of NAFLD [36] Additionally, low levels of ADAMTS13 have also been reported in NASH and may play a role in abnormal primary hemostasis [57]. More studies focusing on the role of both vWF and ADAMTS13 in NAFLDare presently needed to further clarify the role of each in the thrombophilic state of NAFLD.
5. SECONDARY HEMOSTASIS
In NAFLD, there are multiple abnormalities in secondary hemostasis, namely involving increased activity of Factors VII, VIII, IX, XI, and XII [58, 62]. In their cohort of 98 subjects, Kotronen et al. demonstrated the activity of circulating Factors VIII, IX, XI, and XII that were consistently higher independent of age, gender and BMI in subjects with NAFLD compared to those without NAFLD [58]. Similarly, increased factor VII activity has also been reported in otherwise healthy men with hepatic steatosis compared to healthy men without liver steatosis [62] Utilizing endogenous thrombin potential (ETP) measurement, Tripodi et al. demonstrated further evidence that NAFLD is a thrombophilic state [45]. ETP is a measurement of thrombus formation utilizing area under the thrombin generation curve to quantify the capacity of thrombin generation in plasma after stimulation of the clotting cascade [23]. Thus, ETP is a tool that is useful in assessing the forces of pro- and anti-coagulation in plasma [24]. In patients with NAFLD, Tripodi et al. found increasing ETP ratios with worsening severity of NAFLD; the greatest ETP ratio wasobserved for patients with NASH cirrhosis. Additionally, low levels of anticoagulants antithrombin, protein C and protein S have been found in similar dose-dependent fashion with the lowest levels in the most advanced stages of NAFLD [45, 58, 61]. These findings suggest that the procoagulant imbalance worsens as NAFLD progresses [45].
6. TERTIARY HEMOSTASIS
Alterations in tertiary hemostasis are also present in NAFLD independent of cirrhosis. PAI-1, which plays a featured role in tertiary hemostasis, and is a strong procoagulant driver in NAFLD [34]. Levels of PAI-1 are elevated in patients with NAFLD and increase with the severity of steatosis [36]. In their series of 273 subjects, Verrijken et al. found levels of PAI-1 increased with the NASH Activity Score as well as with fibrosis stage in subjects with biopsy-proven NASH [36]. Elevation of PAI-1 levels, in addition to decreases in tissue activating factor antigen and tPA, lead to a state of chronic hypofibrinolysis and prothrombotic potential [44, 62, 63]. Elevated PAI-1 levels lead to longer clot lysis times by inhibiting breakdown of fibrin based clots through physiologic inhibition of plasminogen activators [65].
One major source of PAI-1 is adipose tissue. PAI-1 functions as an adipocytokine, a secretory protein derived from visceral fat, and modulates inflammation [44]. As elevated PAI-1 levels lead to the alteration of normal regulation of the fibrinolytic system, this results in increased CVD risk [60]. Weight loss and dietary restriction lowers PAI-1 level as do certain anti-diabetic agents (e.g., thiazolidinediones and metformin) [60]. Additionally, elevated PAI-1 may accelerate liver disease progression due to local tissue ischemia stemming from intrahepatic thrombi, known as parenchymal extinction, however, further study confirm the requirement at this time [36].
7. CLINICAL IMPLICATIONS- ARTERIAL THROMBOSIS AND CARDIOVASCULAR DISEASE
Many NAFLD risk factors overlap with those that predispose to CVD. Natural history studies have shown patients with NAFLD and NASH have an overall lower survival, [67, 73] largely due to increased rates of CVD [67, 74, 75]. While the majority of NAFLD and NASH patients will die from CVD, only 1% of patients with NASH will die from a liver-related event [75]. The longest longitudinal experience of 33years by Ekstedt et al. demonstrated that patients with NAFLD had a 55% increased risk of CVD [68]. This confirmed their earlier interim analysis at 13.7 years where 43% of deaths were due to CVD, followed by non-hepatic malignancy (15%), and liver-related causes including hepatocellular carcinoma (8%) [67]. Multiple studies have shown that NAFLD is independently associated with non-fatal CVD events [10, 76, 77]. When compared to traditional CVD risk factors, NASH is the strongest adjusted independent predictor of CVD in liver transplant candidates with over a three-fold increased risk. Liver transplant recipients with NASH have significantly greater post-transplant fatal and non-fatal CV events when compared to recipients without NASH [78–80].
Proposed mechanisms for the development of CVD in patients with NAFLD include a genetic predisposition, chronic inflammation, endothelial dysfunction, oxidative stress, as well as hemostatic alterations in the balance between procoagulant and anticoagulant factors [14]. Of all these mechanistic factors, endothelial dysfunction is perhaps the best characterized. Endothelial dysfunction leads to abnormal blood flow and development of an arterial plaque with a fibrous cap, lipid core, and de novo atherosclerosis. Over time, this stable plaque progresses, leading to intimal narrowing. If uncorrected, this progresses to an unstable plaque at risk of rupture and arterial thrombosis [81, 82]. Flow-mediated dilation of the brachial artery is an effective, non-invasive measure of endothelial function [83, 84]. A recent meta-analysis of 5,547 subjects found that after adjusting for confounding factors, for each 1% decrease in brachial flow-mediated dilation, there was a 13% increased risk of adverse CV events [85].
Independent of traditional CVD risk factors (e.g., obesity, insulin resistance, visceral adiposity), endothelial dysfunction is found globally in NAFLD in both systemic and portal venous systems [86–90]. While the exact mechanism in which NAFLD results in endothelial dysfunction is unknown, what is known is that this strongly contributes to increased CVD risk. It is hypothesized that overwhelmed lipid processing and trafficking lead to production of pro-inflammatory cytokines (Interleukin-6, Tumor necrosis factor alpha) and resultant continuous low-grade inflammation. This culminates in inefficient endothelial vasodilation and may accelerate liver fibrosis through local tissue hypoxemia (hypoperfusion), apoptosis, and fibrogenesis [91–93].
