

Abstract
Viral pandemics, such as the one caused by SARS-CoV-2, pose an imminent threat to humanity. Because of its recent emergence, there is a paucity of information regarding viral behavior and host response following SARS-CoV-2 infection. Here we offer an in-depth analysis of the transcriptional response to SARS-CoV-2 compared with other respiratory viruses. Cell and animal models of SARS-CoV-2 infection, in addition to transcriptional and serum profiling of COVID-19 patients, consistently revealed a unique and inappropriate inflammatory response. This response is defined by low levels of type I and III interferons juxtaposed to elevated chemokines and high expression of IL-6. We propose that reduced innate antiviral defenses coupled with exuberant inflammatory cytokine production are the defining and driving features of COVID-19.
Keywords: SARS-CoV-2, COVID-19, interferon, Coronavirus, transcriptomics, virus-host interactions, chemokines, IL6, ferret
Graphical Abstract
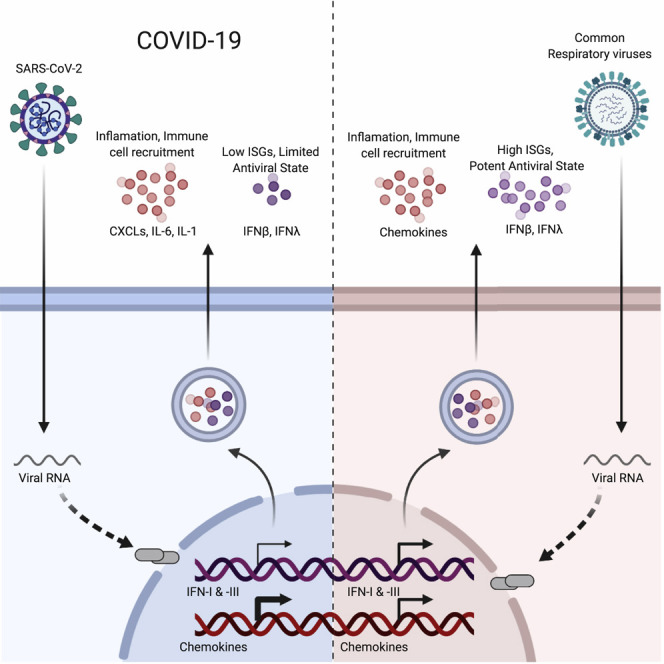
Highlights
-
•
SARS-CoV-2 infection induces low IFN-I and -III levels with a moderate ISG response
-
•
Strong chemokine expression is consistent across in vitro, ex vivo, and in vivo models
-
•
Low innate antiviral defenses and high pro-inflammatory cues contribute to COVID-19
In comparison to other respiratory viruses, SARS-CoV-2 infection drives a lower antiviral transcriptional response that is marked by low IFN-I and IFN-III levels and elevated chemokine expression, which could explain the pro-inflammatory disease state associated with COVID-19.
Introduction
Coronaviruses are a diverse group of single-stranded positive-sense RNA viruses with a wide range of vertebrate hosts (Cui et al., 2019). Four common coronavirus genera (alpha, beta, gamma, and delta) circulate among vertebrates and cause mild upper respiratory tract illnesses in humans and gastroenteritis in animals (Weiss and Navas-Martin, 2005). However, in the past two decades, three highly pathogenic human betacoronaviruses have emerged from zoonotic events (Amanat and Krammer, 2020). In 2002–2003, severe acute respiratory syndrome-related coronavirus 1 (SARS-CoV-1) infected ∼8,000 people worldwide with a case fatality rate of ∼10%, followed by Middle East respiratory syndrome-related coronavirus (MERS-CoV), which has infected ∼2,500 people with a case fatality rate of ∼36% since 2012 (de Wit et al., 2016). At present, the world is suffering from a pandemic of SARS-CoV-2, which causes coronavirus disease 2019 (COVID-19) and has a global mortality rate that remains to be determined (Wu et al., 2020, Zhu et al., 2020). SARS-CoV-2 infection is characterized by a range of symptoms including fever, cough, and general malaise in the majority of cases (Chen et al., 2020). More severe cases of COVID-19 develop acute respiratory distress syndrome and acute lung injury, leading to morbidity and mortality caused by damage to the alveolar lumen leading to inflammation and pneumonia (Wolfel et al., 2020, Xu et al., 2020).
The physiological response to virus infection is generally initiated at the cellular level following replication (tenOever, 2016). After virus entry, the infected cell detects the presence of virus replication through any one of a number of pattern recognition receptors (PRRs) (Janeway and Medzhitov, 2002). These receptors serve as sentinels for a variety of microbes inside and outside of the cell by physically engaging distinct structures that are shared among different pathogens. In the case of virus infection, cellular detection of replication is largely mediated by a family of intracellular PRRs that sense aberrant RNA structures that often form during virus replication (Janeway and Medzhitov, 2002). Engagement of virus-specific RNA structures culminates in oligomerization of these receptors and activation of downstream transcription factors, most notably interferon regulator factors (IRFs) and nuclear factor κB (NF-κB) (Hur, 2019). Transcriptional activation of IRFs and NF-κB results in the launch of two general antiviral programs. The first is engagement of cellular antiviral defenses, which is mediated by transcriptional induction of type I and III interferons (IFN-I and IFN-III, respectively) and subsequent upregulation of IFN-stimulated genes (ISGs) (Lazear et al., 2019). The second arm of the antiviral response involves recruitment and coordination of specific subsets of leukocytes, which is orchestrated primarily by chemokine secretion (Proudfoot, 2002, Sokol and Luster, 2015).
This broad antiviral response puts selective pressure on viruses and has resulted in the evolution of countless viral countermeasures (García-Sastre, 2017). Therefore, the host response to a virus is generally not uniform, and infections can inflict different degrees of morbidity and mortality. The current pandemic of COVID-19 is an acute and rapidly developing global health crisis. To better understand the molecular basis of the disease, we sought to characterize the transcriptional response to infection in a variety of model systems, including in vitro tissue culture, ex vivo infection of primary cells, and in vivo samples derived from COVID-19 patients and animals. We chose to characterize the transcriptional response to SARS-CoV-2 and determine how it compares with common respiratory viruses, including influenza A virus (IAV). These two respiratory viruses encode a variety of different antagonists to the IFN-I and -III response (Frieman and Baric, 2008, García-Sastre, 2017). For the closely related SARS-CoV-1, IFN antagonism has been attributed to ORF3B, ORF6, and the nucleocapsid (N) gene products (Frieman et al., 2010, Kopecky-Bromberg et al., 2007). SARS-CoV-1 also encodes nsp1, a nuclease that has been implicated in cleaving host mRNA to prevent ribosomal loading and causing host shutoff (Kamitani et al., 2006). Similar to SARS-CoV-1, IAV also encodes the IFN-I and -III antagonist nonstructural protein 1 (NS1), which blocks initial detection by the PRR through binding and masking aberrant RNA produced during infection (García-Sastre et al., 1998).
Here we compare the transcriptional response of SARS-CoV-2 with other respiratory viruses to identify transcriptional signatures that may underlie COVID-19 biology. These data demonstrate that the overall transcriptional induction to SARS-CoV-2 is aberrant. Despite virus replication, the host response to SARS-CoV-2 fails to launch a robust IFN-I and -III response while simultaneously inducing high levels of chemokines needed to recruit effector cells. Because a waning immune response would enable sustained viral replication, these findings may explain why serious cases of COVID-19 are more frequently observed in individuals with comorbidities.
Results
Defining the Transcriptional Response to SARS-CoV-2 Relative to Other Respiratory Viruses
To compare the transcriptional response of SARS-CoV-2 with other respiratory viruses, including MERS-CoV, SARS-CoV-1, human parainfluenza virus 3 (HPIV3), respiratory syncytial virus (RSV), and IAV, we first chose to focus on infection in a variety of respiratory cell lines (Figure 1 ). To this end, we collected poly(A) RNA from infected cells and performed RNA sequencing (RNA-seq) to estimate viral load. These data show that virus infection levels ranged from 0.1% to more than 50% of total RNA reads (Figure 1A). In agreement with others (Harcourt et al., 2020), we found A549 lung alveolar cells to be relatively non-permissive to SARS-CoV-2 replication, in contrast to Calu-3 cells (0.1% versus 15% total reads, respectively). The low rate of infection in A549 cells is postulated to be the result of low expression of the viral receptor ACE2 (Harcourt et al., 2020, Hoffmann et al., 2020). To bypass this restriction, we supplemented A549 cells with a vector expressing mCherry or ACE2 (Figures 1B–1D). In low-MOI infections (MOI, 0.2), exogenous expression of ACE2 enabled SARS-CoV-2 to replicate and comprise ∼54% of the total reads mapping more than 300× coverage across the ∼30-kb genome (Figures 1A and 1B). Western blot analyses corroborated these RNA-seq data, showing Nucleocapsid (N) expression only in cells supplemented with ACE2 (Figure 1C). Furthermore, qPCR analyses of these cells demonstrated that the levels of Envelope (E) and non-structural protein 14 (nsp14) were more than three orders of magnitude higher in the presence of ACE2 (Figure 1D). It is noteworthy that, despite this dramatic increase in viral load, we observed neither activation of TBK1, the kinase responsible for IFN-I and IFN-III expression, nor induction of STAT1 and MX1, IFN-I-stimulated genes (Figure S1 A; Sharma et al., 2003). The lack of IFN-I and -III engagement in ACE2-expressing A549 cells could, however, be overcome by using a 10-fold increase in virus (MOI, 2) despite the fact that total viral reads after 24 h of replication where comparable with low-MOI conditions (Figures 1A, 1B, and 1F).
Figure 1.

