

Abstract
Background:
L-asparaginase is a bacterial enzyme used in the treatment of acute lymphoblastic leukemia. In the ongoing U.S. Drug-Induced Liver Injury Network (DILIN) prospective study, standard and pegylated asparaginase were the most frequent cause of liver injury with jaundice among anti-cancer agents (8 of 40: 20%). The unique features of this hepatotoxicity are described.
Methods:
Eight cases from 5 DILIN centers were reviewed for clinical course, laboratory values, imaging, and histopathology.
Results:
Seven women, ages 29 to 59 years, and one 8-year old boy, all with leukemia, developed jaundice within 9 to 21 days (median 15 days) of starting asparaginase or pegaspargase, during the first (n=6) or second (n=2) cycle. Prominent symptoms were jaundice (n=8), fatigue (6), abdominal pain (6) but rarely pruritus (1). Initial median ALT level was 284 U/L (range 83 to 1076), Alk P 159 U/L (64 to 452), and bilirubin 4.4 mg/dL (3.7 to 8.4). Bilirubin levels rose thereafter in all patients to median peak of 17.5 mg/dL (11.7 to 25.7), INR rose to 1.1 to 1.7 and serum albumin fell to 1.5 to 2.6 g/dL. Hepatic imaging revealed fatty liver in all patients. Liver biopsy showed steatosis but minimal hepatocyte necrosis. One patient restarted on pegaspargase re-developed less severe injury.
Conclusion:
Asparaginase is a common cause of antineoplastic-induced liver injury with jaundice, typically with short latency, marked steatosis, and prolonged jaundice, which can lead to delays in antileukemic therapy. The cause of injury is likely direct inhibition of hepatic protein synthesis caused by asparagine depletion.
Keywords: hepatotoxicity, leukemia, antineoplastic agents, jaundice, cholestasis, fatty liver
Introduction
L-asparaginase is a bacterial enzyme derived from Escherichia coli B (E. coli) or Erwinia chrysanthemi that is used as an antineoplastic agent, largely for acute lymphoblastic leukemia (ALL) [1,2]. Standard asparaginase is typically given intramuscularly or intravenously three times weekly for the first two weeks of 4-week cycles. A pegylated formulation of asparaginase (pegaspargase) is given intravenously or intramuscularly no more often than every 2 weeks. The bacterial enzyme hydrolyzes L-asparagine to aspartic acid and ammonia resulting in a selective depletion of asparagine in serum [3]. Leukemic cells are particularly sensitive to asparagine deficiency as they frequently lack asparagine synthetase activity and rely upon serum levels of asparagine [4]. In contrast, normal bone marrow cells and cells in other organs are less affected probably due to a rich supply of intracellular asparagine synthetase. The relative lack of bone marrow suppression by asparaginase makes it particularly attractive in combination with chemotherapeutic regimens that usually include severely myelosuppressive agents.
Asparaginase does have systemic toxicities including hypersensitivity reactions, pancreatitis, thrombosis, hyperglycemia, neurologic dysfunction, nephropathy, and hepatotoxicity [1,5–10]. In an ongoing multicenter prospective study of drug-induced liver injury from the United States [11], asparaginase and pegaspargase were found to be the most frequent cause of clinically apparent liver injury with jaundice among antineoplastic agents. The unique clinical features, course and outcome of eight consecutive cases of asparaginase hepatoxicity from the ongoing Drug-Induced Liver Injury Network (DILIN) are described.
Methods
Initiated in 2004, the DILIN Prospective Study (NCT00345930) has enrolled individuals with suspected drug-induced liver injury at 5 to 8 designated clinical centers across the United States. The inclusion and exclusion criteria, evaluation for competing etiologies, follow-up, and causality and severity assessment have been described in previous publications [11–12] and a full list of sites and participants are listed in the Supplementary Table 1. The DILIN Prospective Study was approved by the Institutional Review Boards of the enrolling clinical centers, and all participants provided written informed consent. In addition, the protocol and consent form were approved, and the study was monitored by an independent data and safety monitoring board appointed by the National Institutes of Health.
The analysis consisted of all eight individuals enrolled between 2004 and 2017 who were judged by the DILIN Causality Committee to have definite, highly likely, or probable drug-induced liver injury from asparaginase or pegaspargase [13]. Patients’ DILIN records were systematically evaluated and relevant data were extracted. When data was missing, the primary center was contacted, and the patient’s medical records were re-reviewed to obtain missing data. HLA typing was done by high resolution genotyping by locus-specific PCR amplification on genomic DNA [14] through the Immunogenetics and Single Cell Technologies Core at Vanderbilt University Medical Center and the Institute for Immunology and Infectious Diseases, Murdoch Western Australia. Allele frequencies were compared to racial and ethnicity matched populations from publicly available datasets found on www.allelefrequencies.net. As a part of this analysis, a search request was made to the FDA Adverse Event Reporting System (FAERS) for cases of liver injury reported with the use of asparaginase products including standard (E. coli and Erwinia derived) and pegylated formulations.
Demographic and clinical data for subjects enrolled in the DILIN Prospective Study were extracted on November 13th 2017. Descriptive statistics, such as means, medians, and frequency distributions, were used to characterize the cohort. The R ratio was calculated as the serum ALT level divided by the serum Alk P level (both expressed as fold x ULN).Statistics were performed by using Excel (version 16.13.1; Microsoft Corp, Redmond, WA and R (version 3.0.2).