8. CLINICAL IMPLICATIONS- VENOUS THROMBOSIS
The prohemostatic environment in patients with NAFLD leads to an increased risk of clinically significant thrombotic events in both the portal venous as well as the systemic circulation. Consequently, liver transplant recipients with NASH have a greater risk of PVT, DVT and PE [15, 17, 94, 95]. Independent of metabolic comorbidities, liver transplant recipients with NASH have a 55% greater risk of PVT. The odds of PVT increase exponentially for candidates who have high-risk NASH (age >60 years with diabetes, hypertension and obesity) [OR 2.1 (95% CI 1.6–2.8)]. Furthermore, the thrombotic state of NASH is not only localized to the portal venous system, but also includes the systemic circulation given the risk of PE and/or DVT is nearly two and a half-fold greater when comparing hospitalized patients with NASH cirrhosis to all other etiologies of liver disease.16 Additionally, in their case-control study of 414 subjects with PE or DVT, DiMinno et al. found that 81% of VTE cases had NAFLD compared to 30% in age, sex and BMI matched controls without VTE (RR 2.7, 95% CI 2.2–3.2, p<0.001).96 When adjusting for inherited thrombophilia, NAFLD was still associated with increased rates of VTE (OR 1.8, 95% CI 1.2–2.7, p<0.001). The importance of this cannot be overstated as patients with cirrhosis have a higher 30-day mortality following DVT, PE, or PVT [116].
Elevated levels of PAI-1 also pose risk for the development of VTE due to decreased fibrinolytic activity [64, 65, 66] Multiple studies have documented PAI-1 to be an independent predictor of VTE when adjusting extensively for prothrombotic confounders [65, 66, 97]. In fact, PAI-1 is one of the strongest independent risk factors for PVT [OR 6.4 (95% CI 2.5–16.1) [97]. Papatheodorois et al. found that patients with NASH have higher levels of IgG anti-cardiolipin antibodies compared to patients with NAFL. In 56% of NASH patients, one or more thrombotic risk factors was isolated compared to only 8% in patient with NAFL [67]. Furthermore, the authors demonstrate that the presence of at least one thrombotic risk factor was associated with a nearly two-fold fibrosis stage increase in NASH, confirming earlier observational reports correlating thrombotic risk factors to the extent of hepatic fibrosis [61].
9. ROLE OF OBESITY AND THROMBOSIS
Across the medical and surgical literature, obesity is a well-established VTE risk factor [98, 99]. Despite multiple basic science, translational, epidemiologic and clinical studies suggesting a hypercoagulable state existing in patients with NAFLD independent of obesity, a recent report by Potze et al. challenges this paradigm [59]. In this study, the authors found very similar hemostatic profiles when comparing subjects with non-cirrhotic biopsy proven NAFLD to controls without NAFLD with several notable exceptions, such as increased PAI-1 levels, less fibrinolysis and a different fibrin clot infrastructure were present in subjects with NAFLD. However, obese controls also had elevations in PAI-1, impaired fibrinolysis and similar fibrin clot changes, leading the authors to conclude that obesity, not NAFLD, was the main driver of prothrombotic risk [59]. Given the conflicting evidence, further studies are necessary to better define the complex interaction between obesity, NAFLD and thrombosis risk. As BMI is a poor marker in patients with end-stage liver disease, it is suggested that measurement of body composition may be a more direct approach to determine the role of adiposity and the production of prothrombotic adipokines, especially given the role of fat mass and adipose tissue in VTE development in populations without NAFLD [100].
10. ROLE OF EXERCISE IN THROMBOSIS
Exercise has a favorable effect on the coagulation system across all three phases of hemostasis [101–103]. Habitual exercise improves primary hemostasis via endothelial-dependent vasodilation and nitric oxide production leading to less platelet activation and aggregation [104]. Moderate intensity exercise improves hemostasis efficiency by activating fibrinolysis in concert with improving coagulation [101, 102]. Specifically, chronic aerobic based training leads to improved fibrinolytic activity in both healthy subjects and those with CVD [101, 102, 105–108]. Reductions in PAI-1 following aerobic exercise programs lasting 3–8 months range from 23–37% [105, 107–109]. When comparing subjects who are aerobically trained to those who are not, further benefit has been observed in fibrinolytic activity via skeletal muscle tPA efficiency [106]. Patients with higher baseline PAI-1 experience the greatest benefit from exercise through both weight and fat loss [105, 108, 110]. Resistance training also appears to improve the efficiency of the fibrinolytic system in the immediate post-exercise period following short-term strength-training routines (<2 weeks) [104, 111]. However, the effect of chronic resistance training on coagulation and fibrinolysis remains unknown. Furthermore, while the current standard of care for NAFLD is lifestyle changes through diet and exercise where guidelines recommend ≥30 minutes of moderateintensity physical activity 3–5 times/week, the benefit of exercise in modulating coagulation and improving the prothrombotic state of NAFLD also remains unknown [112, 113].
CONCLUSION
In conclusion, NAFLD is the leading cause of liver disease worldwide and has expansive extrahepatic morbidity and mortality including increased rates of both CVD and VTE. Derangements in primary, secondary and tertiary hemostasis are found in NAFLD independent of those ascribed to end-stage liver disease. These hemostatic abnormalities explain the increased rates of clinically relevant thrombotic events including PE, DVT and PVT, which on an epidemiologic basis appear to be independent of obesity and other traditional VTE risk factors. However, given the complex interaction between obesity, body composition and NAFLD and the potential for exercise to improve all three, more research is needed to further define the role of each in the prothrombotic state of NAFLD.
