

Abstract
Inflammatory disorders of the pancreas are divided into acute (AP) and chronic (CP) forms. Both states of pancreatitis are a result of pro-inflammatory mediators, including reactive oxygen species (ROS). One of the sources of ROS is NADPH oxidase (Nox). The rodent genome encodes Nox1–4, Duox1 and Duox2. Our purpose was to assess the extent to which Nox enzymes contribute to the pathogenesis of both AP and CP using Nox-deficient mice. Using RT-PCR, Nox1 was found in both isolated mouse pancreatic acini and pancreatic stellate cells (PaSCs). Subsequently, mice with genetically deleted Nox1 were further studied and showed that the histo-morphologic characteristics of caerulein-induced CP, but not caerulein-induced AP, was ameliorated in Nox1 KO mice. We also found that the lack of Nox1 impaired caerulein-induced ROS generation in PaSCs. Using Western blotting, we found that AKT mediates the fibrotic effect of Nox1 in a mouse model of CP. We also found a decrease in phospho-ERK and p38MAPK levels in Nox1 KO mice with CP, but not with AP. Both CP-induced TGF-β up-regulation and NF-ĸB activation were impaired in pancreas from Nox1 KO mice. Western blotting indicated increases in proteins involved in fibrosis and acinar-to-ductal metaplasia in WT mice with CP. No change in those proteins were observed in Nox1 KO mice. The lack of Nox1 lowered mRNA levels of CP-induced matrix metalloproteinase MMP-9 and E-cadherin repressor Twist in PaSCs.
Conclusion:
Nox1-derived ROS in PaSCs mediate the fibrotic process of CP by activating the downstream redox-sensitive signaling pathways AKT and NF-ĸB, up-regulating MMP-9 and Twist, and producing α-smooth muscle actin and collagen I and III.
Keywords: Chronic pancreatitis, NADPH oxidase 1, MMP-9, Twist, NF-ĸB
1. Introduction
Inflammation of the pancreas can occur in two distinct states, either in the acute form or the chronic form. Acute pancreatitis (AP) is a short-term inflammation that develops suddenly. By contrast, chronic pancreatitis (CP) manifests from a long-term inflammation, which results in a significant replacement of the parenchyma by extracellular matrix (ECM)-rich connective tissue (i.e. fibrosis) and permanent organ damage [1]. In 2014, the National Ambulatory Medical Care Survey (NAMCS) and the National Hospital Ambulatory Medical Care Survey (NHAMCS) estimated 562,048 annual office visits and 195,113 annual emergency department visits for both AP and CP [1]. Based on weighted national estimates from HCUP Nationwide Emergency Department Sample (NEDS) 2006 and 2014, there were 279,145 admissions (15% increase from 2005) for AP and 12,770 admissions (42% decrease from 2005) for CP [1]. In 2016, 0.7/100,000 women and 1.1/100,000 men died from AP [1]. In 2015, the total expense for pancreatic disorders was $2386 million in the United States [1]. Thus, a better understanding of the mechanism underlying the pathogenesis of AP and CP is necessary in order to develop more effective the rapeutic options to attenuate the progression of these diseases.
There are several mouse models of AP; the most common models are those induced by administering supraphysiologic doses of caerulein, an orthologue of cholecystokinin-8 (CCK), or by treating with bile acids (e.g., taurocholate). Both treatments lead to activation of inflammatory pathways and a premature intracellular activation of trypsinogen, which is the inactive precursor of the digestive protease trypsin, causing a “digestion” of the acinar cells and pancreatic damage [2,3]. The most common mouse model of CP consists of repetitive inductions of caerulein-induced AP, which lead to increasing damage of the pancreas that eventually results in atrophy and replacement of the parenchyma by fibrotic tissue [4].
The inflammatory response in both states of pancreatitis involves pro-inflammatory mediators, including reactive oxygen species (ROS). A number of ROS sources (e.g., xanthine oxidase (XO), uncoupled nitric oxide synthase (NOS), mitochondria, NADPH oxidase (Nox) enzymes [5]) contribute to the pathogenesis of AP. For example, the XO inhibitor allopurinol attenuates pancreatic damage in canine [6], rat [7] and mouse [8] models of caerulein-induced AP. XO promotes intracellular trypsinogen activation and zymogen granule damage in isolated rat pancreatic acini [9]. XO was also found to be mainly responsible for the glutathione oxidation that occurs at later stages of taurocholate-induced AP [10]. Uncoupled NOS plays a deleterious role in caerulein-induced AP, probably through the decrease in the availability of endogenous tetrahydrobiopterin, which is a cofactor for the synthesis of NO [11]. NO has been shown to play a protective role against injury caused by pancreatitis [12]. Mitochondrial ROS levels increased in the presence of supramaximal CCK and taurocholate in acinar cells [13]. However, among these sources, the main source of ROS during episodes of inflammation is the Nox family [5]. Nox enzymes transfer electrons across membranes to reduce oxygen to superoxide (O −2), which, spontaneously or through a reaction catalyzed by superoxide dismutase (SOD), dismutates to hydrogen peroxide (H2O2), a stable and cell-per-meant ROS responsible for initiating autocrine and paracrine signaling mechanisms. The rodent genome encodes six Nox enzymes: Nox 1–4, Duox1 and Duox2. Support for Nox enzymes in the acute inflammatory process of the pancreas has come from studies using antisense oligo-nucleotides (AS ODN) of Nox subunits p22phox and p47phox, the flavoenzyme inhibitor diphenyleneiodonium (DPI) and p47phox-deficient mice. p47phox is the organizer subunit for Nox2 and is required for Nox2 activation, while p22phox is a Nox subunit that interacts with Nox1, Nox2, Nox3 and Nox4 and stabilizes them [5]. Because Duox enzymes do not require activator or organizer subunits [5], we did not further study the participation of these enzymes in the inflammatory process of the pancreas. Yu et al. [14], found Nox1, p22phox, p47phox, and p67phox in the rat pancreatoma cell line AR42J. When caerulein-treated AR42J cells were pre-incubated with a calcium chelator BAPTA-AM, or transfected with AS ODN of p22phox and p47phox, the activation of Nox was inhibited, and ROS production [14] and apoptosis was suppressed [15]. Furthermore, transfection with AS ODN of p22phox and p47phox or pre-treatment with DPI impaired IL-6 up-regulation and apoptosis of caerulein-stimulated AR42J cells [16]. Caerulein-induced activation of JAK/STAT3 and up-regulation of both MAPKs and tumor growth factor β (TGF-β) were also inhibited by either AS ODN for p22phox and p47phox or pre-treatment with DPI in AR42J cells [17]. Among these previous findings, the source of Nox-derived ROS in pancreatitis is still unclear, with no consensus reached. Because caerulein-induced trypsin activation has decreased in both neutrophils depleted rats and p47phox-deficient mice [18], Nox-derived ROS in pancreatitis was thought to be essentially derived from Nox2 due to phagocyte invasion, but not from pancreatic acini as previously reported [14–17]. These findings have been reviewed in Ref. [19]. However, pancreatic acini, which synthesize and store digestive enzymes [3], and pancreatic stellate cells (PaSCs), which are found in periacinar spaces and play a main role in pancreatic fibrosis [20], have been shown to secrete a variety of inflammatory mediators in response to pancreatitis-causing stimulus and contribute to the robust inflammatory response. In the last years, the development of Nox isoform specific-deficient mice has provided a new approach to study the source and the role of Nox enzymes in the inflammatory process of pancreas. Therefore, the goal of this study was to determine the source and role of Nox enzymes in the pathogenesis of AP and CP by using Nox isoform specific-deficient mice.
2. Materials and methods
Ethical approval.
All experiments were conducted in accordance with the National Institute of Health Guidelines for the Care and Use of Laboratory Animals and approved by the Augusta University Institutional Animal Care and Use Committee (IACUC) (protocol # 2017–0878).
Materials.
Caerulein (#C9026), trypsin inhibitor (SBTI) (#T9003), protease (#P5147) were purchased from Sigma Aldrich (St Louis, MO), collagenase (CLSPA) was purchased from Worthington Biochemical Co. (Lakewood, NJ), collagenase P (11213857001), DNase I recombinant (04716728001), leupeptin and aprotinin were purchased from Roche Diagnostics (Indianapolis, IN), bovine albumin serum fraction V (BSA) from MP Biomedicals (Solon, OH), Phadebas Amylase Assay kit was purchased form Magle Life Sciences (Cambridge, MA), Boc-Gln-Ala-Arg-AMC from BachemAG (Bubendrof, Switzerland), Dulbecco’s minimal essential medium (DMEM)/high glucose from HyClone (Logan, Utah), TRIzol, and 3,3-diaminobenzidine tetrahydrochloride (DAB) from Thermo Scientific (Rockford, IL). Protein determination reagent was purchased from Bio-Rad Life Science Research (Hercules, CA). Supersignal West Femto Chemiluminescent Substrate was purchased from Thermo Fisher Scientific (Rockford, IL).
Mice.
Seven to eight-week old male mice were used for this study. C57BL/6 mice. C57BL/6 mice (WT) were purchased from Envigo (Indianapolis, IN). Whole body Nox1tm1Kkr mice. Nox1tm1Kkr mice (Nox1 KO) were bought from Jackson Laboratory. The development of Nox1 KO mice has previously described [21].
Antibodies.
Antibodies against the following proteins were used: rabbit polyclonal antibodies to collagen 1A1 (#84336), c-Jun-N-terminal kinase (SAPK/JNK) (#9252), AKT (pan) (#4691), rabbit monoclonal antibodies to vimentin (D21H3)XP® (#5741), NF-ĸB p65 (#8242), p44/42 MAPK (ERK1/2) (#4695), p38MAPK (#8690), phospho-p44/42 MAPK (Erk1/2) (#4370), α-smooth muscle actin (α-SMA) (#19245), transforming growth factor- β (TGF-β) (#3709), phospho-AKT (Ser473) (#4060), phospho-SAPK/JNK (#4668), mouse monoclonal antibodies to α-tubulin (#3873) were provided by Cell Signaling Technology (Beverly, MA); rabbit antibody against α-amylase (A8273) was purchased from Sigma Aldrich (St Louis, MO), rat monoclonal against cytokeratin 19 (TROMA-III) from Developmental Studies Hybridoma Bank, goat anti-mouse IgG-horseradish peroxidase (HRP) (sc-2005) and goat anti-rabbit IgG-HRP (sc-2004) were provided by Santa Cruz Biotechnology (Santa Cruz, CA).
Induction of AP.
AP was induced by repetitive injections of caerulein (50 μg/Kg body weight/h) six times hourly as previously described [22]. Mice were euthanized 1 h after the last injection. Control mice received PBS injections.
Induction of CP.
CP was induced by repetitive injections of caerulein (50 μg/Kg body weight/h) three times a week, six times a day for 7 weeks as previously described [4]. Mice were euthanized four days after the last injection. Control mice were given PBS injections.
Pancreas weight/body weight (PW/BW) ratio.
The pancreas weight was recorded when harvested and reported as pancreas weight/body weight ratio as previously described [23].
Determination of pancreatic edema:
Pancreatic edema was quantified by weighing the freshly harvested pancreas (wet weight) and comparing that to the same sample after desiccation to a constant weight at 90 °C for 36 h (dry weight). The results were calculated using the formula: wet weight - dry weight/wet weight × 100 as previously described [22].
Serum lipase and amylase determination.
Blood was collected after euthanization of mice. Blood was allowed to clot, then centrifuged at 10,000×g for 5 min to separate serum from blood cells, and serum was collected in a separate tube. Lipase activity was determined as previously described [24]. Lipa-zyme buffer (70 mM TRIS, pH9.3 ± 0.05, 8.7 mM sodium deoxycholate), Lipa-zyme substrate (0.8% w/v olive oil in ethanol) and the serum were incubated, and the absorbance was determined at 546 nm for 5 min using a Biotek Synergy 2 multi-mode detection microplate reader. Gen5 software was used to perform a kinetic assay, and the final lipase activity (U/L) was calculated. The factor for converting the absorbance change into U/L is 2000 with olive oil as substrate. Amylase levels were determined through the Phadebas amylase test (Magle Life Sciences, Lund, Sweden) as previously described [25]. The Gen5 software was used to perform an endpoint assay using a Biotek Synergy 2 multi-mode detection microplate reader, and the serum amylase levels were determined with the use of a standard curve.
Isolation of pancreatic acini.
Pancreatic acini from WT and Nox1 KO mice were prepared by enzymatic digestion of pancreas with purified collagenase (code: CLSPA) followed by mechanical shearing, filtration through 150-μm Nitex mesh and purification by sedimentation through 4% bovine serum albumin (BSA) as previously described [25]. Freshly digested cultured acini were stimulated with caerulein (0.01,0.1, 1, 10, and 100 nM) for 30 min in DMEM/high glucose medium containing 5 mg/ml BSA and 0.1 mg/ml trypsin inhibitor.
Quantification of amylase secretion.
Samples were centrifuged for 30 s in a microcentrifuge, and the supernatant was assayed for amylase activity with Phadebas reagents (Magle Life Sciences, Lund, Sweden) as previously described [25]. Results were expressed as a percentage of initial acinar amylase content.
Quantification of trypsin activation.