Results
Between 2004 and 2017, 1919 cases of suspected drug-induced liver injury were enrolled in the DILIN Prospective Study, of which 1633 cases were fully adjudicated by the DILIN Causality Committee and 1321 scored as definite, highly likely or probable (Figure 1). Among 75 cases that were attributed to antineoplastic agents, 15 were being treated for non-malignant conditions (such as inflammatory bowel disease or psoriasis). Thus, in only 60 cases (4.5%) were drugs used to treat cancer the suspected cause of liver injury, and only 40 of the cases were associated with jaundice (serum total bilirubin ≥ 2.5 mg/dL). Eight of the 60 cases were attributed to asparaginase or pegaspargase, a frequency only matched by imatinib as a cause of DILI. Importantly, the asparaginase/pegaspargase cases were all associated with hyperbilirubinemia, making it the single most common antineoplastic agent associated with cholestatic liver injury (8 of 40: 20%).
Figure 1: Consort Diagram.
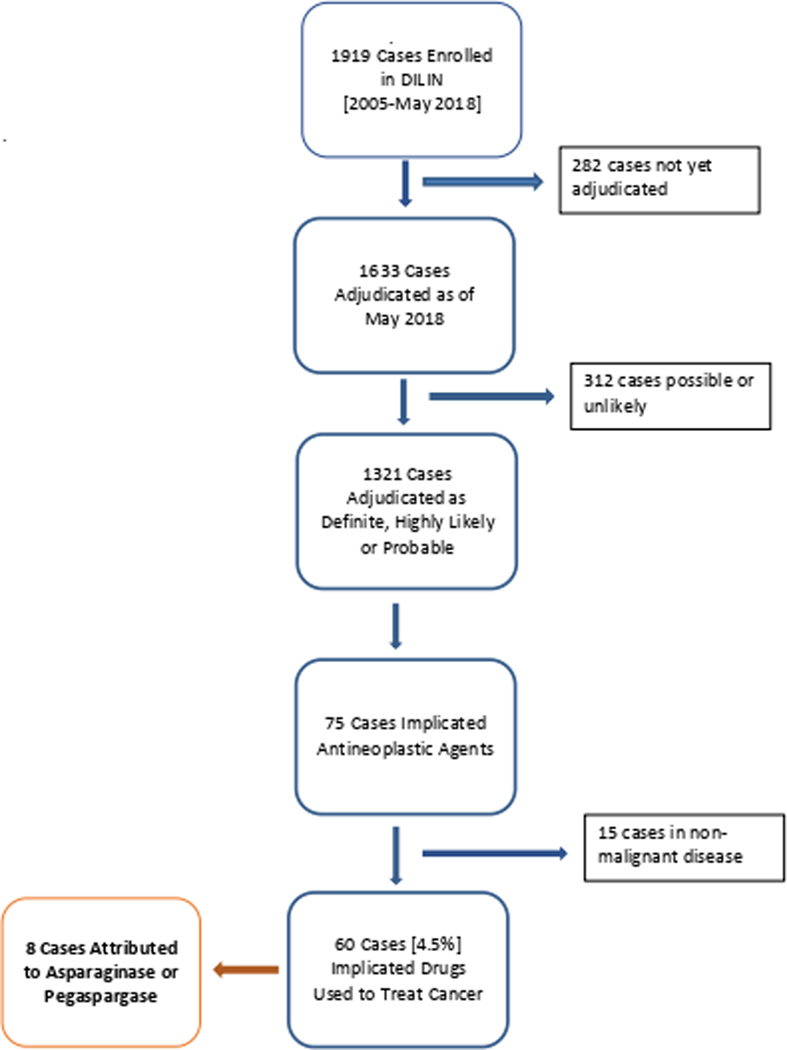
A total of 1919 cases have been enrolled in DILIN from 2005-May of 2018. Out of these, 75 cases have implicated antineoplastic agents as definite, highly likely, or probably due to the drug by the adjudication committee. A total of 8 cases were attributed to asparaginase or pegaspargase.
The clinical features of the 8 cases are summarized in Table 1. The cohort consisted of 7 women, aged 29 to 59 years, and one 8-year old boy. One patient was African American and 7 were Caucasian, one of whom was Hispanic. One patient received standard asparaginase, 6 pegaspargase, and one received both. In all cases, E. coli derived asparaginase was used. All patients were being treated for acute or chronic lymphoblastic leukemia. Three patients received asparaginase or pegaspargase alone while the remaining 5 also received vincristine (n=5), daunorubicin (n=4), cytarabine (n=3), methotrexate (n=3), cyclophosphamide (n=2) and dasatinib (n=1). Other medications taken by these 8 patients during the 2 months before onset are listed in Supplementary Table 2. Three patients were obese (BMI >30 kg/m2), 2 were overweight (BMI >25 kg/m2), 3 had a history of type 2 diabetes, 2 had hypertension, and 1 hyperlipidemia. None were known to have pre-existing liver disease.