GRANTS AND FINANCIAL SUPPORT:
This research was funded in part by NIH grant L30 DK118601
This project is also funded, in part, under a grant with the Pennsylvania Department of Health using Tobacco CURE Funds. The Department specifically disclaims responsibility for any analyses, interpretations or conclusion
LIST OF ABBREVIATIONS
- ADAMTS13
A Disintegrin and Metalloproteinase with a ThromboSpondin type 1 motif, member 13
- BMI
body mass index
- CVD
cardiovascular disease
- DVT
deep vein thrombosis
- ETP
endogenous thrombin potential
- MPV
mean platelet volume
- NAFL
nonalcoholic fatty liver
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- PAI
plasminogen activator inhibitor
- PE
pulmonary embolus
- PVT
portal vein thrombosis
- tPA
tissue plasminogen activator
- uPA
urokinase-type plasminogen activator
- US
United States
- VTE
venous thromboembolism
- vWF
vonWillebrand factor
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
REFERENCES
- [1].Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-Metaanalytic assessment of prevalence, incidence, and outcomes. Hepatology 2016; 64(1): 73–84. [DOI: 10.1002/hep.28431]. 10.1002/hep.28431 [DOI] [PubMed] [Google Scholar]
- [2].Rinella ME. Nonalcoholic fatty liver disease: a systematic review. JAMA 2015; 313(22): 2263–73. [DOI: 10.1001/jama.2015.5370]. 10.1001/jama.2015.5370 [DOI] [PubMed] [Google Scholar]
- [3].Health United States, 2017: Obesity and Overweight. Maryland: Hyatsville; 2017. [Google Scholar]
- [4].Wong RJ, Aguilar M, Cheung R, et al. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology 2015; 148(3): 547–55. [DOI: 10.1053/j.gastro.2014.11.039]. 10.1053/j.gastro.2014.11.039 [DOI] [PubMed] [Google Scholar]
- [5].Cholankeril G, Wong RJ, Hu M, et al. Liver Transplantation for Nonalcoholic Steatohepatitis in the US: Temporal Trends and Outcomes. Dig Dis Sci 2017; 62(10): 2915–22. [DOI: 10.1007/s10620-017-4684-x]. 10.1007/s10620-017-4684-x [DOI] [PubMed] [Google Scholar]
- [6].Noureddin M, Vipani A, Bresee C, et al. NASH Leading Cause of Liver Transplant in Women: Updated Analysis of Indications For Liver Transplant and Ethnic and Gender Variances. Am J Gastroenterol 2018; 113(11): 1649–59. [DOI: 10.1038/s41395-018-0088-6]. 10.1038/s41395-018-0088-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Younossi ZM, Blissett D, Blissett R, et al. The economic and clinical burden of nonalcoholic fatty liver disease in the United States and Europe. Hepatology 2016; 64(5): 1577–86. [DOI: 10.1002/hep.28785]. 10.1002/hep.28785 [DOI] [PubMed] [Google Scholar]
- [8].Luo J, Xu L, Li J, Zhao S. Nonalcoholic fatty liver disease as a potential risk factor of cardiovascular disease. Eur J Gastroenterol Hepatol 2015; 27(3): 193–9. [DOI: 10.1097/MEG.0000000000000254]. 10.1097/MEG.0000000000000254 [DOI] [PubMed] [Google Scholar]
- [9].Hamaguchi M, Kojima T, Takeda N, et al. Nonalcoholic fatty liver disease is a novel predictor of cardiovascular disease. World J Gastroenterol 2007; 13(10): 1579–84. [DOI: 10.3748/wjg.v13.i10.1579]. 10.3748/wjg.v13.i10.1579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zeb I, Li D, Budoff MJ, et al. Nonalcoholic Fatty Liver Disease and Incident Cardiac Events: The Multi-Ethnic Study of Atherosclerosis. J Am Coll Cardiol 2016; 67(16): 1965–6. [DOI: 10.1016/j.jacc.2016.01.070]. 10.1016/j.jacc.2016.01.070 [DOI] [PubMed] [Google Scholar]
- [11].Armstrong MJ, Adams LA, Canbay A, Syn WK. Extrahepatic complications of nonalcoholic fatty liver disease. Hepatology 2014; 59(3): 1174–97. [DOI: 10.1002/hep.26717]. 10.1002/hep.26717 [DOI] [PubMed] [Google Scholar]
- [12].Musso G, Gambino R, Cassader M, Pagano G. Meta-analysis: natural history of non-alcoholic fatty liver disease (NAFLD) and diagnostic accuracy of non-invasive tests for liver disease severity. Ann Med 2011; 43(8): 617–49. [DOI: 10.3109/07853890.2010.518623]. 10.3109/07853890.2010.518623 [DOI] [PubMed] [Google Scholar]
- [13].Kim D, Kim WR, Kim HJ, Therneau TM. Association between noninvasive fibrosis markers and mortality among adults with nonalcoholic fatty liver disease in the United States. Hepatology 2013; 57(4): 1357–65. [DOI: 10.1002/hep.26156]. 10.1002/hep.26156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Francque SM, van der Graaff D, Kwanten WJ. Non-alcoholic fatty liver disease and cardiovascular risk: Pathophysiological mechanisms and implications. J Hepatol 2016; 65(2): 425–43. [DOI: 10.1016/j.jhep.2016.04.005]. 10.1016/j.jhep.2016.04.005 [DOI] [PubMed] [Google Scholar]
- [15].Stine JG, Shah NL, Argo CK, Pelletier SJ, Caldwell SH, Northup PG. Increased risk of portal vein thrombosis in patients with cirrhosis due to nonalcoholic steatohepatitis. Liver Transpl 2015; 21(8): 1016–21. [DOI: 10.1002/lt.24134]. 10.1002/lt.24134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Stine JG, Niccum BA, Zimmet AN, et al. Increased risk of venous thromboembolism in hospitalized patients with cirrhosis due to non-alcoholic steatohepatitis. Clin Transl Gastroenterol 2018; 9(3): 140 [DOI: 10.1038/s41424-018-0002-y]. 10.1038/s41424-018-0002-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Stine JG, Argo CK, Pelletier SJ, Maluf DG, Caldwell SH, Northup PG. Advanced non-alcoholic steatohepatitis cirrhosis: A high-risk population for pre-liver transplant portal vein thrombosis. World J Hepatol 2017; 9(3): 139–46. [DOI: 10.4254/wjh.v9.i3.139]. 10.4254/wjh.v9.i3.139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bunn HF, Furie B. Overview of Hemostasis (Chapter 13) Pathophysiology of blood disorders. Second Edition 2017. [Google Scholar]