Host Transcriptional Response to Respiratory Infection in Human Lung Epithelium-Derived Cell Lines
(A) Virus replication levels in infected cells. RNA-seq was performed on poly(A)-enriched total RNA, and the percentage of virus-aligned reads (over total reads) is indicated for each sample. Error bars represent standard deviation from three independent biological replicates (except for IAV infection, where data are representative of independent biological duplicates). The cell types used for each infection is indicated (+) at the bottom of the figure. All infections were performed at a high MOI (MOI, 2–5), except for ∗, which indicates an MOI of 0.2.
(B) Read coverage along the SARS-CoV-2 genome for mCherry- or ACE2-expressing A549 cells. The graph indicates the number of viral reads per position of the virus genome in A549 cells transduced with adenovirus (AdV)-based vectors expressing mCherry (MOI, 0.2; light blue) or ACE2 (MOI 0.2, salmon (∗); MOI 2, dark red). A scaled model of the SARS-CoV-2 genome and its genes is depicted below (generated in BioRender).
(C) Western blot analysis of mCherry- or ACE2-expressing A549 cells infected with SARS-CoV-2. Whole-cell lysates were analyzed by SDS-PAGE and blotted for ACE2, SARS-CoV-2 nucleocapsid (N), and actin.
(D) qRT-PCR analysis of mCherry- or ACE2-expressing A549 cells infected with SARS-CoV-2 (MOI, 0.2). The graph depicts the relative amount of SARS-CoV-2 Envelope (E), non-structural protein 14 (nsp14), and human IFNB transcripts normalized to human α-tubulin. Error bars represent the standard deviation of the mean log2(fold change) of three independent biological replicates.
(E) Principal-component analysis (PCA) for the global transcriptional response to respiratory viruses. Sparse PCA depicts global transcriptome profiles of the samples in (A). Cell types used for infection are represented by different shapes (circle, A549; square, A549-ACE; diamond, Calu-3; triangle, MRC5).
(F) Heatmap depicting the expression levels of differentially expressed genes (DEGs) of the samples in (A) belonging to the indicated GO biological processes (GO: 0034097, GO: 0045087, GO: 0009615, GO: 0006954). The graph depicts the log2(fold change) of DEGs of infected compared with mock-treated cells. The included genes have a log2(fold change) of more than 2 and a p-adjusted value of less than 0.05. Data from SARS-CoV-1 and MERS-CoV infections correspond to GEO: GSE56192.
Figure S1.

Role of IFN Response in Infection with SARS-CoV-2, Related to Figure 1
(A) Western blot analysis of WT or ACE2-expressing A549 cells mock-treated or infected with SARS-CoV-2 or Sendai virus. Whole cell lysates were analyzed by SDS-PAGE and blotted for SARS-CoV-2 spike, phospho-TBK1, MX1, STAT1 and actin. (B) qRT-PCR analysis of Vero E6 cells infected with SARS-CoV-2 and treated with IFNβ 2 h post infection as indicated. The graph depicts the relative amount of SARS-CoV-2 envelope (E) and non-structural protein 14 (nsp14) normalized to human α-Tubulin. Error bars represent the standard deviation of the mean fold change of three independent biological replicates. Statistical significance calculated by Student's t test corrected for multiple comparisons using Holm-Sidak method (∗∗∗) p value < 0.001. (C) Western blot analysis of conditions as in (B). Whole cell lysates were analyzed by SDS-PAGE and blotted for SARS-CoV-2 spike and GAPDH. (D) Western blot analysis of ACE2-expressing A549 cells infected with SARS-CoV-2 with or without Ruxolitinib. Whole cell lysates were analyzed by SDS-PAGE and blotted for SARS-CoV-2 spike and GAPDH. (E) Virus replication levels in SARS-CoV-2-infected A549-ACE2 cells treated with or without Ruxolitinib. RNA-seq was performed on polyA enriched total RNA and the percentage of virus-aligned reads (over total reads) is indicated for each sample. Error bars represent standard deviation from three independent biological replicates. Infections were preformed at high MOI (MOI: 2). (F-G) Expression levels of (F) ISGs or (G) cytokines and chemokines in conditions as in (E). Scatterplot of the log2(Fold Change) of individual genes in SARS-CoV-2-infected A549-ACE2 cells treated with or without Ruxolitinib. Linear regression line and confidence interval (95%) is shown as a red line and gray shaded area, respectively. Dotted diagonal represent no changes between conditions. (H). Heatmap depicting the expression levels of ISGs as in (F).
To determine whether SARS-CoV-2 is sensitive to IFN-I, we next treated cells with universal IFNβ and assessed viral levels at the RNA and protein levels (Figures S1B and S1C). These data demonstrate that addition of IFN-I resulted in a dramatic reduction in virus replication, in agreement with the findings of others (Lokugamage et al., 2020). We also observed no increase in viral Spike levels (or a considerable effect on viral reads) when IFN-I signaling was blocked by addition of ruxolitinib, a JAK1 and 2 kinase inhibitor, despite significantly preventing induction of ISGs (Figures S1D–S1H). In contrast, ruxolitinib treatment had a minimal effect on induction of cytokines and chemokines, indicating that the high induction of these genes in SARS-CoV-2 infection is independent of IFN-I and -III signaling (Figure S1G).
To next determine how each of these in vitro infections alters the host transcriptional landscape, we first performed a differential expression analysis, comparing infected cell conditions with their respective mock conditions. These analyses indicate that the transcriptional response in cells that allows high replication of SARS-CoV-2 is significantly different from the host response of all other viruses tested (Figure 1E). Moreover, SARS-CoV-2 infection in unmodified A549 cells shows a unique response compared with SARS-CoV-1 despite comparable levels of viral load (Figures 1A and 1E). Last, MERS-CoV infection, which approaches ∼50% of total reads at 24 hours post-infection (hpi), clusters together with SARS-CoV-1 and IAV, reflecting an overall repression of the host antiviral response (Figures 1E and 1F). Conversely, HPIV3 and RSV comprise a unique cluster denoted by high expression of IFNs and ISGs (Figures 1E and 1F). Interestingly, low-MOI SARS-CoV-2-infected A549 cells expressing ACE2 (A549-ACE2) show no significant IFN-I or IFN-III expression but instead display moderate levels of a subset of ISGs and a unique proinflammatory cytokine signature (Figure 1F). This signature is also present in high-MOI infections of SARS-CoV-2 in A549-ACE2 and Calu-3 cells, together with more than 6,000 other differentially expressed genes, further explaining their extreme coordinates on the principal-component analysis (PCA) (Figures 1E and 1F; Table S1). Furthermore, high-MOI infection in these cells also led to high induction of IFNs and ISGs observed for HPIV3 and RSV, despite remarkable differences in viral replication (∼60% total reads in A549-ACE2 cells compared with ∼15% in Calu-3 cells) (Figures 1A and 1F). The discrepancy between the levels of viral replication and IFN production/signaling suggests that, although SARS-CoV-2 is capable of engaging the IFN-I and IFN-III systems, this response is prevented by an antagonist that is rendered ineffective under high-MOI conditions. Alternatively, these data may instead indicate that high-MOI conditions in cell culture result in formation of pathogen-associated molecular patterns (PAMPs), which may or may not reflect physiological conditions in vivo.
SARS-CoV-2 in Primary Cells Induces a Limited IFN-I and III Response
Given the disparate results of our in vitro cell culture systems, we next sought to determine how normal human bronchial epithelial (NHBE) cells respond to SARS-CoV-2 infection, in contrast to treatment with IFN-I alone or infection with wild-type (WT) IAV or a mutant IAV lacking its antiviral antagonist (IAVΔNS1) (Figure 2 ). Treatment of NHBE cells with IFN-I resulted in significant induction of 381 genes, most of these also differentially expressed in IAVΔNS1 infection, together outlining a robust innate immune response in these cells (Figure 2A). In contrast, and despite different levels of replication, the transcriptional response to infection with SARS-CoV-2 and WT IAV are similar in magnitude but different in nature, with only 8 shared significantly induced genes, including interleukin-6 (IL-6), IRF9, ICAM1, and tumor necrosis factor (TNF) (Figure 2A; Figure S2 A). To further understand the global host response as it pertained to each of these conditions, we grouped these samples in a PCA space (Figure 2B). This analysis shows progressive transcriptional perturbations along principal component one, which accounts for more than 60% of sample variation. In this space, SARS-CoV-2 elicits the most modest transcriptional changes, followed by IAV, IFNβ treatment, and IAVΔNS1 (Figure 2B).
Figure 2.

Host Transcriptional Response to IAV and SARS-CoV-2 in Primary Human Bronchial Epithelial Cells
(A) Shared DEGs in IFNβ-treated, SARS-CoV-2- or IAV-infected NHBE cells. The Venn diagram depicts genes shared and/or unique between each comparison.
(B) Sparse PCA depicting global transcriptional profiles of the samples in (A).
(C) Dotplot visualization of enriched GO terms in NHBE cells. Gene enrichment analyses were performed using STRING against the GO dataset for biological processes. The color of the dots represents the false discovery rate (FDR) value for each enriched GO term, and size represents the percentage of genes enriched in the total gene set.
(D) Heatmap indicating the expression levels of DEGs involved in IFN-I responses.
(E) Heatmap as in (D) for genes belonging to GO annotations for cytokine activity and chemokine activity (GO: 0005125, GO: 0008009). The graphs depict the log2(fold change) of DEGs of infected compared with mock-treated cells. Genes included have a log2(fold change) of more than 1 and a p-adjusted value of less than 0.05.
Figure S2.

Infectivity and Host Response to SARS-CoV-2 Infection in NHBE Cells, Related to Figure 2
(A) Virus replication levels in infected cells. RNA-seq was performed on polyA enriched total RNA and the percentage of virus-aligned reads (over total reads) is indicated for each sample. Error bars represent standard deviation from four independent biological replicates (except for SARS-CoV-2 infection where data are representative of independent biological triplicates). (B) Heatmap depicting the expression levels of Interferon transcripts in the indicated conditions. Colors representing transcripts per million (TPMs) in RNA-seq experiments.