Acini were lysed with homogenization buffer (5 mM MES, pH 6.4, 250 mM sucrose, 1 mM MgSO4) in the Dounce homogenizer. To a 96-well plate, assay buffer (5 mM TRIS HCl, pH 8, 150 mM NaCl, 1 mM CaCl2, 0.1 mg/ml BSA) and the homogenate were added. Trypsin activity was determined using Boc-Gln-Ala-Arg-AMC (50 μM) as a substrate as previously described [18]. The fluorescence (380/440 nm) was recorded for 5 min using a Biotek Synergy 2 multi-mode detection microplate reader.
Histology.
Pancreatic tissue was fixed with 10% formalin and embedded in paraffin as previously described [23]. Hematoxylin and eosin (H&E) staining was performed at the Augusta University Histology Core. Images were captured with an Olympus CK2 inverted light microscope (Olympus America, Inc., Melville, NY) with a X40 objective lens. A Canon digital SLR camera was connected to the microscope.
Isolation of pancreatic stellate cells (PaSCs).
Pancreas from WT and Nox1 KO mice was isolated and digested with collagenase P (3 mg), protease (1.4 mg) and DNase I recombinant (8.75 μl) in 10 ml of GBSS plus salt medium. Pancreata were disrupted by pipetting and filtered through a 250 μm mesh. PaSCs were purified using a 28.7% solution of Nycodenz (2.295 g in 8 ml GBSS without salt). PaSCs were resuspended in IMDM GlutaMax medium with 10% fetal bovine serum (FBS) and 100 U/ml penicillin and 100 μg/streptomycin as previously described [26]. PaSCs were cultured in 24-well plates until confluence (1 week) and then transferred to 12-well plates (PaSCs were only passed once). IMDM GlutaMax medium was changed every two days.
Immunohistochemistry (IHC):
IHC for α-SMA, NF-ĸB and p-AKT in whole pancreas: Cells positive for these proteins were determined using 3,3-diaminobenzidine tetrahydrochloride (DAB) as a chromogen (color: brown). Briefly, slides were incubated first with blocking buffer (3% BSA in PBS with 0.05% Tween-20) for 1 h at room temperature and then with antibody against α-SMA, NF-ĸB or p-AKT overnight at 4 °C. Slides were washed with PBS with 0.05% Tween-20 and incubated with HRP-labeled secondary antibody for 30 min. After washing, the DAB solution was added to the slides, incubated for 10 min, and then washed again. The slides were mounted and a coverslip was added to the section. IHC for collagen in whole pancreas: collagen I and III fibers were examined using Picro Sirius Red as a chromogen (color: red). Briefly, slides were incubated with Picro-Sirius Red solution (Abcam, Cambridge, MA) for 60 min, rinsed quickly with 0.5% acetic acid solution and then with absolute ethanol. The slides were mounted and a coverslip was added to the section.
Determination of ROS production in PaSCs and isolated pancreatic acini:
2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) fluorescence: Production of ROS was measured using the cell-permeant H2DCFDA, which is a chemically reduced form of fluorescein. H2O2 produced by cells oxidizes the non-fluorescent form H2DCFDA to 2,7-dichlorofluorescein (DCF), the fluorescent form of which is proportional to the H2O2 produced. The excitation and emission wavelengths for DCF are ~492–495 and 517–527 nm, respectively. PaSCs were grown to confluence in a 24-well plate. Then, they were transferred to a 12-well plate. At confluence, cells were treated with vehicle or caerulein (10 pM and 100 nM) or TGF-β (10 ng/ml) for 30 min. Isolated pancreatic acini were cultured in a 12-well plate and stimulated with caerulein (100 nM) or TGF-β (10 ng/ml) for 30 min. At the end of the incubation time, the cells were loaded for 20 min with 10 μM H2DCFDA at 37 °C in the dark. Fluorescence was measured using a BD Accuri C6 flow cytometer.
2-OH-E+ fluorescence: Intracellular O −2 generation was measured using dihydroethidium (DHE), which is oxidized by superoxide to form 2-hydroxyethidium (2-OH-E+). We plated PaSCs in 12-well plates and preincubated them with 10 μM DHE for 30 min at 37 °C. Cells were then treated with caerulein (100 nM) for 30 min, washed twice with ice-cold PBS, fixed in 2% paraformaldehyde, and fluorescence was determined (excitation/emission for 2-OH-E+: 518 nm/605 nm) using a BD Accuri C6 flow cytometer.
Isolation of peritoneal macrophages from mice with CP:
It was done as previously described [27]. WT and Nox1 KO mice with CP were euthanized using a CO2 chamber, a 10-ml syringe with a 20-G needle was inserted through the peritoneal wall and 10 ml of PBS without calcium and magnesium was injected into the peritoneal cavity. Using the same syringe and needle, the fluid from the peritoneum was aspirated and centrifuged at 400×g for 10 min at 4 °C. The cell pellet was resuspended in DMEM/F12 with 10% FBS and 100 U/ml recombinant mouse macrophage colony stimulating factor (M-CSF) (#cyt-439), Prospec Tany TechnoGene Ltd).
2.1. Determination of gene expression at mRNA level
1). RNA isolation and reverse transcription-PCR:
A small piece of mouse pancreas (40 mg) was cut and immediately placed in 1 ml of RNAlater solution (Invitrogen by Thermo Fisher Scientific, Rockford,IL). Total RNA was isolated from mouse pancreas using Trizol and RNeasy® Mini kit (Qiagen, Inc. USA, Valencia, CA). RNeasy® Mini kit was utilized to extract total RNA from isolated mouse pancreatic acini, PaSCs and peritoneal macrophages. Integrity and quality of RNA was assessed by recording the OD 260/280 ratio measured using NanoDrop™ One (Thermo Scientific, Waltham, MA), and agarose gel electrophoresis. First-strand complementary DNA was synthesized with TaqMan Reverse Transcription Reagents (Thermo Fisher Scientific, Waltham, MA). Five μg of cDNA was used in each PCR reaction. Amplification with Taq DNA polymerase from Expand High Fidelity Enzyme System (Roche Diagnostics, Indianapolis, IN) was conducted using specific primers from GeneCopoeia (Rockville, MD) and as previously described [28]. The PCR primers were listed in Table 1. The efficiency of the primers was checked beforehand by using control DNA provided by GeneCopoeia, Inc.
Table 1.
Primers used for analysis of mRNA expression using reverse transcriptase (RT) and real-time quantitative PCR. Primers of Nox family were obtained from GeneCopoeia™ (Rockville, MD) and as previously described [28]. Primers of genes mediating EMT were designed with NIH primer blast (https://www.ncbi.nlm.nih. gov/tools/primer-blast) based on genes sequences obtained from the Gen Bank NCBI sequence Viewer.
| Gene | Forward Primer (5’ to 3’) | Reverse Primer (5’ to 3’) | Accession Number | PCR product size |
|---|---|---|---|---|
| Nox enzymes | ||||
| Nox1 | GGAGTGGCATCCCTTCACTC | GGCATTGGTGAGTGCTGTTG | NM_172203.2 | 127 bp |
| Nox1 (GeneCopoeia) | MQP097901 | MQP097901 | NM_172203.2 | 132 bp |
| Nox2 | CGAGTCACGGCCACATACA | TGAATGCCAGAGTCGGGATTT | FJ168469.1 | 198 bp |
| Nox3 | CTCGTTGCCTACGGGATAGC | CCTTCAGCATCCTTGGCCT | NM_198958.2 | 184 bp |
| Nox4 | MQP078234 | MQP078234 | NM_001285835 | 147 bp |
| Genes mediating EMT | ||||
| Tgf-P | MQP030343 | MQP030343 | NM_011577.1 | 125 bp |
| Slug or snail family zinc finger 2 (Snai2) | CTGTATGGACATCGTCGGCA | ACTTACACGCCCCAAGGATG | NM_011415.3 | 94 bp |
| Zinc finger E-box binding homeobox 1 (Zeb1), transcript variant 1 | GGTACAGTGGAGGTTGGAGC | CTCTCGCTTTCGCTCTCCAT | NM_011546.3 | 85 bp |
| Zinc finger E-box binding homeobox 2 (Zeb2), transcript variant 2 | CATGCCCAACCATGAGTCCT | TTCTGGCCCCATTGCATCAT | NM_015753.4 | 177 bp |
| Twist basic helix-loop-heUx transcription factor 1 (twist1) | ATTCAGACCCTCAAACTGGCG | TCTTGGAGTCCAGCTCGTCG | NM_011658.2 | 78 bp |
| Matrix metalloproteinase 2 (MMP-2) | TGTACAGACACTGGTCGCAG | AAACAAGGCTTCATGGGGGC | NM_008610.3 | 101 bp |
| Matrix metalloproteinase 9 (MMP-9) | CGACTTTTGTGGTCTTCCCCA | AGGTTTGGAATCGACCCACG | NM_013599.4 | 254 bp |
| Snail family zinc finger 1 (Snai1) | ACCTCCAAACCCACTCGGAT | GACATGCGGGAGAAGGTTCG | NM_011427.3 | 69 bp |
| Housekeeping gene | ||||
| 18S rRNA | GTAACCCGTTGAACCCCATT | CCATCCAATCGGTAGTAGCG | NR_003278.3 | 150 bp |
2). Real-time Quantitative PCR:
The relative expression of genes in mouse pancreas and PaSCs from WT and Nox1 KO mice was evaluated by real-time quantitative PCR analysis using Absolute Blue SYBR Green ROX mix (Thermo Fisher Scientific) and a real-time quantitative PCR machine (LightCycler 96 system, Roche). The PCR primers were listed in Table 1. Results from real-time quantitative PCR were evaluated using the 2−ΔΔCq method as previously described [29]. This method uses the equation ΔΔCq=(Cq, Target gene – Cq, reference) Time × – (Cq, Control – Cq, reference)Time × to make the Cq values relative between WT and Nox1 KO mice. The primers were designed with NIH primer designing tool based on gene sequences obtained from the GeneBankTM NCBI sequence viewer. 18S rRNA was used as a reference.
Determination of gene expression at protein level.
Western blotting analysis was carried out as previously described [25,30]. Cells were lysed using lysis buffer containing 50 mM TRIS HCl, pH 7.5, 150 mM sodium chloride, 5 mM EDTA, 10 mM sodium pyrophosphate, 25 mM β-glycerolphosphate, 0.1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 1 mM sodium vanadate, 25 mM sodium fluoride and 1 mM dithiothreitol. Immunodetection of proteins was carried out using SDS-polyacrylamide gel, which was transferred to a nitrocellulose membrane (Bio-Rad Laboratories, Inc.). The membrane was blocked in 5% nonfat milk dissolved in Tris-buffered saline containing 0.1% (v/v) Tween X-20 for 1 h at room temperature. Corresponding primary antibodies were diluted 1:1000 in 5% bovine serum albumin (BSA), and the membrane was incubated with the antibody overnight at 4 °C followed by treatment with a HRP-conjugated secondary antibody (1:5000 in 5% nonfat milk) for 1 h at room temperature. After washing with Tris-buffered saline containing 0.1% (v/v) Tween X-20, peroxidase activity was visualized using the SuperSignal West Femto sensitivity substrate kit (Pierce). Images were taken using Foto/Analyst Luminary FX Imaging Workstation (Fotodyne Incorporated, Hartland, WI).
2.2. Statistical analysis
Results were expressed as means ± SE. Statistical analysis was carried out using Instat Graphpad software (La Jolla, CA). Statistical significance was determined employing ANOVA followed by Student-Newman-Keuls post hoc test. p < 0.05 was considered to be the minimal level of statistical significance.
3. Results
Nox1 is expressed in isolated mouse pancreatic acini.
Because intrapancreatic activation of digestive enzymes is a primary event in the cell injury of pancreatitis [3], we first tested whether or not the Nox enzymes Nox 1–4 are expressed in isolated pancreatic acini. Conflicting results exist regarding the expression of Nox enzymes in pancreatic acini. Some studies indicate that Nox enzymes are expressed in pancreatic acinar cells [15–17,31], whereas others indicate that Nox enzymes are not [18,32]. Using RT-PCR, we found the expression of Nox1 mRNA, but not Nox2, Nox3 or Nox4 mRNA, in isolated mouse pancreatic acini (Suppl. Fig. 1).
Nox1 does not mediate the inflammatory response in AP.
Because we found that Nox1 is expressed in isolated mouse pancreatic acini, we studied the participation of Nox1 in the inflammatory process of AP using whole body Nox1-defficent (Nox1 KO) mice. The lack of Nox1 in Nox1 KO mice was confirmed by RT-PCR (Suppl. Fig. 2). In in vitro studies, we isolated pancreatic acini from WT mice and Nox1 KO mice and stimulated them with caerulein (a CCK analogue) (0.01, 0.1, 1, 10, and 100 nM) for 30 min as previously described [33]. We found that the lack of Nox1 did not affect caerulein-induced amylase secretion or trypsin activation (Suppl. Fig. 3).
In in vivo studies, we used a mouse model of AP [22]. We found that the lack of Nox1 does not impair the histo-morphologic changes of AP (Suppl. Fig. 4A and B). A slight increase in the PW/BW ratio was observed in both WT mice with AP and Nox1 KO mice with AP and no changes in histological features of AP were observed in pancreas from Nox1 KO mice (Suppl. Fig. 4B).
The increase in water content (an indication of edema) and serum levels of digestive enzymes (amylase and lipase) was observed in WT and Nox1 KO mice in which AP was induced (Suppl. Fig. 5). In conclusion, Nox1 did not mediate the inflammatory process in AP.