- [19].Konkle B. Bleeding and Thrombosis (Chapter 61) In: Harrison’s Principles of Internal Medicine. 20th Edition. [Google Scholar]
- [20].Gale AJ. Continuing education course #2: current understanding of hemostasis. Toxicol Pathol 2011; 39(1): 273–80. 10.1177/0192623310389474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Cesari M, Pahor M, Incalzi RA. Plasminogen activator inhibitor-1 (PAI-1): a key factor linking fibrinolysis and age-related subclinical and clinical conditions. Cardiovasc Ther 2010; 28(5): e72–91. [DOI: 10.1111/j.1755-5922.2010.00171.x]. 10.1111/j.1755-5922.2010.00171.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chapin JC, Hajjar KA. Fibrinolysis and the control of blood coagulation. Blood Rev 2015; 29(1): 17–24. [DOI: 10.1016/j.blre.2014.09.003]. 10.1016/j.blre.2014.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Tripodi A Thrombin Generation Assay and Its Application in the Clinical Laboratory. Clin Chem 2016; 62(5): 699–707. [DOI: 10.1373/clinchem.2015.248625]. 10.1373/clinchem.2015.248625 [DOI] [PubMed] [Google Scholar]
- [24].Chantarangkul V, Clerici M, Bressi C, Giesen PL, Tripodi A. Thrombin generation assessed as endogenous thrombin potential in patients with hyper- or hypo-coagulability. Haematologica 2003; 88(5): 547–54. [PubMed] [Google Scholar]
- [25].Takaya H, Uemura M, Fujimura Y, et al. ADAMTS13 activity may predict the cumulative survival of patients with liver cirrhosis in comparison with the Child-Turcotte-Pugh score and the Model for End-Stage Liver Disease score. Hepatol Res 2012; 42(5): 459–72. [DOI: 10.1111/j.1872-034X.2011.00950.x]. 10.1111/j.1872-034X.2011.00950.x [DOI] [PubMed] [Google Scholar]
- [26].Tripodi A Liver Disease and Hemostatic (Dys)function. Semin Thromb Hemost 2015; 41(5): 462–7. [DOI: 10.1055/s-0035-1550440]. 10.1055/s-0035-1550440 [DOI] [PubMed] [Google Scholar]
- [27].Hollestelle MJ, Geertzen HG, Straatsburg IH, van Gulik TM, van Mourik JA. Factor VIII expression in liver disease. Thromb Haemost 2004; 91(2): 267–75. [DOI: 10.1160/TH03-05-0310]. 10.1160/TH03-05-0310 [DOI] [PubMed] [Google Scholar]
- [28].Terraube V, O’Donnell JS, Jenkins PV. Factor VIII and von Willebrand factor interaction: biological, clinical and therapeutic importance. Haemophilia 2010; 16(1): 3–13. [DOI: 10.1111/j.1365-2516.2009.02005.x]. 10.1111/j.1365-2516.2009.02005.x [DOI] [PubMed] [Google Scholar]
- [29].Tripodi A, Primignani M, Lemma L, Chantarangkul V, Mannucci PM. Evidence that low protein C contributes to the procoagulant imbalance in cirrhosis. J Hepatol 2013; 59(2): 265–70. [DOI: 10.1016/j.jhep.2013.03.036]. 10.1016/j.jhep.2013.03.036 [DOI] [PubMed] [Google Scholar]
- [30].Tripodi A, Primignani M, Chantarangkul V, et al. An imbalance of pro- vs anti-coagulation factors in plasma from patients with cirrhosis. Gastroenterology 2009; 137(6): 2105–11. [DOI: 10.1053/j.gastro.2009.08.045]. 10.1053/j.gastro.2009.08.045 [DOI] [PubMed] [Google Scholar]
- [31].Tripodi A, Primignani M, Lemma L, et al. Detection of the imbalance of procoagulant versus anticoagulant factors in cirrhosis by a simple laboratory method. Hepatology 2010; 52(1): 249–55. [DOI: 10.1002/hep.23653]. 10.1002/hep.23653 [DOI] [PubMed] [Google Scholar]
- [32].Blanchard RA, Furie BC, Jorgensen M, Kruger SF, Furie B. Acquired vitamin K-dependent carboxylation deficiency in liver disease. N Engl J Med 1981; 305(5): 242–8. [DOI: 10.1056/NEJM198107303050502]. 10.1056/NEJM198107303050502 [DOI] [PubMed] [Google Scholar]
- [33].Leebeek FW, Rijken DC. The Fibrinolytic Status in Liver Diseases. Semin Thromb Hemost 2015; 41(5): 474–80. [DOI: 10.1055/s-0035-1550437]. 10.1055/s-0035-1550437 [DOI] [PubMed] [Google Scholar]
- [34].Northup PG, Argo CK, Shah N, Caldwell SH. Hypercoagulation and thrombophilia in nonalcoholic fatty liver disease: mechanisms, human evidence, therapeutic implications, and preventive implications. Semin Liver Dis 2012; 32(1): 39–48. [DOI: 10.1055/s-0032-1306425]. 10.1055/s-0032-1306425 [DOI] [PubMed] [Google Scholar]
- [35].Targher G, Bertolini L, Rodella S, et al. NASH predicts plasma inflammatory biomarkers independently of visceral fat in men. Obesity (Silver Spring) 2008; 16(6): 1394–9. [DOI: 10.1038/oby.2008.64]. 10.1038/oby.2008.64 [DOI] [PubMed] [Google Scholar]
- [36].Verrijken A, Francque S, Mertens I, et al. Prothrombotic factors in histologically proven nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology 2014; 59(1): 121–9. [DOI: 10.1002/hep.26510]. 10.1002/hep.26510 [DOI] [PubMed] [Google Scholar]