Gene enrichment analyses of differentially expressed transcripts illustrate a diminished IFN-I signaling biology for SARS-CoV-2 and IAV infections (Figures 2C and 2D). In both examples, IFN-I and IFN-III are undetectable, but a very small subset of ISGs is induced (Figures 2C, 2D, and S2B; Table S2). In the case of IAV, this diminished antiviral response is mediated by expression of NS1 because IAVΔNS1 infections result in robust IFNB and IFNL1-3 induction (Figures 2C, 2D, and S2B; Table S2). Despite a complete lack of IFN expression, the response to SARS-CoV-2 in NHBE cells still elicited a strong chemotactic and inflammatory response, indicated by expression of CCL20, CXCL1, IL-1B, IL-6, CXCL3, CXCL5, CXCL6, CXCL2, CXCL16, and TNF (Figures 2C and 2E; Table S2). In addition to the modest IFN-I response, SARS-CoV-2 in NHBE cells also triggers some unique pathways, including a response to IFN-II (which is also observed in response to IAVΔNS1), and significant enrichment in chemokine signaling (Figure 2C).
Longitudinal Ferret Studies Mirror the Imbalanced In Vitro Response to SARS-CoV-2
To determine whether the limited response to SARS-CoV-2 observed so far was a by-product of cell culture, we next pursued an in vivo longitudinal study in animals. To this end, we chose to perform SARS-CoV-2 infections in ferrets because this has been described as an appropriate animal model (Kim et al., 2020). Ferrets were infected intranasally with SARS-CoV-2 or influenza A/California/04/2009 and monitored by nasal wash, which generates a small pellet of cells from the upper respiratory tract. RNA-seq was performed on these cells, enabling us to quantify viral load over time. Reads from nasal wash 1 day after infection revealed a low level of virus replication comprising 0.006% of total reads (Figure 3 A). Three days after infection, virus replication peaked at 1.2% of the total sequencing reads before decreasing to 0.05% of total reads on day 7 and completely clearing the virus by day 14 (Figure 3A). As a comparison, a sublethal infection of IAV comprised less than 0.03% of total reads on day 7 from the same sample type (Figure S3 A). The presence of virus in the nasal passage would further suggest that these ferrets had the potential to transmit the virus, in agreement with the findings of others (Kim et al., 2020, Varble et al., 2014).
Figure 3.

Longitudinal Analysis of the Host Response to SARS-CoV-2 in Ferrets
(A) Read coverage along the SARS-CoV-2 genome. The graph indicates the number of viral reads per each position of the virus genome identified in RNA extracted from nasal washes of ferrets 1 (gray), 3 (red), 7 (blue), and 14 (green) days after infection (ND, not detected).
(B) Volcano plots indicating DEGs of ferrets along the course of a SARS-CoV-2 infection as in (A). DEGs (p-adjusted < 0.05) with a |log2(fold change)| of more than 2 are indicated in red. Non-significant DEGs with a |log2(fold change)| of more than 2 are indicated in green.
(C) Heatmap depicting the expression levels of a subset of cytokines differentially expressed in nasal washes collected from ferrets infected with the indicated viruses at specific times.
(D) Heatmap depicting the expression levels of lymphoblast-related genes differentially expressed in trachea samples collected from ferrets infected with the indicated viruses after 3 days. The graphs show the log2(fold change) of DEGs of infected compared with mock-infected animals. Genes included have a log2(fold change) of more than 2 and a p-adjusted value of less than 0.05. Ferrets were randomly assigned to the different treatment groups (naive, n = 2; SARS-CoV-2 infection, n = 6; IAV [pH1N1] infection, n = 2; IAV [H3N2] infection, n = 2).
Figure S3.

Transcriptional Response to SARS-CoV-2 and IAV in Ferrets, Related to Figure 3
(A) Read coverage along the IAV genome. Graph indicates the number of viral reads per each position of the IAV virus genome identified in RNA extracted from nasal washes of ferrets at 7 days post infection. Scaled model of the concatenated IAV segments is depicted below. (B) Volcano plots indicating differentially expressed genes of ferrets infected with SARS-CoV-2 or IAV for 7 days. Differentially expressed genes (p-adjusted value < 0.05) with a |Log2(Fold Change)| > 2 are indicated in red. Non-significant differentially expressed Genes with a |Log2(Fold Change)| > 2 are indicated in green. (C) Cellular profiling from a subset of genes selectively enriched in response to SARS-CoV-2 compared to IAV, as determined by the Immunological Genome Project.
To characterize the response to SARS-CoV-2 over time, upper respiratory cell populations were compared with mock-treated ferrets. On day 1 after infection, we observed very little transcriptional difference correlating to the amount of virus detection at this time (Figure 3B). By day 3, we observed the beginning of a cytokine response marked by CCL8 and CXCL9, consistent with what was observed in cell culture. By day 7, despite waning levels of virus, the cytokine response continued to expand and included CCL2, CCL8, and CXCL9, among others (Figure 3B; Table S3). Moreover, we noted evidence of mixed leukocyte infiltration with significant upregulation in CD163, CD226, CCR5, CCR6, CXCR1, CXCR2, and CXCR7 (Figure 3C). Overall, the magnitude of this transcriptional response in the upper respiratory tract was significantly lower compared with a comparable IAV infection (Figure S3B). However, although IAV induces a greater number of genes, SARS-CoV-2 generates a unique gene signature enriched for cell death and leukocyte activation, including transcripts such as IL1A and CXCL8 (GO: 0008219 and GO: 0045431; Table S3). In contrast, the transcriptional footprint of IAV as it pertains to the cellular antiviral response was strikingly greater in magnitude than that observed for SARS-CoV-2 and included the IFN signature genes MX1, ISG20, OASL, and Tetherin (Figure S3B; Table S3). By day 14, we detected no viral reads for SARS-CoV-2, and the observed cytokines returned to baseline, with the exception of IL-6 and IL1RN or IL1RA, which remained elevated, similar to results observed with MERS (Pascal et al., 2015; Figures 3B and 3C).
Last, to investigate how the host response to SARS-CoV-2 and IAV affected the respiratory tract, we next performed parallel infections and examined the trachea on day 3. With both infections, we observed very low levels of virus but a robust transcriptional response (Table S3). Gene enrichment analysis of differentially expressed transcripts implicated two populations of immune cell signatures (Figure 3D). The first population included common markers for monocytes and lymphocytes, and the induction of these genes was comparable between SARS-CoV-2 and IAV (Figures 3D and S3C). Intriguingly, unique gene signatures from SARS-CoV-2-infected trachea that were largely absent in response to IAV align with those of progenitor cells from the hematopoietic lineage, suggesting that infection may be inducing hematopoiesis (Figures 3D and S3C) (Lefrançais et al., 2017, Yoshida et al., 2019). Additional research in this area will be required to ascertain whether this is a contributing factor toward the development of COVID-19.
COVID-19 Patients Present Low IFN-I and -III and High Chemokine Signatures
Following the characterization of SARS-CoV-2 infection in ferrets, we next sought to correlate these results with natural human infections. To this end, we first compared post-mortem lung samples from COVID-19-positive patients with biopsied healthy lung tissue from uninfected individuals. Transcriptional profiling of these samples, all derived from males older than 60 years (n = 2 for each group), demonstrated ∼2,000 differentially expressed genes with enrichment for the innate and humoral responses (Figures 4 A and S4 A). Genes significantly induced in response to SARS-CoV-2 included a subset of ISGs with no IFN-I or IFN-III detected by RNA-seq or semiquantitative PCR (Figure S4B; Table S4). In addition to genes implicated in innate antiviral immunity, SARS-CoV-2 also induced robust levels of chemokines, including CCL2, CCL8, and CCL11 (Figure 4A). Despite the limited number of patients analyzed, these data corroborate our findings in NHBE and ferrets (Figure S4A).
Figure 4.
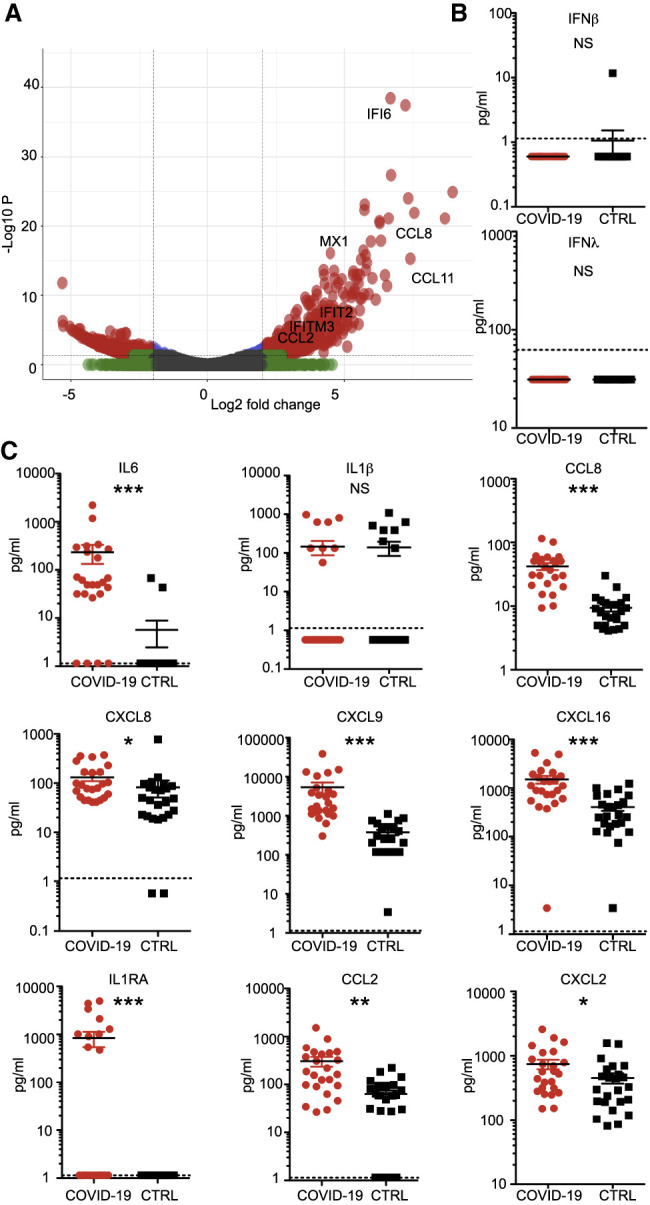
Transcriptional and Serological Profile of Clinical COVID-19 Patients
(A) Volcano plot depicting DEGs in post-mortem lung samples of two COVID-19 patients compared with healthy lung biopsies. DEGs (p-adjusted < 0.05) with a |log2(fold change)| of more than 2 are indicated in red. Non-significant DEGs with a |log2(fold change)| of more than 2 are indicated in green.