Table 1:
Clinical features of 8 cases of asparaginase/pegaspargase hepatotoxicity
| Case | Age [Sex/Race] | Drug | Year | Latency from first dose (days) | Latency from last dose (days) | R ratio at onset | Peak Bilirubin (mg/dL) | Peak ALT (U/L) | Peak Alk P (U/L) | Peak INR | Lowest Albumin (g/dL) | Recovery* (days) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 59 [F/C] | Asp | 2008 | 9 | 2 | 4.1 | 12.0 | 455 | 840 | 1.3 | 2.3 | <30 |
| 2 | 50 [F/C] | Peg-Asp | 2009 | 12 | 4 | 1.7 | 23.5 | 451 | 730 | 1.5 | 1.8 | Unk |
| 3 | 48 [F/C] | Peg-Asp | 2012 | 11 | 11 | 13.6 | 19.5 | 625 | 261 | 1.2 | 2.2 | <180 |
| 4 | 8 [M/C] | Peg-Asp | 2014 | 16 | 16 | 19.6 | 11.7 | 954 | 145 | 1.2 | 2.6 | <30 |
| 5 | 56 [F/C] | Asp and Peg-Asp | 2007 | 17 | 1 | 5.3 | 15.5 | 1016 | 1796 | 1.4 | 2.3 | <30 |
| 6 | 33 [F/AA] | Peg-Asp | 2012 | 21 | 21 | 14.6 | 12.8 | 2331 | 1018 | 1.1 | 1.5 | <30 |
| 7 | 29 [F/C-H] | Peg-Asp | 2015 | 9 | 9 | 2.6 | 25.7 | 340 | 1083 | 1.3 | 1.7 | <60 |
| 8 | 44 [F/C] | Peg-Asp | 2011 | 19 | 5 | 3.0 | 20.1 | 564 | 445 | 1.7 | 2.2 | <180 |
| Median | 46 | 15 | 8 | 4.7 | 17.5 | 595 | 785 | 1.31 | 2.1 |
Abbreviations: C, Caucasian or White; AA, African-American or Black; H, Hispanic; Asp, asparaginase; Peg-Asp, pegaspargase; ALT, alanine aminotransferase; Alk P, alkaline phosphatase; INR, international normalized ratio of prothrombin time.
Latency (time to onset) as measured as days from date first or last drug exposure to date that serum total bilirubin reached 2.5 mg/dL or above; Recovery as measured by estimated time until serum total bilirubin returns to less than 2.5 mg/dL.
R ratio: is the ALT divided by the Alk P level, both expressed as multiples of the upper limit of normal.
The latency to onset of hepatic injury was relatively rapid. Asparaginase is typically given 3 times weekly for 2 weeks, while pegaspargase is given once every 2 weeks. Jaundice with symptoms of liver injury arose during or at the end of the first course, 9 to 21 days (median = 15 days) after the initial dose, and 1 to 19 days (median = 8 days) after the final dose of asparaginase or pegaspargase. Jaundice (n=8), dark urine (n=6), fatigue (n=6) and abdominal pain (n=6) were the most frequently reported symptoms. Only one patient mentioned pruritus, and none had rash, fever, or eosinophilia. Test results for acute hepatitis A, B, C, and E were negative in all patients. Two patients had moderate titers of antinuclear antibody (ANA 1:80 and 1:160), but all 8 were negative for antismooth muscle and antimitochondrial antibodies. No patients abused alcohol or had a history of recent sepsis, shock or hypotension.
At the onset of injury, median serum ALT level was 284 U/L (range: 83 to 1076), Alk P 159 U/L (64 to 452), and total bilirubin 4.4 mg/dL (3.7 to 8.4). The R ratio at the onset ranged widely, from cholestatic (1.7) to very hepatocellular (19.6), the median being in the range of “mixed” (4.7). Serum total bilirubin levels rose for 3 to 18 days and ALT for 3 to 20 days after onset. The median peak ALT was 595 U/L (range 340 to 2331 U/L), Alk P 785 U/L (145 to 1796 U/L), and total bilirubin 17.5 mg/dL (11.7 to 25.7 mg/dL). In half of the cohort, jaundice persisted for less than one month (Figure 2). Serum albumin levels fell in all patients usually starting before the onset of jaundice, and the nadir in levels ranged from 1.5 to 2.6 g/dL between 1 to 16 days after onset coinciding with the period of peak serum total bilirubin levels. In the same time period, INR values increased, peak values ranging from 1.1 to 1.7 (median 1.3).
Figure 2: Duration of Total Bilirubin Elevations in 8 patients with Asparaginase Hepatotoxicity.