- [37].Chang ML, Hsu CM, Tseng JH, et al. Plasminogen activator inhibitor-1 is independently associated with non-alcoholic fatty liver disease whereas leptin and adiponectin vary between genders. J Gastroenterol Hepatol 2015; 30(2): 329–36. [DOI: 10.1111/jgh.12705]. 10.1111/jgh.12705 [DOI] [PubMed] [Google Scholar]
- [38].Tripodi A Tests of coagulation in liver disease. Clin Liver Dis 2009; 13(1): 55–61. [DOI: 10.1016/j.cld.2008.09.002]. 10.1016/j.cld.2008.09.002 [DOI] [PubMed] [Google Scholar]
- [39].Van Thiel DH, George M, Mindikoğlu AL, Baluch MH, Dhillon S. Coagulation and fibrinolysis in individuals with advanced liver disease. Turk J Gastroenterol 2004; 15(2): 67–72. [PubMed] [Google Scholar]
- [40].Van Thiel DH, George M, Fareed J. Low levels of thrombin activatable fibrinolysis inhibitor (TAFI) in patients with chronic liver disease. Thromb Haemost 2001; 85(4): 667–70. 10.1055/s-0037-1615651 [DOI] [PubMed] [Google Scholar]
- [41].Gresele P, Binetti BM, Branca G, et al. TAFI deficiency in liver cirrhosis: relation with plasma fibrinolysis and survival. Thromb Res 2008; 121(6): 763–8. [DOI: 10.1016/j.thromres.2007.08.011]. 10.1016/j.thromres.2007.08.011 [DOI] [PubMed] [Google Scholar]
- [42].Chandler WL. A kinetic model of the circulatory regulation of tissue plasminogen activator. Thromb Haemost 1991; 66(3): 321–8. 10.1055/s-0038-1646415 [DOI] [PubMed] [Google Scholar]
- [43].Papatheodoridis GV, Chrysanthos N, Cholongitas E, et al. Thrombotic risk factors and liver histologic lesions in non-alcoholic fatty liver disease. J Hepatol 2009; 51(5): 931–8. [DOI: 10.1016/j.jhep.2009.06.023]. 10.1016/j.jhep.2009.06.023 [DOI] [PubMed] [Google Scholar]
- [44].Potze W, Siddiqui MS, Sanyal AJ. Vascular Disease in Patients with Nonalcoholic Fatty Liver Disease. Semin Thromb Hemost 2015; 41(5): 488–93. [DOI: 10.1055/s-0035-1550433]. 10.1055/s-0035-1550433 [DOI] [PubMed] [Google Scholar]
- [45].Tripodi A, Fracanzani AL, Primignani M, et al. Procoagulant imbalance in patients with non-alcoholic fatty liver disease. J Hepatol 2014; 61(1): 148–54. [DOI: 10.1016/j.jhep.2014.03.013]. 10.1016/j.jhep.2014.03.013 [DOI] [PubMed] [Google Scholar]
- [46].Anstee QM, Dhar A, Thursz MR. The role of hypercoagulability in liver fibrogenesis. Clin Res Hepatol Gastroenterol 2011; 35(8–9): 526–33. [DOI: 10.1016/j.clinre.2011.03.011]. 10.1016/j.clinre.2011.03.011 [DOI] [PubMed] [Google Scholar]
- [47].Park Y, Schoene N, Harris W. Mean platelet volume as an indicator of platelet activation: methodological issues. Platelets 2002; 13(5–6): 301–6. [DOI: 10.1080/095371002220148332]. 10.1080/095371002220148332 [DOI] [PubMed] [Google Scholar]
- [48].Bath PM, Butterworth RJ. Platelet size: measurement, physiology and vascular disease. Blood Coagul Fibrinolysis 1996; 7(2): 157–61. 10.1097/00001721-199603000-00011 [DOI] [PubMed] [Google Scholar]
- [49].Ozhan H, Aydin M, Yazici M, et al. Mean platelet volume in patients with non-alcoholic fatty liver disease. Platelets 2010; 21(1): 29–32. [DOI: 10.3109/09537100903391023]. 10.3109/09537100903391023 [DOI] [PubMed] [Google Scholar]
- [50].Celikbilek M, Gürsoy S, Deniz K, Karaman A, Zararsiz G, Yurci A. Mean platelet volume in biopsy-proven non-alcoholic fatty liver disease. Platelets 2013; 24(3): 194–9. [DOI: 10.3109/09537104.2012.688898]. 10.3109/09537104.2012.688898 [DOI] [PubMed] [Google Scholar]
- [51].Khaspekova SG, Ziuriaev IT, Iakushkin VV, et al. [Mean platelet volume: interactions with platelet aggregation activity and glycoprotein IIb-IIIa and Ib expression levels]. Biomed Khim 2014; 60(1): 94–108. [Mean platelet volume: interactions with platelet aggregation activity and glycoprotein IIb-IIIa and Ib expression levels]. 10.18097/pbmc20146001094 [DOI] [PubMed] [Google Scholar]
- [52].Haschek and Rooseaux’s Handbook of Toxicologic Pathology. 3rd ed. 2013. [Google Scholar]
- [53].Madan SA, Fida N, Barman P, et al. Frailty Assessment in Advanced Heart Failure. J Card Fail 2016; 22(10): 840–4. [DOI: 10.1016/j.cardfail.2016.02.003]. 10.1016/j.cardfail.2016.02.003 [DOI] [PubMed] [Google Scholar]
- [54].Braekkan SK, Mathiesen EB, Njølstad I, Wilsgaard T, Størmer J, Hansen JB. Mean platelet volume is a risk factor for venous thromboembolism: the Tromsø Study, Tromsø, Norway. J Thromb Haemost 2010; 8(1): 157–62. [DOI: 10.1111/j.1538-7836.2009.03498.x]. 10.1111/j.1538-7836.2009.03498.x [DOI] [PubMed] [Google Scholar]
- [55].Mitchell O, Feldman DM, Diakow M, Sigal SH. The pathophysiology of thrombocytopenia in chronic liver disease. Hepat Med 2016; 8: 39–50. [DOI: 10.2147/HMER.S74612]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Sansanayudh N, Anothaisintawee T, Muntham D, McEvoy M, Attia J, Thakkinstian A. Mean platelet volume and coronary artery disease: a systematic review and meta-analysis. Int J Cardiol 2014; 175(3): 433–40. [DOI: 10.1016/j.ijcard.2014.06.028]. 10.1016/j.ijcard.2014.06.028 [DOI] [PubMed] [Google Scholar]
- [57].Lombardi AM, Fabris R, Berti de Marinis G, et al. Defective ADAMTS13 synthesis as a possible consequence of NASH in an obese patient with recurrent thrombotic thrombocytopenic purpura. Eur J Haematol 2014; 92(6): 497–501. [DOI: 10.1111/ejh.12273]. 10.1111/ejh.12273 [DOI] [PubMed] [Google Scholar]