(B and C) Cytokine profiles of COVID-19 patients. Sera of 24 COVID-19 patients and 24 SARS-CoV-2-negative controls were analyzed by ELISA for the protein levels of (B) IFN-I and IFN-III or (C) a broad panel of cytokines. The dotted line depicts the limit of detection. Statistical significance was calculated by Mann-Whitney non-parametric t test. NS, non-significant; ∗p < 0.05, ∗∗p < 0.005, ∗∗∗p < 0.0001.
Figure S4.

Unique and Shared Biological Processes between Different Models of SARS-CoV-2 Infection, Related to Figure 4
(A) Dotplot visualization of enriched GO terms in NHBE cells, ferrets and COVID-19 patients. Gene enrichment analyses were performed using STRING against the GO dataset for biological processes. The color of the dots represents the false discovery rate (FDR) value for each enriched GO term and its size represents the percentage of genes enriched in the total gene set. (B) Semiquantitative PCR analysis of healthy and COVID-19 derived lung tissues. Image indicates the relative expression of SARS-CoV-2 nsp14, IFNB and tubulin transcripts in healthy human biopsies and biological replicates of lung tissue from COVID-19 patients. Additionally cDNA of A549 cells infected with IAVΔNS1 are included as controls.
Next we wished to further validate our findings with a larger cohort of patients through direct detection of circulating cytokines induced by SARS-CoV-2 infection. To this end, we obtained serum from two cohorts of individuals from the Kaiser Santa Clara testing facility (Santa Clara, CA). These two cohorts tested positive for SARS-CoV-2 by nasopharyngeal swabs or were admitted to the hospital for non-COVID-19-related respiratory issues (n = 24 for each group). During initial analyses, these serum samples consistently tested negative for IFNβ and the IFNλ family of IFNs (Figure 4B). Moreover, analyses of cytokines and chemokines quantified in individual serum samples revealed enhancement of generalized inflammation among COVID-19 patients, marked by a significant increase in circulating IL-6, IL1RA, CCL2, CCL8 CXCL2, CXCL8, CXCL9, and CXCL16 levels (Figure 4C). Significant elevation of CXCL9 and CXCL16 (chemoattractants of T or natural killer (NK) cells, respectively), CCL8 and CCL2 (which recruit monocytes and/or macrophages), and CXCL8 (a classic neutrophil chemoattractant) suggest that the presence of these cells may be a primary driver of the signature pathology observed in COVID-19 patients (Proudfoot, 2002). Although this sample size is not necessarily representative of the whole population of infected COVID-19 patients, our data are consistent with what we observe using our other model systems. Additional sampling will be required to validate these findings.
Discussion
In the present study, we focus on defining the host response to SARS-CoV-2 and other human respiratory viruses in cell lines, primary cell cultures, ferrets, and COVID-19 patients. In general, our data show that the overall transcriptional footprint of SARS-CoV-2 infection was distinct in comparison with other highly pathogenic coronaviruses and common respiratory viruses such as IAV, HPIV3, and RSV. It is noteworthy that, despite a reduced IFN-I and -III response to SARS-CoV-2, we observed a consistent chemokine signature. One exception to this observation is the response to high-MOI infection in A549-ACE2 and Calu-3 cells, where replication was robust and an IFN-I and -III signature could be observed. In both of these examples, cells were infected at a rate to theoretically deliver two functional virions per cell in addition to any defective interfering particles within the virus stock that were not accounted for by plaque assays. Under these conditions, the threshold for PAMP may be achieved prior to the ability of the virus to evade detection through production of a viral antagonist. Alternatively, addition of multiple genomes to a single cell may disrupt the stoichiometry of viral components, which, in turn, may itself generate PAMPs that would not form otherwise. These ideas are supported by the fact that, at a low-MOI infection in A549-ACE2 cells, high levels of replication could also be achieved, but in the absence of IFN-I and -III induction. Taken together, these data suggest that, at low MOIs, the virus is not a strong inducer of the IFN-I and -III system, as opposed to conditions where the MOI is high. These dynamics are also likely to contribute to development of COVID-19 during the course of infection (Wolfel et al., 2020).
A recurrent observation in each of our systems is a robust production of cytokines and their subsequent transcriptional response. According to our longitudinal in vivo data, this response starts as early as 3 days after infection and continues beyond clearance of the virus. A recent study analyzing severe versus mild cases of COVID-19 showed that peripherally derived macrophages predominated in the lungs of severe cases (Liao et al., 2020). Consistent with this, we found, in all of our systems, significant induction of monocyte-associated chemokines such as CCL2 and CCL8. In addition, our data suggest that neutrophils could also contribute to the disease observed in COVID-19 patients, as demonstrated by CXCL2 and CXCL8 induction. This is consistent with data showing elevated circulating neutrophil levels among COVID-19 patients (Chen et al., 2020, Qin et al., 2020), which may have prognostic value for identifying individuals at risk for developing severe disease. It is also noteworthy that two of the cytokines uniquely elevated in response to SARS-CoV-2 are IL-6 and IL1RA, suggesting that there might be a parallel between COVID-19 and cytokine release syndrome (CRS), a complication commonly seen following CAR T-cell treatment (Giavridis et al., 2018). Should this be true, drugs such as tocilizumab and anakinra may prove to be beneficial for treatment of COVID-19 (Norelli et al., 2018). Future studies will be needed to address this formally.
Like SARS-CoV-2, the clinical manifestation of SARS-CoV-1 has been proposed to stem from a dysregulated immune response in patients and delayed expression of IFN-I (Channappanavar et al., 2016, Law et al., 2005, Menachery et al., 2014). Based on animal models, SARS-CoV-1 has been found to induce a robust cytokine response that generally shows a delay in IFN-I, culminating in improper recruitment of inflammatory monocyte-macrophage populations (Channappanavar et al., 2016). This dynamic seems to be in line with what we observed with SARS-CoV-2 because low levels of IFN-I and -III are likely produced in response to infection. Given the moderate viral replication levels observed in vivo, one explanation for the low IFN expression could be that a small subset of cells is refractory to the antagonistic mechanism of SARS-CoV-2, producing sufficient amounts of IFN-I and/or IFN-III to guide immune cell activation and ISG induction.
What makes SARS-CoV-2 distinct from other viruses used in this study is the propensity to selectively induce morbidity and mortality in older populations (Novel Coronavirus Pneumonia Emergency Response Epidemiology Team, 2020). The physiological basis for this morbidity is believed to be selective death of type II pneumocytes, which results in loss of air exchange and fluid leakage into the lungs (Qian et al., 2013, Xu et al., 2020). Although it remains to be determined whether the inappropriate inflammatory response to SARS-CoV-2 is responsible for the abnormally high lethality in older populations, it does explain why the virus is generally asymptomatic in young people with healthy and robust immune systems (Lu et al., 2020). Given the results here, it is tempting to speculate that an already restricted immune response in the aging population prevents successful inhibition of viral spread at early stages of infection, further exacerbating the morbidity and mortality observed for this age group (Jing et al., 2009, Montecino-Rodriguez et al., 2013).