Studies showing that Nox1-deficient mice have less hepatic fibrosis than WT mice after CCl4 injection or bile duct ligation [34,35], and that Nox1 is found in samples from patients with hepatic fibrosis [36] led us to hypothesize that Nox1-derived ROS play a role in the pathogenesis of CP. In addition to these findings, in a previous study, using DPI as a means to reduce the activity of Nox, Nox enzymes were suggested to develop pancreatic fibrosis in rats with dibutyltin dichloride-induced CP [37].
Nox1 contributes to histo-morphologic features of CP.
PW/BW ratio: CP causes pancreatic atrophy [38]. Based on the PW/BW ratio, we found a remarkable atrophy of the pancreas in WT mice with CP. However, Nox1 KO mice in which CP was induced showed no change in this ratio (Fig. 1A). Histology: We observed the most common features of CP, including moderate acinar loss with distended lumen and loss of apical granulations, fibrosis, microscopic duct dilations, and inflammatory infiltration in WT mice with CP. The lack of Nox1 reduced fibrosis and other features of CP (Fig. 1B). Activated PaSCs are fibrogenic and proliferate in CP [26,39]. Because activated PaSCs express α-SMA and collagen type I and III [26], we studied the activation of PaSCs by IHC analysis of these proteins. In healthy WT and Nox1 KO mice, positive α-SMA staining was found in the periacinar space, as well as perivascular and periductal regions of the pancreas, which are places where PaSCs reside [20]. In WT mice with CP, but not in Nox1 KO mice, activated PaSCs proliferated and released α-SMA (Fig. 2A). Staining for collagen I and III with Sirius Red showed an intense presence of collagen in pancreas (perivascular and periacinar) from WT mice with CP (Fig. 2B). In pancreatic tissues from Nox1 KO mice with or without CP, the staining of collagen was mainly perivascular (it was similar to healthy WT mice) (Fig. 2B). These results were confirmed by Western-blotting using collagen 1A1 antibody (Fig. 2C).
Fig. 1A. The absence of Nox1 impairs the effect of CP on pancreas weight. Left panel:
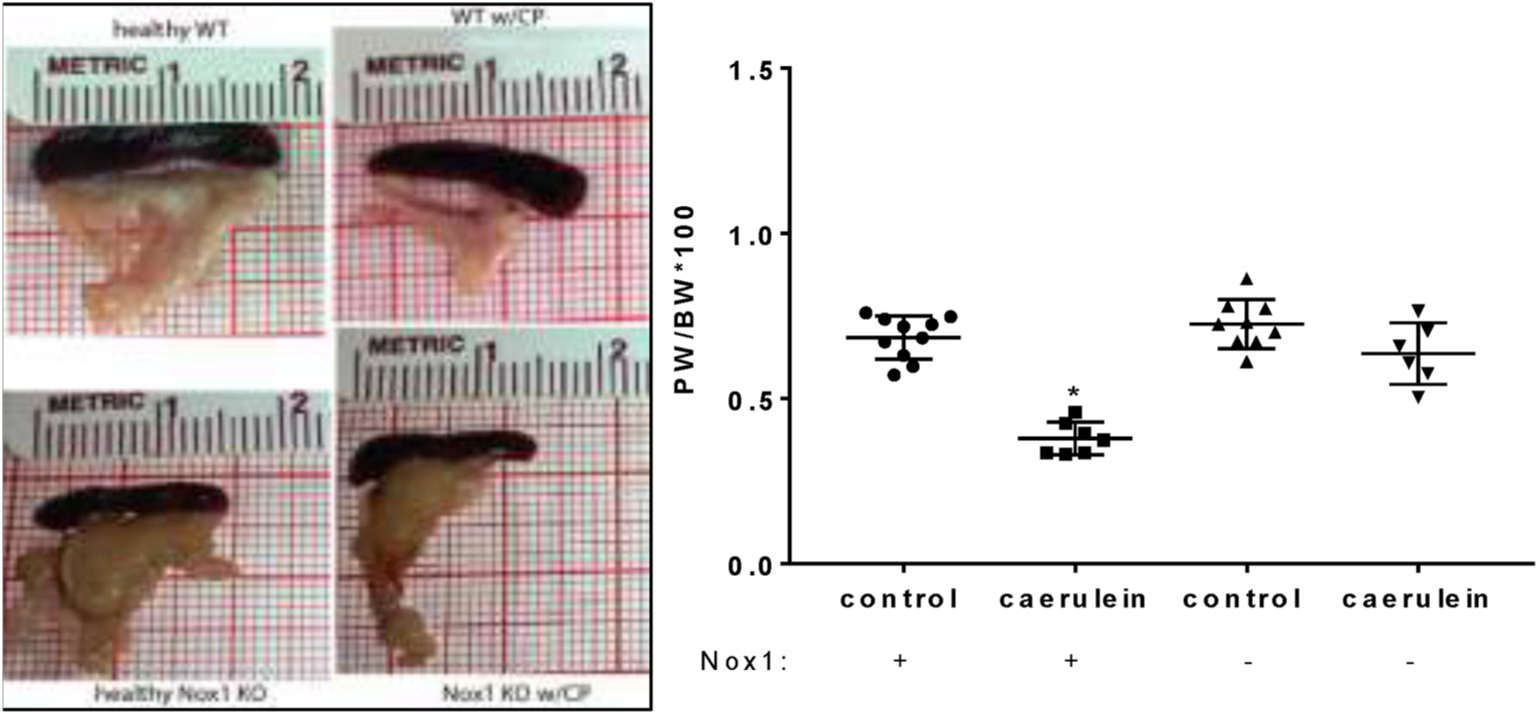
Representative pictures demonstrating pancreas atrophy in WT mice with CP, but not in Nox1 KO mice with CP. Right panel: Comparison of PW/BW ratio. Mean ± SE. n: WT: 10; WT w/CP: 7; Nox1 KO: 9; Nox1 KO w/CP: 6. *:p < 0.05 vs control.
Fig. 1B. The absence of Nox1 impairs the histological feature characteristics of CP.
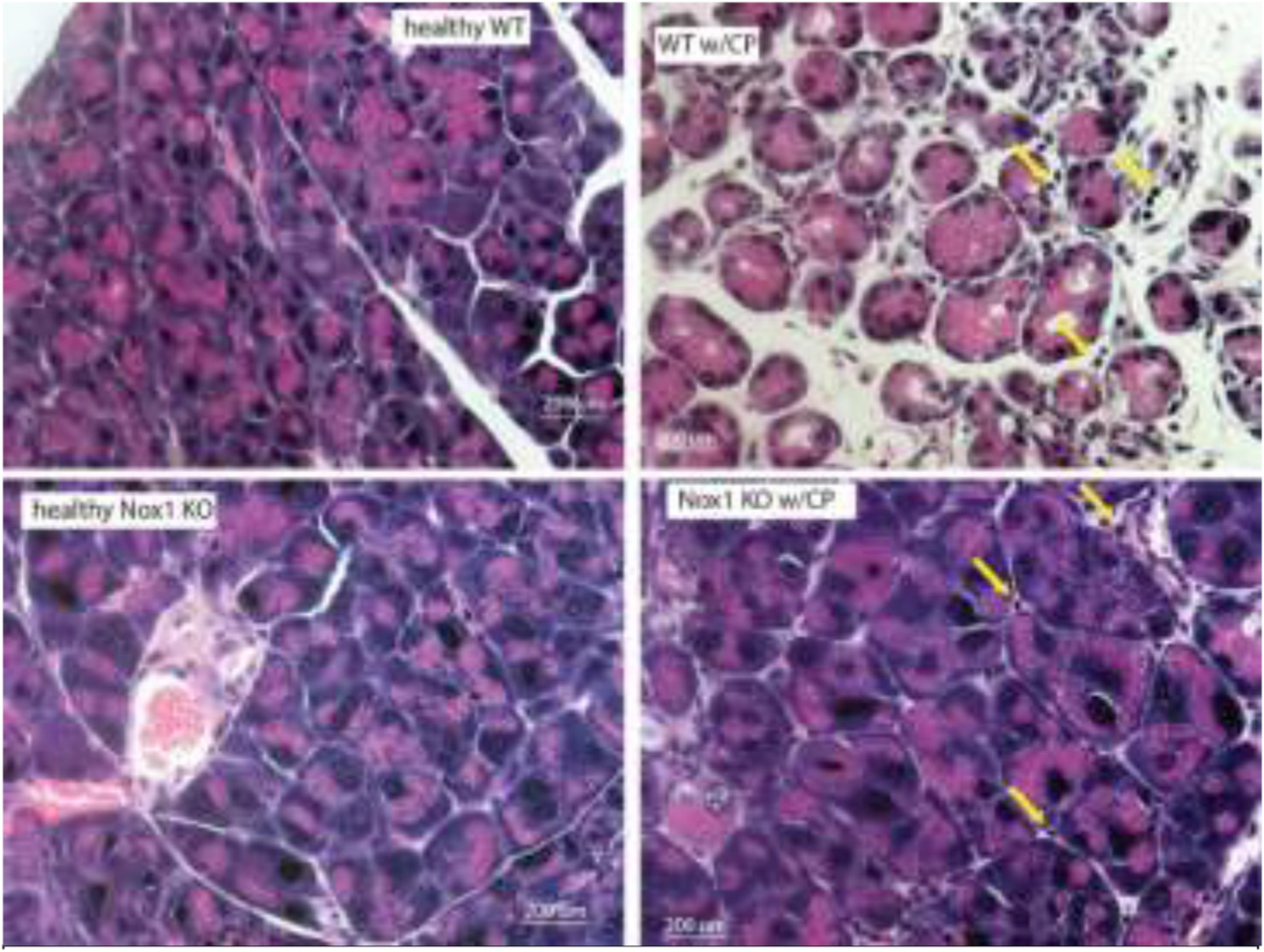
Representative hematoxylin and eosin (H&E)-stained sections (total magnification: 400x) show moderate acinar loss, fibrosis, and inflammatory infiltration (arrows) in pancreas from caerulein-treated WT mice, while normal cytoarchitecture of the pancreas and lack of fibrosis in pancreas from caerulein-treated Nox1 KO mice. n: 3.
Fig. 2A. The lack of Nox1 reduces fibrosis in CP.
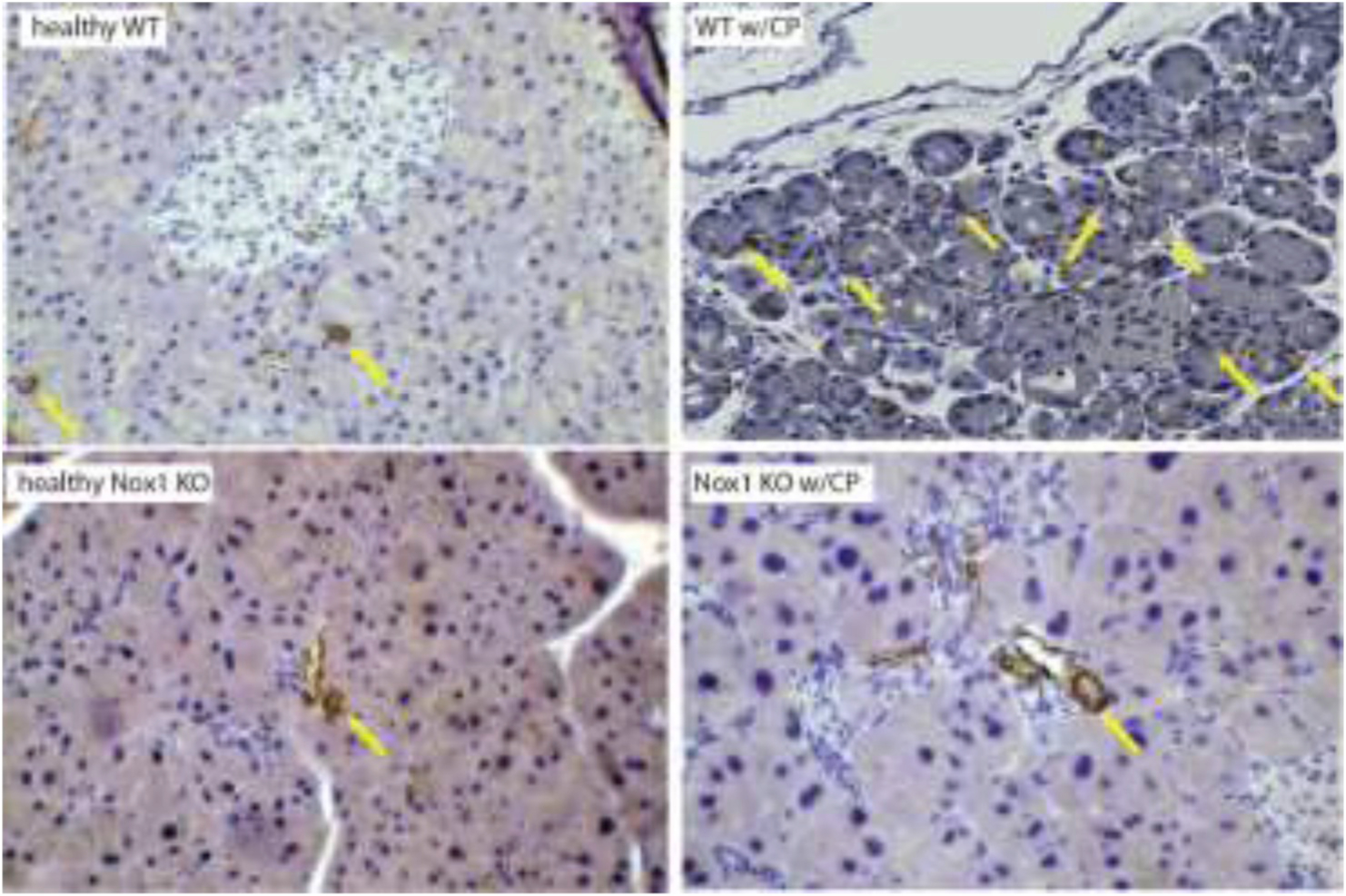
Pancreatic tissue were fixed with 10% formalin and embedded in paraffin. Following incubation with antibody against α-SMA, positive cells were visualized using 3, 3-diaminobenzi-dine tetrahydrochloride (DAB) as a chromogen (color: brown). (Arrows: α-SMA). Total magnification: 200X. n: 3.