- [58].Kotronen A, Joutsi-Korhonen L, Sevastianova K, et al. Increased coagulation factor VIII, IX, XI and XII activities in non-alcoholic fatty liver disease. Liver Int 2011; 31(2): 176–83. [DOI: 10.1111/j.1478-3231.2010.02375.x]. 10.1111/j.1478-3231.2010.02375.x [DOI] [PubMed] [Google Scholar]
- [59].Potze W, Siddiqui MS, Boyett SL, et al. Preserved hemostatic status in patients with non-alcoholic fatty liver disease. J Hepatol 2016; 65(5): 980–7. [DOI: 10.1016/j.jhep.2016.06.001]. 10.1016/j.jhep.2016.06.001 [DOI] [PubMed] [Google Scholar]
- [60].Skurk T, Hauner H. Obesity and impaired fibrinolysis: role of adipose production of plasminogen activator inhibitor-1. Int J Obes Relat Metab Disord 2004; 28(11): 1357–64. [DOI: 10.1038/sj.ijo.0802778]. 10.1038/sj.ijo.0802778 [DOI] [PubMed] [Google Scholar]
- [61].Assy N, Bekirov I, Mejritsky Y, Solomon L, Szvalb S, Hussein O. Association between thrombotic risk factors and extent of fibrosis in patients with non-alcoholic fatty liver diseases. World J Gastroenterol 2005; 11(37): 5834–9. [DOI: 10.3748/wjg.v11.i37.5834]. 10.3748/wjg.v11.i37.5834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Cigolini M, Targher G, Agostino G, Tonoli M, Muggeo M, De Sandre G. Liver steatosis and its relation to plasma haemostatic factors in apparently healthy men--role of the metabolic syndrome. Thromb Haemost 1996; 76(1): 69–73. 10.1055/s-0038-1650524 [DOI] [PubMed] [Google Scholar]
- [63].Stine JG, Northup PG. Coagulopathy Before and After Liver Transplantation: From the Hepatic to the Systemic Circulatory Systems. Clin Liver Dis 2017; 21(2): 253–74. [DOI: 10.1016/j.cld.2016.12.003]. 10.1016/j.cld.2016.12.003 [DOI] [PubMed] [Google Scholar]
- [64].Meltzer ME, Lisman T, Doggen CJ, de Groot PG, Rosendaal FR. Synergistic effects of hypofibrinolysis and genetic and acquired risk factors on the risk of a first venous thrombosis. PLoS Med 2008; 5(5)e97 [DOI: 10.1371/journal.pmed.0050097]. 10.1371/journal.pmed.0050097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Meltzer ME, Lisman T, de Groot PG, et al. Venous thrombosis risk associated with plasma hypofibrinolysis is explained by elevated plasma levels of TAFI and PAI-1. Blood 2010; 116(1): 113–21. [DOI: 10.1182/blood-2010-02-267740]. 10.1182/blood-2010-02-267740 [DOI] [PubMed] [Google Scholar]
- [66].Prins MH, Hirsh J. A critical review of the evidence supporting a relationship between impaired fibrinolytic activity and venous thromboembolism. Arch Intern Med 1991; 151(9): 1721–31. 10.1001/archinte.1991.00400090023006 [DOI] [PubMed] [Google Scholar]
- [67].Ekstedt M, Franzén LE, Mathiesen UL, et al. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology 2006; 44(4): 865–73. [doi: 10.1002/hep.21327]. 10.1002/hep.21327 [DOI] [PubMed] [Google Scholar]
- [68].Ekstedt M, Hagström H, Nasr P, Fredrikson M, Stål P, Kechagias S, et al. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology 2015; 61(5): 1547–54. 10.1002/hep.27368 [DOI] [PubMed] [Google Scholar]
- [69].Satapathy SK, Sanyal AJ. Epidemiology and Natural History of Nonalcoholic Fatty Liver Disease. Semin Liver Dis 2015; 35(3): 221–35. [DOI: 10.1055/s-0035-1562943]. 10.1055/s-0035-1562943 [DOI] [PubMed] [Google Scholar]
- [70].Söderberg C, Stål P, Askling J, et al. Decreased survival of subjects with elevated liver function tests during a 28-year follow-up. Hepatology 2010; 51(2): 595–602. [DOI: 10.1002/hep.23314]. 10.1002/hep.23314 [DOI] [PubMed] [Google Scholar]
- [71].Kim D, Choi SY, Park EH, et al. Nonalcoholic fatty liver disease is associated with coronary artery calcification. Hepatology 2012; 56(2): 605–13. [DOI: 10.1002/hep.25593]. 10.1002/hep.25593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Romero-Gómez M, Zelber-Sagi S, Trenell M. Treatment of NAFLD with diet, physical activity and exercise. J Hepatol 2017; 67(4): 829–46. [DOI: 10.1016/j.jhep.2017.05.016]. 10.1016/j.jhep.2017.05.016 [DOI] [PubMed] [Google Scholar]
- [73].Adams LA, Lymp JF, St Sauver J, et al. The natural history of nonalcoholic fatty liver disease: a population-based cohort study. Gastroenterology 2005; 129(1): 113–21. [Epub 2005/07/14]. 10.1053/j.gastro.2005.04.014 [DOI] [PubMed] [Google Scholar]
- [74].Kotronen A, Yki-Järvinen H. Fatty liver: a novel component of the metabolic syndrome. Arterioscler Thromb Vasc Biol 2008; 28(1): 27–38. [Epub 2007/08/11. doi: 10.1161/atvbaha.107.147538]. [DOI] [PubMed] [Google Scholar]
- [75].VanWagner LB, Rinella ME. Extrahepatic Manifestations of Nonalcoholic Fatty Liver Disease. Current hepatology reports 2016; 15(2): 75–85. 10.1007/s11901-016-0295-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Ong JP, Pitts A, Younossi ZM. Increased overall mortality and liver-related mortality in non-alcoholic fatty liver disease. J Hepatol 2008; 49(4): 608–12. [Epub 2008/08/07]. [doi: 10.1016/j.jhep.2008.06.018]. 10.1016/j.jhep.2008.06.018 [DOI] [PubMed] [Google Scholar]