Taken together, the data presented here suggest that the response to SARS-CoV-2 is imbalanced with regard to controlling virus replication versus activation of the adaptive immune response. Given this dynamic, treatments for COVID-19 have less to do with the IFN response and more to do with controlling inflammation. Because our data suggest that numerous chemokines and ILs are elevated in COVID-19 patients, future efforts should focus on U.S. Food and Drug Administration (FDA)-approved drugs that can be rapidly deployed and have immunomodulating properties.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-Actin (Pan) | Thermo Fisher Scientific | Cat# MS-1295, RRID:AB_63314 |
| Rabbit monoclonal anti-GAPDH, Clone 14C10 | Cell Signaling Technology | Cat# 2118, RRID:AB_561053 |
| Rabbit monoclonal anti-ACE2 | Abcam | ab239924 |
| Rabbit monoclonal anti-Phospho-TBK1(Ser172) | Cell Signaling Technology | Cat# 5483, RRID:AB_10693472 |
| Mouse monoclonal anti-STAT1 | BD Biosciences | Cat# 558537, RRID:AB_647231 |
| Rabbit polyclonal anti-MX1 | Abcam | ab207414 |
| Mouse monoclonal anti-SARS-CoV-2 Spike [2B3E5] | Center for Therapeutic Antibody Discovery at the Icahn School of Medicine at Mount Sinai | N/A |
| Mouse monoclonal anti-SARS-CoV-2 Nucleocapsid [1C7C7] | Center for Therapeutic Antibody Discovery at the Icahn School of Medicine at Mount Sinai | N/A |
| IRDye 680RD Goat anti-Rabbit IgG | LI-COR Biosciences | Cat# 926-68071, RRID:AB_10956166 |
| IRDye 680RD Goat anti-Mouse IgG | LI-COR Biosciences | Cat# 926-68070, RRID:AB_10956588 |
| IRDye 800CW Goat anti-Rabbit IgG | LI-COR Biosciences | Cat# 926-32211, RRID:AB_621843 |
| IRDye 800CW Goat anti-Mouse IgG | LI-COR Biosciences | Cat# 926-32210, RRID:AB_621842 |
| Bacterial and Virus Strains | ||
| Ad-mCherry | Vector Biolabs | Cat# 1767 |
| Ad-GFP-h-ACE2 | Vector Biolabs | Cat# ADV-200183 |
| SARS-CoV-2 Isolate USA-WA1/2020 | BEI Resources | Cat# NR-52281 |
| Influenza A/Puerto Rico/8/1934 (H1N1) virus | Langlois et al., 2013 | N/A |
| Influenza A/California/04/2009 (pH1N1) virus | Langlois et al., 2013 | N/A |
| Influenza A/Texas/71/2017 (H3N2) virus | Langlois et al., 2013 | N/A |
| Influenza A/Puerto Rico/8/1934-ΔNS1 (H1N1) virus | García-Sastre et al., 1998 | N/A |
| rgRSV[224] (strain A2) | Hallak et al., 2000 | N/A |
| rHPIV3JS-GlucP2AeGFP | Blanco-Melo et al., 2020 | N/A |
| Biological Samples | ||
| Healthy human lung tissue | Mount Sinai Institutional Biorepository and Molecular Pathology Shared Resource Facility | N/A |
| COVID-19 human lung tissue | Weill Cornell Medicine | N/A |
| Human patient sera | Kaiser Santa Clara testing facility | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Ruxolitnib | ACT Chemical | Cat# ACT06813 |
| Universal Type I IFN | R&D Systems | Cat# 11200-2 |
| Human IFNβ | BEI Resources | Cat# NR-3080 |
| TRIzol Reagent | Thermo Fisher Scientific | Cat# 15596026 |
| Critical Commercial Assays | ||
| Direct-zol RNA MiniPrep kit | Zymo Research | Cat# R2051 |
| TruSeq RNA Library Prep Kit v2 | Illumina | Cat# RS-122-2001 |
| TruSeq Stranded mRNA Library Prep Kit | Illumina | Cat# 20020594 |
| KAPA SYBR FAST qPCR Master Mix Kit Universal | Kapa Biosystems | Cat# KK4601 |
| VeriKine-HS human IFN-β serum ELISA kit | PBL Interferon Source | Cat# 41415-1 |
| VeriKine-HS human IFN-λ 1/2/3 serum ELISA kit | PBL Interferon Source | Cat# 61840-1 |
| Deposited Data | ||
| Raw and analyzed data | This paper | GEO: GSE147507 |
| SARS-CoV-1 and MERS-CoV data | NCBI GEO | GEO: GSE56192 |
| Experimental Models: Cell Lines | ||
| A549 | ATCC | Cat# CCL-185, RRID:CVCL_0023 |
| Calu-3 | ATCC | Cat# HTB-55, RRID:CVCL_0609 |
| Hep-2 | ATCC | Cat# CCL-23, RRID:CVCL_1906 |
| MDCK | ATCC | Cat# CCL-34, RRID:CVCL_0422 |
| MDCK-NS1 | García-Sastre et al., 1998 | MDCK-NS1 |
| Vero E6 | ATCC | Cat# CRL-1586, RRID:CVCL_0574 |
| NHBE | Lonza | Cat# CC-2540, #580580 |
| Experimental Models: Organisms/Strains | ||
| Ferrets | Triple F. Farm | Fitch ferrets |
| Oligonucleotides | ||
| h-aTubulin_F: GCCTGGACCACAAGTTTGAC | This paper | N/A |
| h-aTubulin_R: TGAAATTCTGGGAGCATGAC | This paper | N/A |
| h-IFNb_F: GTCAGAGTGGAAATCCTAAG | This paper | N/A |
| h-IFNb_R: ACAGCATCTGCTGGTTGAAG | This paper | N/A |
| SARSCoV2-nsp14_F: TGGGGYTTTACRGGTAACCT | Chu et al., 2020 | N/A |
| SARSCoV2-nsp14_R: AACRCGCTTAACAAAGCACTC | Chu et al., 2020 | N/A |
| SARSCoV2-E_F: ACAGGTACGTTAATAGTTAATAGCGT | Corman et al., 2020 | N/A |
| SARSCoV2-E_R: ATATTGCAGCAGTACGCACACA | Corman et al., 2020 | N/A |
| Software and Algorithms | ||
| Prism8 | GraphPad | http://www.graphpad.com |
| ImageStudio | LI-COR | https://www.licor.com/bio/image-studio/ |
| BaseSpace | Ilumina | http://basespace.illumina.com/dashboard |
| RNA-Seq Aligment App v2.0.2 | Ilumina | http://basespace.illumina.com/dashboard |
| RNA-Express v1.1.10 | Ilumina | http://basespace.illumina.com/dashboard |
| DESeq2 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| STRING | Szklarczyk et al., 2019 | https://string-db.org/ |
| gplots | CRAN | https://cran.r-project.org/web/packages/gplots/index.html |
| PMA | Witten et al., 2009 | https://cran.r-project.org/web/packages/PMA/index.html |
| ggplot2 | Tidyverse | https://ggplot2.tidyverse.org/ |
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| ImmGen | Yoshida et al., 2019 | http://www.immgen.org/ |
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Benjamin tenOever (benjamin.tenoever@mssm.edu).
Materials Availability
All materials and reagents will be made available upon instalment of a material transfer agreement (MTA).
Data and Code Availability
The raw sequencing datasets generated during this study are available on the NCBI Gene Expression Omnibus (GEO) server under the accession number GSE147507. The original sequencing datasets for SARS-CoV-1 and MERS-CoV infections can be found on the NCBI Gene Expression Omnibus (GEO) server under the accession number GSE56192.
Experimental Model and Subject Availability
Cell cultures and primary cells
Human adenocarcinomic alveolar basal epithelial (A549) cells (ATCC, CCL-185), human adenocarcinomic lung epithelial (Calu-3) cells (ATCC, HTB-55), human HEp-2 cells (ATCC, CCL-23), Madin-Darby Canine Kidney (MDCK) cells (ATCC, CCL-34), MDCK-NS1 cells (García-Sastre et al., 1998) and African green monkey kidney epithelial Vero E6 cells (ATCC, CRL-1586) were maintained at 37°C and 5% CO2 in Dulbecco’s Modified Eagle Medium (DMEM, GIBCO) supplemented with 10% Fetal Bovine Serum (FBS, Corning). Undifferentiated normal human bronchial epithelial (NHBE) cells (Lonza, CC-2540 Lot# 580580) were isolated from a 79-year-old Caucasian female and were maintained in bronchial epithelial growth media (Lonza, CC-3171) supplemented with BEGM SingleQuots as per the manufacturer’s instructions (Lonza, CC-4175) at 37°C and 5% CO2.
Animal studies
Outbred 4-month old castrated male Fitch ferrets were purchased from Triple F. Farm (North Rose, NY). All animals were confirmed to be seronegative for circulating influenza A (H1N1) viruses, influenza A (H3N2) viruses and influenza B viruses prior to purchase. Ferrets were housed in cages in the enhanced BSL-3 facility of the Emerging Pathogens Institute at the Icahn School of Medicine at Mount Sinai. All animal experiments were performed according to protocols approved by the Institutional Animal Care and Use Committee (IACUC) and Institutional Biosafety Committee of the Icahn School of Medicine at Mount Sinai (NY, USA). Ferrets were randomly assigned to the different experimental groups.
Human studies
For RNA analysis, two COVID19 human subjects were deceased upon tissue acquisition and were provided from Weill Cornell Medicine as fixed samples. For semiquantitative PCR analyses, two additional lung samples were derived post-mortem from males over 60 years of age. The uninfected human lung samples (n = 2) were obtained post surgery through the Mount Sinai Institutional Biorepository and Molecular Pathology Shared Resource Facility (SRF) in the Department of Pathology. The Biorepository operates under a Mount Sinai Institutional Review Board (IRB) approved protocol and follows guidelines set by HIPAA. Sera were obtained from the Kaiser Santa Clara testing facility (Santa Clara, CA). Sera were from subjects with a CoV PCR+ nasopharyngeal swab (n = 24) or from subjects who were not being tested for CoV infection (n = 24). The study was reviewed and approved by the Stanford University institutional review board (PI TTW). Experiments using samples from human subjects were conducted in accordance with local regulations and with the approval of the institutional review board at the Icahn School of Medicine at Mount Sinai under protocol HS#12-00145.