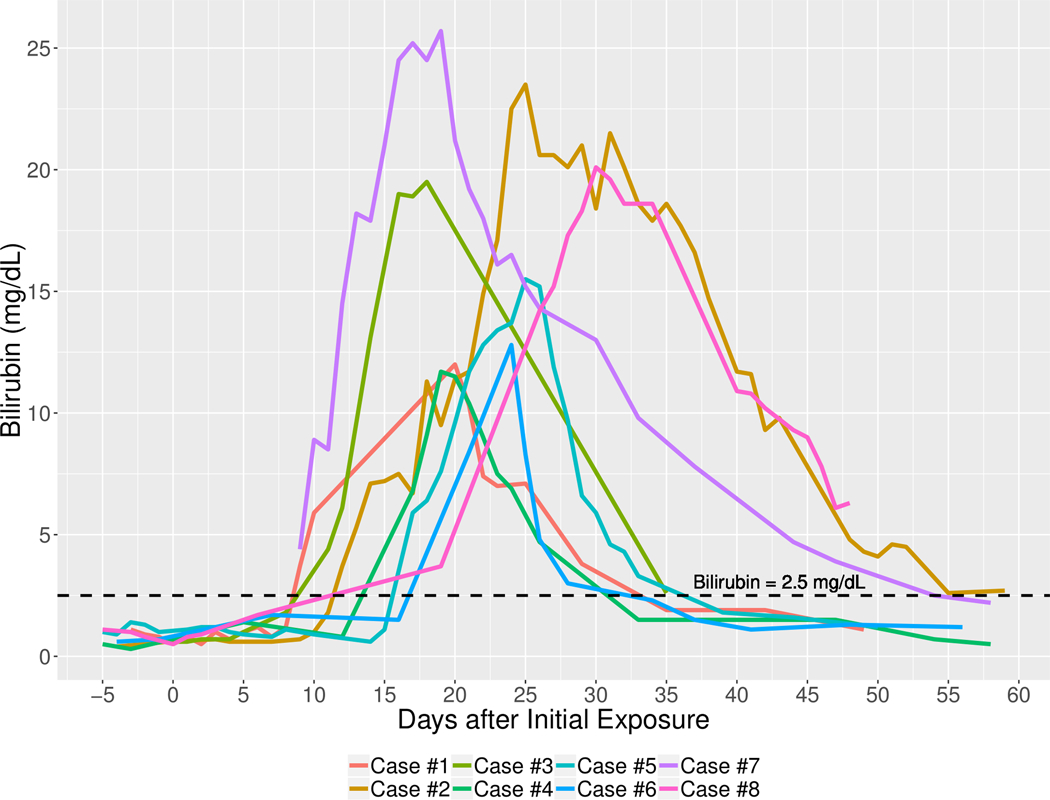
Each line represents total serum bilirubin level by time after onset of asparaginase/pegaspargase injury.
All patients recovered clinically from their drug-induced liver injury. Mildly abnormal serum enzymes were identified in 3 subjects during follow up 6 months or more after onset, but these 3 individuals had restarted anti-leukemic chemotherapy without asparaginase and the abnormalities were attributed to the other antineoplastic agents (methotrexate, mercaptopurine) or graft-vs-host disease after hematopoietic cell transplantation.
Seven patients had hepatic imaging before starting asparaginase and all 8 had imaging at the time of acute injury. Within a month after starting asparaginase, all patients had evidence of diffuse fatty liver by abdominal ultrasound or CT scan, which was present in only 3 of 7 with imaging before chemotherapy. Two subjects also had ascites and 1 hepatomegaly at the time of acute injury. Of the 4 patients who had evidence of no hepatic steatosis before treatment, 3 continued to have evidence of steatosis on imaging done more than 2 months after asparaginase therapy while the fourth was not re-tested until 9 months post therapy, at which time imaging of the liver was normal.
Liver biopsy was done on only one patient (#5) and showed diffuse, wide-spread macrovesicular steatosis and minimal intrahepatic cholestasis, hepatocyte necrosis, and inflammation (Figure 3). There was no evidence of bile duct injury, portal inflammation, ballooning degeneration, Mallory bodies, fibrosis or sinusoidal obstruction.
Figure 3: Histopathology of Asparaginase-induced Liver Injury (Case 5).
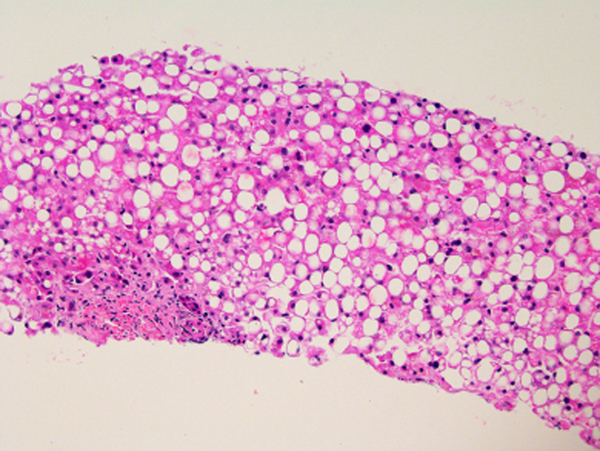
H&E Stain, 40x Magnification. There is diffuse macrovesicular steatosis affecting more than 90% of hepatocytes but no obvious hepatocyte necrosis or ballooning degeneration and scant lobular and portal inflammation. Cholestasis is not observed despite a serum total bilirubin of 13.4 mg/dL on the day of biopsy, 6 days after onset.
There were no cases of mortality or liver transplantation directly due to asparaginase-induce liver injury. Nevertheless, the liver injury delayed re-initiation of chemotherapy in all subjects. Two subjects died within 6 months of DILI onset, but death was attributed to progression of leukemia in both. Ascites developed in only one patient, and serum albumin levels fell below 3.0 in all patients, none developed evidence of frank hepatic failure, encephalopathy, or variceal hemorrhage. One patient (#6) was re-treated using the same pegaspargase dose and re-developed liver injury with a similar latency period, but a milder presentation and course (peak bilirubin 3.8 mg/dL) (Figure 4).