- [77].Targher G, Arcaro G. Non-alcoholic fatty liver disease and increased risk of cardiovascular disease. Atherosclerosis 2007; 191(2): 235–40. [Epub 2006/09/15. doi: 10.1016/j.atherosclerosis.2006.08.021]. 10.1016/j.atherosclerosis.2006.08.021 [DOI] [PubMed] [Google Scholar]
- [78].Wang X, Li J, Riaz DR, Shi G, Liu C, Dai Y. Outcomes of liver transplantation for nonalcoholic steatohepatitis: a systematic review and meta-analysis. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association 2014; 12(3): 394–402.e1. 10.1016/j.cgh.2013.09.023 [DOI] [PubMed] [Google Scholar]
- [79].Vanwagner LB, Bhave M, Te HS, Feinglass J, Alvarez L, Rinella ME. Patients transplanted for nonalcoholic steatohepatitis are at increased risk for postoperative cardiovascular events. Hepatology 2012; 56(5): 1741–50. [Epub 2012/05/23. doi: 10.1002/hep.25855]. 10.1002/hep.25855 [DOI] [PubMed] [Google Scholar]
- [80].Malik SM, deVera ME, Fontes P, Shaikh O, Ahmad J. Outcome after liver transplantation for NASH cirrhosis. Am J Transplant 2009; 9(4): 782–93. [Epub 2009/04/07. doi: 10.1111/j.1600-6143.2009.02590.x]. 10.1111/j.1600-6143.2009.02590.x [DOI] [PubMed] [Google Scholar]
- [81].Lee SB, Park GM, Lee JY, et al. Association between nonalcoholic fatty liver disease and subclinical coronary atherosclerosis: An observational cohort study. J Hepatol 2018; 68(5): 1018–24. [Epub 2017/12/24. doi: 10.1016/j.jhep.2017.12.012]. 10.1016/j.jhep.2017.12.012 [DOI] [PubMed] [Google Scholar]
- [82].Green DJ, Jones H, Thijssen D, Cable NT, Atkinson G. Flow-mediated dilation and cardiovascular event prediction: does nitric oxide matter? Hypertension (Dallas, Tex : 1979) 2011; 57(3): 363–9. 10.1161/HYPERTENSIONAHA.110.167015 [DOI] [PubMed] [Google Scholar]
- [83].Yeboah J, Sutton-Tyrrell K, McBurnie MA, Burke GL, Herrington DM, Crouse JR. Association between brachial artery reactivity and cardiovascular disease status in an elderly cohort: the cardiovascular health study. Atherosclerosis 2008; 197(2): 768-. 10.1016/j.atherosclerosis.2007.07.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Herrington DM, Fan L, Drum M, et al. Brachial flow-mediated vasodilator responses in population-based research: methods, reproducibility and effects of age, gender and baseline diameter. J Cardiovasc Risk 2001; 8(5): 319–28. 10.1177/174182670100800512 [DOI] [PubMed] [Google Scholar]
- [85].Inaba Y, Chen JA, Bergmann SR. Prediction of future cardiovascular outcomes by flow-mediated vasodilatation of brachial artery: a meta-analysis. Int J Cardiovasc Imaging 2010; 26(6): 631–40. 10.1007/s10554-010-9616-1 [DOI] [PubMed] [Google Scholar]
- [86].Gonzalez-Paredes FJ, Hernández Mesa G, Morales Arraez D, et al. Contribution of Cyclooxygenase End Products and Oxidative Stress to Intrahepatic Endothelial Dysfunction in Early Non-Alcoholic Fatty Liver Disease. PLoS One 2016; 11(5)e0156650 10.1371/journal.pone.0156650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Pugh CJ, Spring VS, Kemp GJ, et al. Exercise training reverses endothelial dysfunction in nonalcoholic fatty liver disease. Am J Physiol Heart Circ Physiol 2014; 307(9): H1298–306. 10.1152/ajpheart.00306.2014 [DOI] [PubMed] [Google Scholar]
- [88].Pugh CJ, Sprung VS, Jones H, et al. Exercise-induced improvements in liver fat and endothelial function are not sustained 12 months following cessation of exercise supervision in nonalcoholic fatty liver disease. International journal of obesity 2016; 40(12): 1927–30. 10.1038/ijo.2016.123 [DOI] [PubMed] [Google Scholar]
- [89].Sapmaz F, Uzman M, Basyigit S, et al. Steatosis Grade is the Most Important Risk Factor for Development of Endothelial Dysfunction in NAFLD. Medicine (Baltimore) 2016; 95(14)e3280 10.1097/MD.0000000000003280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Thakur ML, Sharma S, Kumar A, et al. Nonalcoholic fatty liver disease is associated with subclinical atherosclerosis independent of obesity and metabolic syndrome in Asian Indians. Atherosclerosis 2012; 223(2): 507–11. 10.1016/j.atherosclerosis.2012.06.005 [DOI] [PubMed] [Google Scholar]
- [91].Federico A, Dallio M, Masarone M, Persico M, Loguercio C. The epidemiology of non-alcoholic fatty liver disease and its connection with cardiovascular disease: role of endothelial dysfunction. Eur Rev Med Pharmacol Sci 2016; 20(22): 4731–41. [PubMed] [Google Scholar]
- [92].Wanless IR, Shiota K. The pathogenesis of nonalcoholic steatohepatitis and other fatty liver diseases: a four-step model including the role of lipid release and hepatic venular obstruction in the progression to cirrhosis. Semin Liver Dis 2004; 24(1): 99–106. 10.1055/s-2004-823104 [DOI] [PubMed] [Google Scholar]
- [93].Wanless IR, Wong F, Blendis LM, Greig P, Heathcote EJ, Levy G. Hepatic and portal vein thrombosis in cirrhosis: possible role in development of parenchymal extinction and portal hypertension. Hepatology 1995; 21(5): 1238–47. [PubMed] [Google Scholar]
- [94].Stine JG, Intagliata N, Northup PG, Caldwell SH. Nonalcoholic fatty liver disease, portal vein thrombosis and coagulation: more questions than answers? Transplantation 2017; 101(8): e281–2. 10.1097/TP.0000000000001807 [DOI] [PubMed] [Google Scholar]