Viruses
Influenza A/Puerto Rico/8/1934 (H1N1) virus (NCBI:txid183764), influenza A/California/04/2009 (pH1N1) virus and influenza A/Texas/71/2017 (H3N2) virus were grown in MDCK cells (Langlois et al., 2013). Influenza A/Puerto Rico/8/1934 (H1N1) virus lacking the NS1 gene (IAVΔNS1, (García-Sastre et al., 1998)) was grown in MDCK-NS1 cells. Influenza viruses were grown in EMEM supplemented with 0.35% bovine serum albumin (BSA, MP Biomedicals), 4 mM L-glutamine, 10 mM HEPES, 0.15% NaHCO3 and 1 μg/ml TPCK-trypsin (Sigma-Aldrich). Infectious titers of influenza A viruses were determined by plaque assay in MDCK or MDCK-NS1 cells, accordingly. Recombinant GFP-expressing human respiratory syncytial virus (RSV), strain A2 (rgRSV[224]) was generously provided by Dr. M. Peeples (OSU) and was described previously (Hallak et al., 2000). rgRSV[224] was grown in Hep-2 cells in in DMEM supplemented with 2% FBS, 4.5 g/L D-glucose and 4 mM L-glutamine. SARS-related coronavirus 2 (SARS-CoV-2), Isolate USA-WA1/2020 (NR-52281) was deposited by the Center for Disease Control and Prevention and obtained through BEI Resources, NIAID, NIH. SARS-CoV-2 was propagated in Vero E6 cells in DMEM supplemented with 2% FBS, 4.5 g/L D-glucose, 4 mM L-glutamine, 10 mM Non-Essential Amino Acids, 1 mM Sodium Pyruvate and 10 mM HEPES. Infectious titers of SARS-CoV-2 were determined by plaque assay in Vero E6 cells in Minimum Essential Media supplemented with 2% FBS, 4 mM L-glutamine, 0.2% BSA, 10 mM HEPES and 0.12% NaHCO3 and 0.7% agar. eGFP/ GLuc-expressing human parainfluenza virus 3, strain JS (rHPIV3JS-GlucP2AeGFP) was described previously (Blanco-Melo et al., 2020). HPIV3-eGFP/GLuc was grown in HeLa cells at 32°C in DMEM supplemented with 10% FBS and 1ug/mL TPCK-trypsin (Millipore Sigma, Burlington MA, USA) and titers in Vero E6 cells as previously described. All work involving live SARS-CoV-2 was performed in the CDC/USDA-approved BSL-3 facility of the Icahn School of Medicine at Mount Sinai in accordance with institutional biosafety requirements.
Method Details
RNA-Seq of viral infections
Approximately 5 × 105 A549 or Calu-3 cells were infected with influenza A/Puerto Rico/8/1934 (H1N1) virus (IAV), human respiratory syncytial virus (RSV), human parainfluenza virus 3 (HPIV3) or SARS-CoV-2 as indicated. Infections with IAV were performed at a multiplicity of infection of 5 for 9 h in DMEM supplemented with 0.3% BSA, 4.5 g/L D-glucose, 4 mM L-glutamine and 1 μg/ml TPCK-trypsin. Infections with RSV and HPIV3 were performed at an MOI of 2 for 24 h in DMEM supplemented with 2% FBS, 4.5 g/L D-glucose and 4 mM L-glutamine. Infections with SARS-CoV-2 were performed at an MOI of 0.2 or 2 for 24 h in DMEM supplemented with 2% FBS, 4.5 g/L D-glucose, 4 mM L-glutamine, 10 mM Non-Essential Amino Acids, 1 mM Sodium Pyruvate and 10 mM HEPES. Approximately 5 × 105 NHBE cells were infected with either SARS-CoV-2 at an MOI of 2 for 24 h or influenza A/Puerto Rico/8/1934 (H1N1) virus or influenza A/Puerto Rico/8/1934 (H1N1) virus lacking the NS1 gene at an MOI of 3 for 12 h in bronchial epithelial growth media supplemented with BEGM SingleQuots. As a comparison to viral infection, NHBE cells were treated with 100 units / mL of IFNβ for 4 – 12 h. Total RNA from infected and mock infected cells was lysed in TRIzol (Invitrogen) and extracted and DNase I treated using Direct-zol RNA Miniprep kit (Zymo Research) according to the manufacturer’s instructions. RNA-seq libraries of polyadenylated RNA were prepared using the TruSeq RNA Library Prep Kit v2 (Illumina) or TruSeq Stranded mRNA Library Prep Kit (Illumina) according to the manufacturer’s instructions. Sequencing libraries were sequenced on an Illumina NextSeq 500 platform.
Adenovector transductions
Approximately 5 × 105 A549 cells were transduced with Adenovectors purchased from Vector Biolabs at an MOI of 500 to induce expression of mCherry (Ad-mCherry) and human ACE2 (Ad-GFP-h-ACE2). 48 h post-transduction, efficient gene expression of delivered fluorescent proteins was confirmed by fluorescent microscopy using an EVOS M5000 Imaging System.
Drug treatments
Approximately 5 × 105 Vero E6 cells were infected with SARS-CoV-2 at an MOI of 0.05 in DMEM supplemented with 2% FBS, 4.5 g/L D-glucose, 4 mM L-glutamine, 10 mM Non-Essential Amino Acids, 1 mM Sodium Pyruvate and 10 mM HEPES. Vero E6 cells were treated with 100 units of universal Type-I IFNB 2 h post-infection. NHBE cells were treated with 100 units of human IFNβ as indicated. Cells were harvested for RNA and protein analysis 24 hpi as described below. Approximately 2.5 × 105 A549 cells transduced with an ACE2 Adenovector were pre-treated with 500 nM Ruxolitinib or DMSO control for 1 h in infection media before infection with SARS-CoV-2 at an MOI of 2 for 24 h. Cells were harvested for protein analysis as described below.
Western blot
Protein was extracted from cells in Radioimmunoprecipitation assay (RIPA) lysis buffer containing 1X cOmplete Protease Inhibitor Cocktail (Roche) and 1X Phenylmethylsulfonyl fluoride (Sigma Aldrich) prior to safe removal from the BSL-3 facility. Samples were analyzed by SDS-PAGE and transferred onto nitrocellulose membranes. Proteins were detected using mouse monoclonal anti-Actin (Thermo Scientific, MS-2295), rabbit monoclonal anti-GAPDH (Cell Signaling, 2118), rabbit monoclonal anti-ACE2 (Abcam, ab239924), rabbit monoclonal phospho-TBK1(Ser172) (Cell Signaling, D52C2), mouse monoclonal STAT1 (BD Biosciences, 558537), rabbit polyclonal MX1 (Abcam, ab207414), as well as mouse monoclonal anti-SARS-CoV-2 Nucleocapsid [1C7C7] and Spike [2B3E5] protein (a kind gift by Dr. T. Moran, Center for Therapeutic Antibody Discovery at the Icahn School of Medicine at Mount Sinai). Primary antibodies were detected using Fluorophore-conjugated secondary goat anti-mouse (IRDye 680RD, 926-68070; IRDye 800CW, 926-32210) and goat anti-rabbit (IRDye 680RD, 926-68071; IRDye 800CW, 926-32211) antibodies. Fluorescent signal was detected using a LI-COR Odyssey CLx imaging system and analyzed by Image Studio software (LI-COR).
Quantitative real-time and semiquantitative PCR analysis
RNA was reverse transcribed into cDNA using oligo d(T) primers using SuperScript II Reverse Transcriptase (Thermo Fisher). Quantitative real-time PCR was performed on a LightCycler 480 Instrument II (Roche) using KAPA SYBR FAST qPCR Master Mix Kit (Kapa Biosystems) and primers specific for SARS-CoV-2 E and nsp14 transcripts as described previously (Chu et al., 2020, Corman et al., 2020) as well as human IFNB and α-Tubulin transcripts (Table S5). Delta-delta-cycle threshold (ΔΔCT) was determined relative to mock infected samples. Viral RNA levels were normalized to α-Tubulin and depicted as fold change over mock infected samples. Error bars indicate the standard deviation from three biological replicates. Semiquantitative PCR analysis of cDNA was performed using GoTaq Green MasterMix (Promega) and analyzed by 1.5% agarose gel electrophoresis in TAE buffer.
Cytokine and Chemokine Protein Analysis
Serum levels of IFNβ were measured using the VeriKine-HS human IFN-β serum ELISA kit (PBL Interferon Source, NJ). Serum levels of IFNλ were measured using the IFNλ ELISA kit (PBL Interferon Source, NJ). The following cytokines/chemokines were evaluated using multiplex ELISA: CCL2/monocyte chemoattractant protein (MCP-1), CCL8/MCP-2, CXCL8/interleukin 8 (IL-8), CXCL9/monokine induced by IFN-γ (MIG), CXCL16, interleukin 1β (IL-1β), interleukin 1 receptor antagonist (IL-1RA), interleukin 4, and interleukin 6 (IL-6). All antibodies and cytokine standards were purchased as antibody pairs from R&D Systems (Minneapolis, Minnesota) or Peprotech (Rocky Hill, New Jersey). Individual magnetic Luminex bead sets (Luminex Corp, CA) were coupled to cytokine-specific capture antibodies according to the manufacturer’s recommendations. The assays were read on a MAGPIX platform. The median fluorescence intensity of these beads was recorded for each bead and was used for analysis using a custom R script and a 5P regression algorithm.
Ferret infections
All procedures are described in our previous study (Liu et al., 2019). Ferrets were randomly assigned to the different treatment groups (naive, n = 2; SARS-CoV-2 infection, n = 6; influenza A virus (pH1N1) infection, n = 2; influenza A virus (H3N2) infection, n = 2). Both influenza A virus and SARS-CoV-2 infections of ferrets were performed simultaneously in the BSL-3 facility. For influenza A virus infections, all naive ferrets were infected intranasally with 105 PFU of influenza A/California/04/2009 (pH1N1) virus or 106 PFU of influenza A/Texas/71/2017 (H3N2) virus. Nasal washes were collected from anesthetized ferrets challenged with influenza A/California/04/2009 (pH1N1) virus on day 7 post infection and preserved at −80°C. Trachea were collected from euthanized ferrets challenged with influenza A/Texas/71/2017 (H3N2) virus on day 3 post infection and preserved at −80°C. For SARS-CoV-2 virus infections, all naive ferrets were infected with 5 × 104 PFU of SARS-CoV-2 isolate USA-WA1/2020. Nasal washes were collected from anesthetized ferrets on days 1, 3, 7 and 14 post-infection and trachea were collected from euthanized ferrets on day 3 post infection and preserved at −80°C. At the end of the study, anesthetized ferrets were euthanized by exsanguination followed by intracardiac injection of euthanasia solution (Sodium Pentobarbital). Total RNA from nasal washes and trachea was extracted using TRIzol (Invitrogen) and analyzed by RNA-Seq as described above.