Figure 4: Course of Liver Injury After Initial and Re-exposure to Asparaginase (Case #6).
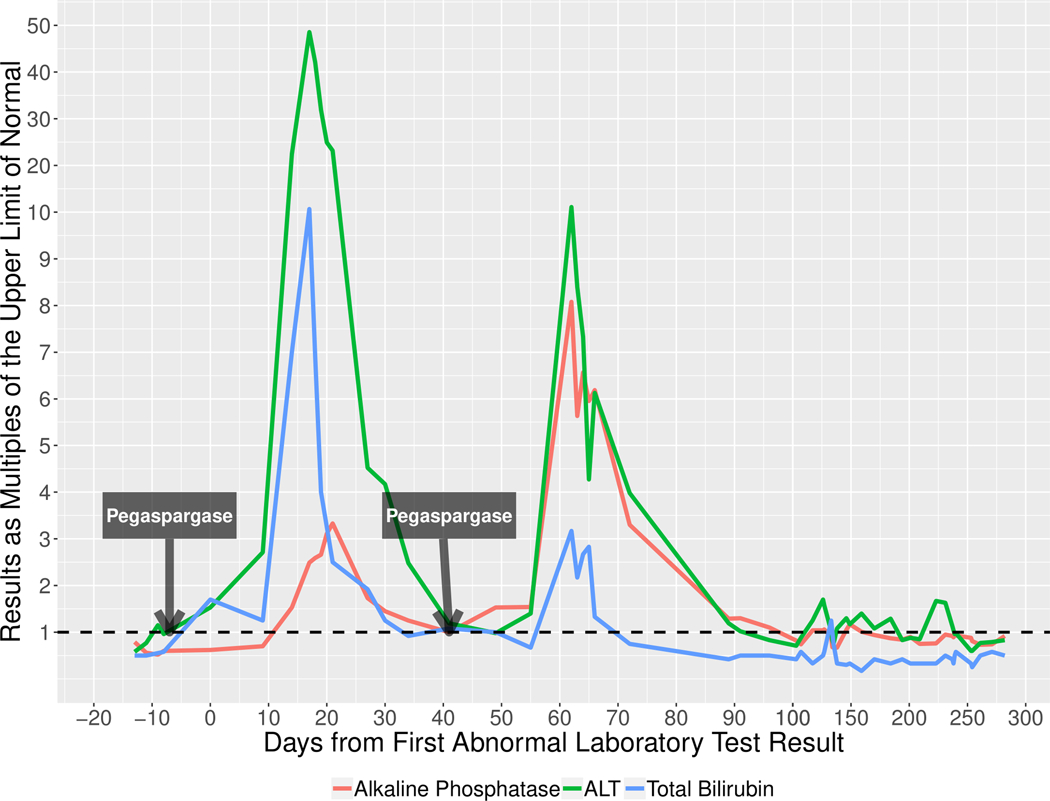
The red line represents the pattern of alkaline phosphatase elevation after initial exposure and re-exposure to pegaspargase in one patient. The green line represents the pattern of alanine aminotransferase (ALT) elevation after initial exposure and re-exposure to pegaspargase in one patient. The blue line represents the pattern of total bilirubin elevation after initial exposure and re-exposure to pegaspargase in one patient.
HLA testing was carried out on patients enrolled in DILIN who consented to genetic testing [14]. Seven of the 8 cases had complete HLA assignment (Table 2). The only HLA type that was overrepresented was HLA-A*02:01 (allele frequency of 0.50, with expected frequency among US Caucasians of 0.27 and African Americans of 0.12.). This allele is common in the general population and has been published to be over-represented in adult populations with chronic lymphoblastic leukemia [15,16].
Table 2:
HLA typing in cases of asparaginase/pegaspargase hepatotoxicity*
| Case† | Allele | HLA-A | HLA-B | HLA-C | HLA-DPB1 | HLA-DQA1 | HLA-DQB1 | HLA-DRB1 |
|---|---|---|---|---|---|---|---|---|
| 1 [C] |
1 2 |
02:01 11:01 |
44:02 44.02 |
05:01 07:04 |
04:01 04:01 |
01:01 03:01 |
03:01 05:03 |
04:01 14:01 |
| 2 [C] |
Not Done | |||||||
| 3 [C] |
1 2 |
02:01 03:01 |
08:01 40.01 |
03:04 07:01 |
02:01 04:02 |
03:01 05:01 |
03:01 03:01 |
04:01 11:01 |
| 4 [C] |
1 2 |
01:01 02:01 |
37:01 41:02 |
06:02 17:01 |
01:01 04:01 |
05:01 05:01 |
02:01 02:01 |
03:01 03:01 |
| 5 [C] |
1 2 |
02:01 02:01 |
07:02 15:01 |
03:03 07:02 |
02:01 04:01 |
01:02 03:01 |
03:01 06:02 |
04:08 15:01 |
| 6 [AA] |
1 2 |
02:01 66:02 |
50:01 58:01 |
06:02 07:01 |
02:01 85:01 |
01:02 01:02 |
06:02 06:02 |
11:01 15:03 |
| 7 [C-H] |
1 2 |
02:01 31:01 |
15:01 35:17 |
03:03 04:01 |
02:01 11:01 |
04:01 05:01 |
03:01 04:02 |
08:02 11:02 |
| 8 [C] |
1 2 |
11:01 31:01 |
07:02 51:01 |
02:02 07:02 |
04:01 17:01 |
03:01 05:01 |
03:01 03:02 |
04:04 11:01 |
: Race and ethnicity [in brackets]: C, Caucasian or White; AA, African-American or Black; H, Hispanic.