Fig. 2B. The lack of Nox1 reduces the amount of collagen I and III fibers in CP.
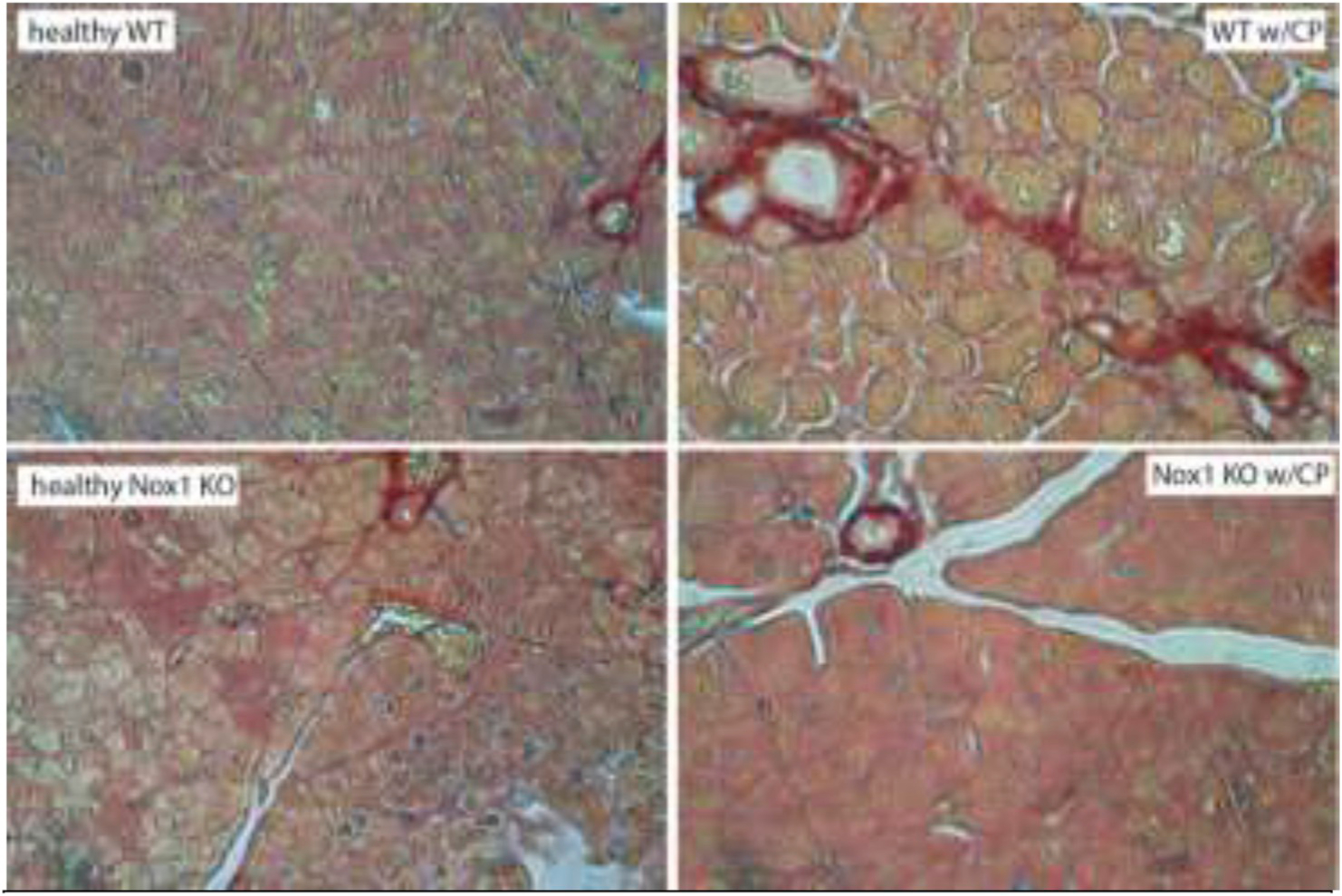
Pancreatic tissue were fixed with 10% formalin and embedded in paraffin. Cryostat sections of the tissue fixed were put onto glass slides, which were incubated with Picro-Sirius Red solution and proceeded according to the instruction provided by the manufacturer (Abcam, Cambridge, MA). Total magnification: 400X. (color: red). n: 3.
Fig. 2C. The lack of Nox1 reduces CP-increased collagen 1A levels in whole pancreatic tissues.
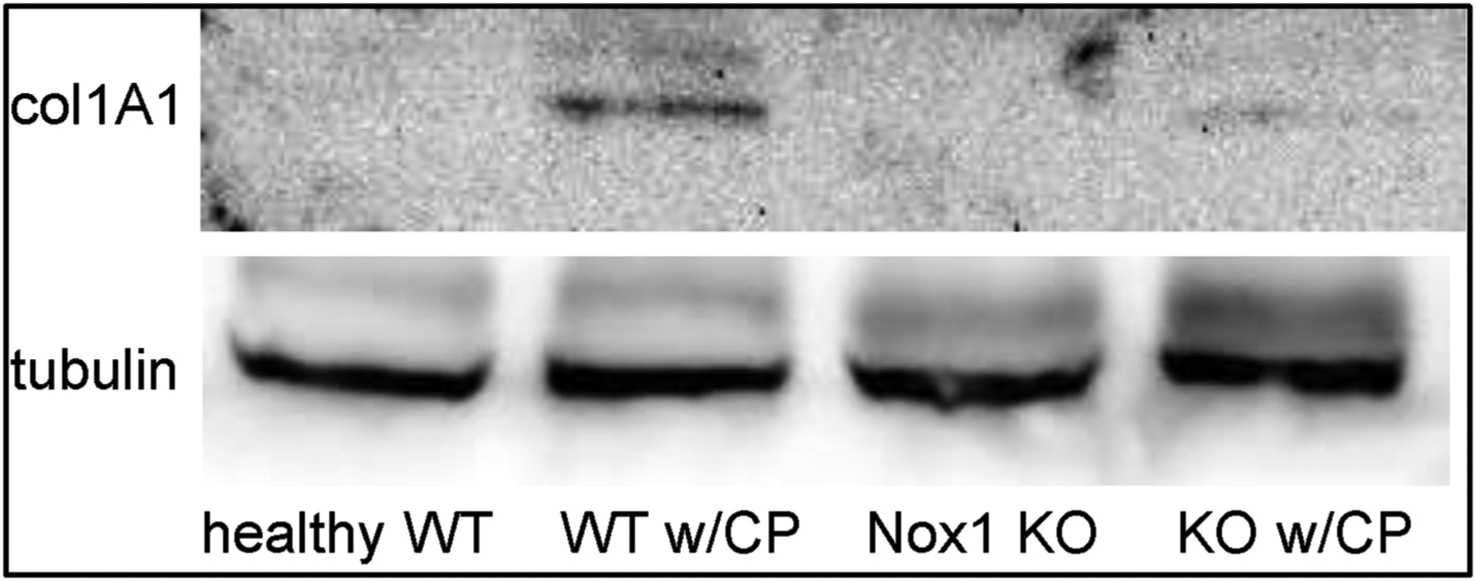
Whole pancreatic lysates were obtained from WT and Nox1 KO mice with or without CP. Representative immunoblots for collagen 1A1 (220 kDa) shows increased levels of collagen 1A1 in whole pancreatic lysates from WT mice with CP, but not from Nox1 KO mice with CP. A representative immunoblot for tubulin (52 kDa) indicated that the same amount of proteins was loaded in each well. (n: 4).
Nox1 evokes nuclear translocation of NF-ĸB in CP.
Dysregulation of NF-ĸB leading to chronic NF-ĸB activation is necessary for the pathogenesis in CP since it induces the expression of TGF-β, Cox-2 and IL-1B, which are drivers of chronic inflammation [40]. Using IHC, we found that caerulein-induced CP caused activation of NF-ĸB, and as a consequence, its nuclear translocation as previously shown [40]. The lack of Nox1 impaired the nuclear staining for p65 NF-ĸB in mouse model of CP (Fig. 3A). We confirmed these results by Western-blotting (Fig. 3B).
Fig. 3A. The lack of Nox1 impairs nuclear translocation of NF-ĸB in CP. Left:
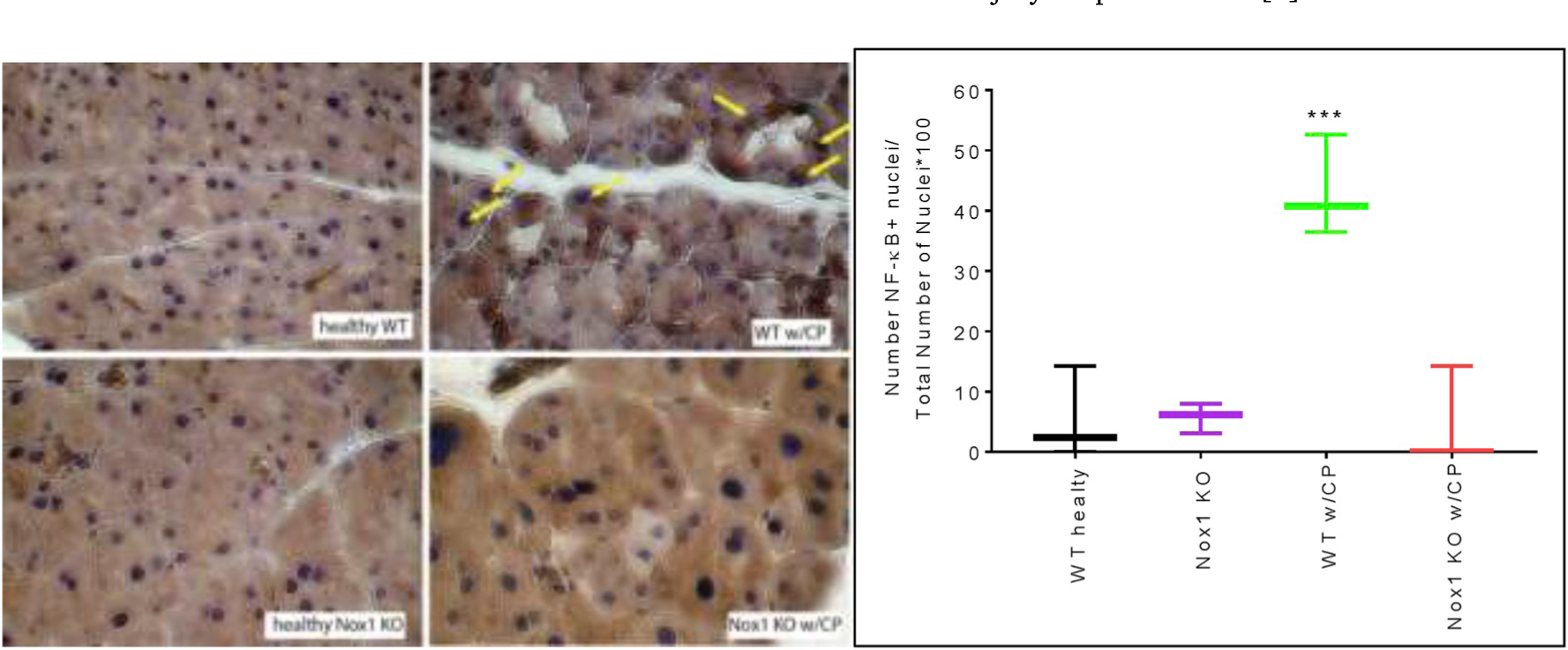
NF-ĸB was visualized using DAB as a chromogen (color: brown). Arrows: NF-ĸB positive nuclei. Total magnification: 400X. Right: Quantitative analysis of nuclear translocation of NF-ĸB is displayed as Box and whiskers plot. Values are means ± SE (n: 4 experiments). ***p < 0.001 vs. heathy WT mice.
Fig. 3B. The lack of Nox1 impairs CP-induced NF-ĸB activation in whole pancreatic tissues.