- [95].Stine JG, Wang J, Shah PM, et al. Decreased Portal Vein Velocity is Predictive of the Development of Portal Vein Thrombosis: a Matched Case-Control Study. Liver international : official journal of the International Association for the Study of the Liver 2017. 10.1111/liv.13500: 28632958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Di Minno MN, Tufano A, Rusolillo A, Di Minno G, Tarantino G. High prevalence of nonalcoholic fatty liver in patients with idiopathic venous thromboembolism. World journal of gastroenterology : WJG 2010; 16(48): 6119–22. 10.3748/wjg.v16.i48.6119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Balta G, Altay C, Gurgey A. PAI-1 gene 4G/5G genotype: A risk factor for thrombosis in vessels of internal organs. Am J Hematol 2002; 71(2): 89–93. 10.1002/ajh.10192 [DOI] [PubMed] [Google Scholar]
- [98].Stein PD, Beemath A, Olson RE. Obesity as a risk factor in venous thromboembolism. Am J Med 2005; 118(9): 978–80. 10.1016/j.amjmed.2005.03.012 [DOI] [PubMed] [Google Scholar]
- [99].Yang G, De Staercke C, Hooper WC. The effects of obesity on venous thromboembolism: A review. Open J Prev Med 2012; 2(4): 499–509. 10.4236/ojpm.2012.24069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Severinsen MT, Kristensen SR, Johnsen SP, Dethlefsen C, Tjønneland A, Overvad K. Anthropometry, body fat, and venous thromboembolism: a Danish follow-up study. Circulation 2009; 120(19): 1850–7. 10.1161/CIRCULATIONAHA.109.863241 [DOI] [PubMed] [Google Scholar]
- [101].El-Sayed MS, El-Sayed Ali Z, Ahmadizad S. Exercise and training effects on blood haemostasis in health and disease: an update. Sports Med 2004; 34(3): 181–200. 10.2165/00007256-200434030-00004 [DOI] [PubMed] [Google Scholar]
- [102].Womack CJ, Nagelkirk PR, Coughlin AM. Exercise-induced changes in coagulation and fibrinolysis in healthy populations and patients with cardiovascular disease. Sports Med 2003; 33(11): 795–807. 10.2165/00007256-200333110-00002 [DOI] [PubMed] [Google Scholar]
- [103].van Stralen KJ, Le Cessie S, Rosendaal FR, Doggen CJ. Regular sports activities decrease the risk of venous thrombosis. J Thromb Haemost 2007; 5(11): 2186–92. 10.1111/j.1538-7836.2007.02732.x [DOI] [PubMed] [Google Scholar]
- [104].Kupchak BR, Creighton BC, Aristizabal JC, et al. Beneficial effects of habitual resistance exercise training on coagulation and fibrinolytic responses. Thromb Res 2013; 131(6): e227–34. 10.1016/j.thromres.2013.02.014 [DOI] [PubMed] [Google Scholar]
- [105].Killewich LA, Macko RF, Montgomery PS, Wiley LA, Gardner AW. Exercise training enhances endogenous fibrinolysis in peripheral arterial disease. J Vasc Surg 2004; 40(4): 741–5. 10.1016/j.jvs.2004.07.030 [DOI] [PubMed] [Google Scholar]
- [106].Francis RM, Romeyn CL, Coughlin AM, Nagelkirk PR, Womack CJ, Lemmer JT. Age and aerobic training status effects on plasma and skeletal muscle tPA and PAI-1. Eur J Appl Physiol 2014; 114(6): 1229–38. 10.1007/s00421-014-2857-2 [DOI] [PubMed] [Google Scholar]
- [107].Stratton JR, Chandler WL, Schwartz RS, et al. Effects of physical conditioning on fibrinolytic variables and fibrinogen in young and old healthy adults. Circulation 1991; 83(5): 1692–7. 10.1161/01.CIR.83.5.1692 [DOI] [PubMed] [Google Scholar]
- [108].de Geus EJ, Kluft C, de Bart AC, van Doornen LJ. Effects of exercise training on plasminogen activator inhibitor activity. Med Sci Sports Exerc 1992; 24(11): 1210–9. 10.1249/00005768-199211000-00004 [DOI] [PubMed] [Google Scholar]
- [109].el-Sayed MS. Effects of high and low intensity aerobic conditioning programs on blood fibrinolysis and lipid profile. Blood coagulation & fibrinolysis : an international journal in haemostasis and thrombosis 1996; 7(4): 484–90. 10.1097/00001721-199606000-00009 [DOI] [PubMed] [Google Scholar]
- [110].Sudi KM, Gallistl S, Tröbinger M, et al. The influence of weight loss on fibrinolytic and metabolic parameters in obese children and adolescents. J Pediatr Endocrinol Metab 2001; 14(1): 85–94. 10.1515/JPEM.2001.14.1.85 [DOI] [PubMed] [Google Scholar]
- [111].Nagelkirk PR, Scalzo R, Harber M, Kaminsky LA. The influence of acute resistance training and body composition on coagulation and fibrinolytic activity in low-risk women. Int J Sports Med 2010; 31(7): 458–62. 10.1055/s-0030-1249623 [DOI] [PubMed] [Google Scholar]
- [112].Pollock ML, Franklin BA, Balady GJ, et al. AHA Science Advisory. Resistance exercise in individuals with and without cardiovascular disease: benefits, rationale, safety, and prescription: An advisory from the Committee on Exercise, Rehabilitation, and Prevention, Council on Clinical Cardiology, American Heart Association; Position paper endorsed by the American College of Sports Medicine. Circulation 2000; 101(7): 828–33. 10.1161/01.CIR.101.7.828 [DOI] [PubMed] [Google Scholar]
- [113].Chalasani N, Younossi Z, Lavine JE, et al. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018; 67(1): 328–57. 10.1002/hep.29367 [DOI] [PubMed] [Google Scholar]