HLA alleles were called using the IMGT HLA allele database release: v3290, (http://www.ebi.ac.uk/ipd/imgt/hla/) as the reference library.
The 8 cases reported here were attributed to E. coli derived products, 2 from standard asparaginase [Elspar] reported in 2007 and 2008, and 6 from pegylated asparaginase [Oncaspar] reported between 2009 and 2017, but none from Erwinia derived enzyme [Erwinase]. An analysis of MedWatch reports from a FAERS search request identified more than 2000 reports in which asparaginase was mentioned. Some reports were duplications, and most were not well documented or arose in patients receiving other potentially hepatotoxic agents and had other possible reasons for liver injury. However, instances of hepatic injury with jaundice and hepatic failure were found with all three formulations.
Discussion
Clinical characterization of 8 cases of asparaginase/pegaspargase hepatotoxicity demonstrated a consistent clinical phenotype of injury, with rapid onset (9 to 21 days) of marked jaundice and diffuse hepatic steatosis. Strikingly, all patients developed serum hypoalbuminemia with albumin levels falling below 3.0 g/dL within 20 to 30 days of first receipt of asparaginase, but only one developed ascites. INR values also rose but only two were 1.5 or above, and no patient developed encephalopathy or variceal bleeding. Hepatic steatosis persisted in some for several months even while serum enzyme levels improved. While jaundice was marked, there was little evidence of hepatic failure and all patients ultimately recovered. The major adverse effect of asparaginase-induced liver injury was delay in resuming chemotherapy and elimination of asparaginase in the chemotherapy regimen, which has important clinical implications in view of the documented benefit of asparaginase in chemotherapy regimens for ALL [17].
Interestingly, in the one patient who was retreated liver injury recurred, but without an accelerated onset or greater severity, as occurs typically with re-challenge after idiosyncratic liver injury. These clinical features have been noted in previous publications [5–10] including the lack of severe recurrence upon restarting the agent after recovery from hepatotoxicity [18].
The clinical features of asparaginase hepatotoxicity also suggest direct hepatotoxicity as shown by the short latency, lack of allergic or autoimmune features, rapid decrease in serum proteins synthesized in the liver, and early development of relatively bland steatosis. Furthermore, this direct hepatotoxicity was different from the typical injury associated with direct toxins (high doses of acetaminophen, methotrexate, niacin, amphetamines), in the variable pattern of serum aminotransferase and alkaline phosphatase elevations and absence of confluent or massive hepatocyte necrosis on liver biopsy. Liver biopsy in one patient (and data from previous cases [19,20]) revealed diffuse macrovesicular steatosis in up to 95% of cells with scant inflammation, necrosis or cholestasis despite ALT levels above 1000 U/L and bilirubin of 13.7 mg/dL. There was also no ballooning necrosis or perisinusoidal fibrosis as occurs in non-alcoholic steatohepatitis and in most forms of drug-induced fatty liver (due to methotrexate, tamoxifen, bleomycin or irinotecan). As shown in this study and in several previous reports, the steatosis from asparaginase injury can persist for months after clinical recovery but eventually resolves [19,20]. This pattern of prolonged steatosis with scant inflammation and necrosis is somewhat reminiscent of the fatty liver of kwashiorkor [21].
The mechanism of hepatotoxicity due to asparaginase is best explained by substrate depletion of asparagine and possibly glutamine [1,4]. In animal models, asparaginase can cause diffuse steatosis and rapid decrease in serum protein levels. In one study in rats, infusions of glutamine blocked this effect, and some degree of glutaminase activity was identified in bacterially-derived asparaginase. These studies suggest that asparaginase causes substrate depletion and selective amino acid deficiency in the liver which might lead to loss of protein and enzyme functions responsible for lipid and bilirubin transport and secretion and synthesis of the major serum proteins made by the liver. If this hypothesis is correct, possible therapies for asparaginase hepatotoxicity would be replacement of amino acids or their precursors.
Recent isolated case reports have suggested that infusions of levocarnitine and vitamin B complex shorten the course of hepatic injury after use of asparaginase [22–25]. None of the patients in this report received levocarnitine or amino acid supplements. Controlled trials of such interventions have not been done and would be difficult in humans with drug-induced liver injury. Nevertheless, studies in animal models of asparagine and amino acid replacement as well as carnitine and antioxidants could provide the necessary impetus to assess promising regimens and select the proper dose and timing, allowing for comparison to historical controls to demonstrate sufficient evidence of efficacy from what is likely benign interventions.
The possible role of asparagine and/or glutamine depletion in the pathogenesis of hepatotoxicity of asparaginase may also provide clues to the varying susceptibility of leukemic patients to this injury. Previous studies suggest that 5% to 10% of patients receiving asparaginase chemotherapy develop liver injury with jaundice [1,5–9], which most likely represents differences in susceptibility rather than idiosyncrasy. Indeed, previous studies have indicated that adults, obese and overweight patients and those with diabetes are more susceptible to asparaginase toxicity than children and normal weight adults without diabetes [6,26–29]. These differences may reflect variations in metabolic status and susceptibility to a relative and select amino acid deficiency rather than a genetic or pharmacologic difference in asparaginase metabolism.