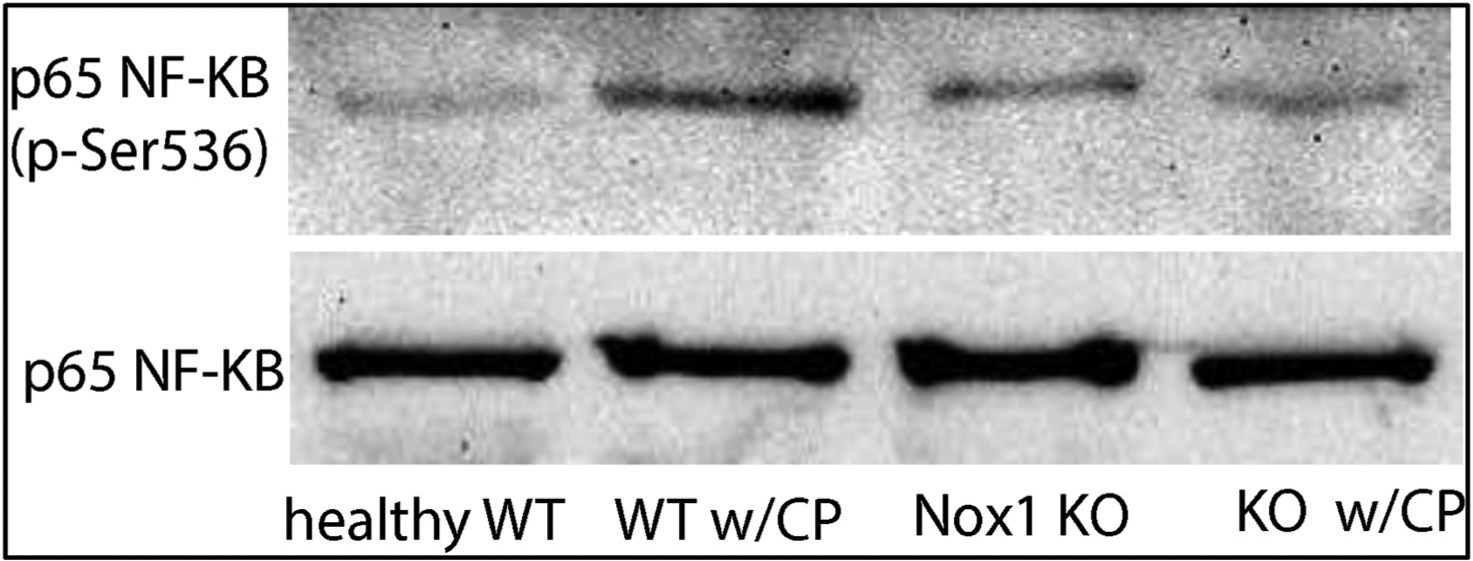
Whole pancreatic lysates were obtained from WT and Nox1 KO mice with or without CP. Representative immunoblots for phospho–NF–ĸB (65 kDa) shows an activation of p65 NF-ĸB in whole pancreatic lysates from WT mice with CP, but not from Nox1 KO mice with CP. A representative immunoblot for total p65 NF-ĸB (65 kDa) indicated that the same amount of proteins was loaded in each well. (n: 4).
Nox1 is involved in CP-induced TGF-β up-regulation.
The NF-ĸB-dependent fibrogenic factor TGF-β is up-regulated in caerulein-induced CP [41,42]. It was therefore of interest to examine the effect of Nox1 on the CP-induced up-regulation of TGF-β. Using quantitative real-time PCR, we found that the lack of Nox1 impaired CP-induced TGF-β up-regulation in the whole pancreas (Fig. 4A).
Fig. 4A. The absence of Nox1 abolishes caerulein-induced TGF-β up-regulation.
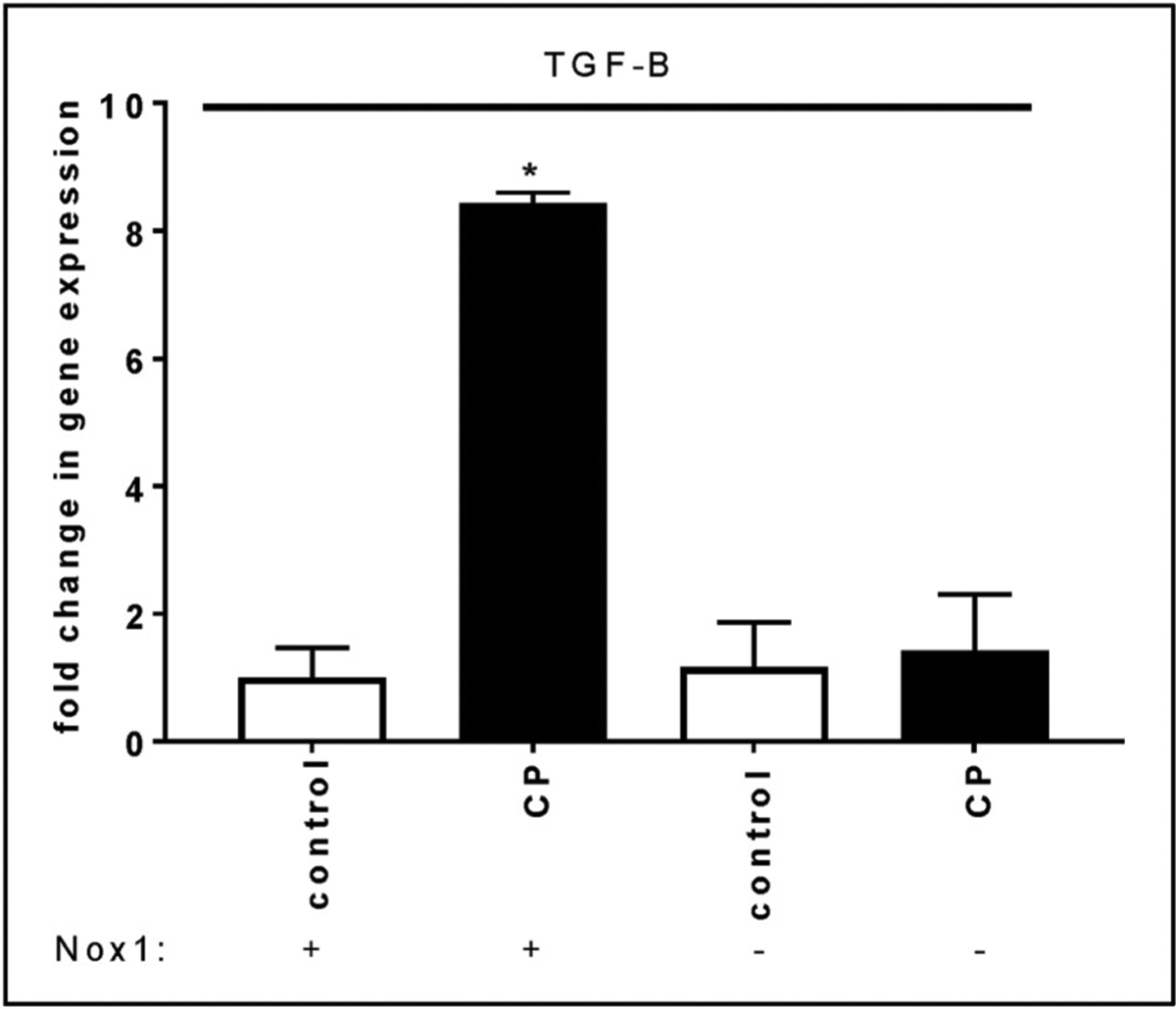
Total RNA was isolated from mouse pancreas using Trizol and RNeasy® Mini kit. Data are expressed as fold change in gene expression (mean ± SE). 18S rRNA was used as a reference. n: 5 mice. *:p < 0.05 vs control.
Nox1 induces PaSC expansion and acinar-to-ductal metaplasia (ADM) in CP.
In light of the importance of Nox1 in the fibrotic process of CP, we tested whether Nox1 mediates the expansion of PaSCs observed in CP [20] using Western-blotting. Using amylase as a marker of acinar cells, and the intermediate filament vimentin and α SMA as markers of PaSC expansion, we observed that repetitive injections of caerulein cause increased levels of vimentin and α SMA and decreased levels of amylase in WT mice with CP, supporting the idea of a possible contribution of Nox1 to the PaSC expansion in CP. In Nox1 KO mice, caerulein-induced increased levels of vimentin and α SMA were impaired (Fig. 4B, left panel).
Fig. 4B. Left panel: Nox1 mediates caerulein-induced ADM.
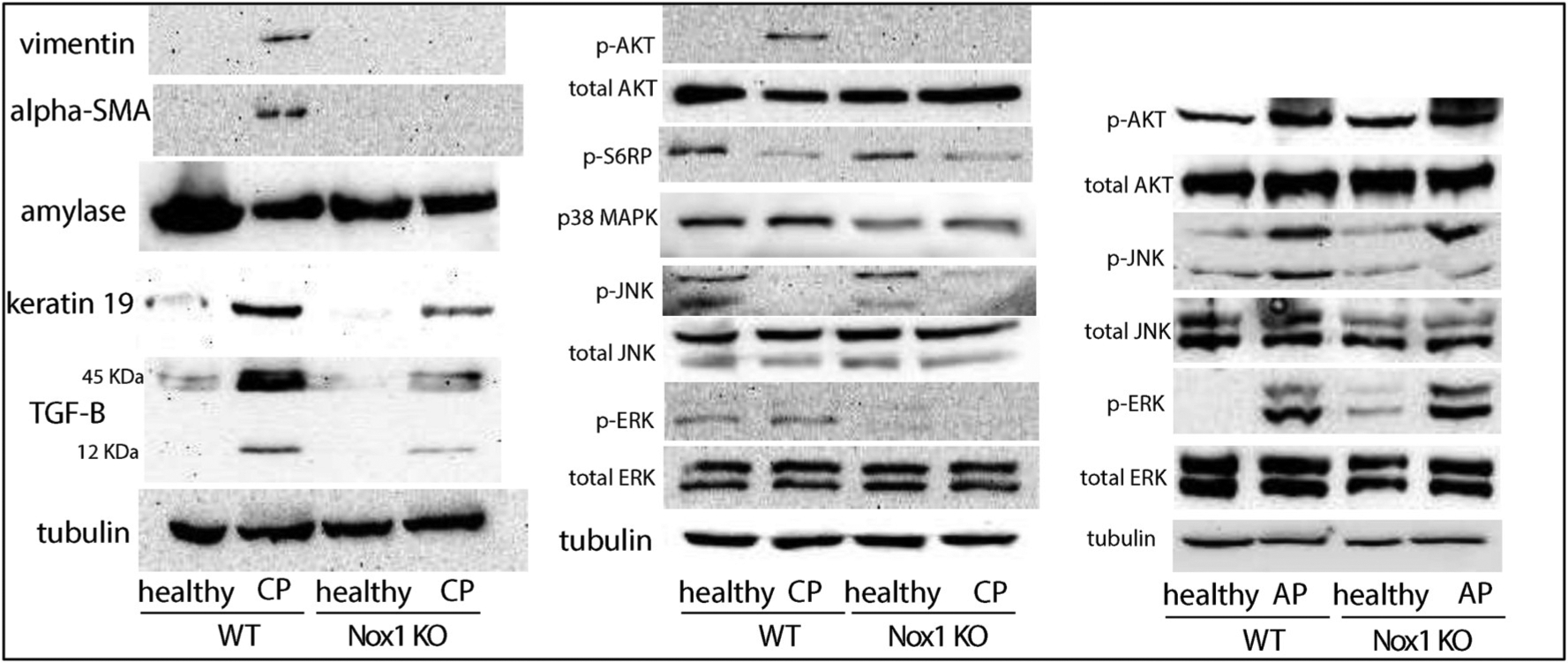
Representative immunoblots for vimentin (57 kDa), α-SMA (42 kDa), amylase (45 kDa), keratin 19(44.5 kDa), and TGF-β (12 kDa, 45 kDa) are shown. Equivalent loading was confirmed using α-tubulin (52 kDa). n: 3. Right panels: Nox1 induces phosphorylation of AKT in a mouse model of CP, but not in a mouse model of AP. Representative immunoblots for p-AKT (60 kDa), total AKT (60 kDa), phospho-S6RP (32 kDa), p38MAPK (40 kDa), p-JNK (46, 54 kDa), total JNK (46, 54 kDa), p-ERK (44, 42 kDa), and total ERK (44, 42 kDa) are shown. Equivalent loading was confirmed using α-tubulin (52 kDa). n: 3.
Caerulein-induced CP leads to formation of tubular complexes (TCs) during the ADM [43]. We also found that Nox1 participates in the formation of TC during the ADM in caerulein-induced CP. Using keratin-19 as a tubular marker [23], we found that repetitive injections of caerulein cause the formation of TC in WT mice and, to a lesser extent, Nox1 KO mice (Fig. 4B; left panel). Using Western blotting, we also confirmed that Nox1 mediates the caerulein-induced TGF-β up-regulation (Fig. 4B; left panel).
Nox1 induces phosphorylation of AKT in a mouse model of CP.