Quantification and Statistical Analysis
Bioinformatic analyses
Raw reads were aligned to the human genome (hg19) using the RNA-Seq Aligment App on Basespace (Illumina, CA), following differential expression analysis using DESeq2 (Love et al., 2014). To diminish the noise introduced by variables inherent to the use of different cell types and systems, our differential expression analyses were always performed by matching each experimental condition with a corresponding mock treated sample that counted for the cell type, collection time, concurrent animal controls, etc. The raw sequencing data (fastq files) for the SARS-CoV-1 and MERS infections were downloaded from GEO (GSE56192, including their corresponding mock-treated controls) and processed in the same way as the rest of our experimental conditions. In order to capture the whole breadth of the response to IFNβ treatment we pooled samples from 4, 6 and 12 hr post treatment and compare them together to mock treated cells. Differentially expressed genes (DEGs) were characterized for each sample (|L2FC| > 1, p-adjusted-value < 0.05) and were used as query to search for enriched biological processes (Gene ontology BP) and network analysis of protein interactions using STRING (Szklarczyk et al., 2019). Heatmaps of gene expression levels were constructing using heatmap.2 from the gplot package in R (https://cran.r-project.org/web/packages/gplots/index.html). Sparse principal component analysis (sPCA) was performed on Log2(Fold Change) values using SPC from the PMA package in R (Witten et al., 2009). Volcano plots, dot plots, scatterplots and linear regressions were constructed using ggplot2 (Wickham, 2016) and custom scripts in R. Heatmap of Type-I IFN responses was constructed on DEGs belonging to the following GO annotations: GO:0035457, GO:0035458, GO:0035455, GO:0035456, GO:0034340. Alignments to viral genomes was performed using bowtie2 (Langmead and Salzberg, 2012). Cell lineage profiling from SARS-CoV-2 unique gene signatures was generated using the Immunological Genome Project (https://www.immgen.org/; Yoshida et al., 2019). The genomes used for this study were: SARS-CoV-2 (GenBank: NC_045512.2), SARS-CoV-1 (GenBank: NC_004718.3), MERS-CoV (GenBank: NC_038294.1), HPIV3 (GenBank: Z11575.1), RSV (GenBank: NC_001803.1), IAV PR8 (GenBank: AF389115.1, AF389116.1, AF389117.1, AF389118.1, AF389119.1, AF389120.1, AF389121.1, AF389122.1) and IAV A/California/VRDL6/2010(H1N1) (GenBank: CY064994, CY064993, CY064992, CY064987, CY064990, CY064989, CY064988, CY064991). All RNA-Seq data performed in this paper can be found on the NCBI Gene Expression Omnibus (GEO) under accession number GSE147507. All non-RNA-seq statistical analyses were performed as indicted in figure legends using prism 8 (GraphPad Software, San Diego, California USA; https://www.graphpad.com/).
Acknowledgments
This work was funded by generous support from the Marc Haas Foundation, the National Institutes of Health, and DARPA’s PREPARE Program (HR0011-20-2-0040). The views, opinions, and/or findings expressed are those of the author and should not be interpreted as representing the official views or policies of the Department of Defense or the U.S. government. We are also indebted to the Kaiser Santa Clara testing facility, which provided the clinical serum samples. We thank Julie Parsonnet and Jeffrey M. Schapiro for provision of clinical samples and Dr. D. Bogunovic for kindly sharing the JAK1/2 inhibitor ruxolitinib. Support for T.T.W. was received from Stanford University, the Chan Zuckerberg Biohub, and the Searle Scholars Program. The research reported in this publication was supported in part by National Institutes of Health 5U19AI111825-07 (to T.T.W.) and 5R01AI139119-02 (to T.T.W.). In addition, partial salary support was provided by the National Institutes of Health (R01DK121072 to R.E.S., R01AIAI145882 and R01AI110575 to B.T., and R21AI149033 to J.K.L.). R.A.A. and W.C.L. were supported by CRIP (Center for Research on Influenza Pathogenesis), a NIAID-funded Center of Excellence for Influenza Research and Surveillance (CEIRS; contract HHSN272201400008C). D.H. was supported in part by USPHS Institutional Research Training Award T32-AI07647. Postdoctoral fellowship support for T.X.J. is provided by the NIH (R01 AI123155). D.B.-M. is an Open Philanthropy Fellow of the Life Sciences Research Foundation. We also thank Dr. T. Moran for providing the SARS-CoV-2 Spike and N antibodies used in this study.
Author Contributions
Conceptualization, D.B.-M., B.E.N.-P., and B.R.T.; Methodology, D.B.-M., B.E.N.-P., W.-C.L., J.K.L., R.A.A., and B.R.T.; Software, D.B.-M. and D.S.; Validation, B.E.N.-P., S.U., D.H., and J.K.L.; Formal Analysis, D.B.-M., D.S., and B.R.T.; Investigation, D.B.-M., B.E.N.-P., W.-C.L., S.U., D.H., R.M., T.X.J., K.O., M.P., D.S., J.K.L., and R.A.A.; Resources, W.-C.L., T.T.W., R.E.S., J.K.L., and R.A.A.; Data Curation, D.B.-M.; Writing – Original Draft, D.B.-M., B.E.N.-P., B.T.O.; Writing – Review & Editing, D.B.-M., B.E.N.-P., J.K.L., and B.R.T.; Visualization, D.B.-M., D.S., B.E.N.-P., S.U., D.H., and B.R.T.; Supervision, R.A.A. and B.R.T.; Project Administration, R.A.A. and B.R.T.; Funding, B.R.T.
Declaration of Interests
The authors declare no competing interests.
Published: May 15, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.cell.2020.04.026.
Supplemental Information
References
- Amanat F., Krammer F. SARS-CoV-2 Vaccines: Status Report. Immunity. 2020;52:583–589. doi: 10.1016/j.immuni.2020.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Melo D., Nilsson-Payant B.E., Uhl S., Escudero-Pèrez B., Olschewski S., Thibault P., Panis M., Rosenthal M., Muñoz-Fontela C., Lee B. An inability to maintain the ribonucleoprotein genomic structure is responsible for host detection of negative-sense RNA viruses. bioRxiv. 2020 doi: 10.1101/2020.2003.2012.989319. [DOI] [Google Scholar]
- Channappanavar R., Fehr A.R., Vijay R., Mack M., Zhao J., Meyerholz D.K., Perlman S. Dysregulated Type I Interferon and Inflammatory Monocyte-Macrophage Responses Cause Lethal Pneumonia in SARS-CoV-Infected Mice. Cell Host Microbe. 2016;19:181–193. doi: 10.1016/j.chom.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen N., Zhou M., Dong X., Qu J., Gong F., Han Y., Qiu Y., Wang J., Liu Y., Wei Y. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020;395:507–513. doi: 10.1016/S0140-6736(20)30211-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu D.K.W., Pan Y., Cheng S.M.S., Hui K.P.Y., Krishnan P., Liu Y., Ng D.Y.M., Wan C.K.C., Yang P., Wang Q. Molecular Diagnosis of a Novel Coronavirus (2019-nCoV) Causing an Outbreak of Pneumonia. Clin. Chem. 2020;66:549–555. doi: 10.1093/clinchem/hvaa029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corman V.M., Landt O., Kaiser M., Molenkamp R., Meijer A., Chu D.K.W., Bleicker T., Brünink S., Schneider J., Schmidt M.L. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Euro Surveill. 2020;25:25. doi: 10.2807/1560-7917.ES.2020.25.3.2000045. 2000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J., Li F., Shi Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019;17:181–192. doi: 10.1038/s41579-018-0118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit E., van Doremalen N., Falzarano D., Munster V.J. SARS and MERS: recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016;14:523–534. doi: 10.1038/nrmicro.2016.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frieman M., Baric R. Mechanisms of severe acute respiratory syndrome pathogenesis and innate immunomodulation. Microbiol. Mol. Biol. Rev. 2008;72:672–685. doi: 10.1128/MMBR.00015-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frieman M.B., Chen J., Morrison T.E., Whitmore A., Funkhouser W., Ward J.M., Lamirande E.W., Roberts A., Heise M., Subbarao K., Baric R.S. SARS-CoV pathogenesis is regulated by a STAT1 dependent but a type I, II and III interferon receptor independent mechanism. PLoS Pathog. 2010;6:e1000849. doi: 10.1371/journal.ppat.1000849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Sastre A. Ten Strategies of Interferon Evasion by Viruses. Cell Host Microbe. 2017;22:176–184. doi: 10.1016/j.chom.2017.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Sastre A., Egorov A., Matassov D., Brandt S., Levy D.E., Durbin J.E., Palese P., Muster T. Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology. 1998;252:324–330. doi: 10.1006/viro.1998.9508. [DOI] [PubMed] [Google Scholar]
- Giavridis T., van der Stegen S.J.C., Eyquem J., Hamieh M., Piersigilli A., Sadelain M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat. Med. 2018;24:731–738. doi: 10.1038/s41591-018-0041-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallak L.K., Collins P.L., Knudson W., Peeples M.E. Iduronic acid-containing glycosaminoglycans on target cells are required for efficient respiratory syncytial virus infection. Virology. 2000;271:264–275. doi: 10.1006/viro.2000.0293. [DOI] [PubMed] [Google Scholar]