The overrepresentation of HLA-A*02:01 in these 8 cases is interesting considering previous associations of this class I allele with other leukemias (15) and the potential for asparaginase to alter amino acid pools. However, HLA-A*02:01 is common in European populations, and the lack of immune features on laboratory testing and histopathology and the subsequent tolerance of asparaginase in patients upon re-exposure (18) argues against an HLA class I-restricted adaptive immune response to asparaginase as a mechanism of the liver injury.
In summary, review of the clinical course of asparaginase hepatotoxicity has confirmed its distinctive and reproducible phenotypic features. The clinical, biochemical and histologic features suggest that asparaginase is a direct hepatotoxin, the mechanism of injury being substrate depletion of secondary necessary amino acids. These conclusions provide hypotheses that would be appropriate for studies in vivo and in vitro (bedside to bench) as well as possible means of therapy and prevention of this unique form of liver injury.
Supplementary Material
Acknowledgements
The authors would like to thank Dr. Huiman Barnhart of the Duke Clinical Research Institute for her help in retrieving and analyzing data from the DILIN database and Drs. Connie Cheng and Allen Brinker of The Division of Pharmacovigilance, Office of Surveillance and Epidemiology, Center for Drug Evaluation and Review, FDA for their help in retrieving and analyzing data from the FDA FAERS database.
Funding: The DILIN Network is structured as a U01 cooperative agreement with funds provided by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) under grants: 2U01-DK065176-06 (Duke), 2U01-DK065201-06 (UNC), 2U01-DK065184-06 (Michigan), 2U01-DK065211-06 (Indiana), 5U01DK065193-04 (UConn), 5U01-DK065238-08 (UCSF/CPMC), 1U01-DK083023-01 (UTSW), 1U01-DK083027-01 (TJH/UPenn), 1U01-DK082992-01 (Mayo), 1U01-DK083020-01 (USC). Additional funding is provided by CTSA grants: UL1 RR025761 (Indiana), UL1TR000083 (UNC), UL1 RR024134 (UPenn), UL1 RR024986 (Michigan), UL1 RR024982 (UTSW), UL1 RR024150 (Mayo) and in part by the Intramural Research Program of the NIDDK and the National Cancer Institute. EP reports grants from NHMRC of Australia and from the NIH: P50GM115305, P30AI110527, R21AI139021, R34AI136815.
Abbreviations:
- ALT
Alanine aminotransferase
- Alk P
Alkaline phosphatase
- ALL
Acute lymphoblastic leukemia
- AST
Aspartate aminotransferase
- BMI
Body mass index
- DILIN
Drug-Induced Liver Injury Network
- DILI
Drug-induced liver injury
- E.coli
Escherichia coli
- RUCAM
Roussel Uclaf Causality Assessment Method
Footnotes
Compliance with Ethical Requirements
Conflict of Interest / Financial Disclosures:
Drs. Kamal has nothing to disclose
Dr. Koh has nothing to disclose
Dr. Samala has nothing to disclose
Dr. Stolz has nothing to disclose
Dr. Hayashi has nothing to disclose
Dr. Wang has nothing to disclose
Dr. Hoofnagle has nothing to disclose
Dr. Durazo receives consulting fees from Intercept Pharmaceuticals
Dr. Fontana receives grant support from Abbvie, Gilead Sciences and Bristol-Myers-Squibb and consulting fees from Sanofi-Aventis.
Dr. Phillips receives personal fees from UpToDate and Biocryst and holds patent for HLA-B*57:01 testing for abacavir hypersensitivity without financial remuneration.
Informed Consent in Studies with Human Subjects
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2008 (5). Informed consent was obtained from all patients for being included in the study.