The next goal was to determine which intracellular pathway mediates the effect of Nox1-derived ROS in a mouse model of CP. A number of intracellular pathways have been activated following Nox1-derived ROS, including JNK, p38 MAP kinase, AKT, and ERK1/2 [44]. The same pathways are activated by CCK [45]. We found that caerulein-induced CP caused phosphorylation of AKT and decreased phosphorylation of JNK in WT mice. No changes were observed in the levels of phospho-ERK and p38 MAPK in WT mice with or without CP. In Nox1 KO mice, caerulein-induced AKT phosphorylation was impaired (Fig. 4B; right panel). The participation of AKT in Nox1 signaling was confirmed by IHC (Fig. 4C). Next, we test whether ribosomal protein S6 (S6RP), which stimulates protein synthesis in pancreatic acini [46], is a target of phosphorylated AKT. Unlike phosphorylated AKT, phosphorylated S6RP decreased in WT mice with CP (Fig. 4B; right panel). Thus, S6RP is not a downstream target of phosphorylated AKT. Moreover, the basal phosphorylated levels of S6RP was not affected by the lack of Nox1 (Fig. 4B right panel). Unlike Mitsushita et al. [47], we found that the lack of Nox1 impaired the phosphorylation of ERK1/2 (Fig. 4B; right panel). The lack of Nox1 also down-regulated p38MAPK (Fig. 4B; right panel). Re-probing the membrane with an antibody against total AKT, total JNK and total ERK demonstrated that the total expression of protein of interest was not affected by either the induction of CP or the lack of Nox1, and that only the phosphorylated form was affected.
Fig. 4C. Nox1 induces phosphorylation of AKT in a mouse model of CP.
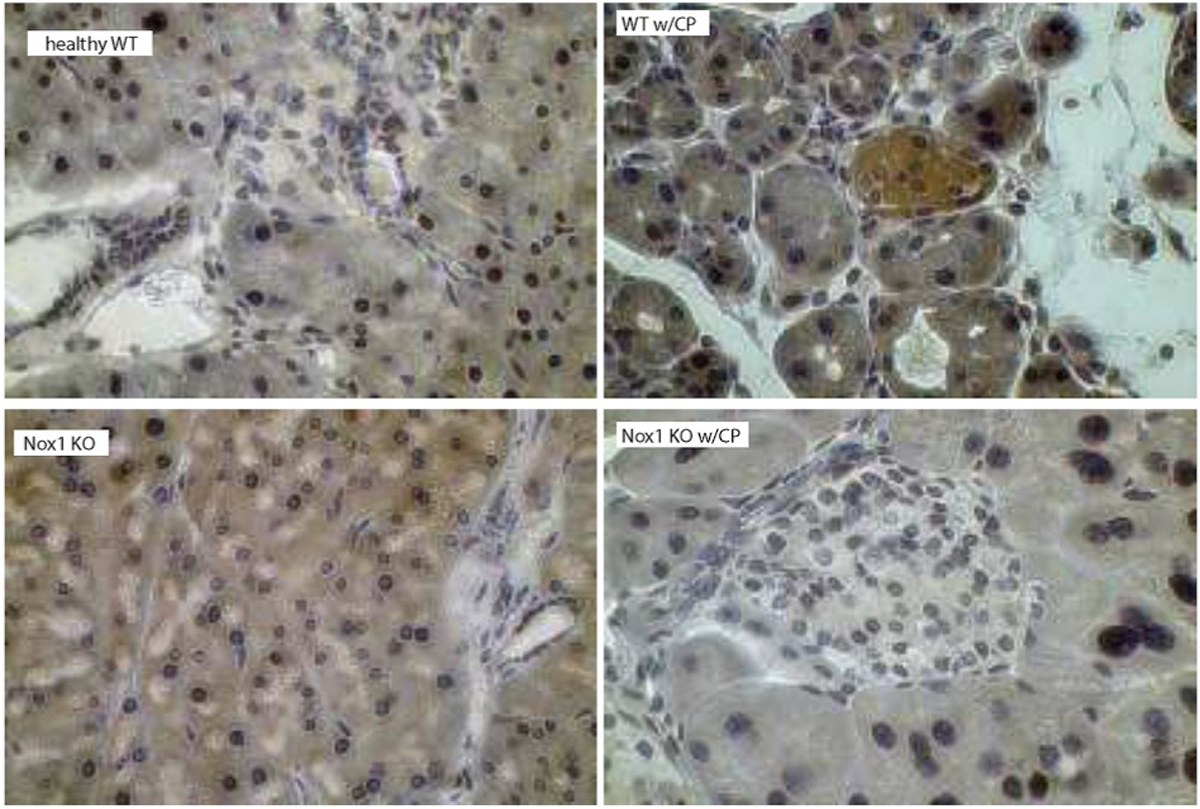
p-AKT was visualized using DAB as a chromogen (color: brown). The staining displayed a cytoplasmic localization of p-AKT in pancreatic acini and islets of Langerhans from WT mice with CP, but not from Nox1 KO mice with CP. Total magnification: 400X.
Caerulein-induced AP caused an increase in the phosphorylation of AKT, JNK and ERK (Fig. 4B; right panel). The lack of Nox1 did not affect AP-induced phosphorylation of any of these proteins (Fig. 4B; right panel; Suppl. Fig. 6).
Nox1, Nox3 and Nox4 are expressed in PaSCs.
In light of the importance of Nox1 in the fibrotic process of CP, we tested the expression of Nox1 in PaSCs, which are cells that initiate pancreatic fibrosis in CP [20] and macrophages, which are the predominant inflammatory cells in CP [48] and have promoted pancreatic fibrosis in CP through the activation of PaSCs [49]. Using RT-PCR, we found that Nox1 is expressed in primary culture of mouse activated PaSCs. Two other Nox enzymes, Nox3 and Nox4, were also expressed (Suppl. Fig. 7; left panel). Peritoneal macrophages only expressed Nox2 (Suppl. Fig. 7; right panel).
The lack of Nox1 in PaSCs impaired caerulein-induced ROS generation.
Because Nox1 is expressed in both pancreatic acini and PaSCs, and mediated the fibrogenic process in caerulein-induced CP, we next evaluated whether caerulein is able to generate ROS in both cell types, and whether this effect is impaired with the lack of Nox1. We observed that, although a physiologic concentration of caerulein (10 pM) did not modify the levels of ROS in PaSCs (data not shown), a supraphysiologic concentration of caerulein (100 nM) increased H2O2 and O −2 formation in primary cultures of activated PaSCs (Fig. 5; left and right panels). The growth factor TGF-β (10 ng/ml), which activates PaSCs and was involved in the effect of Nox1 on CP in vivo (Fig. 4A and B; left panel), increased the levels of H2O2 (Fig. 5; left panel). The lack of Nox1 abolished the effect of caerulein-induced H2O2 and O2− generation, but it did not modify exogenous TGF-β-induced H2O2 generation (Fig. 5; left panel). Unlike in PaSCs, in isolated pancreatic acini, caerulein (100 nM) or TGF-β (10 ng/ml) did not affect H2O2 generation (Suppl. Fig. 8; only caerulein is shown).
Fig. 5. Nox1 mediates caerulein-induced ROS generation in PaSCs. Left panel:
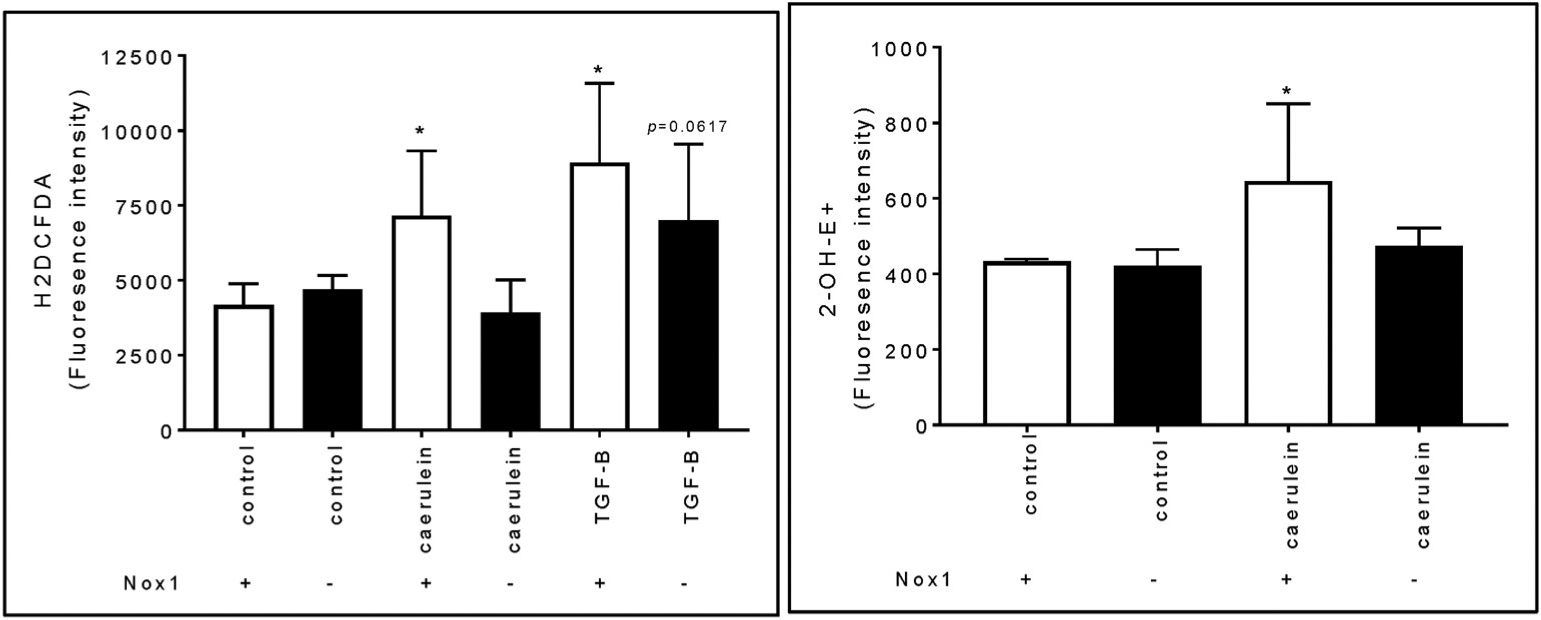
H2O2 levels were tested using fluorescence by H2DCFDA. n: 6 experiments. Right panel: Intracellular levels of O−2 were determined using fluorescence by DHE. n: 3 experiments. (mean ± SE). *: p < 0.05 vs. control PasCS from WT mice.
Nox1 causes the expression of metalloproteinase MMP-9 and epithelial-to-mesenchymal transition (EMT) hallmark gene Twist in activated PaSCs.
Because stellate Nox1, but not acinar Nox1, caused ROS generation in presence of caerulein, we further evaluated the importance of Nox1 in activated PaSCs. In response to pancreatic inflammation, activated PaSCs, through the transcription factor NF-ĸB, express enzymes that are associated with remodeling ECM, including MMP [50] and mediators of EMT [51]. Because Nox1 mediated CP-induced NF-ĸB activation, we studied the relative expression of a number of NF-ĸB-induced genes in PaSCs from WT mice with or without CP compared with PaSCs from Nox1 KO mice with or without CP using quantitative real-time PCR analysis. We previously found that Nox1 is involved in CP-induced TGF-β up-regulation using mRNA from whole pancreas. In these sets of experiments, we found that TGF-β is also up-regulated in PaSCs from WT mice with CP. The lack of Nox1 impaired this effect (Table 2). A family of zinc finger transcription factors (ZF-TFs), including Snail1, Zeb1, Zeb2, and Slug, and the basic helix-loop-helix (bHLH) transcription factor Twist are regulated by NF-ĸB and implicated in repression of genes that promote an epithelial phenotype, thereby inducing a mesenchymal phenotype [51]. The transcription factor Zeb1 was up-regulated in PaSCs from both WT and Nox1 KO mice with CP (Table 2). Transcription factor Twist’s mRNA levels were elevated in PaSCs from WT mice with CP. The lack of Nox1 abolished the CP-induced up-regulation of Twist in PaSCs (Table 2). No significant changes in the expression of Slug, Snail and Zeb2 were observed in different groups. We also studied the expression of two MMPs secreting by PaSCs: gelatinases MMP-2 and MMP-9 [50]. MMP-9 mRNA levels were elevated in PaSCs from WT mice with CP (Table 2), while no change in the expression of MMP-2 was observed (data not shown). The lack of Nox1 reduced the CP-induced up-regulation of MMP-9 in PaSCs.
Table 2. Fold increase in the expression of genes in PaSCs from WT and Nox1 KO mice with or without CP.
Data are expressed as fold change in gene expression (mean ± SE). 18S rRNA was used as a reference. n: healthy WT: 6; Nox1 KO: 5; WT w/CP: 5; Nox1 KO w/CP: 4.
| WT | KO | WT w/CP | KO w/CP | |
|---|---|---|---|---|
| TGF-B | 1.41 ± 0.82 | 1.57 ± 0.70 | 9.91 ± 4.99*** | 0.87 ± 0.55 |
| Slug | 1.68 ± 0.80 | 0.85 ± 0.54 | 2.40 ± 1.60 | 1.63 ± 0.32 |
| Zebl | 1.49 ± 0.56 | 0.26 ± 0.15 | 6.57 ± 1.32* | 8.43 ± 1.61* |
| Zeb2 | 1.76 ± 0.75 | 1.53 ± 0.92 | 1.60 ± 0.51 | 1.90 ± 0.07 |
| Twist | 1.16 ± 0.32 | 0.31 ± 0.15* | 87.81 ± 33.66*** | 0.93 ± 0.56††† |
| MMP-9 | 2.46 ± 1.29 | 1.47 ± 1.32 | 4244 ± 2209** | 24 ± 17†† |
| Snail | 1.18 ± 0.28 | 1.93 ± 0.98 | 2.48 ± 0.77 | 0.84 ± 0.31 |
p < 0.05,
p < 0.01,
p < 0.001 vs PaSCs from WT mice;
p < 0.01
p < 0.001 vs PaSCs from WT mice with CP.