- Harcourt J., Tamin A., Lu X., Kamili S., Sakthivel S.K., Murray J., Queen K., Tao Y., Paden C.R., Zhang J. Severe Acute Respiratory Syndrome Coronavirus 2 from Patient with 2019 Novel Coronavirus Disease, United States. Emerg. Infect. Dis. 2020 doi: 10.3201/eid2606.200516. Published online March 11, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.H., Nitsche A. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020;181:271–280.e8. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur S. Double-Stranded RNA Sensors and Modulators in Innate Immunity. Annu. Rev. Immunol. 2019;37:349–375. doi: 10.1146/annurev-immunol-042718-041356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janeway C.A., Jr., Medzhitov R. Innate immune recognition. Annu. Rev. Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- Jing Y., Shaheen E., Drake R.R., Chen N., Gravenstein S., Deng Y. Aging is associated with a numerical and functional decline in plasmacytoid dendritic cells, whereas myeloid dendritic cells are relatively unaltered in human peripheral blood. Hum. Immunol. 2009;70:777–784. doi: 10.1016/j.humimm.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamitani W., Narayanan K., Huang C., Lokugamage K., Ikegami T., Ito N., Kubo H., Makino S. Severe acute respiratory syndrome coronavirus nsp1 protein suppresses host gene expression by promoting host mRNA degradation. Proc. Natl. Acad. Sci. USA. 2006;103:12885–12890. doi: 10.1073/pnas.0603144103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y.I., Kim S.G., Kim S.M., Kim E.H., Park S.J., Yu K.M., Chang J.H., Kim E.J., Lee S., Casel M.A.B. Infection and rapid transmission of SARS-CoV-2 in ferrets. Cell Host Microbe. 2020 doi: 10.1016/j.chom.2020.03.023. S1931-3128(20)30187-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopecky-Bromberg S.A., Martínez-Sobrido L., Frieman M., Baric R.A., Palese P. Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists. J. Virol. 2007;81:548–557. doi: 10.1128/JVI.01782-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langlois R.A., Albrecht R.A., Kimble B., Sutton T., Shapiro J.S., Finch C., Angel M., Chua M.A., Gonzalez-Reiche A.S., Xu K. MicroRNA-based strategy to mitigate the risk of gain-of-function influenza studies. Nat. Biotechnol. 2013;31:844–847. doi: 10.1038/nbt.2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law H.K., Cheung C.Y., Ng H.Y., Sia S.F., Chan Y.O., Luk W., Nicholls J.M., Peiris J.S., Lau Y.L. Chemokine up-regulation in SARS-coronavirus-infected, monocyte-derived human dendritic cells. Blood. 2005;106:2366–2374. doi: 10.1182/blood-2004-10-4166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazear H.M., Schoggins J.W., Diamond M.S. Shared and Distinct Functions of Type I and Type III Interferons. Immunity. 2019;50:907–923. doi: 10.1016/j.immuni.2019.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefrançais E., Ortiz-Muñoz G., Caudrillier A., Mallavia B., Liu F., Sayah D.M., Thornton E.E., Headley M.B., David T., Coughlin S.R. The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature. 2017;544:105–109. doi: 10.1038/nature21706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao M., Liu Y., Yuan J., Wen Y., Xu G., Zhao J., Chen L., Li J., Wang X., Wang F. The landscape of lung bronchoalveolar immune cells in COVID-19 revealed by single-cell RNA sequencing. medRxiv. 2020 doi: 10.1101/2020.02.23.20026690. [DOI] [Google Scholar]
- Liu W.C., Nachbagauer R., Stadlbauer D., Solórzano A., Berlanda-Scorza F., García-Sastre A., Palese P., Krammer F., Albrecht R.A. Sequential Immunization With Live-Attenuated Chimeric Hemagglutinin-Based Vaccines Confers Heterosubtypic Immunity Against Influenza A Viruses in a Preclinical Ferret Model. Front. Immunol. 2019;10:756. doi: 10.3389/fimmu.2019.00756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lokugamage K., Hage A., Schindewolf C., Rajsbaum R., Menachery V.D. SARS-CoV-2 is sensitive to type I interferon pretreatment. bioRxiv. 2020 doi: 10.1101/2020.03.07.982264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X., Zhang L., Du H., Zhang J., Li Y.Y., Qu J., Zhang W., Wang Y., Bao S., Li Y. SARS-CoV-2 Infection in Children. N. Engl. J. Med. 2020 doi: 10.1056/NEJMc2005073. Published online March 18, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menachery V.D., Eisfeld A.J., Schäfer A., Josset L., Sims A.C., Proll S., Fan S., Li C., Neumann G., Tilton S.C. Pathogenic influenza viruses and coronaviruses utilize similar and contrasting approaches to control interferon-stimulated gene responses. MBio. 2014;5 doi: 10.1128/mBio.01174-14. e01174–e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montecino-Rodriguez E., Berent-Maoz B., Dorshkind K. Causes, consequences, and reversal of immune system aging. J. Clin. Invest. 2013;123:958–965. doi: 10.1172/JCI64096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norelli M., Camisa B., Barbiera G., Falcone L., Purevdorj A., Genua M., Sanvito F., Ponzoni M., Doglioni C., Cristofori P. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat. Med. 2018;24:739–748. doi: 10.1038/s41591-018-0036-4. [DOI] [PubMed] [Google Scholar]
- Novel Coronavirus Pneumonia Emergency Response Epidemiology Team The epidemiological characteristics of an outbreak of 2019 novel coronavirus diseases (COVID-19) in China. Zhonghua Liu Xing Bing Xue Za Zhi. 2020;41:145–151. [Google Scholar]
- Pascal K.E., Coleman C.M., Mujica A.O., Kamat V., Badithe A., Fairhurst J., Hunt C., Strein J., Berrebi A., Sisk J.M. Pre- and postexposure efficacy of fully human antibodies against Spike protein in a novel humanized mouse model of MERS-CoV infection. Proc. Natl. Acad. Sci. USA. 2015;112:8738–8743. doi: 10.1073/pnas.1510830112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proudfoot A.E. Chemokine receptors: multifaceted therapeutic targets. Nat. Rev. Immunol. 2002;2:106–115. doi: 10.1038/nri722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Z., Travanty E.A., Oko L., Edeen K., Berglund A., Wang J., Ito Y., Holmes K.V., Mason R.J. Innate immune response of human alveolar type II cells infected with severe acute respiratory syndrome-coronavirus. Am. J. Respir. Cell Mol. Biol. 2013;48:742–748. doi: 10.1165/rcmb.2012-0339OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin C., Zhou L., Hu Z., Zhang S., Yang S., Tao Y., Xie C., Ma K., Shang K., Wang W. Dysregulation of immune response in patients with COVID-19 in Wuhan, China. Clin. Infect. Dis. 2020 doi: 10.1093/cid/ciaa248. Published online March 12, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S., tenOever B.R., Grandvaux N., Zhou G.P., Lin R., Hiscott J. Triggering the interferon antiviral response through an IKK-related pathway. Science. 2003;300:1148–1151. doi: 10.1126/science.1081315. [DOI] [PubMed] [Google Scholar]
- Sokol C.L., Luster A.D. The chemokine system in innate immunity. Cold Spring Harb. Perspect. Biol. 2015;7 doi: 10.1101/cshperspect.a016303. a016303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D., Gable A.L., Lyon D., Junge A., Wyder S., Huerta-Cepas J., Simonovic M., Doncheva N.T., Morris J.H., Bork P. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47(D1):D607–D613. doi: 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- tenOever B.R. The Evolution of Antiviral Defense Systems. Cell Host Microbe. 2016;19:142–149. doi: 10.1016/j.chom.2016.01.006. [DOI] [PubMed] [Google Scholar]
- Varble A., Albrecht R.A., Backes S., Crumiller M., Bouvier N.M., Sachs D., García-Sastre A., tenOever B.R. Influenza A virus transmission bottlenecks are defined by infection route and recipient host. Cell Host Microbe. 2014;16:691–700. doi: 10.1016/j.chom.2014.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss S.R., Navas-Martin S. Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus. Microbiol. Mol. Biol. Rev. 2005;69:635–664. doi: 10.1128/MMBR.69.4.635-664.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H. Springer; 2016. ggplot2: Elegant Graphics for Data Analysis. [Google Scholar]
- Witten D.M., Tibshirani R., Hastie T. A penalized matrix decomposition, with applications to sparse principal components and canonical correlation analysis. Biostatistics. 2009;10:515–534. doi: 10.1093/biostatistics/kxp008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfel R., Corman V.M., Guggemos W., Seilmaier M., Zange S., Muller M.A., Niemeyer D., Jones T.C., Vollmar P., Rothe C. Virological assessment of hospitalized patients with COVID-2019. Nature. 2020 doi: 10.1038/s41586-020-2196-x. Published online April 1, 2020. [DOI] [PubMed] [Google Scholar]
- Wu F., Zhao S., Yu B., Chen Y.M., Wang W., Song Z.G., Hu Y., Tao Z.W., Tian J.H., Pei Y.Y. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z., Shi L., Wang Y., Zhang J., Huang L., Zhang C., Liu S., Zhao P., Liu H., Zhu L. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020;8:420–422. doi: 10.1016/S2213-2600(20)30076-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H., Lareau C.A., Ramirez R.N., Rose S.A., Maier B., Wroblewska A., Desland F., Chudnovskiy A., Mortha A., Dominguez C. The cis-Regulatory Atlas of the Mouse Immune System. Cell. 2019;176:897–912.e20. doi: 10.1016/j.cell.2018.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R., China Novel Coronavirus Investigating and Research Team A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020;382:727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw sequencing datasets generated during this study are available on the NCBI Gene Expression Omnibus (GEO) server under the accession number GSE147507. The original sequencing datasets for SARS-CoV-1 and MERS-CoV infections can be found on the NCBI Gene Expression Omnibus (GEO) server under the accession number GSE56192.