References
- 1.Egler RA, Ahuja SP, Matloub Y. L-asparaginase in the treatment of patients with acute lymphoblastic leukemia. J Pharmacol Pharmacother 2016; 7: 62–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Raetz EA, Salzer WL. Tolerability and efficacy of L-asparaginase therapy in pediatric patients with acute lymphoblastic leukemia. J Pediatr Hematol Oncol 2010; 32: 554–63. [DOI] [PubMed] [Google Scholar]
- 3.Horowitz B, Madras BK, Meister A, et al. Asparagine synthetase activity of mouse leukemias. Science 1968; 160: 533–5. [DOI] [PubMed] [Google Scholar]
- 4.Broome JD. Studies on the mechanism of tumor inhibition by L-asparaginase. Effects of the enzyme on asparagine levels in the blood, normal tissues, and 6C3HED lymphomas of mice: differences in asparagine formation and utilization in asparaginase-sensitive and -resistant lymphoma cells. J Exp Med; 127: 1055–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Incidence Earl M. and management of asparaginase-associated adverse events in patients with acute lymphoblastic leukemia. Clin Adv Hematol Oncol 2009; 7: 600–6. [PubMed] [Google Scholar]
- 6.Oettgen HF, Stephenson PA, Schwartz MK, et al. Toxicity of E. Coli L-asparaginase in man. Cancer 1970; 25: 261–253-78. [DOI] [PubMed] [Google Scholar]
- 7.Zubrod CG. The clinical toxicities of L-asparaginase: in treatment of leukemia and lymphoma. Pediatrics 1970; 45: 555–9. [PubMed] [Google Scholar]
- 8.Cairo MS. Adverse reactions of L-asparaginase. Am J Pediatr Hematol Oncol 1982; 4: 335–9. [PubMed] [Google Scholar]
- 9.Hijiya N, van der Sluis IM. Asparaginase-associated toxicity in children with acute lympholastic leukemia. Leuk Lymphoma 2016; 57: 748–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Horvat TZ, Pecoraro JJ, Daley RJ, et al. The use of Erwinia asparaginase for adult patients with acute lymphoblastic leukemia after pegaspargase intolerance. Leukemia Research 2016; 50: 17–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chalasani N, Bonkovsky HL, Fontana R, et al. United States Drug Induced Liver Injury Network. Features and outcomes of 899 patients with drug-induced liver injury: the DILIN Prospective Study. Gastroenterology 2015; 148: 1340–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fontana RJ, Watkins PB, Bonkovsky HL, et al. Drug-Induced Liver Injury Network (DILIN) prospective study: rationale, design and conduct. Drug Saf. 2009; 32: 55–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rockey DC, Seeff LB, Rochon J, et al. ; US Drug-Induced Liver Injury Network. Causality assessment in drug-induced liver injury using a structured expert opinion process: comparison to the Roussel-Uclaf causality assessment method. Hepatology 2010; 51: 2117–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karnes JH, Shaffer CM, Bastarache L, et al. Comparison of HLA allelic imputation programs. PLOS One 2017; 12: 30172444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gragert L, Fingerson S, Albrecht M, et al. Fine-mapping of HLA associations with chronic lymphocytic leukemia in US populations. Blood 2014; 124: 2657–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Urayama KY, Thompson PD, Taylor M, et al. Genetic variation in the extended major histocompatibility complex and susceptibility to childhood acute lymphoblastic leukemia: a review of the evidence. Font Oncol 2013; 3: 300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Silverman LB, Gelber RD, Dalton VK, et al. Improved outcome for children with acute lymphoblastic leukemia: results of Dana-Farber Consortium Protocol 91–01. Blood 2001; 97: 1211–8. [DOI] [PubMed] [Google Scholar]
- 18.Burke PW, Aldoss I, Lunning MA, et al. Pegaspargase-related high-grade hepatotoxicity in a pediatric-inspired adult acute lymphoblastic leukemia regimen does not predict recurrent hepatotoxicity with subsequent doses. Leukemia Research 2018; 66: 49–56. [DOI] [PubMed] [Google Scholar]
- 19.Pratt CB, Johnson WW. Duration and severity of fatty metamorphosis of the liver following L-asparaginase therapy. Cancer 1971; 28: 361–4. [DOI] [PubMed] [Google Scholar]
- 20.Sahoo S, Hart J. Histopathological features of L-asparaginase-induced liver disease. Seminar Liver Dis 2003; 23: 295–9. [DOI] [PubMed] [Google Scholar]
- 21.Webber BL, Freiman I. The liver in kwashiorkor. A clinical and electron microscopic study. Arch Pathol 1974; 98: 400–8. [PubMed] [Google Scholar]
- 22.Roesmann A, Afify M, Panse J, et al. L-carnitine ameliorates L-asparaginase-induced acute liver toxicity in steatotic rat livers. Chemotherapy 2013; 59: 167–75. [DOI] [PubMed] [Google Scholar]
- 23.Alshiekh-Nasany R, Douer D. L-carnitine for treatment of pegaspargase-induced hepatotoxicity. Acta Haematol 2016; 135: 208–10. [DOI] [PubMed] [Google Scholar]
- 24.Al-Nawakil C, Willems L, Mauprivez C, et al. Successful treatment of L-asparaginase-induced severe acute hepatotoxicity using mitochondrial cofactors. Leukemia Lymphoma 2014; 55: 1670–4. [DOI] [PubMed] [Google Scholar]
- 25.Blackman A, Boutin A, Shimanovsky A, et al. Levocarnitine and vitamin B complex for the treatment of pegaspargase-induced hepatotoxicity: a case report and review of the literature. J Oncol Pharm Practice 2018; 24: 393–7. [DOI] [PubMed] [Google Scholar]
- 26.Jenkins R, Perlin E. Severe hepatotoxicity from Escherichia coli L-asparaginase. J Natl Med Assoc 1987; 79: 775, 779. [PMC free article] [PubMed] [Google Scholar]
- 27.Bodmer M, Sulz M, Stadlmann S. Fatal liver failure in an adult patient with acute lymphoblastic leukemia following treatment with L-asparaginase. Digestion 2006; 74: 28–32. [DOI] [PubMed] [Google Scholar]
- 28.Aldoss I, Douer D, Behrendt CE, et al. Toxicity profile of repeated doses of PEG-asparaginase incorporated into a pediatric-type regimen for adult acute lymphoblastic leukemia. Eur J Haematol 2016; 96: 375–80. (25%; Age and obesity) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stock W, Douer D, DeAngelo DJ, et al. Prevention and management of asparaginase/pegasparaginase-associated toxicities in adults and older adolescents: recommendations of an expert panel. Leukemia Lymphoma 2011; 52: 2237–53. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.