4. Discussion
The objective of this study was to identify the Nox enzymes that are involved in pathogenesis of both states of pancreatitis (i.e., AP and CP) using Nox-deficient mice. In order to do this, using RT-PCR, we first studied the expression of Nox enzymes in isolated pancreatic acini because intrapancreatic activation of digestive enzyme is a primary event in the cell injury of pancreatitis [3]. Because we found that isolated pancreatic acini only express Nox1, we further studied the participation Nox1 in the pathophysiology of both states of pancreatitis using Nox1-defficient mice.
To study the participation of Nox1 in the inflammatory process of AP, we carried out both in vitro and in vivo studies. In in vitro studies, using isolated pancreatic acini, the lack of Nox1 did not affect either caerulein-induced amylase secretion or trypsin activation. In in vivo studies, using a mouse model of caerulein-induced AP, the lack of Nox1 did not modify the histo-morphologic and biochemical features of AP (e.g., intracellular vacuoles, inflammatory infiltration, and serum levels of digestive enzymes). To study the participation of Nox1 in the pathogenesis of CP, we induced CP in mice using repetitive injections of caerulein. We found that Nox1-deficient mice were less susceptible to caerulein-induced CP because the lack of Nox1 impaired the histo-morphologic features of CP (e.g., atrophy, moderate acinar loss and fibrosis). ADM is another histological feature of CP [43]. Using Western blotting, we observed that Nox1 mediates CP-induced ADM because the levels of keratin 19 (a ductal marker) increased in pancreas from WT mice with CP, but not in pancreas from Nox1 KO in which CP was induced. Therefore, Nox1 is implicated in the fibrotic process of CP, while Nox1 did not mediate the effects of acute treatment with caerulein.
We also found that Nox1 possibly contributes to the PaSC expansion in CP. Using IHC and Western blotting for αSMA, we discovered that in the whole pancreas from WT mice with CP, but not from Nox1 KO with CP, PaSCs proliferated and released α-SMA. Using another approach, we also found that the protein level of vimentin, a marker of activated PaSCs, increased, whereas the protein level of amylase, a marker of acinar cells, decreased in whole pancreatic lysates from WT mice with CP. These effects were not observed in whole pancreatic lysates from Nox1 KO mice (i.e., the levels of amylase in pancreatic lysate was similar between healthy and CP-induced Nox1 KO mice, while vimentin was not expressed in any of these two groups of Nox1 KO mice).
In light of the importance of Nox1 in the fibrotic process of CP, we tested the expression of Nox1 in two other cell types: PaSCs, which are cells that initiate pancreatic fibrosis in CP [20], and macrophages, which are the predominant inflammatory cells in CP [48] and have promoted pancreatic fibrosis in CP through the activation of PaSCs [49]. We found that activated PaSCs express Nox1. In addition to Nox1, PaSCs also expressed Nox3 and Nox4. Unlike rat PaSCs [37], Nox2 was not expressed in mouse PaSCs. Peritoneal macrophages only expressed Nox2.
Next, we studied the source of Nox1-derived ROS in response to caerulein. Using a fluorescent probe, H2DCFDA, we found that the effect of a supraphysiologic concentration of caerulein (100 nM) on ROS generation was impaired in PaSCs isolated from Nox1 KO mice. In pancreatic acini, conflicting results exist regarding whether or not either CCK or caerulein increases ROS generation. For instance, a study using lucigenin and DCFDA indicated that caerulein stimulates the activation of Nox and, subsequently, ROS production within rat pancreatoma cell line AR42J [52]. Other studies using CM-H2DCF-DA showed that CCK (10 nM) evokes ROS formation in isolated pancreatic acini [53,54]. In contrast, two studies using luminol and DCFDA showed no increase in ROS production following CCK (100 nM) [18] and caerulein (0.1 and 100 nM) [55] stimulation in pancreatic acini. Using H2DCFDA, we also found that caerulein (100 nM) did not induce ROS generation in isolated pancreatic acini. This observation can be due to much lower expression of Nox1 in pancreatic acini compared to PaSCs. Therefore, we concluded that caerulein can directly activate PaSCs (“quiescent” state to “myofibroblastic” state) and induce Nox-1 derived ROS generation. A persistent activation and proliferation of PaSCs leads to the production of collagen and other extracellular matrix proteins (e.g., SMA), which contribute to pancreatic fibrosis in CP. Because neutrophilic Nox2 mediates the inflammatory process in AP [18] and macrophages promote pancreatic fibrosis in CP through the activation of PaSCs [49], our findings cannot rule out the contribution of macrophagic Nox2-derived ROS in the fibrogenic process of CP.
Once ROS are generated, a number of downstream redox-sensitive signaling pathways (e,g., MAP kinase activation) and transcription factors (e.g., NF-ĸB) [5] become activated and can regulate cell growth, proliferation, differentiation, and apoptosis. We found that caerulein-induced CP causes phosphorylation of AKT and that Nox1-evoked ROS promotes CP-induced phosphorylation of AKT. The participation of the AKT pathway in the action of Nox1 has been previously reported since Nox1 via AKT pathway has promoted proliferation of fibrotic hepatic stellate cells (hyperplasia) [35]. One of the target of phosphorylated AKT is S6RP. In rat pancreatic acini, AKT-induced phosphorylation of S6RP is required for CCK-induced stimulation of protein synthesis [46]. However, phosphorylation of AKT in CP was not associated with an increase in protein synthesis because S6RP was not a downstream target of phosphorylated AKT. We also found that the mouse model of caerulein-induced CP promotes cell survival and prevents apoptosis because it caused dephosphorylation of pro-apoptotic JNK [56]. No changes in the levels of pro-apoptotic phospho-JNK was observed when Nox1 was not present. Caerulein-induced CP did not affect the levels of phosphorylated ERK and total p38 MAPK. However, the lack of Nox1 reduced the basal levels of phosphorylated form of pro-mitotic ERK and total levels of p38MAPK.
An increase in ROS generation results in the expression of transcription factor NF-ĸB in acinar cells [57], which is a key mediator for chronic inflammation because it stimulates the transcription of cytokines (e.g., TGF-β) and other pro-inflammatory and fibrogenic genes [40,41]. Using IHC, we found that Nox1 activity mediates the translocation of NF-ĸB to the nucleus because, in pancreas from Nox1 KO mice, the staining of NF-ĸB in the nucleus was impaired. Because Nox1 has binding sites for NF-κB [58], the binding of NF-ĸB to the promoter region of Nox1 genes can induce transcription of Nox1. Then, NF-ĸB can create a positive feedback loop in which Nox1 can generate more ROS that, in turn, induces the translocation of NF-κB to the nucleus and causes an increase in the expression of cytokines to initiate the next cycle and exacerbate the fibrosis.
TGF-β is a chronic pro-inflammatory cytokine that potently activates PaSCs and promotes fibrosis by stimulating the production of extracellular matrix (ECM) proteins (e.g., αSMA, collagen I and III) from these cells [42]. We found that TGF-β mediates the effect of Nox1 in pancreas because TGF-β at both mRNA and protein levels was elevated in whole pancreas from WT mice with CP and that effect was abolished in whole pancreas from Nox1 KO mice in which CP was induced. Because TGF-β was up-regulated in caerulein-induced CP and the lack of Nox1 did not modify exogenous TGF-β-induced ROS generation, Nox1 is likely to be located upstream of TGF-β in PaSCs. When TGF-β is administered exogenously, TGF-β is able to cause ROS formation without the need for Nox1 activity. Therefore, stellate Nox1-evoked ROS caused the activation of transcription factor NF-ĸB, an up-regulation of the fibrogenic factor TGF-β and promoted local proliferation of PaSCs, leading to fibrosis via phosphorylation of AKT.
Fibrosis involves a balance between ECM production and degradation. PaSCs can regulate ECM composition through the production of two MMPs, MMP-2 and MMP-9 [50]. We found that Nox1 is required for the expression of the gelatinase MMP-9, but not MMP-2, in PaSCs. Interestingly, MMP-9, but not MMP-2, has a binding site for NF-ĸB [59]. Therefore, in an inflamed pancreas, it is likely that Nox1-derived ROS, via NF-ĸB, increases the release of MMP-9 from PaSCs, which could result in increased collagen IV degradation, and in turn, could facilitate the deposition of collagen I and III, leading to fibrosis.
Other genes induced by NF-ĸB are genes implicated in EMT [51]. We identified the E-cadherin repressor gene Twist as the most important transcription factor induced by Nox1-derived ROS in PaSCs because Twist was down-regulated in PaSCs from Nox1 KO mice with or without CP. Because PaSCs express mesenchymal markers [20], therefore, Nox1 is necessary to maintain the expression E-cadherin repressor genes in activated PaSCs, which allow them to have more migratory and stem cell characteristics, as well as gain the ability to prevent apoptosis [60]. The transcription factor Zeb1 was up-regulated in PaSCs from mice with CP, but this effect did not require Nox1 activation.
5. Conclusion
Repetitive acute pancreatic injury induced by caerulein leads to a persistent activation and proliferation of PaSCs. Activation of PaSCs by caerulein generates Nox1-derived ROS. Increased stellate Nox1-derived ROS activate downstream redox-sensitive signaling pathways (e.g, AKT and NF-ĸB), increasing the expression of fibrogenic genes (e.g, TGF-β), the metalloproteinase MMP-9 and the epithelial repressor gene Twist. The outcome is mesenchymal PaSCs, which release α-SMA, collagen I and III, and other ECM proteins and replace the necrotic pancreatic parenchyma with fibrotic tissue (Fig. 6).
Fig. 6.
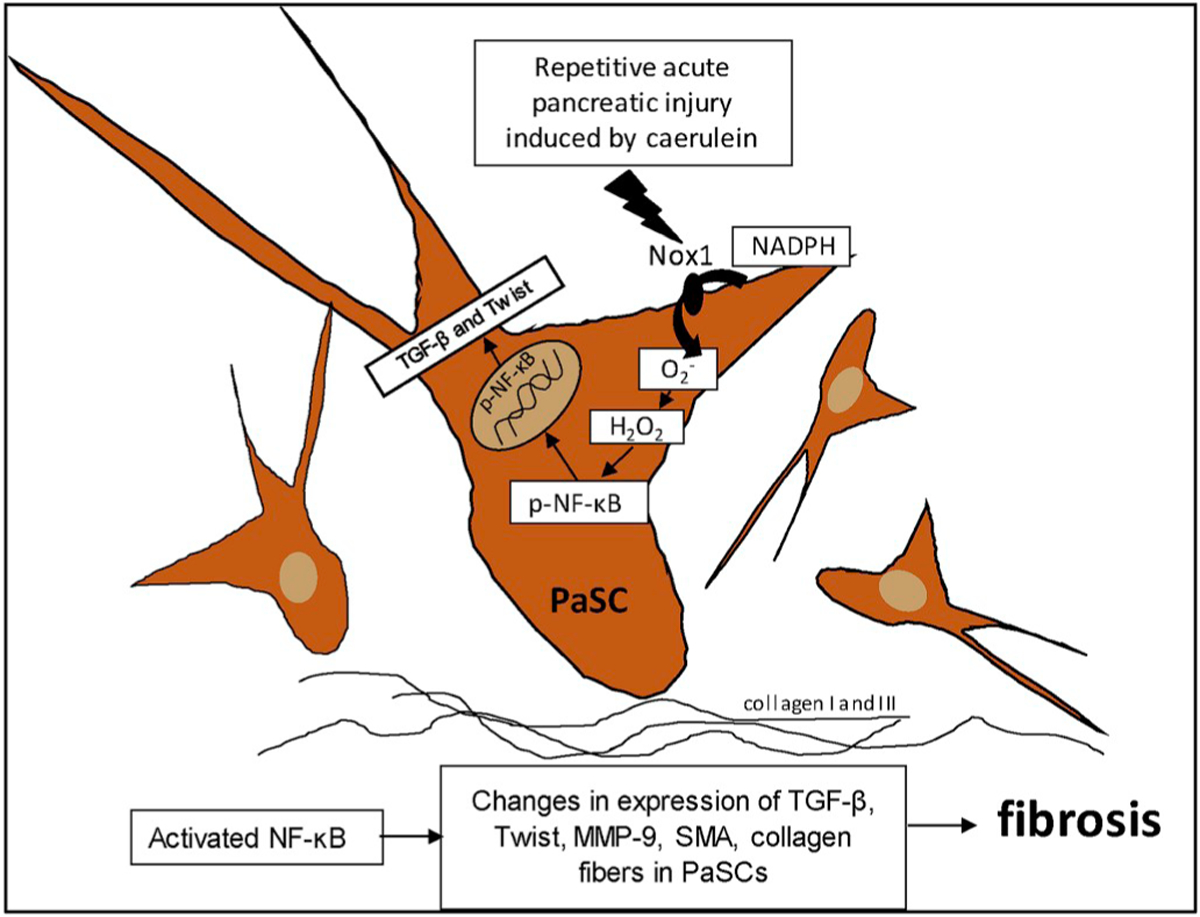
A schematic model proposed for the role of Nox1 in the fibrotic process of CP.
Until now, there are no specific therapies to inhibit pancreatic fibrosis. These findings in both in vivo and in vitro studies propose Nox1 inhibitors as candidates for a novel treatment to ameliorate the fibrogenic process of CP.
Supplementary Material
Funding
This work was supported in part by the Augusta University Provost’s office, and the Translational Research Program of the Department of Medicine, Medical College of Georgia at Augusta University awarded to M.E.S., National Institutes of Health grants (4R00HL114648-03 and 1R01HL139562-01A1) awarded to G.C and American Heart Association Postdoctoral Fellowship (17POST33661254) given to B.S.
Footnotes
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.freeradbiomed.2019.11.034.
References
- [1].Peery AF, et al. , Burden and Cost of Gastrointestinal, Liver, and Pancreatic Diseases in the United States: Update 2018, (2018) Gastroenterology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Habtezion A, Gukovskaya AS, Pandol SJ, Acute pancreatitis: a multifaceted set of organelle and cellular interactions, Gastroenterology 156 (7) (2019) 1941–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Saluja A, et al. , Early intra-acinar events in pathogenesis of pancreatitis, Gastroenterology 156 (7) (2019) 1979–1993. [DOI] [PubMed] [Google Scholar]
- [4].Neuschwander-Tetri BA, et al. , Repetitive self-limited acute pancreatitis induces pancreatic fibrogenesis in the mouse, Dig. Dis. Sci 45 (4) (2000) 665–674. [DOI] [PubMed] [Google Scholar]
- [5].Bedard K, Krause KH, The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology, Physiol. Rev 87 (1) (2007) 245–313. [DOI] [PubMed] [Google Scholar]
- [6].Sanfey H, Bulkley GB, Cameron JL, The pathogenesis of acute pancreatitis. The source and role of oxygen-derived free radicals in three different experimental models, Ann. Surg 201 (5) (1985) 633–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Wisner JR, Renner IG, Allopurinol attenuates caerulein induced acute pancreatitis in the rat, Gut 29 (7) (1988) 926–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Devenyi ZJ, Orchard JL, Powers RE, Xanthine oxidase activity in mouse pancreas: effects of caerulein-induced acute pancreatitis, Biochem. Biophys. Res. Commun 149 (3) (1987) 841–845. [DOI] [PubMed] [Google Scholar]
- [9].Niederau C, et al. , Oxidative injury to isolated rat pancreatic acinar cells vs. isolated zymogen granules, Free Radic. Biol. Med 20 (7) (1996) 877–886. [DOI] [PubMed] [Google Scholar]
- [10].Escobar J, et al. , Oxidative and nitrosative stress in acute pancreatitis. Modulation by pentoxifylline and oxypurinol, Biochem. Pharmacol 83 (1) (2012) 122–130. [DOI] [PubMed] [Google Scholar]
- [11].Sugiyama Y, et al. , Pathogenic role of endothelial nitric oxide synthase (eNOS/NOS-III) in cerulein-induced rat acute pancreatitis, Dig. Dis. Sci 51 (8) (2006) 1396–1403. [DOI] [PubMed] [Google Scholar]
- [12].Werner J, et al. , On the protective mechanisms of nitric oxide in acute pancreatitis, Gut 43 (3) (1998) 401–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gonzalez A, et al. , Changes in mitochondrial activity evoked by cholecystokinin in isolated mouse pancreatic acinar cells, Cell. Signal 15 (11) (2003) 1039–1048. [DOI] [PubMed] [Google Scholar]
- [14].Yu JH, et al. , NADPH oxidase and apoptosis in cerulein-stimulated pancreatic acinar AR42J cells, Free Radic. Biol. Med 39 (5) (2005) 590–602. [DOI] [PubMed] [Google Scholar]
- [15].Yu JH, Kim KH, Kim H, Role of NADPH oxidase and calcium in cerulein-induced apoptosis: involvement of apoptosis-inducing factor, Ann. N. Y. Acad. Sci 1090 (2006) 292–297. [DOI] [PubMed] [Google Scholar]
- [16].Yu JH, et al. , NADPH oxidase mediates interleukin-6 expression in cerulein-stimulated pancreatic acinar cells, Int. J. Biochem. Cell Biol 37 (7) (2005) 1458–1469. [DOI] [PubMed] [Google Scholar]
- [17].Ju KD, et al. , Potential role of NADPH oxidase-mediated activation of Jak2/Stat3 and mitogen-activated protein kinases and expression of TGF-beta1 in the pathophysiology of acute pancreatitis, Inflamm. Res 60 (8) (2011) 791–800. [DOI] [PubMed] [Google Scholar]
- [18].Gukovskaya AS, et al. , Neutrophils and NADPH oxidase mediate intrapancreatic trypsin activation in murine experimental acute pancreatitis, Gastroenterology 122(4) (2002) 974–984. [DOI] [PubMed] [Google Scholar]
- [19].Cao WL, et al. , Potential role of NADPH oxidase in pathogenesis of pancreatitis, World J. Gastrointest. Pathophysiol 5 (3) (2014) 169–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Omary MB, et al. , The pancreatic stellate cell: a star on the rise in pancreatic diseases, J. Clin. Investig 117 (1) (2007) 50–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Gavazzi G, et al. , Decreased blood pressure in NOX1-deficient mice, FEBS Lett. 580(2) (2006) 497–504. [DOI] [PubMed] [Google Scholar]
- [22].Adler G, Rohr G, Kern HF, Alteration of membrane fusion as a cause of acute pancreatitis in the rat, Dig. Dis. Sci 27 (11) (1982) 993–1002. [DOI] [PubMed] [Google Scholar]
- [23].Sans MD, et al. , Secretin is not necessary for exocrine pancreatic development and growth in mice, Am. J. Physiol. Gastrointest. Liver Physiol 301 (5) (2011) G791–G798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Burlina A, Galzigna L, Turbidimetric procedure for determination of lipase activity, Clin. Chem 19 (4) (1973) 384–386. [PubMed] [Google Scholar]
- [25].Sabbatini ME, Williams JA, Cholecystokinin-mediated RhoGDI phosphorylation via PKCalpha promotes both RhoA and Rac1 signaling, PLoS One 8 (6) (2013) e66029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Apte MV, et al. , Periacinar stellate shaped cells in rat pancreas: identification, isolation, and culture, Gut 43 (1) (1998) 128–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zhang X, Goncalves R, Mosser DM, The isolation and characterization of murine macrophages, Curr. Protoc. Im November (2008) 1–18 (Chapter: Unit 14–1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Singla B, et al. , PKCdelta-mediated Nox2 activation promotes fluid-phase pinocytosis of antigens by immature dendritic cells, Front. Immunol 9 (2018) 537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Livak KJ, Schmittgen TD, Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method, Methods 25 (4) (2001) 402–408. [DOI] [PubMed] [Google Scholar]
- [30].Sabbatini ME, et al. , Adenylyl cyclase 6 mediates the action of cyclic AMP-dependent secretagogues in mouse pancreatic exocrine cells via protein kinase A pathway activation, J. Physiol 591 (15) (2013) 3693–3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Fujimori N, et al. , Vasoactive intestinal peptide reduces oxidative stress in pancreatic acinar cells through the inhibition of NADPH oxidase, Peptides 32 (10) (2011) 2067–2076. [DOI] [PubMed] [Google Scholar]
- [32].Booth DM, et al. , Reactive oxygen species induced by bile acid induce apoptosis and protect against necrosis in pancreatic acinar cells, Gastroenterology 140 (7) (2011) 2116–2125. [DOI] [PubMed] [Google Scholar]
- [33].Saluja AK, et al. , Cerulein-induced in vitro activation of trypsinogen in rat pancreatic acini is mediated by cathepsin B, Gastroenterology 113 (1) (1997) 304–310. [DOI] [PubMed] [Google Scholar]
- [34].Paik YH, et al. , The nicotinamide adenine dinucleotide phosphate oxidase (NOX) homologues NOX1 and NOX2/gp91(phox) mediate hepatic fibrosis in mice, Hepatology 53 (5) (2011) 1730–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Cui W, et al. , NOX1/nicotinamide adenine dinucleotide phosphate, reduced form (NADPH) oxidase promotes proliferation of stellate cells and aggravates liver fibrosis induced by bile duct ligation, Hepatology 54 (3) (2011) 949–958. [DOI] [PubMed] [Google Scholar]
- [36].Bataller R, et al. , NADPH oxidase signal transduces angiotensin II in hepatic stellate cells and is critical in hepatic fibrosis, J. Clin. Investig 112 (9) (2003) 1383–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Masamune A, et al. , NADPH oxidase plays a crucial role in the activation of pancreatic stellate cells, Am. J. Physiol. Gastrointest. Liver Physiol 294 (1) (2008) G99–G108. [DOI] [PubMed] [Google Scholar]
- [38].Kleeff J, et al. , Chronic pancreatitis, Nat. Rev. Dis. Primers 3 (2017) 17060. [DOI] [PubMed] [Google Scholar]
- [39].Bachem MG, et al. , Identification, culture, and characterization of pancreatic stellate cells in rats and humans, Gastroenterology 115 (2) (1998) 421–432. [DOI] [PubMed] [Google Scholar]
- [40].Sah RP, et al. , Cerulein-induced chronic pancreatitis does not require intra-acinar activation of trypsinogen in mice, Gastroenterology 144 (5) (2013) 1076–1085 e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Huang H, et al. , Activation of nuclear factor-kappaB in acinar cells increases the severity of pancreatitis in mice, Gastroenterology 144 (1) (2013) 202–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Menke A, Adler G, TGFbeta-induced fibrogenesis of the pancreas, Int. J. Gastrointest. Cancer 31 (1–3) (2002) 41–46. [DOI] [PubMed] [Google Scholar]
- [43].Strobel O, et al. , In vivo lineage tracing defines the role of acinar-to-ductal transdifferentiation in inflammatory ductal metaplasia, Gastroenterology 133 (6) (2007) 1999–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Brown DI, Griendling KK, Nox proteins in signal transduction, Free Radic. Biol. Med 47 (9) (2009) 1239–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Williams JA, Cholecystokinin (CCK) regulation of pancreatic acinar cells: physiological actions and signal transduction mechanisms, Comp. Physiol 9 (2) (2019) 535–564. [DOI] [PubMed] [Google Scholar]
- [46].Bragado MJ, Groblewski GE, Williams JA, Regulation of protein synthesis by cholecystokinin in rat pancreatic acini involves PHAS-I and the p70 S6 kinase pathway, Gastroenterology 115 (3) (1998) 733–742. [DOI] [PubMed] [Google Scholar]
- [47].Mitsushita J, Lambeth JD, Kamata T, The superoxide-generating oxidase Nox1 is functionally required for Ras oncogene transformation, Cancer Res. 64 (10) (2004) 3580–3585. [DOI] [PubMed] [Google Scholar]
- [48].Sumimoto H, Miyano K, Takeya R, Molecular composition and regulation of the Nox family NAD(P)H oxidases, Biochem. Biophys. Res. Commun 338 (1) (2005) 677–686. [DOI] [PubMed] [Google Scholar]
- [49].Xue J, et al. , Alternatively activated macrophages promote pancreatic fibrosis in chronic pancreatitis, Nat. Commun 6 (2015) 7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Phillips PA, et al. , Rat pancreatic stellate cells secrete matrix metalloproteinases: implications for extracellular matrix turnover, Gut 52 (2) (2003) 275–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Min C, et al. , NF-kappaB and epithelial to mesenchymal transition of cancer, J. Cell. Biochem 104 (3) (2008) 733–744. [DOI] [PubMed] [Google Scholar]
- [52].Yu JH, et al. , Diphenyleneiodonium suppresses apoptosis in cerulein-stimulated pancreatic acinar cells, Int. J. Biochem. Cell Biol 39 (11) (2007) 2063–2075. [DOI] [PubMed] [Google Scholar]
- [53].Granados MP, et al. , Generation of ROS in response to CCK-8 stimulation in mouse pancreatic acinar cells, Mitochondrion 3 (5) (2004) 285–296. [DOI] [PubMed] [Google Scholar]
- [54].Binker MG, et al. , Inhibition of Rac1 decreases the severity of pancreatitis and pancreatitis-associated lung injury in mice, Exp. Physiol 93 (10) (2008) 1091–1103. [DOI] [PubMed] [Google Scholar]
- [55].Muller S, et al. , Age-dependent effects of UCP2 deficiency on experimental acute pancreatitis in mice, PLoS One 9 (4) (2014) e94494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Szegezdi E, et al. , Mediators of endoplasmic reticulum stress-induced apoptosis, EMBO Rep. 7 (9) (2006) 880–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Perez S, et al. , Redox signaling in acute pancreatitis, Redox Biol. 5 (2015) 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Kuwano Y, et al. , Interferon-gamma activates transcription of NADPH oxidase 1 gene and upregulates production of superoxide anion by human large intestinal epithelial cells, Am. J. Physiol. Cell Physiol 290 (2) (2006) C433–C443. [DOI] [PubMed] [Google Scholar]
- [59].Spinale FG, Myocardial matrix remodeling and the matrix metalloproteinases: influence on cardiac form and function, Physiol. Rev 87 (4) (2007) 1285–1342. [DOI] [PubMed] [Google Scholar]
- [60].Thiery JP, et al. , Epithelial-mesenchymal transitions in development and disease, Cell 139 (5) (2009) 871–890. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.